 Yang Shen1Mao Ding1Zhaohong Xie2
Yang Shen1Mao Ding1Zhaohong Xie2 Xiangtian Liu1Hui Yang2
Xiangtian Liu1Hui Yang2 Suqin Jin2
Suqin Jin2 Shunliang Xu2Zhengyu Zhu2Yun Wang2Dewei Wang2Linlin Xu2Xiaoyan Zhou2
Shunliang Xu2Zhengyu Zhu2Yun Wang2Dewei Wang2Linlin Xu2Xiaoyan Zhou2 Ping Wang2*
Ping Wang2* Jianzhong Bi2*
Jianzhong Bi2*- 1Medicine School, Shandong University, Jinan, China
- 2Department of Neurology Medicine, Second Hospital of Shandong University, Jinan, China
Alzheimer disease (AD) is the most common form of dementia. Amyloid β-peptide (Aβ) deposition is a major neuropathologic feature of AD. When unfolded or misfolded proteins accumulate in mitochondria, the unfolded protein responses (UPRmt) is initiated. Numerous lines of evidence show that AD pathogenesis involves mitochondrial dysfunction. However little is known about whether the UPRmt is engaged in the process of AD development. In this study, we investigated the UPRmt in mouse and cell models of AD. We found that UPRmt was activated in the brain of 3 and 9 months old APP/PS1 mice, and in the SHSY5Y cells after exposure to Aβ25–35, Aβ25–35 triggered UPRmt in SHSY5Y cells could be attenuated upon administration of simvastatin or siRNA for HMGCS-1 to inhibit the mevalonate pathway, and or upon knocking down Serine palmitoyltransferase long chain subunit 1 (SPTLC-1) to lower sphingolipid biosynthesis. We observed that inhibition of UPRmt aggravated cytotoxic effects of Aβ25–35 in SHSY5Y cells. Our research suggests that the UPRmt activation and two pathways necessary for this response, and further provides evidence for the cytoprotective effect of UPRmt during the AD process.
Introduction
Alzheimer disease (AD) is the most common form of dementia in the elderly population and characterized by progressive deterioration of cognitive and functional abilities (Pleckaityte, 2010). Extracellular amyloid plaques, consisting of polymers of amyloid-β peptides (Aβ), and intracellular neurofibrillary tangles, formed mainly by hyperphosphorylated protein tau, are the main neuropathology of AD (Swerdlow, 2007). Diverse lines of evidence support that oligomeric species of Aβ are the causative agent of AD. Aβ is a general term to define 38–43 amino acid peptides generated from the sequential cleavage of the Amyloid Precursor Protein (APP) by β- and γ-secretase (Da Costa Dias et al., 2011). While Aβ1–40 and Aβ1–42 are the main forms of Aβ peptides deposited in extracellular amyloid plaques, Aβ1–42 is more prone to aggregation than Aβ1–40 (Selkoe, 2001). The APPsw/PS1dE9 double transgenic mouse line is widely used for investigating the pathogenic mechanisms of AD for its property of effective production of Aβ1–42 (Borchelt et al., 1996). Aβ25–35, a synthetic peptide of 11 amino acids, retains physical and biological properties of full-length Aβ and is often employed for generating acute AD models (Kang et al., 1987; Harkany et al., 2000).
Mitochondria are crucial cellular organelles essential for numerous cellular functions including energy homeostasis, metabolism, and apoptosis (Wang and Youle, 2009; Blackstone, 2015). Disruption of mitochondrial protein folding homeostasis results in the accumulation of unfolded or misfolded proteins and induce a mitochondria-to-nuclear signal transduction pathway termed the unfolded protein responses (UPRmt), through which upregulated expression of mitochondrial molecular chaperones and proteases is induced to re-establish protein homeostasis (Zhao et al., 2002; Tatsuta and Langer, 2008; Papa and Germain, 2011; Pellegrino et al., 2013). The accumulation of misfolded proteins has been described as a pathological hallmark of numerous neurodegenerative diseases, including AD, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis (Skovronsky et al., 2006). Although Aβ accumulation in mitochondria has long been known to exist in brain of AD patients as well as TgAPP mice (Lustbader et al., 2004; Caspersen et al., 2005; Manczak et al., 2006), little is known about whether the UPRmt is involved in the pathogenesis of AD. A couple of lines of evidence suggest a role for the UPRmt in the development of AD pathology. Levels of HtrA2/Omi (high temperature requirement protein A2/Omi), a protein operating in the process of UPRmt, are reduced in AD frontal cortex, yet its enzymatic activity is significantly increased in the same samples (Westerlund et al., 2011). In addition, there exists increased transcription and translation of UPRmt associated genes in the brain of familial and sporadic AD patients, e.g., upregulation of hspd1 (HSP60) and clpp (CLPP mitochondrial protease) genes, but not lon1p1 (the LONP1 mitochondrial protease; Beck et al., 2016).
The mevalonate pathway produces isoprenoids, which are vital for diverse cellular functions (Goldstein and Brown, 1990), and has been demonstrated to be required for the activation of the UPRmt in C. elegans. 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase is the rate-limiting enzyme in the mevalonate pathway; statins inhibit this enzyme to lower plasma cholesterol levels. Upon treatment with statins, C. elegans fail to sense mitochondrial damage and to activate the UPRmt (Liu et al., 2014; Ranji et al., 2014; Oks et al., 2018). Inactivation of the hmgs-1 gene, which encodes HMG-CoA synthase, renders C. elegans to lose the capability to respond to mitochondrial dysfunction and to inhibit antimycin-induced UPRmt induction (Liu et al., 2014). Human hydroxymethylglutaryl-CoA synthase 1 (HMGCS-1) is the ortholog of C. elegans HMGS-1 protein and mediates the first committed step of the mevalonate pathway (Sapir et al., 2014). We hypothesize that the mevalonate pathway participates in the activation of UPRmt in the process of AD development.
Sphingolipids are a class of lipids that are highly enriched in the central nervous system and play important functions in membrane structure, signal transduction, and a variety of biological processes (Spiegel and Merrill, 1996; Mielke and Haughey, 2012). Alterations in the sphingolipids metabolism are thought to be concerned with AD development. The rate-limiting enzyme of sphingolipid biosynthesis is serine palmitoyltransferase (SPT), a multiprotein complex catalyzing the first step of sphingolipid de novo synthesis pathway (Hanada, 2003; Hornemann et al., 2007). Serine palmitoyltransferase long chain subunit 1 (SPTLC-1) is one subunit of SPT (Hanada, 2003; Hornemann et al., 2006). Inactivation of the sptl-1 gene causes C. elegans unresponsive to mitochondrial dysfunction and inhibits antimycin-induced UPRmt induction (Liu et al., 2014). Hence, we speculate that the sphingolipids metabolism pathway also takes part in the UPRmt activation during the AD process.
In this study, we examined UPRmt related proteins levels in APP/PS1 mouse and SHSY5Y cells treated with Aβ to determine if the UPRmt contributes to AD pathogenesis. We exploited chemical drugs and small interfering RNAs to manipulate the mevalonate and sphingolipids biogenesis pathways, using drug or siRNA to evidence the involvement of these pathways in activating UPRmt.
Materials and Methods
Reagents and Preparation of Drugs
Amyloid β protein fragment 25–35 (Aβ25–35A4559) was purchased from Sigma-Aldrich. The Aβ25–35 was first dissolved in tri-distilled water to 1 mM and then incubated at 37°C for 7 days. The solution was aliquoted and stored at −20°C, until use. Simvastatin was purchased from MedChem Express (MCE, HY-17502).
Antibodies
Antibodies specific for APP (126732), HtrA2/Omi (75982), β-Actin (11132), CLPP (124822), GAPDH (9485), respectively, were purchased from Abcam. Antibodies for LONP1 (15440), Hsp60 (15282), HMGCS-1 (17643), SPTLC-1 (15376) were obtained from Proteintech.
Animals and Tissues
All procedures regarding the use of animals were conducted according to the guidelines and approved by the Ethical Committee for Animal Experiments of Shandong University. We used the APPsw/PS1dE9 double transgenic mice at the age of 3 and 9 months, age-matched C57BL/6 mice as a control. All mice were male (n = 10 per group) and purchased from Beijing HFK Bioscience Co., Limited (Beijing, China). When reaching the age of 3 and 9 months, mice were anesthetized by 10% of chloral hydrate and then sacrificed by cervical dislocation and decapitation. Fresh hippocampal tissues were harvested and stored at −80°C until use for studies. All applicable Shandong University and the ethical committee of the Second Hospital of Shandong University guidelines for the care and use of animals were followed.
Cell Culture and Treatments
The human neuroblastoma cell line SHSY5Y was obtained from the Cell Resource Center, IBMS, CAMS/PUMC. Cells were cultured in RPMI-1640 medium (HyClone) supplemented with 10% fetal bovine serum (Gibco) at 37°C and 5% C02. Cells at 50–70% confluence were treated with Aβ25–35, and or simvastatin. Control cells were cultured under normal conditions.
Cell Viability Assay
Cells were seeded into 96-well plates. After culturing for 24 h, cells were treated with simvastatin or SPTLC-1 siRNA for 48 h and then treated with Aβ25–35 for another 4 h. After treatments, cells in each well were incubated with Cell Counting Kit 8 (CCK8) solution (MedCjemExpress) at 37°C for 3 h and then used for measuring the absorbance at 450 nm with a microplate reader (Thermo, Multiskan MK3, USA). Experiments were repeated for at least three times.
SDS-PAGE and Western Blotting
The Hippocampal tissues and cells after treatments were homogenized in RIPA lysis buffer (Beyotime Biotechnology). Cell debris and nuclei were discarded after a centrifugation at 4°C 12,000 g for 10 min. The supernatants were collected and the protein concentration was measured with the bicinchoninic acid (BCA) method. After SDS-PAGE, proteins were blotted onto polyvinylidene difluoride (PVDF) membranes. Blots were blocked in 5% skimmed milk powder in TBST and incubated with corresponding primary antibodies at 4°C for 12–16 h. After extensive washes in TBST, blots were incubated with HRP conjugated secondary antibodies for 1–2 h at room temperature. Proteins were detected with ECL (enhanced chemiluminescence) regents.
Transmission Electron Microscopy
Cells were collected and fixed in pre-cooled glutaraldehyde for at least 2 h. After postfixation in osmic acid solutions and sequential dehydration, cells were embedded in EPON812 resins. Ultrathin sections were cut and collected onto grids, stained with uranium and lead citrate, and observed under a JEM-1200EX electron microscope (JEOL, Tokyo, Japan).
Measurement of ROS Levels
Intracellular reactive oxygen species (ROS) levels were detected using DCFH-DA (Beyotime, S0033), which is non-fluorescent and generates fluorescent signals after being oxidized into DCF in the presence of intracellular ROS. The day before experiments, cells were planted into 12-well plates and cultured for 24 h. After brief washes in PBS, cells were incubated with DCFH-DA (10 μmol/L) at 37°C for 20 min, then washed three times with RPMI-1640 medium. Fluorescence images were captured using a fluorescence microscope (OLYMPUS BX43) at excitation and emission wavelengths of 488 and 525 nm. The amount of ROS was quantified with the use of a fluorescence microplate reader. All experiments were repeated for at least three times.
Transfection of siRNAs
SHSY5Y cells were transfected with target siRNAs using INTERFRERin (Polyplus-transfection Inc., New York, NY, USA). Corresponding scrambled siRNAs were used as controls. After transfection with siRNA for 48 h, cells were treated with or without Aβ as indicated and then collected cells for examinations.
Statistical Analysis
All data were expressed as mean ± SEM. One-way analysis of variance (ANOVA) or Two-tailed student’s t-test was used for determining statistical significance. Data were analyzed using SPSS 20.0 (SPSS Inc., Chicago, IL, USA). A p-value of less than 0.05 was considered to be statistically significant.
Results
Aβ Activates UPRmt Responses
To determine whether Aβ25–35 treatment can cause the above UPRmt reactions, we treated SHSY5Y cells with different concentrations of Aβ25–35 for 24 h and then examined expression levels of UPRmt related proteins. We found that expression levels of mitochondrial matrix chaperone Hsp60, AAA proteases CLPP and the IMS-localized quality control protease HtrA2/Omi were increased after exposure to 2.5, 10 and 20 μM Aβ25–35 for 24 h. Levels of Hsp60 and CLPP, but not HtrA2/Omi, were also significantly increased in cells treated with 5 μM Aβ25–35 for 24 h. Under the same conditions, the upregulation of the LONP1 protein was detected only in cells exposed to 10 and 20 μM Aβ25–35 (Figure 1A). We then investigated whether treatment with 20 μM Aβ25–35 for a shorter time period could sufficiently activate UPRmt. Our data showed that the expression levels of all the four proteins mentioned above were elevated in SHSY5Y cells after exposure to 20 μM Aβ25–35 for 4 h (Figure 1B).
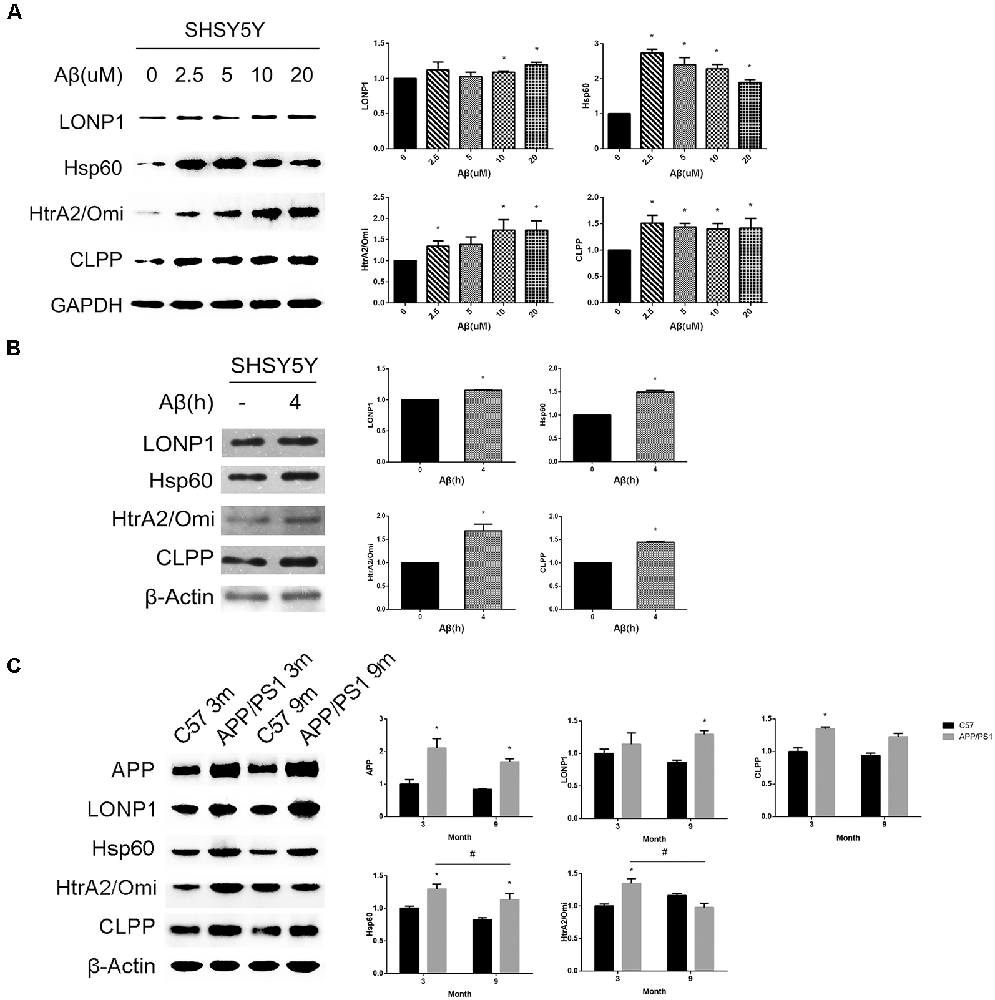
Figure 1. UPRmt response activated during the Alzheimer disease (AD) process in vitro and in vivo. (A) Western blot analysis of UPRmt related proteins level in SHSY5Y cells treated with different concentrations of Aβ25–35 for 24 h and quantification, GAPDH served as the internal control. (B) Western blot analysis of UPRmt related proteins level in SHSY5Y cells treated with 20 μM Aβ25–35 for 4 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. control group). (C) Western blot analysis of UPRmt related proteins level in C57 and APP/PS1 transgenic mice at 3 and 9 months, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 10, *p < 0.05 vs. age-matched C57 mice group, #p < 0.05).
To determine whether the UPRmt occurred in the brain of APPsw/PS1dE9 double transgenic mice, we first conducted biochemical studies to compare expression levels of UPRmt related proteins in hippocampi of 3 and 9 months old wildtype (WT) and APPsw/PS1dE9 transgenic mice, levels of APP in hippocampal lysates of both 3 and 9 months old transgenic mice was significantly increased compared with those in hippocampal lysates of age-matched WT mice (Figure 1C). Western blot analysis revealed that expression levels of Hsp60, CLPP and HtrA2/Omi were significantly increased in the hippocampus of 3 months old APPsw/PS1dE9 transgenic mice relative to those of 3 months old WT mice (Figure 1C). Under the same conditions, levels of the LONP1 protein were comparable in hippocampi of 3 months old WT and transgenic mice. However, levels of HtrA2/Omi and CLPP in hippocampal lysates of 9 months old transgenic mice were not different from those in hippocampal lysates of 9 months old WT mice, whereas levels of LONP1 and Hsp60 in the hippocampus of 9 months old transgenic mice were increased compared with those of age-matched control mice (Figure 1C). We also found that the expression levels of Hsp60 and HtrA2/Omi were significantly decreased in the hippocampus of 9 months old APPsw/PS1dE9 transgenic mice relative to those of 3 months old transgenic mice. Taken together, our in vitro and in vivo data suggest that UPRmt occurs in the process of AD pathological development.
The Mevalonate Pathway is Involved in Aβ25–35 Evoked UPRmt in SHSY5Y Cells
There was upregulated expression of HMGCS-1, an enzyme that mediates the first committed step of the mevalonate pathway, in cells treated with 20 μM Aβ25–35 for 4 h (Figure 2A). This observation led us to speculate that the mevalonate pathway might be involved in the activation of the UPRmt in cells treated with Aβ. To test this possibility, we treated SHSY5Y cells with HMGCS-1 siRNAs or with simvastatin to manipulate the mevalonate pathway. The expression level of HMGCS-1 was decreased in cells transfected with HMGCS-1 siRNAs for 48 h, compared with the HMGCS-1 level in cells transfected with scrambled siRNA. As shown in Figure 2B, in the absence of Aβ, there was no statistically significant difference in UPRmt related protein levels between cells transfected with scrambled siRNA and target siRNA. However, we found that after transfected with HMGCS-1 siRNAs, the expression levels of LONP1, Hsp60, HtrA2/Omi, and CLPP were significantly down-regulated in cells treated with 20 μM Aβ for 4 h (Figure 2C). We then detected whether simvastatin treatment could similarly dampen the effect of Aβ25–35 on the activation of the UPRmt reaction. We first treated cells with or without 1 μM or 10 μM of simvastatin for 2 h, 6 h, and 24 h, and then added 20 μM Aβ25–35 to cell culture media to treat cells for 4 h. The data in Figure 2D showed that except for the treatment with 1 μM simvastatin for 24 h, all other treatment conditions significantly decreased the expression of Hsp60 and HtrA2/Omi compared with the vehicle treatment. Compared with those in vehicle-treated cells, levels of CLPP in all of the simvastatin treatment conditions were decreased. Except for the group that 1 μM simvastatin treatment for 6 h and 24 h, expression of Lonp1 protein in other groups were decreased compared with control (Figure 2D). From the above, we can see that inhibition of the mevalonate pathway induced by HMGCS-1 siRNAs or simvastatin reduce the expression of UPRmt related proteins in SHSY5Y cells treated with Aβ25–35.
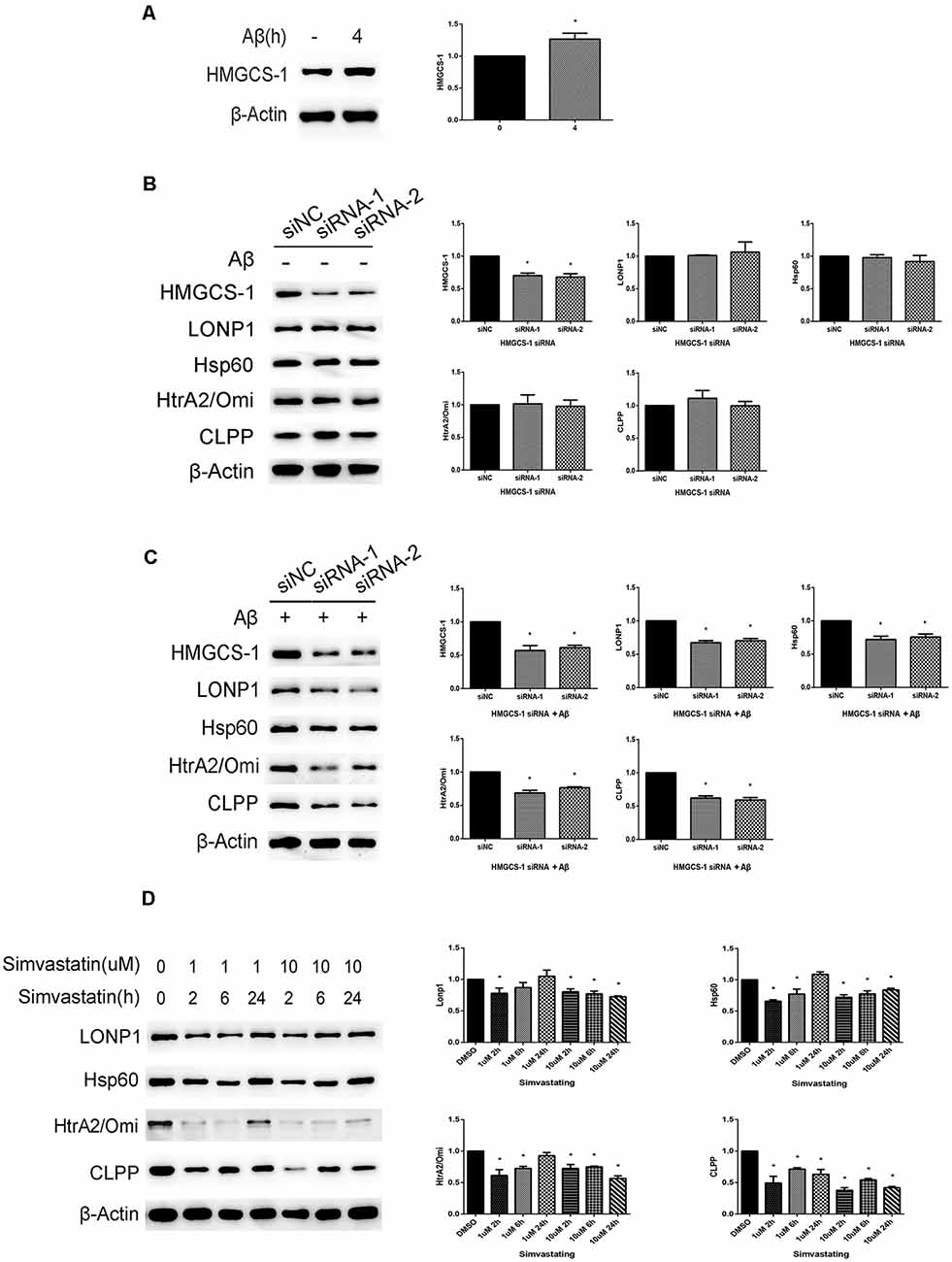
Figure 2. Inhibition of the mevalonate pathway by HMGCS-1 siRNAs or simvastatin reduces the UPRmt reaction induced by Aβ25–35 in SHSY5Y cells. (A) Western blot analysis of HMGCS-1 protein level in SHSY5Y cells treated with 20 μM Aβ25–35 for 4 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. control group). (B) Western blot analysis of HMGCS-1 and UPRmt related proteins level in SHSY5Y cells transfected with scramble or HMGCS-1 siRNAs for 48 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. siNC group). (C) Western blot analysis of HMGCS-1 and UPRmt related proteins level in SHSY5Y cells transfected with scramble or HMGCS-1 siRNAs for 48 h and 20 μM Aβ25–35 treatment for 4 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. siNC group). (D) After treated with 1 μM and 10 μM simvastatin for different time courses, we detected the UPRmt related proteins level in SHSY5Y cell with 20 μM Aβ25–35 by western blot analysis, β-Actin served as the internal control.The data are mean ± SEM (n = 3, *p < 0.05 vs. DMSO group).
Simvastatin Treatments Change the Cell Morphology and Mitochondrial Structure, Increase the Intracellular ROS Level and Aggravate the Cytotoxic Effect of Aβ25–35 in SHSY5Y Cells
During experiments, we observed a phenomenon with a microscope, that 24 h treatment of 10 μM simvastatin changed the cell’s morphology and promoted the apoptosis of cells treated with Aβ25–35 (Figure 3A). The electron micrograph also showed that the simvastatin treatment aggravates the morphological changes of mitochondrial, such as mitochondrial vacuolation, disorganization, and reduction of the crista (Figure 3B). We further tested the intracellular ROS level by use of the ROS-sensitive fluorescent probe DCFH-DA, the fluorescence images and the amount of ROS showed that simvastatin treatment aggravated the increased intracellular ROS level induced by Aβ25–35 (Figure 3C). The cells viability was tested with the CCK8 method. As shown in Figure 3D, pretreatment with 10 μM simvastatin for 24 h aggravated the decrease of cell viability induced by Aβ25–35.
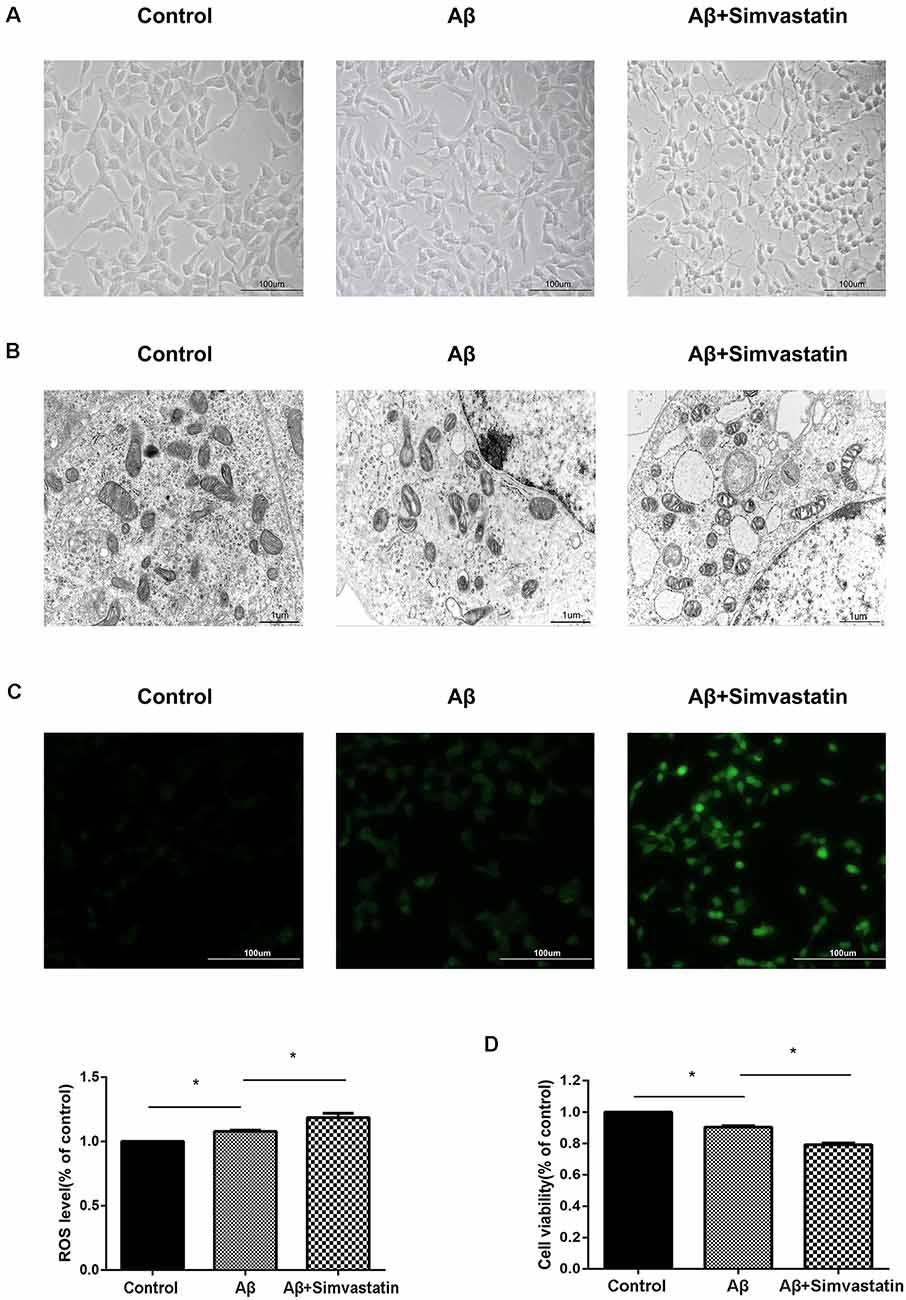
Figure 3. Simvastatin treatment changes the cell morphology and mitochondrial structure, increases the intracellular reactive oxygen species (ROS) level and aggravates the cytotoxic effect of Aβ25–35 in SHSY5Y cells. After treated with 10 μM simvastatin for 24 h and 20 μM Aβ25–35 for 4 h, the morphology of SHSY5Y cells were observed by a light microscope (A), the mitochondrial structures were observed by an electron microscope (B), the intracellular ROS level was measured by DCFH (C), viability of the cells was examined by Cell Counting Kit 8 (CCK8) assay (D). The data are mean ± SEM (n = 3, *p < 0.05).
The Sphingolipid Biosynthesis Pathway is Involved in Aβ25–35 Evoked UPRmt in SHSY5Y Cells
In a previous study, RNAi mediated inactivation of the sptl-1 gene, which encodes SPT, disrupted response to mitochondrial dysfunction in C. elegans. To detect whether it happens in SHSY5Y cells treated with Aβ25–35, we used the SPTLC-1 siRNAs to hinder sphingolipid biosynthesis. As shown in Figure 4A, treatment of cells with 20 μM Aβ25–35 for 4 h led to the upregulation of SPTLC-1. The expression level of SPTLC-1 was decreased in cells transfected with SPTLC-1 siRNAs for 48 h, compared with the SPTLC-1 level in cells transfected with scrambled siRNA. As shown in Figure 4B, in the absence of Aβ, there was no statistically significant difference in UPRmt related protein levels between cells transfected with scrambled siRNA and target siRNA. However, we found that after transfected with SPTLC-1 siRNAs, the protein levels of LONP1, Hsp60, HtrA2/Omi and CLPP were both decreased in cells treated with 20 μM Aβ for 4 h (Figure 4C). Thus, the Inhibition of the sphingolipid biosynthesis pathway by SPTLC-1 siRNAs decreases the UPRmt related proteins level in SHSY5Y cells treated with Aβ25–35.
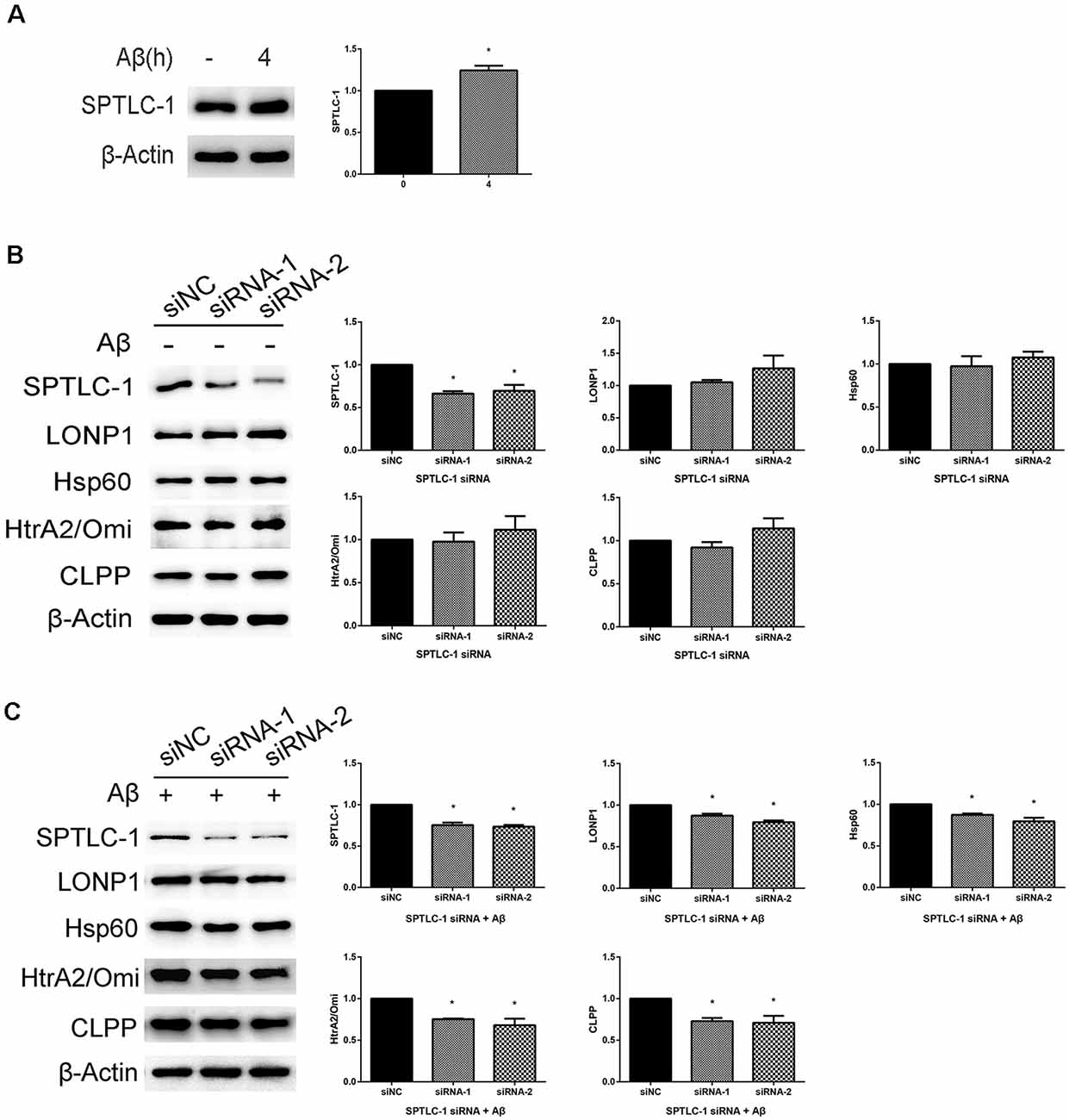
Figure 4. Inhibition of the sphingolipid biosynthesis pathway by serine palmitoyltransferase long chain subunit 1 (SPTLC-1) siRNAs reduces the UPRmt reaction induced by Aβ25–35 in SHSY5Y cells. (A) Western blot analysis of SPTLC-1 protein level in SHSY5Y cells treated with 20 μM Aβ25–35 for 4 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. control group). (B) Western blot analysis of SPTLC-1 and UPRmt related proteins level in SHSY5Y cells transfected with scramble or SPTLC-1 siRNAs for 48 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. siNC group). (C) Western blot analysis of SPTLC-1 and UPRmt related proteins level in SHSY5Y cells transfected with scramble or SPTLC-1 siRNAs for 48 h and 20 μM Aβ25–35 treatment for 4 h, quantification and β-Actin served as the internal control. The data are mean ± SEM (n = 3, *p < 0.05 vs. siNC group).
Inhibition of the Sphingolipid Biosynthesis Pathway by SPTLC-1 siRNAs Changes the Mitochondrial Structure, Increases the Intracellular ROS Level and Aggravates the Cytotoxic Effect of Aβ25–35 in SHSY5Y Cells
To determine whether inhibition of the sphingolipid biosynthesis pathway can cause a similar effect like simvastatin, we tested the cells’ mitochondrial structure, intracellular ROS level, and cell viability after transfected with SPTLC-1 siRNAs. As shown in Figure 5A, the Inhibition of the sphingolipid biosynthesis pathway by SPTLC-1 siRNAs aggravated the abnormalities in mitochondrial morphology, such as mitochondrial vacuolation and reduction of the crista. The fluorescence images and the amount of ROS also showed that Aβ25–35 increased the intracellular ROS level and SPTLC-1 siRNAs transfection aggravated the ROS accumulation (Figure 5B). The cells viability tested by the CCK8 method also showed that SPTLC-1 siRNAs transfection aggravated the decrease of cell viability induced by Aβ25–35 (Figure 5C).
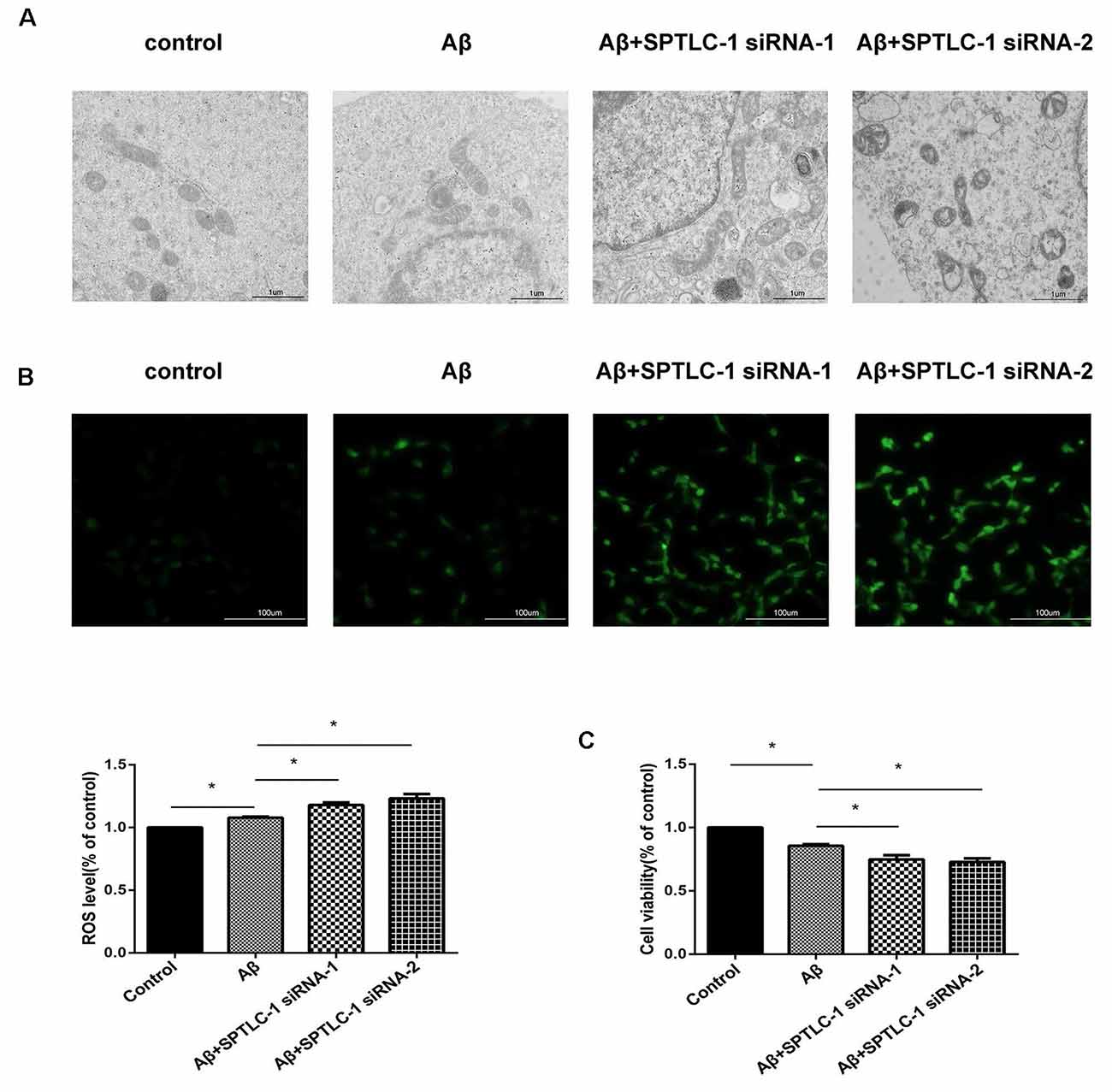
Figure 5. Inhibition of the sphingolipid biosynthesis pathway by SPTLC-1 siRNAs changes the mitochondrial structure, increases the intracellular ROS level and aggravates the cytotoxic effect of Aβ25–35 in SHSY5Y cells. After transfected with SPTLC-1 siRNAs for 48 h and 20 μM Aβ25–35 for 1 h, the mitochondrial structure of SHSY5Y cells was observed by a electron microscope (A), the intracellular ROS level was measured by DCFH (B), viability of the cells was examined by CCK8 assay (C). The data are mean ± SEM (n = 3, *p < 0.05).
Discussion
This study showed that the UPRmt was activated in SHSY5Y cells with Aβ25–35 treatment and APPsw/PS1dE9 transgenic mice, and inhibition of the mevalonate and sphingolipid biosynthesis pathway disrupted the activation of UPRmt in SHSY5Y cells induced by Aβ25–35. We further found that inhibition of these two pathways through simvastatin and SPTLC-1 siRNAs aggravated the mitochondrial injury, ROS accumulation and the decrease of cell viability in SHSY5Y cells induced by Aβ25–35.
Numerous diseases are associated with UPRmt, including spastic paraplegia, Parkinson’s disease, Friedreich’s ataxia and cancer (Haynes and Ron, 2010). Most reports about the unfolded protein response in AD were focused on the endoplasmic reticulum, but studies about the UPRmt were little except for the research mentioned above (Beck et al., 2016). So we tested the UPRmt reaction in cell and mice models of AD in this research. In cell models, all of the four UPRmt related proteins were upregulated in SHSY5Y cells treated with 10 μM or 20 μM Aβ25–35 for 24 h. As shown in results, the UPRmt related proteins Hsp60, HtrA2/Omi and CLPP were upregulated in APPsw/PS1dE9 transgenic mice at 3 months, while to 9 months, Hsp60 and LONP1 proteins were increased compared with age-matched WT mice. Thus, we conclude that the UPRmt response was activated during the AD process in vitro and in vivo.
Among the UPRmt related proteins, LONP1 as a mitochondrial matrix protease, maintain the mitochondrial homeostasis by degrading misfolded or oxidized polypeptides. As shown in Figure 1A, the LONP1 protein level was unchanged after treated the SHSY5Y cell with 2.5 or 5 μM Aβ25–35 for 24 h, but increased after 10 or 20 μM Aβ25–35 treatment. Combined with the results in the mouse model, the LONP1 protein level was unchanged in APPsw/PS1dE9 transgenic mice at 3 months but increased at 9 months compared with age-matched WT mice. Hence we speculate that the activation of LONP1 protein may need a prolonged or a large dose of Aβ stimulation, but further research is needed to confirm this. From the result in Figure 1C, we found that the expression levels of Hsp60 and HtrA2/Omi were significantly decreased in the hippocampus of 9 months old APPsw/PS1dE9 transgenic mice relative to those of 3 months old transgenic mice. Compared with age-matched WT mice, the UPRmt related proteins HtrA2/Omi and CLPP were upregulated in APPsw/PS1dE9 transgenic mice at 3 month but unchanged at 9 month, which suggest that the UPRmt activation decreased with age during theAD process. This is consistent with Sorrentino’s research that several UPRmt genes were down-regulated in the cortex samples of 9 months old AD mice compared with those of 6 months old AD mice (Sorrentino et al., 2017). Another research about the endoplasmic reticulum UPR response in Down syndrome also found that the UPR was selectively activated at 3 months, but restored to basal controls at 9 months (Lanzillotta et al., 2018). So we conclude that the UPRmt response is activated during the AD process in mice and cell models, but levels are altered in AD mice at different time points.
The mevalonate pathway is a key metabolic cascade that catalyzes the synthesis of sterol and nonsterol isoprenoids, which plays important effects on many cellular processes and physiological states (Goldstein and Brown, 1990). The HMG-CoA synthase HMGCS-1 and the HMG-CoA reductase HMGCR mediate this pathway as mentioned above, whereas stains as cholesterol-lowering drugs act by inhibiting the HMGCR. In our study, the HMGCS-1 protein level was increased in SHSY5Y cells after treated with 20 μM Aβ25–35 for 4 h, and after inhibiting the mevalonate pathway by HMGCS-1 siRNA or simvastatin in SHSY5Y cells treated with Aβ25–35, the expression of UPRmt related proteins were decreased. This is consistent with the researches in C. elegans that the UPRmt upregulates the expression of HMGS-1 (Oks et al., 2018), and inhibition of the mevalonate pathway through the hmgr-1 deletion mutant, hmgs-1 gene inactivation or statins prevents the activation of UPRmt (Liu et al., 2014; Ranji et al., 2014). We also found that inactivation of HMGCS-1 by siRNAs has no effect on the UPRmt reaction in SHSY5Y cells without Aβ treatment. From the above, our research provides evidence that the mevalonate pathway is necessary to the activation of UPRmt induced by Aβ and this effect of the mevalonate pathway was Aβ-dependent.
Several reports have revealed a novel connection between the sphingolipid biosynthesis and the endoplasmic reticulum UPR activation (Spassieva et al., 2009; Lépine et al., 2011; Epstein et al., 2012), but none was about the correlations between induction of UPRmt and the ceramide synthesis pathway in AD. As shown in our results, the SPTLC-1 protein level is increased in SHSY5Y cells after treated with 20 μM Aβ25–35 for 4 h. Inhibition of the ceramide synthesis by SPTLC-1 siRNA decreased the UPRmt related proteins levels in SHSY5Y cells treated with Aβ25–35 transiently, which provides evidence for an involvement of ceramide synthase SPTLC-1 in the activation of UPRmt response in AD cell models. The result is consistent with the researches in C. elegans that the UPRmt upregulates the transcript level of sptl-1, and inhibition of the sphingolipid biosynthesis pathway through the sptl-1 gene inactivation prevents the activation of UPRmt (Liu et al., 2014). Ceramide, which is the core structure of all complex sphingolipids, serves as the second messenger that regulates diverse cellular processes such as growth, differentiation, and apoptosis (Pettus et al., 2002). Ceramide levels can be reduced by inhibition of SPT both in vitro and in vivo (Hojjati et al., 2005; Holland et al., 2007). It have been proved that ceramide levels were increased at the earliest stage during the AD process (Han et al., 2002; Katsel et al., 2007; Mielke et al., 2010). Studies in patients with sporadic AD also showed that the levels of ceramide and SPT protein including SPTLC-1 were significantly elevated compared with control (Geekiyanage and Chan, 2011). SPT activity has proved increasing in dealing with various stimuli in cell research (Perry et al., 2000; Scarlatti et al., 2003). We also found that inactivation of SPTLC-1 by siRNAs has no effect on the UPRmt reaction in SHSY5Y cells without Aβ treatment. Thus, we conclude that Aβ25–35 treatment increases the protein levels of SPTLC-1, and the sphingolipid biosynthesis pathway is necessary to the activation of UPRmt induced by Aβ and this effect of the mevalonate pathway was Aβ-dependent.
As shown in our results, inhibition of the mevalonate pathway or the sphingolipid biosynthesis pathway can prevent the activation of UPRmt, change the mitochondrial structure, increase the intracellular ROS level and aggravate the cytotoxic effect of Aβ25–35 in SHSY5Y cells, which suggest that the UPRmt can protect SHSY5Y cells from the damage of Aβ25–35. The UPRmt has been proved cytoprotective effects in several reports (Baker et al., 2012; Runkel et al., 2013; Lamech and Haynes, 2015), and activation of the UPRmt response can also extend lifespan (Durieux et al., 2011; Houtkooper et al., 2013). Therefore, our research provides new evidence for the UPRmt cytoprotective effect during the AD process. It also provides a new sight for us whether enhancing or artificial activation of UPRmt can protect cells from the cytotoxic effect of Aβ and works on the treatment of AD. But several reports have revealed that the prolonged or over activation of the UPRmt has an adverse effect like shortening the lifespan of C. elegans (Bennett et al., 2014), perturb mitochondrial function (Lamech and Haynes, 2015; Lin et al., 2016). So further research is needed on whether enhanced UPRmt activation can be used in the treatment of AD because the intensity and duration of the response should be considered.
Together, these observations imply that the UPRmt activated in AD cell and mice models, but the levels vary according to the ages of mice. We further observed that the mevalonate and sphingolipid biosynthesis pathways were necessary for the activation of UPRmt in AD. When inhibiting the two pathways, the cytotoxic effects of Aβ25–35 in SHSY5Y cells were aggravated. Hence our research helps us understanding the role of UPRmt in the pathogenesis of AD, which may provide new sights for the treatment of AD.
Data Availability Statement
All datasets generated for this study are included in the article.
Ethics Statement
The animal study was reviewed and approved by the Ethical Committee for Animal Experiments of the Second Hospital of Shandong University.
Author Contributions
JB, PW, and YS designed the research. YS performed the experiments. MD, ZX, and XL provided technical support and helped with mouse dissections. HY, SJ, SX, ZZ, YW, DW, LX, and XZ analyzed the data. JB, PW, and YS wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Grant Nos. 81571052, 81870848 and 81401052), The Fundamental Research Funds of Chinese Academy of Medical Sciences (Grant No. 2019-RC-HL-026), the Key Research and Development Program of Shandong Province (Grant Nos. 2015GSF118056, 2017GSF218036 and 2017GSF218046) and The Fundamental Research Funds of Shandong University (Grant No. 2016JC022).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AD, Alzheimer disease; Aβ, Amyloid β-peptide; APP, Amyloid Precursor Protein; UPRmt, Mitochondrial unfolded protein response; HtrA2/Omi, High-temperature requirement protein A2/Omi; Hsp60, Heat shock protein 60; CLPP, Caseinolytic protease P; LONP1, Lon peptidase 1; HMG-CoA, 3-hydroxy-3-methyl-glutaryl-CoA; HMGCS-1, Human hydroxymethylglutaryl-CoA synthase 1; SPT, serine palmitoyltransferase; SPTLC-1, Serine palmitoyltransferase long chain subunit 1; CCK8, Cell Counting Kit 8; BCA, Bicinchoninic acid; PVDF, Polyvinylidene difluoride; ROS, Reactive oxygen species; WT, Wildtype; IMS, Intermembrane space.
References
Baker, B. M., Nargund, A. M., Sun, T., and Haynes, C. M. (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8:e1002760. doi: 10.1371/journal.pgen.1002760
Beck, J. S., Mufson, E. J., and Counts, S. E. (2016). Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 13, 610–614. doi: 10.2174/1567205013666151221145445
Bennett, C. F., Vander Wende, H., Simko, M., Klum, S., Barfield, S., Choi, H., et al. (2014). Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 5:3483. doi: 10.1038/ncomms4483
Blackstone, N. W. (2015). The impact of mitochondrial endosymbiosis on the evolution of calcium signaling. Cell Calcium 57, 133–139. doi: 10.1016/j.ceca.2014.11.006
Borchelt, D. R., Thinakaran, G., Eckman, C. B., Lee, M. K., Davenport, F., Ratovitsky, T., et al. (1996). Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013. doi: 10.1016/s0896-6273(00)80230-5
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Da Costa Dias, B., Jovanovic, K., Gonsalves, D., and Weiss, S. F. (2011). Structural and mechanistic commonalities of amyloid-β and the prion protein. Prion 5, 126–137. doi: 10.4161/pri.5.3.17025
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. doi: 10.1016/j.cell.2010.12.016
Epstein, S., Kirkpatrick, C. L., Castillon, G. A., Muniz, M., Riezman, I., David, F. P., et al. (2012). Activation of the unfolded protein response pathway causes ceramide accumulation in yeast and INS-1E insulinoma cells. J. Lipid Res. 53, 412–420. doi: 10.1194/jlr.m022186
Geekiyanage, H., and Chan, C. (2011). MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 31, 14820–14830. doi: 10.1523/jneurosci.3883-11.2011
Goldstein, J. L., and Brown, M. S. (1990). Regulation of the mevalonate pathway. Nature 343, 425–430. doi: 10.1038/343425a0
Han, X., Holtzman, D. M., Mckeel, D. W. Jr., Kelley, J., and Morris, J. C. (2002). Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J. Neurochem. 82, 809–818. doi: 10.1046/j.1471-4159.2002.00997.x
Hanada, K. (2003). Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta 1632, 16–30. doi: 10.1016/s1388-1981(03)00059-3
Harkany, T., Abraham, I., Timmerman, W., Laskay, G., Toth, B., Sasvari, M., et al. (2000). β-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur. J. Neurosci. 12, 2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x
Haynes, C. M., and Ron, D. (2010). The mitochondrial UPR—protecting organelle protein homeostasis. J. Cell Sci. 123, 3849–3855. doi: 10.1242/jcs.075119
Hojjati, M. R., Li, Z., Zhou, H., Tang, S., Huan, C., Ooi, E., et al. (2005). Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J. Biol. Chem. 280, 10284–10289. doi: 10.1074/jbc.m412348200
Holland, W. L., Brozinick, J. T., Wang, L. P., Hawkins, E. D., Sargent, K. M., Liu, Y., et al. (2007). Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat- and obesity-induced insulin resistance. Cell Metab. 5, 167–179. doi: 10.1016/j.cmet.2007.01.002
Hornemann, T., Richard, S., Rutti, M. F., Wei, Y., and Von Eckardstein, A. (2006). Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J. Biol. Chem. 281, 37275–37281. doi: 10.1074/jbc.m608066200
Hornemann, T., Wei, Y., and von Eckardstein, A. (2007). Is the mammalian serine palmitoyltransferase a high-molecular-mass complex? Biochem. J. 405, 157–164. doi: 10.1042/bj20070025
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Katsel, P., Li, C., and andHaroutunian, V. (2007). Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 32, 845–856. doi: 10.1007/s11064-007-9297-x
Lamech, L. T., and Haynes, C. M. (2015). The unpredictability of prolonged activation of stress response pathways. J. Cell Biol. 209, 781–787. doi: 10.1083/jcb.201503107
Lanzillotta, C., Tramutola, A., Meier, S., Schmitt, F., Barone, E., Perluigi, M., et al. (2018). Early and selective activation and subsequent alterations to the unfolded protein response in down syndrome mouse models. J. Alzheimers Dis. 62, 347–359. doi: 10.3233/jad-170617
Lépine, S., Allegood, J. C., Park, M., Dent, P., Milstien, S., and Spiegel, S. (2011). Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 18, 350–361. doi: 10.1038/cdd.2010.104
Lin, Y. F., Schulz, A. M., Pellegrino, M. W., Lu, Y., Shaham, S., and Haynes, C. M. (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. doi: 10.1038/nature17989
Liu, Y., Samuel, B. S., Breen, P. C., and Ruvkun, G. (2014). Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508, 406–410. doi: 10.1038/nature13204
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Mielke, M. M., Bandaru, V. V., Haughey, N. J., Rabins, P. V., Lyketsos, C. G., and Carlson, M. C. (2010). Serum sphingomyelins and ceramides are early predictors of memory impairment. Neurobiol. Aging 31, 17–24. doi: 10.1016/j.neurobiolaging.2008.03.011
Mielke, M. M., and Haughey, N. J. (2012). Could plasma sphingolipids be diagnostic or prognostic biomarkers for Alzheimer’s disease? Clin. Lipidol. 7, 525–536. doi: 10.2217/clp.12.59
Oks, O., Lewin, S., Goncalves, I. L., and Sapir, A. (2018). The UPRmt protects Caenorhabditis elegans from mitochondrial dysfunction by upregulating specific enzymes of the mevalonate pathway. Genetics 209, 457–473. doi: 10.1534/genetics.118.300863
Papa, L., and Germain, D. (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 124, 1396–1402. doi: 10.1242/jcs.078220
Pellegrino, M. W., Nargund, A. M., and Haynes, C. M. (2013). Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416. doi: 10.1016/j.bbamcr.2012.02.019
Perry, D. K., Carton, J., Shah, A. K., Meredith, F., Uhlinger, D. J., and Hannun, Y. A. (2000). Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J. Biol. Chem. 275, 9078–9084. doi: 10.1074/jbc.275.12.9078
Pettus, B. J., Chalfant, C. E., and Hannun, Y. A. (2002). Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta 1585, 114–125. doi: 10.1016/s1388-1981(02)00331-1
Pleckaityte, M. (2010). [Alzheimer’s disease: a molecular mechanism, new hypotheses and therapeutic strategies]. Medicina 46, 70–76. doi: 10.3390/medicina46010011
Ranji, P., Rauthan, M., Pitot, C., and Pilon, M. (2014). Loss of HMG-CoA reductase in C. elegans causes defects in protein prenylation and muscle mitochondria. PLoS One 9:e100033. doi: 10.1371/journal.pone.0100033
Runkel, E. D., Liu, S., Baumeister, R., and Schulze, E. (2013). Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 9:e1003346. doi: 10.1371/journal.pgen.1003346
Sapir, A., Tsur, A., Koorman, T., Ching, K., Mishra, P., Bardenheier, A., et al. (2014). Controlled sumoylation of the mevalonate pathway enzyme HMGS-1 regulates metabolism during aging. Proc. Natl. Acad. Sci. U S A 111, E3880–E3889. doi: 10.1073/pnas.1414748111
Scarlatti, F., Sala, G., Somenzi, G., Signorelli, P., Sacchi, N., and Ghidoni, R. (2003). Resveratrol induces growth inhibition and apoptosis in metastatic breast cancer cells via de novo ceramide signaling. FASEB J. 17, 2339–2341. doi: 10.1096/fj.03-0292fje
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Skovronsky, D. M., Lee, V. M., and Trojanowski, J. Q. (2006). Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu. Rev. Pathol. 1, 151–170. doi: 10.1146/annurev.pathol.1.110304.100113
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193. doi: 10.1038/nature25143
Spassieva, S. D., Mullen, T. D., Townsend, D. M., and Obeid, L. M. (2009). Disruption of ceramide synthesis by CerS2 down-regulation leads to autophagy and the unfolded protein response. Biochem. J. 424, 273–283. doi: 10.1042/bj20090699
Spiegel, S., and Merrill, A. H. Jr. (1996). Sphingolipid metabolism and cell growth regulation. FASEB J. 10, 1388–1397. doi: 10.1096/fasebj.10.12.8903509
Tatsuta, T., and Langer, T. (2008). Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306–314. doi: 10.1038/sj.emboj.7601972
Wang, C., and Youle, R. J. (2009). The role of mitochondria in apoptosis*. Annu. Rev. Genet. 43, 95–118. doi: 10.1146/annurev-genet-102108-134850
Westerlund, M., Behbahani, H., Gellhaar, S., Forsell, C., Belin, A. C., Anvret, A., et al. (2011). Altered enzymatic activity and allele frequency of OMI/HTRA2 in Alzheimer’s disease. FASEB J. 25, 1345–1352. doi: 10.1096/fj.10-163402
Keywords: Alzheimer disease, UPRmt, mevalonate pathway, sphingolipid biosynthesis pathway, simvastatin
Citation: Shen Y, Ding M, Xie Z, Liu X, Yang H, Jin S, Xu S, Zhu Z, Wang Y, Wang D, Xu L, Zhou X, Wang P and Bi J (2020) Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Front. Cell. Neurosci. 13:568. doi: 10.3389/fncel.2019.00568
Received: 03 September 2019; Accepted: 09 December 2019;
Published: 08 January 2020.
Edited by:
Miriam L. Greenberg, Wayne State University, United StatesReviewed by:
Xing Guo, Nanjing Medical University, ChinaJinsoo Seo, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South Korea
Copyright © 2020 Shen, Ding, Xie, Liu, Yang, Jin, Xu, Zhu, Wang, Wang, Xu, Zhou, Wang and Bi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Wang, d3BpbmcwMTA4QDE2My5jb20=; Jianzhong Bi, Ymp6QHNkdS5lZHUuY24=