
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci., 26 February 2020
Sec. Cellular Neurophysiology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00033
This article is part of the Research TopicCellular and Molecular Targets In Epileptogenesis Focusing on Disease PreventionView all 13 articles
 Felix Schulze1†
Felix Schulze1† Steffen Müller1†
Steffen Müller1† Xiati Guli1Lukas Schumann1
Xiati Guli1Lukas Schumann1 Hannes Brehme1
Hannes Brehme1 Till Riffert1
Till Riffert1 Marco Rohde1Doreen Goerss2,3Simone Rackow1Anne Einsle1
Marco Rohde1Doreen Goerss2,3Simone Rackow1Anne Einsle1 Timo Kirschstein1,4*†
Timo Kirschstein1,4*† Rüdiger Köhling1,4*†
Rüdiger Köhling1,4*†Purpose: Epilepsy therapy is currently based on anti-seizure drugs that do not modify the course of the disease, i.e., they are not anti-epileptogenic in nature. Previously, we observed that in vivo casein kinase 2 (CK2) inhibition with 4,5,6,7-tetrabromotriazole (TBB) had anti-epileptogenic effects in the acute epilepsy slice model.
Methods: Here, we pretreated rats with TBB in vivo prior to the establishment of a pilocarpine-induced status epilepticus (SE) in order to analyze the long-term sequelae of such a preventive TBB administration.
Results: We found that TBB pretreatment delayed onset of seizures after pilocarpine and slowed down disease progression during epileptogenesis. This was accompanied with a reduced proportion of burst firing neurons in the CA1 area. Western blot analyses demonstrated that CA1 tissue from TBB-pretreated epileptic animals contained significantly less CK2 than TBB-pretreated controls. On the transcriptional level, TBB pretreatment led to differential gene expression changes of KCa2.2, but also of HCN1 and HCN3 channels. Thus, in the presence of the HCN channel blocker ZD7288, pretreatment with TBB rescued the afterhyperpolarizing potential (AHP) as well as spike frequency adaptation in epileptic animals, both of which are prominent functions of KCa2 channels.
Conclusion: These data indicate that TBB pretreatment prior to SE slows down disease progression during epileptogenesis involving increased KCa2 function, probably due to a persistently decreased CK2 protein expression.
Temporal lobe epilepsy (TLE) is the most common focal symptomatic epileptic disorder in the adulthood and typically difficult to treat (Kwan and Brodie, 2000; Blümcke et al., 2013). Moreover, all currently available drugs used to treat focal symptomatic epilepsy are merely anti-seizure drugs, i.e., they are anti-ictogenic in nature, but lack any anti-epileptogenic potential (Schmidt and Sillanpää, 2016). In the last decades, several rodent post-status epilepticus (SE) models such as the pilocarpine animal model have provided valuable insights into the latent period between the initial insult and the subsequent development of chronic TLE (Leite et al., 2002). During this period of epileptogenesis, marked changes in synaptic and intrinsic properties take place that may facilitate the occurrence of seizures—most profoundly and studied at best—within the hippocampal network (Leite et al., 2002).
Among these changes, it appears that channels that are active near the resting membrane potential (RMP) such as K+ or hyperpolarization-activated cyclic nucleotide-gated non-selective (HCN) channels are particularly intriguing candidates involved in epileptogenesis. With respect to the CA1 area in the pilocarpine model, there is evidence of persistent downregulation of Kv4.2 (Bernard et al., 2004), KCa2.2 (Schulz et al., 2012), and HCN1 (Jung et al., 2007, 2010). Importantly, these transcriptional changes partly seem to involve the activation of protein kinases or phosphatases (Bernard et al., 2004; Jung et al., 2010). In turn, protein kinase and/or phosphatase activation also seem to be involved in acute epileptic conditions (Jung et al., 2010; Kernig et al., 2012) suggesting that neuronal hyperactivity may trigger these enzymatic pathways.
Casein kinase 2 (CK2) is a widely expressed enzyme with a substantial number of possible targets; however, one of its major targets is calmodulin (CaM) and KCa2.2 (Bildl et al., 2004; Allen et al., 2007). We recently demonstrated that oral administration of the CK2 blocker 4,5,6,7-tetrabromotriazole (TBB) enhanced K+ currents mediating the afterhyperpolarizing potential (AHP) in the pilocarpine model, and even more intriguing, blocked the occurrence of spontaneous epileptic activity in the acute slice model with Mg2+ removal (Brehme et al., 2014). This compound obviously exerted an anti-epileptogenic potential in the in vitro slice preparation, but is not an anti-seizure drug in this model in vivo (Bajorat et al., 2018). Therefore, we asked whether TBB might have disease modifying effects in the pilocarpine model in vivo, too. To this end, we treated the rats with TBB prior to pilocarpine-induced SE and analyzed the animals in their chronically epileptic stage.
Male Wistar rats (27–30 days; Charles River, Sulzfeld, Germany) were treated with the CK2 inhibitor TBB (Tocris Bioscience, Bristol, United Kingdom or Sigma–Aldrich, Taufkirchen, Germany) 3 days before SE by intraperitoneal (i.p.) injection of 6 μmol/kg body weight [stock solution 6 mmol/l, dissolved in dimethylsulfoxide (DMSO), 1 μl/g body weight]. This was repeated for three times every 24 h (i.e., four consecutive daily injections, Figure 1A). The same amount of vehicle (DMSO, 1 μl/g body weight) was injected as control. Allocation of animals to the TBB or vehicle pretreatment group was performed in randomized fashion. Following the last of four injections, the animals were allowed to recover for one hour, before pilocarpine was injected to induce chronic epilepsy. During the TBB pretreatment period, the weight gain was monitored and did not differ significantly between TBB-treated (from 105 ± 18 to 131 ± 19 g, mean ± SD) and vehicle-treated (from 107 ± 19 to 133 ± 18 g, mean ± SD) animals (ANOVA).
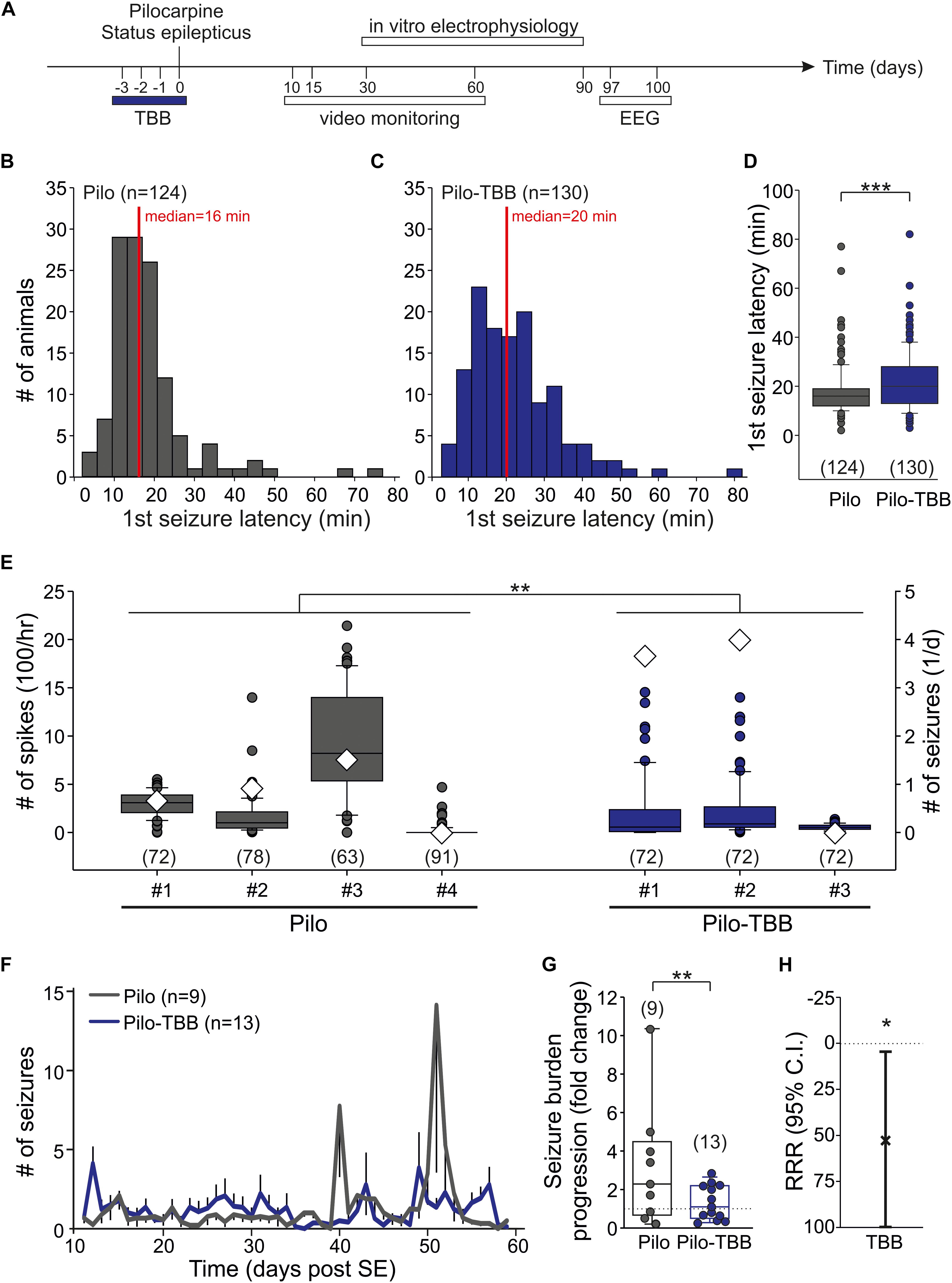
Figure 1. CK2 inhibition exerts disease modifying properties. (A) Time frame of the experiment. TBB was administered four times in vivo on days “−3” (i.e., 3 days before status epilepticus) through “0” (i.e., day of pilocarpine-induced status epilepticus). All experiments thereafter were performed during the chronic stage of recurrent seizures. (B–D) Latency to the first stage 4 or 5 seizure after pilocarpine injection in vehicle-pretreated (B) and TBB-pretreated (C) rats. The median latency was significantly (***P < 0.001, Mann-Whitney U-test) prolonged in the TBB-pretreated group (D). (E) Number of interictal spikes per hour (left y-axis) in the electroencephalogram (EEG) during 3 days (97–100 post-SE). Note that the number of daily seizures during this period is also depicted (diamonds, right y-axis). There was a significant group effect of TBB pretreatment (**P < 0.01, two-way ANOVA with Tukey post hoc test). (F) Number of generalized seizures (at least stage 3) at daylight (from 06:00 to 18:00 h) for both animal groups as grand average for the entire recording period. (G) Seizure burden progression as fold change of seizure rates during epileptogenesis. The variability was significantly reduced in TBB-pretreated epileptic animals (**P < 0.01, F-test). (H) Relative risk reduction (RRR) of seizure progression by TBB pretreatment was significant (*P < 0.05, t-test).
Chronic epilepsy was induced by systemic pilocarpine injection 1 h after the last TBB application. As described previously (Kirschstein et al., 2007; Bajorat et al., 2011; Müller et al., 2013), rats received N-methyl scopolamine nitrate (1 mg/kg, i.p.) in order to reduce peripheral cholinergic effects, and then pilocarpine hydrochloride (340 mg/kg, i.p.). All pilocarpine-injected animals were carefully monitored to detect spontaneous motor seizures with progression into limbic SE. All stage 4 or 5 seizures (Racine, 1972) were registered for each individual animal to detect the latency to the first stage 4 or 5 seizure after pilocarpine. The onset of SE was determined when an animal had a stage 4 or 5 seizure (Racine, 1972) that was followed by continuous seizure motor activity without showing any reaction to sensory stimuli such as gently touching against the whiskers. SE was terminated after 40 min by administration of diazepam solution (Ratiopharm, Ulm, Germany, 5 mg/ml, i.p.). Finally, the rats were fed with 5% glucose solution for 1 day and kept in separate cages. Experimenters were blinded for all in vivo applications and observations, thus were unaware as to whether the animal was pretreated with TBB or vehicle (DMSO) before.
All procedures were performed according to national and international guidelines on the ethical use of animals (European Council Directive 86/609/EEC, approval of local authority LALLF M-V/TSD/7221.3-1.1-013/10 and 7221.3-1.1-019/13), and all efforts were made to minimize animal suffering and to reduce the number of animals used.
Epileptiform potentials in the electroencephalogram (EEG) were recorded using a telemetric system [Neuroscore 2.0, Data Science International (DSI), Arden Hills, MN, United States]. Electrode implantation (TA10EA-F20 or TA11CA-F40 from DSI) was performed as previously described (Bajorat et al., 2011, 2016, 2018). Video-EEG monitoring was performed in three TBB-treated pilocarpine-treated epileptic animals and four vehicle-treated epileptic rats under environmentally controlled conditions (12 h light/dark cycles, lights switched on from 6 a.m. to 6 p.m., 22 ± 2°C, and 40–60% relative humidity in an isolated room). Pilocarpine-treated rats were housed in individual cages and continuous video-EEG monitoring was performed using a telemetric system (Dataquest A.R.T. 4.2., DSI) in combination with a light and dark network camera (Axis 223M, Axis communications, Lund, Sweden). Night recordings were performed with a small blue night light for residual light intensification. The EEG was screened for epileptiform potentials (spikes, sharp waves, spike-and-wave-complexes, sharp-and-slow-wave-complexes) and seizures were detected by concomitant analysis of the video signal (Bajorat et al., 2011, 2016). The EEG experimenter was blinded to the treatment (TBB versus vehicle).
In addition, we performed video analysis in order to detect generalized seizures (13 TBB-treated and nine vehicle-treated epileptic rats) for 7 weeks (10–60 days after pilocarpine-induced SE, i.e., 5–12 weeks of age). All generalized seizures (stage 4–5, Racine, 1972) at daylight (from 6 a.m. to 6 p.m.) were detected in a blinded fashion, i.e., without knowledge about the treatment (TBB versus vehicle). Control animals, i.e., animals that received saline instead of pilocarpine, were repeatedly reported to get no spontaneous seizures at all (Bajorat et al., 2011, 2016) and were therefore not included in these in vivo studies. The seizure burden progression was calculated as the ratio of seizure rate day 35–59 over the seizure rate day 11–20. The relative risk reduction (RRR) of seizure burden progression was calculated as 1 – the relative risk (seizure burden progression in the TBB-pretreated animal over the seizure burden progression in the vehicle-pretreated group).
Hippocampal slices were prepared using 8–12-week-old animals (i.e., 4–8 weeks after SE). At this age, chronically epileptic animals show on average 4.5 seizures per day (Bajorat et al., 2011). After deep anesthesia with diethyl ether (Merck, Darmstadt, Germany), rats were decapitated and the brain was rapidly removed and submerged into oxygenated ice-cold dissection solution containing (in mM) 125 NaCl, 26 NaHCO3, 3 KCl, 1.25 NaH2PO4, 0.2 CaCl2, 5 MgCl2, and 13 D-glucose (95% O2, 5% CO2; pH 7.4; osmolality 306–314 mosmol/kg H2O). Hippocampal horizontal brain slices (400 μm) were prepared from the brain using a vibratome (Leica VT1200S, Wetzlar, Germany), and these slices were gently transferred into a holding chamber containing artificial cerebrospinal fluid (ACSF) of the following composition: (in mM) 125 NaCl, 26 NaHCO3, 3 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.3 MgCl2, and 13 D-glucose (95% O2, 5% CO2; pH 7.4; osmolality 306–314 mosmol/kg H2O).
For electrophysiological experiments, hippocampal slices were allowed to recover at room temperature (20–22°C) for at least 1 h before being transferred into recording chamber. For quantitative real-time reverse transcriptase PCR (RT-PCR) analyses, hippocampal slices were immediately dissected into mini-slices. For Timm staining, hippocampal slices were immediately immersed into Na2S solution.
After recovery, hippocampal slices were placed into an interface chamber continuously bathed with ACSF at a flow rate of 2 ml/min using a volumetric infusion pump (MCM-500, MC Medicine technique GmbH, Alzenau, Germany). Bath temperature was maintained at 32 ± 1°C by (npi electronic GmbH, Tamm, Germany), and all experiments started after an equilibration time of at least 30 min. Schaffer collateral fibers were stimulated using bipolar platinum wire electrodes connected to an A365 stimulus isolator in constant-current mode (Word Precision Instruments, Friedberg, Germany) and driven by the Master-8 stimulator (A.M.P.I., Jerusalem, Israel). The stimulating electrode was placed into stratum radiatum at the border between CA2 and CA1. Field excitatory postsynaptic potentials (fEPSPs) from the CA1 area were recorded using borosilicate glass pipettes (2–3 MΩ, pulled with PIP5 from HEKA Elektronik, Lambrecht, Germany; filled with ACSF). Data were acquired using a Micro1401 A/D converter and signal 2.16 software (CED, Cambridge Electronic Design, Cambridge, United Kingdom). The fEPSP slope was determined as the local minimum of the differentiated fEPSP curve indicating the maximal negative gradient during its falling phase. With these extracellular recordings, we obtained the input–output curve, the paired-pulse ratio (PPR) with an interstimulus interval of 40 ms and short-term plasticity data.
Patch pipettes with a tip resistance 3–6 MΩ were pulled from borosilicate glass capillaries (GB150-8P; Science Products GmbH, Hofheim, Germany) using a horizontal electrode puller (P-97 Micropipette Puller; Sutter Instrument Company, Novato, CA, United States) and filled with an intracellular solution (K+ gluconate 140, MgCl2 2, HEPES 10, phosphocreatine 10, Na2-ATP 2, Na2-GTP 0.4, pH 7.3 adjusted with KOH, osmolality 290–300 mosmol/kg H2O). Whole-cell patch-clamp recordings from visualized CA1 pyramidal cells were performed in the slice as previously described (Müller et al., 2013) at room temperature using an upright microscope (Nikon Eclipse FN1, Düsseldorf, Germany) and a standard amplifier (ELC-03XS, npi electronic, Tamm Germany, run with signal 2.16 software, CED).
Signals were low-pass filtered at 2 kHz and digitized at a sampling frequency of 4 kHz. Only cells with an RMP of −50 mV or more negative were included in this study. The AHP-mediating K+ current and charge transfer were measured in the voltage-clamp mode as previously described (Brehme et al., 2014). In brief, the recording solution contained 500 nM tetrodotoxin (TTX) and 1 mM tetraethylammonium (TEA) (both from Tocris Bioscience, Bristol, United Kingdom) to block voltage-activated Na+ and K+ channels, respectively. AHP-mediating currents were elicited by a depolarizing voltage command from the holding potential −60 to +60 mV for 100 ms, followed by a step to −120 mV for 2 ms and a return to −60 mV for 2800 ms. The medium AHP (mAHP) was determined as the peak outward current during the first 400 ms following the short hyperpolarizing step to −120 mV (2 ms) and normalized to cell capacitance. The slow component of the AHP (sAHP) was assessed by determining the charge transfer, i.e., the area under the curve (AUC), for the interval between 400 and 2400 ms (i.e., 2000 ms), also normalized to cell capacitance. Low and fast capacitive current transients as well as leak currents were fully compensated and monitored (Cf and Cs). Series resistance ranged from 4 to 15 MΩ and was monitored at regular intervals throughout the experiments.
Intracellular recordings were performed in CA1 pyramidal cells impaled with borosilicate glass microelectrodes (80–120 MΩ, pulled with Sutter P-97 and filled with 3 M potassium acetate and 0.013 M KCl) using an SEC-10LX amplifier (npi electronic). In these recordings, RMP, membrane resistance, and membrane time constant were determined. The membrane resistance was calculated as the slope of the steady-state current–voltage curve obtained by hyperpolarizing current injection (ranging from −1.0 to −0.2 nA for 400 ms). The membrane time constant was calculated as the average time constant during the hyperpolarizing steps. Subsequent depolarizing current injection (from + 0.1 to + 0.5 nA) was used to evoke a train of action potentials (400 ms). The AHP following this prolonged depolarization (referred to as train-AHP) was calculated as the difference between the train-AHP peak amplitude and the RMP (for which the last sweep with + 0.5 nA depolarization was used). In addition, burst firing was tested with short depolarizing current steps (duration 3–7 ms, from + 0.5 to + 2.4 nA). Unless otherwise indicated, chemicals were purchased from Sigma–Aldrich (Taufkirchen, Germany).
Hippocampal horizontal slices were immediately transferred into ice-cold phosphate-buffered saline (PBS, pH = 7.4, PAA Laboratories GmbH, Austria), and the CA1 region was dissected out (referred to as mini-slice) under a binocular microscope (Leica). All mini-slices from one animal (n = 6–8) were pooled and immediately frozen in liquid nitrogen. For mRNA isolation, TRIZOL reagent was used, and total RNA was reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (final concentration [Cf] = 10 U/μl) and RNasin Plus RNase inhibitor (Cf = 2 U/μl, both Promega Corporation, Madison, WI, United States) in the presence of random hexamers (Cf = 0.01 μg/μl) and dNTP Mix (Cf = 0.5 nmol/μl each, Invitrogen, Carlsbad, CA, United States). For the real-time PCR of the target genes (KCa2.2, HCN1, HCN2, HCN3, and HCN4) as well as three standard reference genes (phosphoglycerate kinase 1, S18 ribosomal protein, 18S rRNA), we used the QuantiFast SYBR Green PCR Kit (concentration as recommended by the manufacturer, Qiagen Inc., Valencia, CA, United States). The mastermix was aliquoted, cDNA (Cf = 0.5 ng/μl) and primers (Cf = 0.5 pmol/μl; TIB Molbiol, Berlin, Germany) were added.
Real-time PCRs were performed using the ep mastercycler (software realplex 2.2, Eppendorf, Hamburg, Germany) with cycling parameters as follows: 95°C for 5 min once, followed by 95°C for 15 s and 60°C for 15 s, with normalized fluorescence read at 68°C (530 nm) for 40 cycles. To confirm single product amplification, melting curve analysis and gel electrophoresis were done. Expression levels of ion channel mRNA were normalized to all three housekeeping genes (TIB Molbiol, Berlin, Germany) and analyzed by 2–ΔΔCt method (i.e., normalized to control levels).
For CK2 western blot analysis, six control and six epileptic TBB-pretreated animals were used at age 133/134 days. Pools of mini-slices, each for CA1, CA3, and DG area from individual animals were subjected to protein extraction with RIPA buffer [50 mM Tris, 150 mM NaCl, 1% (v/v) TritonX-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA] in the presence of Complete protease inhibitor cocktail (Roche). After centrifugation at 14,000 g at 4°C for 20 min, 10 μg of total proteins from each sample were separated by 10% SDS-PAGE and blotted onto a PVDF membrane (Immobilon-FL; Millipore). Primary antibody incubation (mouse monoclonal anti-CK2, 1:2000) was done overnight at 4°C and secondary antibody incubation (goat anti-mouse IRDye 680RD, Odyssey, 1:5000) was done for 30 min at room temperature. A single specific protein band was visualized with the Odyssey infrared imaging scanner from Li-cor.
For quantification purposes, GAPDH was used as an internal control for CK2 normalization. Briefly: after CK2 detection, membranes were washed 15 min at 55°C with a buffer (pH 2.2) containing 1.5% glycine, 0.1% SDS, 1% Tween 20 to remove any bound anti-CK2 antibody. Then, according to CK2 immunoreaction, the rabbit monoclonal anti-GAPDH primary antibody (Cell Signaling; 1:5000) and a secondary antibody (goat anti-rabbit IRDye 800CW, Odyssey; 1:5000) was loaded. For each sample, CK2 and GAPDH fluorescence signals were determined with Image Studio lite software (Li-cor) and the ratio between CK2 and GAPDH was calculated.
Timm staining (Timm, 1958) which typically demonstrates the zinc-containing mossy fibers was done for confirmation of mossy fiber sprouting (MFS) using a previously published protocol (Müller et al., 2014). After incubation the brain section in disodium sulfide solution (9.7 mM Na2S, 19.8 mM NaH2PO4, in 500 ml distilled H2O) the sections were fixed in glutaraldehyde solution (100 ml PBS 0.15 M; 1.2 ml 25% glutaraldehyde) for 10 min and subsequently placed in a 30% sucrose solution at 4°C until the brain pieces sank to the bottom of the chamber. Transversal cryosections were cut at 20 μm using a freezing microtome (Leica CM 3050S, Leica Microsystems, Wetzlar, Germany). After washing for 15 min with PBS, the sections were processed in the dark for 30 min in developer solution (80 ml distilled H2O; 120 ml gum arabicum; 132.7 mM citric acid; 120.7 mM sodium citrate; 154.4 mM hydroquinone; only in the dark: 0.16 mM AgNO3 in distilled H2O) and washed with distilled H2O. After short incubation with the stop solution (63.25 mM Na2S2O3 in distilled H2O), the sections were again washed with distilled H2O. Finally, the nuclei were counter-stained (8.17 mM toluidine blue and 49.7 mM Na2B4O7 in distilled H2O). Following washing, the sections from the hippocampi were dehydrated in alcohol and mounted on slides with slide mounting medium (Fluoromount-GTM, Beckman-Coulter, Fullerton, CA, United States).
Mossy fiber sprouting was assessed as previously reported (Müller et al., 2014). Briefly, we used a semi-quantitative classification scheme performed by one investigator (TR) blinded with respect to other data as well as to the vehicle or TBB pretreatment (0 = no staining, 1 = patches of staining in the inner molecular layer, 2 = confluent staining, 3 = massive staining). Second, a morphometric analysis measuring the brownish-colored area within the inner molecular layer (MFS area) normalized to the granule cell area in the same slice to produce an MFS ratio (mean values of three sections per animal).
Unless otherwise indicated, data were expressed as mean values ± the standard error of the mean (SEM). For statistical comparisons, however, data were tested for normal distribution, and then analyses were performed using the appropriate test as indicated. The level of significance was set to P < 0.05, and significant differences were indicated with asterisks in all figures (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001).
The aim of this study was to investigate the acute and long-term effect of CK2 inhibition in vivo on the pilocarpine-induced SE and the post-SE chronic epilepsy. To this end, we administered TBB three days before pilocarpine-induced SE, i.e., four consecutive injections (Figure 1A). One hour after the last TBB injection, we started the pilocarpine procedure. Roughly 85% of the animals developed stage 4 or 5 seizures after a single pilocarpine injection. The proportions of rats developing seizures were not influenced by TBB pretreatment (Table 1). In contrast, the median latency of the first stage 4 or 5 seizure after pilocarpine was significantly prolonged from 16 min (Figure 1B) in vehicle-pretreated rats to 20 min in TBB-pretreated animals (P < 0.001, Mann–Whitney U-test; Figures 1C,D). Other parameters such as number of seizures before SE, diazepam dose, SE rate, and mortality did not differ between both groups (Table 1). Hence, TBB pretreatment prolonged the latency to the first generalized seizure, but did not alter SE rate or mortality.

Table 1. Status epilepticus (SE) in vivo data in vehicle-pretreated and TBB-pretreated epileptic animals.
Next, we asked whether pretreatment with TBB could influence epileptogenesis and thus we analyzed animals in their chronic stages of TLE, i.e., at a time-point of fully established epileptogenesis. To this end, we implanted a telemetric EEG system and counted the number of spikes/hour within 3 days (97–100 days post-SE; see timescale in Figure 1A) of four vehicle-pretreated and three TBB-pretreated epileptic animals. Overall, we found 314 ± 25 spikes/hour in vehicle-pretreated rats (in 304 animal hours), which was significantly higher than 168 ± 19 spikes/hour in TBB-pretreated rats (in 216 animal hours; P < 0.01, two-way ANOVA followed by Tukey test; Figure 1E). During this period, we detected 32 generalized seizures (23 in TBB-pretreated and nine in vehicle-pretreated animals; see diamonds in Figure 1E). Hence, the interictal spike counts were quite representative and not taken from a period with increased seizure rate. Since the clinical course of chronic epilepsy in this model is often associated with an increasing seizure rate and also with seizure clustering (Bajorat et al., 2011, 2016, 2018), this short-term analysis of EEG-detected seizures cannot reflect seizure burden and hence is insufficient to study the TBB effect.
Therefore, we performed a long-term video analysis of daytime seizures from nine vehicle-pretreated and 13 TBB-pretreated littermates for 7 weeks covering the full period of epileptogenesis (10–60 days after SE; Figures 1F–H). Over this period, vehicle-pretreated animals showed 1.3 ± 1.0 seizures/12 h (n = 9) during daylight, i.e., between 6 a.m. and 6 p.m., consistent with previous data (Bajorat et al., 2011, 2016, 2018). TBB-pretreated littermates showed the same overall seizure rates (1.3 ± 1.3 seizures/12 h,n = 13), but developed gross changes in seizure rates during the second half of the observation period (Figure 1F). Seizure clusters were present in 6/9 vehicle-pretreated animals, but only in 4/13 TBB-pretreated epileptic animals (P = 0.096, χ2 test). Disease progression assessed as the ratio of seizure rate during the second half of observation period over the baseline seizure rate was substantially higher in the vehicle-pretreated group as compared to TBB-pretreated animals (314 ± 99% versus 133 ± 24%, P = 0.062, t-test). Moreover, and probably more importantly, seizure rate variability was significantly reduced by TBB pretreatment (P < 0.01, F-test; Figure 1G), indicating that the seizure burden was stabilized in the cohort of TBB-treated epileptic rats. Overall, we observed a significant RRR of seizure burden by 58% (95% confidence interval 4–100%, P < 0.05, t-test, Figure 1H) indicating that disease progression following SE was slowed down in TBB-pretreated epileptic animals.
Next, we asked whether this disease modifying effect was associated with a reduced MFS, which is a pathological hallmark of TLE and commonly observed in this model (Schulz et al., 2012). However, MFS in TBB-pretreated epileptic rats did not differ from MFS in vehicle-pretreated epileptic animals (Figure 2).
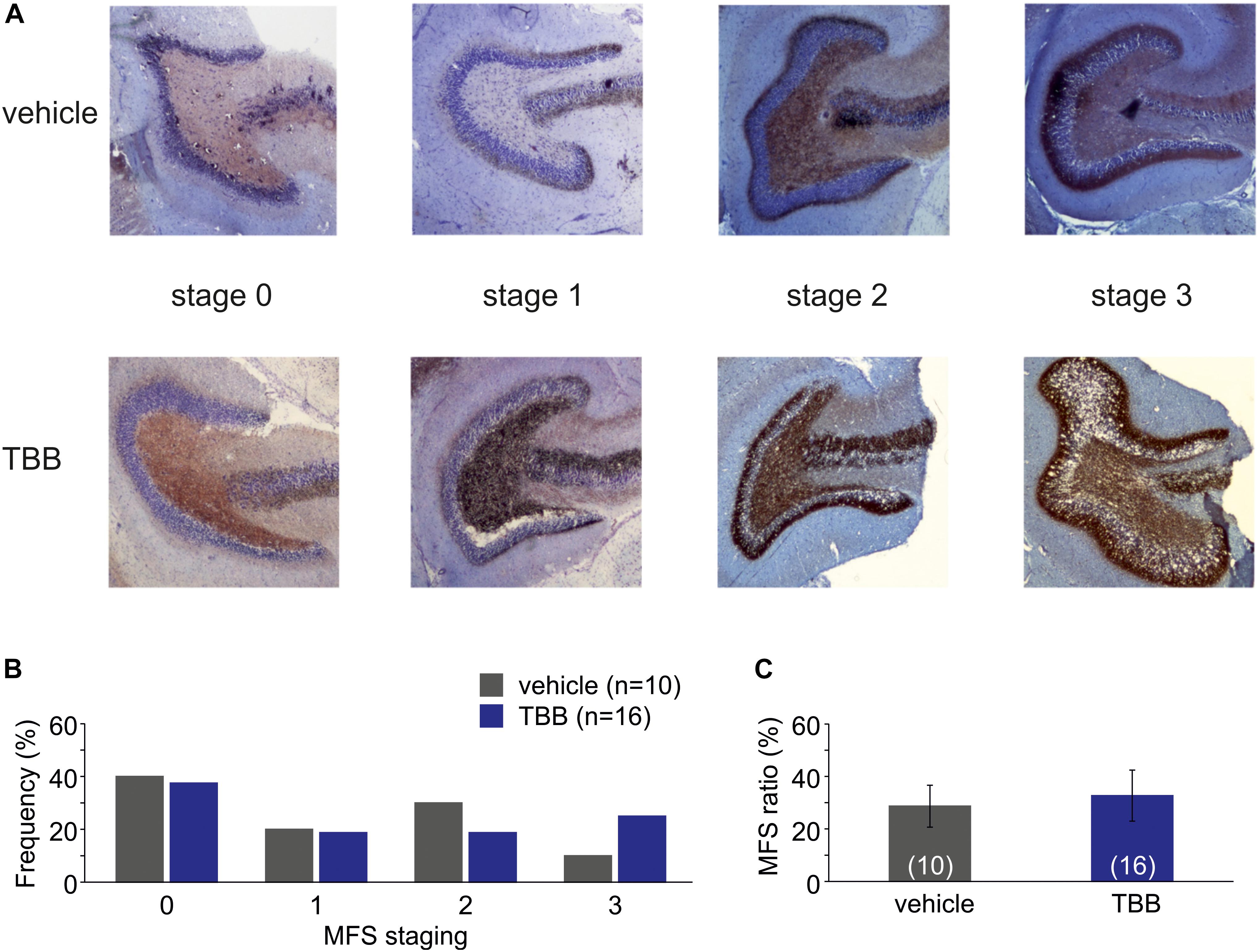
Figure 2. Mossy fiber sprouting (MFS). (A) Photomicrographs from vehicle-pretreated epileptic animals (upper row) and TBB-pretreated epileptic animals (lower row) demonstrate the MFS stages 0–3. (B) Relative frequencies of MFS stages 0–3 in vehicle- and TBB-pretreated animals. (C) MFS ratio (MFS area divided by granule cell area) in both experimental groups.
Our data so far indicate that in vivo TBB pretreatment affects pilocarpine-induced SE and exerts disease modifying properties during epileptogenesis. Since TBB was not continued after SE, we hypothesized that TBB leads to persistent changes in the epileptic, but probably also in the control tissue. In order to address this issue, we first analyzed extracellular field potentials in control and post-SE tissue, both taken from either vehicle- or TBB-pretreated animals. Basic properties of synaptic transmission such as input–output curves or paired-pulse plasticity were not altered by TBB (Figures 3A–C). In contrast, short-term plasticity protocols of five stimuli at increasing frequencies showed significantly higher potentiation in TBB-pretreated epileptic animals as compared to both vehicle-pretreated epileptic tissue as well as tissue from control rats (both comparisons: P < 0.05, three-way ANOVA followed by Holm–Sidak test; Figure 3D; no significant interaction between stimulus number and animal group or between stimulus number and pretreatment). After delivering the short-term plasticity protocols, the field potentials were significantly increased in tissue from TBB-pretreated epileptic animals (P < 0.05 versus baseline, Wilcoxon signed rank test; Figure 3E) indicating that some degree of long-term plasticity might have been elicited by these short protocols. Concomitantly with this potentiation, the PPR was significantly reduced (P < 0.05, Wilcoxon signed rank test; Figure 3F) suggesting presynaptic mechanism for this plasticity-facilitating effect of TBB in tissue from chronically epileptic rats. These results demonstrate that TBB may lead to persistent changes in the synaptic network of the CA1 area which may be even more pronounced in epileptic than in control tissue.
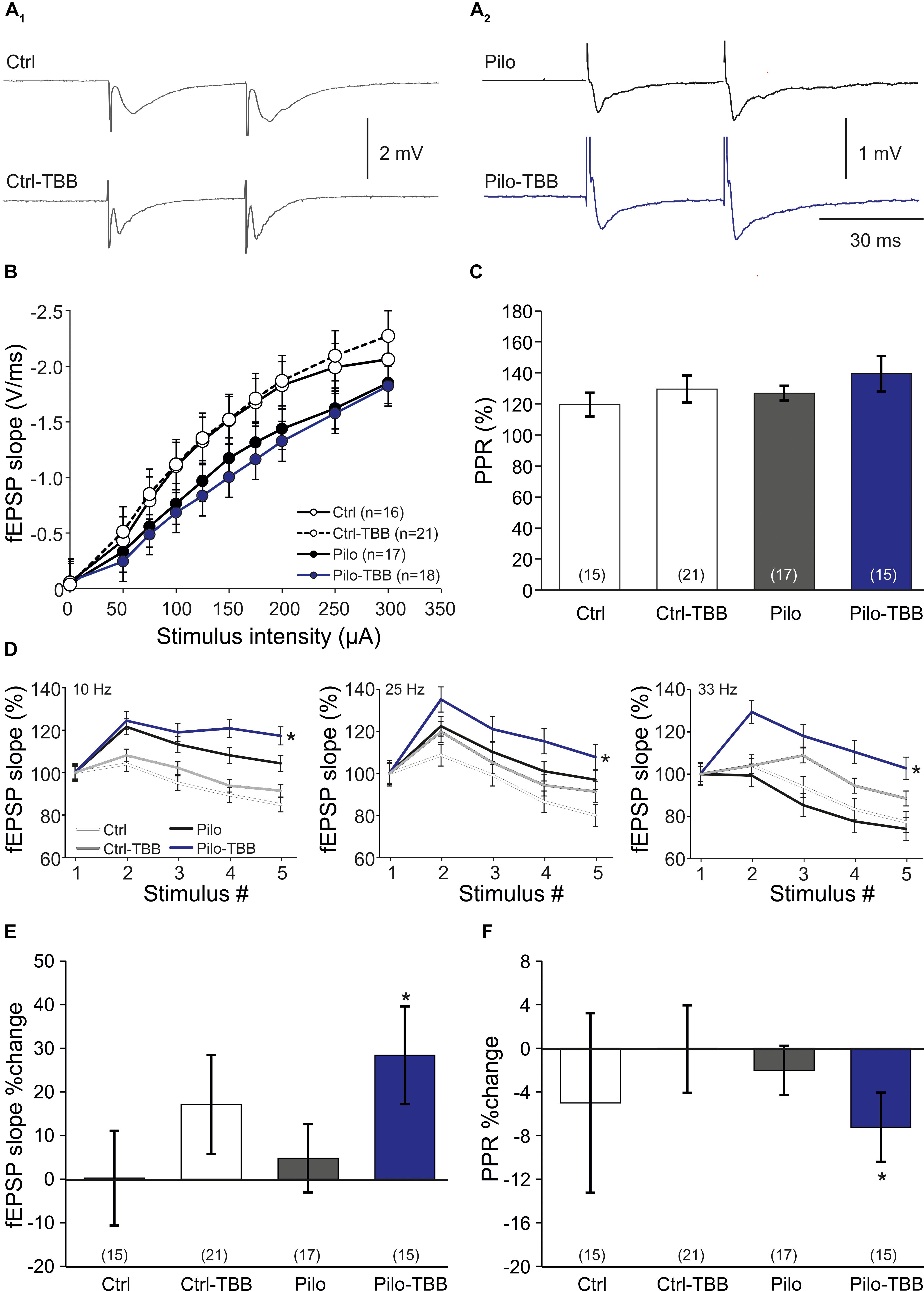
Figure 3. CK2 inhibition facilitates CA1 plasticity. (A) Sample traces of CA1 stratum radiatum field excitatory postsynaptic potentials (fEPSP) following Schaffer collateral stimulation in control (A1) and epileptic animals (A2). (B) Input–output curves of all four groups without significant group differences. (C) Paired-pulse ratios (PPRs) were similar among all four experimental groups. Short-term plasticity during five stimuli at frequencies of 10, 25, and 33 Hz (D), and subsequent changes of fEPSP slope (E) and PPR (F) after this short-term plasticity paradigm. Only slices from the TBB-pretreated epileptic group showed significantly facilitated plasticity. *P < 0.05.
One hallmark of chronically epileptic tissue is the presence of burst firing neurons, especially in CA1 (Chen et al., 2011). To study this issue in more detail, we performed intracellular recordings from CA1 neurons in vehicle- or TBB-pretreated control and epileptic animals. A typical burst firing behavior is depicted in Figure 4A, obtained from a neuron of a vehicle-pretreated animal (upper trace in gray), showing three action potentials for the lowest suprathreshold depolarizing current injection. In contrast, a regular firing behavior is shown in the lower trace in Figure 4A (blue trace), taken from a TBB-pretreated animal. Burst firing was observed in 4/9 neurons from vehicle-pretreated epileptic animals, but in 0/7 neurons from TBB-pretreated epileptic animals (P < 0.05, χ2 test; Figure 4B). In control rats (either vehicle- or TBB-pretreated), no burst firing neurons were observed (Figure 4B).
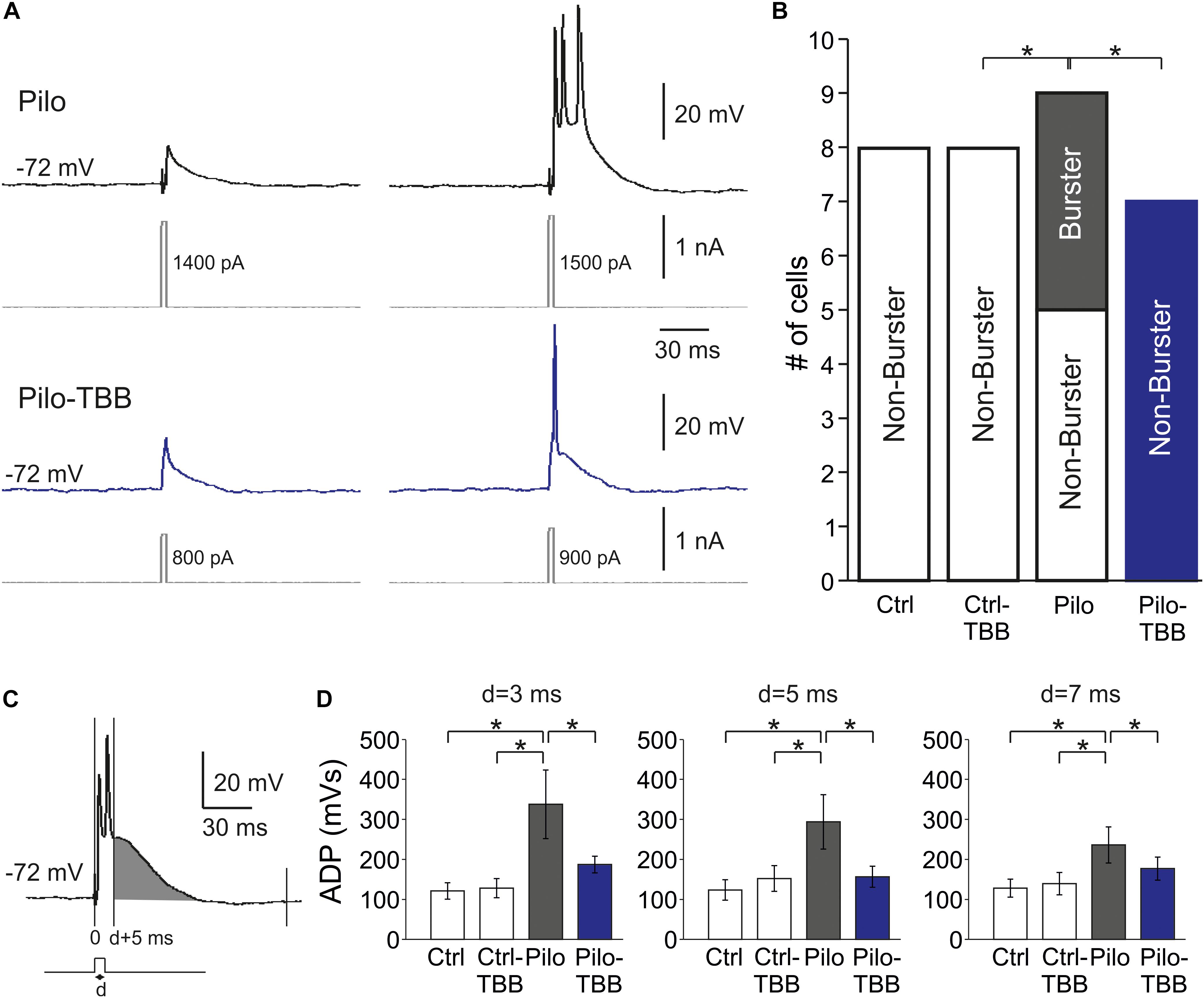
Figure 4. CK2 inhibition reduces bursting in CA1. (A) Sample traces of CA1 pyramidal cell intracellular recordings from a vehicle-pretreated epileptic rat (Pilo, black traces) and a TBB-pretreated epileptic rat (Pilo-TBB, blue traces). Note the bursting of the upper traces upon just suprathreshold stimulation. (B) Proportions of burster and non-burster neurons in the four experimental groups (*P < 0.05). (C,D) Afterdepolarizing potential (ADP) with different pulse durations (d = 3, 5, 7 ms). The area under curve of the ADP (starting from d + 5 ms, indicated in gray in C) of cells from vehicle-pretreated epileptic animals (Pilo) was significant against the ADP of all other experimental groups (*P < 0.05, three-way ANOVA with Tukey post hoc test).
Bursting behavior may be the consequence of a prolonged afterdepolarizing potential (ADP; Chen et al., 2011). We therefore determined the AUC of the ADP starting at 5 ms after the depolarizing step (Figure 4C). The duration of the depolarizing step (“d” in Figures 4C,D) was set to 3, 5, and 7 ms, and the ADP was significantly larger in the vehicle-pretreated epileptic group as compared to both TBB-pretreated epileptic and control groups (P < 0.05, three-way ANOVA; Figure 4D). Taken together, pilocarpine-induced SE caused a significant increase in ADP size probably involved in burst firing behavior which in turn was restored by TBB pretreatment in vivo prior to SE.
The significant reduction of the number of burst firing neurons in the epileptic CA1 after TBB pretreatment following ADP normalization is an attractive molecular mechanism for the observed disease modifying properties in vivo. Since we previously demonstrated that KCa2.2 was downregulated in isolated CA1 neurons from chronically epileptic tissue and TBB administered during chronic epileptic stages enhanced KCa2.2 levels in CA1 from post-SE animals (Schulz et al., 2012; Bajorat et al., 2018), we first performed patch-clamp recordings from visualized CA1 pyramidal neurons in hippocampal slices and confirmed the impaired function of KCa2 by recording the mAHP-mediating current density (0.26 ± 0.06 pA/pF,n = 7 versus 1.19 ± 0.38 pA/pF,n = 4, P < 0.05, unpaired t-test, Figure 5A) as previously reported in isolated cells (Schulz et al., 2012). Pretreatment with TBB prior to SE significantly enhanced the mAHP current density (0.92 ± 0.18 pA/pF,n = 14, P < 0.05, unpaired t-test; Figure 5A) suggesting that TBB might indeed enhance KCa2 function. We next analyzed the sAHP, for which the underlying ion channels are less clearly determined, but do not seem to involve KCa2 (Bond et al., 2004; Villalobos et al., 2004; Pedarzani et al., 2005). Consistent with this, the sAHP charge transfer as assessed by determining the AUC of the sAHP-mediating current density was significantly reduced in CA1 neurons from chronically epileptic rats as compared to control tissue (P < 0.05, two-way ANOVA), but without any significant TBB effect (Figure 5B).
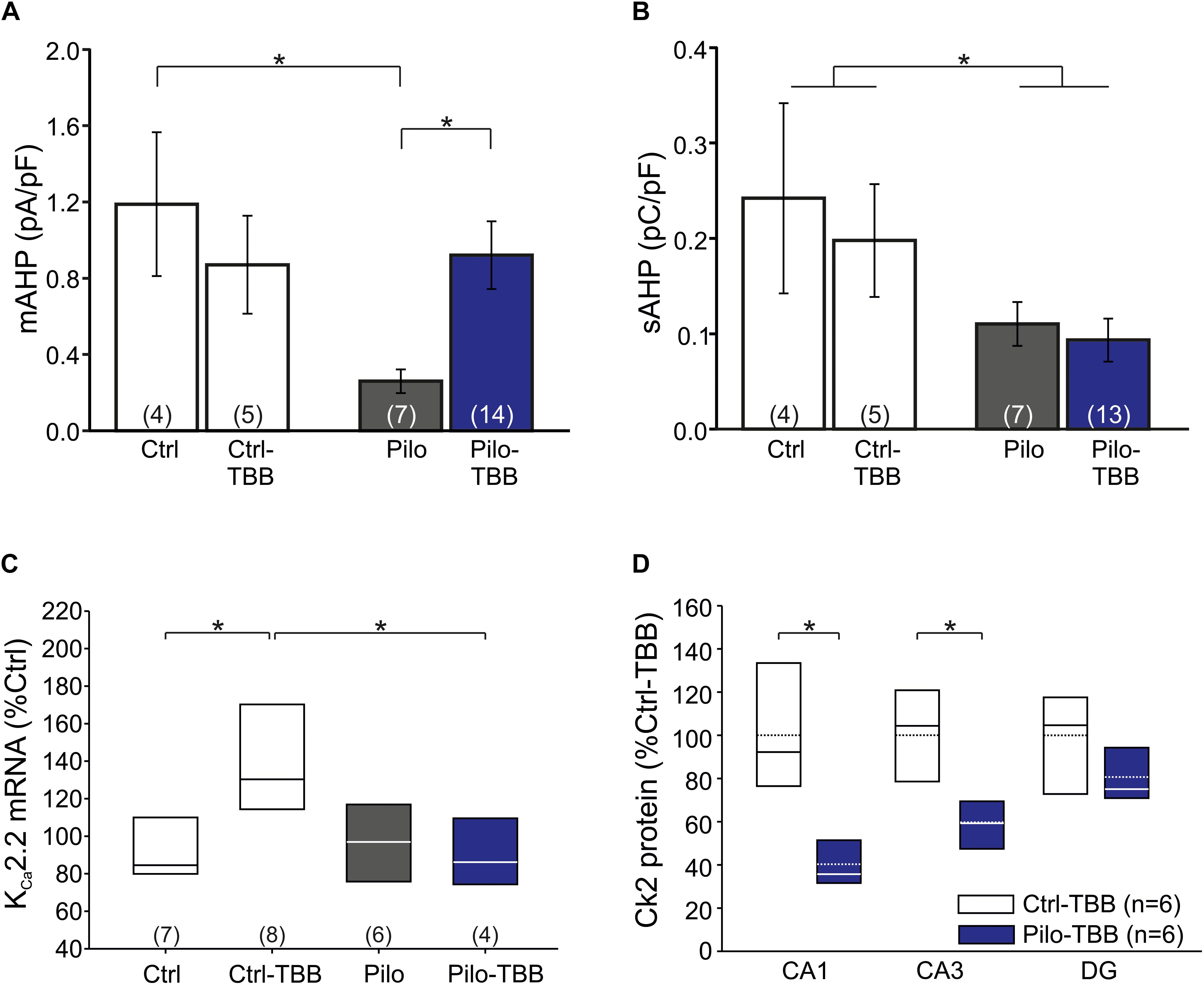
Figure 5. CK2 inhibition modifies KC2 function. (A) Bar graph of the mAHP-mediating current in all four experimental groups. CA1 cells from vehicle-pretreated epileptic animals (Pilo) showed significantly less mAHP currents than control cells and cells from TBB-pretreated epileptic animals. (B) The sAHP charge transfer is significantly smaller in epileptic animals than in controls. TBB has no effect on the sAHP charge transfer. (C) Quantitative RT-PCR analysis of the CA1 subfield reveals that tissue from TBB-pretreated control animals contained significantly more KCa2.2 mRNA than vehicle-pretreated controls and TBB-pretreated epileptic rats. (D) Western blot analysis of CK2 protein in TBB-pretreated tissue. Note the region-specific differences between control and epileptic tissues. *P < 0.05.
From these findings so far, it is conceivable that KCa2 function is reduced in chronically epileptic CA1 neurons, and we speculated that TBB might upregulate KCa2.2 gene expression (Bajorat et al., 2018). We, therefore, performed real-time RT-PCR and, as expected, KCa2.2 mRNA was significantly upregulated in TBB-pretreated control animals (P < 0.05, ANOVA and Duncan post hoc test; Figure 5C). More importantly, in TBB-pretreated animals, SE was associated with a significant reduction of KCa2.2 expression as previously observed (Schulz et al., 2012; P < 0.05, ANOVA and Duncan post hoc test; Figure 5C). However, mRNA levels of KCa2.2 were not different between Pilo and Pilo-TBB groups. Hence, the TBB effect on the mAHP in epileptic tissue cannot be explained by simple upregulation of KCa2.2 in CA1 neurons. Based on the idea that KCa2.2 function is reduced by CK2 activity (Bildl et al., 2004; Allen et al., 2007), we performed Western blotting of CK2 protein in six control and six epileptic TBB-pretreated animals (Figure 5D). These analyses indeed showed a region-specific lack of CK2 protein in epileptic animals which was most pronounced in CA1 (40 ± 5% of Ctrl-TBB tissue, P < 0.002, t-test; Figure 5D) suggesting a persistent downregulation of this enzyme in this area as a potential mechanism for the disease modifying effect.
The findings so far point to an impaired function of KCa2 in the epileptic CA1 area. Nonetheless, some data appeared inconclusive. Therefore, we asked whether the AHP which is a prominent KCa2.2 function (Hotson and Prince, 1980; Bond et al., 2004) might be altered following SE and TBB pretreatment. To this end, we performed intracellular recordings from CA1 pyramidal cells in vehicle- and TBB-pretreated epileptic and control animals (Figure 6A). Basic passive membrane properties such as RMP, membrane resistance and time constant were not different in these four groups (Table 2). With respect to the AHP, we found that the AHP following a train of action potentials (train-AHP) was significantly reduced in epileptic CA1 neurons (−4.2 ± 0.6 mV,n = 10 versus −6.8 ± 1.0 mV,n = 9, P < 0.05, two-way ANOVA with Tukey post hoc test; Figures 6A,B) consistent with a previous report (Schulz et al., 2012). However, TBB pretreatment had no significant effect on these values (Figure 6B). Instead, we observed a prominent voltage sag in the individual voltage traces in cells from controls (gray traces in Figure 6A) as well as TBB-pretreated epileptic rats (blue traces in Figure 6A), but not from vehicle-pretreated epileptic rats (black traces in Figure 6A) suggesting that TBB might modify HCN channel function. Indeed, there was a significant enhancement of the voltage sag following TBB pretreatment in both control (3.0 ± 1.4 mV,n = 10 versus 2.1 ± 1.0 mV,n = 10, P < 0.05, two-way ANOVA with Tukey post hoc test; Figure 6B) and epileptic animals (3.1 ± 2.9 mV,n = 9 versus 2.0 ± 2.1 mV,n = 10, two-way ANOVA with Tukey post hoc test; Figure 6B). Thus, we repeated the gene expression analysis with all HCN isoforms (HCN1-4; Figures 6C–F). In control animals, TBB led to differential alteration of HCN transcription with a significant upregulation of HCN1 (288 ± 27%,n = 8, P < 0.05, ANOVA with Duncan post hoc test; Figure 6C) and concomitantly, a significant downregulation of HCN3 (51 ± 10%,n = 8, P < 0.05, ANOVA with Duncan post hoc test; Figure 6E). Interestingly, epileptic animals showed similar differential alterations when compared to controls (HCN1 up-regulated to 187 ± 9% and HCN3 downregulated to 28 ± 8%,n = 6, P < 0.05, ANOVA with Duncan post hoc test; Figures 6C,E). In epileptic animals, however, TBB pretreatment before SE did not significantly alter HCN1 or HCN3 expression levels (Figures 6C,E), and for the other two isoforms, HCN2 and HCN4, no significant changes were observed at all (Figures 6D,F). Taken together, this RT-PCR analysis indicates that TBB leads to differential expression changes of HCN channels which might have contaminated our analyses on KCa2 channel function.
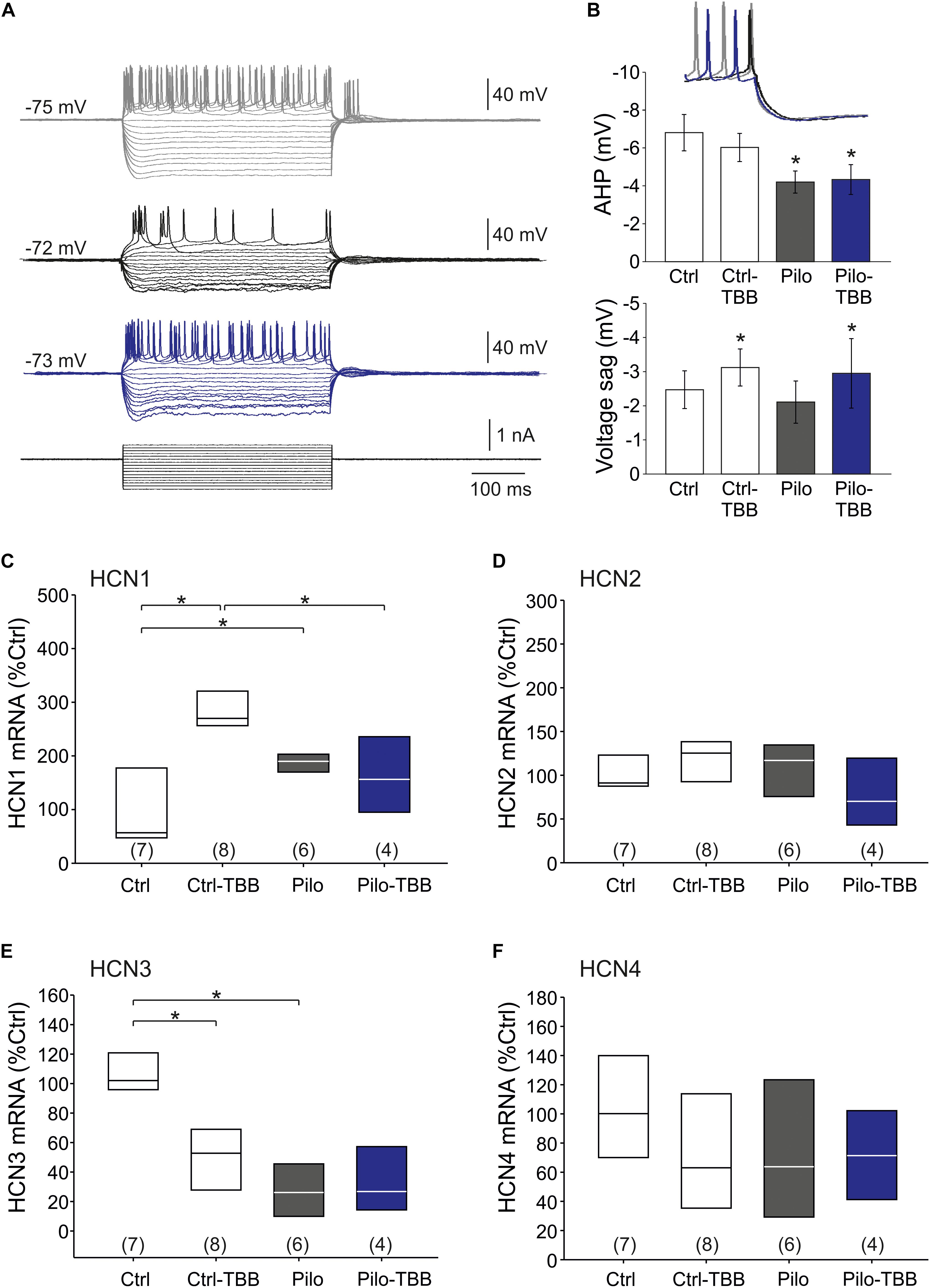
Figure 6. CK2 inhibition modifies HCN channel function. (A) Sample traces of the membrane potential following hyperpolarizing and depolarizing pulse steps in intracellular recordings from CA1 neurons (gray: control, black: vehicle-pretreated epileptic, blue: TBB-pretreated epileptic). (B) Bar graphs of the train-AHP amplitude and voltage sag obtained from recordings shown in A. The train-AHP was significantly smaller in epileptic tissues regardless of the treatment with TBB. In contrast, the voltage sag was enhanced in TBB-pretreated tissue from both control and epileptic animals. Quantitative RT-PCR analysis of the CA1 subfield of all four HCN channel isoforms revealed significant and inverse expression changes for HCN1 (C) and HCN3 (E), but no changes for HCN2 (D) and HCN4 (F). *P < 0.05.

Table 2. Intrinsic cellular parameters.
In order to rule out contamination of differential HCN expression changes following TBB pretreatment, we repeated intracellular recordings in the presence of the HCN channel blocker ZD7288 (20 μM, Figure 7). Under these conditions, the proportion of burst firing neurons in the epileptic CA1 area was reduced, but still present (two of six cells) and no longer significant when compared to CA1 neurons from TBB-pretreated epileptic rats (zero of six cells, Figure 7A) suggesting that both HCN channel and KCa2 function contribute to bursting behavior. However, the power of this analysis is insufficient to dissect a differential TBB effect on those channels with respect to bursting, in particular with respect to other candidate mechanisms such as persistent sodium currents (Chen et al., 2011). Intriguingly, in contrast to the train-AHP analysis above in the absence of ZD7288, prolonged depolarization led to a markedly increased train-AHP in TBB-pretreated epileptic animals under HCN-blocking conditions (see blue traces and blue arrow in Figure 7B). On average, the train-AHP in this group was −5.7 ± 1.5 mV (n = 6, blue bar in Figure 7C) and thus matched with control levels (−5.8 ± 1.0 mV,n = 5). The statistical comparison by three-way ANOVA revealed a significant animal effect (Ctrl versus Pilo, P < 0.05, ANOVA with Holm–Sidak post hoc test) and significant pretreatment effect (vehicle versus TBB, P < 0.05, ANOVA with Holm–Sidak post hoc test), but no significant effect of ZD7288 (Figure 7C). However, there was a significant interaction between ZD7288 and pretreatment with TBB or vehicle (P < 0.05, three-way ANOVA) indicating that the ZD7288 effect depends on the type of pretreatment. The sample traces in Figure 7B also suggested an impaired spike frequency adaptation in CA1 neurons from vehicle-treated epileptic animals when studied in the presence of ZD7288 (black traces in Figure 7B). As shown in Figure 7D, the mean number of action potentials during the 400 ms pulse was substantially higher in this group, and two-way ANOVA showed a trend effect by animal (Ctrl versus Pilo, P = 0.08, indicated by “+,” ANOVA with Tukey post hoc test) and a significant effect by treatment (vehicle versus TBB, P < 0.05, ANOVA with Tukey post hoc test). Hence, TBB appears to modify both KCa2 and HCN channel function with partly opposing effects. We conclude that under HCN-blocking conditions, TBB treatment prior to SE leads to persistent increase of the train-AHP amplitude and spike frequency adaptation in epileptic animals, both of which are prominent KCa2 functions.
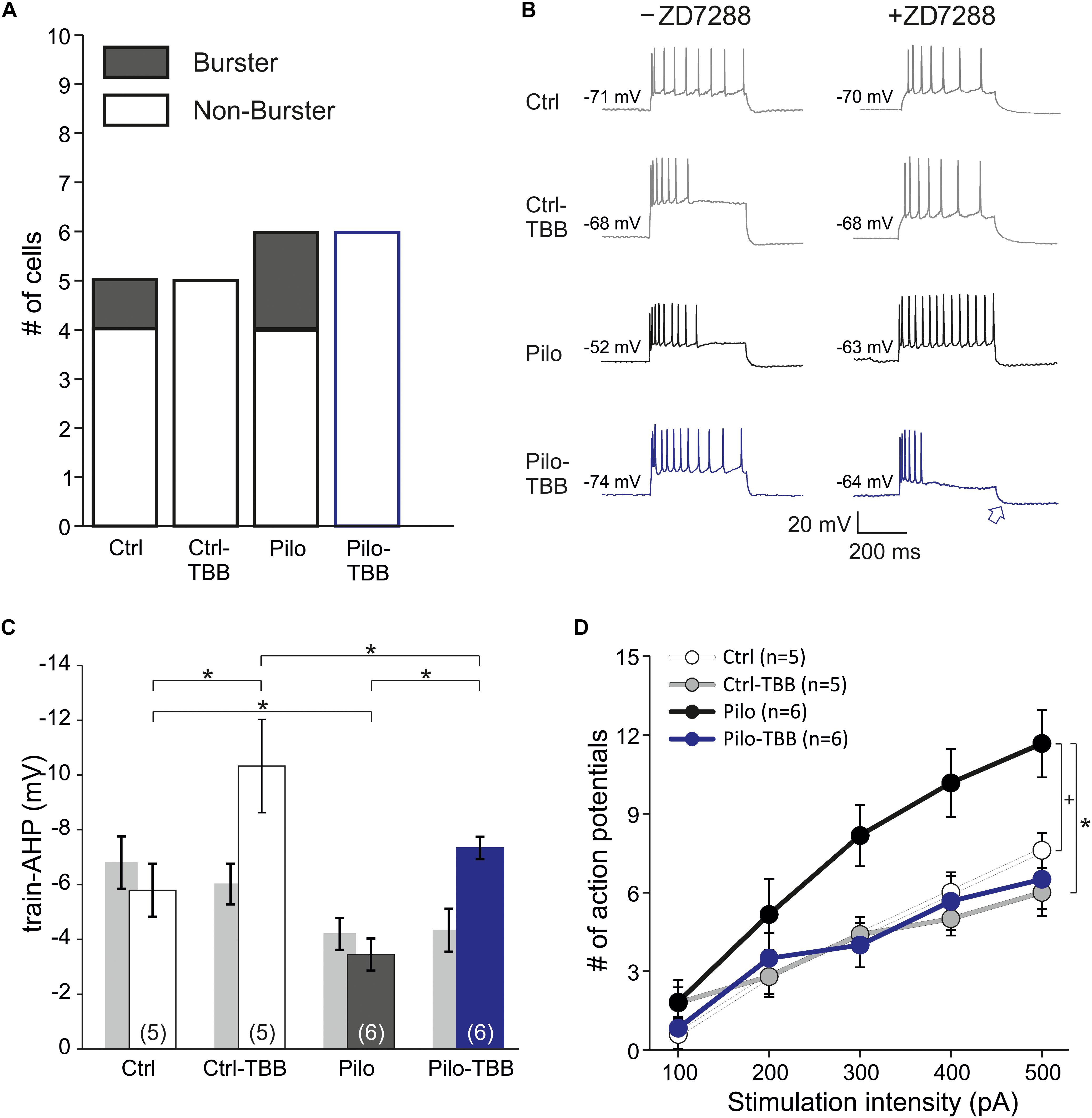
Figure 7. CK2 inhibition enhances train-AHP and reduces excitability in HCN-blocking conditions. (A) Proportions of burster and non-burster neurons in the four experimental groups in the presence of the HCN channel blocker ZD7288. (B) Sample traces of intracellular recordings following prolonged depolarization (600 ms) used for train-AHP amplitude (e.g., see blue arrow) and number of action potentials. For comparison, sample traces in the absence of ZD7288 were also given (left side). (C) Bar graph of the train-AHP amplitude shows a significantly increased train-AHP in cells from TBB-pretreated epileptic rats (blue bar graph). For the sake of clarity, the train-AHP in the absence of ZD7288 (gray bar graphs, values from Figure 5B) are also shown. Significant effects were detected between animal groups (Ctrl versus Pilo, *P < 0.05) and pretreatment groups (vehicle versus TBB, *P < 0.05), but not for the ZD7288 effect by three-way ANOVA with Holm–Sidak post hoc tests. However, there was a significant interaction between ZD7288 and pretreatment with TBB or vehicle (*P < 0.05). (D) Number of action potentials during 600 ms depolarization (from 100 to 500 pA). Two-way ANOVA with Tukey post hoc tests showed a trend effect between animal groups (Ctrl versus Pilo, +P = 0.08) and a significant effect treatment groups (vehicle versus TBB, *P < 0.05).
In the present study, we investigated the effects of a short-term in vivo CK2 inhibition with TBB prior to pilocarpine-induced SE. First, we found that the first generalized seizure after pilocarpine injection was delayed, and moreover, the epileptic phenotype as assessed by EEG spike and video seizure rate analyses was altered in TBB-pretreated epileptic animals. Interestingly, the typical seizure cluster pattern observed in almost half of the chronically epileptic rats in this model (Bajorat et al., 2011, 2016, 2018) was less frequently detected in the TBB-pretreated group, and TBB slowed down disease progression during epileptogenesis. Since TBB was not effective as an anti-seizure drug when administered after epileptogenesis (Bajorat et al., 2018), these data indicate that TBB has potential disease modifying properties. In addition, Western blot analysis demonstrated that CK2 protein abundance was significantly reduced in TBB-pretreated epileptic tissue, especially in CA1. Interestingly, water maze training significantly reduced CK2 activity in the CA1 area, but not in the dentate gyrus (Chao et al., 2007) indicating region-specific regulation of this enzyme. This might be relevant since CA1 long-term potentiation was found to be increased after water maze training (Rehberg et al., 2017).
If CK2 is less abundant, what might be the consequences in the epileptic CA1 neuron? CaM is phosphorylated by CK2 and, in turn, dephosphorylated by protein phosphatase 2A (Bildl et al., 2004; Allen et al., 2007). The phosphomimetic T80D-CaM species decreased Ca2+ sensitivity and reduced KCa2 function (Allen et al., 2007). Hence, decreased CK2 activity and concomitantly predominant activity of protein phosphatase 2A would increase KCa2 function without necessarily altering transcription. In the present study, we observed that TBB increased KCa2.2 transcription in control (Bajorat et al., 2018), but not in epileptic animals. This is an intriguing finding probably indicating that epileptogenesis could have reversed the KCa2.2 upregulation. Although we have not performed a serial analysis of KCa2.2 transcription directly after SE as well as during epileptogenesis, we assume that ongoing epileptic activity might decrease KCa2 function since chronically epileptic rats have significantly less KCa2.2 protein (Schulz et al., 2012). However, we did not detect a significant transcriptional downregulation of KCa2.2 in vehicle-pretreated epileptic rats as found in our previous paper (Schulz et al., 2012), because we used three different housekeeping genes and averaged the results. Hence, the TBB-induced upregulation of KCa2.2 in control tissue was indeed confirmed (Bajorat et al., 2018), but reduced KCa2.2 protein levels in chronically epileptic tissue (Schulz et al., 2012) are probably due to internalization and degradation observed at potentiated synapses (Lin et al., 2008) as well as after epileptiform activity (Kernig et al., 2012; Müller et al., 2018) rather than transcriptional downregulation. Furthermore, it is important to note that the KCa2.3 isoform which may also be relevant in CA1 and dentate gyrus (Ballesteros-Merino et al., 2014) was not found to be reduced in the chronically epileptic CA1 (Schulz et al., 2012) suggesting that the presumed reduction of CK2 activity should enhance KCa2.3 function as well. Taken together, our findings are consistent with a persistently diminished CK2 activity leading to enhanced KCa2 function in CA1 as a potentially relevant mechanism for the disease modifying effect of TBB.
Are there alternative molecular pathways for the disease modifying effect of TBB? On the one hand, we found an increased number of burst-spiking neurons in CA1 of vehicle-pretreated, but not in CA1 of TBB-pretreated epileptic rats. The ADP and burst-firing behavior have been recognized as common intrinsic properties of epileptic tissue, especially in the subiculum and CA1 (Magee and Carruth, 1999; Su et al., 2001; Yue and Yaari, 2004; van Welie et al., 2006; Jarsky et al., 2008). Based on this literature, the ADP was demonstrated to be due to persistent Na+ and Ni+-sensitive Ca2+ currents, rather than Ca2+-activated K+ currents (Magee and Carruth, 1999; Su et al., 2001). However, pharmacological inhibition of KCNQ/M-type K+ currents increased spike ADP and burst-spiking in CA1 (Yue and Yaari, 2004) suggesting that K+ currents can substantially modify these intrinsic firing properties. Thus, the effect of TBB pretreatment to prevent burst-firing behavior might indicate that Ca2+-activated K+ channels and probably further ion channels have been modified by TBB pretreatment. In this respect, it is noteworthy that functional HCN3 upregulation by phosphatidylinositol-4,5-bisphosphate facilitated low-threshold burst-firing in thalamic neurons (Ying et al., 2011). Hence, one might speculate whether the HCN3 downregulation found in TBB-pretreated epileptic tissue could have exerted the opposite effect and inhibited the development of burst-firing. On the other hand, we observed at least one control burster cell in the presence of ZD7288, and increased excitability has been associated with downregulation of HCN channels in chronically epileptic CA1 (Adams et al., 2009). Nonetheless, there is also uncertainty about the selectivity of TBB which will inhibit other kinases, too (Pagano et al., 2008). CK2 has a pretty large number of substrates (Meggio and Pinna, 2003), and recently, CK2 was demonstrated to phosphorylate histone chaperone Spt6 that is associated with RNA polymerase II and therefore promotes DNA transcription (Dronamraju et al., 2018). Since CK2 is an oncogenic enzyme involved in epigenetic processes (reviewed by Gowda et al., 2019), further targets need to be unraveled. Here, we focused on KCa2 channel-mediated mechanisms.
On the network level, we did not observe significant changes of basal synaptic transmission after TBB pretreatment indicating that a potential KCa2-mediated reduction of the EPSP (Hammond et al., 2006) did not occur in our hands because postsynaptic Ca2+ increase during basal transmission was quite low. Instead, short-term plasticity was significantly facilitated and PPRs were changed in potentiated synapses in TBB-pretreated epileptic animals. These findings might point to a presynaptic mechanism, but KCa2.2 channels were excluded to have relevant presynaptic functions (Hammond et al., 2006).
Hence, what is the evidence for an enhanced KCa2 function in TBB-pretreated epileptic CA1? Intrinsic cellular properties such as the mAHP and the spike frequency adaptation are well-established KCa2 functions of CA1 neurons (Bond et al., 2004; Pedarzani et al., 2005; Hammond et al., 2006; Schulz et al., 2012). First, we could demonstrate that mAHP-mediating K+ currents were significantly enhanced by TBB pretreatment in tissue from epileptic animals. On the other hand, the sAHP was also reduced in epileptic CA1 neurons, but not altered by TBB treatment. This is consistent with the view that KCa2 is not involved in sAHP (Stocker et al., 1999; Bond et al., 2004; Villalobos et al., 2004; Pedarzani et al., 2005), but the molecular sAHP-mediating candidates are less clear and may certainly involve HCN channels (Gu et al., 2005; Kaczorowski, 2011). Importantly, we observed that HCN channel function was enhanced in TBB-pretreated epileptic tissue. In addition, the transcriptional analysis revealed HCN1 upregulation and HCN3 downregulation in epileptic CA1. This is in contrast to previous reports showing HCN1 downregulation in the pilocarpine model (Jung et al., 2007, 2010), but more recently, HCN1 immunoreactivity was found be increased, especially in CA1 interneurons (Oh et al., 2012). Moreover, HCN1 upregulation was also found in the epileptic dentate gyrus (Bender et al., 2003; Surges et al., 2012). Hence, HCN isoform expression and protein abundance may depend on the cell type. Nonetheless, since pharmacological HCN channel inhibition by ZD7288 that does not distinguish between HCN isoforms led to an increase of sAHP-mediating currents (Gu et al., 2005), we propose that a net enhancement of HCN channel function by TBB could have antagonized an expected TBB-mediated sAHP increase due to enhanced KCa2 function. In fact, we previously found that the sAHP was increased in control and epileptic tissue when animals were pretreated in vivo with TBB (Brehme et al., 2014). On the one hand, this could be simply due to different recording procedures (isolated cells versus slice experiments) that differ with respect to recording from dendritic compartments expressing high levels of HCN1 (Nolan et al., 2004). On the other hand, however, this discrepancy may indicate that enhanced HCN channel function following CK2 inhibition may occur with delayed time course and, probably, activity-dependent (Shin and Chetkovich, 2007). This is an attractive mechanism to be addressed in further studies.
Second, we addressed KCa2 function under HCN channel-blocking conditions. In the presence of the non-selective inhibitor ZD7288, the train-AHP recorded with intracellular electrodes was significantly reduced in vehicle-treated epileptic tissue, but could be enhanced by TBB pretreatment in both control and epileptic animals. This is an important finding, because HCN inhibition shifts the RMP to hyperpolarizing values which should rather decrease the driving force for KCa2 channels. On the other hand, membrane resistance also depends on HCN channel function (Surges et al., 2004), and ZD7288 increased the resistance—and thus could have counteracted the probably reduced K+ currents. Hence, the enhanced train-AHP recorded intracellularly confirmed the membrane current recordings obtained with the patch-clamp method.
Third, KCa2 channels also play a major role in spike frequency adaptation (Pedarzani et al., 2005). In the present study, we studied spike frequency adaptation in HCN channel-blocking conditions and could demonstrate that this phenomenon was impaired in chronically epileptic tissue confirming previous results in isolated neurons (Schulz et al., 2012). Moreover, TBB restored spike frequency adaptation in epileptic tissue which can be viewed as an important antiepileptic phenomenon. In summary, our data strongly suggest that CA1 neurons from TBB-pretreated epileptic animals show a significantly enhanced KCa2 function that likely contributes to disease modification.
Our experiments demonstrated that TBB pretreatment prior to SE led to reduced epileptogenesis and to differential gene expression changes of KCa2.2, but also of HCN1 and HCN3 channels. Under HCN channel blocking conditions, pretreatment with TBB could significantly enhance KCa2 channel functions such as the AHP and the spike frequency adaptation in epileptic animals, probably due to a persistently decreased CK2 protein abundance.
The datasets generated for this study are available on request to the corresponding author.
The animal study was reviewed and approved by Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern (numbers M-V/TSD/7221.3-1.1-013/10 and 7221.3-1.1-019/13).
FS, SM, XG, LS, HB, and TR performed the experiments. FS, TK, and RK contributed to conception and design of the study. FS, SR, AE, and RK organized the database. FS, SM, LS, MR, DG, TK, and RK performed the statistical analysis. TK wrote the first draft of the manuscript. FS, SM, MR, and RK wrote the sections of the manuscript. LS, MR, and DG contributed to manuscript preparation. All authors contributed to manuscript revision and read and approved the final version of this manuscript for submission.
This work was supported by the German Research Foundation (KO 1779/14-1 and KI 1283/5-1) and the Intramural Research Program of the Medical Faculty of the University of Rostock.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Tina Sellmann, Katrin Porath, Hanka Schmidt, Bernd Memmener, and Andreas Prestel for their excellent technical assistance.
Adams, B. E., Reid, C. A., Myers, D., Ng, C., Powell, K., Phillips, A. M., et al. (2009). Excitotoxic-mediated transcriptional decreases in HCN2 channel function increase network excitability in CA1. Exp. Neurol. 219, 249–257. doi: 10.1016/j.expneurol.2009.05.030
Allen, D., Fakler, B., Maylie, J., and Adelman, J. P. (2007). Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J. Neurosci. 27, 2369–2376. doi: 10.1523/jneurosci.3565-06.2007
Bajorat, R., Goerss, D., Brenndörfer, L., Schwabe, L., Köhling, R., and Kirschstein, T. (2016). Interplay between interictal spikes and behavioral seizures in young, but not aged pilocarpine-treated epileptic rats. Epilepsy Behav. 57, 90–94. doi: 10.1016/j.yebeh.2016.01.014
Bajorat, R., Porath, K., Kuhn, J., Goßla, E., Goerss, D., Sellmann, T., et al. (2018). Oral administration of the casein kinase 2 inhibitor TBB leads to persistent KCa2.2 channel up-regulation in the epileptic CA1 area and cortex, but lacks anti-seizure efficacy in the pilocarpine epilepsy model. Epilepsy Res. 147, 42–50. doi: 10.1016/j.eplepsyres.2018.08.012
Bajorat, R., Wilde, M., Sellmann, T., Kirschstein, T., and Köhling, R. (2011). Seizure frequency in pilocarpine-treated rats is independent of circadian rhythm. Epilepsia 52, e118–e122. doi: 10.1111/j.1528-1167.2011.03200.x
Ballesteros-Merino, C., Watanabe, M., Shigemoto, R., Fukazawa, Y., Adelman, J. P., and Luján, R. (2014). Differential subcellular localization of SK3-containing channels in the hippocampus. Eur. J. Neurosci. 39, 883–892. doi: 10.1111/ejn.12474
Bender, R. A., Soleymani, S. V., Brewster, A. L., Nguyen, S. T., Beck, H., Mathern, G. W., et al. (2003). Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J. Neurosci. 23, 6826–6836. doi: 10.1523/jneurosci.23-17-06826.2003
Bernard, C., Anderson, A., Becker, A., Poolos, N. P., Beck, H., and Johnston, D. (2004). Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305, 532–535. doi: 10.1126/science.1097065
Bildl, W., Strassmaier, T., Thurm, H., Andersen, J., Eble, S., Oliver, D., et al. (2004). Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron 43, 847–858. doi: 10.1016/j.neuron.2004.08.033
Blümcke, I., Thom, M., Aronica, E., Armstrong, D. D., Bartolomei, F., Bernasconi, A., et al. (2013). International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia 54, 1315–1329. doi: 10.1111/epi.12220
Bond, C. T., Herson, P. S., Strassmaier, T., Hammond, R., Stackman, R., Maylie, J., et al. (2004). Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J. Neurosci. 24, 5301–5306. doi: 10.1523/jneurosci.0182-04.2004
Brehme, H., Kirschstein, T., Schulz, R., and Köhling, R. (2014). In vivo treatment with the casein kinase 2 inhibitor 4,5,6,7- tetrabromotriazole augments the slow afterhyperpolarizing potential and prevents acute epileptiform activity. Epilepsia 55, 175–183. doi: 10.1111/epi.12474
Chao, C. C., Ma, Y. L., and Lee, E. H. (2007). Protein kinase CK2 impairs spatial memory formation through differential cross talk with PI-3 kinase signaling: activation of Akt and inactivation of SGK1. J. Neurosci. 27, 6243–6248. doi: 10.1523/jneurosci.1531-07.2007
Chen, S., Su, H., Yue, C., Remy, S., Royeck, M., Sochivko, D., et al. (2011). An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J. Neurophysiol. 105, 117–129. doi: 10.1152/jn.00184.2010
Dronamraju, R., Kerschner, J. L., Peck, S. A., Hepperla, A. J., Adams, A. T., Hughes, K. D., et al. (2018). Casein kinase II phosphorylation of Spt6 enforces transcriptional fidelity by maintaining Spn1-Spt6 interaction. Cell Rep. 25:3476-3489.e5. doi: 10.1016/j.celrep.2018.11.089
Gowda, C., Song, C., Ding, Y., Iyer, S., Dhanyamraju, P. K., McGrath, M., et al. (2019). Cellular signaling and epigenetic regulation of gene expression in leukemia. Adv. Biol. Regul. doi: 10.1016/j.jbior.2019.100665 [Epub ahead of print].
Gu, N., Vervaeke, K., Hu, H., and Storm, J. F. (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J. Physiol. 566, 689–715. doi: 10.1113/jphysiol.2005.086835
Hammond, R. S., Bond, C. T., Strassmaier, T., Ngo-Anh, T. J., Adelman, J. P., Maylie, J., et al. (2006). Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J. Neurosci. 26, 1844–1853. doi: 10.1523/jneurosci.4106-05.2006
Hotson, J. R., and Prince, D. A. (1980). A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. J. Neurophysiol. 43, 409–519.
Jarsky, T., Mady, R., Kennedy, B., and Spruston, N. (2008). Distribution of bursting neurons in the CA1 region and the subiculum of the rat hippocampus. J. Comp. Neurol. 506, 535–547. doi: 10.1002/cne.21564
Jung, S., Bullis, J. B., Lau, I. H., Jones, T. D., Warner, L. N., and Poolos, N. P. (2010). Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signaling. J. Neurosci. 30, 6678–6688. doi: 10.1523/JNEUROSCI.1290-10.2010
Jung, S., Jones, T. D., Lugo, J. N. Jr., Sheerin, A. H., Miller, J. W., D’Ambrosio, R., et al. (2007). Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J. Neurosci. 27, 13012–13021. doi: 10.1523/jneurosci.3605-07.2007
Kaczorowski, C. C. (2011). Bidirectional pattern-specific plasticity of the slow afterhyperpolarization in rats: role for high-voltage activated Ca2+ channels and Ih. Eur. J. Neurosci. 34, 1756–1765. doi: 10.1111/j.1460-9568.2011.07899.x
Kernig, K., Kirschstein, T., Würdemann, T., Rohde, M., and Köhling, R. (2012). The afterhyperpolarizing potential following a train of action potentials is suppressed in an acute epilepsy model in the rat Cornu Ammonis 1 area. Neuroscience 201, 288–296. doi: 10.1016/j.neuroscience.2011.11.008
Kirschstein, T., Bauer, M., Müller, L., Rüschenschmidt, C., Reitze, M., Becker, A. J., et al. (2007). Loss of metabotropic glutamate receptor-dependent long-term depression via downregulation of mGluR5 after status epilepticus. J. Neurosci. 27, 7696–7704. doi: 10.1523/jneurosci.4572-06.2007
Kwan, P., and Brodie, M. J. (2000). Early identification of refractory epilepsy. N. Engl. J. Med. 342, 314–319. doi: 10.1056/nejm200002033420503
Leite, J. P., Garcia-Cairasco, N., and Cavalheiro, E. A. (2002). New insights from the use of pilocarpine and kainate models. Epilepsy Res. 50, 93–103. doi: 10.1016/s0920-1211(02)00072-4
Lin, M. T., Luján, R., Watanabe, M., Adelman, J. P., and Maylie, J. (2008). SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat. Neurosci. 11, 170–177. doi: 10.1038/nn2041
Magee, J. C., and Carruth, M. (1999). Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 82, 1895–1901. doi: 10.1152/jn.1999.82.4.1895
Meggio, F., and Pinna, L. A. (2003). One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–368. doi: 10.1096/fj.02-0473rev
Müller, L., Müller, S., Sellmann, T., Groeneweg, L., Tokay, T., Köhling, R., et al. (2014). Effects of oxygen insufflation during pilocarpine-induced status epilepticus on mortality, tissue damage and seizures. Epilepsy Res. 108, 90–97. doi: 10.1016/j.eplepsyres.2013.10.017
Müller, L., Tokay, T., Porath, K., Köhling, R., and Kirschstein, T. (2013). Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 54, 183–193. doi: 10.1016/j.nbd.2012.12.011
Müller, S., Guli, X., Hey, J., Einsle, A., Pfanz, D., Sudmann, V., et al. (2018). Acute epileptiform activity induced by gabazine involves proteasomal rather than lysosomal degradation of KCa2.2 channels. Neurobiol. Dis. 112, 79–84. doi: 10.1016/j.nbd.2018.01.005
Nolan, M. F., Malleret, G., Dudman, J. T., Buhl, D. L., Santoro, B., Gibbs, E., et al. (2004). A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119, 719–732. doi: 10.1016/s0092-8674(04)01055-4
Oh, Y. J., Na, J., Jeong, J. H., Park, D. K., Park, K. H., Ko, J. S., et al. (2012). Alterations in hyperpolarization-activated cyclic nucleotidegated cation channel (HCN) expression in the hippocampus following pilocarpine-induced status epilepticus. BMB Rep. 45, 635–640. doi: 10.5483/bmbrep.2012.45.11.091
Pagano, M. A., Bain, J., Kazimierczuk, Z., Sarno, S., Ruzzene, M., Di Maira, G., et al. (2008). The selectivity of inhibitors of protein kinase CK2: an update. Biochem. J. 415, 353–365. doi: 10.1042/BJ20080309
Pedarzani, P., McCutcheon, J. E., Rogge, G., Jensen, B. S., Christophersen, P., Hougaard, C., et al. (2005). Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current I(AHP) and modulates the firing properties of hippocampal pyramidal neurons. J. Biol. Chem. 280, 41404–41411. doi: 10.1074/jbc.m509610200
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294. doi: 10.1016/0013-4694(72)90177-0
Rehberg, M., Kirschstein, T., Guli, X., Müller, S., Rohde, M., Franz, D., et al. (2017). Functional metaplasticity of hippocampal schaffer collateral-CA1 synapses is reversed in chronically epileptic rats. Neural Plast. 2017:8087401. doi: 10.1155/2017/8087401
Schmidt, D., and Sillanpää, M. (2016). Prevention of Epilepsy: Issues and Innovations. Curr. Neurol. Neurosci. Rep. 16:95. doi: 10.1007/s11910-016-0695-9
Schulz, R., Kirschstein, T., Brehme, H., Porath, K., Mikkat, U., and Köhling, R. (2012). Network excitability in a model of chronic temporal lobe epilepsy critically depends on SK channel-mediated AHP currents. Neurobiol. Dis. 45, 337–347. doi: 10.1016/j.nbd.2011.08.019
Shin, M., and Chetkovich, D. M. (2007). Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J. Biol. Chem. 282, 33168–33180. doi: 10.1074/jbc.m703736200
Stocker, M., Krause, M., and Pedarzani, P. (1999). An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 4662–4667. doi: 10.1073/pnas.96.8.4662
Su, H., Alroy, G., Kirson, E. D., and Yaari, Y. (2001). Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J. Neurosci. 21, 4173–4182. doi: 10.1523/jneurosci.21-12-04173.2001
Surges, R., Freiman, T. M., and Feuerstein, T. J. (2004). Input resistance is voltage dependent due to activation of Ih channels in rat CA1 pyramidal cells. J. Neurosci. Res. 76, 475–480. doi: 10.1002/jnr.20075
Surges, R., Kukley, M., Brewster, A., Rüschenschmidt, C., Schramm, J., Baram, T. Z., et al. (2012). Hyperpolarization-activated cation current Ih of dentate gyrus granule cells is upregulated in human and rat temporal lobe epilepsy. Biochem. Biophys. Res. Commun. 420, 156–160. doi: 10.1016/j.bbrc.2012.02.133
Timm, F. (1958). Histochemistry of heavy metals; the sulfide-silver procedure [Zur Histochemie der Schwermetalle. Das Sulfid-Silberverfahren]. Dtsch. Z. Gesamte Gerichtl. Med. 46, 706–711.
van Welie, I., Remme, M. W., van Hooft, J. A., and Wadman, W. J. (2006). Different levels of Ih determine distinct temporal integration in bursting and regular-spiking neurons in rat subiculum. J. Physiol. 576, 203–214. doi: 10.1113/jphysiol.2006.113944
Villalobos, C., Shakkottai, V. G., Chandy, K. G., Michelhaugh, S. K., and Andrade, R. (2004). SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J. Neurosci. 24, 3537–3542. doi: 10.1523/jneurosci.0380-04.2004
Ying, S. W., Tibbs, G. R., Picollo, A., Abbas, S. Y., Sanford, R. L., Accardi, A., et al. (2011). PIP2-mediated HCN3 channel gating is crucial for rhythmic burst firing in thalamic intergeniculate leaflet neurons. J. Neurosci. 31, 10412–10423. doi: 10.1523/JNEUROSCI.0021-11.2011
Keywords: temporal lobe epilepsy, patch-clamp, intracellular recording, field potential recording, video-EEG monitoring, Timm stain
Citation: Schulze F, Müller S, Guli X, Schumann L, Brehme H, Riffert T, Rohde M, Goerss D, Rackow S, Einsle A, Kirschstein T and Köhling R (2020) CK2 Inhibition Prior to Status Epilepticus Persistently Enhances KCa2 Function in CA1 Which Slows Down Disease Progression. Front. Cell. Neurosci. 14:33. doi: 10.3389/fncel.2020.00033
Received: 08 August 2019; Accepted: 04 February 2020;
Published: 26 February 2020.
Edited by:
Tycho Hoogland, Erasmus Medical Center, NetherlandsReviewed by:
Fang Zheng, University of Arkansas for Medical Sciences, United StatesCopyright © 2020 Schulze, Müller, Guli, Schumann, Brehme, Riffert, Rohde, Goerss, Rackow, Einsle, Kirschstein and Köhling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timo Kirschstein, dGltby5raXJzY2hzdGVpbkB1bmktcm9zdG9jay5kZQ==; Rüdiger Köhling, cnVlZGlnZXIua29laGxpbmdAdW5pLXJvc3RvY2suZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.