 Frederick Bonsack
Frederick Bonsack Sangeetha Sukumari-Ramesh
Sangeetha Sukumari-Ramesh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 14 May 2019
Sec. Cellular Neuropathology
Volume 13 - 2019 | https://doi.org/10.3389/fncel.2019.00157
Intracerebral hemorrhage (ICH) is a devastating sub-type of stroke with no proven treatment. Given the emerging role of Galectin-1 and Galectin-3 in neuroimmune responses, the objective of the current manuscript is to elucidate hemorrhagic-injury induced modulation and cellular expression of Galectin-1 and Galectin-3 in the brain in a pre-clinical model of ICH. To address this, ICH was induced in male CD1 mice by collagenase injection method. Western blotting as well as Immunofluorescence staining was performed to characterize the temporal expression pattern as well as cellular localization of Galectin-1 and Galectin-3 after ICH. Further, genetic studies were conducted to assess the functional role of Galectin-1 and Galectin-3 in inflammatory response employing a murine macrophage cell line, RAW 264.7. Galectin-1 and Galectin-3 exhibited very profound and increased expression from day 3 to day 7-post-injury, in the perihematomal brain region after ICH in comparison to Sham. Further, Galectin-1 expression was mostly observed in GFAP-positive astrocytes whereas Galectin-3 expression was observed mostly in Iba1-positive microglia/macrophages as well as CD16/32 (M1 microglial/macrophage marker)-positive cells. Moreover, genetic studies revealed a negative regulatory role of both Galectin-1 and Galectin-3 in the release of a proinflammatory cytokine, IL-6 from RAW 264.7 cells depending on the stimulus. Altogether, the present manuscript demonstrates for the first time, increased expression as well as cellular localization of Galectin-1 and Galectin-3 in the perihematomal brain regions after ICH. In addition, the manuscript raises the potential of Galectin-1 and Galectin-3 in modulating glial responses and thereby brain injury after ICH, warranting further investigation.
Intracerebral hemorrhage is a fatal stroke subtype (Qureshi et al., 2001a) that accounts for an in-hospital mortality rate and a disability rate of 40 and 80%, respectively (van Asch et al., 2010). ICH is responsible for 10–15% of all strokes, and the worldwide incidence of ICH is 2 million cases per year (van Asch et al., 2010) with approximately 120,000 cases per year in the United States (Ribo and Grotta, 2006; Broderick et al., 2007; Aguilar and Freeman, 2010). However, the incidence is expected to have doubled by 2050 (Qureshi et al., 2001b) due to aging and the spreading use of anticoagulants (Wang, 2010). Notably, there is no effective treatment for ICH, and the pathophysiology of the disease is poorly defined.
Neuroinflammation characterized by the activation of microglia, the neuroimmune cells of the CNS, is a key contributor of ICH-induced secondary brain injury and loss of neurological function (Wang and Dore, 2007; Carmichael et al., 2008; Wang, 2010). The introduction of blood components, including thrombin, Hb and Hb metabolites such as hemin into the brain creates the basis for neuroinflammatory responses after ICH (Wang and Dore, 2007; Carmichael et al., 2008; Robinson et al., 2009; Wang, 2010; Cai et al., 2011; Dang et al., 2011; Babu et al., 2012; Lin et al., 2012; Weng et al., 2015; Min et al., 2017). Notably, the proinflammatory activation of microglia after ICH correlates with blood-brain barrier damage, brain swelling/edema, hematoma expansion, neurological deterioration, and poor functional recovery (Platt et al., 1998; Hickenbottom et al., 1999; Leira et al., 2004; Zhao et al., 2007). Furthermore, inflammatory response after ICH also regulates the brain recruitment of blood-derived monocytes/macrophages that are known to regulate ICH-induced brain injury and thereby functional recovery (Tessier et al., 1997; Shiratori et al., 2010; Starossom et al., 2012).
Galectins are a family of evolutionary conserved carbohydrate-binding proteins (Barondes et al., 1994a,b; Kasai and Hirabayashi, 1996) involved in cell activation, differentiation, proliferation, migration and apoptosis (Perillo et al., 1995; Yang et al., 1996; Perillo et al., 1998; Moiseeva et al., 1999; Vespa et al., 1999; Yamaoka et al., 2000; Goldring et al., 2002). Among the various galectin family members, emerging evidences implicate key roles of Galectin-1 and Galectin-3 in neuroimmune responses in several neuropathological conditions (Jeon et al., 2010; Starossom et al., 2012; Parikh et al., 2015). However, there exists a critical knowledge gap in the understanding of their cellular expression and function after ICH. The objective of the current manuscript is to elucidate hemorrhagic-injury induced modulation and cellular expression of Galectin-1 and Galectin-3 expression in the brain in a preclinical model of ICH.
Intracerebral hemorrhage was induced in adult male CD-1 mouse (8–12 weeks; n = 43), as reported previously (Sukumari-Ramesh et al., 2012a,b, 2016; Bonsack et al., 2016; Sukumari-Ramesh and Alleyne, 2016; Ahmad et al., 2017; Chen-Roetling et al., 2017). Briefly, mouse was anesthetized (ketamine and xylazine) and a small incision was made to expose the skull. Using a high-speed drill, a burr hole (0.5 mm) was made on the skull approximately 2.2 mm lateral to bregma. Then the mouse was placed on to a stereotaxic head frame and a 26-G Hamilton Syringe was used to inject 0.04U of bacterial type IV collagenase (Sigma, St. Louis, MO, United States) in 0.5 μL Phosphate Buffer Saline (pH 7.4; PBS) into the left striatum (3.0 mm) under stereotaxic guidance. Upon removal of the needle, bone wax was used to seal the burr hole. Mice were kept at 37 ± 0.5°C using a small animal temperature controller throughout the procedure. The temporal pattern of hematoma after ICH is provided (Supplementary Figure S1).
Mice were anesthetized and transcardially perfused with PBS. Ipsilateral brain tissue (both hematomal and peri-hematomal brain regions) was collected in RIPA buffer containing protease and phosphatase inhibitors and subjected to sonication. The samples were then centrifuged at 14,000 rpm for 5 min at 4°C to collect the supernatant. Using a BCA protein assay kit (Pierce, Rockford, IL, United States) protein concentrations were estimated, and 30–50 micrograms of samples were run on a 4–20% sodium dodecyl sulfate–polyacrylamide gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. Blots were incubated with the respective primary antibody, [Galectin-1 (1:1000,), R&D systems, Minneapolis, MN, United States), Galectin-3 (1:1000, Abcam, Cambridge, MA, United States), or β-actin (1:3000; Sigma, St Louis, MO, United States)] overnight at 4°C. This was followed by a 2-h incubation with a corresponding Alexa Fluor tagged secondary antibody. Blots were read using a Li-Cor Odyssey near-infrared imaging system and quantification was done using Quantity One software (Bio-Rad, Foster City, CA, United States).
Mice were anesthetized and transcardially perfused with PBS. Brains were collected and placed in 4% paraformaldehyde overnight at 4°C, and then snap frozen. Brains were then cut into 25-mm coronal sections using a cryostat and mounted onto glass slides. Sections were incubated for 2 h in 10% normal donkey serum at room temperature. This was followed by an overnight incubation with the respective primary antibody at 4°C. After washing, the sections were incubated with the corresponding Alexa Fluor-tagged secondary antibody for 1 h at room temperature. Immunofluorescence was determined using a Zeiss LSM510 Meta confocal laser microscope and cellular co-localization was determined, as described earlier (Laird et al., 2010). We analyzed three non- consecutive sections per animal and a minimum of 3 random fields around the hematoma.
RAW 264.7, a murine macrophage cell line, were plated on a 24 well plate and allowed to incubate for 48 h in DMEM (Dulbecco’s Modified Eagle Medium) containing 5% Fetal Bovine Serum, 5% Bovine Growth Serum, and 1% Penicillin/Streptomycin. Cells were then incubated with mouse recombinant Galectin-1 (6.25 or 12.5 μg/ml; R&D Systems, Minneapolis, MN, United States) for 1 h and it was followed by an 18-h treatment with LPS (100 ng/ml) or hemin (30 μg/ml). The supernatant was collected and used for the detection of IL-6, by ELISA as per manufacturer’s instructions (Biolegend, San Diego, CA, United States). Briefly, a 96 well plate was coated overnight at 4°C with a specific capture antibody. Following a 1-h blocking, the cell culture supernatant was added to the wells and incubated for 2 h at room temperature. Any unbound materials were removed by washing and a detection antibody solution was added to the wells and allowed to incubate for 1 h at room temperature. After further washing, 100 μl of Avidin- Horse Radish Peroxidase (HRP) solution was added to each well for 30 min at room temperature. The substrate solution was added to the wells after washing for color development. A stop solution was used, and the plate was read at 450 nm using a microtiter plate reader (Bio-Tek, Epoch).
RAW 264.7 cells were transfected with either control siRNA (ON-TARGET plus Non-targeting Pool; GE Dharmacon) or Galectin-3 siRNA (ON-TARGET plus Mouse Lgals3 siRNA; GE Dharmacon) using HiPerFect Transfection Reagent (QIAGEN) according to manufacturer’s instructions. Galectin-3 knockdown was verified 48h post- transfection by western blotting as described earlier.
The data were analyzed using t-test or one-way analysis of variance followed by Student–Newman–Keuls post hoc test and was expressed as mean ± Standard Error (SE). A p-value of < 0.05 was considered to be significant.
To evaluate whether hemorrhagic-injury results in modulation of Galectin-1 and Galectin-3 expression in the brain, ICH or Sham was induced in mice using the collagenase injection method. Given the emerging role of Galectin-1 and Galectin-3 in neuroimmune responses, the ipsilateral brain sections from sham or ICH mice were subjected to evaluation employing both western blotting and immunohistochemistry analysis at various time points ranging from day 1 through day 7 post surgery, a post-injury time period, which exhibited remarkable induction of both pro- as well as anti-inflammatory activation of microglia/macrophages as well as astrocytes after ICH (Sukumari-Ramesh et al., 2012b, 2016; Bonsack et al., 2016).
Notably, brain sections from sham or contralateral brain areas from ICH exhibited very marginal or undetectable expression of Galectin-1 and Galectin-3 whereas augmented expression of Galectin-1 and Galectin-3 was observed at day 3, day 5, and day 7-post ICH (Figures 1, 2). Along these lines, the number of Galectin-1 immunopositive cells significantly increased by approximately 4, 6 and 4-fold on day 3, day 5, and day 7 post -ICH, respectively, in comparison to sham (Figure 1B). This observation was further validated using western blotting analysis, which revealed a remarkable and significant induction of Galectin-1 starting from day 3-post ICH in comparison to sham (Figures 1C,D). Further, the induction of Galectin-3 (Figure 2) mirrored the Galectin-1 expression after ICH and Galectin-3 immunopositive cells were approximately 15, 28 and 24- fold higher on day 3, day 5, and day 7 post-ICH (Figure 2B) in comparison to sham and the western blotting followed by densitometry analysis confirmed the injury-induced increased expression of Galectin-3 after ICH (Figures 2C,D).
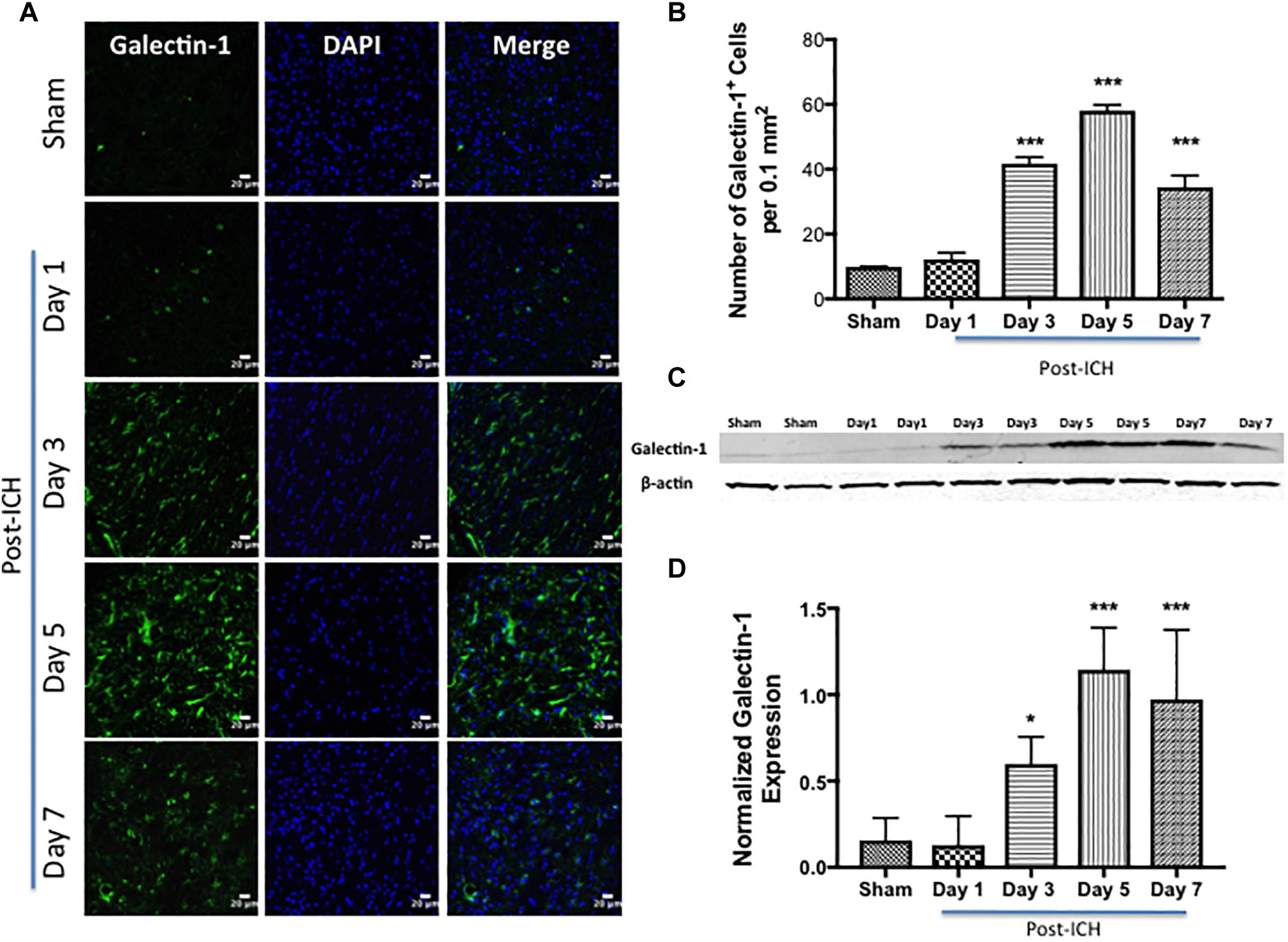
Figure 1. Increased expression of Galectin-1 after ICH. (A) Confocal images depicting the temporal expression pattern of Galectin-1 after ICH (scale bar = 20 μm; n = 3–5 per group). (B) The average number of Galectin-1-positive cells per 0.1 mm2 in the ipsilateral striatum. (C) Increased expression of Galectin-1 was confirmed by western blotting of brain tissue from the ipsilateral striatum. (D) Densitometry analysis (n = 3–5 per group) of the western blotting data. ∗p < 0.05, ∗∗∗p < 0.001 vs. sham.
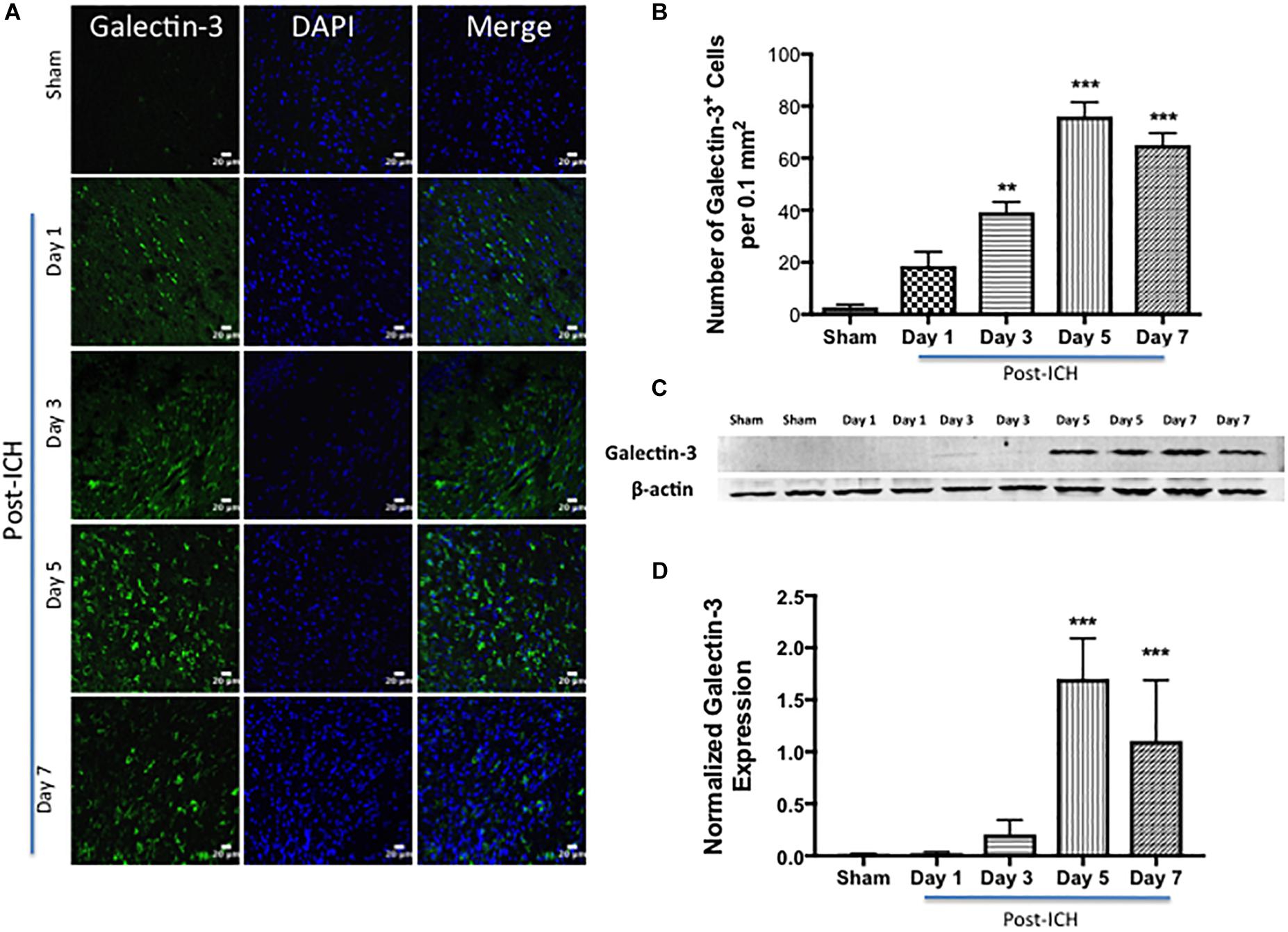
Figure 2. Augmented expression of Galectin-3 after ICH. (A) Temporal expression pattern of Galectin-3 in the perihematomal area after ICH. The representative confocal images show a remarkable increase in Galectin-3 expression after ICH in comparison to Sham (scale bar = 20 μm; n = 3–5 per group). (B) The average number of Galectin-3 positive cells per 0.1 mm2 in the ipsilateral striatum. (C) Temporal expression of Galectin-3 was further confirmed using western blotting. (D) Densitometry analysis (n = 3–5 per group) of the western blotting data. ∗∗p < 0.01, ∗∗∗p < 0.001 vs. sham.
To determine the cellular localization of Galectin-1 and Galectin-3 after ICH, the brain sections were subjected to dual label immunostaining. Galectin-1 expression was mostly observed in GFAP-positive astrocytes (Figure 3A) and stereotactic cell counting revealed that 85% of Galectin-1 positive cells co-expressed GFAP after ICH. In addition, Galectin-1 expression was also observed in Iba1- positive microglia/macrophages after ICH (Figure 3B) but only 12% of Galectin-1 positive cells co-expressed Iba1.
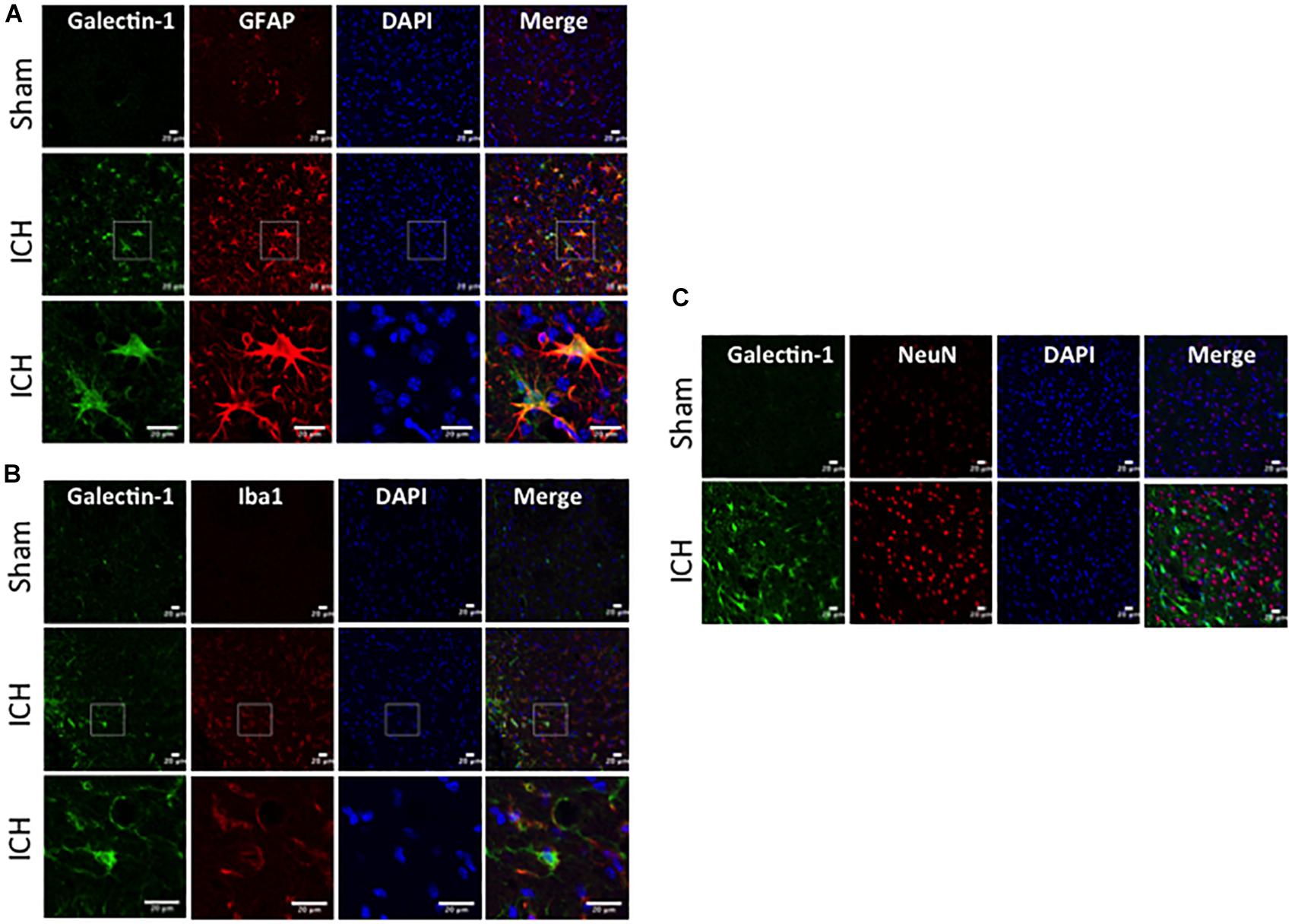
Figure 3. Galectin-1 expression is mostly observed in glial cells after ICH. Brain sections were double immunostained for (A) Galectin-1 and GFAP, (B) Galectin-1 and Iba1, (the lowest panel depicts the high magnification images) and (C) Galectin-1 and NeuN (Scale bar = 20 μm; n = 3 per group). Galectin-1 expression was observed mostly in GFAP-positive cells and in a subset of Iba1-positive cells. NeuN positive cells didn’t express Galectin-1 (n = 3 per group).
In contrast, expression of Galectin-3 was mostly confined to Iba1-positive cells (Figure 4A) and Galectin-3 expression was absent in GFAP-positive cells (Figure 5A) indicating differential cellular expression of Galectin-1 and Galectin-3 after ICH. Further, the expression of Galectin-3 was observed in proinflammatory, M1 microglial or macrophage marker, CD16/32-positive cells (Figure 4B) implicating a novel role of Galectin-3 in neuroinflammatory responses after ICH. Notably, 88 and 92% of Galectin-3 positive cells coexpressed Iba1 and CD16/32-positive cells, respectively. Of note, Galectin-3 expressing microglia or macrophages exhibited phagocytic phenotype (Figures 4A,B) implicating its unexplored role in microglial or macrophage mediated phagocytosis after ICH. Further, NeuN-positive cells didn’t express either Galectin-1 or Galectin-3 (Figures 3C, 5B).
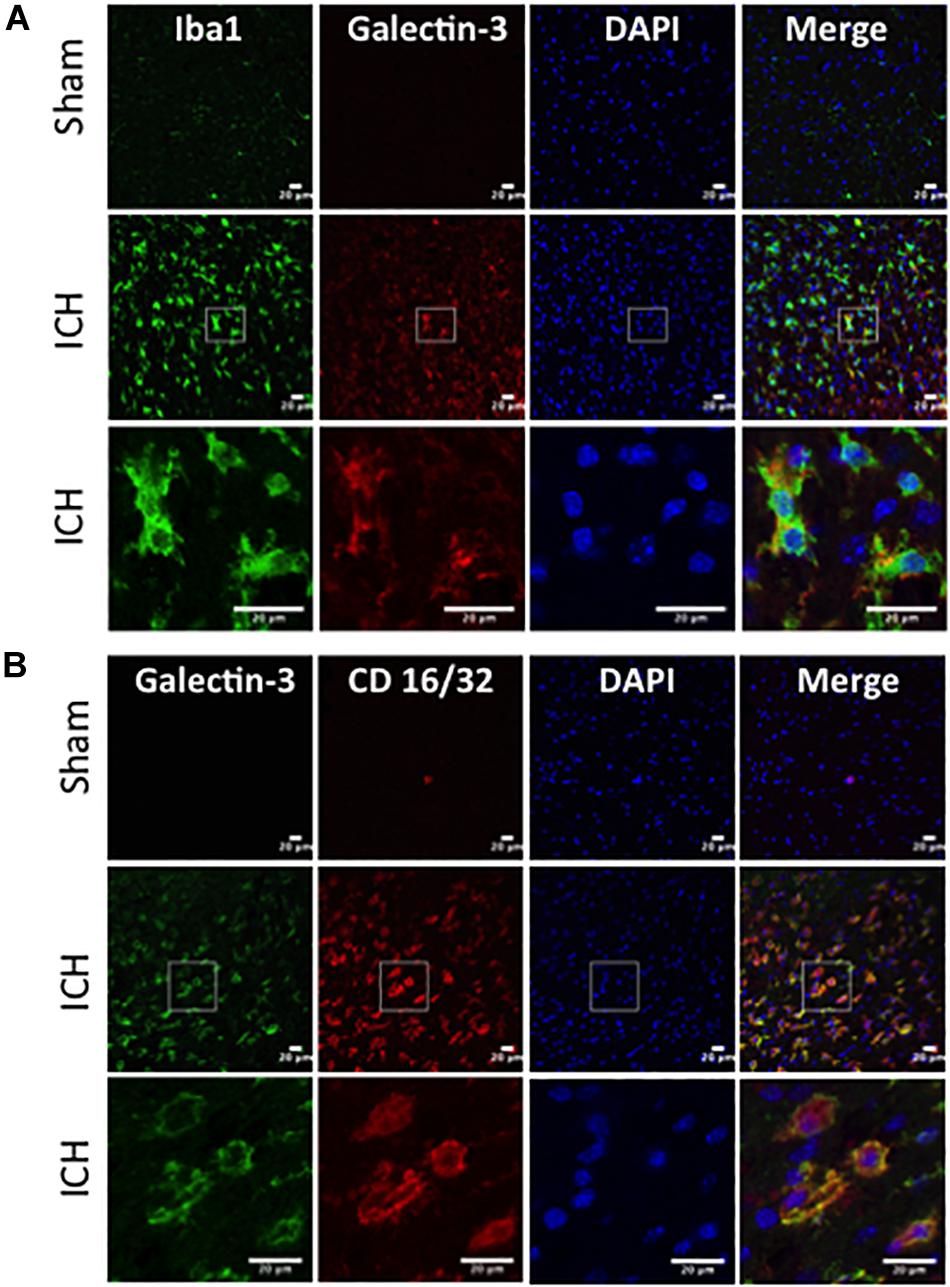
Figure 4. Galectin-3 expression in microglia/macrophages after ICH. Brain sections were immunostained for (A) Galectin-3 and Iba1, and (B) Galectin-3 and CD 16/32. Both Iba1 (a marker of microglia/macrophages) as well as CD16/32 (a marker of proinflammatory M1 microglia or macrophages)-positive cells, co expressed Galectin-3 (n = 3 per group).
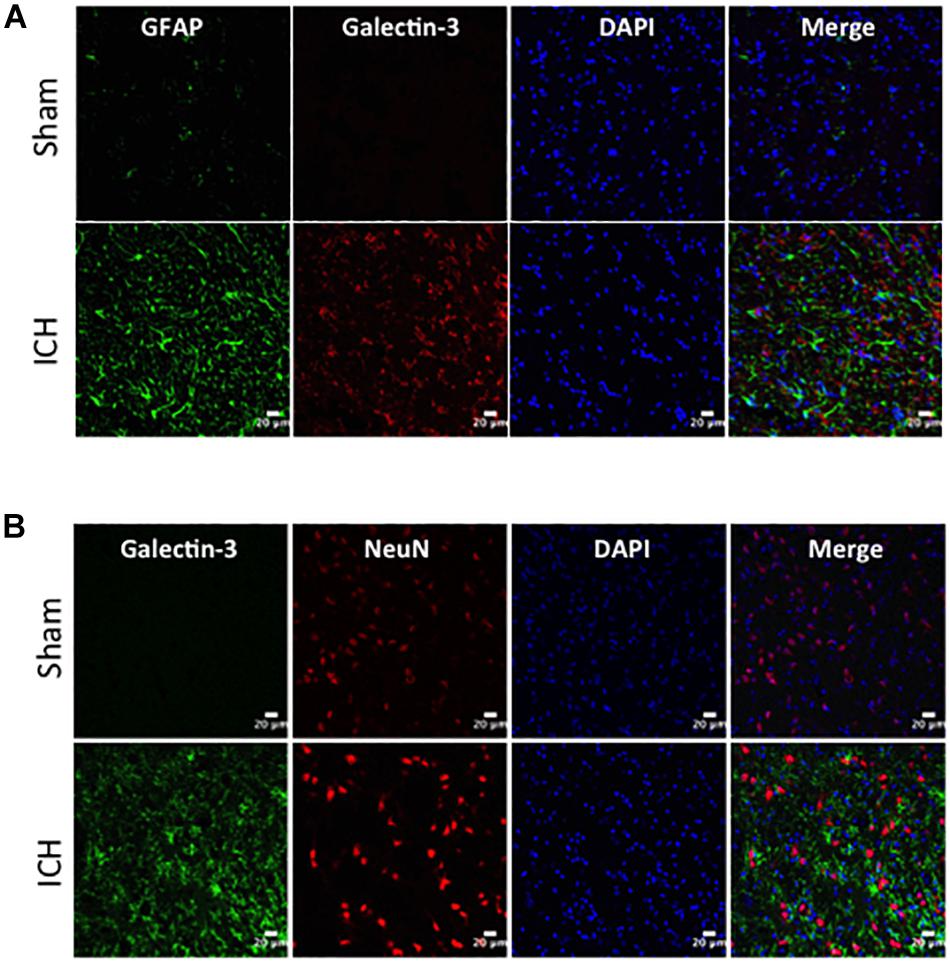
Figure 5. Cellular localization of Galectin-3 after ICH. Brain sections were double immunostained with (A) Galectin-3 and GFAP, and (B) Galectin-3 and NeuN. Galectin-3 expression was absent in either GFAP-positive or NeuN-positive cells. Scale bar = 20 μm; n = 3 per group.
To establish the possible functional role of Galectin-1 and Galectin-3 after ICH, we performed in vitro studies. Recombinant Galectin-1 (6.25 and 12.5 μg/ml) significantly attenuated LPS-induced release of a proinflammatory cytokine, Interleukin -6 (IL-6) from RAW 264.7 cells as estimated by ELISA in comparison to controls (Figure 6) implicating a negative regulatory role of Galectin-1 in inflammation.
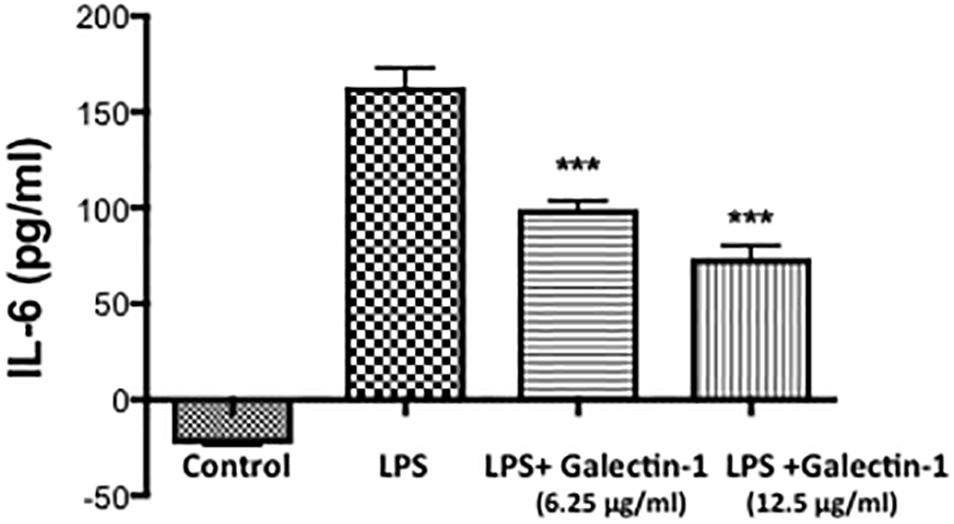
Figure 6. Recombinant Galectin-1 and inflammatory response. Prior to LPS (100 ng/ml) stimulation, Raw 264.7 cells were treated with recombinant Galectin-1, and the release of IL-6 was measured using ELISA, as detailed in methods. Recombinant Galectin-1 significantly reduced LPS-induced release of IL-6 from RAW 246.7 cells (n = 4 per group). ∗∗∗p < 0.001 vs. LPS.
To test the functional role of Galectin-3, we performed siRNA-mediated genetic knockdown of Galectin-3 in RAW 264.7 cells and examined the inflammatory response. Gene silencing with siRNA significantly reduced the Galectin-3 expression in RAW 264.7 cells by 52.95% as evidenced by western blotting (Figures 7A,B). Notably, siRNA medicated genetic knockdown of Galectin-3 did not modulate LPS-induced release of IL- 6 (Figure 7C) whereas the genetic knock down of Galectin-3 significantly augmented hemin (a hemoglobin metabolite that accumulates at high concentration in intracranial hematomas) –induced the release of IL-6 (Figure 7D) implicating an unexplored role of Galectin-3 in modulating inflammatory response after ICH.
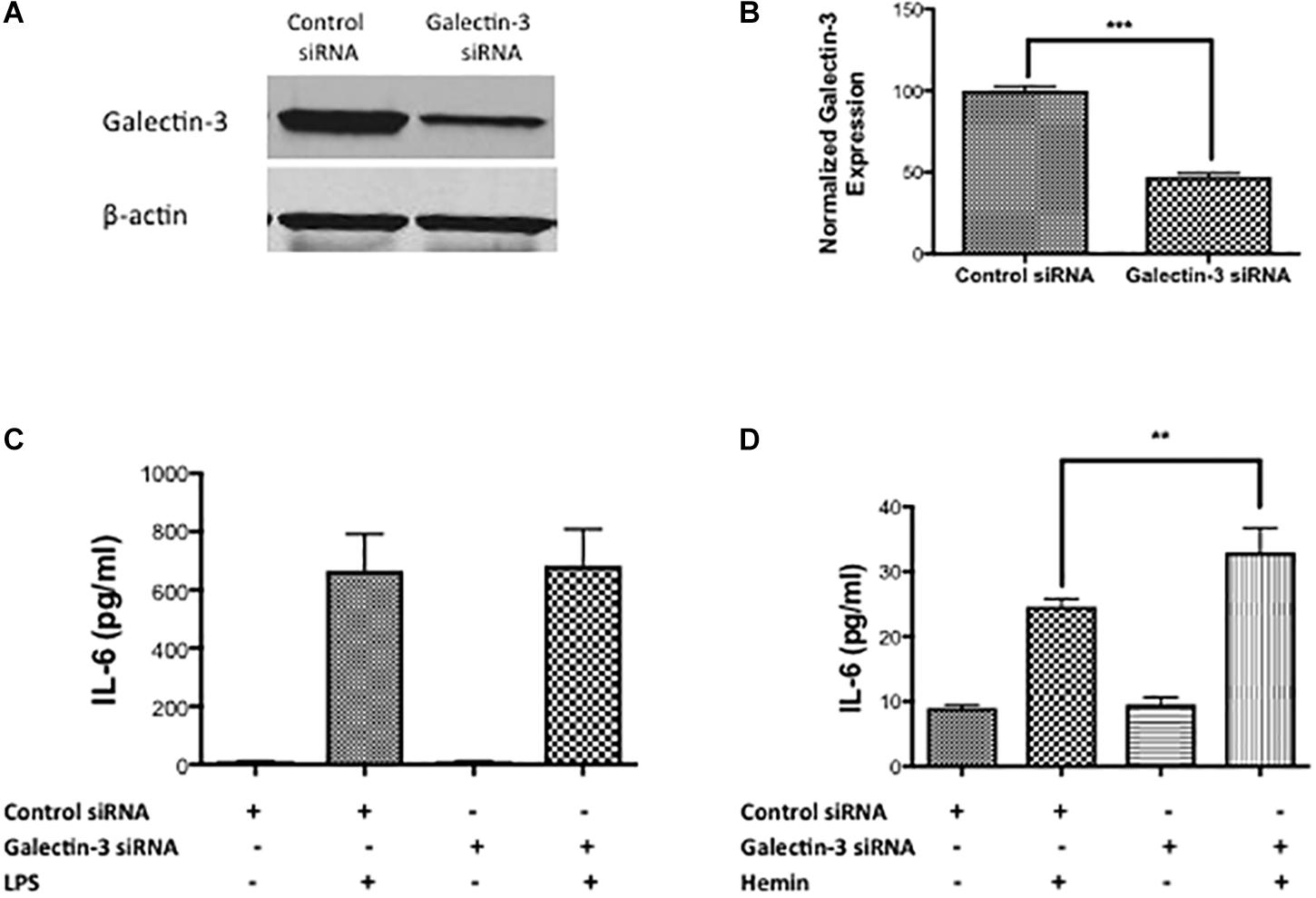
Figure 7. Galectin-3 and inflammatory response (A) RAW 246.7 cells were treated with either control siRNA or Galectin-3 siRNA as detailed in methods and the genetic knockdown of Galectin-3 was verified using (A) western blotting followed by (B) densitometry analysis. ∗∗∗p < 0.001 vs. control siRNA. (C) Galectin-3 knockdown didn’t modulate LPS-induced release of IL-6 (D) whereas significantly unregulated hemin–induced release of IL-6 from RAW 246.7 cells in comparison to control (n = 3 per group). ∗∗p < 0.01 vs. hemin.
Galectins are a family of endogenous carbohydrate-binding proteins that play critical roles in both physiological and pathological conditions by interacting with glycosylated receptors on the cell surface and modulating intracellular signaling pathways (Perillo et al., 1995, 1998; Yang et al., 1996; Moiseeva et al., 1999; Vespa et al., 1999; Yamaoka et al., 2000; Goldring et al., 2002; Laaf et al., 2019). Galectins exhibit significant sequence similarity in their carbohydrate-recognition domain (CRD) with an enhanced affinity toward β-galactosides and are originally defined by their ability to recognize the disaccharide N-acetyllactosamine (Barondes et al., 1994a,b; Kasai and Hirabayashi, 1996). However, recent studies demonstrate substantial differences in their carbohydrate binding properties (Hirabayashi et al., 2002; Leffler et al., 2002; Carlsson et al., 2007).
Galectin-1, the most ubiquitously expressed member of the galectin family (Stillman et al., 2006) has been implicated in the regulation of innate and adaptive immunity and is present in both intracellular and extracellular locations (Verschuere et al., 2014). The extracellular functions of Galectin-1 rely largely on the carbohydrate-binding properties while the intracellular functions involve mainly carbohydrate-independent interactions (Verschuere et al., 2014). Consistent with the role of Galectin-1 in immune response in the periphery, Galectin-1 is known to suppress macrophage activation (Barrionuevo et al., 2007), promotes selective apoptosis of T cells (Toscano et al., 2007), induces the secretion of anti-inflammatory cytokine, IL-10 (van der Leij et al., 2004; Cedeno-Laurent et al., 2012), and attenuates nitric oxide (NO) production by macrophages (Correa et al., 2003).
Galectin-1 is expressed widely in nervous tissues at embryonic stages but becomes restricted mainly to peripheral tissues upon maturation (Horie and Kadoya, 2002). Consistently, uninjured brain striatum exhibited marginal expression of Galectin-1. However, upon hemorrhagic brain injury very remarkable Galectin-1 expression was observed in GFAP-positive astrocytes. Along these lines, Galectin-1 is implicated in astrocyte differentiation and subsequent release of BDNF (Brain Derived Neurotrophic factor) after a brain injury implicating a role of Galectin-1 in neuroprotection (Sasaki et al., 2004; Qu et al., 2010). Further, Galectin-1 is one of the key regulators of adult neurogenesis through its carbohydrate-binding ability and promotes functional recovery after stroke (Ishibashi et al., 2007). Galectin-1 administration reduced apoptosis of neurons, decreased brain infarction volume and improved neurological function induced by brain ischemia (Qu et al., 2011). Also, native and recombinant galectin-1 protected mouse and rat cerebellar neurons from the neurotoxic effects of glutamate (Lekishvili et al., 2006). Of note, Galectin-1 deactivates inflammatory microglia and protects from inflammation-induced neurodegeneration (Starossom et al., 2012). Further, our studies demonstrated that recombinant Galectin-1 attenuates the release of a proinflammatory cytokine, IL-6 from LPS-stimulated murine macrophages, RAW 264.7 in comparison to controls implicating a negative regulatory role of Galectin-1 in inflammation.
Galectin-1 is one of the endogenous ligands of CD45 (Walzel et al., 1999), which regulates microglia/macrophage activation. In addition, Galectin-1 interaction with CD45 leads to the retention of this glycoprotein on the plasma membrane and augmenting its phosphatase activity. Recent studies demonstrated that CD45 negatively regulates proinflammatory M1 microglia activation but promotes anti-inflammatory, M2 phenotype through modulation of the mitogen-activated protein kinase p38 (p38MAPK), cAMP response element binding (CREB), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) signaling pathways (Starossom et al., 2012). This effect involved binding of Galectin-1 to core 2 O-glycans on CD45 suggesting that the expression of glycan moieties on activated microglial/macrophages is required for Galectin-1 binding and function (Starossom et al., 2012). In addition, Galectin-1 suppressed methamphetamine-induced neuroinflammation in human brain microvascular endothelial cells (Parikh et al., 2015) and Galectin-1 is suggested to be involved in neurite outgrowth and synaptic connectivity. Altogether, the data suggest that Galectin-1 induction in reactive astrocytes after ICH could be an intercellular communication mechanism facilitating astrocyte-mediated regulation of neuroprotection after ICH warranting further investigation.
Galectin-3 is a 25–35 kDa chimeric type protein with functions tightly depend on the localization (Thomas and Pasquini, 2018). The expression of Galectin-3 has been found in the nucleus, and cytoplasm (Liu et al., 2002). Further, macrophages and activated microglia can release Galectin-3 in the extracellular space leading to extracellular matrix remodeling and altered inflammatory response, respectively (Li et al., 2008; Jeon et al., 2010). Instead of the classical endoplasmic reticulum/Golgi secretion pathway, Galectin-3 follows an alternative secretory pathway for secretion and export (Mehul and Hughes, 1997) and upon release, Galectin-3 interacts with several extracellular receptors. Though, Galectin-3 is closely linked to the inflammatory cascade of reactions; the precise functional role of Galectin-3 in neuroinflammation is largely controversial. However, it is reported that galectin-3 released by microglia acted as an endogenous TLR-4 (Toll Like Receptor-4) ligand (Burguillos et al., 2015). Further, the genetic deletion of Galectin-3 reduced neuronal loss and administration of Galectin-3 antibody exerted neuroprotective effects in a preclinical model of traumatic brain injury (Yip et al., 2017) together implicating a detrimental role of Galectin-3 after a brain injury. In contrast, targeted deletion of Galectin-3 exacerbated ischemic brain injury and neurodegeneration after cerebral ischemia (Lalancette-Hebert et al., 2012) suggesting a neuroprotective role of Gelectin-3 after brain damage. In addition, Galectin-3 contributes to angiogenesis and neurogenesis implicating its possible role in post-ischemic brain repair (Yan et al., 2009). Galectin-3 also promoted oligodendroglia differentiation, contributing to functional recovery following demyelinating disorders (Pasquini et al., 2011). These conflicting functional roles of Galectin-3 after neuropathology could be due to the differential subcellular expression of Galectin-3 or due to the difference in the pathophysiology of brain disorders warranting further investigation.
Consistent with other neuropathological conditions, we observed elevated expression of Galectin-3 after ICH and expression was predominantly observed in Iba1 positive cells, the inflammatory cells of the CNS. Iba1 positive cells after ICH could be either microglia or infiltrating macrophages, which play roles in innate immune response. Recent studies demonstrate that microglia and macrophages may have differential roles after brain pathology (Gao et al., 2017). Along these lines, studies with primary microglial culture document a proinflammatory role of Galectin-3 (Burguillos et al., 2015), whereas studies with macrophages demonstrate an anti-inflammatory role (MacKinnon et al., 2008) warranting further investigation. Moreover, genetic knockdown of Galectin-3 in RAW 246.7 cells augmented hemin- induced release of IL-6, a proinflammatory cytokine implicating a role of Galectin-3 in inflammatory responses after ICH. Besides, Galectin-3 expressing microglia or macrophages exhibited phagocytic phenotype implicating its unexplored role in microglial or macrophage mediated phagocytosis, which plays a key role in hematoma resolution and subsequent brain recovery after ICH. Consistently, recent reports suggest that macrophages that accumulate in the CNS during parasite infection abundantly express Galectin-3 (Quenum Zangbede et al., 2018) and activated microglia phagocytose cells via Galectin-3 (Nomura et al., 2017). In addition, elevated plasma Galectin-3 levels were strongly associated with inflammation, severity and poor outcomes in patients with acute ICH (Yan et al., 2016). Therefore, further studies are needed elucidating the functional roles of Galectin-3 after ICH.
Galectin-1 and Galectin-3 exhibited very profound and increased expression from day 3 to day 7-post-injury, in the perihematomal brain region after ICH in comparison to Sham. Further, Galectin-1 expression was mostly observed in GFAP- positive astrocytes whereas Galectin-3 expression was observed mostly in Iba1 as well as CD16/32-positive cells, the inflammatory cells of the CNS. Moreover, genetic studies revealed a negative regulatory role of both Galectin-1 and Galectin-3 in the release of a proinflammatory cytokine, IL-6 depending on the stimulus. Altogether, the data suggest that Galectin-1 and Galectin-3 could be targeted in modulating glial responses and thereby brain injury after ICH, warranting further investigation.
Animal studies were reviewed and approved by the Committee on Animal Use for Research and Education at Augusta University, in compliance with NIH and USDA guidelines.
FB carried out the immunohistochemical, and cell culture studies and western blotting and participated in the data analysis. SS-R conceived and designed the experiments, conducted the animal surgeries, genetic studies, and data analysis, and drafted the manuscript. Both authors read and approved the final manuscript.
This work was supported by grants from the National Institutes of Health (R01NS107853) and American Heart Association (14SDG18730034) to SS-R.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00157/full#supplementary-material
FIGURE S1 | The temporal pattern of hematoma after ICH.
CD16/32, Cluster of Differentiation 16/32; CD45: Cluster of Differentiation 45; CNS, central nervous system; ELISA, enzyme linked immunosorbent assay; GFAP, glial fibrillary acidic protein; Hb, hemoglobin; Iba1, ionized calcium binding adaptor molecule 1; ICH, Intracerebral Hemorrhage; LPS, lipopolysaccharide; NeuN, neuronal nuclei; PBS, phosphate buffer saline; RIPA, radioimmunoprecipitation.
Aguilar, M. I., and Freeman, W. D. (2010). Spontaneous intracerebral hemorrhage. Semin. Neurol. 30, 555–564.
Ahmad, A. S., Mendes, M., Hernandez, D., and Dore, S. (2017). Efficacy of laropiprant in minimizing brain injury following experimental intracerebral hemorrhage. Sci. Rep. 7:9489.
Babu, R., Bagley, J. H., Di, C., Friedman, A. H., and Adamson, C. (2012). Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg. Focus 32:E8.
Barondes, S. H., Castronovo, V., Cooper, D. N., Cummings, R. D., Drickamer, K., Feizi, T., et al. (1994a). Galectins: a family of animal beta-galactoside-binding lectins. Cell 76, 597–598. doi: 10.1016/0092-8674(94)90498-7
Barondes, S. H., Cooper, D. N., Gitt, M. A., and Leffler, H. (1994b). Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem. 269, 20807–20810.
Barrionuevo, P., Beigier-Bompadre, M., Ilarregui, J. M., Toscano, M. A., Bianco, G. A., Isturiz, M. A., et al. (2007). A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J. Immunol. 178, 436–445. doi: 10.4049/jimmunol.178.1.436
Bonsack, F. T., Alleyne, C. H. Jr., and Sukumari-Ramesh, S. (2016). Augmented expression of TSPO after intracerebral hemorrhage: a role in inflammation? J. Neuroinflamm. 13:151.
Broderick, J., Connolly, S., Feldmann, E., Hanley, D., Kase, C., Krieger, D., et al. (2007). Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American heart association/american stroke association stroke council, high blood pressure research council, and the quality of care and outcomes in research interdisciplinary working group. Circulation 116,e391–e413.
Burguillos, M. A., Svensson, M., Schulte, T., Boza-Serrano, A., Garcia-Quintanilla, A., Kavanagh, E., et al. (2015). Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 10, 1626–1638. doi: 10.1016/j.celrep.2015.02.012
Cai, Y., Cho, G. S., Ju, C., Wang, S. L., Ryu, J. H., Shin, C. Y., et al. (2011). Activated microglia are less vulnerable to hemin toxicity due to nitric oxide-dependent inhibition of JNK and p38 MAPK activation. J. Immunol. 187, 1314–1321. doi: 10.4049/jimmunol.1002925
Carlsson, S., Oberg, C. T., Carlsson, M. C., Sundin, A., Nilsson, U. J., Smith, D., et al. (2007). Affinity of galectin-8 and its carbohydrate recognition domains for ligands in solution and at the cell surface. Glycobiology 17, 663–676. doi: 10.1093/glycob/cwm026
Carmichael, S. T., Vespa, P. M., Saver, J. L., Coppola, G., Geschwind, D. H., Starkman, S., et al. (2008). Genomic profiles of damage and protection in human intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 28, 1860–1875. doi: 10.1038/jcbfm.2008.77
Cedeno-Laurent, F., Opperman, M., Barthel, S. R., Kuchroo, V. K., and Dimitroff, C. J. (2012). Galectin-1 triggers an immunoregulatory signature in Th cells functionally defined by IL-10 expression. J. Immunol. 188, 3127–3137. doi: 10.4049/jimmunol.1103433
Chen-Roetling, J., Kamalapathy, P., Cao, Y., Song, W., Schipper, H. M., and Regan, R. F. (2017). Astrocyte heme oxygenase-1 reduces mortality and improves outcome after collagenase-induced intracerebral hemorrhage. Neurobiol. Dis. 102, 140–146. doi: 10.1016/j.nbd.2017.03.008
Correa, S. G., Sotomayor, C. E., Aoki, M. P., Maldonado, C. A., and Rabinovich, G. A. (2003). Opposite effects of galectin-1 on alternative metabolic pathways of L-arginine in resident, inflammatory, and activated macrophages. Glycobiology 13, 119–128. doi: 10.1093/glycob/cwg010
Dang, T. N., Robinson, S. R., Dringen, R., and Bishop, G. M. (2011). Uptake, metabolism and toxicity of hemin in cultured neurons. Neurochem. Int. 58, 804–811. doi: 10.1016/j.neuint.2011.03.006
Gao, H., Danzi, M. C., Choi, C. S., Taherian, M., Dalby-Hansen, C., Ellman, D. G., et al. (2017). Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep. 18, 198–212. doi: 10.1016/j.celrep.2016.11.083
Goldring, K., Jones, G. E., Thiagarajah, R., and Watt, D. J. (2002). The effect of galectin-1 on the differentiation of fibroblasts and myoblasts in vitro. J. Cell Sci. 115(Pt 2), 355–366.
Hickenbottom, S. L., Grotta, J. C., Strong, R., Denner, L. A., and Aronowski, J. (1999). Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke 30, 2472–2477.
Hirabayashi, J., Hashidate, T., Arata, Y., Nishi, N., Nakamura, T., Hirashima, M., et al. (2002). Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta 1572, 232–254. doi: 10.1016/s0304-4165(02)00311-2
Horie, H., and Kadoya, T. (2002). Galectin-1 plays essential roles in adult mammalian nervous tissues. Roles of oxidized galectin-1. Glycoconjugate J. 19, 479–489. doi: 10.1023/b:glyc.0000014077.84016.52
Ishibashi, S., Kuroiwa, T., Sakaguchi, M., Sun, L., Kadoya, T., Okano, H., et al. (2007). Galectin-1 regulates neurogenesis in the subventricular zone and promotes functional recovery after stroke. Exp. Neurol. 207, 302–313. doi: 10.1016/j.expneurol.2007.06.024
Jeon, S. B., Yoon, H. J., Chang, C. Y., Koh, H. S., Jeon, S. H., and Park, E. J. (2010). Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J. Immunol. 185, 7037–7046. doi: 10.4049/jimmunol.1000154
Kasai, K., and Hirabayashi, J. (1996). Galectins: a family of animal lectins that decipher glycocodes. J. Biochem. 119, 1–8. doi: 10.1093/oxfordjournals.jbchem.a021192
Laaf, P., Elling, L., and Kren, V. (2019). Galectin-carbohydrate interactions in biomedicine and biotechnology. Trends Biotechnol. 37, 402–415.
Laird, M. D., Sukumari-Ramesh, S., Swift, A. E., Meiler, S. E., Vender, J. R., and Dhandapani, K. M. (2010). Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? J. Neurochem. 113, 637–648. doi: 10.1111/j.1471-4159.2010.06630.x
Lalancette-Hebert, M., Swarup, V., Beaulieu, J. M., Bohacek, I., Abdelhamid, E., Weng, Y. C., et al. (2012). Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 32, 10383–10395. doi: 10.1523/jneurosci.1498-12.2012
Leffler, H., Carlsson, S., Hedlund, M., Qian, Y., and Poirier, F. (2002). Introduction to galectins. Glycoconjugate J. 19, 433–440. doi: 10.1023/b:glyc.0000014072.34840.04
Leira, R., Davalos, A., Silva, Y., Gil-Peralta, A., Tejada, J., Garcia, M., et al. (2004). Early neurologic deterioration in intracerebral hemorrhage: predictors and associated factors. Neurology 63, 461–467. doi: 10.1212/01.wnl.0000133204.81153.ac
Lekishvili, T., Hesketh, S., Brazier, M. W., and Brown, D. R. (2006). Mouse galectin-1 inhibits the toxicity of glutamate by modifying NR1 NMDA receptor expression. Eur. J. Neurosci. 24, 3017–3025. doi: 10.1111/j.1460-9568.2006.05207.x
Li, Y., Komai-Koma, M., Gilchrist, D. S., Hsu, D. K., Liu, F. T., Springall, T., et al. (2008). Galectin-3 is a negative regulator of lipopolysaccharide-mediated inflammation. J. Immunol. 181, 2781–2789. doi: 10.4049/jimmunol.181.4.2781
Lin, S., Yin, Q., Zhong, Q., Lv, F. L., Zhou, Y., Li, J. Q., et al. (2012). Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 9:46.
Liu, F. T., Patterson, R. J., and Wang, J. L. (2002). Intracellular functions of galectins. Biochim. Biophys. Acta 1572, 263–273. doi: 10.1016/s0304-4165(02)00313-6
MacKinnon, A. C., Farnworth, S. L., Hodkinson, P. S., Henderson, N. C., Atkinson, K. M., Leffler, H., et al. (2008). Regulation of alternative macrophage activation by galectin-3. J. Immunol. 180, 2650–2658. doi: 10.4049/jimmunol.180.4.2650
Mehul, B., and Hughes, R. C. (1997). Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J. Cell Sci. 110( Pt 10), 1169–1178.
Min, H., Choi, B., Jang, Y. H., Cho, I. H., and Lee, S. J. (2017). Heme molecule functions as an endogenous agonist of astrocyte TLR2 to contribute to secondary brain damage after intracerebral hemorrhage. Mol. Brain 10:27.
Moiseeva, E. P., Spring, E. L., Baron, J. H., and de Bono, D. P. (1999). Galectin 1 modulates attachment, spreading and migration of cultured vascular smooth muscle cells via interactions with cellular receptors and components of extracellular matrix. J. Vasc. Res. 36, 47–58. doi: 10.1159/000025625
Nomura, K., Vilalta, A., Allendorf, D. H., Hornik, T. C., and Brown, G. C. (2017). Activated microglia desialylate and phagocytose cells via neuraminidase. Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 198, 4792–4801. doi: 10.4049/jimmunol.1502532
Parikh, N. U., Aalinkeel, R., Reynolds, J. L., Nair, B. B., Sykes, D. E., Mammen, M. J., et al. (2015). Galectin-1 suppresses methamphetamine induced neuroinflammation in human brain microvascular endothelial cells: neuroprotective role in maintaining blood brain barrier integrity. Brain Res. 1624, 175–187. doi: 10.1016/j.brainres.2015.07.033
Pasquini, L. A., Millet, V., Hoyos, H. C., Giannoni, J. P., Croci, D. O., Marder, M., et al. (2011). Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death. Diff. 18, 1746–1756. doi: 10.1038/cdd.2011.40
Perillo, N. L., Marcus, M. E., and Baum, L. G. (1998). Galectins: versatile modulators of cell adhesion, cell proliferation, and cell death. J. Mol. Med. (Berl.) 76, 402–412. doi: 10.1007/s001090050232
Perillo, N. L., Pace, K. E., Seilhamer, J. J., and Baum, L. G. (1995). Apoptosis of T cells mediated by galectin-1. Nature 378, 736–739. doi: 10.1038/378736a0
Platt, N., da Silva, R. P., and Gordon, S. (1998). Recognizing death: the phagocytosis of apoptotic cells. Trends Cell Biol. 8, 365–372. doi: 10.1016/s0962-8924(98)01329-4
Qu, W. S., Wang, Y. H., Ma, J. F., Tian, D. S., Zhang, Q., Pan, D. J., et al. (2011). Galectin-1 attenuates astrogliosis-associated injuries and improves recovery of rats following focal cerebral ischemia. J. Neurochem. 116, 217–226. doi: 10.1111/j.1471-4159.2010.07095.x
Qu, W. S., Wang, Y. H., Wang, J. P., Tang, Y. X., Zhang, Q., Tian, D. S., et al. (2010). Galectin-1 enhances astrocytic BDNF production and improves functional outcome in rats following ischemia. Neurochem. Res. 35, 1716–1724. doi: 10.1007/s11064-010-0234-z
Quenum Zangbede, F. O., Chauhan, A., Sharma, J., and Mishra, B. B. (2018). Galectin-3 in M2 macrophages plays a protective role in resolution of neuropathology in brain parasitic infection by regulating neutrophil turnover. J. Neurosci. 38, 6737–6750. doi: 10.1523/jneurosci.3575-17.2018
Qureshi, A. I., Ling, G. S., Khan, J., Suri, M. F., Miskolczi, L., Guterman, L. R., et al. (2001a). Quantitative analysis of injured, necrotic, and apoptotic cells in a new experimental model of intracerebral hemorrhage. Crit. Care Med. 29, 152–157. doi: 10.1097/00003246-200101000-00030
Qureshi, A. I., Tuhrim, S., Broderick, J. P., Batjer, H. H., Hondo, H., and Hanley, D. F. (2001b). Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 344, 1450–1460.
Ribo, M., and Grotta, J. C. (2006). Latest advances in intracerebral hemorrhage. Curr. Neurol. Neurosci. Rep. 6, 17–22. doi: 10.1007/s11910-996-0004-0
Robinson, S. R., Dang, T. N., Dringen, R., and Bishop, G. M. (2009). Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep. 14, 228–235. doi: 10.1179/135100009x12525712409931
Sasaki, T., Hirabayashi, J., Manya, H., Kasai, K., and Endo, T. (2004). Galectin-1 induces astrocyte differentiation, which leads to production of brain-derived neurotrophic factor. Glycobiology 14, 357–363. doi: 10.1093/glycob/cwh043
Shiratori, M., Tozaki-Saitoh, H., Yoshitake, M., Tsuda, M., and Inoue, K. (2010). P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J. Neurochem. 114, 810–819. doi: 10.1111/j.1471-4159.2010.06809.x
Starossom, S. C., Mascanfroni, I. D., Imitola, J., Cao, L., Raddassi, K., Hernandez, S. F., et al. (2012). Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 37,249–263. doi: 10.1016/j.immuni.2012.05.023
Stillman, B. N., Hsu, D. K., Pang, M., Brewer, C. F., Johnson, P., Liu, F. T., et al. (2006). Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J. Immunol. 176, 778–789. doi: 10.4049/jimmunol.176.2.778
Sukumari-Ramesh, S., and Alleyne, C. H. Jr. (2016). Post-injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J. Mol. Neurosci. MN 58, 525–531. doi: 10.1007/s12031-016-0722-y
Sukumari-Ramesh, S., Alleyne, C. H. Jr., and Dhandapani, K. M. (2012a). Astrocyte-specific expression of survivin after intracerebral hemorrhage in mice: a possible role in reactive gliosis? J. Neurotrauma 29, 2798–2804. doi: 10.1089/neu.2011.2243
Sukumari-Ramesh, S., Alleyne, C. H. Jr., and Dhandapani, K. M. (2012b). Astrogliosis: a target for intervention in intracerebral hemorrhage? Transl. Stroke Res. 3(Suppl. 1), 80–87. doi: 10.1007/s12975-012-0165-x
Sukumari-Ramesh, S., Alleyne, C. H. Jr., and Dhandapani, K. M. (2016). The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) confers acute neuroprotection after intracerebral hemorrhage in mice. Transl. Stroke Res. 7, 141–148. doi: 10.1007/s12975-015-0421-y
Tessier, P. A., Naccache, P. H., Clark-Lewis, I., Gladue, R. P., Neote, K. S., and McColl, S. R. (1997). Chemokine networks in vivo: involvement of C-X-C and C-C chemokines in neutrophil extravasation in vivo in response to TNF-alpha. J. Immunol. 159, 3595–3602.
Thomas, L., and Pasquini, L. A. (2018). Galectin-3-mediated glial crosstalk drives oligodendrocyte differentiation and (Re)myelination. Front. Cell. Neurosci. 12:297. doi: 10.3389/fncel.2018.00297
Toscano, M. A., Bianco, G. A., Ilarregui, J. M., Croci, D. O., Correale, J., Hernandez, J. D., et al. (2007). Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 8, 825–834. doi: 10.1038/ni1482
van Asch, C. J., Luitse, M. J., Rinkel, G. J., van der Tweel, I., Algra, A., and Klijn, C. J. (2010). Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol. 9, 167–176. doi: 10.1016/s1474-4422(09)70340-0
van der Leij, J., van den Berg, A., Blokzijl, T., Harms, G., van Goor, H., Zwiers, P., et al. (2004). Dimeric galectin-1 induces IL-10 production in T-lymphocytes: an important tool in the regulation of the immune response. J. Pathol. 204, 511–518. doi: 10.1002/path.1671
Verschuere, T., Toelen, J., Maes, W., Poirier, F., Boon, L., Tousseyn, T., et al. (2014). Glioma-derived galectin-1 regulates innate and adaptive antitumor immunity. Int. J. Cancer 134, 873–884. doi: 10.1002/ijc.28426
Vespa, G. N., Lewis, L. A., Kozak, K. R., Moran, M., Nguyen, J. T., Baum, L. G., et al. (1999). Galectin-1 specifically modulates TCR signals to enhance TCR apoptosis but inhibit IL-2 production and proliferation. J. Immunol. 162, 799–806.
Walzel, H., Schulz, U., Neels, P., and Brock, J. (1999). Galectin-1, a natural ligand for the receptor-type protein tyrosine phosphatase CD45. Immunol. Lett. 67, 193–202. doi: 10.1016/s0165-2478(99)00012-7
Wang, J. (2010). Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 92, 463–477. doi: 10.1016/j.pneurobio.2010.08.001
Wang, J., and Dore, S. (2007). Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 27, 894–908.
Weng, X., Tan, Y., Chu, X., Wu, X. F., Liu, R., Tian, Y., et al. (2015). N-methyl-D-aspartic acid receptor 1 (NMDAR1) aggravates secondary inflammatory damage induced by hemin-NLRP3 pathway after intracerebral hemorrhage. Chin. J. Traumatol. 18, 254–258. doi: 10.1016/j.cjtee.2015.11.010
Yamaoka, K., Mishima, K., Nagashima, Y., Asai, A., Sanai, Y., and Kirino, T. (2000). Expression of galectin-1 mRNA correlates with the malignant potential of human gliomas and expression of antisense galectin-1 inhibits the growth of 9 glioma cells. J. Neurosci. Res. 59, 722–730. doi: 10.1002/(sici)1097-4547(20000315)59:6<722::aid-jnr4>3.0.co;2-h
Yan, X. J., Yu, G. F., Jie, Y. Q., Fan, X. F., Huang, Q., and Dai, W. M. (2016). Role of galectin-3 in plasma as a predictive biomarker of outcome after acute intracerebral hemorrhage. J. Neurol. Sci. 368, 121–127. doi: 10.1016/j.jns.2016.06.071
Yan, Y. P., Lang, B. T., Vemuganti, R., and Dempsey, R. J. (2009). Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 1288, 116–124. doi: 10.1016/j.brainres.2009.06.073
Yang, R. Y., Hsu, D. K., and Liu, F. T. (1996). Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 6737–6742. doi: 10.1073/pnas.93.13.6737
Yip, P. K., Carrillo-Jimenez, A., King, P., Vilalta, A., Nomura, K., Chau, C. C., et al. (2017). Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 7:41689.
Keywords: ICH, Galectin-1, Galectin-3, astrocytes, microglia
Citation: Bonsack F and Sukumari-Ramesh S (2019) Differential Cellular Expression of Galectin-1 and Galectin-3 After Intracerebral Hemorrhage. Front. Cell. Neurosci. 13:157. doi: 10.3389/fncel.2019.00157
Received: 03 January 2019; Accepted: 08 April 2019;
Published: 14 May 2019.
Edited by:
Dirk M. Hermann, University of Duisburg-Essen, GermanyReviewed by:
Anatol Manaenko, University Hospital Erlangen, GermanyCopyright © 2019 Bonsack and Sukumari-Ramesh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangeetha Sukumari-Ramesh, c3JhbWVzaEBhdWd1c3RhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.