 Shudong Chen
Shudong Chen Ruimin Tian
Ruimin Tian Dan Luo1
Dan Luo1 Dingkun Lin
Dingkun Lin- 1The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, China
- 2Guangdong Provincial Academy of Chinese Medical Sciences, Guangzhou, China
- 3School of Basic Medical Sciences, Guangzhou University of Chinese Medicine, Guangzhou, China
Spinal cord ischemia is a severe clinical complication induced by thoracoabdominal aortic surgery, severe trauma, or compression to the spinal column. As one of the most important functional cells in the spinal cord, spinal motor neurons (SMNs) suffer most during the process since they are vulnerable to ischemic injury due to high demands of energy. Previous researches have tried various animal models or organotypic tissue experiments to mimic the process and get to know the pathogenesis and mechanism. However, little work has been performed on the cellular model of spinal cord ischemia, which has been hampered by the inability to obtain a sufficient number of pure primary SMNs for in vitro study. By optimizing the isolation and culture of SMNs, our laboratory has developed an improved culture system of primary SMNs, which allows cellular models and thus mechanism studies. In the present study, by establishing an in vitro model of spinal cord ischemia, we intended to observe the dynamic time-course changes of SMNs and investigate the role of autophagy in SMNs during the process. It was found that oxygen-glucose deprivation (OGD) resulted in destruction of neural networks and decreased cell viability of primary SMNs, and the severity increased with the prolonging of the OGD time. The OGD treatment enhanced autophagy, which reached a peak at 5 h. Further investigation demonstrated that inhibition of autophagy exacerbated the injury, evidencing that autophagy plays a protective role during the process.
Introduction
Spinal cord ischemia is a devastating complication following thoracoabdominal aortic surgery (Drinkwater et al., 2010; Shimizu and Yozu, 2011; Etz et al., 2014), and it frequently occurs as well when the spinal cord suffers from direct trauma during spinal cord injury (Rivlin and Tator, 1978; Aslan et al., 2009), or compression from vertebral stenosis and various other spinal lesions (Gooding et al., 1975; Griffiths et al., 1979; Yang et al., 2015), which may lead to various degrees of disability or even paraparesis/paraplegia (Coselli et al., 1997; Etz et al., 2008; Suarez et al., 2013). During the spinal cord ischemia, the blood flow of the spinal cord is reduced, and the tissue auto-regulation is disrupted (Kise et al., 2015; Weidauer et al., 2015), which takes an essential part in the pathophysiological mechanisms. During the past decades, numerous studies, by adopting various animal models (in rats, mice, rabbits, cats, pigs, and primates; Griffiths et al., 1979; Kato et al., 1997; Kolenda et al., 2003; Wang et al., 2010; Hwang et al., 2012, 2017; Nazli et al., 2015; Yang et al., 2015) and tissue (organotypic spinal cord slices) experiments (Turner and Johnson, 2011; Esposito et al., 2012) of spinal cord ischemia, have been studied for the pathogenesis and developed some methods to reduce the injury; however, up until now, the treatment options remain strongly controversial and limited, and the concrete mechanisms and roles of certain types of cells involved in this process are still far from clear. Besides in vivo or organotypic models, cellular models of a certain disease to mimic a certain cell type phenotype hold the key to understand the pathogenesis of a disease; nevertheless, progress in performing researches about spinal cord ischemia have been impeded by the inability to gain sufficient number with well uniformity of certain types of cells in vitro.
In eukaryotic cells, the maintenance of normal metabolic balance relies on two major protein catabolism pathways: the ubiquitin–proteasome pathway (UPS) and autophagy lysosomal pathway. A number of neurological disorders have been discovered to be associated with autophagy. Autophagy (means “self-eating” in Greek, “auto” oneself, “phagy” to eat), a term which was first coined by Christian de Duve in 1963 (De Duve, 1963; Klionsky, 2008), is a highly regulated process in which protein aggregates and damaged organelles are degraded via the lysosomal pathway. It is an evolutionary conserved catabolic system where unnecessary or dysfunctional cellular components, including cytosolic proteins and organelles, are detained in a double-membrane vesicle, and the resulting vacuoles (autophagosomes) are degraded after they are transmitted to the lysosomal compartment (Klionsky and Emr, 2000; Mizushima and Komatsu, 2011; Feng et al., 2014). It serves like a cellular housekeeper, to keep the amino acid/energy recycling and try to mitigate various metabolic stresses (Levine and Kroemer, 2008; Wirawan et al., 2012). Although some of abovementioned functions overlap with those of the UPS, autophagy mainly contributes to the turnover of long-lived proteins and the maintenance of amino acid pools in the setting of cellular stresses (Ciechanover et al., 2000; Nedelsky et al., 2008; Lu et al., 2013). This process is considered to be adaptive and essential for survival, differentiation, development, and homeostasis under both physiological and pathological conditions. In various neurological diseases, autophagy may be either upregulated or downregulated or even impaired (Komatsu et al., 2006; Lee et al., 2010; Lynch-Day et al., 2012; Gu et al., 2013; Martin et al., 2015). Previous in vivo studies have indicated that autophagy is implicated in the spinal cord ischemic injury (Kanno et al., 2009a; Fan et al., 2014; Fujita et al., 2015; Fang et al., 2016), whereas the concrete mechanisms during the process are controversial, and the impact of autophagy in primary spinal neurons has not been fully understood. Accumulating evidence suggested that autophagy may act as a “double-edged sword” with regard to central nervous system injury. The role of autophagy varies with the type or degree of injuries. Mounting studies showed that autophagy activation may be involved in neuroprotection in cerebral or spinal cord injury (Sheng et al., 2010; Mariño et al., 2011; Wang P. et al., 2012; Sun et al., 2016), but some investigators reported that the ischemic injury may induce “autophagic cell death,” and inhibition of autophagy can prevent neuron death after ischemic injury (Yu et al., 2004; Rami et al., 2008; Uchiyama et al., 2008; Kanno et al., 2009b). Some in vivo studies have also documented that, in the model of cerebral ischemia, autophagy is activated in various cell types—neurons, astrocytes, and vascular endothelial cells, while in the model of spinal cord injury (Kanno et al., 2009a; Fang et al., 2016), autophagesomes mainly accumulate in neurons, microglia, or oligodendrocytes, rather than in astrocytes. Nevertheless, the concrete change in a certain cell type during the process of spinal cord injury has never been reported, and the underlying mechanisms deserve further investigation.
Spinal motor neurons (SMNs), the nerve cells that connect the ventral horn of the spinal cord to directly or indirectly control muscles or glands, act as one of the most important neurons in the spinal cord. Existing studies have demonstrated that SMNs are vulnerable to ischemic injury due to their high demands of energy (Kanno et al., 2009a; Fujita et al., 2015; Fang et al., 2016). The ischemia induces changes in SMNs, which can, in turn, affect the process. Nevertheless, little is known about the dynamic time-course changes and the function of autophagy in SMNs during the process. By optimizing the isolation and culture of SMNs, our laboratory has developed an improved culture system of SMNs, which allows establishing cellular models and performing mechanism studies. Based on this culture system, we also tried some in vitro models of SMNs to mimic the metabolic perturbation occurring in vivo during the spinal cord ischemic injury, which may include the state of hypoxia, aglycemia, acidosis, oxidative stress, etc. Oxygen-glucose deprivation (OGD), which has been widely utilized to study the cerebral ischemia injury or ischemia/reperfusion injury, is a commonly used model to mimic an ischemic milieu in vitro (Fontella et al., 2005; Cimarosti et al., 2012; Wang R. et al., 2012; Tasca et al., 2015). Performing OGD-induced SMNs injury model can well mimic the extracellular condition in spinal ischemia and results in neuronal insult; however, unlike cerebral ischemia injury and ischemia/reperfusion injury, which have been studied by numerous researches, investigations about the spinal neurons are far from enough.
To date, no studies have addressed the time-course changes and the role of autophagy in primary SMNs subjected to OGD conditions. In the current study, by establishing a cellular model of spinal cord ischemia in vitro, we intended to investigate the potential involvement of autophagy and its time course in primary SMNs. Furthermore, by treating with the autophagy inhibitor, the role of autophagy during the process was clearly observed.
Materials and Methods
Reagents and Chemicals
NeurobasalTM medium, B-27TM supplement, HBSS, PBS, and trypsin were purchased from Gibco. Brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF), glutamate, L-glutamine, Dnase, poly-D-lysine (PDL), paraformaldehyde (PFA), and 4′,6-diamidino-2-phenylindole (DAPI) were all obtained from Sigma-Aldrich. RIPA Lysis and Extraction Buffer, as well as PierceTM BCA Protein Assay Kit were purchased from Thermo Scientific. The protease inhibitor cocktail was from Bimake. Primary antibodies against NGF receptor (p75NTR; ab6172), CHAT (ab6168), and β-actin (ab8226) were provided by Abcam. Antibody against SMI 32 (NE1023), Cy3-conjugated sheep anti-rabbit IgG secondary antibody (AC111C), and polyvinylidene difluoride (PVDF) membranes were purchased from EMD Millippore. Primary antibody against light chain 3 (LC3; M152-3) was purchased from MBL International. Primary antibodies against Beclin1 (#3738), SQSTM1/p62 (#5114), as well as horseradish peroxidase (HRP)-linked secondary antibodies (#7074, #7076) were obtained from Cell Signaling Technology. Alexa fluor 488-conjugated donkey anti-mouse IgG secondary antibody (A-21202) was bought from Invitrogen. ClarityTM Western ECL Substrate was obtained from Bio-rad. CCK8 kit were purchased from Dojindo. Bafilomycin A1 (Baf-A1) and 3-Methyladenine (3-MA) were purchased from Selleck Chemicals.
Isolation and Culture of Primary Spinal Motor Neurons (SMNs)
Pregnant Sprague–Dawley rats were used in the experiments in accordance with internationally accepted standard guidelines for animal use and care, and our study protocol was reviewed and approved by the ethical committee of Guangzhou University of Chinese Medicine. SMNs were prepared from 14- to 16-day embryonic rats as we have reported previously (Chen et al., 2018). Briefly, the spinal cords from embryonic rats were dissected, with vessels and menings gently removed. Then, tissues were washed and cut into small slices and transferred a new dish for trypsinization (0.25% trypsin and 0.4% Dnase) for 20 min at 37°C, followed by adding complete media to inactivate the trypsin, Later, the tissues were gently triturated using Gilson blue pipette tips and a 100-mesh filter to obtain the single-cell suspension. Cells were harvested by centrifugation, re-suspended by NeurobasalTM medium supplemented with 2% B27, and plated on panning dishes, which had been coated with affinity-purified goat anti-mouse IgG in Tris-HCl buffer at 4°C overnight, washed, and then incubated with p75NTR antibody (1–10 μg/ml in PBS). After immunopanning, panning dishes were gently washed to remove those loosely attached cells. Adherent cells (phase bright and quite big under the inverted phase-contrast microscope) were collected by trypsinization and centrifugation, then re-suspended by plating media, whose components were Neurobasal medium supplemented with 2% B27 and 25 μM Glutamate. An aliquot (2 μl) was used for counting before cells were seeded at a proper density (2 × 104/ml–5 × 104/ml) on Petri dishes or a 96-well plate, which had been pre-treated with PDL (20 μg/ml, for 30 min at room temperature). 24 hours after plating, media was fully replaced by growth media, whose components were Neurobasal medium supplemented with 2% B27, 2 mM of L-Glutamine, 10 ng/ml of BDNF, and 10 ng/ml of GDNF. Neurons were maintained at 37°C in a humidified 5% CO2 incubator, and half volume of the medium was changed with fresh media every 3 days. Experiments were performed after day in vitro (DIV) 7–14.
Oxygen-Glucose Deprivation (OGD) Exposure of SMNs
OGD has been utilized as a simulation model of hypoxic insult in vitro. OGD experiments were performed between DIV 7 and DIV 14, at which time SMNs represented at least 95% of the population as assessed by SMN marker and DAPI staining. To initiate OGD, the cultured primary SMNs were rinsed with HBSS and then incubated with Earle’s balanced salt solution (EBSS), whose components were (in mg/L): 6,800 NaCl, 400 KCl, 200 CaCl2, 200 MgSO4–7H2O, 140 NaH2PO4–H2O, 2,200 NaHCO3, pH 7.4, and placed in a tri-gas incubator (Thermo Fisher Scientific), which was set at 5% O2 and 5% CO2 at 37°C for certain hours to mimic ischemic insult. The experimental schedule is shown in Figure 1. Control cells were cultured in normal culture medium (Neurobasal medium supplemented with 2% B27 and 2 mM L-Glutamine) and placed in the regular incubator (95% air and 5% CO2 at 37°C).
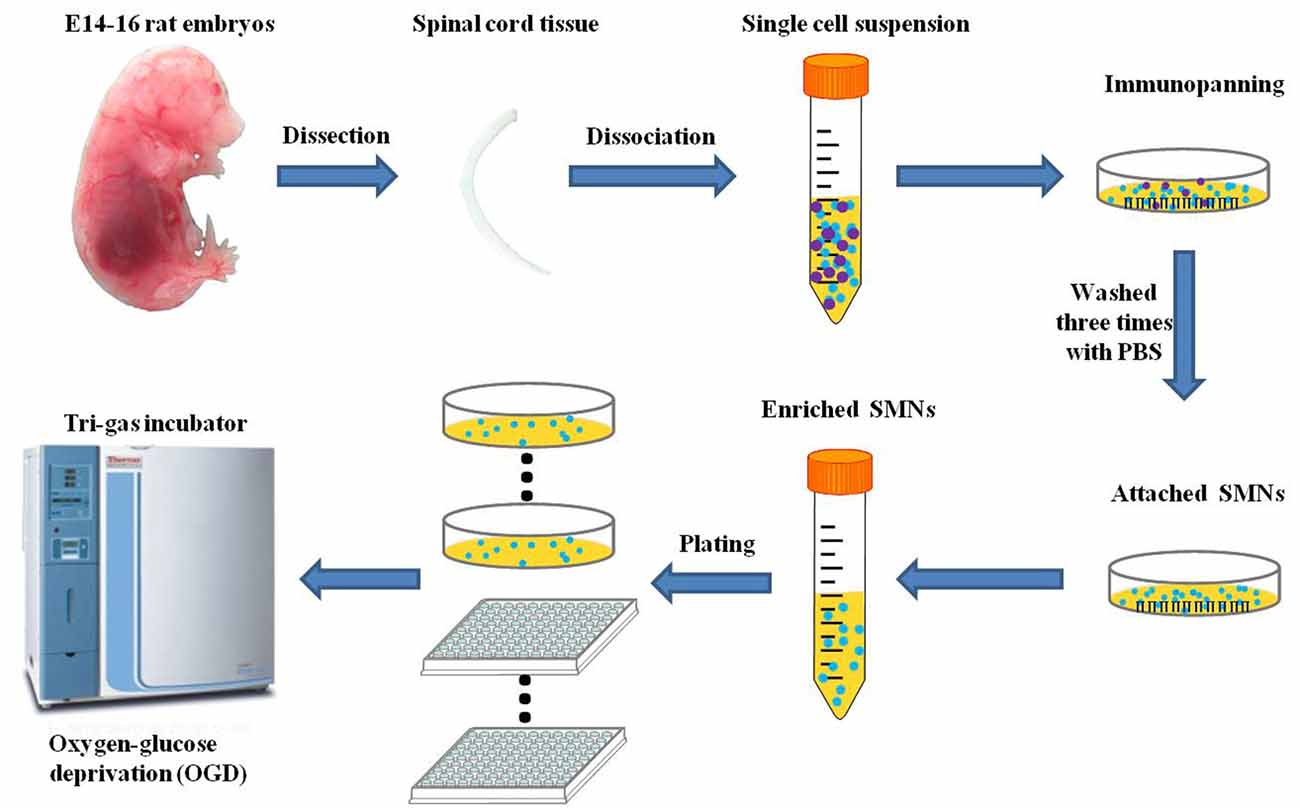
Figure 1. Schematic diagram of the isolation and purification of primary spinal motor neurons (SMNs) and oxygen-glucose deprivation (OGD) modeling process.
Quantification of Cell Viability With CCK8 Assay
Cell viability of cultured SMNs was tested with a nonradioactive cell counting kit (CCK8) following the manufacturer’s instructions. In brief, the primary SMNs were seeded on 96-well plates at a density of 2.5 × 103 per well. After cells were subjected to OGD for 0.5, 1, 2, 3, 5, and 7 h, 10 μl of CCK8 was added to each well, and the mixture was incubated for 4 h at 37°C. Then, absorbance was determined at 450 nm by a microplate reader. Results were expressed as the percentage of CCK8 reduction, and the absorbance of control cells was set at 100%.
Immunofluorescence Assay
Isolated SMNs were plated on confocal dishes for immunofluorescence assay. Cells were washed with ice-cold PBS, fixed with 4% paraformaldehyde solution for 15 min at room temperature, permeabilized by 0.2% Triton X-100 (in PBS, pH 7.4) and further blocked in 5% goat serum for 1 h at room temperature. Cells were then incubated with appropriate primary antibodies at 4°C overnight. For double immunofluorescence, different primary antibodies from different species were simultaneously incubated. For the identification of SMNs, mouse anti-SMI 32 (1:500) and/or rabbit anti-CHAT (1:500) were used. While for the identification of autophagic marker, mouse anti-LC3 (1:500) was used. After being washed, cells were incubated with Alexa fluor 488-conjugated donkey anti-mouse IgG (1:1,000) or/and Cy3 conjugated sheep anti-rabbit secondary antibody (1:1,000) for 1 h in dark. For nuclear counterstaining, DAPI was used. Labeled cells were identified using Olympus FX-70 fluorescence microscope or Zeiss LSM 510 META laser scanning confocal microscope, and digital images were recorded by Adobe Photoshop software and Zeiss LSM Image Examiner software.
Protein Isolation, Quantification, and Western Blot Analysis
Isolated SMNs were plated on Petri dishes for Western blot analysis. After OGD treatment, cells were harvested and lysed in ice-cold RIPA lysis buffer with protease inhibitor cocktail. Protein concentrations were detected using BCA protein assay kit, and proteins in each group were adjusted to the same concentrations. Equivalent amounts of protein were loaded and electrophoretically on 12% or 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and blotted onto PVDF membranes. Then, the nonspecific binding was blocked by incubating membranes in 5% skimmed milk (in TBS containing 0.05% Tween 20) for 1 h. The membranes were incubated with primary antibodies diluted in TBST as follows: mouse anti-LC3 (1:1,000), rabbit anti-Beclin1 (1:1,000), rabbit anti-SQSTM1/p62 (1:1,000), and mouse anti-β-actin (1:3,000) overnight at 4°C. The next day, membranes were washed three times with TBST for 5 min each wash, followed by incubating with anti-mouse IgG HRP-linked antibody (1:2,000) or anti-rabbit IgG HRP-linked antibody (1:2,000) for 1 h at room temperature. Immunoreactivity was detected with ClarityTM Western ECL Substrate, and reacting bands were captured by Bio-rad ChemiDocTM XRS+ system, and analyzed for final determination of protein expression with Image Lab and normalized by β-actin as internal controls.
Electron Microscopy
The neurons were harvested and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 8 h, followed by treatment with 1% osmium tetroxide for an additional 1 h, and dehydrated by gradient ethanol and acetone. Samples were immersed in resin, hardened, and sectioned (50–70 nm). Later, the ultrathin sections were stained with lead citrate and uranyl acetate and observed by electron microscopy (Zeiss EM910).
Statistical Analysis
All experiments were performed at least three times independently. Statistical analysis of all data was performed using SPSS 22.0 and GraphPad Prism 7. Results were presented as mean ± standard deviation (SD). Statistical comparisons between two groups were determined using unpaired Student’s t-test. Differences among groups were evaluated with one-way analysis of variance (ANOVA) followed by Tukey’s or Dunnett’s post hoc test when appropriate, or two-way ANOVA followed by Sidak’s multiple comparisons test. A difference was considered statistically significant when p < 0.05.
Results
Characterization, Identification, and Purity of Primary SMNs
Within 24 h after plating, most cells were adherent to the bottom, and some even grew tiny neurites, but all cells were solitary. After the first media replacement (24 h) to remove the floating cells and change some media components, larger cells appeared, cell outgrowths grew longer and formed multiple branches. After 7 days in culture, cells grew mature with plump soma and high refraction, and formed interconnected neural networks diffusely (Figure 2).
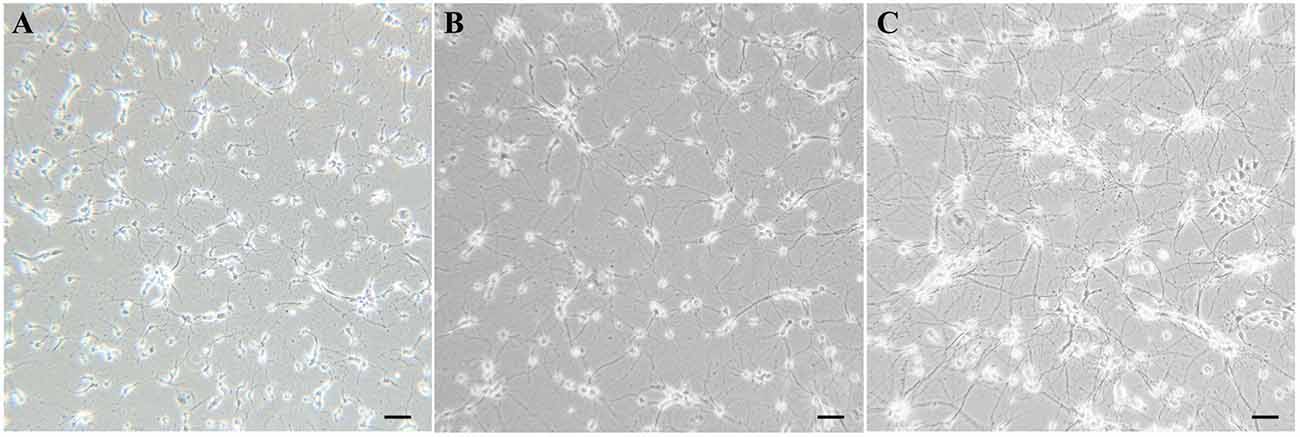
Figure 2. Visual characterization of cultured primary SMNs. (A) Morphology of cultured cells after plating for 2 days. (B) SMNs developed large cell bodies with long neurites. (C) Mature SMNs formed vastly interconnected neurite networks.
In order to identify motor neurons in the culture system, SMI-32 and CHAT antibodies were both used. As shown in Figure 3, the SMI-32 and CHAT stainings showed cell bodies, dendrites, and large axons. On the 10th day, as signs for maturation and high differentiation, the cultured SMNs developed large cell bodies with long neurites, which formed interconnected neurite networks and showed prominent arborization. The purity of SMNs was calculated by motor neuron marker (CHAT and SMI-32) immunofluorescence and DAPI staining. The results showed that the proportion of SMNs reached over 95%, which can meet the requirements of subsequent experiments.
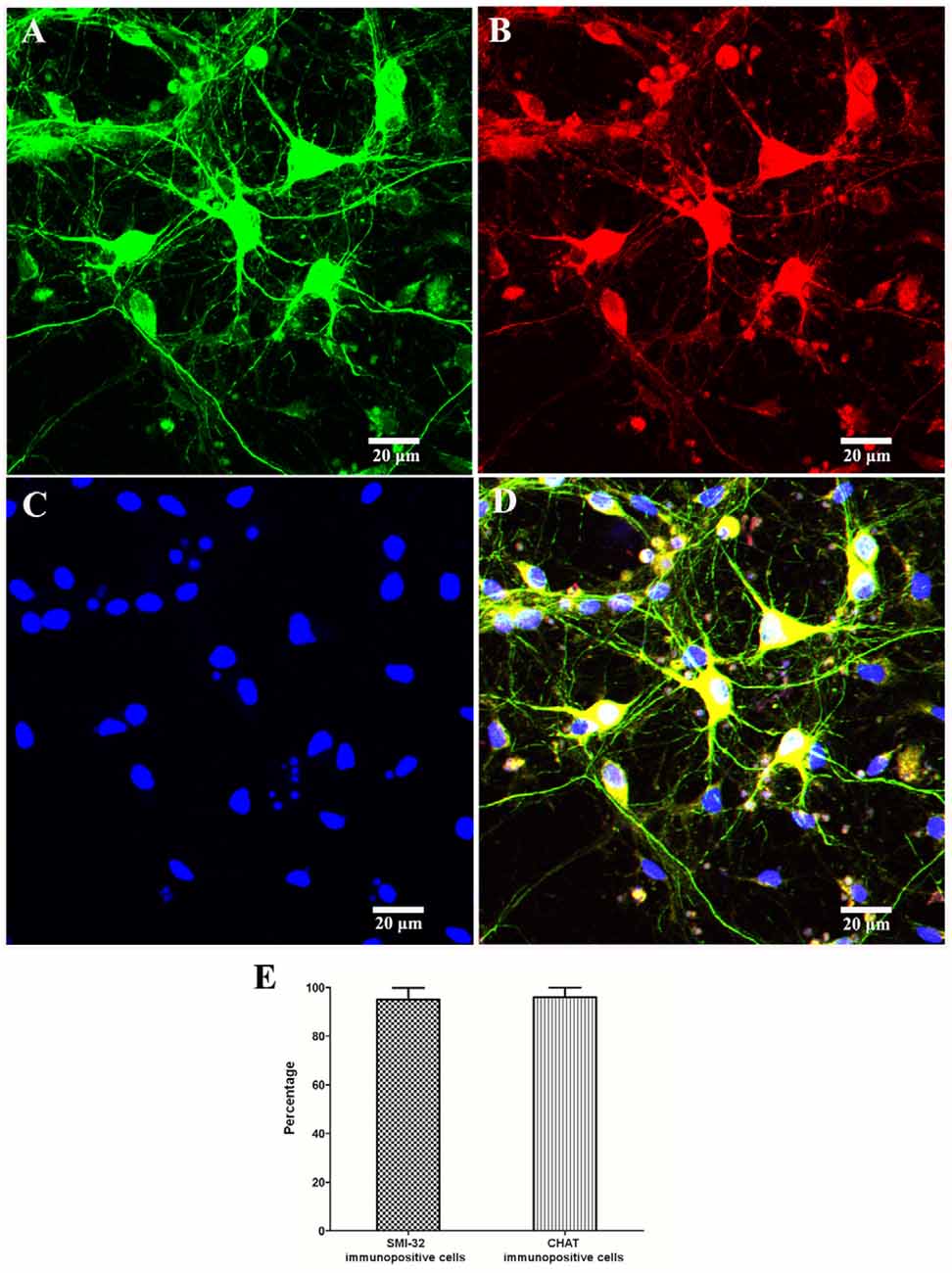
Figure 3. Identification and purity of SMNs by immunofluorescence with motor neuron-specific markers against SMI-32 and CHAT. The immunofluorescence assay of motor neuron marker SMI-32 (green, A) and CHAT (red, B) showed cell bodies, dendrites, and large axons, with 4′,6-diamidino-2-phenylindole (DAPI) staining (blue, C) showing the nuclei. (D) Merged image. (E) The percentage of SMI-32 or CHAT-immuopositive cells were calculated. Data were presented as mean ± SD (n = 5 independent experiments). At least 10 random fields from one sample were averaged in each independent experiment. Scale bars represent 20 μm.
Morphological Changes of Primary SMNs Under OGD at Different Time Points
OGD-induced SMN injury acts as a model to mimic spinal cord ischemia injury in vitro and results in neuronal insult. In this experiment, primary SMNs were cultured under OGD condition for 1, 2, 3, 5, 7, and 24 h, with motor neurons cultured in normal media and normoxia serving as control. As shown in Figures 4A–F, it seems that cells showed no obvious morphological changes within 1 h; however, most SMNs exhibited atrophic cyton and disrupted neurofilament after 3 h, and the injury continued to aggravate as time prolonged, with apparent impaired neural networks after 7 h and even massive cell loss for 24 h.
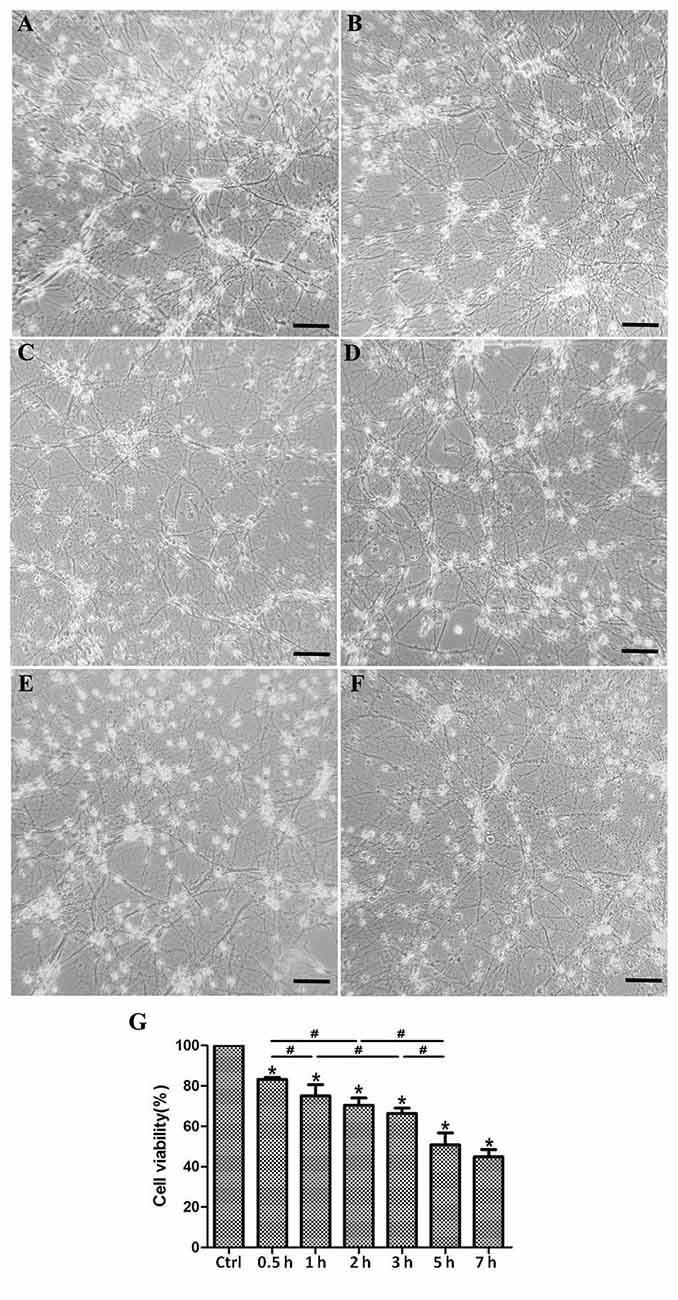
Figure 4. Morphological changes and cell viability of cultured SMNs under OGD for different time. Primary SMNs were cultured normally (A) or subjected to OGD for various periods of time (B: 1 h, C: 3 h, D: 5 h, E: 7 h, or F: 24 h, respectively), and the representative phase photomicrographs showed the morphologic changes (×250). (A–F) OGD resulted in destruction of neural networks, and the severity increased with the prolonging of OGD time. (G) Cell viability was reduced under OGD in a time-dependent manner. The cell viability was assessed using the CCK8 kit, and results were expressed as the percentage of CCK8 reduction. The absorbance of control cells was set at 100%. Data were presented as mean ± SD (n = 5 independent experiments). Statistical comparisons were carried out with one-way analysis of variance (ANOVA). *p < 0.05 compared with control group. #p < 0.05 between groups.
Cell Viability Is Reduced Under OGD in a Time-Dependent Manner
We assessed the survival rate of SMNs after OGD insult. It was reduced under OGD in a time-dependent manner. According to Figure 4G, the cell viability declined sharply when cells were initially subjected to OGD (1 h), with significant changes every half an hour (control vs. 0.5 h, 0.5 vs. 1 h), but later declined relative slowly within 3 h (without significant change 1 vs. 2 h, and 2 vs. 3 h). Until 5 h upon OGD stress, viability declined markedly compared with 3 h. As time prolonged, cell viability declined more slowly, reaching a nearly 50% viability decrease in 7 h, but with no significant difference with 5 h.
OGD Treatment Enhances Autophagy in Primary SMNs, and Autophagy Reaches a Peak at 5 h
To determine whether autophagy was involved in SMNs under OGD conditions, we first examined the classical autophagy makers by Western blot assays. The microtubule-associated protein LC3, a mammalian homol of the yeast ATG8 gene (Aut7/Apg8), serves as the markers for autophagy. As shown in Figure 5, the bands showed the time course of autophagy induction under OGD, and the ratio of LC3-II/LC3-I was seen as a dramatic increase in SMNs after OGD for 3 h and reached a peak at 5 h. Immunofluorescence assay revealed strong and punctate LC3 staining in the OGD group (Figure 6), whose trends were consistent with that of Western blot analysis. Intriguingly, we can also observe that strong, punctate LC3 appeared much more in soma than in axons.
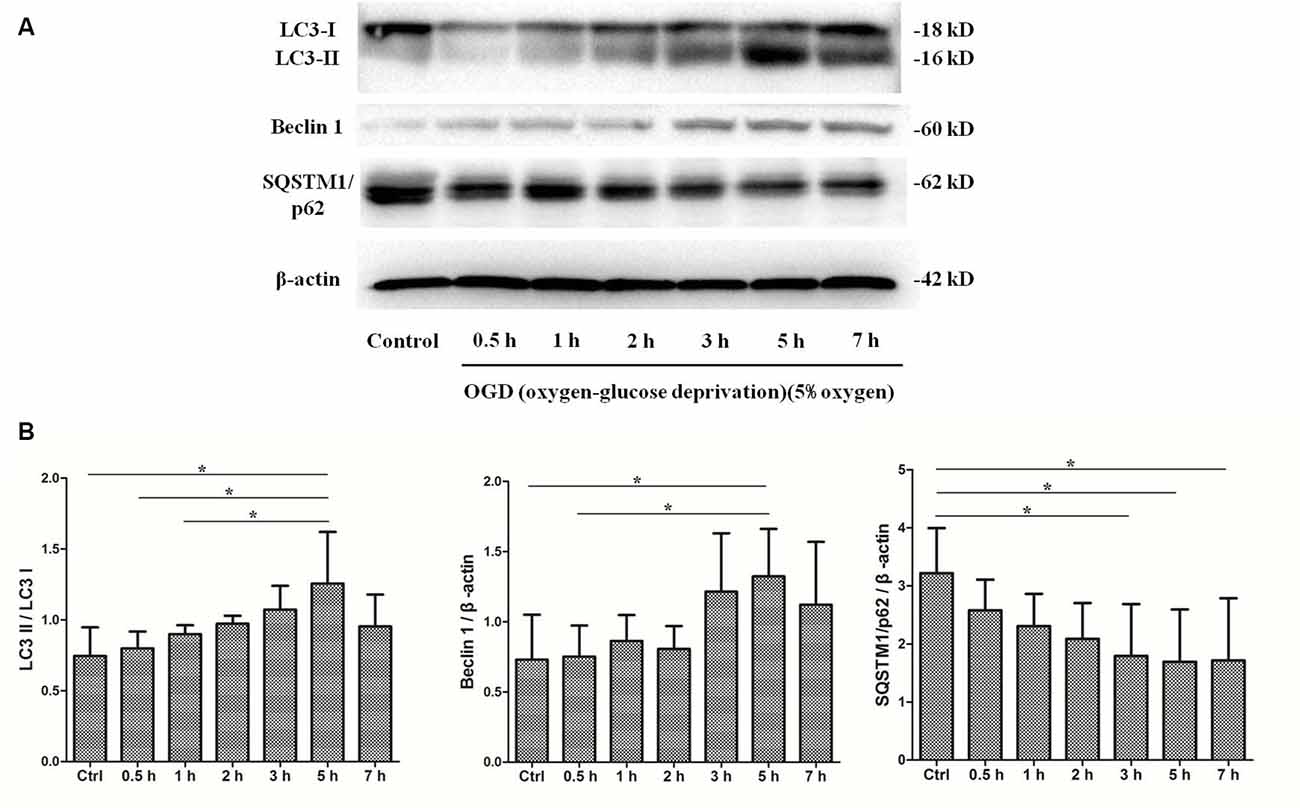
Figure 5. OGD treatment induces autophagy activation. SMNs were exposed to OGD for and indicated time, and the expression of light chain 3 (LC3) I, LC3 II, Beclin1, and SQSTM1/p62 were detected by Western blot (A). (B) The quantitative results are shown after normalizing with β-actin as an internal control. Data were presented as mean ± SD (n = 3 independent experiments). Statistical comparisons were carried out with one-way ANOVA. *p < 0.05.
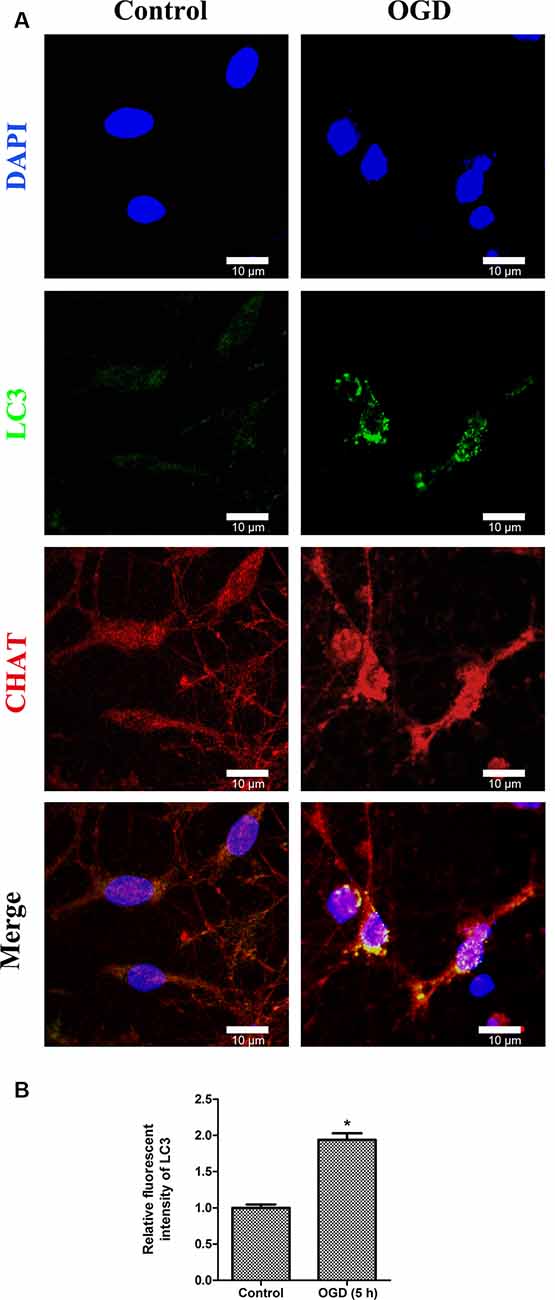
Figure 6. Double immunofluorescence stainings show the LC3-positive puncta in primary SMNs under OGD. (A) Representative fluorescent confocal images of cells counterstained with DAPI (blue, nuclei), autophagy-specific marker—LC3 (green) as well as motor neuron marker—CHAT (red) are shown (n = 3 independent experiments). Strong, punctate LC3 staining can be observed in primary SMNs under OGD. Scale bar presents 10 μm. (B) The relative fluorescent intensity of LC3 was analyzed using ImageJ. Data were presented as mean ± SD (n = 3 independent experiments). Statistical comparisons were carried out with unpaired Student’s t-test. *p < 0.05.
P62, also called sequestosome 1 (SQSTM1), is the selective cargo receptor for autophagy to degenerate misfolded proteins and serves as the marker for autophagic degradation. We detected the expression of SQSTM1/p62 and found that its level was diminished significantly after OGD for 3 h, which corresponded to the results of LC3. Additionally, Beclin1, the mammalian homolog of the yeast Agt6, forms a protein complex with class III phosphatidyl inositol-3 kinase within the autophagosome. Results showed that Beclin 1 expression was upregulated markedly and continually after OGD within 7 h.
OGD Treatment Induces Autophagic Flux in Primary SMNs
Given that autophagy was involved in primary SMNs upon OGD stress, we next decided to confirm that the enhancement of autophagy markers during the process was due to induction of autophagy or blockage of autophagosome maturation. For this purpose, lysosome inhibitor Bafilomycin A1 (Baf-A1) needs to be added in the culture medium to assess the responses of SMNs, which is a classical approach to observing the real state of autophagic flux. Baf A1 is a late-phase autophagy inhibitor, which acts by inhibiting vacuolar H + ATPase (V-ATPase) and thus prevents the maturation of autophagic vacuole fusion between autophagosomes and lysosomes (Gómez-Sánchez et al., 2015; Klionsky et al., 2016). Results in Figure 7 demonstrated that, in the presence of Baf-A1, LC3 II increased and accumulated, LC3-II/LC3-I displayed a much higher level when SMNs were treated with OGD plus Baf-A1 than OGD alone. This indicated that the increase in LC3-II levels by OGD was because of an increase in production rather than the decreased recycling of autophagy.
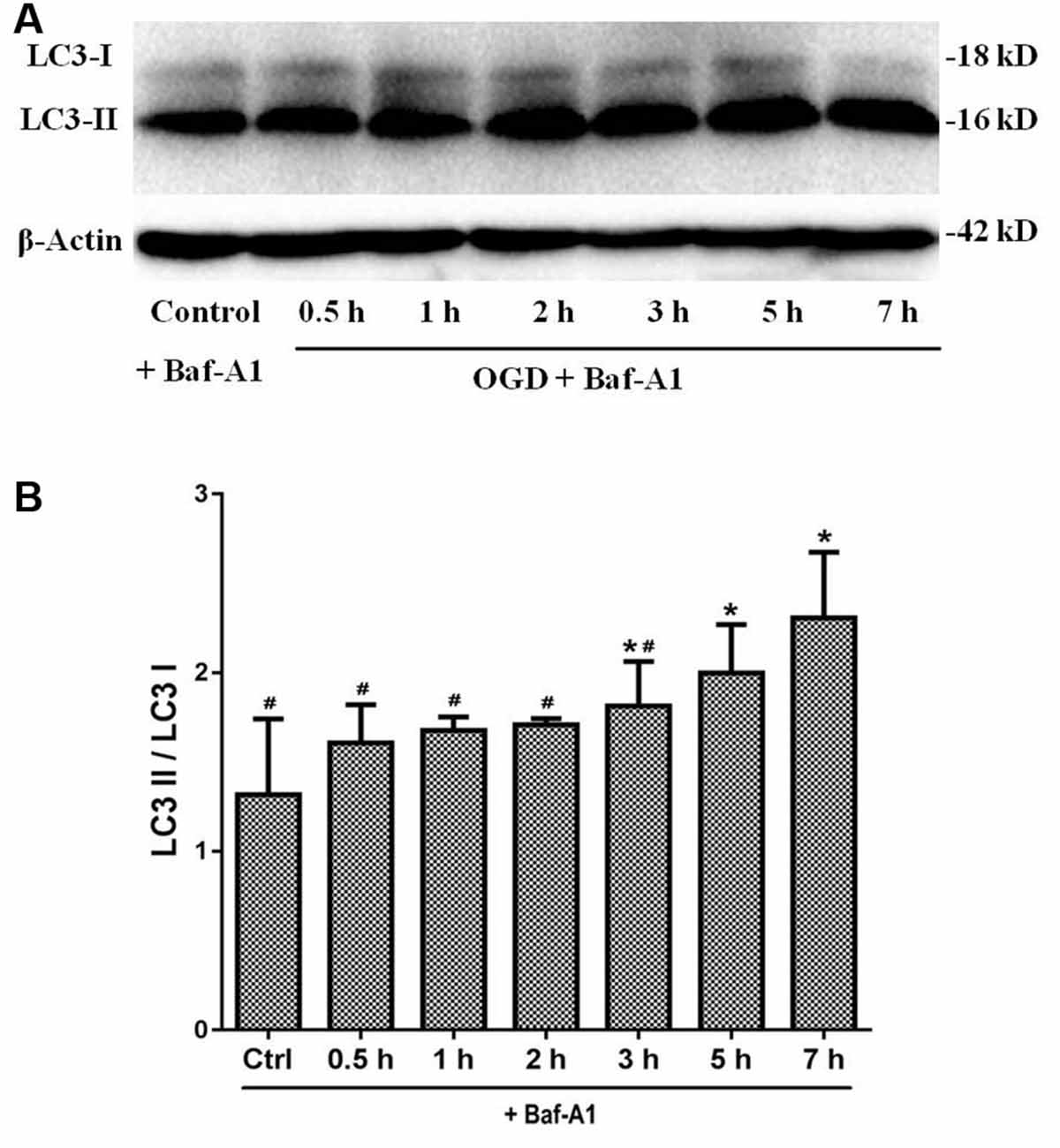
Figure 7. LC3 expression under OGD with Baf-A1 to observe the real state of autophagic flux. (A) The expression of LC3 I and LC3 II were detected by Western blot. In the presence of Baf-A1, LC3 II dramatically increased and accumulated. (B) The quantitative results are shown in the bar chart. Data were presented as mean ± SD (n = 3 independent experiments). Statistical comparisons were carried out with one-way ANOVA. *p < 0.05 compared with the control group. #p < 0.05 compared with the 7-h group.
Results of Transmission Electron Microscopy
The transmission electron microscopy (TEM) acts as one of the most important methods for monitoring autophagy (Swanlund et al., 2010). Autophagy was first detected by TEM in the 1950s, and it was originally observed as a focal degradation of cytoplasmic areas performed by lysosomes, which still remains the hallmarks of this process. Up until now, TEM is still the only tool that reveals the morphology of autophagic structures at a resolution in the nm range and shows the cellular substructure during the process, which allows the exact identification (Martinet et al., 2014). Therefore, we used TEM to examine the ultrastructural changes of SMNs subjected to OGD insult. In order to identify the typical differences, we chose the control cells and SMNs after 5 h of OGD. As shown in Figure 8, control cells contained normal-looking organelles, nucleus, and chromatin. The mitochondria had a dense matrix, neatly aligned cristae, and no signs of autophagy were observed. While after OGD insults, SMNs were found to contain many vesicles with typical morphological features of autophagosomes (AP). A number of double or multi-membrane structures, which engulfed cytoplasm fractions and organelles, can be observed in the cytoplasm. When autophagosomes fused with lysosomes, the inner membranes disappeared, and autophagosomes turned to be single-membrane autophagic vacuoles. The mitochondria displayed swelling with partially broken or dilated cristae. The TEM strongly once again suggested that overactivation of autophagy was triggered in primary SMNs subjected to OGD.
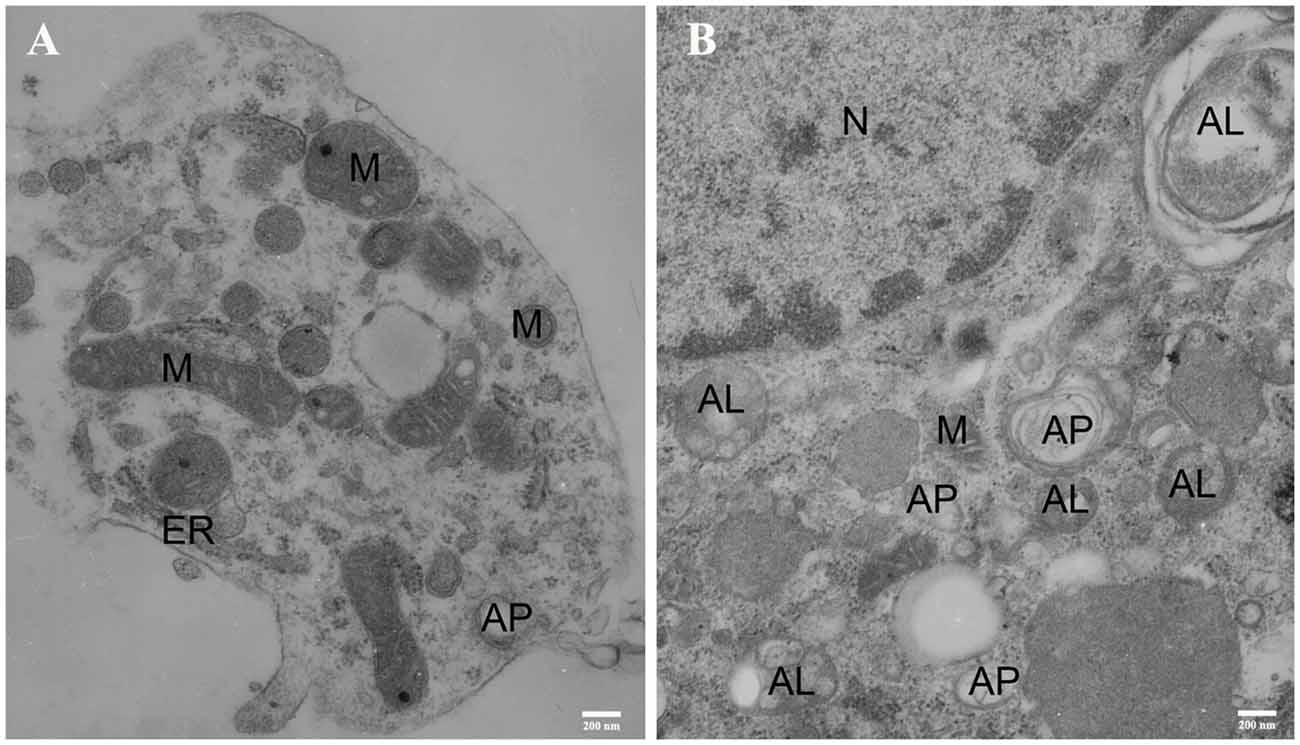
Figure 8. Representative electron micrographs (EM) of primary SMNs in the control (A) and OGD group (B). The ultrastructural changes in primary SMNs after OGD injury are shown, and distinct autophagic structures are marked: autophagosome (AP), autolysosome (AL), mitochondria (M), endoplasmic reticulum (ER), cell nucleus (N). (A) The neuronal soma was plump and showed a normal morphology, with intact nuclear and plasma membranes. In addition, the mitochondria had a dense matrix and neatly aligned cristae, and no signs of autophagy were observed. (B) After OGD insults, crescent-shaped or goblet-like phagophores with double- or multiple-layer membrane structures and formation of autophagosomes can be observed, and autophagic vacuoles fused with lysosomes to form autolysosomes with single-layer membrane structures can also be seen. Images shown are representative examples from three independent experiments. Scale bars presents 200 nm.
Inhibition of Autophagy Exacerbates the Injury of Primary SMNs Exposed to OGD
In order to clarify the effects of autophagy in OGD-induced neuron injury, primary cultured SMNs were treated with autophagy inhibitor 3-MA, whose dosage has been selected to avoid cytotoxity (Figure 9A), and its corresponding inhibition effect was confirmed by Western blot (Figure 9B), followed by OGD for an indicated time, and then the cell viability was measured.
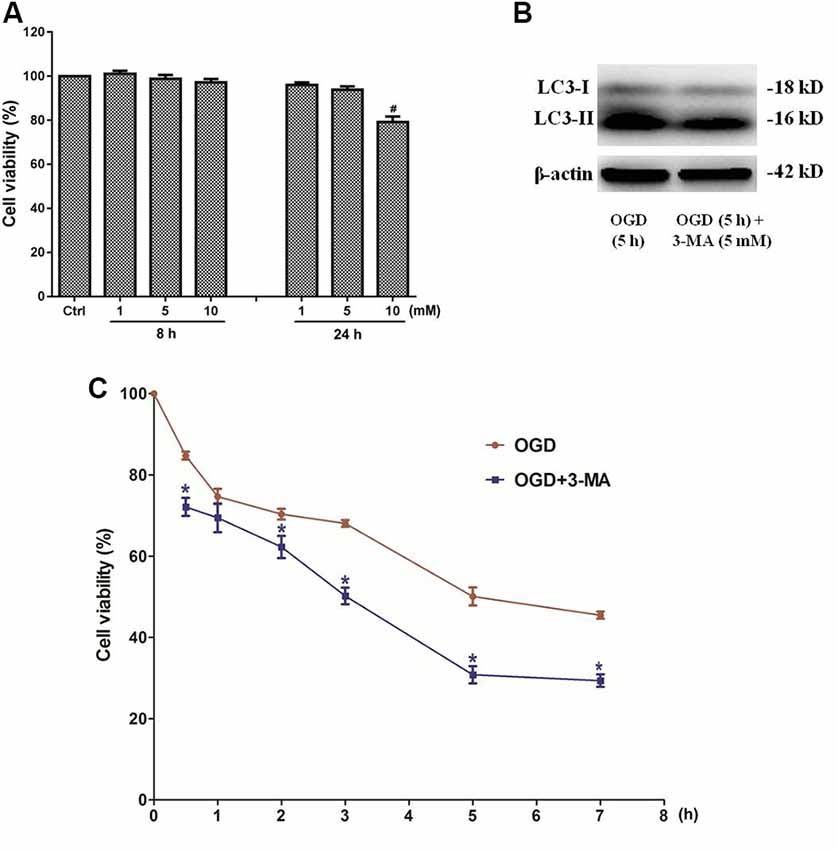
Figure 9. Inhibition of autophagy by 3-methyladenine (3-MA) exacerbates the injury of primary SMNs exposed to OGD. (A) Primary SMNs in normal conditions were treated with different dosages of 3-MA to select its safe dosage. (B) The autophagy inhibition effect of 5 mM 3-MA was confirmed by Western blot. To test it simple and clear, SMNs were subjected to OGD for 5 h in the presence or absence of 3-MA, and their expressions of LC3 I and LC3 II were detected. Results indicated that this dosage can significantly block autophagy in SMNs under OGD conditions. (C) The dosage 5 mM was applied in all the time points, and the cell viability of primary SMNs with or without 3-MA was assessed at different time points using the CCK8 kit. Results were expressed as the percentage of CCK8 reduction, and the absorbance of the control cells was set at 100%. Data were presented as mean ± SD (n = 5 independent experiments). Statistical comparisons were carried out with one-way ANOVA (A) or two-way ANOVA (C). #p < 0.05 compared with the control group. *p < 0.05 compared with cell viability in the same time point under OGD conditions.
3-MA, a well-established inhibitor of autophagy, relatively selectively inhibits the class III phosphatidylinositol kinase (Wu et al., 2010), whose activity is required for autophagosome formation. It has been generally accepted and widely used as an inhibitor of autophagy. Our data showed that the cell viability was even decreased by 3-MA treatment (Figure 9C), indicating that inhibition of autophagy exacerbates the injury of primary SMNs exposed to OGD.
Discussion
In the current study, we identified the dynamic time-course changes and role of autophagy in primary SMNs subjected to OGD. To the best of our knowledge, this report is among the first to clarify the autophagic expression changes in a primary cell model of spinal cord ischemia.
Primary cultures facilitate the limitation of similar in situ counterpart morphology and physiology. However, due to the low yields of motor neuron cultures from spinal cord before, hybrid cell lines of motor neurons (NSC 34 and VSC 4.1) have long been utilized and tacitly regarded as the most stable motor neuron cell line to mimic the pathophysiology of motor neuron disorders (Mosier et al., 1995; Eggett et al., 2000; Samantaray et al., 2006; Maier et al., 2013; Perera et al., 2018). Whereas, with the deepening and development of research, it has been found that there are obvious differences between the hybrid cell lines and primary cells, including the susceptibility of glutamate-induced death and calcium influx (Madji Hounoum et al., 2016). Hence, more and more scholars suggest that primary cells are more suitable as experimental models.
The isolation and culture of primary SMNs had been reported and described well (Schaffner et al., 1987; Martinou et al., 1989; Camu and Henderson, 1992, 1994; Graber and Harris, 2013); however, scarcely rapid and easy protocols have been achieved, let alone cellular models of SMNs. Previous literature mainly focuses on the possibility of successful isolation and culture of SMNs, by density gradient centrifugation, fluorescence-activated cell sorting or immunopanning, or a combination of these methods. Their results are inspiring, but the processes are often complicated, time-consuming, and hard to obtain sufficient quantities of SMNs once, which make it impossible to perform protein or RNA profiling systematically. Rapid and easy protocols facilitate mechanism studies or drug screens by performing downstream analyses (protein or RNA profiling), which require enough quantities of motor neurons in vitro. Therefore, we developed rapid, efficient, and reproducible procedures for the dissection of spinal cords and harvest of SMNs with high survival and purity (Chen et al., 2018), and develop cellular models of SMNs, which allow performing mechanism studies or drug screens.
Autophagy is an indispensable catabolic process in cell survival, differentiation, development, and homeostasis (Levine and Kroemer, 2008; Mizushima and Komatsu, 2011). The dual role of autophagy varies, either to protect cells through adaptive regulation or induce “autophagic cell death.” Accumulating studies have confirmed that autophagy activation is involved in the central nervous system injury and some neurodegenerative diseases (Tan et al., 2014; Martinez-Vicente, 2015; Guo et al., 2018), yet researches on autophagy in spinal cord injury are far from enough. Meanwhile, it is still debatable whether autophagy exerts a protective or destructive role in spinal cord injury. For instance, there are studies documenting that autophagy plays a protective role in spinal ischemia via sustaining autophagy (Fan et al., 2014). However, transient spinal cord ischemia can induce autophagy in motor neurons, which may result in neuronal death (Baba et al., 2009; Fujita et al., 2015). Meanwhile, even some research provided evidence that autophagy exerts opposing impact on early and later stages after spinal I/R injury: early activated autophagy alleviates spinal cord I/R injury by inhibiting apoptosis and inflammation, while later, excessively enhanced autophagy aggravates the injury (Fang et al., 2016). Nonetheless, the abovementioned researches are mainly in vivo studies, which did not show the concrete change of a certain cell type during the process of spinal cord ischemia injury, which we have tried to investigate in our studies.
Lack or deprivation of energy, such as oxygen or glucose, may lead to low physiological function, abnormal metabolism, and even cell death. In our experiment, the cell viability was reduced under OGD (Figure 4). According to the cell viability curve (Figure 4G), we clearly found that the cell viability declined sharply when cells were initially subjected to OGD, but later declined relatively slowly. Correspondingly, we detected the autophagic expression changes during the process (Figure 5). Concomitant with the rate of viability changes, it was interesting to find that autophagy was upregulated and kept increasing in the first few hours, reaching a peak at 5 h, suggesting that cells started to maintain the balance of internal environment after receiving adverse stimulus. However, since the OGD insult existed persistently, the cell viability continued to decline. Morphologically (Figures 4A–F), the SMNs showed slight injury in the first few hours and apparent impaired neural networks after 7 h, indicating that the normal physiological functions were unable to be maintained after stimulated by persistent adverse factors.
The microtubule-associated protein LC3, a mammalian homolog of the yeast ATG8 gene (Aut7/Apg8), takes an important part in the formation of autophagic vacuoles (Klionsky et al., 2016). The cytoplasmic form LC3 (LC3-I) is distributed throughout the cytoplasm diffusely and uniformly, but during autophagy activation, it will be conjugated to phosphatidylethanolamine to form LC3–phosphatidylethanolamine conjugate (LC3-II) and recruited to autophagosomal membranes (Mizushima and Yoshimori, 2007). When associated with autophagosomes, LC3 exhibits a typically apparent mobility in electrophoresis, changing from 18 kD to 16 kD and indicated as “LC3 puncta processing,” which is commonly referred to as LC3-II. Autophagosomes fuse with lysosomes to form autolysosomes, and intra-autophagosomal components are degraded by lysosomal hydrolases. At the same time, LC3-II in autolysosomal lumen is degraded. Therefore, lysosomal turnover of the autophagosomal marker LC3-II can reflect the autophagic activity to some extent, and detecting LC3 by immunoblotting and immunofluorescence have become reliable methods for monitoring autophagy and autophagy-related processes. The level of autophagy can be assessed by the ratio of LC3-II/I by immunoblotting and positive puncta of LC3 by immunofluorescence. In our study, the Western blot analysis revealed a significantly increase in LC3 level (Figure 5), which was further corroborated by immunofluorescence assays (Figure 6) and TEM (Figure 8). Meanwhile, the marker of autophagic degradation (SQSTM1/p62) diminished significantly after OGD for 3 h, and Beclin 1 expression was upregulated markedly and continually after OGD within 7 h (Figure 5).
Besides the fact that immunofluorescence assays confirmed the induction of autophagy during OGD, intriguingly, from the immunofluorescence images, strong LC3 puncta were assembly visualized in soma rather than in axons (Figure 6). Previous studies have demonstrated that retrograde transportation along axons back toward the neuronal cell bodies are critical during the autophagic process of neurons (Maday et al., 2012; Maday and Holzbaur, 2014; Zheng et al., 2019). It has already been proven that the process of retrograde transport along axons back toward the neuronal soma occurs in hippocampal, DRG, and cortical neurons (Lee et al., 2011; Maday et al., 2012; Maday and Holzbaur, 2014; Zheng et al., 2019). Our findings may provide some support for this theory in SMNs.
Detecting LC3 by immunoblotting and immunofluorescence are the basic ways to monitor autophagy and autophagy-related processes; however, these results can hardly represent the real state of autophagic flux since autophagy is a dynamic process with multiple steps. In this regard, lysosome inhibitor Baf-A1 was added to the culture medium to assess the responses of SMNs, which is a classical approach in observing the real state of autophagic flux. Actually, up until now, besides the lysosomal or vacuolar degradation inhibitor present or not in Western blot analysis, an alternative way to detect the autophagic flux is to measure the GFP–LC3 fusion protein with the aid of the GFP–LC3 expressing plasmid. In our experiment, we have tried both of the methods, but limited to the low transfection efficiency of GFP–LC3 expressing plasmid in primary SMNs, we only showed the results of autophagic flux detection by adding Baf-A1. In the presence of Baf-A1, LC3 II increased and accumulated (Figure 7), which indicated the increase in LC3-II levels by OGD owing to an increase in production rather than the decreased recycling of autophagy.
Later, in order to evaluate the role of autophagy, the autophagy inhibitor 3-MA was added in the culture system. It showed that blockade of autophagy activation by 3-MA aggravated cell injury, presenting worse cell viability.
Conclusion
Taken together, by successfully establishing a primary cellular model of spinal cord ischemia in vitro, the dynamic time-course changes of autophagy and autophagy flux in SMNs was clearly investigated. Autophagy was upregulated in primary SMNs subjected to OGD, and autophagy reached a peak at 5 h. Besides, inhibition of autophagy exacerbated the injury of primary SMNs exposed to OGD, suggesting that autophagy plays a protective role during the process. Further studies need to explore the possible pathway and mechanisms underlying the effects of autophagy overactivation.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Ethical Committee of Guangzhou University of Chinese Medicine.
Author Contributions
SC and DLi conceived and designed the experiments. SC, RT, DLu, and ZX performed the experiments, analyzed the data, and wrote the manuscript. HL and DLi revised the manuscript. All authors have read and approved the manuscript for publication.
Funding
This research was funded by the National Natural Science Foundation of China (Nos. 81704095 and 81673992), Administration of Traditional Chinese Medicine of Guangdong Province (No. 20173022), and Science and Technology Project of Guangdong Province (No. 2016A020226008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aslan, A., Cemek, M., Buyukokuroglu, M. E., Altunbas, K., Bas, O., Yurumez, Y., et al. (2009). Dantrolene can reduce secondary damage after spinal cord injury. Eur. Spine J. 18, 1442–1451. doi: 10.1007/s00586-009-1033-6
Baba, H., Sakurai, M., Abe, K., and Tominaga, R. (2009). Autophagy-mediated stress response in motor neuron after transient ischemia in rabbits. J. Vasc. Surg. 50, 381–387. doi: 10.1016/j.jvs.2009.03.042
Camu, W., and Henderson, C. E. (1992). Purification of embryonic rat motoneurons by panning on a monoclonal antibody to the low-affinity NGF receptor. J. Neurosci. Methods 44, 59–70. doi: 10.1016/0165-0270(92)90114-s
Camu, W., and Henderson, C. E. (1994). Rapid purification of embryonic rat motoneurons: an in vitro model for studying MND/ALS pathogenesis. J. Neurol. Sci. 124, 73–74. doi: 10.1016/0022-510x(94)90185-6
Chen, S., Tian, R., Li, H., Chen, M., Zhang, H., and Lin, D. (2018). Optimized methods for rapidly dissecting spinal cords and harvesting spinal motor neurons with high survival and purity from rats at different embryonic stages. J. Spinal Cord Med. 41, 281–291. doi: 10.1080/10790268.2017.1329075
Ciechanover, A., Orian, A., and Schwartz, A. L. (2000). Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 22, 442–451. doi: 10.1002/(sici)1521-1878(200005)22:5<442::aid-bies6>3.0.co;2-q
Cimarosti, H., Ashikaga, E., Jaafari, N., Dearden, L., Rubin, P., Wilkinson, K. A., et al. (2012). Enhanced SUMOylation and SENP-1 protein levels following oxygen and glucose deprivation in neurones. J. Cereb. Blood Flow Metab. 32, 17–22. doi: 10.1038/jcbfm.2011.146
Coselli, J. S., LeMaire, S. A., de Figueiredo, L. P., and Kirby, R. P. (1997). Paraplegia after thoracoabdominal aortic aneurysm repair: is dissection a risk factor? Ann. Thorac. Surg. 63, 28–35; discussion 35–26. doi: 10.1016/s0003-4975(96)01029-6
Drinkwater, S. L., Goebells, A., Haydar, A., Bourke, P., Brown, L., Hamady, M., et al. (2010). The incidence of spinal cord ischaemia following thoracic and thoracoabdominal aortic endovascular intervention. Eur. J. Vasc. Endovasc. Surg. 40, 729–735. doi: 10.1016/j.ejvs.2010.08.013
Eggett, C. J., Crosier, S., Manning, P., Cookson, M. R., Menzies, F. M., McNeil, C. J., et al. (2000). Development and characterisation of a glutamate-sensitive motor neurone cell line. J. Neurochem. 74, 1895–1902. doi: 10.1046/j.1471-4159.2000.0741895.x
Esposito, E., Paterniti, I., Meli, R., Bramanti, P., and Cuzzocrea, S. (2012). GW0742, a high-affinity PPAR-delta agonist, mediates protection in an organotypic model of spinal cord damage. Spine 37, E73–E78. doi: 10.1097/brs.0b013e3182276d88
Etz, D. C., Luehr, M., Aspern, K. V., Misfeld, M., Gudehus, S., Ender, J., et al. (2014). Spinal cord ischemia in open and endovascular thoracoabdominal aortic aneurysm repair: new concepts. J. Cardiovasc. Surg. 55, 159–168.
Etz, C. D., Luehr, M., Kari, F. A., Bodian, C. A., Smego, D., Plestis, K. A., et al. (2008). Paraplegia after extensive thoracic and thoracoabdominal aortic aneurysm repair: does critical spinal cord ischemia occur postoperatively? J. Thorac. Cardiovasc. Surg. 135, 324–330. doi: 10.1016/j.jtcvs.2007.11.002
Fan, J., Zhang, Z., Chao, X., Gu, J., Cai, W., Zhou, W., et al. (2014). Ischemic preconditioning enhances autophagy but suppresses autophagic cell death in rat spinal neurons following ischemia-reperfusion. Brain Res. 1562, 76–86. doi: 10.1016/j.brainres.2014.03.019
Fang, B., Li, X. Q., Bao, N. R., Tan, W. F., Chen, F. S., Pi, X. L., et al. (2016). Role of autophagy in the bimodal stage after spinal cord ischemia reperfusion injury in rats. Neuroscience 328, 107–116. doi: 10.1016/j.neuroscience.2016.04.019
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Fontella, F. U., Cimarosti, H., Crema, L. M., Thomazi, A. P., Leite, M. C., Salbego, C., et al. (2005). Acute and repeated restraint stress influences cellular damage in rat hippocampal slices exposed to oxygen and glucose deprivation. Brain Res. Bull. 65, 443–450. doi: 10.1016/j.brainresbull.2005.02.026
Fujita, S., Sakurai, M., Baba, H., Abe, K., and Tominaga, R. (2015). Autophagy-mediated stress response in motor neurons after hypothermic spinal cord ischemia in rabbits. J. Vasc. Surg. 62, 1312–1319. doi: 10.1016/j.jvs.2014.03.297
Gómez-Sánchez, R., Pizarro-Estrella, E., Yakhine-Diop, S. M., Rodríguez-Arribas, M., Bravo-San Pedro, J. M., Fuentes, J. M., et al. (2015). Routine Western blot to check autophagic flux: cautions and recommendations. Anal. Biochem. 477, 13–20. doi: 10.1016/j.ab.2015.02.020
Gooding, M. R., Wilson, C. B., and Hoff, J. T. (1975). Experimental cervical myelopathy. Effects of ischemia and compression of the canine cervical spinal cord. J. Neurosurg. 43, 9–17. doi: 10.3171/jns.1975.43.1.0009
Graber, D. J., and Harris, B. T. (2013). Purification and culture of spinal motor neurons from rat embryos. Cold Spring Harb. Protoc. 2013, 319–326. doi: 10.1101/pdb.prot074161
Griffiths, I. R., Trench, J. G., and Crawford, R. A. (1979). Spinal cord blood flow and conduction during experimental cord compression in normotensive and hypotensive dogs. J. Neurosurg. 50, 353–360. doi: 10.3171/jns.1979.50.3.0353
Gu, Z., Sun, Y., Liu, K., Wang, F., Zhang, T., Li, Q., et al. (2013). The role of autophagic and lysosomal pathways in ischemic brain injury. Neural Regen. Res. 8, 2117–2125. doi: 10.3969/j.issn.1673-5374.2013.23.001
Guo, F., Liu, X., Cai, H., and Le, W. (2018). Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 28, 3–13. doi: 10.1111/bpa.12545
Hwang, J., Huh, J., Kim, J., Jeon, Y., Cho, S., and Han, S. (2012). Pretreatment with erythropoietin attenuates the neurological injury after spinal cord ischemia. Spinal Cord 50, 208–212. doi: 10.1038/sc.2011.136
Hwang, J. Y., Sohn, H. M., Kim, J. H., Park, S., Park, J. W., Lim, M. S., et al. (2017). Reproducible motor deficit following aortic occlusion in a rat model of spinal cord ischemia. J. Vis. Exp. 125:e55814. doi: 10.3791/55814
Kanno, H., Ozawa, H., Sekiguchi, A., and Itoi, E. (2009a). The role of autophagy in spinal cord injury. Autophagy 5, 390–392. doi: 10.4161/auto.5.3.7724
Kanno, H., Ozawa, H., Sekiguchi, A., and Itoi, E. (2009b). Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol. Dis. 33, 143–148. doi: 10.1016/j.nbd.2008.09.009
Kato, H., Kanellopoulos, G. K., Matsuo, S., Wu, Y. J., Jacquin, M. F., Hsu, C. Y., et al. (1997). Neuronal apoptosis and necrosis following spinal cord ischemia in the rat. Exp. Neurol. 148, 464–474. doi: 10.1006/exnr.1997.6707
Kise, Y., Kuniyoshi, Y., Inafuku, H., Nagano, T., Hirayasu, T., and Yamashiro, S. (2015). Directly measuring spinal cord blood flow and spinal cord perfusion pressure via the collateral network: correlations with changes in systemic blood pressure. J. Thorac. Cardiovasc. Surg. 149, 360–366. doi: 10.1016/j.jtcvs.2014.09.121
Klionsky, D. J. (2008). Autophagy revisited: a conversation with Christian de Duve. Autophagy 4, 740–743. doi: 10.4161/auto.6398
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. doi: 10.1126/science.290.5497.1717
Kolenda, H., Steffens, H., Hagenah, J., and Schomburg, E. D. (2003). Different susceptibility of facilitatory and inhibitory spinal pathways to ischemia in the cat. Neurosci. Res. 47, 357–366. doi: 10.1016/j.neures.2003.07.001
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Lee, S., Sato, Y., and Nixon, R. A. (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 31, 7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Lu, H., Li, G., Liu, L., Feng, L., Wang, X., and Jin, H. (2013). Regulation and function of mitophagy in development and cancer. Autophagy 9, 1720–1736. doi: 10.4161/auto.26550
Lynch-Day, M. A., Mao, K., Wang, K., Zhao, M., and Klionsky, D. J. (2012). The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009357. doi: 10.1101/cshperspect.a009357
Maday, S., and Holzbaur, E. L. (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. doi: 10.1016/j.devcel.2014.06.001
Maday, S., Wallace, K. E., and Holzbaur, E. L. (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417. doi: 10.1083/jcb.201106120
Madji Hounoum, B., Vourc’h, P., Felix, R., Corcia, P., Patin, F., Gueguinou, M., et al. (2016). NSC-34 motor neuron-like cells are unsuitable as experimental model for glutamate-mediated excitotoxicity. Front. Cell. Neurosci. 10:118. doi: 10.3389/fncel.2016.00118
Maier, O., Böhm, J., Dahm, M., Brück, S., Beyer, C., and Johann, S. (2013). Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem. Int. 62, 1029–1038. doi: 10.1016/j.neuint.2013.03.008
Mariño, G., Madeo, F., and Kroemer, G. (2011). Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 23, 198–206. doi: 10.1016/j.ceb.2010.10.001
Martin, D. D., Ladha, S., Ehrnhoefer, D. E., and Hayden, M. R. (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35. doi: 10.1016/j.tins.2014.09.003
Martinet, W., Timmermans, J. P., and De Meyer, G. R. (2014). Methods to assess autophagy In situ—transmission electron microscopy versus immunohistochemistry. Methods Enzymol. 543, 89–114. doi: 10.1016/b978-0-12-801329-8.00005-2
Martinez-Vicente, M. (2015). Autophagy in neurodegenerative diseases: from pathogenic dysfunction to therapeutic modulation. Semin. Cell Dev. Biol. 40, 115–126. doi: 10.1016/j.semcdb.2015.03.005
Martinou, J. C., Le Van Thai, A., Cassar, G., Roubinet, F., and Weber, M. J. (1989). Characterization of two factors enhancing choline acetyltransferase activity in cultures of purified rat motoneurons. J. Neurosci. 9, 3645–3656. doi: 10.1523/JNEUROSCI.09-10-03645.1989
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mizushima, N., and Yoshimori, T. (2007). How to interpret LC3 immunoblotting. Autophagy 3, 542–545. doi: 10.4161/auto.4600
Mosier, D. R., Baldelli, P., Delbono, O., Smith, R. G., Alexianu, M. E., Appel, S. H., et al. (1995). Amyotrophic lateral sclerosis immunoglobulins increase Ca2+ currents in a motoneuron cell line. Ann. Neurol. 37, 102–109. doi: 10.1002/ana.410370119
Nazli, Y., Colak, N., Alpay, M. F., Uysal, S., Uzunlar, A. K., and Cakir, O. (2015). Neuroprotective effect of atorvastatin in spinal cord ischemia-reperfusion injury. Clinics 70, 52–60. doi: 10.6061/clinics/2015(01)10
Nedelsky, N. B., Todd, P. K., and Taylor, J. P. (2008). Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699. doi: 10.1016/j.bbadis.2008.10.002
Perera, N. D., Sheean, R. K., Lau, C. L., Shin, Y. S., Beart, P. M., Horne, M. K., et al. (2018). Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 14, 534–551. doi: 10.1080/15548627.2017.1385674
Rami, A., Langhagen, A., and Steiger, S. (2008). Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol. Dis. 29, 132–141. doi: 10.1016/j.nbd.2007.08.005
Rivlin, A. S., and Tator, C. H. (1978). Regional spinal cord blood flow in rats after severe cord trauma. J. Neurosurg. 49, 844–853. doi: 10.3171/jns.1978.49.6.0844
Samantaray, S., Ray, S. K., Ali, S. F., and Banik, N. L. (2006). Calpain activation in apoptosis of motoneurons in cell culture models of experimental parkinsonism. Ann. N Y Acad. Sci. 1074, 349–356. doi: 10.1196/annals.1369.034
Schaffner, A. E., St John, P. A., and Barker, J. L. (1987). Fluorescence-activated cell sorting of embryonic mouse and rat motoneurons and their long-term survival in vitro. J. Neurosci. 7, 3088–3104. doi: 10.1523/JNEUROSCI.07-10-03088.1987
Sheng, R., Zhang, L. S., Han, R., Liu, X. Q., Gao, B., and Qin, Z. H. (2010). Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 6, 482–494. doi: 10.4161/auto.6.4.11737
Shimizu, H., and Yozu, R. (2011). Current strategies for spinal cord protection during thoracic and thoracoabdominal aortic aneurysm repair. Gen. Thorac. Cardiovasc. Surg. 59, 155–163. doi: 10.1007/s11748-010-0705-9
Suarez, N. C., Levi, R., and Bullington, J. (2013). Regaining health and wellbeing after traumatic spinal cord injury. J. Rehabil. Med. 45, 1023–1027. doi: 10.2340/16501977-1226
Sun, Y., Liu, D., Su, P., Lin, F., and Tang, Q. (2016). Changes in autophagy in rats after spinal cord injury and the effect of hyperbaric oxygen on autophagy. Neurosci. Lett. 618, 139–145. doi: 10.1016/j.neulet.2016.02.054
Swanlund, J. M., Kregel, K. C., and Oberley, T. D. (2010). Investigating autophagy: quantitative morphometric analysis using electron microscopy. Autophagy 6, 270–277. doi: 10.4161/auto.6.2.10439
Tan, C. C., Yu, J. T., Tan, M. S., Jiang, T., Zhu, X. C., and Tan, L. (2014). Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol. Aging 35, 941–957. doi: 10.1016/j.neurobiolaging.2013.11.019
Tasca, C. I., Dal-Cim, T., and Cimarosti, H. (2015). In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 1254, 197–210. doi: 10.1007/978-1-4939-2152-2_15
Turner, S. M., and Johnson, S. M. (2011). Delta-opioid receptor activation prolongs respiratory motor output during oxygen-glucose deprivation in neonatal rat spinal cord in vitro. Neuroscience 187, 70–83. doi: 10.1016/j.neuroscience.2011.04.059
Uchiyama, Y., Koike, M., and Shibata, M. (2008). Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy 4, 404–408. doi: 10.4161/auto.5598
Wang, P., Guan, Y. F., Du, H., Zhai, Q. W., Su, D. F., and Miao, C. Y. (2012). Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 8, 77–87. doi: 10.4161/auto.8.1.18274
Wang, Z., Yang, W., Britz, G. W., Lombard, F. W., Warner, D. S., and Sheng, H. (2010). Development of a simplified spinal cord ischemia model in mice. J. Neurosci. Methods 189, 246–251. doi: 10.1016/j.jneumeth.2010.04.003
Wang, R., Zhang, X., Zhang, J., Fan, Y., Shen, Y., Hu, W., et al. (2012). Oxygen-glucose deprivation induced glial scar-like change in astrocytes. PLoS One 7:e37574. doi: 10.1371/journal.pone.0037574
Weidauer, S., Nichtweiß, M., Hattingen, E., and Berkefeld, J. (2015). Spinal cord ischemia: aetiology, clinical syndromes and imaging features. Neuroradiology 57, 241–257. doi: 10.1007/s00234-014-1464-6
Wirawan, E., Vanden Berghe, T., Lippens, S., Agostinis, P., and Vandenabeele, P. (2012). Autophagy: for better or for worse. Cell Res. 22, 43–61. doi: 10.1038/cr.2011.152
Wu, Y. T., Tan, H. L., Shui, G., Bauvy, C., Huang, Q., Wenk, M. R., et al. (2010). Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861. doi: 10.1074/jbc.M109.080796
Yang, T., Wu, L., Wang, H., Fang, J., Yao, N., and Xu, Y. (2015). Inflammation level after decompression surgery for a rat model of chronic severe spinal cord compression and effects on ischemia-reperfusion injury. Neurol. Med. Chir. 55, 578–586. doi: 10.2176/nmc.oa.2015-0022
Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., et al. (2004). Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502. doi: 10.1126/science.1096645
Keywords: spinal motor neurons (SMNs), spinal cord ischemia, cellular model, oxygen-glucose deprivation (OGD), autophagy
Citation: Chen S, Tian R, Luo D, Xiao Z, Li H and Lin D (2020) Time-Course Changes and Role of Autophagy in Primary Spinal Motor Neurons Subjected to Oxygen-Glucose Deprivation: Insights Into Autophagy Changes in a Cellular Model of Spinal Cord Ischemia. Front. Cell. Neurosci. 14:38. doi: 10.3389/fncel.2020.00038
Received: 01 August 2019; Accepted: 07 February 2020;
Published: 20 March 2020.
Edited by:
Luis B. Tovar-y-Romo, National Autonomous University of Mexico, MexicoReviewed by:
James N. Sleigh, University College London, United KingdomLourdes Massieu, National Autonomous University of Mexico, Mexico
Copyright © 2020 Chen, Tian, Luo, Xiao, Li and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingkun Lin, lindingkun_tcm@163.com
† These authors have contributed equally to this work