 Camila Fabiani
Camila Fabiani Silvia S. Antollini
Silvia S. Antollini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 18 July 2019
Sec. Cellular Neurophysiology
Volume 13 - 2019 | https://doi.org/10.3389/fncel.2019.00309
This article is part of the Research Topic Emerging Mechanisms in Neuronal Signaling: from Cell Biology to Pathogenesis View all 34 articles
Biological membranes show lateral and transverse asymmetric lipid distribution. Cholesterol (Chol) localizes in both hemilayers, but in the external one it is mostly condensed in lipid-ordered microdomains (raft domains), together with saturated phosphatidyl lipids and sphingolipids (including sphingomyelin and glycosphingolipids). Membrane asymmetries induce special membrane biophysical properties and behave as signals for several physiological and/or pathological processes. Alzheimer’s disease (AD) is associated with a perturbation in different membrane properties. Amyloid-β (Aβ) plaques and neurofibrillary tangles of tau protein together with neuroinflammation and neurodegeneration are the most characteristic cellular changes observed in this disease. The extracellular presence of Aβ peptides forming senile plaques, together with soluble oligomeric species of Aβ, are considered the major cause of the synaptic dysfunction of AD. The association between Aβ peptide and membrane lipids has been extensively studied. It has been postulated that Chol content and Chol distribution condition Aβ production and posterior accumulation in membranes and, hence, cell dysfunction. Several lines of evidence suggest that Aβ partitions in the cell membrane accumulate mostly in raft domains, the site where the cleavage of the precursor AβPP by β- and γ- secretase is also thought to occur. The main consequence of the pathogenesis of AD is the disruption of the cholinergic pathways in the cerebral cortex and in the basal forebrain. In parallel, the nicotinic acetylcholine receptor has been extensively linked to membrane properties. Since its transmembrane domain exhibits extensive contacts with the surrounding lipids, the acetylcholine receptor function is conditioned by its lipid microenvironment. The nicotinic acetylcholine receptor is present in high-density clusters in the cell membrane where it localizes mainly in lipid-ordered domains. Perturbations of sphingomyelin or cholesterol composition alter acetylcholine receptor location. Therefore, Aβ processing, Aβ partitioning, and acetylcholine receptor location and function can be manipulated by changes in membrane lipid biophysics. Understanding these mechanisms should provide insights into new therapeutic strategies for prevention and/or treatment of AD. Here, we discuss the implications of lipid-protein interactions at the cell membrane level in AD.
Biological membranes were, are, and will be complex, dynamic and controversial. Several different theories/models were postulated until the fluid-mosaic model was proposed by Singer and Nicolson (1972). This description of a biological membrane was very well accepted and gave light about membrane structure and membrane function. Although a lot of new information appeared in the following 40 years, the model was able to survive by absorbing some modifications, as it was emphasized by Nicolson (2014). Table 1 details and compares the most important features of the original fluid-mosaic model membrane (Singer and Nicolson, 1972) and the current vision of a membrane (Engelman, 2005; Bagatolli, 2010; Goñi, 2014; Nicolson, 2014).

Table 1. Comparison of the main membrane characteristics proposed by the Singer and Nicolson (1972) model and the current cell membrane vision (based on Engelman (2005), Bagatolli (2010), Goñi (2014), Nicolson (2014), and references there in).
Nowadays, a membrane is thought of as an increasingly complex crowded structure of a great variety of lipid and protein arrangements with lateral and transverse asymmetry, variable patchiness, variable thickness, and higher protein occupancy (Engelman, 2005; Nicolson, 2014). It is universally accepted that biological membranes behave as barriers separating two fluid media and avoiding contact with each other. But being a physical barrier is not its only or main function. Many of the biochemical reactions essential for cell life (metabolic and signaling reactions involving G-protein coupled receptors as the rhodopsin or muscarinic receptor and ion channels as nicotinic, histamine, GABA or glutamate receptors among others transmembrane proteins) occur in the cell membranes, making them a truly important agent in almost all cellular physiological and pathological processes. These reactions imply molecular communication, which involves protein–protein and also protein–lipid interactions. Lipid membranes are not just a “sea” where proteins are embedded, as it was initially postulated by Singer and Nicolson. Lipids (including fatty acids, cholesterol, endocannabinoids, arachidonic acid metabolites as prostaglandins, leukotrienes, and epoxyeicosatrienoic acids, etc.) are active molecules with important implications. Lipids such as chol, cardiolipin, PIP2 and glycolipids condition the function of several transmembrane proteins, a fact reflected in the thousands of research papers that report the effect of these lipids on protein functions (Lee, 2003; Barenholz, 2004; Barrera et al., 2013). Here, we will discuss the implications of lipid-protein interactions at the cell membrane level in AD.
Alzheimer’s disease is the most prevalent neurodegenerative disorder in the elderly, and is characterized by progressive cognitive decline. The main pathophysiological characteristics include extracellular accumulation of β-amyloid senile plaques and intracellular accumulation of neurofibrillary tangles (hyperphosphorylated microtubule-associated tau protein) (Feng and Wang, 2012; Kumar et al., 2015). A disruption of the cholinergic pathways that contribute to the cognitive impairment of AD patients is described in the cerebral cortex and in the basal forebrain. AD implicates the formation of extracellular insoluble peptides derived from the action of two transmembrane enzymes, a β-secretase (β-site APP-cleaving transmembrane aspartic protease, BACE 1) and a γ-secretase (an imprecise multimeric protein complex), on the membrane-bound APP. Aβ peptides of different lengths, containing 39–42 amino acid residues, are produced. 1–40 Aβ is produced more frequently while 1–42 Aβ is the predominant species in senile plaques (Iwatsubo et al., 1994). They are amphiphilic peptides with residues 1–28 constituting a hydrophilic domain and residues 29 up to 42 (which correspond to part of the transmembrane domain of APP), a hydrophobic one (Ji et al., 2002). Whereas low concentrations of 1–40 Aβ are related to neurotrophic properties (Yankner et al., 1990; Zou et al., 2002, 2003), 1–42 Aβ, produced in low amounts under physiological conditions, has a much higher tendency to form oligomers, protofibrils and fibrils, which are the ones that constitute AD brain plaques (Jarrett et al., 1993; Gu and Guo, 2013). The structural-activity relation between these assemblies and the differences between 1–40 Aβ and 1–42 Aβ are under continuous investigation and exceed the aim of this review. Alternatively, APP can be cleaved by another membrane enzyme (α-secretase) between amino acids 16 and 17 of the Aβ region, avoiding Aβ peptides generation and producing a neurotrophic and neuroprotective soluble AβPP (sAβPPα) through a non-amyloidogenic pathway (Thornton et al., 2006; Wang et al., 2016). In neurons, amyloidogenic and non-amyloidogenic pathways compete with each other, jumping between neuroprotection and neurodegeneration (Vetrivel and Thinakaran, 2006; Tan and Gleeson, 2019). Furthermore, in normal brains, 1–42 Aβ is produced in low picomolar concentrations and, as it will be explained later, these low, non-toxic concentrations have physiological implications in synaptic plasticity and memory, among others (Plant et al., 2003; Puzzo et al., 2008, 2011, 2015). In fact, physiological 1–42 Aβ binds to several target molecules as apoE, the receptor for advanced glycosylation end products (RAGE), serpin–enzyme complex receptor (SEC-R) and nicotinic acetylcholine (nACh) receptors (Turner et al., 2003). Thus, although during a person’s lifetime there is a continuous formation of all these peptides, the deregulation of the enzymatic equilibrium with the consequent accumulation of insoluble peptides is characteristic of AD. 1–42 Aβ is the most hydrophobic peptide that forms soluble oligomeric intermediates before aggregating as insoluble plaques with cytotoxic properties in the AD brains. It induces iron and cooper reduction in the brain triggering oxidative stress and damage, it causes calcium homeostasis deregulation probably through lipid perturbation at the cell membrane, and it causes oxy-radicals formation and finally neurodegeneration (Butterfield et al., 2013; Fonseca et al., 2015; Cheignon et al., 2018). Amyloidogenic and non-amyloidogenic pathways are thought to occur in different cellular compartments depending on secretases localization. γ-secretase complex is present in multiple compartments: near 6% in the plasma membrane and the rest in intracellular organelles such as endoplasmic reticulum, late Golgi/trans-Golgi network and endosomes (Vetrivel et al., 2004; Chyung et al., 2005). However, α- and β-secretases are more compartmentalized. α-cleavage occurs at the cell surface (Parvathy et al., 1999; Haass et al., 2012; Sun and Roy, 2018). APP is released to the plasma membrane through the secretory pathway and stays there for a short time. Therefore, during this short time, APP is proteolytically processed by α-secretase. Anyway, near 70% of APP is internalized by endocytosis. A fraction of this APP is recycled to the cell surface and another one is degraded in lysosomes. BACE 1 is localized late in the Golgi/trans-Golgi network and endosomes and cleaves APP during the endocytic/recycling cycle (Koo and Squazzo, 1994); thus, β-cleavage depends on endocytosis (Koo and Squazzo, 1994; Perez et al., 1999; Huse et al., 2000; Daugherty and Green, 2001; Kamal et al., 2001; Ehehalt et al., 2003) and Aβ is produced mainly in the trans-Golgi network during the recycling pathway (Vetrivel and Thinakaran, 2006). Additionally, it was suggested that 1–42 Aβ is produced mainly in the endoplasmatic reticulum whereas 1–40 Aβ is produced in the trans-Golgi network (Annaert et al., 1999; Greenfield et al., 1999).
APP cleavage by secretases always happens in a membrane, independently of the subcellular compartment. To understand the importance of this fact, it is recommended to read the general commentary by Lukiw (2013) in Frontiers in Physiology titled “Alzheimer’s disease (AD) as a disorder of the plasma membrane,” whereas the author pointed out the implication that the membrane has in the physiopathology of this disease. Several studies postulated that membrane components condition the APP enzymatic processing. Particularly, Chol is a key element in the membrane and it has been related to AD in several ways. Lahdo and De La Fournière-Bessueille (2004) studied the minimum lipid requirements of a monolayer for the insertion of APP. They concluded that APP insertion depends on the Chol content, the Chol/PC and the Chol/SM ratios, and the monolayer membrane order. They identified a critical inflection point at near 30% Chol: at a lower ratio APP localizes in the membrane surface mainly in a β-sheet conformation, whereas as this Chol percentage increases, APP can insert spontaneously into the membrane changing its conformation (Ji et al., 2002; Lahdo and De La Fournière-Bessueille, 2004). Consequently, once APP is confined to the interior of the membrane it can perturb the biophysical properties of this membrane and the activity of several transmembrane or associated-membrane proteins. The Chol concentration and Chol location in brain plasma membranes change throughout a person’s life. At early ages, about 87% of the Chol is localized mainly in the inner layer of the brain plasma membrane, but during aging, the percentage of Chol increases in the outer layer losing the initial transmembrane asymmetry and reaching at least 30 mol%, the critical value with respect to APP membrane insertion (Igbavboa et al., 1996; Wood et al., 2002). In another work, it was suggested that modifications of Chol compartmentation and the equilibrium free cholesterol/cholesteryl esters through acyl-coenzyme A:Chol acyltransferase (ACAT) activation, instead of variations of total membrane Chol, are the determinant of Aβ accumulation and cell dysfunction (Puglielli et al., 2001).
The importance of the amount of Chol for APP insertion leads us to think that APP would probably prefer raft domains (Cordy et al., 2006; Reid et al., 2007). Initial studies in the brain showed that both APP and Aβ reside in detergent-insoluble glycolipid-enriched membrane domains (DIG) (Lee et al., 1998), suggesting that those domains are the place in the membrane where the APP processing occurs. The non-amyloidogenic pathway through α-secretase is thought to occur in non-raft domains (Kojro et al., 2001; Reid et al., 2007). On the other hand, although there is no consensus about the localization of APP and BACE 1, there is agreement that APP cleavage by β- and γ-secretase occurs in raft domains (see Table 1 for a detailed explanation of raft domains). Experiments in culture cells showed that overexpressed APP and both secretases enzymes localize in Chol-rich domains (Burns and Duff, 2002; Ehehalt et al., 2003), and that Chol depletion by Chol synthesis inhibition or Chol membrane extraction resulted in a reduction of Aβ production (Simons et al., 1998; Fassbender et al., 2001; Ehehalt et al., 2003). Several studies suggest that APP is present in two cellular pools: one in raft domains and another in non-raft domains. Ehehalt et al. (2003) concluded that this APP membrane compartmentation explains how the same protein could be processed in two different ways (generating Aβ in raft domains and being cleaved by α-secretase in non-raft domains). Furthermore, they said that although BACE 1 is present in both raft and non-raft domains it needs to be in raft domains to be functional, outside these domains the enzyme is inactive (Figure 1a). That is the reason why, when Chol diminishes, Aβ production also diminishes but increases αCTF (C-terminal fragment) or C83, which is a direct product of α-secretase. Thus, Chol regulates the access of α or β secretase to APP (Ehehalt et al., 2003). On the other hand, immediately after this study, a study in human hippocampal membranes showed that the vast majority of APP is located in non-raft domains, while β-secretase BACE 1 is found in two cellular pools: one in raft domains and another in non-raft domains (Abad-Rodriguez et al., 2004). These authors gave an explanation opposite to the previous one: when Chol diminishes, which is what happens in the membrane from AD patients (Mason et al., 1992; Roth et al., 1995), BACE 1 increases in non-raft domains and then an enhancement of amyloid peptide production occurs. They concluded that BACE 1 in raft domains corresponds to an inactive pool that needs to relocate to non-raft domains to perform its activity, and that it is the Chol which directly conditions APP processing by “allowing” BACE 1 to exit or not from neighboring domains (Figure 1b). However, they distinguished between a mild membrane Chol reduction (less than 25%), which results in an increase of APP processing, and a drastic membrane Chol reduction (more than 35%), where an overall disruption of membrane integrity occurs concomitantly with a lower Aβ production. Working with primary cultures of rat hippocampal neurons infected with recombinant Semliki Forest virus (SFV) carrying APP, Simons et al. (1998) arrived to a different conclusion. They showed that depletion of Chol up to 60–70% did not affect the amount of APPsec (the main processed form of APP in neurons obtained by direct α-cleavage), but drastically decreased the amount of Aβ. Therefore, Chol depletion appears to redirect the APP processing from amyloidogenic processing to non-amyloidogenic cleavage. One possible explanation for this is that the small raft-resident pool of APP and BACE 1 is the active one and that it generates C99 to be processed by γ-secretase (Rushworth and Hooper, 2011). Another explanation considers that the amount of both proteins in rafts is so small that the APP processing by BACE 1 is effective once a clustering of raft domains occurs during endocytosis, meanwhile, in the plasma membrane, APP will be mainly cleaved by α-secretase through a non-amyloidogenesis pathway (Ehehalt et al., 2003). Thus, it is possible that APP processing can be altered by membrane lipid composition perturbations. Eckert et al. (2003) showed that Chol depletion decreases the amount of APP in raft domains and, consequently, the production of Aβ. On the other side, Chol increment as in Niemann Pick type C model cells, causes an APP augmentation in raft domains.
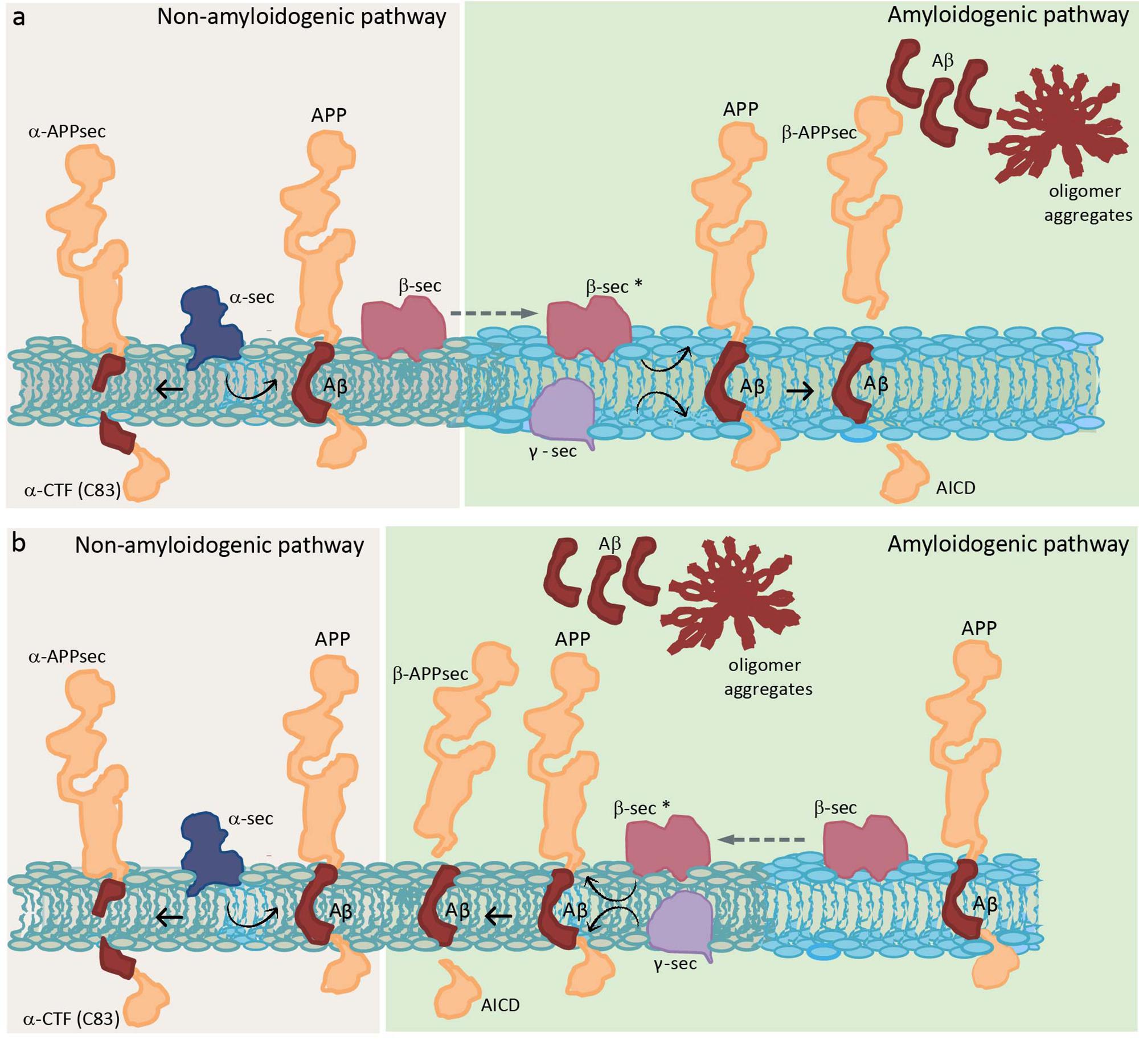
Figure 1. Schematic diagram showing two distinct hypotheses of APP processing, which differ in the membrane location of the whole process. Two different colors are used to represent a raft domain and a liquid-disordered domain (light blue and gray, respectively). (a) Hypothesis where β sec is present in both raft and non-raft domains but needs to be in raft domains to be functional (represented as β sec*) (Ehehalt et al., 2003). (b) Hypothesis where β sec in raft domains corresponds to an inactive pool that needs to relocate to non-raft domains to be functional (Abad-Rodriguez et al., 2004). APP, amyloid precursor protein; α-CTF, C-terminal fragment obtained by α-secretase; α-APPsec, soluble N-terminal APP fragment obtained by α-secretase; Aβ, amyloid β peptide; β-APPsec, soluble N-terminal APP fragment obtained by β-secretase; AICD, APP intracellular domain obtained by the action of γ-secretase on β-CTF or C99 (intermediate peptide that is not shown and corresponds to Aβ plus AICD, obtained in the first step by the action of β-secretase); α-sec, α-secretase; β-sec, β-secretase; and γ-sec, γ-secretase.
A controversial point is where are secretases located, especially β-secretase, and where they function in the membrane. With respect to γ-secretase, however, there is broad consensus. It is postulated that this enzyme is localized in raft domains confirming that the last step in the generation of Aβ occurs in those domains (Vetrivel et al., 2005). These authors postulated that once APP is cleaved by β-secretase, the CTFs (or C99) produced are recruited or sequestered into raft domains where cleavage by γ-secretase takes place. They indicated that ∼20% of BACE 1, less than 5% of APP and more than 70% of CTFs reside in raft domains; and, based on previous work, they assume that all cleavage occurs in these rigid domains (Vetrivel et al., 2005).
By magnetic nuclear resonance of C99, Beel et al. (2010) identified a short sequence of 5 amino acids (VGSNK) between the extracellular segment and the transmembrane domain that interacts with Chol, probably through hydrogen bonds. These authors recognize that although C99 is in raft domains, APP, which has the same loop, localizes mainly in non-raft domains, concluding that one possibility is that APP and C99 have different affinities for Chol. This Chol interaction site is also present in Aβ peptides, thus explaining the reported Chol-Aβ peptides interactions that trigger oligomerization, fibrillization, etc. (Beel et al., 2010) which will be discussed below.
Chol is not only crucial for APP processing in the membrane by compartmentalizing the location of both APP and secretases, but also for modulating the secretases activity. Briefly, Chol positively modulates BACE 1 and γ-secretase activities, and negatively modulates α-secretase (Bodovitz and Klein, 1996; Simons et al., 1998; Frears et al., 1999; Kojro et al., 2001; Wahrle et al., 2002; Ghribi et al., 2006). In lysates from human brain and in cultured cells, a certain amount of Chol stimulated β and γ-secretase activities, but at 20 μM Chol γ-secretase activity was inhibited. It is probable that high Chol can directly stabilize the activities of the enzymes to the maximum level in the correct lipid domain or can reduce enzymes degradation increasing Aβ production (Xiong et al., 2008). Furthermore, APP processing can be modulated by Chol conditioning membrane biophysical properties (Kojro et al., 2001; Fukaya et al., 2007; Kogel et al., 2008; Peters et al., 2009; Yang et al., 2010; Askarova et al., 2011). For example, substitution of Chol by lanosterol or polyunsaturated free fatty acids (PUFAs) induced an increment of membrane fluidity, which was related to an enhancement of α-secretase activity (Kojro et al., 2001; Yang et al., 2010; Askarova et al., 2011).
Chol is also important for Aβ peptide action/effect (Eckert et al., 2003; Wood et al., 2014). Aβ peptide can adopt different conformations: a random-coil conformation in aqueous solution, an antiparallel β-sheet in the core of the amyloid plaques, and an α-helix in membranes containing Chol (Ji et al., 2002). It can exist as monomers, oligomers or as amyloid fibrils (Klein et al., 2004; Reid et al., 2007). Several studies indicate that Chol directly binds to APP as was described above (Beel et al., 2010), and also to C99 and monomeric Aβ peptides (Barrett et al., 2012), to oligomers (Ashley et al., 2006), to aggregates (Avdulov et al., 1997), and to fibrils (Harris, 2008). Considering that the mechanism by which Aβ produces brain dysfunction in AD patients is still unknown, these evidences turned the view of Aβ peptides pathogenesis from extracellular plaques (the “amyloid theory”: extracellular amyloid plaques are the responsible for cell death; Hardy and Higgins, 1992; Haass, 1996; Rushworth and Hooper, 2011; Serrano-Pozo et al., 2011) to Aβ peptides interaction with the plasma membrane (Relini et al., 2014). A more recent explanation indicates that Aβ monomers or small oligomers are responsible of neuronal death rather than amyloid plaques as it was previously thought (Irvine et al., 2008; Shankar et al., 2008). Furthermore, amyloid plaques reduce neuronal death by sequestering the dangerous Aβ monomers/small oligomers (Arbor et al., 2016) (Figure 2). This explanation is contrary to a previous one that considered that Aβ aggregation in β-sheet conformation, which will finally end as neurotoxic fibrils, is reduced by Aβ insertion as α-helix after interaction with Chol-containing membranes (Ji et al., 2002). They showed that both 1–40 Aβ and 1–42 Aβ peptides prefer Chol enriched LDM and that while in healthy humans the amount of the former peptide is more than twice the second one, the progression of 1–42 Aβ deposition runs in parallel with an increase of this peptide in LDM domains (Oshima et al., 2001; Ji et al., 2002). The authors concluded that Chol enrichment would be beneficial for reducing fibrils, a membrane condition opposite to the one found in AD brains that show a drastic decrease of membrane Chol content and hence do not have the needed conditions for Aβ insertion, favoring the dangerous pathway of Aβ aggregation (Ji et al., 2002). In the case of AD patients, isolated brain membranes showed a significant decrease in membrane Chol, disfavoring the insertion of Aβ into the membrane (Mason et al., 1992; Roth et al., 1995). Thus, Aβ remains in the membrane surface with a great tendency to aggregation and, ultimately, to plaque formation (Ji et al., 2002). By confocal laser microscopy and fluorescence anisotropy, it was shown that 1–42 Aβ peptides interact with raft domains and that there is an inverse correlation between Chol content and membrane perturbation (Cecchi et al., 2009). It was further indicated that a Chol increment decreases amyloid-induced membrane perturbations at lipid rafts by altering the physicochemical properties of the domain (Cecchi et al., 2009). Specific interactions that induce changes in the lipid bilayer conducing to membrane disruption were described between lipids and Aβ peptides (Qiang et al., 2014). Different kinds of interactions were proposed in the last few years. Vestergaard et al. (2010) performed studies of Aβ interactions with model biomimetic membranes and showed that immediately after peptide addition, membrane fluctuations/morphological changes occur. They suggested that both Chol levels and lipid composition affect how Aβ oligomers interact with the membrane. X-ray diffraction studies of the interaction between a 25–35 Aβ peptide and anionic membranes enabled the identification of immiscible Chol plaques when more than 30 mol% Chol was added. The peptide interacts with the bilayers sequestering more Chol molecules into the plaques and, hence, decreasing the amount of Chol in the membrane (Dies et al., 2014).
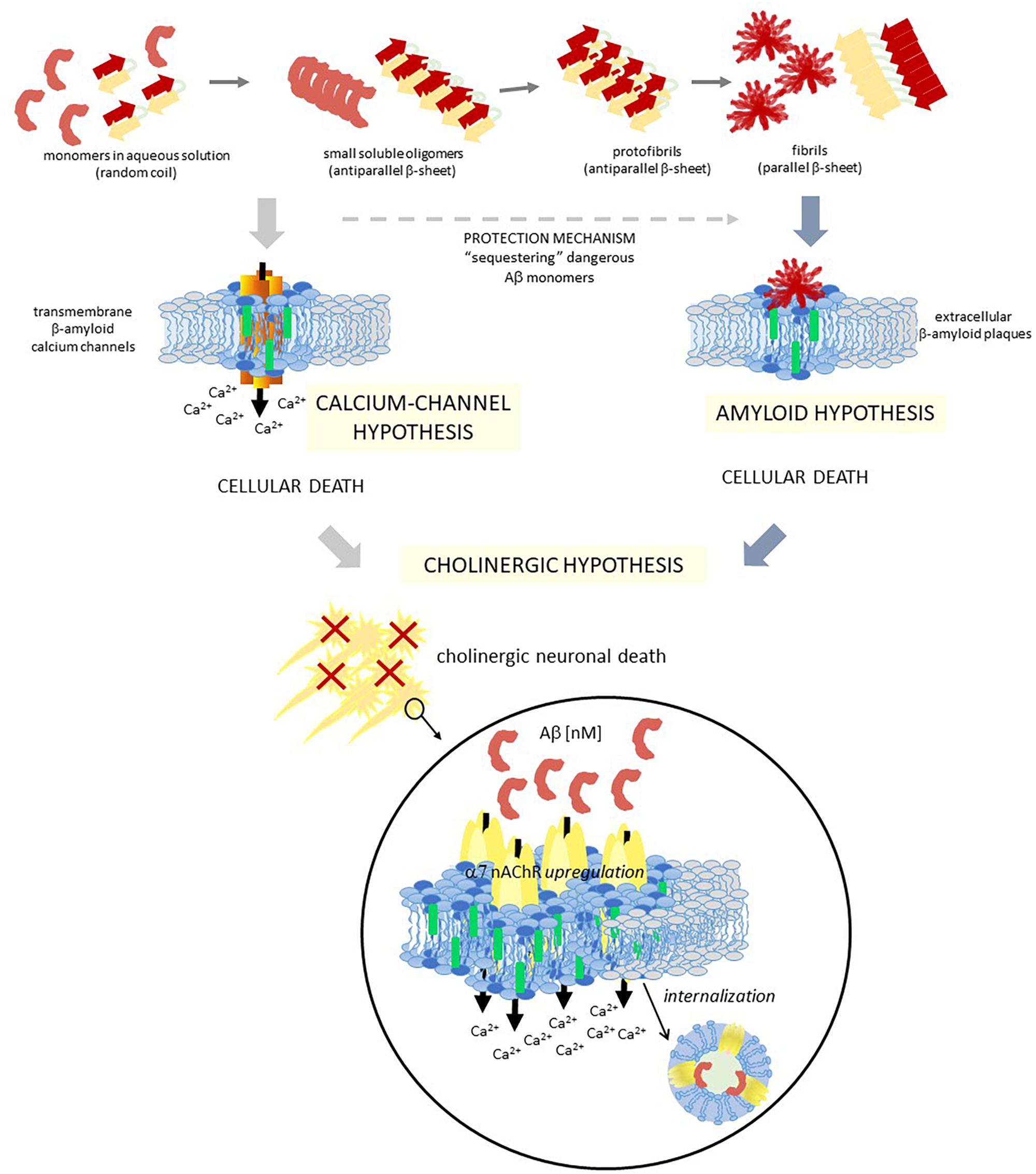
Figure 2. Schematic diagram showing three different hypotheses of AD, which are closely related. Two different colors are used to represent a raft domain and a liquid-disordered domain (light blue and gray, respectively), and also within raft domains, two central lipids are identified for these hypotheses with different colors: chol and GM1 (green and blue, respectively). Aβ, amyloid β peptide; α7 nAChR, α7 nicotinic acetylcholine receptor.
Chol is not the only important lipid in the Aβ-membrane interactions, there is also GM1, which is a resident lipid of raft domains (Lin et al., 2008). High Chol levels facilitate gangliosides clustering, which is postulated to modulate Aβ oligomerization. These clusters interact with Aβ peptides in a concentration dependent manner inducing Aβ aggregation in β-sheet rich structures with a high Aβ/ganglioside relationship (McLaurin et al., 1998; Ariga et al., 2001; Kakio et al., 2001, 2002; Matsuzaki, 2007). The binding of Aβ to a GM1 cluster favors a conformation transition that depends on the protein density of the membrane. At low peptide/lipid ratios a transition from random coil to α-helix conformation occurs, whereas at high peptide/lipid ratios a β-sheet rich structure appears, which ends in fibrils formation (Matsuzaki et al., 2010; Fukunaga et al., 2012). Significant alterations in the lipid composition of raft domains in frontal cortex of AD patients were described (Martín et al., 2010; Kosicek and Hecimovic, 2013; Fabelo et al., 2014). A detailed study of the lipid composition of DRM from temporal and frontal cortex of AD brains indicated that there was an increment of GM1 and GM2 in both areas of the brain studied (Molander-Melin et al., 2005). This difference, which was considered an early event in the progression of AD, was not observed between samples from brains of different ages or gender (Molander-Melin et al., 2005). Other studies agree with an age-dependent high-density GM1 clustering in synaptosomes (Gylys et al., 2007; Yamamoto et al., 2008) and specific Aβ peptides and GM1 complexes in early AD brains. GM1-Aβ interactions (GAβ) were described in the brain and associated with AD pathology (Yanagisawa et al., 1995; Choo-Smith et al., 1997; Yanagisawa and Ihara, 1998; Kakio et al., 2001, 2002; Yamamoto et al., 2004, 2005; Wakabayashi et al., 2005). Thus, GM1 is postulated as the seed for the formation of amyloid fibrils (Staneva et al., 2018), and several studies considered raft platforms as the site where these interactions happen. Sasahara et al. (2013) showed that the interaction and aggregation of the peptide enhances lipid phase separation because of the GM1 relation with Aβ aggregates. A two-step phase was postulated to occur in membranes of AD patients: at early stages the proportion of GM1 in raft domains increases accelerating Aβ plaques formation and triggering a gradual raft disruption and perturbation of the cellular function that involves these membrane domains; at a later stage, there is also a decrement in Chol content which prevents Aβ aggregation and increases neurotoxicity (Molander-Melin et al., 2005). A more detailed study of the mechanism of Aβ interaction with GM1 indicates that Aβ oligomers, which have increased hydrophobicity compared to Aβ monomers, primarily bind to GM1 initiating progressive alterations such as membrane biophysical and ion permeability perturbations that end in the well-known synaptotoxic effects of Aβ (Hong et al., 2014).
Although all data points to a direct implication of gangliosides on Aβ oligomerization, a ganglioside-independent Aβ oligomerization mechanism was also observed, suggesting that other lipid components or carbohydrates in raft domains would be also implicated (Kim et al., 2006).
One important consequence of these raft domains/Aβ peptide interaction is the occurrence of Aβ peptide aggregation and Ca+2 channels formation in raft domains (Lin et al., 2001). In hippocampal cell membranes this process was related to neurotoxicity (Sepúlveda et al., 2014). Di Scala et al. (2013) identified a Chol binding domain in a 20–35 fragment of 1–42 Aβ, which is also present in other peptides with high Chol affinity. Interestingly, although both APP and 1–42 Aβ interact with Chol, they have distinct binding domains [17–40 Aβ for APP (Barrett et al., 2012) and 20–35 Aβ for 1–42 Aβ (Di Scala et al., 2013; Fantini et al., 2014)]. By physicochemical and in silico experiments, it was demonstrated that this 20–35 Aβ domain forms oligomeric Ca2+ channels in the plasma membrane in a Chol dependent manner (Di Scala et al., 2014). The high interaction with Chol of this 20–35 Aβ domain triggers the helix to an adequate tilted orientation inside the membrane, which allows accurate peptide-peptide interactions and the formation of the circular channel. This oligomeric channel is formed by eight 20–35 Aβ subunits and eight Chol molecules, with a pore size and an external diameter of 1.46 and 4.4 nm, respectively. The formation of these channels could help explain the neurotoxic properties of 1–42 Aβ (Figure 2). Similar in silico studies performed with 1–40 Aβ showed that the interactions between Chol and peptide are different to those observed with 1–42 Aβ (Di Scala et al., 2013, 2014). Since the initial proposal of the existence of transmembrane ion channels formed by Aβ peptides (Arispe et al., 1993a,b), lots of studies deepened in the “β-amyloid calcium channel hypothesis” (Pollard et al., 1995; Kagan et al., 2002; Kawahara, 2010). The first step for this ion channel formation must be the contact between the peptides and the membrane. It was demonstrated that both the lipid composition of the outer membrane and the structural conformation of the Aβ peptide are crucial for this interaction. In solution, it was possible to find Aβ as β-sheet, α-helix or random coil conformations, being the conformational balance dependent on its concentration. It is postulated that the presence of certain lipids can shift the equilibrium to one preferred conformation. Particularly, it was demonstrated that negatively charged lipids take contact with the peptide by specific electrostatic interactions (Hertel et al., 1997; McLaurin and Chakrabartty, 1997; Terzi et al., 1997). Aβ selectively recognizes and accumulates on GM1-rich membrane domains (Yanagisawa et al., 1995; Wakabayashi et al., 2005; Yanagisawa, 2005), and Aβ insertion into the membrane is critically dependent on the Chol/phospholipids ratio (Ji et al., 2002), as it was detailed above. More recent works showed that the formation of Ca2+ pores (“annular protofibrils,” Lashuel et al., 2002) in the plasma membrane is a mechanism dependent on both gangliosides and Chol. As it was described above, amyloid monomers or soluble oligomers interact with a ganglioside at the cell surface, with a specificity that responds to a ganglioside-binding domain for each amyloid protein (common amino acid residues at specific locations, with specific variations for each ganglioside), being the Ca2+ pores significantly diminished in ganglioside deprived cells (Di Scala et al., 2016). Based on this “calcium-channel hypothesis” of the AD, a chimeric peptide formed with a minimal ganglioside-binding domain of α-synuclein and two contiguous His residues as in 1–42 Aβ (Yahi and Fantini, 2014) avoid pore formation by 1–42 Aβ. Treatment of WT 5XFAD mice with a sialic-specific lectin (LFA, Limax flavus agglutinin) significantly reduced amyloid depositions in the brain, probably by interfering with the binding of amyloid peptides to gangliosides (Dukhinova et al., 2019). Furthermore, Cascella et al. (2017) showed that different oligomer conformations can perturb Ca2+ cell-permeation by both a channel-independent mechanism as annular protofibrils, or by a channel-dependent one (through NMDA-R and AMPA-R).
Aβ peptides that stay in the membrane surface are in a β-sheet conformation, and once inside the membrane they turn their conformation to an α-helix (Yu and Zheng, 2012). Other studies suggested that 1–40 Aβ interacts with the membrane in two sequential steps. The first one involves the formation of a pore-like structure and membrane permeation, and the second one involves subsequent growth of aggregates with fibril formation and lipid clustering around the fiber which implies lipid extraction, membrane fragmentation, and loss of membrane integrity (Engel et al., 2008; Stefani, 2010; Milanesi et al., 2012; Sciacca et al., 2012; Relini et al., 2013; Kotler et al., 2014).
Recently, Rondelli et al. (2016) described more in detail the interactions between cell membranes and Aβ peptides. Those interactions depend on peptide conformation: structural oligomers are imbibed in the outer hemilayer of the membrane triggering more Aβ addition and further elongation; on the other hand, early labile oligomers in equilibrium with monomers are incorporated as monomers deeply in the membrane coming up to the inner hemilayer, whereas Aβ organization leads to pore formation.
A study of the changes induced by 1–42 Aβ on the morphology and the mechanical stability of model membranes with different Chol content indicates that Chol drives 1–42 Aβ toward rafts domains and that at high Chol concentration the presence of the amyloid peptide did not alter any membrane property, thus assigning a protective effect against membrane destabilization by 1–42 Aβ to the presence of Chol (Seghezza et al., 2014). Recently, Staneva et al. (2018) deepened this idea. They observed that 1–42 Aβ has a higher affinity for liquid-disordered (ld) than ordered (lo) phases, confirming previous results (Ahyayauch et al., 2012). They concluded that the fraction of Aβ in lo domains, probably the functionally important one, might be smaller. While in a lo phase 1–42 Aβ induces practically no changes in the lipid packing, a significant perturbation of the lipid packing by its presence was observed in a ld phase. They focus on the presence of GM1 as a crucial lipid. In ld phases without GM1, the peptide penetrates and messes up the neighboring lipids. However, in the presence of GM1 the peptide interacts with the headgroup of several GM1 promoting a condensing effect and an increased lipid packing and decreases Aβ penetration. The presence of GM1 could affect the line tension between lo and ld domains which in turn affects the kinetics of domains formation, growth, shape and size. Thus, although it cannot be discarded that the functional peptide, or at least a minority of it, binds directly to lo domains, the authors suggested that the fibrillation of Aβ peptides in raft domains is the consequence of a reorganization modulated through Aβ peptides in non-raft domains (Staneva et al., 2018).
Not only specific lipid raft characteristics are necessary for Aβ insertion into the membrane, but also its insertion has consequences on the membrane (Chang et al., 2017). Several studies analyzed the membrane biophysical perturbations caused by Aβ interaction, which could be considered the first step of its biological effect (Kanfer et al., 1999; Chochina et al., 2001; Eckert et al., 2003). A decrease in the fluidity of mouse brain membranes, human lymphocyte membranes and membranes from rat cortex, hippocampus and striatum was observed in the presence of 25–35 Aβ and 1–40 Aβ, and in all cases the effect was dependent on peptide concentration (Müller et al., 1995). Low concentrations of Aβ significantly perturb membrane fluidity by specifically altering the acyl-chain mobility of brain membranes, an effect dependent on peptide length, with almost no effect at the polar head groups (Müller et al., 2001). Lately, it was observed that monomeric 1–40 Aβ has no effect on membrane fluidity, while oligomeric forms do (Peters et al., 2009). Contrary to these results, by exposition of hippocampal neurons to nanomolar concentrations of Aβ oligomers for 24 h we could not observe changes in membrane fluidity tested with three different fluorescence probes (Uranga et al., 2017). Peters et al. (2009) showed that membrane perturbation by Aβ is a consequence of Aβ complexing with GM1; thus, it is possible that in our experiments the cell membrane did not have the correct GM1/Chol relationship. A previous study of the interaction of 1–42 Aβ with planar bilayers had already demonstrated that the Chol content is directly correlated with Aβ assembly on the membrane surface, that during this process membrane changes occur, and that all this process is governed by lipid bilayer composition (Yip et al., 2002). Thus, membrane lipid environment modulates Aβ production and at the same time Aβ causes a membrane perturbation that positively feedbacks its own production (Peters et al., 2009). Moreover, Aβ insertion into the membrane not only potentiates Aβ production but also unspecifically activates a variety of membrane processes which could eventually end in neuronal cell death (Kanfer et al., 1999). 25–35 Aβ peptide interacts with phospholipids through electrostatic interactions favoring peptide aggregation which causes perturbations at the lipid-water interphase of the membrane (Martínez-Senac et al., 1999). Mass spectroscopy studies showed that Aβ inserts into model membranes containing Chol, but not in the absence of Chol (Ji et al., 2002). This study also indicated that the membrane insertion is initiated by the C-terminus of the Aβ peptide which has the hydrophobic domain.
Brain membranes from middle aged mice were more susceptible to Aβ perturbations than membranes from aged mice; and in vitro studies showed that a decrease in membrane Chol content enhanced Aβ effect, while an increase in membrane Chol strongly decreased the perturbation effect (Kirsch et al., 2002), suggesting that Chol protects neuronal membranes from Aβ perturbations and neurotoxicity (Eckert et al., 2003). However, they also observed in vivo that a reduction of Chol levels by approx. 30% by treatment with lovastatin (HMG-CoA-reductase inhibitor) resulted in moderated membrane alterations without acyl chain flexibility perturbations and reduced Aβ bulk fluidity perturbation (Kirsch et al., 2002). A possible explanation is that Chol membrane modification involves different membrane Chol pools with different sensitivity for Aβ perturbations whether it is in vitro or in vivo, with the one at the membrane acyl-chain being the most receptive (Kirsch et al., 2003).
Another important consequence of an enhanced Aβ production linked to lipid membrane is oxidative stress with an excess of lipid peroxidation and increased lipid susceptibility to oxidative damage, which exacerbates Aβ toxicity in the membrane (Behl et al., 1994; Opazo et al., 2002; Cutler et al., 2004; Boyd-Kimball et al., 2005; Wu and Luo, 2005). It is reported that Aβ prefers to interact with membranes with high oxidatively damaged phospholipids (Zampagni et al., 2010), particularly in raft domains, and that these membranes promote misfolding and aggregations of Aβ peptides into fibrils (Shringarpure et al., 2000; Magni et al., 2002; Zhang et al., 2004; Bieschke et al., 2005; Lee et al., 2006; Murray et al., 2007), whereas the misfolded peptides promote more oxidative damage in the membrane, conducing to a positive feedback (Murray et al., 2007). Aβ increases 4-hydroxy-2-nonenal (HNE) production which promotes oxidative damage and also induces Aβ to form β-structure and amyloid fibrils (Mark et al., 1997; Lauderback et al., 2001; Murray et al., 2005, 2007).
Even though it is not a topic of interest for this review, it is important to remember that just as Chol is a crucial lipid molecule for Aβ processing and Aβ membrane effects, the round trip is also valid since Aβ has an impact on Chol homeostasis (Koudinova et al., 2000; Michikawa et al., 2001; Gong et al., 2002; Michikawa, 2003; Koudinov and Koudinova, 2004; Grimm et al., 2005, 2007). This ultimate effect suggests that Aβ down-regulates Chol content and also raft content (Beel et al., 2010). Thus, the peptide behaves as a Chol sensor: when there is high Chol content in a membrane, the amyloidogenic pathway is favored and, thus, an enhancement takes place in Aβ processing, which in turn reduces both Chol uptake and biosynthesis, following up a negative feedback mechanism (Beel et al., 2010).
The basis of AD pathogenesis is still controversial today, even though several hypotheses try to explain it, such as the Aβ amyloid cascade (Hardy and Allsop, 1991; Hardy and Higgins, 1992), the hyperphosphorylated microtubule-associated tau (Götz et al., 2004), abnormalities of the cholinergic system (Bartus et al., 1982), oxidative stress (Butterfield and Boyd-Kimball, 2005), etc. Even though the amyloid hypothesis is the most popular explanation for the mechanism of AD, it fails to explain several aspects of this multifactorial etiopathology (Herrup, 2015). In addition, until now, the majority of clinical trials conducted to diminish the amount of Aβ did not give good results (Puzzo et al., 2015; Maia and Sousa, 2019). Although these failures are not enough to discard the amyloid hypothesis (see for example Rosenblum, 2014), attention is now focused on the cholinergic hypothesis since it became the main therapeutic strategy for this disease (Figure 2). As we will work out in the following paragraphs, these two hypotheses are highly linked.
The cholinergic system involves two families of receptors, nAChR and muscarinic acetylcholine receptors (mAChR). Although both types of receptors are related with cognitive processes (Ghoneim and Mewaldt, 1977; Petersen, 1977; Sarter and Paolone, 2011) and are affected in AD, only the relation between nAChR and AD has been largely studied (Lombardo and Maskos, 2014).
The nAChR is an integral membrane protein that belongs to the Cys-loop superfamily of ligand-gated ion channels (Karlin and Akabas, 1995; Le Novère and Changeux, 1995; Changeux and Edelstein, 1998; Paterson and Nordberg, 2000). The binding of its natural agonist acetylcholine triggers a conformational change that ends in the opening of a channel and the flux of positive ions across the membrane, causing membrane depolarization and a subsequence intracellular cascade of events (Lindstrom, 2003; Brown, 2006; McKay et al., 2007; Pohanka, 2012). The nAChR presents a pentameric arrangement, with each subunit having a large N-terminal extracellular domain, four transmembrane segments (M1–M4), a small cytoplasmic domain between M3 and M4, and a short C-terminal extracellular domain. To this day, 16 different nAChR subunits (including: α1-7, α9-10, β1-4, γ, δ, and ε) that form homologous and heterologous receptors with distinct structures, functions and locations are known (Champtiaux et al., 2003; Dajas-Bailador and Wonnacott, 2004; Fucile, 2004; Giniatullin et al., 2005; Gotti et al., 2006a, 2007, 2009; Albuquerque et al., 2009; Shen and Yakel, 2009). The muscle-type nAChR of the electric organ of Torpedo, first receptor described and still the prototype of the family, is formed by α12β1δγ (similar to embryonic muscle nAChR of vertebrates, which change to α12β1εγ in adult). Two receptor subtypes are highly expressed in the central nervous system: the heteropentamer α4β2 nAChR and the homopentamer α7 nAChR (Schmidt and Freeman, 1980; Sargent et al., 1991; Clarke, 1992; Sargent and Garrett, 1995; Cooper et al., 1999; Nashmi et al., 2003; Scholze et al., 2011). The latter is particularly important in AD (Ma and Qian, 2019). It is present in high density in the striatum, thalamus, neocortex, and limbic system suggesting a central role in normal cognition and, hence, in age-related cognitive decline (Bigl et al., 1982; Mesulam et al., 1983; Muir et al., 1993; Sarter and Bruno, 1997; Wenk, 1997; Woolf, 1998; Guillem et al., 2011). It was shown that α7 nAChR is important for growth, development and aging, regulating the plasticity of the neural circuit, neuronal differentiation, proliferation, apoptosis and clearance of aged neurons (Nees, 2015). The levels of this receptor change during development and adult stage, and in AD patients, they decrease significantly (Bowen et al., 1976; Perry et al., 1981, 1985, 1987, 1988; Whitehouse et al., 1981, 1982, 1986; Coyle et al., 1983; Shimohama et al., 1986; Nordberg et al., 1995; Paterson and Nordberg, 2000; Auld et al., 2002; Gotti et al., 2006b; Kim et al., 2013; Ma and Qian, 2019).
Activation of the α7 nAChR opens a high permeability Ca++ channel that consequently activates voltage-dependent Ca++ channels (Perry et al., 1992; Sharma and Vijayaraghavan, 2001) and triggers an intracellular signaling cascade through activation of a protein kinase. In the case of activation of presynaptic α7 nAChR, the final event is the fusion of vesicles loaded with neurotransmitters (glutamic acid, norepinephrine, acetylcholine, dopamine and GABA) to the presynaptic membrane and the massive release of these neurotransmitters to the synaptic cleft (Wonnacott et al., 2006; Ma and Qian, 2019). Postsynaptic α7 nAChR depolarize the postsynaptic membrane and participate in signal transduction (Messi et al., 1997; Morley and Happe, 2000; Berg and Conroy, 2002). ACE metabolizes acetylcholine after its release to the synaptic cleft ending the cholinergic stimulus (Bowen et al., 1976; Davies and Maloney, 1976; Coyle et al., 1983; Auld et al., 2002).
The cholinergic hypothesis of AD focuses on the fact that in brains of AD patients there is a decrease in the total amount of nAChRs (Whitehouse et al., 1982; Banerjee et al., 2000), which is an outcome of progressive death of forebrain cholinergic neurons with an extended cholinergic presynaptic denervation (Bartus et al., 1982; Court et al., 2000; Graham et al., 2002; Contestabile, 2011; Hampel et al., 2018). This is considered a consequence of enhanced Aβ production (Liu and Wu, 2006). Banerjee et al. (2000) observed that in the remaining cholinergic neurons there was a higher amount of nAChR, which suggests a possible compensatory mechanism. Many efforts were performed to ameliorate this loss. However, current approved pharmacological agents, such as physostigmine, tacrine, donepezil, rivastigmine and galantamine (Martorana et al., 2010), are targeted to inhibit ACE function increasing the amount of acetylcholine at the synapse cleft and ameliorating the clinical symptoms of AD without halting the progress of the disease.
The affinity of α7 nAChR for 1–42 Aβ is in the low picomolar concentration, a range estimated to occur in healthy brains, while the affinity of α4β2 nAChR for 1–42 Aβ is between 100 to 5000 times lower (Wang et al., 2000a); thus, it is expected that both α7 nAChR and 1–42 Aβ could associate under physiological conditions. Puzzo et al. (2015) hypothesized that under physiological conditions a positive feedback mechanism occurs: synaptic activity induces Aβ release that acts as an endogenous ligand and modulates α7 nAChR, which in turn induces release of neurotransmitters and enhances synaptic plasticity and memory. Under pathological conditions, abnormal accumulation of Aβ (nanomolar concentration, Näslund et al., 1994, 2000; Tapiola et al., 2000; Andreasen et al., 2003) induces a negative feedback mechanism which implies inhibition and internalization of α7 nAChR, leading to synaptic dysfunction and memory loss. The Aβ-α7 complex influences tau hyperphosphorylation (Wang et al., 2003) and its internalization leads to plaque formation (Nagele et al., 2003, 2004; Dineley, 2007).
It is thought that the soluble form of Aβ interacts with α7 nAChR with apparently high affinity (Wang et al., 2000a) regulating its function (Dineley et al., 2001; Liu et al., 2001; Pettit et al., 2001). However, there is no consensus about the nature and consequences of this interaction (Farhat and Ahmed, 2017). While several studies propose an agonist-like effect for presynaptic nicotinic receptors (Dineley et al., 2002; Dougherty et al., 2003; Wu et al., 2007; Puzzo et al., 2008; Mehta et al., 2009; Lilja et al., 2011; Arora et al., 2013), others propose an inhibitory action (Dineley et al., 2001; Liu et al., 2001; Pettit et al., 2001; Tozaki et al., 2002; Lee and Wang, 2003; Wu et al., 2004; Wang et al., 2009; Parri et al., 2011), and others a concentration-dependent relationship with a stimulatory effect at picomolar Aβ concentration and an inhibitory effect at high nanomolar Aβ concentration (Puzzo et al., 2008). The variability between all the performed studies is so large in terms of in vitro and in vivo models, Aβ concentrations and Aβ preparations/conformations, and other conditions, that it is difficult to find a rule for the data obtained. Khan et al. (2010) gave a possible explanation for these inconsistency centered in a different Aβ effect on pre or postsynaptic receptors. Aβ induces a rapid stabilization of an inactive/desensitized state of postsynaptic receptors, resulting in an antagonist effect, and a slower desensitization of presynaptic receptors resulting in an agonist-like effect. The authors pointed to differences in the lipid microenvironment of the pre and postsynaptic α7 nAChR for these different desensitization rates. Presynaptic terminals have abundance of raft domains, and experimental disruption of these domains dramatically attenuates Aβ evoked α7 nAChR currents (Khan et al., 2010). With respect to the concentration-dependent effect, it is important to take into consideration that in a normal central nervous system 1–42 Aβ is found, although at low picomolar concentrations. Under this condition, it is postulated that Aβ exerts a positive effect on synaptic plasticity and memory formation (Phinney et al., 2003; Plant et al., 2003; Puzzo et al., 2008, 2011, 2015; Puzzo and Arancio, 2013). However, in a pathological condition, Aβ cannot exert its physiological function and hence a feedback mechanism induces more Aβ production, leading to an enhancement of the peptide with the subsequent reduction of α7 nAChR with Aβ removal and synaptic alteration and memory loss (Phinney et al., 2003; Puzzo et al., 2015).
Interaction of Aβ with α7 nAChR increases Aβ internalization (Nagele et al., 2002; D’Andrea and Nagele, 2006) and accumulation in lysosomes causing an excessive intraneuronal 1-42 Aβ accumulation. The majority of the amyloid plaques proceed from the lysis of degenerated, Aβ-overburdened neurons (Wang et al., 2000b; Gyure et al., 2001; Langui et al., 2004; Nunomura et al., 2010; Palop and Mucke, 2010; Li et al., 2011; Deutsch et al., 2014, 2016). Additionally, the formation of the Aβ-α7 nAChR complex may influence the membrane lipid and membrane protein organization (Deutsch et al., 2014, 2016; Ma and Qian, 2019). At the same time, the internalization of the Aβ-α7 nAChR complex triggers an upregulation of the α7 nAChR and magnifies the toxicity of the pathology (Molinari et al., 1998; Xiu et al., 2005; Yu et al., 2005; Liu et al., 2013, 2015; Shen and Wu, 2015) (Figure 2). In AD patients and preclinical AD models, a high expression of α7 nAChR was described (Hellström-Lindahl et al., 1999, 2004a,b; Dineley et al., 2002; Jones et al., 2006; Counts et al., 2007; Ikonomovic et al., 2009). Chronic exposure to Aβ enhances the expression of α7 nAChR in neuron and glia cells (Yu et al., 2005; Liu et al., 2013). Also, an age-dependent increase of cell surface α7 nAChR was observed in 5xFAD mice, a model that rapidly develops amyloid pathology (Jin et al., 2015). Several studies contributed to this hypothesis. Treatment of PC12 cells with 1–42 Aβ increased cell surface α7 nAChR, suggesting that the peptide induces translocation of the receptor toward the plasma membrane (Jin et al., 2015). They observed that the agonist nicotine prevented Aβ induced cell death, whereas the competitive antagonist α-bungarotoxin potentiates the peptide effect, indicating that α7 nAChR plays a role in protecting neuronal cells from Aβ 1–42 peptide (Dziewczapolski et al., 2009; Wang et al., 2009; Jin et al., 2015). Contrary to this, Liu et al. (2015) concluded that upregulation of α7 nAChR induced by Aβ is necessary to mediate peptide neurotoxicity, both in hippocampal neurons and differentiated cholinergic SH-SY5Ycells. α7 nAChR function, which is exacerbated by its upregulation, may be necessary for the toxicity of Aβ aggregates; this effect was prevented by α7 nAChR inhibition or deletion. Previous studies showed that the blockade of α7 nAChR significantly ameliorated attentional deficits (Levin et al., 2013; Burke et al., 2014). Likewise, the deletion of α7 nAChR gene was correlated with an improvement in synaptic plasticity and a reduction in cognitive deficiency (Dziewczapolski et al., 2009). Two possible cytotoxic α7 nAChR-mediated mechanisms were proposed: one considers that the α7 nAChR increment in the membrane conduces to a high calcium permeability, which could be the ultimate responsible for cell toxicity, and the other that the high Aβ-α7 nAChR complex internalization and intracellular accumulation leads to neurotoxicity (Liu et al., 2015). Thus, while several studies point to α7 nAChR activation as a beneficial treatment, others suggest that a function inhibition for a beneficial effect is necessary.
A different hypothesis about Aβ and α7 nAChR relationship was postulated by Small et al. (2007). They concluded that Aβ does not bind directly to α7 nAChR but to the lipids of the plasma membrane, and that the perturbation of the structure or fluidity of the lipid microenvironment of the receptor could be the responsible for toxicity through an alteration of the receptor function. Their conclusion is supported by previous evidence that showed that Aβ binds strongly to lipids (Subasinghe et al., 2003; Hou et al., 2005). We will return to this issue later.
We here described the most relevant information about the interaction between Aβ and α7 nAChR, and its final consequences, focusing on the events that occur through the membrane. However, not only the interactions between Aβ and α7 nAChR are important. Other proteins that interact with α7 nAChR including Lynx proteins, NMDA-receptors and the Wnt/β-catenin pathway are important as well. All those interactions that modulate receptor function are specifically altered in AD and can lead to differences in the clinical effect of nAChR ligands in AD (Thomsen et al., 2016). It is also important to take into account that there is an internal cascade of signaling downstream α7 nAChR activation that involves several other active molecules, such as glycogen synthase kinase-3β (GSK-3β), phosphoinosite 3-kinase (PI3K)-Akt, Wnt and the mitogen-activated protein kinase (MAPK) signaling pathway, which are also altered in AD (see Ma and Qian, 2019 for a further explanation).
The last step in cholinergic signaling is the degradation of acetylcholine by the enzyme ACE to end the synaptic transmission. ACE is a globular non-transmembrane protein that can exist in different molecular forms, depending on the splicing of the ACE gene (Henderson et al., 2010). Although all ACE molecular forms and variants have similar catalytic activity, they also have other non-catalytic, non-classical functions, which depend on the multiple molecular forms of this enzyme and on cell types and cellular compartments (Small et al., 1996; Grisaru et al., 1999; Massoulié, 2002; Hicks et al., 2011). In non-neuronal tissues, ACE regulates cell proliferation, differentiation, apoptosis and cell–cell interaction, which is important to take into consideration when ACE inhibitors for AD are designed (Lazarevic-Pasti et al., 2017). ACET is the predominant form in central nervous system, which has a C-terminal α-helix peptide of 40 amino acids named T peptide. Through disulphure bondings between these peptides they can be found as homodimers and homotetramers of ACET. Also, the T peptide binds to hydrophobic proline-rich domains of membrane anchoring-proteins (like collagen-like Q subunit in NMJ and proline-rich membrane anchor, PRiMA, in the central nervous system; Massoulié et al., 2005). In the central nervous system, the majority of ACE is found as tetrameric ACET (G4) bound to PRiMA (Navaratnam et al., 2000; Perrier et al., 2002; Massoulié et al., 2005), which constitute the functional units at cholinergic synapse (Perrier et al., 2002; Dvir et al., 2010; Henderson et al., 2010; Hicks et al., 2011). PRiMA could bring the membrane-bound ACE together with other proteins in specialized membrane areas, such as raft domains, specifically with α7 nAChR at basal forebrains cholinergic neurons (Henderson et al., 2005; Hicks et al., 2012). A significant proportion of ACET is effectively located in raft domains through a Chol-binding domain of 13 amino acids of PRiMA (a CRAC, Chol recognition amino acid consensus, sequence), and Chol depletion or mutations at this domain reduced the lipid raft-PRiMA association (Xie et al., 2010a,b). A diminution of ACE activity in the cerebral cortex and other areas in AD patients was described, being the G4-PRiMA complex the ACE form markedly altered, whereas the ACE monomeric form was almost preserved (Atack et al., 1983; Fishman et al., 1986; Sáez-Valero et al., 1999). Interactions of PRiMA subunit with presenilin 1 (PS1, the catalytic subunit of γ-secretase), which is an aspartyl protease that cleaves substrates inside membrane, were described to occur in raft domains (García-Ayllón et al., 2014). This interaction could explain, in part, the cellular release of ACE through a shedding mechanism that was postulated to involve a metalloprotease (Hicks et al., 2013a). Furthermore, a direct relationship between PS1 and ACE was observed, with an overexpression of ACE related to higher levels of PS1, ACE knockdown leaded to decreased PS1 and a mutated PS1 was related with decreased ACE in the brain (Silveyra et al., 2008, 2012). At the same time, it was also observed that ACE inhibits AβPP processing through γ-secretase (Niu et al., 2012), perhaps, acting as an inhibitor of the secretase by interacting with PS1 (Campanari et al., 2014). In AD, ACE activity is diminished and hence impedes its potentiality to modulate γ-secretase (Campanari et al., 2014).
Interactions of ACE with Aβ are important in AD (Inestrosa et al., 1996; Wang et al., 2000b; Small et al., 2007), as the peptide alters several ACE properties such as its pH optimum and inhibitor sensitivity (Geula and Mesulam, 1989), making Aβ even more neurotoxic (Inestrosa et al., 1996; Alvarez et al., 1998). Moreover, ACE was detected in amyloid plaques evidencing the high affinity between both molecules and suggesting that ACE could promote Aβ aggregation (Morán et al., 1993; Inestrosa et al., 1996). Even more, in some cerebral areas of AD patients almost all ACE is in these complexes. The binding between Aβ and ACE occurs at the ACE peripheral anionic site (PAS); ACE inhibitors that bind to the anionic site (i.e., propidium), as well as antibodies against it, significantly reduce fibril formation (Reyes et al., 1997; Bartolini et al., 2003). Although the ACE catalytic domain does not participate of this interaction (Inestrosa et al., 1996), new compounds with a dual action (blocking PAS and catalytic site) are being designed, looking for the prevention of fibril aggregation with the aim of reversing the progression of the disease and, at the same time, inhibiting acetylcholine degradation to ameliorate the symptomatology (Alptüzün et al., 2010).
Furthermore, a negative relationship between APP and ACE was observed, as an overexpression of APP repressed ACE transcription with reductions of both ACE levels and ACE activity (Hicks et al., 2013b). A similar negative regulation was observed between APP and PRiMA; however, it is not clear if there is a direct downregulation by APP or if this diminution is a consequence of decreased ACE levels (Hicks et al., 2013b; Nalivaeva and Turner, 2016). The authors proposed that this ACE downregulation could be a novel neuroprotective function of APP.
As we said in the previous section, there are several nAChR subtypes depending on the individual pentameric arrangement. Summing up, all nAChR have two well defined structural domains: the neurotransmitter-binding site extracellular domain and the transmembrane domain containing the ion pore. Whereas the extracellular domain is the site where the agonists or different activators/inhibitors bind, the transmembrane region, besides having the ion pore, exhibits extensive contacts with the surrounding lipids through structural motifs remarkably conserved along phylogenic evolution (Antollini et al., 2005; Unwin, 2005; Jha et al., 2007; Baenziger and Corringer, 2011; Baenziger and daCosta, 2013; Barrantes, 2015). It is well known that a correct allosteric coupling between both domains is crucial for nAChR function, strongly dependent on its surrounding lipid, which modulates the relative proportion of nAChR in its resting or desensitized states (daCosta et al., 2002; Baenziger et al., 2000, 2008, 2015; daCosta and Baenziger, 2009; Barrantes, 2010; Barrantes et al., 2010; Hénault et al., 2015). The most studied nicotinic receptor is the muscle nAChR, which is not only the paradigm of all other nAChR but also of the entire cys-loop superfamily. In the following paragraphs we will discuss the relationship between distinct lipids or raft domains and the muscle nAChR, knowledge that can be extended to other members of the family, in particular to α7 nAChR.
Several years ago, Marsh and Barrantes (1978) described a layer of immobilized lipids that encircle the muscle nAChR with characteristics different from those of bulk lipids. Subsequent studies assigned an important role to these bounded lipids on muscle nAChR (Criado et al., 1982; Ellena et al., 1983; Ochoa et al., 1983; Sunshine and McNamee, 1992, 1994; Narayanaswami et al., 1993; Fernández-Ballester et al., 1994; Dreger et al., 1997; Barrantes, 2002, 2007; Quesada et al., 2016). The presence of both Chol and negatively charged lipids in the nAChR-lipid microenvironment is necessary to stabilize the nAChR in a functional conformation (Criado et al., 1984; Fong and McNamee, 1986; Butler and McNamee, 1993; Méthot et al., 1995; Antollini et al., 1996). However, there is no consensus about if it is the entity/identity of the lipid itself or the fluidity that each lipid confers to the membrane the responsible of this role. In spite of this controversy, the importance of a proper lipid microenvironment for muscle nAChR becomes clear when highly hydrophobic molecules, such as free fatty acids or steroids, perturb nAChR function through the membrane localizing at the lipid-nAChR interphase (Andreasen and McNamee, 1980; Villar et al., 1988; Bouzat et al., 1993; Bouzat and Barrantes, 1996; Nurowska and Ruzzier, 1996, 2002; Minota and Watanabe, 1997; Blanton et al., 1999; Garbus et al., 2001, 2002; Antollini and Barrantes, 2002, 2016; Fernández Nievas et al., 2007, 2008). Working with reconstituted Torpedo nAChR, Jones and McNamee (1988) distinguished two different populations of lipids in the nAChR-lipid microenvironment region: annular and non-annular lipids. Annular lipids interact with the protein in a relatively less specific manner with a fast rate of exchange with bulk lipids. Contrarily, non-annular lipids are in close contact with the protein, probably in between α-helix transmembrane segments or subunits, and can be associated to lipid binding sites with a slow exchange rate with bulk lipids (Lee, 2003). We identified the same two types of lipids in native Torpedo membranes (Antollini and Barrantes, 1998). The entity/identity of non-annular lipids are considered crucial for nAChR function; in the case of annular lipids the biophysical characteristics are more relevant. This is in concordance with other studies that assigned several roles to the lipids in a membrane, two of the main ones being: a collective one, in which they form a viscoelastic lipid “solvent” with the above-mentioned heterogeneities; and an individual one as signaling molecules (Piomelli et al., 2007).
Two annular lipids that are of particular interest are negative lipids and SM. With respect to the requirement of negative lipids, PA is particularly of interest. The segregation of PA domains containing nAChR and the stabilization of a functional conformation of the receptor by PA were described (daCosta et al., 2002, 2004; Poveda et al., 2002, 2008; Wenz and Barrantes, 2005; Dickey and Faller, 2008). SM showed moderated affinity for the nAChR (Bonini et al., 2002) but it is important for proper nAChR stability in the membrane. Its deficit affects the efficiency of the nAChR assembly process and the nAChR targeting to the membrane and increases the rate of turnover (Roccamo et al., 1999; Baier and Barrantes, 2007). Moreover, SM is important for membrane biophysical properties as it is asymmetrically distributed between both membrane hemilayers and it is one of the main actors of lipid raft domains, being both aspects that impact on nAChR (Perillo et al., 2016).
A separate paragraph is for Chol, a key lipid for nAChR (Middlemas and Raftery, 1987). This lipid molecule can be found in every region of a membrane: as a bulk, annular or non-annular lipid. In the first two cases, it probably plays an important function conditioning the physical properties of the environment, mainly because of its participation in raft domain formation and in the maintenance of the asymmetrical membrane condition. As a non-annular lipid, the occurrence of allosteric binding sites is postulated (Addona et al., 1998). It was suggested that the binding domain for Chol is at the nAChR lipid-protein interface, taking contact with the transmembrane subunits αM4, αM1, and γM4 (Corbin et al., 1998); other studies identified interactions of Chol with the transmembrane segments M1, M3, and M4 of each subunit (Hamouda et al., 2006). By fluorescence quenching and energy-transfer measurements of T. californica reconstituted membranes, sites accessible to Chol but not to phospholipids were identified (Narayanaswami and McNamee, 1993). Using Molecular Dynamics simulations of the nAChR structure, Brannigan et al. (2008) identified 15 Chol binding sites, large hydrophobic intersubunit and intrasubunit gaps. The location of Chol molecules at these sites improved nAChR stability; and in the case of intrasubunit sites, occupation of these sites by Chol precludes the nAChR from collapsing. A recent study using coarse-grained molecular dynamics simulations suggested that while long n-3 chains (in this case, docosahexaenoic acid, 22:6) have a high propensity for annular and non-annular sites, displacing Chol and occupying sites even deeper within the bundle, shorter n-6 chains do not displace Chol from non-annular sites as efficiently as long n-3 chains (Sharp et al., 2019).
Considering the intimate and close relationship between Chol and the muscle nAChR, studies looking for a consensus about specific Chol domains in the nAChR subunits were performed. A CRAC sequence in a region immediately adjacent to the M1 transmembrane domain of all the subunits of the muscle nAChR was identified (Baier et al., 2011). These sequences are located exiting the membrane bilayer, which suggests that they are probably not good partners for Chol in the hydrophobic membrane environment. However, a novel Chol recognizing domain was identified by in silico studies, a sequence opposite to a CRAC one (inverted CRAC or “CARC” sequence) at M1, M3, and M4, which is located inside the membrane and is highly preserved in the evolutionary scale, from prokaryotes to humans (Baier et al., 2011; Di Scala et al., 2017). These in silico results were also experimentally confirmed (Fantini et al., 2016). Furthermore, the authors concluded that a CARC sequence generally exhibits more affinity for Chol than a CRAC one (Fantini and Barrantes, 2013), and that it is of high affinity, lipid specific, and saturable (Fantini et al., 2016).
Chol not only conditions nAChR function but also its stability in the plasma membrane. There are some controversies about how the nAChR is organized in the membrane. At the NMJ, supramolecular aggregations of nAChRs (micron-sized two-dimensional clusters) are postulated to occur in Chol-rich lipid microdomains, together with several postsynaptic proteins including rapsyn, MuSK and Src-family kinases. Chol would stabilize NMJ and promote its maturation (Willmann et al., 2006). Depletion of cell-surface Chol produced a marked alteration of the organization of the nAChR (Kellner et al., 2007). One hypothesis for this situation is that after an agrin (extracellular heparan sulfate proteoglycan that aggregates nAChRs on cultured myotubes) stimulus, nAChR and MuSK translocate into raft domains where nAChR clustering occurs, as raft domains concentrate the agrin/MuSK signaling, nAChR and rapsyn. Disruption of these microdomains by Chol depletion inhibits agrin stimulation and formation and maintenance of nAChR clusters (Zhu et al., 2006). A contemporary study suggested that agrin causes the translocation of nAChR into raft domains, which is in agreement with the mentioned hypothesis (Campagna and Fallon, 2006). A slightly different hypothesis indicates that agrin does not reclute nAChRs into raft domains, as they are already in those domains independently of agrin activation, but it triggers the coalescence of raft domains conducing to nAChR clustering and it is also responsible for its maintenance, as Chol is necessary for all this process (Stetzkowski-Marden et al., 2006a,b; Cartaud et al., 2011). A previous study supports this hypothesis where the authors observed that nAChR subunits and rapsyn are cotargeted in the exocytic pathway to the cell surface inserted in Chol-rich microdomains (Marchand et al., 2002). Furthermore, Chol depletion affects the maintenance of the nAChR in the plasma membrane by several mechanisms. Treatments of cells with methyl-β-cyclodextrin, which extracts Chol from the membrane, enhanced nAChR internalization by endocytosis with a marked decrease of the number of nAChR domains, concomitantly with a gain-of-function of the remaining nAChR (Borroni et al., 2007; Borroni and Barrantes, 2011; Kamerbeek et al., 2013). Furthermore, chronic treatments with mevinolin, an inhibitor of 3-hydroxy-3-methyl-glutaryl-CoA reductase and hence of Chol synthesis, inhibited the trafficking of the receptor toward the membrane surface, which caused low nAChR cell-surface expression, and increased the intracellular nAChR pools (Pediconi et al., 2004). Moreover, Chol conditions muscle nAChR cell-surface diffusion (Baier et al., 2010; Mosqueira et al., 2018) and nAChR stability in confined raft domains (Mosqueira et al., 2018).
Different results of the interaction between muscle nAChR and lipid domains were obtained in model systems. We observed that reconstituted Torpedo nAChR in symmetric model membranes with coexistence of liquid-ordered (lo) and liquid-disordered (ld) domains was distributed homogeneously, without preference for any domain (Bermúdez et al., 2010). However, similar experiments with a synthetic peptide corresponding to the γM4 peptide showed a marked preference of this peptide for lo domains (de Almeida et al., 2004; Bermúdez et al., 2010). Thus, although this transmembrane segment could give the nAChR the potentiality to localize in raft domains, it is not sufficient and other conditions must occur which influence nAChR partition profile. One of these mentioned conditions is membrane asymmetry. By increasing SM in the outer hemilayer, we observed an increment of the Torpedo nAChR in lo domains, and the same was observed when specific SM species instead of brain SM were used in symmetric models (Perillo et al., 2016). Recently, by using coarse-grained molecular dynamics simulations of nAChR inserted in a ternary system of DPPC:Chol:PE or PC with PUFA, the authors concluded that nAChR partitioned in ld domains poor in Chol (Sharp et al., 2019). The simulated membrane, despite having lo and ld domains, (a) did not have SM of any species, which is a critical lipid for raft domains in biological membranes, (b) used PUFA which are known to behave as nAChR inhibitors probably by competition with Chol for non-annular sites, as the authors observed in the study, and (c) was symmetric, a condition different to the natural asymmetry of biological membranes. Thus, this work emphasizes that it is not just the presence of an lo domain, but also its physicochemical characteristics and specific lipid components which condition nAChRs agglomeration.
With respect to neuronal nAChR, it was observed that α7 nAChR is associated with Chol-rich microdomains at somatic spine-rich regions of ciliary neurons and that the maintenance of these receptors within these domains is Chol-dependent (Brusés et al., 2001). Furthermore, in PC-12 cells, a rat pheochromocytoma cell line, α7 nAChR location in raft domains is necessary to regulate cAMP signal through the nicotinic activation, signaling that was altered by Chol depletion (Oshikawa et al., 2003). A similar relation between α7 nAChR location at raft domains and efficiently signaling, with a direct Chol influence, was also observed in CG neurons (Liu et al., 2008). Disruption of raft domains in the same CG neurons increased the mobility of α7 nAChRs in the synaptic space (Fernandes et al., 2010). Disruption of raft domains by removal of Chol and/or SM in rat primary hippocampal neurons slowed the kinetics of α7 nAChR desensitization through increasing the rate of recovery from desensitization and increased the agonist affinity and single-channel conductance (Colón-Sáez and Yakel, 2011). The authors observed the effects of raft domains disruption also on α3β2 nAChRs functionality. These results confirm that, as with muscle nAChR, neuronal nAChR functionality is modulated by its lipid microenvironment with the raft domains integrity a critical factor. On the contrary, α7 nAChR at non-neural tissues, in rat arterial endothelial (RAEC) and human venous endothelial (HUVEC), was found to occur in non-raft subcellular membrane fractions (Peña et al., 2011).
Alzheimer’s disease is a progressive neurodegenerative condition, the etiopathogenic mechanisms of which are not totally understood. Due to its multifactorial character, the development of new drugs and effective treatments is still a challenge (Dineley, 2007). Here, we intended to focus only in the processes related to this disease that occur in the cell membrane, which allows to observe the multiple crosslinking between specific lipids and the membrane proteins involved in the amyloid process. In a dry human brain, half of its weight corresponds to lipids, molecules with great chemical diversity and complex dynamical heterogeneities (Piomelli et al., 2007). Thus, it is not surprising that through the years more and more biological functions are being related to them. Raft domains are implicated in several of the events involved in AD. Chol is a very important lipid at synaptic membranes (Barrantes, 2007) and it is also a principal author in AD, together with other lipids such as GM1, SM or PA. It is not surprising that APP, Aβ, nAChR and G4-PRiMA all have Chol-recognition amino acid sequences. Although there are still some controversies, there is no doubt that APP processing, Aβ production and Aβ action are intimately related to raft domains, and that the cholinergic system function is highly conditioned by both raft domains and Aβ. A continuous crosstalk between amyloid processing and cholinergic signaling occurs at physiological and pathological conditions, and shifting from one condition to the other is triggered by an imbalance in Aβ synthesis, being Chol homeostasis intimately implicated (Figure 3). Currently, the only available treatment for AD is a group of drugs that inhibit ACE. A better understanding of Aβ-α7 nAChR interactions and of the implication of Chol in particular, and membrane heterogeneities in general, could allow for a deepening of the understanding of this neurodegenerative pathology and could help define new therapeutic strategies and potential novel molecular targets.
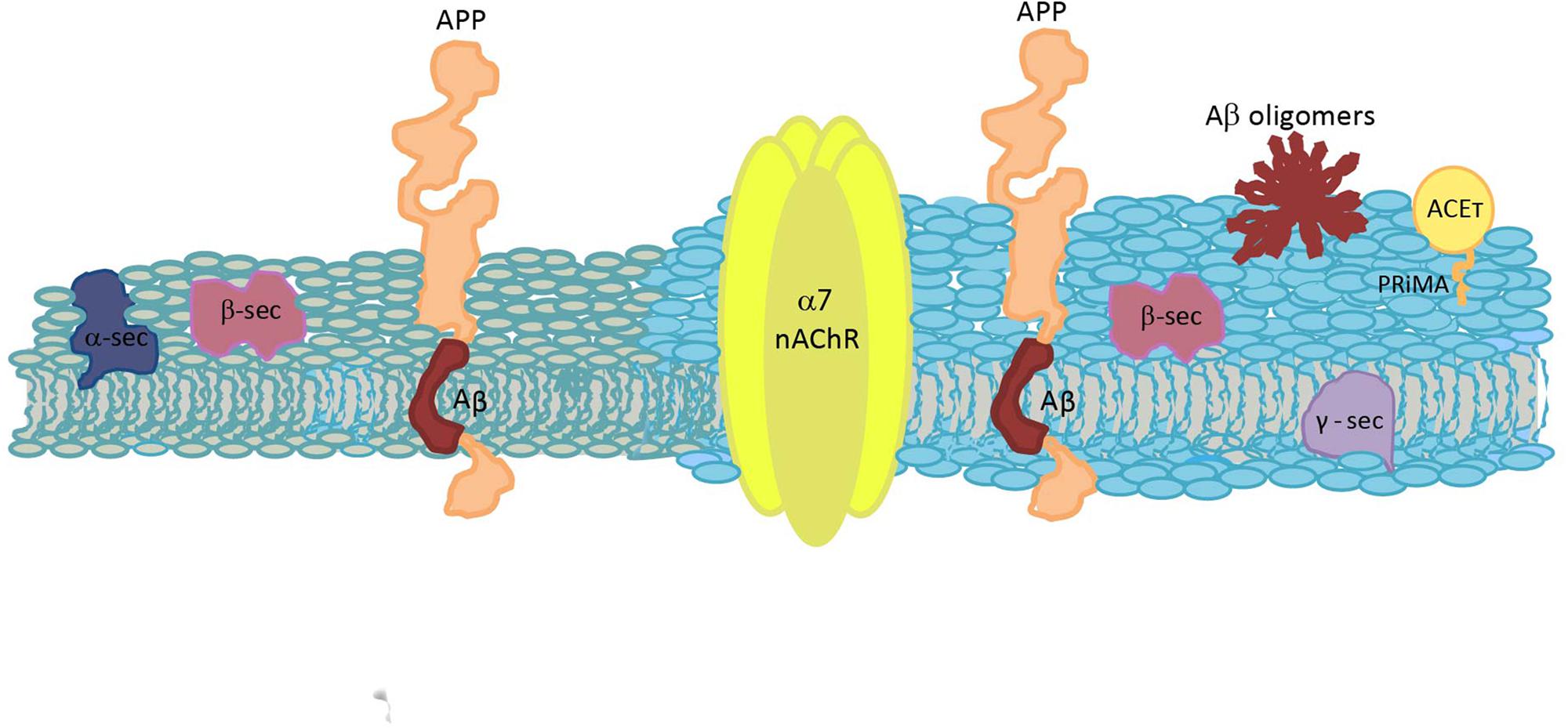
Figure 3. Schematic diagram of a plasma membrane, depicting the spatial relationship between the molecules involved in Aβ synthesis and the cholinergic system. Two different colors are used to represent a raft domain and a liquid-disordered domain (light blue and gray, respectively). APP, amyloid precursor protein; Aβ, amyloid β peptide; α7 nAChR, α7 nicotinic acetylcholine receptor; ACET, tetrameric acetylcholinesterase; α-sec, α-secretase; β-sec, β-secretase; γ-sec, γ-secretase; and PRiMA, proline-rich membrane anchor.
The World Health Organization (WHO) declared dementia as a public health priority (in Priority Medicines for Europe and the World “A Public Health Approach to Innovation” by Saloni Tanna). The number of people worldwide with this condition is in continuous growth: whereas in 2010 this number was estimated to be 35.6 million, it is expected to be near 115.4 million in 2050, in line with the view that this number nearly doubles every 20 years. AD is the most common form of dementia and, hence, it has become a major public health problem because of the continuous increase in the age of the population (in fact, in 2050 it is expected that 22% of the world population will be aged 60 and over). Thus, it is imperative to count with specific biomarkers for early stages of the disease, to improve detection and evaluation and, of course, with effective therapies. At present, the only treatment available is symptomatic: ACE inhibitors, like physostigmine, tacrine, donepezil, rivastigmine, and galantamine. Although new knowledge is continuously emerging, until now and as suggested in this work, there is no consensus among the different coexisting hypotheses around this subject, several of which are antagonistic. This fact clearly contributes to the current situation: there is not a single specific AD treatment commercially available. A great variety of molecular targets were proposed for AD treatment, a few of which were explained here (like β and γ-secretases, α7 nAChR and ACE), and plenty of studies have been conducted on them. Much effort has been invested in this area, but more is still required. Science is facing a huge challenge. Further studies that contribute to the description and explanation of the AD etiopathology will be crucial for a final consensus on AD. Multitarget-drug design is an interesting strategy as AD involves a large number of different molecules. And finally, it should not be forgotten that membrane lipids are not just a “sea” where proteins function but, as explained in detail above, they are necessary for the proper function of these proteins. Chol, GM1, SM, among others, are important lipids for AChR function, conformation and membrane stabilization, and also for Aβ processing and Aβ-membrane insertion. Thus, lipid membrane perturbation, and hence, raft domains alteration and membrane signal perturbation, directly impact in several hot points of AD etiopathology and, for this reason, they can also be considered as interesting molecular targets for AD.
CF and SA contributed to the design, analysis, interpretation, and writing of the manuscript.
This work was supported by grants from the Universidad Nacional del Sur (UNS) and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) to SA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aβ, amyloid β protein; ACE, acetylcholinesterase; AD, Alzheimer’s disease; APP, amyloid precursor protein; BACE 1, β-site APP-cleaving transmembrane aspartic protease 1; CARC, inversed CRAC; Chol, cholesterol; CRAC, Chol recognition amino acid consensus; DPPC, dipalmitoylphosphatidylcholine; ld, liquid-disorder domain; LDM, low density membrane domain; lo, liquid-ordered domain; M1 to M4, nAChR transmembrane segments; MuSK, muscle specific receptor tyrosine kinase; nAChR, nicotinic acetylcholine receptor; NMJ, neuromuscular junction; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PRiMA, proline-rich membrane anchor; PS1, presenilin 1; PUFA, polyunsaturated fatty acid; SM, sphingomyelin.
Abad-Rodriguez, J., Ledesma, M. D., Craessaerts, K., Perga, S., Medina, M., Delacourte, A., et al. (2004). Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell. Biol. 167, 953–960. doi: 10.1083/jcb.200404149
Addona, G. H., Sandermann, H., and Kloczewiak, M. A. (1998). Where does cholesterol act during activation of the nicotinic acetylcholine receptor? Biochim. Biophys. Acta 1370, 299–309. doi: 10.1016/s0005-2736(97)00280-0
Ahyayauch, H., Raab, M., Busto, J. V., Andraka, N., Arrondo, J. L., Masserini, M., et al. (2012). Binding of β-amyloid (1-42) peptide to negatively charged phospholipid membranes in the liquid-ordered state: modeling and experimental studies. Biophys. J. 103, 453–463. doi: 10.1016/j.bpj.2012.06.043
Albuquerque, E. X., Pereira, E. F. R., Alkondon, M., and Rogers, S. W. (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120. doi: 10.1152/physrev.00015.2008
Alptüzün, V., Prinz, M., Hörr, V., Scheiber, J., Radacki, K., Fallarero, A., et al. (2010). Interaction of (benzylidene-hydrazono)-1,4-dihydropyridines with beta-amyloid, acetylcholine, and butyrylcholine esterases. Bioorg. Med. Chem. 218, 2049–2059. doi: 10.1016/j.bmc.2010.01.002
Alvarez, A., Alarcón, R., Opazo, C., Campos, E. O., Muñoz, F. J., Calderón, F. H., et al. (1998). Stable complexes involving acetylcholinesterase and amyloid-beta peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer’s fibrils. J. Neurosci. 18, 3213–3223. doi: 10.1523/jneurosci.18-09-03213.1998
Andreasen, N., Sjögren, M., and Blennow, K. (2003). CSF markers for Alzheimer’s disease: total tau, phospho-tau and Abeta42. World J. Biol. Psychiatry 4, 147–155. doi: 10.1080/15622970310029912
Andreasen, T. J., and McNamee, M. G. (1980). Inhibition of ion permeability control properties of acetylcholine receptor from Torpedo californica by long-chain fatty acids. Biochemistry 19, 4719–4726. doi: 10.1021/bi00561a027
Annaert, W. G., Levesque, L., Craessaerts, K., Dierinck, I., Snellings, G., Westaway, D., et al. (1999). Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J. Cell. Biol. 147, 277–294. doi: 10.1083/jcb.147.2.277
Antollini, S. S., and Barrantes, F. J. (1998). Disclosure of discrete sites for phospholipid and sterols at the protein-lipid interface in native acetylcholine receptor-rich membrane. Biochemistry 37, 16653–16662. doi: 10.1021/bi9808215
Antollini, S. S., and Barrantes, F. J. (2002). Unique effects of different fatty acid species on the physical properties of the torpedo acetylcholine receptor membrane. J. Biol. Chem. 277, 1249–1254. doi: 10.1074/jbc.M106618200
Antollini, S. S., and Barrantes, F. J. (2016). Fatty Acid Regulation of Voltage- and Ligand-Gated Ion Channel Function. Front. Physiol. 28:573. doi: 10.3389/fphys.2016.00573
Antollini, S. S., Soto, M. A., Bonini de Romanelli, I., Gutiérrez-Merino, C., Sotomayor, P., and Barrantes, F. J. (1996). Physical state of bulk and protein-associated lipid in nicotinic acetylcholine receptor-rich membrane studied by laurdan generalized polarization and fluorescence energy transfer. Biophys. J. 70, 1275–1284. doi: 10.1016/s0006-3495(96)79684-4
Antollini, S. S., Xu, Y., Jiang, H., and Barrantes, F. J. (2005). Fluorescence and molecular dynamics studies of the acetylcholine receptor gammaM4 transmembrane peptide in reconstituted systems. Mol. Membr. Biol. 22, 471–483. doi: 10.1080/09687860500367915
Arbor, S. C., Lafontaine, M., and Cumbay, M. (2016). Amyloid-beta Alzheimer targets -protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 89, 5–21.
Ariga, T., Kobayashi, K., Hasegawa, A., Kiso, M., Ishida, H., and Miyatake, T. (2001). Characterization of high-affinity binding between gangliosides and amyloid beta-protein. Arch. Biochem. Biophys. 388, 225–230. doi: 10.1006/abbi.2001.2304
Arispe, N., Pollard, H. B., and Rojas, E. (1993a). Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc. Natl. Acad. Sci. U.S.A. 90, 10573–10577. doi: 10.1073/pnas.90.22.10573
Arispe, N., Rojas, E., and Pollard, H. B. (1993b). Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U.S.A. 90, 567–571. doi: 10.1073/pnas.90.2.567
Arora, K., Alfulaij, N., Higa, J. K., Panee, J., and Nichols, R. A. (2013). Impact of sustained exposure to β-amyloid on calcium homeostasis and neuronal integrity in model nerve cell system expressing α4β2 nicotinic acetylcholine receptors. J. Biol. Chem. 288, 11175–11190. doi: 10.1074/jbc.M113.453746
Ashley, R. H., Harroun, T. A., Hauss, T., Breen, K. C., and Bradshaw, J. P. (2006). Autoinsertion of soluble oligomers of Alzheimer’s Abeta(1-42) peptide into cholesterol-containing membranes is accompanied by relocation of the sterol towards the bilayer surface. BMC. Struct. Biol. 6:21. doi: 10.1186/1472-6807-6-21
Askarova, S., Yang, X., and Lee, J. C. (2011). Impacts of membrane biophysics in Alzheimer’s Disease: from amyloid precursor protein processing to Aβ peptide-induced membrane changes. Int. J. Alzheimers Dis. 2011:134971. doi: 10.4061/2011/134971
Atack, J. R., Perry, E. K., Bonham, J. R., Perry, R. H., Tomlinson, B. E., Blessed, G., et al. (1983). Molecular forms of acetylcholinesterase in senile dementia of Alzheimer type: selective loss of the intermediate (10S) form. Neurosci. Lett. 40, 199–204. doi: 10.1016/0304-3940(83)90302-6
Auld, D. S., Kornecook, T. J., Bastianetto, S., and Quirion, R. (2002). Alzheimer’s disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68, 209–245. doi: 10.1016/s0301-0082(02)00079-5
Avdulov, N. A., Chochina, S. V., Igbavboa, U., Warden, C. S., Vassiliev, A. V., and Wood, W. G. (1997). Lipid binding to amyloid beta-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J. Neurochem. 69, 1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x
Baenziger, J. E., and Corringer, P. J. (2011). 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology 60, 116–125. doi: 10.1016/j.neuropharm.2010.08.007
Baenziger, J. E., and daCosta, C. J. B. (2013). Molecular mechanisms of acetylcholine receptor-lipid interactions: from model membranes to human biology. Biophys. Rev. 5, 1–9. doi: 10.1007/s12551-012-0078-77
Baenziger, J. E., Hénault, C. M., Therien, J. P. D., and Sun, J. (2015). Nicotinic acetylcholine receptor-lipid interactions: mechanistic insight and biological function. Biochim. Biophys. Acta Biomembr. 1848, 1806–1817. doi: 10.1016/j.bbamem.2015.03.010
Baenziger, J. E., Morris, M., Darsaut, T. E., and Ryan, S. E. (2000). Effect of membrane lipid composition on the conformational equilibria of the Nicotinic acetylcholine receptor. J. Biol. Chem. 275, 777–784. doi: 10.1074/jbc.275.2.777
Baenziger, J. E., Ryan, S. E., Goodreid, M. M., Vuong, N. Q., Sturgeon, R. M., and Corrie, J. B. (2008). Lipid composition alters drug action at the Nicotinic acetylcholine receptor. Mol. Pharmacol. 73, 880–890. doi: 10.1124/mol.107.039008
Bagatolli, L. A. (2010). Microscopy imaging of membrane domains. Biochim. Biophys. Acta 1798:1285. doi: 10.1016/j.bbamem.2010.05.023
Baier, C. J., and Barrantes, F. J. (2007). Sphingolipids are necessary for nicotinic acetylcholine receptor export in the early secretory pathway. J. Neurochem. 101, 1072–1084. doi: 10.1111/j.1471-4159.2007.04561.x
Baier, C. J., Fantini, J., and Barrantes, F. J. (2011). Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 1:69. doi: 10.1038/srep00069
Baier, C. J., Gallegos, C. E., Levi, V., and Barrantes, F. J. (2010). Cholesterol modulation of nicotinic acetylcholine receptor surface mobility. Eur. Biophys 39, 213–227. doi: 10.1007/s00249-009-0521-522
Banerjee, C., Nyengaard, J. R., Wevers, A., de Vos, R. A., Jansen Steur, E. N., Lindstrom, J., et al. (2000). Cellular expression of alpha7 Nicotinic Acetylcholine receptor protein in the temporal cortex in Alzheimer’s and Parkinson’s disease - A stereological approach. Neurobiol. Dis. 7, 666–672. doi: 10.1006/nbdi.2000.0317
Barenholz, Y. (2004). Sphingomyelin and cholesterol: from membrane biophysics and rafts to potential medical applications. Subcell. Biochem. 37, 167–215. doi: 10.1007/978-1-4757-5806-1_5
Barrantes, F. J. (2002). Lipid matters: nicotinic acetylcholine receptor-lipid interactions. Mol. Membr. Biol. 219, 277–284. doi: 10.1080/09687680210166226
Barrantes, F. J. (2007). Cholesterol effects on nicotinic acetylcholine receptor. Neurochem. 103, 72–80. doi: 10.1111/j.1471-4159.2007.04719.x
Barrantes, F. J. (2010). Cholesterol effects on nicotinic acetylcholine receptor: cellular aspects. Subcell. Biochem. 51, 467–487. doi: 10.1007/978-90-481-8622-8_17
Barrantes, F. J. (2015). Phylogenetic conservation of protein-lipid motifs in pentameric ligand-gated ion channels. Biochim. Biophys. Acta 1848, 1796–1805. doi: 10.1016/j.bbamem.2015.03.028
Barrantes, F. J., Borroni, V., and Vallés, S. (2010). Neuronal nicotinic acetylcholine receptor – cholesterol crosstalk in Alzheimer’s disease. FEBS Lett. 584, 1856–1863. doi: 10.1016/j.febslet.2009.11.036
Barrera, N. P., Zhou, M., and Robinson, C. V. (2013). The role of lipids in defining membrane protein interactions: insights from mass spectrometry. Trends. Cell. Biol. 23, 1–8. doi: 10.1016/j.tcb.2012.08.007
Barrett, P. J., Song, Y., Horn, W. D., Van Hustedt, E. J., Johanna, M., Hadziselimovic, A., et al. (2012). The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 336, 1168–1171. doi: 10.1126/science.1219988
Bartolini, M., Bertucci, C., Cavrini, V., and Andrisano, V. (2003). Beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 65, 407–416. doi: 10.1016/s0006-2952(02)01514-9
Bartus, R. T., Dean, R. L., Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 1217, 408–414. doi: 10.1126/science.7046051
Bazzi, M. D., and Nelsestuen, G. L. (1996). Extensive segregation of acidic phospholipids in membranes induced by protein kinase C and related proteins. Biochemistry 32, 7961–7969. doi: 10.1021/bi00246a013
Beel, A. J., Sakakura, M., Barrett, P. J., and Sanders, C. R. (2010). Direct binding of cholesterol to the amyloid precursor protein: an important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta 1801, 975–982. doi: 10.1016/j.bbalip.2010.03.008
Behl, C., Davis, J. B., Lesley, R., and Schubert, D. (1994). Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77, 817–827. doi: 10.1016/0092-8674(94)90131-7
Berg, D. K., and Conroy, W. G. (2002). Nicotinic alpha 7 receptors: synaptic options and downstream signaling in neurons. J. Neurobiol. 53, 512–523. doi: 10.1002/neu.10116
Bermúdez, V., Antollini, S. S., Nievas, G. A. F., Aveldaño, M. I., and Barrantes, F. J. (2010). Partition profile of the nicotinic acetylcholine receptor in lipid domains upon reconstitution. J. Lipid Res. 51, 2629–2641. doi: 10.1194/jlr.M005132
Bieschke, J., Zhang, Q., Powers, E. T., Lerner, R. A., and Kelly, J. W. (2005). Oxidative metabolites accelerate Alzheimer’s amyloidogenesis by a two-step mechanism, eliminating the requirement for nucleation. Biochemistry 44, 4977–4983. doi: 10.1021/bi0501030
Bigl, V., Woolf, N. J., and Butcher, L. L. (1982). Cholinergic projections from the basal forebrain to frontal, parietal, temporal, occipital, and cingulate cortices: a combined fluorescent tracer and acetylcholinesterase analysis. Brain Res. Bull. 8, 727–749. doi: 10.1016/0361-9230(82)90101-0
Blanton, M. P., Xie, Y., Dangott, L. J., and Cohen, J. B. (1999). The steroid promegestone is a noncompetitive antagonist of the Torpedo nicotinic acetylcholine receptor that interacts with the lipid-protein interface. Mol. Pharmacol. 55, 269–278. doi: 10.1124/mol.55.2.269
Bodovitz, S., and Klein, W. L. (1996). Cholesterol Modulates alpha-Secretase Cleavage of Amyloid Precursor Protein. J. Biol. Chem. 271, 4436–4440. doi: 10.1074/jbc.271.8.4436
Bonini, I. C., Antollini, S. S., Gutiérrez-Merino, C., and Barrantes, F. J. (2002). Sphingomyelin composition and physical asymmetries in native acetylcholine receptor-rich membranes. Eur. Biophys. J. 31, 417–427. doi: 10.1007/s00249-002-0230-236
Borroni, V., Baier, C. J., Lang, T., Bonini, I., White, M. M., Garbus, I., et al. (2007). Cholesterol depletion activates rapid internalization of submicron-sized acetylcholine receptor domains at the cell membrane. Mol. Membr. Biol. 24, 1–15. doi: 10.1080/09687860600903387
Borroni, V., and Barrantes, F. J. (2011). Cholesterol Modulates the Rate and Mechanism of Acetylcholine Receptor Internalization. J Biol Chem. 286, 17122–17132. doi: 10.1074/jbc.M110.211870
Bouzat, C., and Barrantes, F. J. (1996). Modulation of muscle nicotinic acetylcholine receptors by the glucocorticoid hydrocortisone. Possible allosteric mechanism of channel blockade. J. Biol. Chem. 271, 25835–25841. doi: 10.1074/jbc.271.42.25835
Bouzat, C. B., Lacorazza, H. D., de Jiménez Bonino, M. B., and Barrantes, F. J. (1993). Effect of chemical modification of extracellular histidyl residues on the channel properties of the nicotinic acetylcholine receptor. Pflugers. Arch. 423, 365–371. doi: 10.1007/bf00374929
Bowen, D. M., Smith, C. B., White, P., and Davison, A. N. (1976). Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 99, 459–496. doi: 10.1093/brain/99.3.459
Boyd-Kimball, D., Castegna, A., Sultana, R., Poon, H. F., Petroze, R., Lynn, B. C., et al. (2005). Proteomic identification of proteins oxidized by Abeta(1-42) in synaptosomes: implications for Alzheimer’s disease. Brain Res. 1044, 206–215. doi: 10.1016/j.brainres.2005.02.086
Brannigan, G., Hénin, J., Law, R., Eckenhoff, R., and Klein, M. L. (2008). Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 14418–14423. doi: 10.1073/pnas.0803029105
Brown, D. A. (2006). Acetylcholine. Br. J. Pharmacol. 147(Suppl. 1), S120–S126. doi: 10.1038/sj.bjp.0706474
Brown, D. A., and London, E. (2000). Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275, 17221–17224. doi: 10.1074/jbc.r000005200
Brusés, J. L., Chauvet, N., and Rutishauser, U. (2001). Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J. Neurosci. 21, 504–512. doi: 10.1523/jneurosci.21-02-00504.2001
Burke, D. A., Heshmati, P., Kholdebarin, E., and Levin, E. D. (2014). Decreasing nicotinic receptor activity and the spatial learning impairment caused by the NMDA glutamate antagonist dizocilpine in rats. Eur. J. Pharmacol. 741, 132–139. doi: 10.1016/j.ejphar.2014.07.030
Burns, M., and Duff, K. (2002). Cholesterol in Alzheimer’s disease and tauopathy. Ann. N. Y. Acad. Sci. 977, 367–375. doi: 10.1111/j.1749-6632.2002.tb04839.x
Butler, D. H., and McNamee, M. G. (1993). FTIR analysis of nicotinic acetylcholine receptor secondary structure in reconstituted membranes. Biochim. Biophys. Acta 11150, 17–24. doi: 10.1016/0005-2736(93)90116-h
Butterfield, D. A., and Boyd-Kimball, D. (2005). The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 1703, 149–156. doi: 10.1016/j.bbapap.2004.10.014
Butterfield, D. A., Swomley, A. M., and Sultana, R. (2013). Amyloid β-Peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease Pathogenesis and Progression. Antioxid. Redox. Signal. 19, 823–835. doi: 10.1089/ars.2012.5027
Campagna, J. A., and Fallon, J. (2006). Lipid rafts are involved in C95 (4,8) agrin fragment-induced acetylcholine receptor clustering. Neuroscience 138, 123–132. doi: 10.1016/j.neuroscience.2005.11.019
Campanari, M. L., García-Ayllón, M. S., Belbin, O., Galcerán, J., Lleó, A., and Sáez-Valero, J. (2014). Acetylcholinesterase modulates presenilin-1 levels and γ-secretase activity. J. Alzheimers Dis. 41, 911–924. doi: 10.3233/JAD-140426
Cartaud, A., Stetzkowski-Marden, F., Maoui, A., and Cartaud, J. (2011). Agrin triggers the clustering of raft-associated acetylcholine receptors through actin cytoskeleton reorganization. Biol. Cell. 103, 287–301. doi: 10.1042/BC20110018
Cascella, R., Evangelisti, E., Bigi, A., Becatti, M., Fiorillo, C., Stefani, M., et al. (2017). Soluble oligomers require a ganglioside to trigger neuronal calcium overload. J. Alzheimers. Dis. 60, 923–938. doi: 10.3233/JAD-170340
Cecchi, C., Nichino, D., Zampagni, M., Bernacchioni, C., Evangelisti, E., Pensal, A., et al. (2009). A protective role for lipid raft cholesterol against amyloid-induced membrane damage in human neuroblastoma cells. Biochim. Biophys. Acta 1788, 2204–2216. doi: 10.1016/j.bbamem.2009.07.019
Champtiaux, N., Gotti, C., Cordero-Erausquin, M., David, D. J., Przybylski, C., Léna, C., et al. (2003). Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J. Neurosci. 23, 7820–7829. doi: 10.1523/jneurosci.23-21-07820.2003
Chang, T., Yamauchi, Y., Hasan, M. T., and Chang, C. (2017). Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid. Res. 58, 2239–2254. doi: 10.1194/jlr.R075630
Changeux, J. P., and Edelstein, S. J. (1998). Allosteric receptors after 30 years. Neuron 21, 959–980. doi: 10.1016/s0896-6273(00)80616-9
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chernomordik, L. V., and Zimmerberg, J. (1995). Bending membranes to the task: structural intermediates in bilayer fusion. Curr. Opin. Struct. Biol. 5, 541–547. doi: 10.1016/0959-440x(95)80041-7
Chochina, S. V., Avdulov, N. A., Igbavboa, U., Cleary, J. P., O’Hare, E. O., and Wood, W. G. (2001). Amyloid beta-peptide1-40 increases neuronal membrane fluidity: role of cholesterol and brain region. J. Lipid Res. 42, 1292–1297.
Choo-Smith, L. P., Garzon-Rodriguez, W., Glabe, C. G., and Surewicz, W. K. (1997). Acceleration of amyloid fibril formation by specific binding of Abeta-(1-40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272, 22987–22990. doi: 10.1074/jbc.272.37.22987
Chyung, J. H., Raper, D. M., and Selkoe, D. J. (2005). Gamma-secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J. Biol. Chem. 280, 4383–4392. doi: 10.1074/jbc.M409272200
Clarke, P. B. (1992). The fall and rise of neuronal alpha-bungarotoxin binding proteins. Trends Pharmacol. Sci. 113, 407–413. doi: 10.1016/0165-6147(92)90125-p
Colón-Sáez, J. O., and Yakel, J. L. (2011). The α7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J. Physiol. 589, 3163–3174. doi: 10.1113/jphysiol.2011.209494
Contestabile, A. (2011). The history of the cholinergic hypothesis. Behav. Brain Res. 221, 334–340. doi: 10.1016/j.bbr.2009.12.044
Cooper, S. T., Harkness, P. C., Baker, E. R., and Millar, N. S. (1999). Up-regulation of cell-surface alpha4beta2 neuronal Nicotinic receptors by lower temperature and expression of chimeric subunits. J. Biol. Chem. 274, 27145–27152. doi: 10.1074/jbc.274.38.27145
Corbin, J., Wang, H. H., and Blanton, M. P. (1998). Identifying the cholesterol binding domain in the nicotinic acetylcholine receptor with [125I] azido-cholesterol. Biochim. Biophys. Acta 1414, 65–74. doi: 10.1016/s0005-2736(98)00153-9
Cordy, J. M., Hooper, N. M., and Turner, A. J. (2006). The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr. Biol. 23, 111–122. doi: 10.1080/09687860500496417
Counts, S. E., He, B., Che, S., Ikonomovic, M. D., DeKosky, S. T., Ginsberg, S. D., et al. (2007). Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch. Neurol. 64, 1771–1776. doi: 10.1001/archneur.64.12.1771
Court, J. A., Piggott, M. A., Lloyd, S., Cookson, N., Ballard, C. G., McKeith, I. G., et al. (2000). Nicotine binding in human striatum: elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson’s disease and Alzheimer’s disease and in relation to neuroleptic medication. Neuroscience 98, 79–87. doi: 10.1016/s0306-4522(00)00071-3
Coyle, J. T., Price, D. L., and DeLong, M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190. doi: 10.1126/science.6338589
Criado, M., Eibl, H., and Barrantes, F. J. (1984). Functional properties of the acetylcholine receptor incorporated in model lipid membranes. Differential effects of chain length and head group of phospholipids on receptor affinity states and receptor-mediated ion translocation. J. Biol. Chem. 259, 9188–9198.
Criado, M., Vaz, W. L., Barrantes, F. J., and Jovin, T. M. (1982). Translational diffusion of acetylcholine receptor (monomeric and dimeric forms) of Torpedo marmorata reconstituted into phospholipid bilayers studied by fluorescence recovery after photobleaching. Biochemistry 21, 5750–5755. doi: 10.1021/bi00266a004
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 2070–2075. doi: 10.1073/pnas.0305799101
daCosta, C. J., Ogrel, A. A., McCardy, E. A., Blanton, M. P., and Baenziger, J. E. (2002). Lipid-protein interactions at the nicotinic acetylcholine receptor. A functional coupling between Nicotinic receptors and Phosphatidic acid-containing lipid bilayers. J. Biol. Chem. 277, 201–208. doi: 10.1074/jbc.M108341200
daCosta, C. J., Wagg, I. D., McKay, M. E., and Baenziger, J. E. (2004). Phosphatidic acid and phosphatidylserine have distinct structural and functional interactions with the nicotinic acetylcholine receptor. J. Biol. Chem. 279, 14967–14974. doi: 10.1074/jbc.M310037200
daCosta, C. J. B., and Baenziger, J. E. (2009). A lipid-dependent uncoupled conformation of the acetylcholine receptor. J. Biol. Chem. 284, 17819–17825. doi: 10.1074/jbc.M900030200
Dajas-Bailador, F., and Wonnacott, S. (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends. Pharmacol. Sci. 25, 317–324. doi: 10.1016/s0165-6147(04)00118-x
Daleke, D. L. (2003). Regulation of transbilayer plasma membrane phospholipid asymmetry. J. Lipid Res. 44, 233–242. doi: 10.1194/jlr.r200019-jlr200
D’Andrea, M. R., and Nagele, R. G. (2006). Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Curr. Pharm. Des. 12, 677–684. doi: 10.2174/138161206775474224
Daugherty, B. L., and Green, S. A. (2001). Endosomal sorting of amyloid precursor protein-P-selectin chimeras influences secretase processing. Traffic 2, 908–916. doi: 10.1034/j.1600-0854.2001.21206.x
Davies, P., and Maloney, A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2:1403. doi: 10.1016/s0140-6736(76)91936-x
de Almeida, R. F., Loura, L. M., Prieto, M., Watts, A., Fedorov, A., and Barrantes, F. J. (2004). Cholesterol modulates the organization of the gammaM4 transmembrane domain of the muscle nicotinic acetylcholine receptor. Biophys. J. 86, 2261–2272. doi: 10.1016/S0006-3495(04)74284-74288
Deutsch, S. I., Burket, J. A., and Benson, A. D. (2014). Targeting the α7 nicotinic acetylcholine receptor to prevent progressive dementia and improve cognition in adults with Down’s syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 254, 131–139. doi: 10.1016/j.pnpbp.2014.05.011
Deutsch, S. I., Burket, J. A., Benson, A. D., and Urbano, M. R. (2016). The 15q13.3 deletion syndrome: Deficient α(7)-containing nicotinic acetylcholine receptor-mediated neurotransmission in the pathogenesis of neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol Psychiatry 64, 09–17. doi: 10.1016/j.pnpbp.2015.08.001
Di Scala, C., Baier, C. J., Evans, L. S., Williamson, P. T. F., Fantini, J., and Barrantes, F. J. (2017). Relevance of CARC and CRAC Cholesterol-Recognition Motifs in the Nicotinic Acetylcholine Receptor and Other Membrane-Bound Receptors. Curr. Top. Membr. 80, 3–23. doi: 10.1016/bs.ctm.2017.05.001
Di Scala, C., Chahinian, H., Yahi, N., Garmy, N., and Fantini, J. (2014). Interaction of Alzheimer’s β-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry 53, 4489–4502. doi: 10.1021/bi500373k
Di Scala, C., Yahi, N., Boutemeur, S., Flores, A., Rodriguez, L., Chahinian, H., et al. (2016). Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 29:28781. doi: 10.1038/srep28781
Di Scala, C., Yahi, N., Lelièvre, C., Garmy, N., Chahinian, H., and Fantini, J. (2013). Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s β amyloid peptide. ACS Chem. Neurosci. 4, 509–517. doi: 10.1021/cn300203a
Dickey, A. N., and Faller, R. (2008). Behavioral differences between Phosphatidic acid and Phosphatidylcholine in the Presence of the Nicotinic Acetylcholine receptor. Biophys. J. 95, 5637–5647. doi: 10.1529/biophysj.108.136895
Dies, H., Toppozini, L., and Rheinstädter, M. C. (2014). The interaction between amyloid-β peptides and anionic lipid membranes containing cholesterol and melatonin. PLoS One 9:e99124. doi: 10.1371/journal.pone.0099124
Dineley, K. T. (2007). Beta-amyloid peptide–nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front. Biosci. 12:5030–5038.
Dineley, K. T., Bell, K. A., Bui, D., and Sweatt, J. D. (2002). Beta -Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J. Biol. Chem. 277, 25056–25061. doi: 10.1074/jbc.M200066200
Dineley, K. T., Westerman, M., Bui, D., Bell, K., Ashe, K. H., and Sweatt, J. D. (2001). Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 21, 4125–4133. doi: 10.1523/jneurosci.21-12-04125.2001
Dougherty, J. J., Wu, J., and Nichols, R. A. (2003). Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740–6747. doi: 10.1523/jneurosci.23-17-06740.2003
Dreger, M., Krauss, M., Herrmann, A., and Hucho, F. (1997). Interactions of the Nicotinic acetylcholine receptor Transmembrane segments with the Lipid Bilayer in Native receptor-rich membranes. Biochemistry 36, 839–847. doi: 10.1021/bi960666z
Dukhinova, M., Veremeyko, T., Yung, A. W. Y., Kuznetsova, I. S., Lau, T. Y. B., Kopeikina, E., et al. (2019). Fresh evidence for major brain gangliosides as a target for the treatment of Alzheimer’s disease. Neurobiol. Aging 77, 128–143. doi: 10.1016/j.neurobiolaging.2019.01.020
Dvir, H., Silman, I., Harel, M., Rosenberry, T. L., and Sussman, J. L. (2010). Acetylcholinesterase: from 3D structure to function. Chem. Biol. Interact. 187, 10–22. doi: 10.1016/j.cbi.2010.01.042
Dziewczapolski, G., Glogowski, C. M., Masliah, E., and Heinemann, S. F. (2009). Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 29, 8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009
Eckert, G. P., Kirsch, C., Leutz, S., Wood, W. G., and Müller, W. E. (2003). Cholesterol Modulates Amyloid Beta-peptide’s Membrane Interactions. Pharmacopsychiatry 36(Suppl. 2), S136–S143. doi: 10.1055/s-2003-43059
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell. Biol. 160, 113–123. doi: 10.1083/jcb.200207113
Ellena, J. F., Blazing, M. A., and McNamee, M. G. (1983). Lipid-protein interactions in reconstituted membranes containing Acetylcholine receptor. Biochemistry 22, 5523–5535. doi: 10.1021/bi00293a012
Engel, M. F., Khemtémourian, L., Kleijer, C. C., Meeldijk, H. J., Jacobs, J., Verkleij, A. J., et al. (2008). Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 6033–6038. doi: 10.1073/pnas.0708354105
Engelman, D. M. (2005). Membranes are more mosaic than fluid. Nature 438, 578–580. doi: 10.1038/nature04394
Epand, R. F., Kraayenhof, R., Sterk, G. J., Wong Fong Sang, H. W., and Epand, R. M. (1996). Fluorescent probes of membrane surface properties. Biochim Biophys Acta 1284, 191–195.
Fabelo, N., Martín, V., Marín, R., Moreno, D., Ferrer, I., and Díaz, M. (2014). Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 35, 1801–1812. doi: 10.1016/j.neurobiolaging.2014.02.005
Fantini, J., and Barrantes, F. J. (2013). How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 4:31. doi: 10.3389/fphys.2013.00031
Fantini, J., Di Scala, C., Evans, L. S., Williamson, P. T. F., and Barrantes, F. J. (2016). Open a mirror code for protein- cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 26:21907. doi: 10.1038/srep21907
Fantini, J., Di Scala, C., Yahi, N., Troadec, J. D., Sadelli, K., and Chahinian, H. (2014). Bxarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer’s β-amyloid peptides. ACS Chem. Neurosci. 5, 216–224. doi: 10.1021/cn400183w
Farhat, S. M., and Ahmed, T. (2017). Neuroprotective and Neurotoxic implications of α7 Nicotinic Acetylcholine receptor and Aβ Interaction: Therapeutic options in Alzheimer’s Disease. Curr. Drug. Targets 18, 1537–1544. doi: 10.2174/1389450117666161005145143
Fassbender, K., Masters, C., and Beyreuther, K. (2001). Alzheimer’s disease: molecular concepts and therapeutic targets. Naturwissenschaften 88, 261–267. doi: 10.1007/s001140100237
Feng, Y., and Wang, X. (2012). Antioxidant therapies for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012:472932. doi: 10.1155/2012/472932
Fernandes, C. C., Berg, D. K., and Gómez-Varela, D. (2010). Lateral mobility of nicotinic acetylcholine receptors on neurons is determined by receptor composition, local domain, and cell type. J. Neurosci. 30, 8841–8851. doi: 10.1523/JNEUROSCI.6236-09.2010
Fernández Nievas, G. A., Barrantes, F. J., and Antollini, S. S. (2007). Conformation-sensitive steroid and fatty acid sites in the transmembrane domain of the nicotinic acetylcholine receptor. Biochemistry 46, 3503–3512. doi: 10.1021/bi061388z
Fernández Nievas, G. A., Barrantes, F. J., and Antollini, S. S. (2008). Modulation of nicotinic acetylcholine receptor conformational state by free fatty acids and steroids. J. Biol. Chem. 283, 21478–21486. doi: 10.1074/jbc.M800345200
Fernández-Ballester, G., Castresana, J., Fernandez, A. M., Arrondo, J. L., Ferragut, J. A., and Gonzalez-Ros, J. M. (1994). Role of cholesterol as a structural and functional effector of the nicotinic acetylcholine receptor. Biochem. Soc. Trans. 122, 776–780. doi: 10.1042/bst0220776
Fishman, E. B., Siek, G. C., MacCallum, R. D., Bird, E. D., Volicer, L., and Marquis, J. K. (1986). Distribution of the molecular forms of acetylcholinesterase in human brain: alterations in dementia of the Alzheimer type. Ann. Neurol. 119, 246–252. doi: 10.1002/ana.410190305
Fong, T. M., and McNamee, M. G. (1986). Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry 25, 830–840. doi: 10.1021/bi00352a015
Fonseca, A. C., Moreira, P. I., Oliveira, C. R., Cardoso, S. M., Pinton, P., and Pereira, C. F. (2015). Amyloid-Beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol. Neurobiol 51, 610–622. doi: 10.1007/s12035-014-8740-8747
Frears, E. R., Stephens, D. J., Walters, C. E., Davies, H., and Austen, B. M. (1999). The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport 10, 1699–1705. doi: 10.1097/00001756-199906030-00014
Fucile, S. (2004). Ca2+ permeability of nicotinic acetylcholine receptors. Cell. Calcium 35, 1–8. doi: 10.1016/j.ceca.2003.08.006
Fukaya, T., Gondaira, T., Kashiyae, Y., Kotani, S., Ishikura, Y., Fujikawa, S., et al. (2007). Arachidonic acid preserves hippocampal neuron membrane fluidity in senescent rats. Neurobiol. Aging 28, 1179–1186. doi: 10.1016/j.neurobiolaging.2006.05.023
Fukunaga, S., Ueno, H., Yamaguchi, T., Yano, Y., Hoshino, M., and Matsuzaki, K. (2012). GM1 cluster mediates formation of toxic Aβ fibrils by providing hydrophobic environments. Biochemistry 51, 8125–8131. doi: 10.1021/bi300839u
Garbus, I., Bouzat, C., and Barrantes, F. J. (2001). Steroids differentially inhibit the nicotinic acetylcholine receptor. Neuroreport 12, 227–231. doi: 10.1097/00001756-200102120-00010
Garbus, I., Roccamo, A. M., and Barrantes, F. J. (2002). Identification of threonine 422 in transmembrane domain alpha M4 of the nicotinic acetylcholine receptor as a possible site of interaction with hydrocortisone. Neuropharmacology 43, 65–73. doi: 10.1016/s0028-3908(02)00068-0
García-Ayllón, M., Campanari, M., Montenegro, M., Cuchillo-ibáñez, I., Belbin, O., Lleó, A., et al. (2014). Neurobiology of aging Presenilin-1 influences processing of the acetylcholinesterase membrane anchor PRiMA. Neurobiol. Aging 35, 1526–1536. doi: 10.1016/j.neurobiolaging.2014.01.147
Geula, C., and Mesulam, M. (1989). Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 498, 185–189. doi: 10.1016/0006-8993(89)90419-8
Ghoneim, M. M., and Mewaldt, S. P. (1977). Studies on human memory: the interactions of diazepam, scopolamine, and physostigmine. Psychopharmacology 52, 1–6. doi: 10.1007/bf00426592
Ghribi, O., Larsen, B., Schrag, M., and Herman, M. M. (2006). High cholesterol content in neurons increases BACE, β -amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp. Neurol. 200, 460–467. doi: 10.1016/j.expneurol.2006.03.019
Gilbert, R. J. (2016). Protein-lipid interactions and non-lamellar lipidic structures in membrane pore formation and membrane fusion. Biochim. Biophys. Acta 1858, 487–499. doi: 10.1016/j.bbamem.2015.11.026
Gilbert, R. J., Dalla Serra, M., Froelich, C. J., Wallace, M. I., and Anderluh, G. (2014). Membrane pore formation at protein-lipid interfaces. Trends Biochem. Sci. 39, 510–516. doi: 10.1016/j.tibs.2014.09.002
Giniatullin, R., Nistri, A., and Yakel, J. L. (2005). Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends. Neurosci. 28, 371–378. doi: 10.1016/j.tins.2005.04.009
Gong, J. S., Sawamura, N., Zou, K., Sakai, J., Yanagisawa, K., and Michikawa, M. (2002). Amyloid beta-protein affects cholesterol metabolism in cultured neurons: implications for pivotal role of cholesterol in the amyloid cascade. Neurosci. Res. 70, 438–446. doi: 10.1002/jnr.10347
Goñi, F. M. (2014). The basic structure and dynamics of cell membranes?: an update of the Singer - Nicolson model. Biochim. Biophys. Acta 1838, 1467–1476. doi: 10.1016/j.bbamem.2014.01.006
Gotti, C., Clementi, F., Fornari, A., Gaimarri, A., Guiducci, S., Manfredi, I., et al. (2009). Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 78, 703–711. doi: 10.1016/j.bcp.2009.05.024
Gotti, C., Moretti, M., Gaimarri, A., Zanardi, A., Clementi, F., and Zoli, M. (2007). Heterogeneity and complexity of native brain nicotinic receptors. Biochem. Pharmacol. 74, 1102–1111. doi: 10.1016/j.bcp.2007.05.023
Gotti, C., Riganti, L., Vailati, S., and Clementi, F. (2006b). Brain neuronal nicotinic receptors as new targets for drug discovery. Curr. Pharm. Des. 12, 407–428. doi: 10.2174/138161206775474486
Gotti, C., Zoli, M., and Clementi, F. (2006a). Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol. Sci. 27, 482–491. doi: 10.1016/j.tips.2006.07.004
Götz, J., Schild, A., Hoerndli, F., and Pennanen, L. (2004). Amyloid-induced neurofibrillary tangle formation in Alzheimer’s disease: insight from transgenic mouse and tissue-culture models. Int. J. Dev. Neurosci. 22, 453–465. doi: 10.1016/j.ijdevneu.2004.07.013
Graham, A. J., Martin-Ruiz, C. M., Teaktong, T., Ray, M. A., and Court, J. A. (2002). Human brain nicotinic receptors, their distribution and participation in neuropsychiatric disorders. Curr. Drug. Targets CNS Neurol. Disord. 4, 387–397. doi: 10.2174/1568007023339283
Greenfield, J. P., Tsai, J., Gouras, G. K., Hai, B., Thinakaran, G., Checler, F., et al. (1999). Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 96, 742–747. doi: 10.1073/pnas.96.2.742
Grimm, M. O., Grimm, H. S., and Hartmann, T. (2007). Amyloid beta as a regulator of lipid homeostasis. Trends. Mol. Med. 13, 337–344. doi: 10.1016/j.molmed.2007.06.004
Grimm, M. O., Grimm, H. S., Pätzold, A. J., Zinser, E. G., Halonen, R., Duering, M., et al. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid- β and presenilin. Nat. Cell. Biol. 7, 1118–1123. doi: 10.1038/ncb1313
Grisaru, D., Sternfeld, M., Eldor, A., Glick, D., and Soreq, H. (1999). Structural roles of acetylcholinesterase variants in biology and pathology. Eur. J. Biochem. 264, 672–686. doi: 10.1046/j.1432-1327.1999.00693.x
Gu, L., and Guo, Z. (2013). Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 126, 305–311. doi: 10.1111/jnc.12202
Guillem, K., Bloem, B., Poorthuis, R. B., Loos, M., Smit, A. B., Maskos, U., et al. (2011). Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science 333, 888–891. doi: 10.1126/science.1207079
Gylys, K. H., Fein, J. A., Yang, F., Miller, C. A., and Cole, G. M. (2007). Increased cholesterol in Abeta-positive nerve terminals from Alzheimer’s disease cortex. Neurobiol. Aging 28, 8–17. doi: 10.1016/j.neurobiolaging.2005.10.018
Gyure, K. A., Durham, R., Stewart, W. F., Smialek, J. E., and Troncoso, J. C. (2001). Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch. Pathol. Lab. Med. 125, 489–492.
Haass, C. (1996). The molecular significance of amyloid beta-peptide for Alzheimer’s disease. Eur. Arch. Psychiatry. Clin. Neurosci. 246, 118–123.
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and Proteolytic Processing of APP. Cold. Spring. Harb. Perspect. Med. 2, 1–25. doi: 10.1101/cshperspect.a006270
Hamouda, A. K., Chiara, D. C., Sauls, D., Cohen, J. B., and Blanton, M. P. (2006). Cholesterol interacts with transmembrane alpha-helices M1, M3, and M4 of the Torpedo nicotinic acetylcholine receptor: photolabeling studies using [3H]Azicholesterol. Biochemistry 45, 976–986. doi: 10.1021/bi051978h
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., and Grossberg, G. T. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. doi: 10.1093/brain/awy132
Hardy, J. A., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends. Pharmacol. 12, 383–388. doi: 10.1016/0165-6147(91)90609-v
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s Disease?: the amyloid cascade hypothesis. Science 256, 184–185.
Harris, J. R. (2008). Cholesterol binding to amyloid-b fibrils: a TEM study. Micron 39, 1192–1196. doi: 10.1016/j.micron.2008.05.001
Hellström-Lindahl, E., Court, J., Keverne, J., Svedberg, M., Lee, M., Marutle, A., et al. (2004a). Nicotine reduces A beta in the brain and cerebral vessels of APPsw mice. Eur. J. Neurosci. 19, 2703–2710. doi: 10.1111/j.0953-816X.2004.03377.x
Hellström-Lindahl, E., Mousavi, M., Ravid, R., and Nordberg, A. (2004b). Reduced levels of Abeta 40 and Abeta 42 in brains of smoking controls and Alzheimer’s patients. Neurobiol. Dis. 15, 351–360. doi: 10.1016/j.nbd.2003.11.024
Hellström-Lindahl, E., Mousavi, M., Zhang, X., Ravid, R., and Nordberg, A. (1999). Regional distribution of nicotinic receptor subunit mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res. Mol. Brain. Res. 661, 94–103. doi: 10.1016/s0169-328x(99)00030-3
Hénault, C. M., Sun, J., Therien, J. P., daCosta, C. J., Carswell, C. L., Labriola, J. M., et al. (2015). The role of the M4 lipid-sensor in the folding, trafficking, and allosteric modulation of nicotinic acetylcholine receptors. Neuropharmacology 296, 157–168. doi: 10.1016/j.neuropharm.2014.11.011
Henderson, Z., Boros, A., Janzso, G., Westwood, A. J., Monyer, H., and Halasy, K. (2005). Somato-dendritic nicotinic receptor responses recorded in vitro from the medial septal diagonal band complex of the rodent. J. Physiol. Lond. 562, 165–182. doi: 10.1113/jphysiol.2004.070300
Henderson, Z., Matto, N., John, D., Nalivaeva, N. N., and Turner, A. J. (2010). Co-localization of PRiMA with acetylcholinesterase in cholinergic neurons of rat brain: an immunocytochemical study. Brain Res. 1344, 34–42. doi: 10.1016/j.brainres.2010.05.022
Herrup, K. (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799. doi: 10.1038/nn.4017
Hertel, C., Terzi, E., Hauser, N., Jakob-Rotne, R., Seelig, J., and Kemp, J. A. (1997). Inhibition of the electrostatic interaction between beta-amyloid peptide and membranes prevents beta-amyloid-induced toxicity. Proc. Natl. Acad. Sci. U.S.A. 94, 9412–9416. doi: 10.1073/pnas.94.17.9412
Hicks, D. A., John, D., Makova, N. Z., Henderson, Z., Nalivaeva, N. N., and Turner, A. J. (2011). Membrane targeting, shedding and protein interactions of brain acetylcholinesterase. J. Neurochem. 116, 742–746. doi: 10.1111/j.1471-4159.2010.07032.x
Hicks, D. A., Makova, N. Z., Gough, M., Parkin, E. T., Nalivaeva, N. N., and Turner, A. J. (2013b). The amyloid precursor protein represses expression of acetylcholinesterase in neuronal cell lines. J. Biol. Chem. 288, 26039–26051. doi: 10.1074/jbc.M113.461269
Hicks, D. A., Makova, N. Z., Nalivaeva, N. N., and Turner, A. J. (2013a). Chemico-Biological Interactions Characterisation of acetylcholinesterase release from neuronal cells. Chem. Biol. Interact. 203, 302–308. doi: 10.1016/j.cbi.2012.09.019
Hicks, D. A., Nalivaeva, N. N., and Turner, A. J. (2012). Lipid rafts and Alzheimer’s disease?: protein-lipid interactions and perturbation of signaling. Front. Physiol. 3:189. doi: 10.3389/fphys.2012.00189
Hong, S., Ostaszewski, B. L., Yang, T., Malley, T. T. O., Jin, M., Yanagisawa, K., et al. (2014). Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron. 82, 308–319. doi: 10.1016/j.neuron.2014.02.027
Hou, X., Richardson, S. J., Aguilar, M. I., and Small, D. H. (2005). Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry 44, 11618–11627. doi: 10.1021/bi050700m
Huse, J. T., Pijak, D. S., Leslie, G. J., Lee, V. M., and Doms, R. W. (2000). Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J. Biol. Chem. 275, 33729–33737. doi: 10.1074/jbc.M004175200
Igbavboa, U., Avdulov, N. A., Schroeder, F., and Wood, W. G. (1996). Increasing age alters transbilayer fluidity and cholesterol asymmetry in synaptic plasma membranes of mice. J. Neurochem. 66, 1717–1725. doi: 10.1046/j.1471-4159.1996.66041717.x
Ikonomovic, M. D., Wecker, L., Abrahamson, E. E., Wuu, J., Counts, S. E., Ginsberg, S. D., et al. (2009). Cortical alpha7 nicotinic acetylcholine receptor and beta-amyloid levels in early Alzheimer disease. Arch. Neurol. 66, 646–651. doi: 10.1001/archneurol.2009.46
Inestrosa, N. C., Alvarez, A., and Calderón, F. (1996). Acetylcholinesterase is a senile plaque component that promotes assembly of amyloid beta-peptide into Alzheimer’s filaments. Mol. Psychiatry 1, 359–361.
Irvine, G. B., El-agnaf, O. M., Shankar, G. M., and Walsh, D. M. (2008). Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’ s diseases. Mol. Med. 14, 451–464. doi: 10.2119/2007-00100
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13, 45–53. doi: 10.1016/0896-6273(94)90458-8
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jha, A., Cadugan, D. J., Purohit, P., and Auerbach, A. (2007). Acetylcholine receptor gating at extracellular transmembrane domain interface: the cys-loop and M2-M3 linker. J. Gen. Physiol. 130, 547–558. doi: 10.1085/jgp.200709856
Ji, S. R., Wu, Y., and Sui, S. F. (2002). Cholesterol is an important factor affecting the membrane insertion of beta-amyloid peptide (A beta 1-40), which may potentially inhibit the fibril formation. J. Biol. Chem. 277, 6273–6279. doi: 10.1074/jbc.M104146200
Jin, Y., Tsuchiya, A., Kanno, T., and Nishizaki, T. (2015). Biochemical and biophysical research communications Amyloid- b peptide increases cell surface localization of a7 ACh receptor to protect neurons from amyloid b -induced damage. Biochem. Biophys. Res. Commun. 468, 157–160. doi: 10.1016/j.bbrc.2015.10.141
Jones, I. W., Westmacott, A., Chan, E., Jones, R. W., Dineley, K., and O’Neill, M. J. (2006). Alpha7 nicotinic acetylcholine receptor expression in Alzheimer’s disease: receptor densities in brain regions of the APP(SWE) mouse model and in human peripheral blood lymphocytes. J. Mol. Neurosci. 30, 83–84. doi: 10.1385/JMN:30:1:83
Jones, O. T., and McNamee, M. G. (1988). Annular and nonannular binding sites for cholesterol associated with the nicotinic acetylcholine receptor. Biochemistry 127, 2364–2374. doi: 10.1021/bi00407a018
Kagan, B. L., Hirakura, Y., Azimov, R., Azimova, R., and Lin, M. (2002). The channel hypothesis of Alzheimer’s disease: current status. Peptides 23, 1311–1315. doi: 10.1016/s0196-9781(02)00067-0
Kakio, A., Nishimoto, S., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. (2002). Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 241, 7385–7390. doi: 10.1021/bi0255874
Kakio, A., Nishimoto, S. I., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. (2001). Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276, 24985–24990. doi: 10.1074/jbc.M100252200
Kamal, A., Almenar-Queralt, A., LeBlanc, J. F., Roberts, E. A., and Goldstein, L. S. (2001). Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414, 643–648. doi: 10.1038/414643
Kamerbeek, C. B., Borroni, V., Pediconi, M. F., Sato, S. B., Kobayashi, T., and Barrantes, F. J. (2013). Antibody-induced acetylcholine receptor clusters inhabit liquid-ordered and liquid-disordered domains. Biophys. J. 150, 1601–1611. doi: 10.1016/j.bpj.2013.08.03
Kanfer, J. N., Sorrentino, G., and Sitar, D. S. (1999). Amyloid beta peptide membrane perturbation is the basis for its biological effects. Neurochem. Res. 24, 1621–1630.
Karlin, A., and Akabas, M. H. (1995). Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron 15, 1231–1244. doi: 10.1016/0896-6273(95)90004-7
Kawahara, M. (2010). Neurotoxicity of β-amyloid protein: oligomerization, channel formation, and calcium dyshomeostasis. Curr. Pharm. Des. 16, 2779–2789. doi: 10.2174/138161210793176545
Kellner, R. R., Baier, C. J., Willig, K. I., Hell, S. W., and Barrantes, F. J. (2007). Nanoscale organization of nicotinic acetylcholine receptors revealed by stimulated emission depletion microscopy. Neuroscience 144, 135–143. doi: 10.1016/j.neuroscience.2006.08.071
Khan, G. M., Tong, M., Jhun, M., Arora, K., and Nichols, R. A. (2010). Beta-Amyloid activates presynaptic alpha7 nicotinic acetylcholine receptors reconstituted into a model nerve cell system: involvement of lipid rafts. Eur. J. Neurosci. 231, 788–796. doi: 10.1111/j.1460-9568.2010.07116.x
Kim, H.-J., Moon, W.-J., and Han, S.-H. (2013). Differential cholinergic pathway involvement in Alzheimer’s disease and subcortical ischemic vascular dementia. J. Alzheimers Dis. 35, 129–136. doi: 10.3233/JAD-122320
Kim, S., Yi, J., and Ko, Y. (2006). Amyloid b Oligomerization Is Induced by Brain Lipid Rafts. J. Cell. Biochem. 99, 878–889. doi: 10.1002/jcb.20978
Kirsch, C., Eckert, G. P., and Mueller, W. E. (2002). Cholesterol attenuates the membrane perturbing properties of beta-amyloid peptides. Amyloid 9, 149–159.
Kirsch, C., Eckert, G. P., and Mueller, W. E. (2003). Statin effects on cholesterol micro-domains in brain plasma membranes. Biochem. Pharmacol. 65, 843–856. doi: 10.1016/s0006-2952(02)01654-4
Klein, W. L., Steine, W. B. Jr., and Teplow, D. B. (2004). Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol. Aging 25, 569–580. doi: 10.1016/j.neurobiolaging.2004.02.010
Kogel, D., Copanaki, E., Hartig, U., Bottner, S., Peters, I., Muller, W. E., et al. (2008). “Modulation of membrane fluidity by omega 3 fatty acids: enhanced generation of sAPPalpha is required for the neuroprotective effects of DHA,” in The 38th Annual Meeting of the Society for Neuroscience, Washington, DC, 15–19.
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. U.S.A. 98, 5815–5820. doi: 10.1073/pnas.081612998
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Kosicek, M., and Hecimovic, S. (2013). Phospholipids and Alzheimer’s disease: alterations, mechanisms and potential biomarkers. Int. J. Mol. Sci. 214, 1310–1322. doi: 10.3390/ijms14011310
Kotler, S. A., Walsh, P., Brender, J. R., and Ramamoorthy, A. (2014). Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 19, 6692–6700. doi: 10.1039/c3cs60431d
Koudinov, A. R., and Koudinova, N. V. (2004). Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J. Neurol. Sci. 229-230, 233–240. doi: 10.1016/j.jns.2004.11.036
Koudinova, N. V., Koudinov, A. R., and Yavin, E. (2000). Alzheimer’s Abeta1-40 peptide modulates lipid synthesis in neuronal cultures and intact rat fetal brain under normoxic and oxidative stress conditions. Neurochem. Res. 25, 653–660.
Kumar, A., Singh, A., and Ekavali. (2015). A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep. 67, 195–203. doi: 10.1016/j.pharep.2014.09.004
Lahdo, R., and De La Fournière-Bessueille, L. (2004). Insertion of the amyloid precursor protein into lipid monolayers: effects of cholesterol and apolipoprotein E. Biochem. J. 382, 987–994. doi: 10.1042/BJ20040777
Langui, D., Girardot, N., El, Hachimi KH, Allinquant, B., Blanchard, V., Pradier, L., et al. (2004). Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am. J. Pathol. 165, 1465–1477. doi: 10.1016/s0002-9440(10)63405-0
Lashuel, H. A., Hartley, D., Petre, B. M., Walz, T., and Lansbury, P. T. Jr. (2002). Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature 418:291. doi: 10.1038/418291a
Lauderback, C. M., Hackett, J. M., Keller, J. N., Varadarajan, S., Szweda, L., Kindy, M., et al. (2001). Vulnerability of synaptosomes from apoE knock-out mice to structural and oxidative modifications induced by A beta(1-40): implications for Alzheimer’s disease. Biochemistry 240, 2548–2554. doi: 10.1021/bi002312k
Lazarevic-Pasti, T., Leskovac, A., Momic, T., Petrovic, S., and Vasic, V. (2017). Modulators of Acetylcholinesterase activity: from Alzheimer’s disease to anti-cancer drugs. Curr. Med. Chem. 24, 3283–3309. doi: 10.2174/0929867324666170705123509
Le Novère, N., and Changeux, J. P. (1995). Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J Mol. Evol. 40, 155–172. doi: 10.1007/bf00167110
Lee, A. G. (2003). Lipid - protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta 1612, 1–40. doi: 10.1016/S0005-2736(03)00056-57
Lee, D. H. S., and Wang, H.-Y. (2003). Differential physiologic responses of α7 nicotinic acetylcholine receptors to β-amyloid1–40 and β-amyloid1–42. J. Neurobiol. 55, 25–30. doi: 10.1002/neu.10203
Lee, E. B., Leng, L. Z., Zhang, B., Kwong, L., Trojanowski, J. Q., Abel, T., et al. (2006). Targeting amyloid-β peptide (Aβ) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Aβ precursor protein (APP) transgenic mice. J. Biol. Chem. 281, 4292–4299. doi: 10.1074/jbc.M511018200
Lee, S. J., Liyanage, U., Bickel, P. E., Xia, W., Lansbury, P. T. Jr., and Kosik, K. S. (1998). A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 4, 730–734. doi: 10.1038/nm0698-730
Levin, E. D., Cauley, M., and Rezvani, A. H. (2013). Improvement of attentional function with antagonism of nicotinic receptors in female rats. Eur. J. Pharmacol. 702, 269–274. doi: 10.1016/j.ejphar.2013.01.056
Li, L., Cheung, T., Chen, J., and Herrup, K. (2011). A comparative study of five mouse models of Alzheimer’s disease: cell cycle events reveal new insights into neurons at risk for death. Int. J. Alzheimers Dis. 2011:171464. doi: 10.4061/2011/171464
Lilja, A. M., Porras, O., Storelli, E., Nordberg, A., and Marutle, A. (2011). Functional interactions of fibrillar and oligomeric amyloid-β with alpha7 nicotinic receptors in Alzheimer’s disease. J. Alzheimers Dis. 23, 335–347. doi: 10.3233/JAD-2010-101242
Lin, H., Bhatia, R., and Lal, R. (2001). Amyloid beta protein forms ion channels: implications for Alzheimer’s diseases pathophysiology. FASEB J. 15, 2433–2444. doi: 10.1096/fj.01-0377com
Lin, M. S., Chen, L. Y., Wang, S. S., Chang, Y., and Chen, W. Y. (2008). Examining the levels of ganglioside and cholesterol in cell membrane on attenuation the cytotoxicity of beta-amyloid peptide. Colloids Surf. B. Biointerfaces 65, 172–177. doi: 10.1016/j.colsurfb.2008.03.012
Lindstrom, J. M. (2003). Nicotinic acetylcholine receptors of muscles and nerves: comparison of their structures, functional roles, and vulnerability to pathology. Ann. N.Y. Acad. Sci. 998, 41–52. doi: 10.1196/annals.1254.007
Liu, Q., Kawai, H., and Berg, D. K. (2001). Beta -Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 4734–4739. doi: 10.1073/pnas.081553598
Liu, Q., and Wu, J. (2006). Neuronal nicotinic acetylcholine receptors serve as sensitive targets that mediate beta-amyloid neurotoxicity. Acta. Pharmacol. Sin. 27, 1277–1286. doi: 10.1111/j.1745-7254.2006.00430.x
Liu, Q., Xie, X., Emadi, S., Sierks, M. R., and Wu, J. (2015). A novel nicotinic mechanism underlies b -amyloid-induced neurotoxicity. Neuropharmacology 97, 457–463. doi: 10.1016/j.neuropharm.2015.04.025
Liu, Q., Xie, X., Lukas, R. J., St John, P. A., and Wu, J. (2013). A novel nicotinic mechanism underlies β-amyloid-induced neuronal hyperexcitation. J. Neurosci. 33, 7253–7263. doi: 10.1523/JNEUROSCI.3235-12.2013
Liu, R., Gu, R., Qi, X., Zhang, T., Zhao, Y., and He, Y. (2008). Decreased Nicotinic receptors and cognitive deficit in rats intracerebroventricularly injected with beta-amyloid peptide (1-42) and fed a high-cholesterol diet. J. Neurosci. Res. 86, 183–193. doi: 10.1002/jnr.21463
Lombardo, S., and Maskos, U. (2014). Neuropharmacology Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 96, 255–262. doi: 10.1016/j.neuropharm.2014.11.018
London, E. (2005). How principles of domain formation in model membranes may explain ambiguities concerning lipidraft formation in cells. Biochim. Biophys. Acta 30, 203–220. doi: 10.1016/j.bbamcr.2005.09.002
Lukiw, W. J. (2013). Alzheimer’s disease (AD) as a disorder of the plasma membrane. Front. Physiol. 4:24. doi: 10.3389/fphys.2013.00024
Ma, K., and Qian, Y. (2019). Neuropeptides Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 73, 96–106. doi: 10.1016/j.npep.2018.12.003
Magni, F., Galbusera, C., Tremolada, L., Ferrarese, C., and Kienle, M. G. (2002). Characterisation of adducts of the lipid peroxidation product 4-hydroxy-2-nonenal and amyloid beta-peptides by liquid chromatography/electrospray ionisation mass spectrometry. Rapid. Commun. Mass. Spectrom. 16, 1485–1493. doi: 10.1002/rcm.743
Maia, M. A., and Sousa, E. (2019). BACE-1 and γ-secretase as therapeutic targets for Alzheimer’s disease. Pharmaceuticals 12:41. doi: 10.3390/ph12010041
Marchand, S., Devillers-thie, A., Pons, S., Changeux, J., and Cartaud, J. (2002). Rapsyn escorts the Nicotinic Acetylcholine receptor along the exocytic pathway via association with lipid rafts. J. Neurosci. 22, 8891–8901. doi: 10.1523/jneurosci.22-20-08891.2002
Mark, R., Lovell, M. A., Markesbery, W., Koiiuchida, I., and Mattson, P. (1997). A role for 4-Hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid- beta peptide. J. Neurochem. 68, 255–264. doi: 10.1046/j.1471-4159.1997.68010255.x
Marsh, D., and Barrantes, F. J. (1978). Immobilized lipid in acetylcholine receptor-rich membranes from Torpedo marmorata. Proc. Natl. Acad. Sci. U.S.A. 75, 4329–4333. doi: 10.1073/pnas.75.9.4329
Martín, V., Fabelo, N., Santpere, G., Puig, B., Marín, R., Ferrer, I., et al. (2010). Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimers. Dis. 19, 489–502. doi: 10.3233/JAD-2010-1242
Martínez-Senac, M., Villalaín, J., and Gómez-Fernández, J. C. (1999). Structure of the Alzheimer beta-amyloid peptide (25-35) and its interaction with negatively charged phospholipid vesicles. Eur. J. Biochem. 265, 744–753. doi: 10.1046/j.1432-1327.1999.00775.x
Martorana, A., Esposito, Z., and Koch, G. (2010). Beyond the cholinergic hypothesis: do current drugs work in Alzheimer’s disease? CNS Neurosci. Ther. 16, 235–245. doi: 10.1111/j.1755-5949.2010.00175.x
Mason, R. P., Shoemaker, W. J., Shajenko, L., Chambers, T. E., and Herbette, L. G. (1992). Evidence for changes in the Alzheimer’s disease brain cortical membrane structure mediated by cholesterol. Neurobiol. Aging 13, 413–419. doi: 10.1016/0197-4580(92)90116-f
Massoulié, J. (2002). The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 11, 130–143. doi: 10.1159/000065054
Massoulié, J., Bon, S., Perrier, N., and Falasca, C. (2005). The C-terminal peptides of acetylcholinesterase: cellular trafficking, oligomerization and functional anchoring. Chem. Biol. Interact. 157-158, 3–14. doi: 10.1016/j.cbi.2005.10.002
Matsuzaki, K. (2007). Physicochemical interactions of amyloid β -peptide with lipid bilayers. Biochim. Biophys. Acta 1768, 1935–1942. doi: 10.1016/j.bbamem.2007.02.009
Matsuzaki, K., Kato, K., and Yanagisawa, K. (2010). Abeta polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta 1801, 868–877. doi: 10.1016/j.bbalip.2010.01.008
McKay, B. E., Placzek, A. N., and Dani, J. A. (2007). Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 74, 1120–1133. doi: 10.1016/j.bcp.2007.07.001
McLaurin, J., and Chakrabartty, A. (1997). Characterization of the interactions of Alzheimer beta-amyloid peptides with phospholipid. Eur. J. Biochem. 1245, 355–363. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x
McLaurin, J., Franklin, T., Fraser, P. E., and Chakrabartty, A. (1998). Structural transitions associated with the interaction of Alzheimer beta-amyloid peptides with gangliosides. J. Biol. Chem. 273, 4506–4515. doi: 10.1074/jbc.273.8.4506
Mehta, T. K., Dougherty, J. J., Wu, J., Choi, C. H., Khan, G. M., and Nichols, R. A. (2009). Defining pre-synaptic nicotinic receptors regulated by beta amyloid in mouse cortex and hippocampus with receptor null mutants. J. Neurochem. 109, 1452–1458. doi: 10.1111/j.1471-4159.2009.06070.x
Messi, M. L., Renganathan, M., Grigorenko, E., and Delbono, O. (1997). Activation of alpha7 nicotinic acetylcholine receptor promotes survival of spinal cord motoneurons. FEBS Lett. 411, 32–38. doi: 10.1016/s0014-5793(97)00600-5
Mesulam, M. M., Mufson, E. J., Levey, A. I., and Wainer, B. H. (1983). Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol. 20, 170–197. doi: 10.1002/cne.902140206
Méthot, N., Demers, C. N., and Baenziger, J. E. (1995). Structure of both the ligand- and lipid-dependent channel-inactive states of the nicotinic acetylcholine receptor probed by FTIR spectroscopy and hydrogen exchange. Biochemistry 34, 15142–15149. doi: 10.1021/bi00046a021
Michikawa, M. (2003). The role of cholesterol in pathogenesis of Alzheimer’s disease: dual metabolic interaction between amyloid β-protein and cholesterol. Mol. Neurobiol. 27, 1–12. doi: 10.1385/MN:27:1:1
Michikawa, M., Gong, J. S., Fan, Q. W., Sawamura, N., and Yanagisawa, K. (2001). A novel action of alzheimer’s amyloid beta-protein (Abeta): oligomeric Abeta promotes lipid release. J. Neurosci. 218, 7226–7235.
Middlemas, D. S., and Raftery, M. A. (1987). Identification of Subunits of Acetylcholine Receptor That Interact with a Cholesterol Photoaffinity Probe. Biochemistry 126, 1219–1223. doi: 10.1021/bi00379a003
Milanesi, L., Sheynis, T., Xue, W. F., Orlova, E. V., Hellewell, A. L., Jelinek, R., et al. (2012). Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 20455–20460. doi: 10.1073/pnas.1206325109
Minota, S., and Watanabe, S. (1997). Inhibitory effects of arachidonic acid on nicotinic transmission in bullfrog sympathetic neurons. J. Neurophysiol. 178, 2396–2401. doi: 10.1152/jn.1997.78.5.2396
Molander-Melin, M., Blennow, K., Bogdanovic, N., Dellheden, B., and Fredman, P. (2005). Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent- resistant membrane domains. J. Neurochem. 92, 171–182. doi: 10.1111/j.1471-4159.2004.02849.x
Molinari, E. J., Delbono, O., Messi, M. L., Renganathan, M., Arneric, S. P., Sullivan, J. P., et al. (1998). Up-regulation of human alpha7 nicotinic receptors by chronic treatment with activator and antagonist ligands. Eur. J. Pharmacol. 347, 131–139. doi: 10.1016/s0014-2999(98)00084-3
Morán, M. A., Mufson, E. J., and Gómez-Ramos, P. (1993). Colocalization of cholinesterases with beta amyloid protein in aged and Alzheimer’s brains. Acta Neuropathol. 85, 362–369. doi: 10.1007/bf00334445
Morley, B. J., and Happe, H. K. (2000). Cholinergic receptors: dual roles in transduction and plasticity. Hear. Res. 147, 104–112. doi: 10.1016/s0378-5955(00)00124-6
Mosqueira, A., Camino, P. A., and Barrantes, F. J. (2018). Cholesterol modulates acetylcholine receptor diffusion by tuning confinement sojourns and nanocluster stability. Sci. Rep. 8:11974. doi: 10.1038/s41598-018-30384
Mouritsen, O. G., and Bloom, M. (1984). Mattress model of lipid-protein interactions in membranes. Biophys J. 46, 141–153. doi: 10.1016/s0006-3495(84)84007-2
Muir, J. L., Page, K. J., Sirinathsinghji, D. J., Robbins, T. W., and Everitt, B. J. (1993). Excitotoxic lesions of basal forebrain cholinergic neurons: effects on learning, memory and attention. Behav. Brain Res. 157, 123–131. doi: 10.1016/0166-4328(93)90128-d
Müller, W. E., Kirsch, C., and Eckert, G. P. (2001). Membrane-disordering effects of beta-amyloid peptides. Biochem. Soc. Trans. 29, 617–623. doi: 10.1042/bst0290617
Müller, W. E., Koch, S., Eckert, A., Hartmann, H., and Scheuer, K. (1995). β-amyloid peptide decreases membrane fluidity. Brain Res. 674, 133–136. doi: 10.1016/0006-8993(94)01463-R
Murray, I. V. J., Liu, L., Komatsu, H., Uryu, K., Xiao, G., John, A., et al. (2007). Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J. Biol. Chem. 282, 9335–9345. doi: 10.1074/jbc.M608589200
Murray, J. K., Farooqi, B., Sadowsky, J. D., Scalf, M., Freund, W. A., Smith, L. M., et al. (2005). Efficient synthesis of a beta-peptide combinatorial library with microwave irradiation. J. Am. Chem. Soc. 127, 13271–13280. doi: 10.1021/ja052733v
Nagele, R. G., D’Andrea, M. R., Anderson, W. J., and Wang, H. Y. (2002). Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110, 199–211. doi: 10.1016/s0306-4522(01)00460-2
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., and Wang, H. Y. (2003). Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209. doi: 10.1016/s0006-8993(03)02361-8
Nagele, R. G., Wegiel, J., Venkataraman, V., Imaki, H., Wang, K.-C., and Wegiel, J. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674. doi: 10.1016/j.neurobiolaging.2004.01.007
Nalivaeva, N. N., and Turner, A. J. (2016). Chemico-Biological Interactions AChE and the amyloid precursor protein (APP) -Cross-talk in Alzheimer’s disease. Chem. Biol. Interact. 259, 301–306. doi: 10.1016/j.cbi.2016.04.009
Narayanaswami, V., Kim, J., and McNamee, M. G. (1993). Protein-lipid interactions and Torpedo californica nicotinic acetylcholine receptor function. 1. Spatial disposition of cysteine residues in the γ subunit analyzed by fluorescence-quenching and energy-transfer measurements. Biochemistry 32, 12413–12419. doi: 10.1021/bi00097a020
Narayanaswami, V., and McNamee, M. G. (1993). Protein-lipid interactions and Torpedo californica nicotinic acetylcholine receptor function. 2. Membrane fluidity and ligand-mediated alteration in the accessibility of gamma subunit cysteine residues to cholesterol. Biochemistry 123, 12420–12427. doi: 10.1021/bi00097a021
Nashmi, R., Dickinson, M. E., McKinney, S., Jareb, M., Labarca, C., Fraser, S. E., et al. (2003). Assembly of alpha4beta2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J. Neurosci. 23, 11554–11567. doi: 10.1523/jneurosci.23-37-11554.2003
Näslund, J., Haroutunian, V., Mohs, R., Davis, K. L., Davies, P., Greengard, P., et al. (2000). Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283, 1571–1577.
Näslund, J., Schierhorn, A., Hellman, U., Lannfelt, L., Roses, A. D., Tjernberg, L. O., et al. (1994). Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. U.S.A. 91, 8378–8382. doi: 10.1073/pnas.91.18.8378
Navaratnam, D. S., Fernando, F. S., Priddle, J. D., Giles, K., Clegg, S. M., Pappin, D. J., et al. (2000). Hydrophobic protein that copurifies with human brain acetylcholinesterase: amino acid sequence, genomic organization, and chromosomal localization. J. Neurochem. 74, 2146–2153. doi: 10.1046/j.1471-4159.2000.0742146.x
Nees, F. (2015). The nicotinic cholinergic system function in the human brain. Neuropharmacology 96, 289–301. doi: 10.1016/j.neuropharm.2014.10.021
Nicolson, G. L. (2014). The fluid-mosaic model of membrane structure?: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta. 1838, 1451–1466. doi: 10.1016/j.bbamem.2013.10.019
Niu, X., Zhang, X., Xie, J., and Zhang, X. (2012). Acetylcholinesterase blocks cleavage of APP by γ-secretase in 293 cells and mouse brain. Mol. Neurodegener. 7(Suppl. 1):S11. doi: 10.1186/1750-1326-7-S1-S11
Nordberg, A., Lundqvist, H., Hartvig, P., Lilja, A., and Långström, B. (1995). Kinetic analysis of regional (S)(-)11C-nicotine binding in normal and Alzheimer brains–in vivo assessment using positron emission tomography. Alzheimer Dis. Assoc. Disord. 9, 21–27. doi: 10.1097/00002093-199505000-00006
Nunomura, A., Tamaoki, T., Tanaka, K., Motohashi, N., Nakamura, M., and Hayashi, T. (2010). Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol. Dis. 37, 731–737. doi: 10.1016/j.nbd.2009.12.012
Nurowska, E., and Ruzzier, F. (1996). Corticosterone modifies the murine muscle acetylcholine receptor channel kinetics. Neuroreport 8, 77–80. doi: 10.1097/00001756-199612200-00016
Nurowska, E., and Ruzzier, F. (2002). Modulation of acetylcholine receptor channel kinetics by hydrocortisone. Biochim. Biophys. Acta 1564, 14–20. doi: 10.1016/s0005-2736(02)00356-5
Ochoa, E. L., Dalziel, A. W., and McNamee, M. G. (1983). Reconstitution of acetylcholine receptor function in lipid vesicles of defined composition. Biochim. Biophys. Acta 1727, 151–162. doi: 10.1016/0005-2736(83)90379-6
Opazo, C., Huang, X., Cherny, R. A., Moir, R. D., Roher, A. E., White, A. R., et al. (2002). Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to Neurotoxic H(2)O(2). J. Biol. Chem. 277, 40302–40308. doi: 10.1074/jbc.M206428200
Oshikawa, J., Toya, Y., Fujita, T., Egawa, M., Kawabe, J., Umemura, S., et al. (2003). Nicotinic acetylcholine receptor alpha 7 regulates cAMP signal within lipid rafts. Am. J. Physiol. Cell. Physiol. 285, C567–C574.
Oshima, N., Morishima-kawashima, M., Yamaguchi, H., Yoshimura, M., Schenk, D., and Ihara, Y. (2001). Accumulation of amyloid beta - protein in the low - density membrane domain accurately reflects the extent of beta -amyloid deposition in the Brain. Am. J. Pathol. 158, 2209–2218. doi: 10.1016/s0002-9440(10)64693-7
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Parri, H. R., Hernandez, C. M., and Dineley, K. T. (2011). Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem. Pharmacol. 82, 931–942. doi: 10.1016/j.bcp.2011.06.039
Parvathy, S., Hussain, I., Karran, E. H., Turner, A. J., and Hooper, N. M. (1999). Cleavage of Alzheimer’s amyloid precursor protein by alpha. -secretase occurs at the surface of neuronal cells. Biochemistry 38, 9728–9734. doi: 10.1021/bi9906827
Paterson, D., and Nordberg, A. (2000). Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 61, 75–111. doi: 10.1016/s0301-0082(99)00045-3
Pediconi, M. F., Gallegos, C. E., and Barrantes, F. J. (2004). Metabolic cholesterol depletion hinders cell-surface trafficking of the nicotinic acetylcholine receptor. Neuroscience 128, 239–249. doi: 10.1016/j.neuroscience.2004.06.007
Peña, V. B., Bonini, I. C., Antollini, S. S., Kobayashi, T., and Barrantes, F. J. (2011). A7-Type Acetylcholine Receptor Localization and Its Modulation By Nicotine and Cholesterol in Vascular Endothelial Cells. J. Cell. Biochem. 112, 3276–3288. doi: 10.1002/jcb.23254
Perez, R. G., Soriano, S., Hayes, J. D., Ostaszewski, B., Xia, W., Selkoe, D. J., et al. (1999). Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 274, 18851–18856. doi: 10.1074/jbc.274.27.18851
Perillo, V. L., Peñalva, D. A., Vitale, A. J., Barrantes, F. J., and Antollini, S. S. (2016). Transbilayer asymmetry and sphingomyelin composition modulate the preferential membrane partitioning of the nicotinic acetylcholine receptor in Lo domains. Arch. Biochem. Biophys. 591, 76–86. doi: 10.1016/j.abb.2015.12.003
Perrier, A. L., Massoulié, J., and Krejci, E. (2002). PRiMA: the membrane anchor of acetylcholinesterase in the brain. Neuron. 33, 275–285.
Perry, E. K., Blessed, G., Tomlinson, B. E., Perry, R. H., Crow, T. J., Cross, A. J., et al. (1981). Neurochemical activities in human temporal lobe related to aging and Alzheimer-type changes. Neurobiol. Aging 2, 251–266.
Perry, E. K., Curtis, M., Dick, D. J., Candy, J. M., Atack, J. R., Bloxham, C. A., et al. (1985). Cholinergic correlates of cognitive impairment in Parkinson’s disease: comparisons with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 48, 413–421.
Perry, E. K., Johnson, M., Kerwin, J. M., Piggott, M. A., Court, J. A., Shaw, P. J., et al. (1992). Convergent cholinergic activities in aging and Alzheimer’s disease. Neurobiol. Aging 13, 393–400. doi: 10.1016/0197-4580(92)90113-c
Perry, E. K., Perry, R. H., Smith, C. J., Dick, D. J., Candy, J. M., Edwardson, J. A., et al. (1987). Nicotinic receptor abnormalities in Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 50, 806–809.
Perry, G., Lipphardt, S., Mulvihill, P., Kancherla, M., Mijares, M., Gambetti, P., et al. (1988). Amyloid precursor protein in senile plaques of Alzheimer disease. Lancet 2, 746–751.
Peters, I., Igbavboa, U., Schütt, T., Haidari, S., Hartig, U., Rosello, X., et al. (2009). The interaction of beta-amyloid protein with cellular membranes stimulates its own production. Biochim. Biophys. Acta 1788, 964–972. doi: 10.1016/j.bbamem.2009.01.012
Petersen, R. C. (1977). Scopolamine induced learning failures in man. Psychopharmacology 52, 283–289. doi: 10.1007/BF00426713
Pettit, D. L., Shao, Z., and Yakel, J. L. (2001). Beta-Amyloid(1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J. Neurosci. 21:RC120.
Phinney, A. L., Horne, P., Yang, J., Janus, C., Bergeron, C., and Westaway, D. (2003). Mouse models of Alzheimer’s disease: the long and filamentous road. Neurol. Res. 25, 590–600. doi: 10.1179/016164103101202020
Piomelli, D., Astarita, G., and Rapaka, R. (2007). A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 8, 743–754. doi: 10.1038/nrn2233
Plant, L. D., Boyle, J. P., Smith, I. F., Peers, C., and Pearson, H. A. (2003). The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J. Neurosci. 23, 5531–5535. doi: 10.1523/jneurosci.23-13-05531.2003
Pohanka, M. (2012). Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int. J. Mol. Sci. 13, 2219–2238. doi: 10.3390/ijms13022219
Pollard, H. B., Arispe, N., and Rojas, E. (1995). Ion Channel Hypothesis for Alzheimer Amyloid Peptide Neurotoxicity. Cell. Mol. Neurobiol. 15, 513–526. doi: 10.1007/bf02071314
Poveda, J. A., Encinar, J. A., Fernández, A. M., Mateo, C. R., Ferragut, J. A., and González-Ros, J. M. (2002). Segregation of phosphatidic acid-rich domains in reconstituted acetylcholine receptor membranes. Biochemistry 41, 12253–12262. doi: 10.1021/bi0200099
Poveda, J. A., Fernández, A. M., and Encinar, J. A. (2008). Protein-promoted membrane domains. Biochim. Biophys. Acta 1778, 1583–1590. doi: 10.1016/j.bbamem.2008.01.021
Puglielli, L., Konopka, G., Pack-Chung, E., Ingano, L. A. M., Berezovska, O., Hyman, B. T., et al. (2001). Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat. Cell. Biol. 3, 905–912. doi: 10.1038/ncb1001-905
Puzzo, D., and Arancio, O. (2013). Amyloid-β peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimers Dis. 33, S111–S120.
Puzzo, D., Gulisano, W., Arancio, O., and Palmeri, A. (2015). The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neuroscience 307, 26–36. doi: 10.1016/j.neuroscience.2015.08.039
Puzzo, D., Privitera, L., Fa’, M., Staniszewski, A., Hashimoto, G., Aziz, F., et al. (2011). Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830. doi: 10.1002/ana.22313
Puzzo, D., Privitera, L., Leznik, E., Fà, M., Staniszewski, A., Palmeri, A., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008
Qiang, W., Yau, W. M., and Schulte, J. (2014). Fibrillation of β amyloid peptides in the presence of phospholipid bilayers and the consequent membrane disruption. Biochim. Biophys. Acta 1848, 266–276. doi: 10.1016/j.bbamem.2014.04.011
Quesada, O., González-Freire, C., Ferrer, M. C., Colón-Sáez, J. O., Fernández-García, E., Mercado, J., et al. (2016). Uncovering the lipidic basis for the preparation of functional nicotinic acetylcholine receptor detergent complexes for structural studies. Sci. Rep. 19:32766. doi: 10.1038/srep32766
Quinn, P. J. (2002). Plasma membrane phospholipid asymmetry. Subcell Biochem. 36, 39–60. doi: 10.1007/0-306-47931-1_3
Reid, P. C., Urano, Y., Kodama, T., and Hamakubo, T. (2007). Alzheimer’s Disease?: cholesterol, membrane rafts, isoprenoids and statins. J. Cell. Mol. Med. 11, 383–392. doi: 10.1111/j.1582-4934.2007.00054.x
Relini, A., Marano, N., and Gliozzi, A. (2013). Misfolding of amyloidogenic proteins and their interactions with membranes. Biomolecules 27, 20–55. doi: 10.3390/biom4010020
Relini, A., Marano, N., and Gliozzi, A. (2014). Probing the interplay between amyloidogenic proteins and membranes using lipid monolayers and bilayers. Adv. Colloid Interface Sci. 207, 81–92. doi: 10.1016/j.cis.2013.10.015
Reyes, A. E., Perez, D. R., Alvarez, A., Garrido, J., Gentry, M. K., and Doctor, B. P. (1997). A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem. Biophys. Res. Commun. 232, 652–655. doi: 10.1006/bbrc.1997.6357
Roccamo, A. M., Pediconi, M. F., Aztiria, E., Zanello, L., Wolstenholme, A., and Barrantes, F. J. (1999). Cells defective in sphingolipids biosynthesis express low amounts of muscle nicotinic acetylcholine receptor. Eur. J. Neurosci. 11, 1615–1623. doi: 10.1046/j.1460-9568.1999.00574.x
Rondelli, V., Brocca, P., Motta, S., Messa, M., Colombo, L., Salmona, M., et al. (2016). Amyloid - β Peptides in interaction with raft-mime model membranes: a neutron reflectivity insight. Sci. Rep. 16:20997. doi: 10.1038/srep20997
Rosenblum, W. I. (2014). Why Alzheimer trials fail: removing soluble oligomeric beta amyloid is essential, inconsistent, and difficult. Neurobiol. Aging 35, 969–974. doi: 10.1016/j.neurobiolaging.2013.10.085
Roth, G. S., Joseph, J. A., and Mason, R. P. (1995). Membrane alterations as causes of impaired signal transduction in Alzheimer’s disease and aging. Trends. Neurosci. 18, 203–206. doi: 10.1016/0166-2236(95)93902-a
Rushworth, J. V., and Hooper, N. M. (2011). Lipid rafts: linking Alzheimer’s Amyloid- β production, aggregation, and toxicity at neuronal membranes. Int. J. Alzheimers Dis. 2011:603052. doi: 10.4061/2011/603052
Sáez-Valero, J., Sberna, G., McLean, C. A., and Small, D. H. (1999). Molecular isoform distribution and glycosylation of acetylcholinesterase are altered in brain and cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurochem. 72, 1600–1608. doi: 10.1046/j.1471-4159.1999.721600.x
Sargent, P. B., Bryan, G. K., Streichert, L. C., and Garrett, E. N. (1991). Denervation does not alter the number of neuronal bungarotoxin binding sites on autonomic neurons in the frog cardiac ganglion. J. Neurosci. 11, 3610–3623. doi: 10.1523/jneurosci.11-11-03610.1991
Sargent, P. B., and Garrett, E. N. (1995). The characterization of alpha-bungarotoxin receptors on the surface of parasympathetic neurons in the frog heart. Brain Res. 680, 99–107. doi: 10.1016/0006-8993(95)00250-t
Sarter, M., and Bruno, J. P. (1997). Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res. 23, 28–46. doi: 10.1016/s0165-0173(96)00009-4
Sarter, M., and Paolone, G. (2011). Deficits in attentional control: cholinergic mechanisms and circuitry-based treatment approaches. Behav. Neurosci. 125, 825–835. doi: 10.1037/a0026227
Sasahara, K., Morigaki, K., and Shinya, K. (2013). Effects of membrane interaction and aggregation of amyloid β-peptide on lipid mobility and membrane domain structure. Phys. Chem. Chem. Phys. 15, 8929–8939. doi: 10.1039/c3cp44517h
Schmidt, J. T., and Freeman, J. A. (1980). Electrophysiologic evidence that retinotectal synaptic transmission in the goldfish is nicotinic cholinergic. Brain Res. 187, 129–142. doi: 10.1016/0006-8993(80)90499-0
Scholze, P., Ciuraszkiewicz, A., Groessl, F., Orr-Urtreger, A., McIntosh, J. M., and Huck, S. (2011). α4β2 nicotinic acetylcholine receptors in the early postnatal mouse superior cervical ganglion. Dev. Neurobiol. 71, 390–399. doi: 10.1002/dneu.20870
Sciacca, M. F., Kotler, S. A., Brender, J. R., Chen, J., Lee, D. K., and Ramamoorthy, A. (2012). Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 103, 702–710. doi: 10.1016/j.bpj.2012.06.045
Seghezza, S., Diaspro, A., Canale, C., and Dante, S. (2014). Cholesterol drives aβ(1-42) interaction with lipid rafts in model membranes. Langmuir 30, 13934–13941. doi: 10.1021/la502966m
Sepúlveda, F. J., Fierro, H., Fernandez, E., Castillo, C., Peoples, R. W., Opazo, C., et al. (2014). Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer’s disease. Neurobiol. Aging 5, 472–481. doi: 10.1016/j.neurobiolaging.2013.08.035
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold. Spring. Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Sharma, G., and Vijayaraghavan, S. (2001). Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. U.S.A. 98, 4148–4153. doi: 10.1073/pnas.071540198
Sharp, L., Salari, R., and Brannigan, G. (2019). Boundary lipids of the nicotinic acetylcholine receptor: spontaneous partitioning via coarse-grained molecular dynamics simulation. Biochim. Biophys. Acta. Biomembr. 1861, 887–896. doi: 10.1016/j.bbamem.2019.01.005
Shen, J., and Wu, J. (2015). Nicotinic cholinergic mechanisms in Alzheimer’s disease. Int. Rev. Neurobiol. 124, 275–292. doi: 10.1016/bs.irn.2015.08.00
Shen, J. X., and Yakel, J. L. (2009). Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 30, 673–680. doi: 10.1038/aps.2009.64
Shimohama, S., Taniguchi, T., Fujiwara, M., and Kameyama, M. (1986). Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J. Neurochem. 46, 288–293. doi: 10.1111/j.1471-4159.1986.tb12960.x
Shringarpure, R., Grune, T., Sitte, N., and Davies, K. J. (2000). 4-Hydroxynonenal-modified amyloid-beta peptide inhibits the proteasome: possible importance in Alzheimer’s disease. Cell. Mol. Life Sci. 57, 1802–1809. doi: 10.1007/pl00000660
Silveyra, M. X., Evin, G., Montenegro, M. F., Vidal, C. J., Martínez, S., and Culvenor, J. G. (2008). Presenilin 1 interacts with acetylcholinesterase and alters its enzymatic activity and glycosylation. Mol. Cell. Biol. 28, 2908–2919. doi: 10.1128/MCB.02065-2067
Silveyra, M. X., García-Ayllón, M. S., Serra-Basante, C., Mazzoni, V., García-Gutierrez, M. S., and Manzanares, J. (2012). Changes in acetylcholinesterase expression are associated with altered presenilin-1 levels. Neurobiol. Aging 33, 627–637. doi: 10.1016/j.neurobiolaging.2011.04.006
Simons, K., and Ikonen, E. (1997). Functional rafts in cell membranes. Nature 387, 569–572. doi: 10.1038/42408
Simons, K., and van Meer, G. (1988). Lipid sorting in epithelial cells. Biochemistry 23, 6197–6202. doi: 10.1021/bi00417a001
Simons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464. doi: 10.1073/pnas.95.11.6460
Singer, A. S. J., and Nicolson, G. L. (1972). The Fluid Mosaic Model of the Structure of Cell Membranes. Science 175, 720–731. doi: 10.1126/science.175.4023.720
Small, D. H., Maksel, D., Kerr, M. L., Ng, J., Hou, X., Chu, C., et al. (2007). The b -amyloid protein of Alzheimer’s disease binds to membrane lipids but does not bind to the a 7 nicotinic acetylcholine receptor. J. Neurochem. 101, 1527–1538. doi: 10.1111/j.1471-4159.2006.04444.x
Small, D. H., Michaelson, S., and Sberna, G. (1996). Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 28, 453–483. doi: 10.1016/0197-0186(95)00099-2
Sonnino, S., and Prinetti, A. (2013). Membrane domains and the “lipid raft” concept. Curr Med Chem. 20, 4–21. doi: 10.2174/0929867311320010003
Staneva, G., Puff, N., Stanimirov, S., Tochev, T., Angelova, M. I., and Seigneuret, M. (2018). The Alzheimer’s disease amyloid-β peptide affects the size-dynamics of raft-mimicking Lo domains in GM1-containing lipid bilayers. Soft. Matter. 14, 9609–9618. doi: 10.1039/c8sm01636d
Stefani, M. (2010). Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 277, 4602–4613. doi: 10.1111/j.1742-4658.2010.07889.x
Stetzkowski-Marden, F., Gaus, K., Recouvreur, M., Cartaud, A., and Cartaud, J. (2006a). Agrin elicits membrane lipid condensation at sites of acetylcholine receptor clusters in C2C12 myotubes. J. Lipid. Res. 47, 2121–2133. doi: 10.1194/jlr.M600182-JLR200
Stetzkowski-Marden, F., Recouvreur, M., Camus, G., Cartaud, A., Marchand, S., and Cartaud, J. (2006b). Rafts are required for acetylcholine receptor clustering. J. Mol. Neurosci. 30, 37–38. doi: 10.1385/JMN:30:1:37
Subasinghe, S., Unabia, S., Barrow, C. J., Mok, S. S., Aguilar, M. I., and Small, D. H. (2003). Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J. Neurochem. 84, 471–479. doi: 10.1046/j.1471-4159.2003.01552.x
Sun, J., and Roy, S. (2018). The physical approximation of APP and BACE-1: a key event in alzheimer’s disease pathogenesis. Dev. Neurobiol. 78, 340–347. doi: 10.1002/dneu.22556
Sunshine, C., and McNamee, M. G. (1992). Lipid modulation of nicotinic acetylcholine receptor function: the role of neutral and negatively charged lipids. Biochim. Biophys. Acta 1108, 240–246. doi: 10.1016/0005-2736(92)90031-g
Sunshine, C., and McNamee, M. G. (1994). Lipid modulation of nicotinic acetylcholine receptor function: the role of membrane lipid composition and fluidity. Biochim. Biophys. Acta 1191, 59–64. doi: 10.1016/0005-2736(94)90233-X
Tan, J. Z. A., and Gleeson, P. A. (2019). The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimer’s disease. BBA - Biomembr. 1861, 697–712. doi: 10.1016/j.bbamem.2018.11.013
Tapiola, T., Pirttilä, T., Mehta, P. D., Alafuzofff, I., Lehtovirta, M., and Soininen, H. (2000). Relationship between apoE genotype and CSF beta-amyloid (1-42) and tau in patients with probable and definite Alzheimer’s disease. Neurobiol. Aging 21, 735–740. doi: 10.1016/s0197-4580(00)00164-0
Tenchov, B. G., MacDonald, R. C., and Siegel, D. P. (2006). Cubic phases in phosphatidylcholine-cholesterol mixtures: cholesterol as membrane “fusogen”. Biophys. J. 91, 2508–2516. doi: 10.1529/biophysj.106.083766
Terzi, E., Hölzemann, G., and Seelig, J. (1997). Interaction of Alzheimer beta-amyloid peptide(1-40) with lipid membranes. Biochemistry 36, 14845–14852. doi: 10.1021/bi971843e
Thomsen, M. S., Andreasen, J. T., Arvaniti, M., and Kohlmeier, K. A. (2016). Nicotinic Acetylcholine receptors in the pathophysiology of Al zheimer’s disease: the role of protein-protein interactions in current and future treatment. Curr. Pharm. Des. 22, 2015–2034. doi: 10.2174/1381612822666160127112357
Thornton, E., Vink, R., Blumbergs, P. C., and Van Den Heuvel, C. (2006). Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 1094, 38–46. doi: 10.1016/j.brainres.2006.03.107
Tozaki, H., Matsumoto, A., Kanno, T., Nagai, K., Nagata, T., Yamamoto, S., et al. (2002). The inhibitory and facilitatory actions of amyloid-beta peptides on nicotinic ACh receptors and AMPA receptors. Biochem. Biophys. Res. Commun. 294, 42–45. doi: 10.1016/S0006-291X(02)00429-421
Turner, P. R., O’Connor, K., Tate, W. P., and Abraham, W. C. (2003). Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 70, 1–32. doi: 10.1016/s0301-0082(03)00089-3
Unwin, N. (2005). Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 346, 967–989. doi: 10.1016/j.jmb.2004.12.031
Uranga, R. M., Alza, N. P., Conde, M. A., Antollini, S. S., and Salvador, G. A. (2017). Phosphoinositides: two-path signaling in neuronal response to Oligomeric amyloid β peptide. Mol. Neurobiol. 54, 3236–3252. doi: 10.1007/s12035-016-9885-9883
van Meer, G. (2011). Dynamic transbilayer lipid asymmetry. Cold Spring Harb. Perspect. Biol. 3:004671. doi: 10.1101/cshperspect.a004671
Vestergaard, M., Hamada, T., Morita, M., and Takagi, M. (2010). Cholesterol, Lipids, Amyloid Beta, and Alzheimer’ s. Curr. Alzheimer Res. 7, 262–270. doi: 10.2174/156720510791050821
Vetrivel, K. S., Cheng, H., Kim, S., Chen, Y., Barnes, N. Y., Parent, A. T., et al. (2005). Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 280, 25892–25900. doi: 10.1074/jbc.M503570200
Vetrivel, K. S., Cheng, H., Lin, W., Sakurai, T., Li, T., Nukina, N., et al. (2004). Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954. doi: 10.1074/jbc.M407986200
Vetrivel, K. S., and Thinakaran, G. (2006). Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 66, 69–73. doi: 10.1212/01.wnl.0000192107.17175.39
Villar, M. T., Artigues, A., Ferragut, J. A., and Gonzalez-Ros, J. M. (1988). Phospholipase A2 hydrolysis of membrane phospholipids causes structural alteration of the nicotinic acetylcholine receptor. Biochim. Biophys. Acta 938, 35–43. doi: 10.1016/0005-2736(88)90119-8
Wahrle, S., Das, P., Nyborg, A. C., Mclendon, C., Shoji, M., Kawarabayashi, T., et al. (2002). Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23. doi: 10.1006/nbdi.2001.0470
Wakabayashi, M., Okada, T., Kozutsumi, Y., and Matsuzaki, K. (2005). GM1 ganglioside-mediated accumulation of amyloid beta-protein on cell membranes. Biochem. Biophys. Res. Commun. 328, 1019–1023. doi: 10.1016/j.bbrc.2005.01.060
Wang, H.-Y., Lee, D. H., Davis, C. B., and Shank, R. P. (2000a). Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J. Neurochem. 75, 1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x
Wang, H.-Y., Lee, D. H. S., D’Andrea, M. R., Peterson, P. A., Shank, R. P., and Reitz, A. B. (2000b). β-amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 275, 5626–5632. doi: 10.1074/jbc.275.8.5626
Wang, H. Y., Li, W., Benedetti, N. J., and Lee, D. H. (2003). Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J. Biol. Chem. 278, 1547–1553. doi: 10.1074/jbc.M212532200
Wang, H. Y., Stucky, A., Liu, J., Shen, C., Trocme-Thibierge, C., and Morain, P. (2009). Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J. Neurosci. 229, 10961–10973. doi: 10.1523/JNEUROSCI.6088-08.2009
Wang, Y., Qu, D., and Wang, K. (2016). Therapeutic approaches to Alzheimer’s disease through stimulating of non-amyloidogenic processing of amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 20, 2389–2403.
Wen, P.-C., Mahinthichaichan, P., Trebesch, N., Jiang, T., Zhao, Z., Shinn, E., et al. (2018). Microscopic view of lipids and their diverse biological functions. Curr. Opin. Struct. Biol. 51, 177–186. doi: 10.1016/j.sbi.2018.07.003
Wenk, G. L. (1997). The nucleus basallis magnocellularis cholinergic system: one hundred years of progress. Neurobiol. Learn. Mem. 67, 85–95. doi: 10.1006/nlme.1996.3757
Wenz, J. J., and Barrantes, F. J. (2005). Nicotinic acetylcholine receptor induces lateral segregation of phosphatidic acid and phosphatidylcholine in reconstituted membranes. Biochemistry 44, 398–410. doi: 10.1021/bi048026g
Whitehouse, P. J., Martino, A. M., Antuono, P. G., Lowenstein, P. R., Coyle, J. T., Price, D. L., et al. (1986). Nicotinic acetylcholine binding sites in Alzheimer’s disease. Brain Res. 371, 146–151. doi: 10.1016/0006-8993(86)90819-x
Whitehouse, P. J., Price, D. L., Clark, A. W., Coyle, J. T., and DeLong, M. R. (1981). Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 10, 122–126. doi: 10.1002/ana.410100203
Whitehouse, P. J., Struble, R. G., Clark, A. W., and Price, D. L. (1982). Alzheimer disease: plaques, tangles, and the basal forebrain. Ann. Neurol. 12:494. doi: 10.1002/ana.410120517
Willmann, R., Pun, S., Sadasivam, G., Santos, A. F., Caroni, P., and Fuhrer, C. (2006). Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 25, 4050–4060. doi: 10.1038/sj.emboj.7601288
Wonnacott, S., Barik, J., Dickinson, J., and Jones, I. W. (2006). Nicotinic receptors modulate transmitter cross talk in the CNS: Nicotinic modulation of transmitters. J. Mol. Neurosci. 30, 137–140. doi: 10.1385/JMN:30:1:137
Wood, W. G., Li, L., Müller, W. E., and Eckert, G. P. (2014). Cholesterol as a causative factor in Alzheimer disease: a debatable hypothesis. J.Neurochem. 129, 559–572. doi: 10.1111/jnc.12637
Wood, W. G., Schroeder, F., Igbavboa, U., Avdulov, N. A., and Chochina, S. V. (2002). Brain membrane cholesterol domains, aging and amyloid beta-peptides. Neurobiol. Aging 23, 685–694. doi: 10.1016/s0197-4580(02)00018-0
Woolf, N. J. (1998). A structural basis for memory storage in mammals. Prog. Neurobiol. 55, 59–77. doi: 10.1016/s0301-0082(97)00094-4
Wu, J., Khan, G. M., and Nichols, R. A. (2007). Dopamine release in prefrontal cortex in response to beta-amyloid activation of alpha7 nicotinic receptors. Brain Res. 1182, 82–89. doi: 10.1016/j.brainres.2007.08.079
Wu, J., Kuo, Y. P., George, A. A., Xu, L., Hu, J., and Lukas, R. J. (2004). Beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J. Biol. Chem. 279, 37842–37851. doi: 10.1074/jbc.M400335200
Wu, Y., and Luo, Y. (2005). Transgenic C. elegans as a model in Alzheimer’s research. Curr. Alzheimer. Res. 2, 37–45. doi: 10.2174/1567205052772768
Xie, H. Q., Leung, K. W., Chen, V. P., Chan, G. K. L., Xu, S. L., Guo, A. J. Y., et al. (2010a). Chemico-Biological Interactions PRiMA directs a restricted localization of tetrameric AChE at synapses. Chem. Biol. Interact. 187, 78–83. doi: 10.1016/j.cbi.2010.02.018
Xie, H. Q., Liang, D., Leung, K. W., Chen, V. P., Zhu, K. Y., Chan, W. K. B., et al. (2010b). Targeting Acetylcholinesterase to Membrane Rafts a function mediated by the proline-rich membrane anchor (PRiMA) in neurons. J. Biol. Chem. 285, 11537–11546. doi: 10.1074/jbc.M109.038711
Xiong, H., Callaghan, D., Jones, A., Walker, D. G., Lue, L. F., and Beach, T. G. (2008). Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis. 29, 422–437. doi: 10.1016/j.nbd.2007.10.005
Xiu, J., Nordberg, A., Zhang, J. T., and Guan, Z. Z. (2005). Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the alpha7, alpha4 and beta2 subunits in response to nanomolar concentrations of the beta-amyloid peptide(1-42). Neurochem. Int. 47, 281–290. doi: 10.1016/j.neuint.2005.04.023
Yahi, N., and Fantini, J. (2014). Deciphering the glycolipid code of Alzheimer’s and Parkinson’s amyloid proteins allowed the creation of a universal ganglioside-binding peptide. PLoS One. 9:e104751. doi: 10.1371/journal.pone.0104751
Yamamoto, N., Hasegawa, K., Matsuzaki, K., Naiki, H., and Yanagisawa, K. (2004). Environment- and mutation-dependent aggregation behavior of Alzheimer amyloid beta-protein. J. Neurochem. 90, 62–69. doi: 10.1111/j.1471-4159.2004.02459.x
Yamamoto, N., Matsubara, T., Sato, T., and Yanagisawa, K. (2008). Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β -protein fi brillogenesis. Biochim. Biophys. Acta 1778, 2717–2726. doi: 10.1016/j.bbamem.2008.07.028
Yamamoto, N., Matsuzaki, K., and Yanagisawa, K. (2005). Cross-seeding of wild-type and hereditary variant-type amyloid beta-proteins in the presence of gangliosides. J. Neurochem. 95, 1167–1176. doi: 10.1111/j.1471-4159.2005.03444.x
Yanagisawa, K. (2005). GM1 ganglioside and the seeding of amyloid in Alzheimer’s disease: endogenous seed for Alzheimer amyloid. Neuroscientist 11, 250–260. doi: 10.1177/1073858405275177
Yanagisawa, K., and Ihara, Y. (1998). GM1 ganglioside-bound amyloid beta-protein in Alzheimer’s disease brain. Neurobiol. Aging 19(1 Suppl.), S65–S67. doi: 10.1177/1073858405275177
Yanagisawa, K., Odaka, A., Suzuki, N., and Ihara, Y. (1995). GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer’s disease. Nat. Med. 1, 1062–1066. doi: 10.1038/nm1095-1062
Yang, X., Askarova, S., and Lee, J. C.-M. (2010). Membrane biophysics and mechanics in Alzheimer’s disease. Mol. Neurobiol. 41, 138–148. doi: 10.1007/s12035-010-8121-9
Yankner, B. A., Duffy, L. K., and Kirschner, D. A. (1990). Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250, 279–282. doi: 10.1126/science.2218531
Yip, C. M., Darabie, A. A., and McLaurin, J. (2002). Abeta42-peptide assembly on lipid bilayers. J. Mol. Biol. 318, 97–107. doi: 10.1016/s0022-2836(02)00028-1
Yu, W. F., Guan, Z. Z., Bogdanovic, N., and Nordberg, A. (2005). High selective expression of alpha7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic plaques. Exp. Neurol. 2192, 215–225. doi: 10.1016/j.expneurol.2004.12.015
Yu, X., and Zheng, J. (2012). Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol. 421, 561–571. doi: 10.1016/j.jmb.2011.11.006
Zampagni, M., Evangelisti, E., Cascella, R., Liguri, G., Becatti, M., Pensalfini, A., et al. (2010). Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J. Mol. Med. 88, 597–608. doi: 10.1007/s00109-010-0603-608
Zhang, Q., Powers, E. T., Nieva, J., Huff, M. E., Dendle, M. A., Bieschke, J., et al. (2004). Metabolite-initiated protein misfolding may trigger Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 4752–4757. doi: 10.1073/pnas.0400924101
Zhu, D., Xiong, W. C., and Mei, L. (2006). Lipid rafts serve as a signaling platform for Nicotinic Acetylcholine receptor clustering. J. Neurosci. 26, 4841–4851. doi: 10.1523/JNEUROSCI.2807-05.2006
Zimmerberg, J., and Gawrisch, K. (2006). The physical chemistry of biological membranes. Nat. Chem. Biol. 2, 564–567. doi: 10.1038/nchembio1106-564
Zou, K., Gong, J. S., Yanagisawa, K., and Michikawa, M. (2002). A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 22, 4833–4841. doi: 10.1523/jneurosci.22-12-04833.2002
Keywords: Alzheimer’s disease, Aβ peptide, nicotinic acetylcholine receptor, acetylcholinesterase, cell membranes, lipid rafts, cholesterol
Citation: Fabiani C and Antollini SS (2019) Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell. Neurosci. 13:309. doi: 10.3389/fncel.2019.00309
Received: 14 February 2019; Accepted: 25 June 2019;
Published: 18 July 2019.
Edited by:
Mario Eduardo Guido, Center for Research in Biological Chemistry Córdoba (CIQUIBIC), ArgentinaReviewed by:
Gunter Peter Eckert, University of Giessen, GermanyCopyright © 2019 Fabiani and Antollini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia S. Antollini, c2lsdmlhbnRAY3JpYmEuZWR1LmFy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.