 Rasem Mustafa1,2
Rasem Mustafa1,2 Grzegorz Kreiner3
Grzegorz Kreiner3 Katarzyna Kamińska4,5
Katarzyna Kamińska4,5 Amelia-Elise J. Wood6Joachim Kirsch2
Amelia-Elise J. Wood6Joachim Kirsch2 Kerry L. Tucker6
Kerry L. Tucker6 Rosanna Parlato1,2*
Rosanna Parlato1,2*- 1Institute of Applied Physiology, University of Ulm, Ulm, Germany
- 2Institute of Anatomy and Cell Biology, Medical Cell Biology, University of Heidelberg, Heidelberg, Germany
- 3Department of Brain Biochemistry, Maj Institute of Pharmacology, Polish Academy of Sciences, Kraków, Poland
- 4Department of Pharmacology, Maj Institute of Pharmacology, Polish Academy of Sciences, Kraków, Poland
- 5Jagiellonian Center for Experimental Therapeutics, Jagiellonian University, Kraków, Poland
- 6Department of Biomedical Sciences, Center for Excellence in the Neurosciences, College of Osteopathic Medicine, University of New England, Biddeford, ME, United States
Multiple pathomechanisms triggered by mutant Huntingtin (mHTT) underlie progressive degeneration of dopaminoceptive striatal neurons in Huntington’s disease (HD). The primary cilium is a membrane compartment that functions as a hub for various pathways that are dysregulated in HD, for example, dopamine (DA) receptor transmission and the mechanistic target of rapamycin (mTOR) pathway. The roles of primary cilia (PC) for the maintenance of striatal neurons and in HD progression remain unknown. Here, we investigated PC defects in vulnerable striatal neurons in a progressive model of HD, the mHTT-expressing knock-in zQ175 mice. We found that PC length is affected in striatal but not in cortical neurons, in association with the accumulation of mHTT. To explore the role of PC, we generated conditional mutant mice lacking IFT88, a component of the anterograde intraflagellar transport-B complex lacking PC in dopaminoceptive neurons. This mutation preserved the expression of the dopamine 1 receptor (D1R), and the survival of striatal neurons, but resulted in a mild increase of DA metabolites in the striatum, suggesting an imbalance of ciliary DA receptor transmission. Conditional loss of PC in zQ175 mice did not trigger astrogliosis, however, mTOR signaling was more active and resulted in a more pronounced accumulation of nuclear inclusions containing mHTT. Further studies will be required of aged mice to determine the role of aberrant ciliary function in more advanced stages of HD.
Introduction
Huntington’s disease (HD) is an autosomal dominant progressive neurodegenerative disorder caused by the toxic expansion of CAG trinucleotide repeats at the N-terminus of the Huntingtin gene. The mechanisms underlying selective vulnerability of dopaminoceptive medium spiny neurons (MSNs), resulting in impaired control of voluntary movement in HD, remain elusive (Ghosh and Tabrizi, 2018). The variability of disease onset and progression depends on the CAG number, and on genetic modifiers interacting with the Huntingtin mutation [Genetic Modifiers of Huntington’s Disease (GeMHD) Consortium (2015)]. Multiple signaling pathways and cellular functions are affected by mutant Huntingtin (mHTT), including protein aggregate degradation, which results in the accumulation of toxic proteins (Saudou and Humbert, 2016).
Primary cilia (PC) are single, non-motile microtubule-based organelles resembling a cellular antenna that represents a hub for receptors and components of numerous signaling pathways (Malicki and Johnson, 2017). Lack of HTT results in reduced and aberrant PC growth, and increased mHTT results in increased ciliogenesis (Keryer et al., 2011). Notably, longer PC have been observed in immortalized cellular models of HD in culture, and ependymal cilia in the lateral ventricles are disorganized in a mouse model of HD and in HD human post-mortem brains (Keryer et al., 2011). Another study showed that photoreceptor cilia pathology accounts for their degeneration in the retina of R6/2 transgenic mice overexpressing exon 1 of the human mHTT (Karam et al., 2015). It has been previously proposed that PC altered structure might affect the function of signaling pathways whose components are localized in the PC (Maiuri et al., 2013; Kaliszewski et al., 2015).
Interestingly, increased PC length results in the induction of autophagy by inhibition of the mechanistic target of rapamycin (mTOR) kinase activity (Kaliszewski et al., 2015), and components essential for ciliogenesis are degraded by autophagy (Pampliega et al., 2013; Tang et al., 2013). Because autophagy is altered in HD (Ravikumar et al., 2004), as well as dopamine (DA)-mediated signaling (Chen et al., 2013), it is possible that HD pathophysiology depends, at least in part, on defective cilia. These previous studies investigated neither PC dysfunction in the most vulnerable striatal neurons, nor the impact of defective PC on HD pathogenesis in the striatum. A deeper understanding of the role of PC in mHTT-dependent neurotoxicity might help to identify new determinants modifying HD progression.
To this end, we monitored neuron- and stage-specific changes of PC structure in a full-length progressive mouse model of HD, called zQ175 (Menalled et al., 2012; Carty et al., 2015). This knock-in model carries a chimeric human/mouse HTT exon 1 containing expanded CAG repeats within the murine htt gene and recapitulates several hallmarks of HD pathology (Menalled et al., 2012; Farrar et al., 2014; Carty et al., 2015; Ma et al., 2015). Moreover, we generated a new genetic mouse model of defective ciliary function in striatal neurons, as a tool to investigate the specific impact of PC loss on striatal neuron maintenance and on HD neuropathological hallmarks.
Materials and Methods
Mice
To generate a mutant mouse in which the Ift88 gene is conditionally ablated by the Cre-LoxP system in MSNs, we employed the B6.FVB/N-Tg(D1RCre)Gsc (D1R:Cre) transgene, which expresses the Cre recombinase under the control of the dopamine 1 receptor (D1R) promoter (Lemberger et al., 2007). The D1R:Cre mice were crossed to Ift88tm1.1Bky mice carrying the Ift88 floxed allele (Ift88flox/flox; Haycraft et al., 2007) to generate Ift88flox/flox; D1R:Cre mice (Ift88D1RCre; here abbreviated Ift88 cKO mice) that lack PC in MSNs. Htttmtm1Mfc/190tChdi (zQ175 knock-in) mice were received courtesy of the CHDI Foundation from the Jackson Laboratory. The analysis of the genotype was performed by PCR of tail snips as previously described (Levine et al., 1999). For the experiments reported here, male and female mice were used and wild-type and mutant littermates were analyzed. The zQ175 knock-in mice carry ca. 190 CAG repeats in a chimeric human/mouse exon 1 of the murine huntingtin gene (Menalled et al., 2012). The zQ175 mutation was kept in heterozygosity, to limit toxicity and mimic a genetic situation more relevant for the disease, as it is autosomal dominant (Menalled et al., 2012). These mutant mice were born at the expected Mendelian ratio; they showed normal lifespan and no gross abnormalities (monitored until 1-year-old).
For genotyping of D1R:Cre and floxed Ift88 alleles by PCR the following primer pairs was used: forward-primer/Cre (5′-GGA AAT GGT TTC CCG CAG AAC-3′) and reverse-primer/Cre (5′-ACG GAA ATC CAT CGC TCG ACC-3′), BY919 (5′-GGTCCTAACAAGTAAGCCCAGTGTT-3′) and BY598 (5′-GCCTCCTGTTTCTTGACAACAGTG-3′), respectively. For the zQ175 we used forward-primer/Neo: 5′-GAT CGG CCA TTG AAC AAG ATG– 3′ and reverse-primer/Neo: 5′-AGA GCA GCC GAT TGT CTG TTG– 3′.
Brain Dissection and Tissue Preparation
For histological analysis, brains were either transcardially perfused or post-fixed in 4% paraformaldehyde (PFA; pH 7.2) overnight at 4°C. After washing in PBS (pH 7.2) the brains were cryoprotected by incubating them in 10%, 20%, and 30% sucrose for 3 days at 4°C. The brains were embedded in a coronal orientation (Tissue freezing medium, Leica), frozen by a mixture of liquid nitrogen and dry ice, and stored at −80°C until sectioning (Leica CM3050S cryostat). For the analysis of adult brains, we have used coronal sections from striatum collected serially on Superfrost Ultraplus glass slides (12 μm) and free-floating striatum (30 μm). We analyzed the striatum in the region comprised between Bregma +1.18 mm and −0.34 mm based on the adult mouse brain Atlas (Franklin, 2008). For immunofluorescence on paraffin sections, one brain hemisphere was fixed in 4% PFA in PBS, pH 7.2 overnight at 4°C and paraffin-embedded.
Immunofluorescence
Immunofluorescence (IF) on cryosections was performed according to established protocols (Gazea et al., 2016). Cryosections (12 μm) were pre-incubated with 5% normal pig serum in PBS for 30 min at room temperature before overnight incubation at 4°C with the primary antibodies appropriately diluted in the blocking solution. After washing in PBS the sections were incubated with the secondary antibodies (diluted in 5% pig serum in PBS) for 30 min at room temperature. After further washes in PBS the sections were stained for 10 min with 4′,6′-diamidino-2-pheylindol (DAPI, Thermoscientific) diluted in PBS.
Free-floating cryosections (30 μm) were treated in a similar way; however, PBST (0.2% Triton X-100 in PBS) was used for washing steps and blocking solution. After staining the sections were placed on glass slides. Paraffin sections (7 μm) containing the striatum comprised between Bregma +0.14 mm and −0.98 mm were incubated with primary antibodies overnight at 4°C. Visualization of antigen-bound primary antibodies was carried out following antigen retrieval (HK086-9K, Biogenex). The following primary antibodies were used: EM48 (1:100, MAB5374, Millipore), NeuN (1:100, MAB377, Millipore), adenylate cyclase III (ACIII; 1:500, SC588, Santa Cruz Biotechnology, Dallas, TX, USA), TH (1:500, AB1542, Millipore), D1R (1:500, D2944, Sigma), glial fibrillary acidic protein (GFAP; 1:300, G-3893, Sigma), phospho-S6 (S235/236; 91B2; 1:100, 4857S, Cell Signaling), p62/SQSTM1 (1:100, P0067, Sigma). Incubation with the appropriate secondary antibodies marked with the fluorophores Alexa 594 or Alexa 488 (1:100, Thermoscientific) was followed by DAPI staining.
Confocal Microscopy Imaging and Image Analysis
Images were acquired by a Leica SP8 confocal and a Leica LASX software. For PC number and length quantification, we used a 63× oil-immersion objective maximal intensity projection z-stacked images (1 μm interval). To determine the number of striatal NeuN positive neurons showing PC stained by ACIII antibody eight non-consecutive free-floating cryosections per mouse were analyzed (one every fourth section, each 30 μm thick) in control and mutant mice. Either the number or the length of PC per microscopic field at 63× magnification was counted. In general, about 80 cells per mouse were measured by the ImageJ software after tracing the ACIII signal in line with established protocols (Miyoshi et al., 2014; Parker et al., 2016). For semi-quantitative analysis of D1R and TH immunoreactivity, and of GFAP and phosphoS6 positive cells, we used a 20× oil-immersion objective for single planes. For p62 positive cells, we used a 63× and for the EM48 a 100× oil-immersion objective. To determine D1R and TH immunoreactivity the optical fiber density was measured in cryosections (12 μm) at different ages by ImageJ software. The quantification was performed in eight coronal serial sections per mouse on 8-bit images (grayscale). To measure the mean optical intensity the “Mean Gray Value” was determined. The respective immunoreactivity was measured by subtracting the mean gray value of the respective background from the mean gray value of dorsal striatum. The measurements were limited to the drawn region of interest and the same area was selected in all sections (0.05 mm2). The number of GFAP, pS6 and p62 and EM48 positive cells per microscopic field was counted in at least four independent paraffin sections per mouse in the dorsolateral striatal region. The EM48 signal area is calculated in μm2. We used the Quick LUT view to avoid the acquisition of images with under- and over-saturated pixels. The quantification and counting were performed blind to genotype and age.
HPLC Analysis of Dopamine Content
The tissue levels of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA) and 3-methoxytyramine (3-MT), were measured using high-performance liquid chromatography with electrochemical detection (HPLC-EC). The concentrations of endogenous DA and its metabolites (DOPAC, HVA) were measured using HPLC-EC, according to a previously described method (Sikora et al., 2016). Briefly, the striatum was isolated by the adult mouse brain matrix (coronal slices, World Precision Instruments) in the 2 mm region comprised between Bregma +1.42 mm and −0.58 mm. After weighing the tissue samples were deep-frozen in dry ice and stored at −80°C until further use. Prior to analysis, the samples were homogenized in ice-cold 0.1 M HClO4 and were centrifuged at 10,000 g for 10 min at 4°C. The supernatant (5 μl) was injected into the HPLC system. The chromatography system consisted of an LC-4C amperometric detector with a cross-flow detector cell (BAS, IN, USA), an Ultimate 3000 pump (Thermoscientific, USA) and a Hypersil Gold analytical column (3 μm, 100 × 3 mm, Thermoscientific, USA). The mobile phase consisted of 0.1 M KH2PO4, 0.5 mM Na2EDTA, 80 mg/L sodium 1-octane sulfonate, and a 4% methanol, adjusted to pH 3.8 with an 85% H3PO4. The flow rate was 1 ml/min. The potential of a 3-mm glassy carbon electrode was set at 0.7 V with a sensitivity of 5 nA/V. The temperature of the column was maintained at 30°C. The Chromax 2007 program (Pol-Lab, Warszawa, Poland) was used for data collection and analysis. The external standard consisted of noradrenaline (NA), DA, 5-hydroxytryptamine (serotonin; 5-HT) and 5-hydroxy indole acetic acid (5-HIAA; SIGMA) at a concentration of 50 ng/ml.
Statistical Analysis
All data are expressed as mean ± SEM. Two-tailed unpaired Student’s t-test was used for single comparisons. Multiple comparisons were performed either by one-way ANOVA or by two-way ANOVA with post hoc analyses as indicated in the figure legends (GraphPad Prism Software Inc.).
Results
Altered Primary Cilia Length in the Striatum but Not in Cortex of zQ175 Mice
By IF and confocal analysis of brain sections, we observed the presence of sporadic mHTT inclusions at 4 months in the striatum. From 8 months mHTT accumulated mostly in the striatum but also in the cortex, in agreement with previous results (Carty et al., 2015; Figures 1A–F). To test the hypothesis that altered PC impacts HD neuropathology, we first investigated PC alterations in the heterozygous zQ175 knock-in model of HD. We measured the stage-specific alterations of PC length in both striatum and cortex at 4 and 8 months in control and zQ175 mice, focusing on cells labeled by the neuronal nuclei marker NeuN (Figures 1G–P). PC were identified by ACIII, a major PC marker in many regions of the adult mouse brain (Bishop et al., 2007). We found that PC in dorsolateral striatum of zQ175 mice were shorter than their littermate controls at 4 months, while they were longer than respective controls at 8 months (Figure 1O). Notably, PC length in the striatum of controls, decreased between 4 and 8 months, but this age-dependent decrease did not occur in the zQ175 mice (Figure 1O). Next, we analyzed the cell-specificity of this phenotype by measuring the average PC length in cortical cells (Figures 1K–N). Interestingly, cortical cells of zQ175 mice did not show changes in PC length at any of the considered stages (Figure 1P).
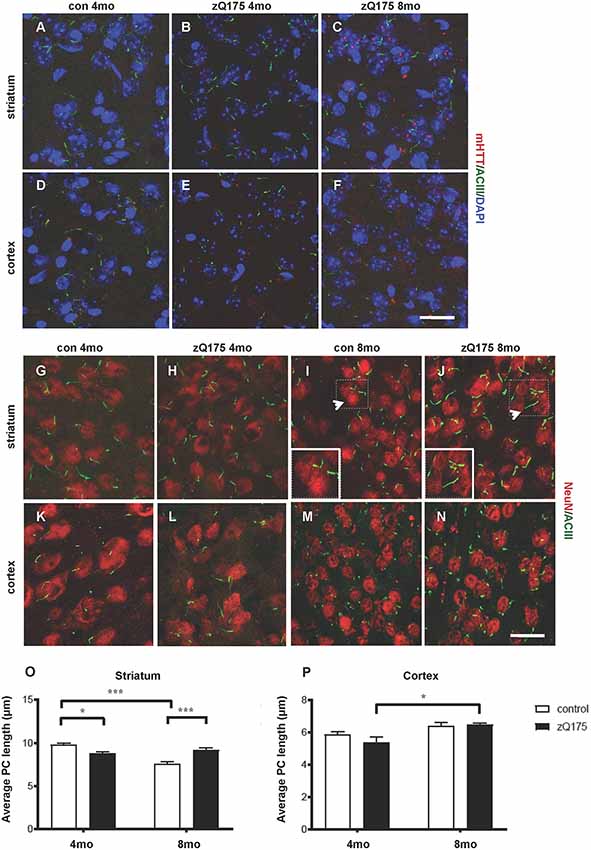
Figure 1. Context-specific changes in primary cilia (PC) length in the zQ175 Huntington’s disease (HD) mouse model are concomitant with mutant Huntingtin (mHTT) accumulation. (A–F) Representative confocal images of mHTT identified by the EM48 antibody (red) and of PC by adenylate cyclase III (ACIII), as a marker of PC (green) on cryosections from control and zQ175 mice in the dorsolateral striatum and cortex. Nuclei are visualized by 4′,6′-diamidino-2-pheylindol (DAPI) staining (blue). Scale bar: 25 μm. (G–N) Representative confocal images of immunofluorescent stainings on cryosections by NeuN, as a neuronal marker (red), ACIII (green), in the striatum (G,J) and cortex (K,N) to identify PC that protrudes from NeuN labeled neurons. Scale bars: 25 μm (G–J, M,N), 12 μm (K,L), 8 μm (insets, I and J). Arrows point to the area in the inset. (O,P) Diagrams showing the analysis of PC average length in striatal and cortical neurons at 4 and 8 months in control and zQ175 mice; N = 3, 5 control, and N = 4, 4 zQ175. Values represent means ± SEM. *p < 0.05, ***p < 0.0005, two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons.
Hence, mHTT accumulation either directly or indirectly affects PC length in a stage- and region-specific fashion, suggesting that altered PC function might contribute to striatal vulnerability in HD.
Conditional Ablation of the Ift88 Gene in Dopaminoceptive Neurons Leads to a Mild Increase of Dopamine Metabolite Levels in the Striatum
To investigate the effects of PC loss on HD neuropathology, we first generated inducible mutant mice conditionally lacking Ift88, a gene encoding a microtubular component essential for the formation of the PC (Haycraft et al., 2007). These mice are characterized by the expression of Cre recombinase under the control of dopaminoceptive D1-receptor (D1R; Lemberger et al., 2007; Figures 2A–D). The conditional ablation of the Ift88 gene resulted in D1R:Cre;Ift88flox/flox (abbreviated as Ift88 cKO) mice lacking PC in most of the striatal neurons stained for neuronal nuclei (NeuN; Figure 2E). The Ift88 cKO mice did not show any gross abnormalities. We monitored changes in body weight (g) over time and we found evidence of increased weight in the Ift88 cKO at 1 year (eight male control, 34.4 ± 6.1 vs. six male mutants, 44.9 ± 4.9; p = 0.004 two-tailed Student’s t-test). This conditional model enabled us to mimic a condition of loss of PC in dopaminoceptive neurons to identify their context-specific function. Next, we analyzed D1R expression as a read-out of the survival of positive neurons in control and Ift88 cKO at different ages (1, 3, 6, and 12 months; Figures 2F–H). To investigate the impact of PC loss on neuronal survival, we analyzed the immunoreactivity of D1R in dorsolateral striatum at different ages in control and Ift88 cKO mice by IF and semi-quantitative analysis of D1R mean signal intensity upon confocal imaging (Figure 2H). This analysis showed no significant differences in D1R immunoreactivity at any of the considered ages in control and Ift88 cKO mutant mice, if at all a tendency to higher D1R immunoreactivity, suggesting that PC is not required for survival of D1R MSNs under basal conditions.
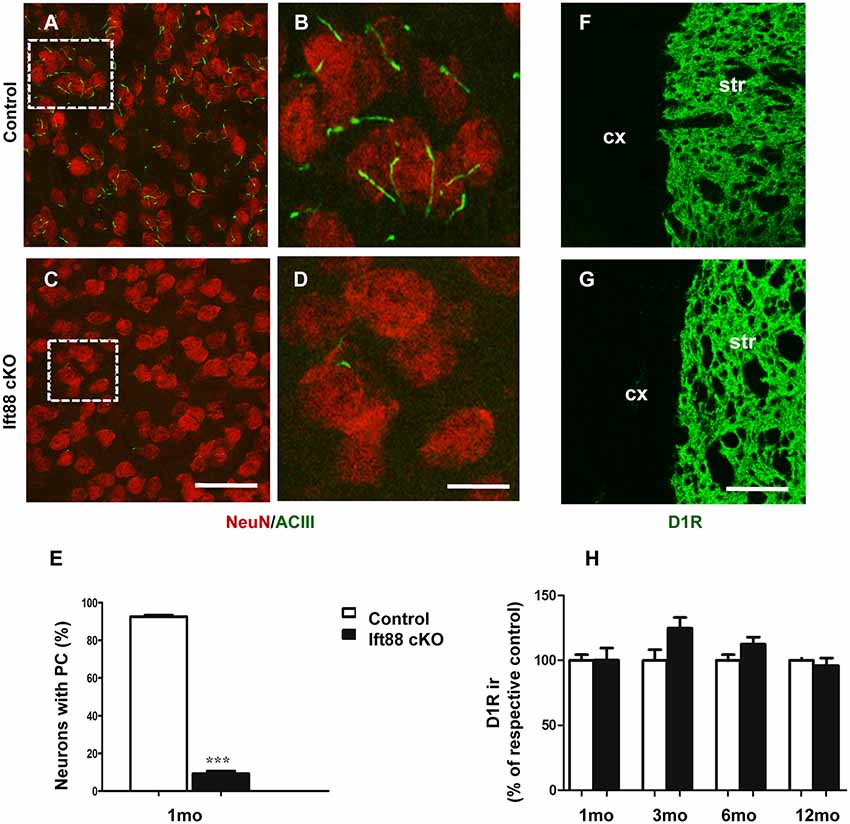
Figure 2. PC is dispensable for the maintenance of dopaminoceptive D1-receptor (D1R) expression in the striatum. (A–D) Representative images of immunofluorescent stainings showing PC stained with ACIII (green) and NeuN (red) in control and Ift88 cKO mice at 1 month. (E) Diagram shows the percentage of NeuN positive cells showing ACIII staining in control and Ift88 cKO at 1 month (N = 3). Error bars represent SEM, ***p < 0.001 based on two-tailed unpaired Student’s t-test. Scale bars represent 30 μm in (A,C) and 12 μm in (B,D). (F,G) Examples of D1R immunostaining (green) on cryosections showing dorsolateral striatum in control and Ift88 cKO at 6 months; str, striatum; cx, cortex. (H) Diagram showing semi-quantitative analysis of D1R immunoreactivity (ir) in dorsolateral striatum at different ages; N = 3–5 controls, N = 3–5 Ift88 cKO. Values represent means ± SEM. Scale bar in (F,G): 100 μm. No significant differences with respective controls by unpaired Student’s t-test.
To further characterize the phenotype of the Ift88 cKO, we investigated dopaminergic input to dopaminoceptive striatal neurons (Figure 3). To this end, we focused on young (1 month) and older (1 year) mice. We measured tyrosine-hydroxylase (TH) immunoreactivity by IF (Figures 3A,B). Interestingly, TH immunoreactivity increased in the mutant at 1 year (Figure 3C). Next, we asked whether the loss of PC in D1R neurons affects DA content in the striatum (Figures 3D–G). High performance liquid chromatography followed by electrochemical detection (HPLC-EC) showed that 1-year-old Ift88 cKO mutant mice are characterized both by a tendency to increased levels of total DA content in the striatum as well as by a mild but significant increase of the two most important DA metabolites DOPAC, and HVA (Figures 3D–F).
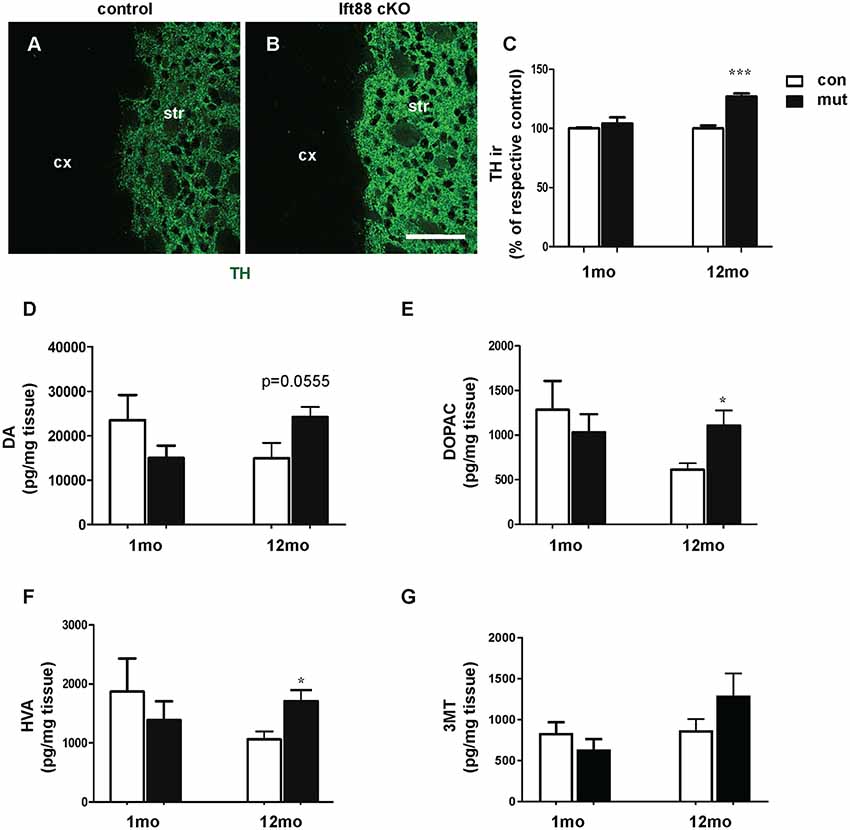
Figure 3. Loss of striatal PC results in a mild increase of dopamine (DA) metabolites in the striatum. (A,B) Examples of tyrosine-hydroxylase (TH) immunofluorescence (IF), as a marker for dopaminergic projections, in cryosections from the dorsolateral striatum of control and Ift88 cKO mice (1-year-old). Scale bar: 100 μm. (C) Semi-quantitative analysis of TH immunoreactivity (ir) at 1 and 12 months; N = 5 per group. (D–G) Levels of DA and its major metabolites [3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 3-methoxytyramine (3-MT)] in the striatum by HPLC-EC; N = 5 per group. Values represent means ± SEM. *p < 0.05, ***p < 0.001 by unpaired Student’s t-test with respective controls.
These observations indicate subtle PC-dependent crosstalk between PC-depleted dopaminoceptive neurons and dopaminergic neurons to cope with a potentially altered dopaminergic neurotransmission.
Loss of PC in the zQ175 HD Mice Results in mTOR Activation and in Larger mHTT Nuclear Inclusions
To study the impact of PC loss on HD neuropathology, we generated zQ175 heterozygous mice lacking PC in striatal but not in cortical neurons in an Ift88 cKO background (double mutants, dm; Supplementary Figure S1). To establish whether PC loss is toxic in combination with the zQ175 mutation and whether the combination of these mutations results in a more severe HD phenotype, we analyzed control, Ift88 cKO, zQ175, and dm at 8 months (Supplementary Figure S2 and Figure 4). We found no significant difference in the number of GFAP positive cells, as a marker of astrogliosis, associated with neurodegeneration, suggesting that there is no induction of massive degeneration at least until this stage (Supplementary Figures S2A–D,I). To monitor the clearance of protein aggregates, we quantified changes in the number of sequestosome 1 (SQSTM1/p62) protein immunopositive cells in the dorsolateral striatum by IF in the four experimental groups above (Supplementary Figures S2E–H,J). p62 is a cargo-binding protein associated with proteotoxic stress (Johansen and Lamark, 2011). As shown in Supplementary Figures S2G,H, the localization of p62 in the zQ175 and in the dm mice was intra-nuclear rather than cytoplasmic, as in controls and Ift88 cKO, however, the number of p62 positive cells was comparable between all groups. Next, we investigated mTOR kinase activity by IF based on the levels of one of its targets, phosphorylated ribosomal protein S6 (phospho-S6, pS6; Figures 4A–E). Interestingly, zQ175 and dm mice showed a higher number of pS6 positive cells in comparison to controls, and this was significantly higher in the dm in comparison to the zQ175 (Figure 4E), suggesting that PC loss in the zQ175 mice promotes mTOR activation. Moreover, we analyzed mHTT nuclear inclusions that are absent in controls and Ift88 cKO, and visible in the zQ175 and dm (Figures 4F–I). At the age examined (8 months), the number of mHTT positive cells was similar in the zQ175 and dm mice (Figure 4J). The area of the mHTT signal in the dm is ca. Thirty percent larger in comparison to the zQ175 mice, indicating that PC limit mHTT accumulation in this model (Figure 4K).
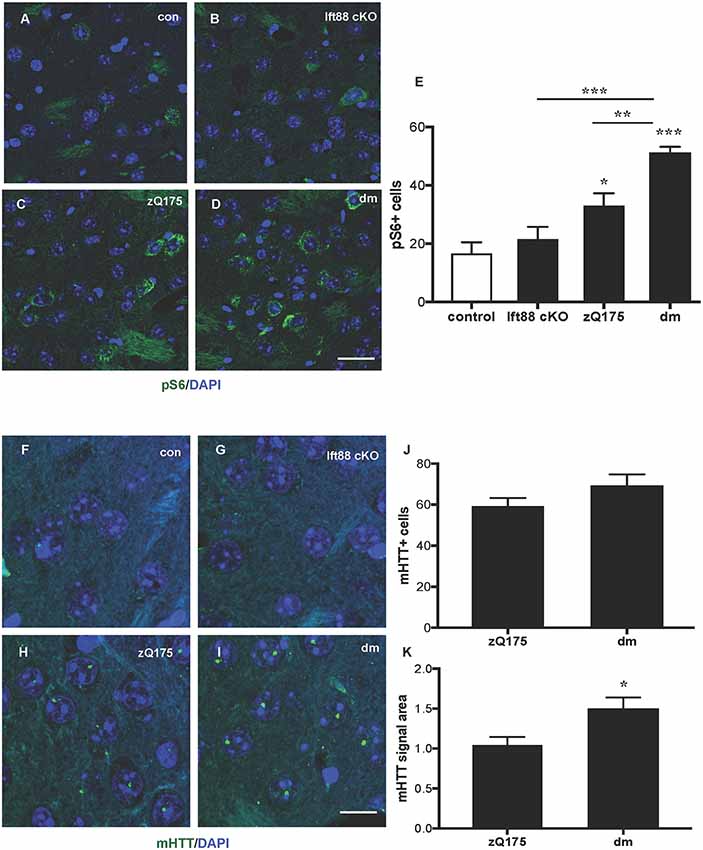
Figure 4. Impact of PC loss in medium spiny neurons (MSNs) of zQ175 HD mice on the mechanistic target of rapamycin (mTOR) pathway and mHTT nuclear inclusions. (A–D) Representative confocal images of phopsho-S6 (pS6) IF (green) and DAPI staining (blue) on paraffin sections showing dorsolateral striatum in control, Ift88 cKO, zQ175, and dm at 8 months. Scale bar: 25 μm. (E) Quantification of the pS6 positive cells expressed as mean values of the number of counted cells per microscopic field. Control vs. zQ175 *p < 0.05; control vs. dm ***p < 0.0001 Ift88 cKO vs. dm ***p < 0.001, zQ175 vs. dm **p < 0.01 by one-way ANOVA followed by Dunnett’s post hoc test for multiple comparisons. Values represent means ± SEM. pS6: control (N = 5), Ift88 cKO (N = 5), zQ175 (N = 6) and dm (N = 5) mice. (F–I) Representative confocal images of mHTT immunostaining (green) by EM48 antibody and DAPI (blue) on paraffin sections from dorsolateral striatum in control, Ift88 cKO, zQ175, and dm at 8 months. Scale bar: 10 μm. (J,K) Quantification of the EM48 positive cells expressed as a percentage of DAPI positive cells and of the mean EM48 signal area (in μm2) per microscopic field. Values represent means ± SEM. *p < 0.05 (p = 0.019) based on two-tailed unpaired Student’s t-test; zQ175 (N = 8) and dm (N = 6) mice.
Discussion
In the present study, we addressed the following three questions: (1) whether PC is specifically altered in different neuronal types and HD stages; (2) what are the consequences of PC disruption in striatal neurons for their survival; and (3) to what extent these contribute to HD neuropathology in a neuronal population vulnerable in HD such as the striatum. We showed that PC length is affected in the striatum in association with mHTT accumulation. PC disruption in MSN does not affect neuronal survival. Subtle compensatory mechanisms might be activated by dopaminergic neurons in response to PC loss on dopaminoceptive neurons; however, a detailed characterization awaits future experiments. PC disruption in striatal neurons of an HD mouse model results in increased mTOR activation and larger mHTT nuclear inclusions, suggesting that PC is required in a pathological context.
Changes in PC length have been reported in previous animal and cellular models expressing mHTT; however, this is the first study focusing on PC pathology in the striatal neurons that are pathologically affected in HD and addressing the impact of loss of PC in a progressive mouse model of HD. Here, we identified changes in PC length that are concomitant with mHTT accumulation: PC length is altered in striatal but not in cortical neurons in the zQ175 mice. Previous work has indicated that HTT is important for the transport of proteins required for ciliogenesis (Keryer et al., 2011). Ciliogenesis is a tightly regulated process, and it is crucial for its proper function that PC is produced in the right size and time (Avasthi and Marshall, 2012). Given the time- and cell-specific impact of mHTT on PC, it will be important to better understand how differences in mHTT state differentially modulate PC maintenance.
Interestingly, various HD models show altered levels of DA and its metabolites (Smith et al., 2014; Koch and Raymond, 2019), and lack of dopaminergic inputs results in longer PC in the striatum (Miyoshi et al., 2014). Hence changes of DA input in the zQ175 mice might alter PC length. Because DA receptors are located in the PC membrane (Marley and von Zastrow, 2010; Domire et al., 2011; Leaf and Von Zastrow, 2015), it will be important to investigate striatal synaptic transmission, and motor and psychiatric phenotypes in the HD mice lacking PC. Future studies should address the behavioral implications of the mild increase in DA metabolite levels and increased TH expression in the striatum. These studies might help to understand the impact of current DA-based symptomatic HD treatments on PC physiology and homeostasis.
The generation of tools enabling targeting of PC function independently of changes in their structural integrity might be necessary to define subtle regulatory functions. The Ift88 cKO approach, in which both PC function and structure are affected, does not allow us to dissect out the specific role of PC signaling from PC structure, however, it does allow us to identify cell-autonomous effects as well as non-cell autonomous responses to ciliary impairment. In aged homozygous zQ175 mice mTOR activity increases (Abd-Elrahman and Ferguson, 2019). We found in the zQ175 and in the dm mice increased phospho-S6 positive cells. Hence, another possible mechanism to explain changes in PC length might be linked to a block of autophagy by mTOR activation (Pampliega et al., 2013). Together, it will be important to analyze older zQ175 and dm mice. Conditional ablation of Ift88 in neural progenitors by the Nestin-Cre transgenic line revealed a significant increase in mTOR pathway activity and phospho-S6 ribosomal protein levels, resulting in hydrocephaly at late embryonic stages (Foerster et al., 2017). In the D1R-Cre driven conditional Ift88 knock-out, phospho-S6 does not increase in comparison to controls. These results indicate the cell-type, developmental stage and age-specific function of PC alterations, and support the need to further address this question in a mature and aging brain.
Diseases primarily caused by ciliary dysfunctions are commonly referred to as ciliopathies. As of yet, we cannot ascribe HD among the primary ciliopathies, nevertheless, PC structure and function are altered in association with a more severe phenotype. Notably, the role of PC in neuronal homeostasis, and in the control of the cellular stress response signaling cascades is emerging in other neurodegenerative disorders. PC pathology has been described for example in Alzheimer’s disease (AD), in Parkinson’s disease (PD) and in spinocerebellar ataxias, another polyglutamine disease, although distinct mechanisms are probably involved (Steger et al., 2017; Bowie et al., 2018; Dhekne et al., 2018; Vorobyeva and Saunders, 2018). Interestingly, PC were elongated in the hippocampus of the APP/PS1 mouse models of AD compared with wild-type mice, and serotonin 5-HT6 receptors playing a critical role in AD development regulate the morphology and function of neuronal PC (Hu et al., 2017). Moreover, in mouse models of PD expressing mutant LRRK2 R1441C, PC was affected in cholinergic neurons while the overall ciliation of neurons in the striatum was not significantly different from wild type (Steger et al., 2017; Dhekne et al., 2018). Nevertheless, defective ciliogenesis in striatal cholinergic neurons might impair a protective mechanism involving non-cell autonomous Sonic hedgehog between cholinergic and dopaminergic neurons (Gonzalez-Reyes et al., 2012). The Spinocerebellar ataxia type 11-associated mutation of the serine/threonine kinase Tau tubulin kinase 2 dominantly interferes with ciliogenesis and cilium stability (Bowie et al., 2018). Shortened primary cilium length and dysregulated Sonic hedgehog signaling were also reported in Niemann-Pick type C1 (NPC1) disease, a neurodegenerative lysosomal storage disorder caused by mutations in the NPC1 gene (Canterini et al., 2017).
In summary, although we obtained similar mean PC length as previously reported for striatum and cortex in mice (Sipos et al., 2018), a systematic quantitative comparison of PC marker expression and length in various neurodegenerative disease models and in human tissues will benefit from the use of automatized segmentation approaches (Vorobyeva and Saunders, 2018), and high content automated image acquisition. These future studies will be important to determine in other disease models which cells and tissues display the cilia defects and at what stages, and try to understand when, whether and how those changes lead to specific neuronal loss in the brain.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
Procedures involving animal care were approved by the Committee on Animal Care and Use (Regierungspräsidium Karlsruhe, Germany) in accordance with the local Animal Welfare Act and the European Communities Council Directives (2010/63/EU and 2012/707/EU; Ref. number: G-252/17).
Author Contributions
RM, GK, KK, and RP: data acquisition. RM, GK, JK, A-EW, KT, and RP: data analysis and interpretation. RP: study concept and design, drafting of the manuscript. All authors revised the submitted manuscript.
Funding
This work was supported by the European Huntington’s Disease Network (EHDN) seed-fund project 0907, by the “Deutsche Forschungsgemeinschaft” (DFG): DFG PA 1529/2-1 to RP, by start-up funds from the Center for Excellence in the Neurosciences at the University of New England (KT), and by the 2017/25/B/NZ7/02406 (Opus13) grant from the National Science Center and statutory funds of the Institute of Pharmacology (PAS) to GK and KK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the CHDI Foundation for providing the zQ175 mice used to establish the colony.
Abbreviations
ACIII, adenylate cyclase III; AD, Alzheimer’s disease; DA, dopamine; DAPI, 4′,6′-diamidino-2-pheylindol; DOPAC, 3,4-dihydroxyphenylacetic acid; D1R, dopaminoceptive D1-receptor; GFAP, glial fibrillary acidic protein; HD, Huntington’s disease; HPLC-EC, High-performance liquid chromatography-electrochemical detection; 5-HT, 5-hydroxytryptamine; HVA, homovanillic acid; IF, immunofluorescence; IFT-B, intraflagellar transport B; IHC, immunohistochemistry; mHTT, mutant Huntingtin; MSNs, medium spiny neurons; mTOR, mechanistic target of rapamycin; NeuN, neuronal nuclei; NPC1, Niemann-Pick type C1; PC, primary cilia; PD, Parkinson’s disease; PFA, paraformaldehyde; phospho-S6, phosphorylated ribosomal protein S6; p62/SQSTM1, p62/sequestosome 1; TH, tyrosine-hydroxylase.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00565/full#supplementary-material.
References
Abd-Elrahman, K. S., and Ferguson, S. S. G. (2019). Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol. Brain 12:35. doi: 10.1186/s13041-019-0456-1
Avasthi, P., and Marshall, W. F. (2012). Stages of ciliogenesis and regulation of ciliary length. Differentiation 83, S30–S42. doi: 10.1016/j.diff.2011.11.015
Bishop, G. A., Berbari, N. F., Lewis, J., and Mykytyn, K. (2007). Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol. 505, 562–571. doi: 10.1002/cne.21510
Bowie, E., Norris, R., Anderson, K. V., and Goetz, S. C. (2018). Spinocerebellar ataxia type 11-associated alleles of Ttbk2 dominantly interfere with ciliogenesis and cilium stability. PLoS Genet. 14:e1007844. doi: 10.1371/journal.pgen.1007844
Canterini, S., Dragotto, J., Dardis, A., Zampieri, S., De Stefano, M. E., Mangia, F., et al. (2017). Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum. Mol. Genet. 26, 2277–2289. doi: 10.1093/hmg/ddx118
Carty, N., Berson, N., Tillack, K., Thiede, C., Scholz, D., Kottig, K., et al. (2015). Characterization of HTT inclusion size, location, and timing in the zQ175 mouse model of Huntington’s disease: an in vivo high-content imaging study. PLoS One 10:e0123527. doi: 10.1371/journal.pone.0123527
Chen, J. Y., Wang, E. A., Cepeda, C., and Levine, M. S. (2013). Dopamine imbalance in Huntington’s disease: a mechanism for the lack of behavioral flexibility. Front. Neurosci. 7:114. doi: 10.3389/fnins.2013.00114
Dhekne, H. S., Yanatori, I., Gomez, R. C., Tonelli, F., Diez, F., Schüle, B., et al. (2018). A pathway for Parkinson’s disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. Elife 7:e40202. doi: 10.7554/eLife.40202
Domire, J. S., Green, J. A., Lee, K. G., Johnson, A. D., Askwith, C. C., and Mykytyn, K. (2011). Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell. Mol. Life Sci. 68, 2951–2960. doi: 10.1007/s00018-010-0603-4
Farrar, A. M., Murphy, C. A., Paterson, N. E., Oakeshott, S., He, D., Alosio, W., et al. (2014). Cognitive deficits in transgenic and knock-in HTT mice parallel those in Huntington’s disease. J. Huntingtons Dis. 3, 145–158. doi: 10.3233/jhd-130061
Foerster, P., Daclin, M., Asm, S., Faucourt, M., Boletta, A., Genovesio, A., et al. (2017). mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 144, 201–210. doi: 10.1242/dev.138271
Gazea, M., Tasouri, E., Tolve, M., Bosch, V., Kabanova, A., Gojak, C., et al. (2016). Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev. Biol. 409, 55–71. doi: 10.1016/j.ydbio.2015.10.033
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. doi: 10.1016/j.cell.2015.07.003
Ghosh, R., and Tabrizi, S. J. (2018). Clinical features of Huntington’s disease. Adv. Exp. Med. Biol. 1049, 1–28. doi: 10.1007/978-3-319-71779-1_1
Gonzalez-Reyes, L. E., Verbitsky, M., Blesa, J., Jackson-Lewis, V., Paredes, D., Tillack, K., et al. (2012). Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron 75, 306–319. doi: 10.1016/j.neuron.2012.05.018
Haycraft, C. J., Zhang, Q., Song, B., Jackson, W. S., Detloff, P. J., Serra, R., et al. (2007). Intraflagellar transport is essential for endochondral bone formation. Development 134, 307–316. doi: 10.1242/dev.02732
Hu, L., Wang, B., and Zhang, Y. (2017). Serotonin 5-HT6 receptors affect cognition in a mouse model of Alzheimer’s disease by regulating cilia function. Alzheimers Res. Ther. 9:76. doi: 10.1186/s13195-017-0304-4
Johansen, T., and Lamark, T. (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296. doi: 10.4161/auto.7.3.14487
Kaliszewski, M., Knott, A. B., and Bossy-Wetzel, E. (2015). Primary cilia and autophagic dysfunction in Huntington’s disease. Cell Death Differ. 22, 1413–1424. doi: 10.1038/cdd.2015.80
Karam, A., Tebbe, L., Weber, C., Messaddeq, N., Morle, L., Kessler, P., et al. (2015). A novel function of Huntingtin in the cilium and retinal ciliopathy in Huntington’s disease mice. Neurobiol. Dis. 80, 15–28. doi: 10.1016/j.nbd.2015.05.008
Keryer, G., Pineda, J. R., Liot, G., Kim, J., Dietrich, P., Benstaali, C., et al. (2011). Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Invest. 121, 4372–4382. doi: 10.1172/jci57552
Koch, E. T., and Raymond, L. A. (2019). Dysfunctional striatal dopamine signaling in Huntington’s disease. J. Neurosci. Res. 97, 1636–1654. doi: 10.1002/jnr.24495
Leaf, A., and Von Zastrow, M. (2015). Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 4:e06996. doi: 10.7554/eLife.06996
Lemberger, T., Parlato, R., Dassesse, D., Westphal, M., Casanova, E., Turiault, M., et al. (2007). Expression of Cre recombinase in dopaminoceptive neurons. BMC Neurosci. 8:4. doi: 10.1186/1471-2202-8-4
Levine, M. S., Klapstein, G. J., Koppel, A., Gruen, E., Cepeda, C., Vargas, M. E., et al. (1999). Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 58, 515–532. doi: 10.1002/(sici)1097-4547(19991115)58:4<515::aid-jnr5>3.3.co;2-6
Ma, Q., Yang, J., Li, T., Milner, T. A., and Hempstead, B. L. (2015). Selective reduction of striatal mature BDNF without induction of proBDNF in the zQ175 mouse model of Huntington’s disease. Neurobiol. Dis. 82, 466–477. doi: 10.1016/j.nbd.2015.08.008
Maiuri, T., Woloshansky, T., Xia, J., and Truant, R. (2013). The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum. Mol. Genet. 22, 1383–1394. doi: 10.1093/hmg/dds554
Malicki, J. J., and Johnson, C. A. (2017). The cilium: cellular antenna and central processing unit. Trends Cell Biol. 27, 126–140. doi: 10.1016/j.tcb.2016.08.002
Marley, A., and von Zastrow, M. (2010). DISC1 regulates primary cilia that display specific dopamine receptors. PLoS One 5:e10902. doi: 10.1371/journal.pone.0010902
Menalled, L. B., Kudwa, A. E., Miller, S., Fitzpatrick, J., Watson-Johnson, J., Keating, N., et al. (2012). Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS One 7:e49838. doi: 10.1371/journal.pone.0049838
Miyoshi, K., Kasahara, K., Murakami, S., Takeshima, M., Kumamoto, N., Sato, A., et al. (2014). Lack of dopaminergic inputs elongates the primary cilia of striatal neurons. PLoS One 9:e97918. doi: 10.1371/journal.pone.0097918
Pampliega, O., Orhon, I., Patel, B., Sridhar, S., Diaz-Carretero, A., Beau, I., et al. (2013). Functional interaction between autophagy and ciliogenesis. Nature 502, 194–200. doi: 10.1038/nature12639
Parker, A. K., Le, M. M., Smith, T. S., Hoang-Minh, L. B., Atkinson, E. W., Ugartemendia, G., et al. (2016). Neonatal seizures induced by pentylenetetrazol or kainic acid disrupt primary cilia growth on developing mouse cortical neurons. Exp. Neurol. 282, 119–127. doi: 10.1016/j.expneurol.2016.05.015
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Sikora, M., Tokarski, K., Bobula, B., Zajdel, J., Jastrębska, K., Cieślak, P. E., et al. (2016). NMDA receptors on dopaminoceptive neurons are essential for drug-induced conditioned place preference. eNeuro 3:ENEURO.0084-15.2016. doi: 10.1523/eneuro.0084-15.2016
Sipos, É., Komoly, S., and Ács, P. (2018). Quantitative comparison of primary cilia marker expression and length in the mouse brain. J. Mol. Neurosci. 64, 397–409. doi: 10.1007/s12031-018-1036-z
Smith, G. A., Rocha, E. M., McLean, J. R., Hayes, M. A., Izen, S. C., Isacson, O., et al. (2014). Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum. Mol. Genet. 23, 4510–4527. doi: 10.1093/hmg/ddu166
Steger, M., Diez, F., Dhekne, H. S., Lis, P., Nirujogi, R. S., Karayel, O., et al. (2017). Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6:e31012. doi: 10.7554/eLife.31012
Tang, Z., Lin, M. G., Stowe, T. R., Chen, S., Zhu, M., Stearns, T., et al. (2013). Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502, 254–257. doi: 10.1038/nature12606
Keywords: primary cilium, dopamine system, Huntington’s disease, mTOR, p62
Citation: Mustafa R, Kreiner G, Kamińska K, Wood A-EJ, Kirsch J, Tucker KL and Parlato R (2019) Targeted Depletion of Primary Cilia in Dopaminoceptive Neurons in a Preclinical Mouse Model of Huntington’s Disease. Front. Cell. Neurosci. 13:565. doi: 10.3389/fncel.2019.00565
Received: 10 July 2019; Accepted: 05 December 2019;
Published: 20 December 2019.
Edited by:
Thomas Fath, Macquarie University, AustraliaReviewed by:
Jiro Kasahara, Tokushima University, JapanErik B. Malarkey, Vertex Pharmaceuticals, United States
Copyright © 2019 Mustafa, Kreiner, Kamińska, Wood, Kirsch, Tucker and Parlato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosanna Parlato, cm9zYW5uYS5wYXJsYXRvQHVuaS11bG0uZGU=