 Maria Calvo-Rodriguez
Maria Calvo-Rodriguez Elena Hernando-Perez1
Elena Hernando-Perez1 Lucia Nuñez
Lucia Nuñez Carlos Villalobos
Carlos Villalobos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 08 February 2019
Sec. Cellular Neurophysiology
Volume 13 - 2019 | https://doi.org/10.3389/fncel.2019.00022
This article is part of the Research Topic Cellular Neurophysiology Editors’ Pick 2021 View all 19 articles
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and strongly associated to aging. AD has been related to excess of neurotoxic oligomers of amyloid β peptide (Aβo), loss of intracellular Ca2+ homeostasis and mitochondrial damage. However, the intimate mechanisms underlying the pathology remain obscure. We have reported recently that long-term cultures of rat hippocampal neurons resembling aging neurons are prone to damage induced by Aβ oligomers (Aβo) while short-term cultured cells resembling young neurons are not. In addition, we have also shown that aging neurons display critical changes in intracellular Ca2+ homeostasis including increased Ca2+ store content and Ca2+ transfer from the endoplasmic reticulum (ER) to mitochondria. Aging also promotes the partial loss of store-operated Ca2+ entry (SOCE), a Ca2+ entry pathway involved in memory storage. Here, we have addressed whether Aβo treatment influences differentially intracellular Ca2+ homeostasis in young and aged neurons. We found that Aβo exacerbate the remodeling of intracellular Ca2+ induced by aging. Specifically, Aβo exacerbate the loss of SOCE observed in aged neurons. Aβo also exacerbate the increased resting cytosolic Ca2+ concentration, Ca2+ store content and Ca2+ release as well as increased expression of the mitochondrial Ca2+ uniporter (MCU) observed in aging neurons. In contrast, Aβo elicit none of these effects in young neurons. Surprisingly, we found that Aβo increased the Ca2+ transfer from ER to mitochondria in young neurons without having detrimental effects. Consistently, Aβo increased also colocalization of ER and mitochondria in both young and aged neurons. However, in aged neurons, Aβo suppressed Ca2+ transfer from ER to mitochondria, decreased mitochondrial potential, enhanced reactive oxygen species (ROS) generation and promoted apoptosis. These results suggest that modulation of ER—mitochondria coupling in hippocampal neurons may be a novel physiological role of Aβo. However, excess of Aβo in the face of the remodeling of intracellular Ca2+ homeostasis associated to aging may lead to loss of ER—mitochondrial coupling and AD.
Alzheimer’s disease (AD) is the most common form of dementia, accounting for 60%–80% of all dementia cases. Despite of all the efforts, the mechanisms underlying the pathology are still unknown. The two main hallmarks of AD are extracellular plaques composed of amyloid β (Aβ) peptide and intracellular neurofibrillary tangles composed of hyper-phosphorylated and misfolded tau. Most AD cases are sporadic (late-onset AD) and affect elderly people, but some cases (around 5%) are inherited in an autosomal dominant fashion, caused by mutations in the genes that encode presenilin1 (PS1), PS2 and amyloid precursor protein (APP; early-onset AD or familial AD). One of the most accepted hypothesis of AD is the “amyloid cascade hypothesis,” which postulates that the deposition of the Aβ is a central event to the pathology of AD (Hardy and Selkoe, 2002). This hypothesis considers that the pathology of AD is caused by an abnormal processing of the protein Aβ, occasioned by a reduction in its clearance or by an increase in its production. Most AD cases are sporadic, being aging the most important risk factor for the disease. During aging and, particularly, in neurodegenerative diseases, the systems that regulate intracellular Ca2+ homeostasis are affected, leading to synaptic dysfunction, plasticity detriment and neuronal degeneration. Consequently, a wide amount of studies has shown that the disturbance of Ca2+ homeostasis is associated with the neurotoxicity that occurs in AD (Mattson, 2007; Bezprozvanny, 2009; Berridge, 2010). The “Ca2+ hypothesis of AD” (Khachaturian, 1989) postulates that the activation of the amyloidogenic pathway induces a remodeling of the neuronal Ca2+ signaling, disturbing the normal Ca2+ homeostasis and therefore, the mechanisms responsible for learning and memory. In addition, oxidative stress, alterations in mitochondrial energetic metabolism and protein aggregation, such as Aβ in AD, affect negatively the intracellular Ca2+ homeostasis. Recent studies have also shown that impaired function of intracellular organelles such as the endoplasmic reticulum (ER) and mitochondria plays an important role in the regulation of Ca2+ during aging and AD (Toescu and Verkhratsky, 2003). In this manner, PSs, located in the membrane of the ER, have been associated to familial AD. They are integral membrane proteins that form the catalytic core of the γ-secretase complex that cleaves APP. Mutations in PSs increase the production of Aβ1–42 form or reduce the production of Aβ1–40 (Borchelt et al., 1996; Lemere et al., 1996). In addition, PSs are believed to form low conductivity channels, being involved in the formation of the leak channels of the ER, releasing Ca2+ in a passive manner from the ER to the cytoplasm (Tu et al., 2006). Mutations in PSs result in an increased release of Ca2+ from the ER (Nelson et al., 2007), probably due to a loss of function of the leak channel. The other key player of intracellular Ca2+ homeostasis, mitochondria, are dynamic organelles that generate ATP and contribute to many cellular functions. They play a main role in apoptotic signaling, lipid synthesis and buffering of intracellular Ca2+. Many reports have also proposed mitochondrial dysfunction in AD. In this manner, dysfunction of mitochondrial bioenergetics (Atamna and Frey, 2007; Yao et al., 2009), increased fission and decreased fusion (Wang et al., 2009; Santos et al., 2010), morphological changes (Hirai et al., 2001; DuBoff et al., 2013; Xie et al., 2013) and redistribution of mitochondria (Kopeikina et al., 2011) have been extensively reported.
ER and mitochondria are connected through the mitochondria-associated ER membranes (MAMs). They play a central role in the processes that occur between ER and mitochondria, such as communication between the two organelles, which includes Ca2+ transport. When the Ca2+ from the ER is released to the cytoplasm, part of this Ca2+ is taken up by mitochondria, acting as a buffer of Ca2+ (Szabadkai et al., 2003). If normal flux is affected, the amount of Ca2+ taken up by mitochondria could be increased. If there is mitochondrial Ca2+ overload, the mitochondrial permeability transition pore (PTPm) will open, mitochondrial membrane potential will collapse and proapoptotic factors such as cytochrome c will be released, activating the caspase pathway and triggering apoptosis. In addition, an excessive Ca2+ increase will enhance the production of reactive oxygen species (ROS), that will also contribute to the opening of the PTPm (Berridge, 2010).
Alteration of the correct MAM function has been previously shown in AD (Müller et al., 2018). In this manner, increased connectivity ER-mitochondria was found in human fibroblasts from patients with familiar AD mutations as well as in fibroblasts from patients with sporadic AD (Area-Gomez et al., 2012; Area-Gomez and Schon, 2017). Our group has also previously reported that Aβ oligomers (Aβo), unlike the fibrils, promote the entry of Ca2+ into the cell, causing mitochondrial Ca2+ overload and cell death by apoptosis (Sanz-Blasco et al., 2008; Calvo-Rodríguez et al., 2016a). Also, exposure of primary hippocampal neurons to conditioned media containing Aβo increased the contact points ER-mitochondria. In addition, these media also increased ER-mitochondria Ca2+ transfer in neuroblastoma cells (Hedskog et al., 2013). However, the effects of oligomers in the context of aging has not been addressed. Moreover, this study has never been performed in primary neurons.
We have previously described an intraorganellar Ca2+ remodeling with aging, involving ER-mitochondria cross-talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons (Calvo-Rodríguez et al., 2016b). Most studies on mechanisms underlying the pathology of AD use transgenic animal models that develop the pathology at early ages (i.e., around 6 months of age), cells from AD patients that are therefore already diagnosed, or cell lines exposed to Aβ peptides. In consequence, the alterations in the intraorganellar Ca2+ remodeling in AD in the context of aging have not been described, and the full spectrum of mechanisms through which Ca2+ homeostasis is altered in AD in aging is still missing. Based on this, we decided to study whether the soluble oligomers of the most toxic peptide involved in AD, Aβ1–42 (Aβo), are able to alter: (1) resting levels of cytosolic free Ca2+ concentration ([Ca2+]cyt); (2) SOCE; (3) the content and release of Ca2+ from the internal stores, as well as; (4) the Ca2+ transfer from ER-to mitochondria; (5) the interaction ER-mitochondria; and (6) the expression of the mitochondrial Ca2+ uniporter (MCU) and inositol trisphosphate receptors (IP3Rs) in young and aged hippocampal neurons. In order to model aging, we have employed an in vitro culture of hippocampal rat neurons maintained for several days in vitro (DIV).
Primary rat hippocampal neurons in long-term culture have been previously used as a model of aging by our group and others (Porter et al., 1997; Brewer et al., 2007; Calvo-Rodríguez et al., 2016b, 2017). Importantly, some of the changes that occur in aging are mimicked by this in vitro model of aging, such as accumulation of ROS, lipofuscin granules, heterochromatic foci, activation of the Jun N-terminal protein kinase (pJNK) and p53/p21 pathways and changes in NMDA receptor expression (Sodero et al., 2011; Calvo et al., 2015a). By using fluorescence Ca2+ imaging of fura-2 loaded cells, we have evaluated the change in the Ca2+ content in the ER, SOCE, the Ca2+ depletion from ER induced by different physiological agonists and the coupling ER-mitochondria, in young (4–8 DIV, short-term) and aged (15–21 DIV, long term) hippocampal neurons cultured in vitro and exposed to Aβo as a model of AD and in vitro aging.
Wistar rat pups (newborn P0–1) were obtained from the Valladolid University animal facility. All animals were handled according to the ethical standard of Valladolid University under protocols approved by the animal housing facility and in accordance with the European Convention 123/Council of Europe and Directive 86/609/EEC. Fura-2/AM and Rhod-5N are from Invitrogen (Barcelona, Spain). Fetal bovine serum (FBS) is from Lonza (Barcelona, Spain). Horse serum, neurobasal medium, HBSS medium, B27, L-glutamine and gentamycin are from Gibco (Barcelona, Spain). Papain solution is from Worthington (Lakewood, NJ, USA). The poly-D-lysine and annexin V are from BD (Madrid, Spain). DNase I and antibody against the MCU are from Sigma (Madrid, Spain). IP3R1 and IP3R2 primary antibodies are from Santa Cruz Biotechnology (Dallas, TX, USA). IP3R3 primary antibody is from BD Transduction Laboratories (Madrid, Spain). ER tracker, mitotracker, tetramethylrhodamine, methyl ester (TMRM) and CM-H2DCFDA are from ThermoFisher Scientific. Aβ1–42 peptide is from Bachem AG (Bubenforf, Switzerland). Other reagents and chemicals were obtained either from Sigma or Merck.
Hippocampal neurons were prepared from Wistar rat pups under sterile conditions as reported by Brewer et al. (1993) with further modifications by Pérez-Otaño et al. (2006). Briefly, newborn rat pups were decapitated and, after brain removal, meninges were discarded and hippocampi were separated from cortex. Hippocampal tissue was cut in small pieces, transferred to papain solution (20 u./ml) and incubated at 37°C for 30 min. After 15 min, DNase I (50 μl/ml) was added. Tissue pieces were washed with Neurobasal medium and cell suspension was obtained using a fire-polished pipette in Neurobasal medium supplemented with 10% FBS. Cells were centrifuged at 160 g for 5 min and pellet was suspended in Neurobasal medium. Hippocampal cells were plated onto poly-D-lysine-coated, 12 mm diameter glass coverslips at 30 × 103 cells/dish (plating density, 169 cells/mm2), and grown in Neurobasal media supplemented with L-glutamine (2 mM), gentamicin (1 μg/ml), 2% B27 and 10% FBS, maintained in a humidified 37°C incubator with 5% CO2 without further media exchange. Cells were cultured for 4–8 DIV for young cultures, and 15–21 DIV for aged cultures. Other details have been reported in detail elsewhere (Calvo et al., 2015a; Calvo-Rodríguez et al., 2015).
Aβ1–42 oligomers (Aβo) were prepared as previously reported by a new procedure (Caballero et al., 2016). Briefly, Aβ1–42 peptide was initially solved at 1 mM concentration in iced cold hexafluoroisopropanol (HFIP), and separated into aliquots in sterile microcentrifuge tubes. The solution was then incubated for 2 h at room temperature (RT) to allow monomerization of the peptide. HFIP was removed under vacuum in a speed vacuum (800 g × 10 min at RT), and the peptide film was stored desiccated at −20°C. For aggregation, the peptide was first suspended in dry DMSO at 5 mM concentration. For complete suspension of the peptide, it was subjected to ultrasounds for 10 min, aliquoted in propylene non-siliconized tubes, and stored at −20°C until use. MEM (Gibco) medium supplemented with 0.5 mg/ml Fe2+, 0.5 mg/ml Cu2+ and 0.5 mg/ml Zn2+ was added to bring the peptide to a concentration of 80 μM. Finally, it was incubated at 37°C for 24 h. For experiments, Aβ1–42 was solved in the culturing media to 1 μM final concentration. Composition of these oligomers has been characterized previously (Caballero et al., 2016; Calvo-Rodríguez et al., 2016b).
For apoptosis, hippocampal neurons were exposed to either vehicle or Aβo 1 μM for 24 h. Cells were then washed with phosphate buffered saline (PBS) and apoptosis was evaluated using annexin V (1:20, 10 min) in annexin binding buffer 1× (in mM) NaCl, 140; CaCl2, 2.5; Hepes, 10 (pH 7.4) and assessed by fluorescence microscopy using a Nikon Eclipse TS100 microscope (objective 40×).
Hippocampal cells were cultured for 4–8 DIV (short-term) or 15–21 DIV (long-term) and exposed to either vehicle (supplemented MEM) or 1 μM Aβo for 24 h before the experiment. The day of the experiment, hippocampal cells were washed in standard external medium (SEM) containing (in mM) NaCl 145, KCl 5, CaCl2 1, MgCl2 1, glucose 10 and Hepes/NaOH 10 (pH 7.4). Then cells were incubated in the same medium containing fura-2 (4 μM) for 60 min at RT in the dark. Then coverslips were placed on the perfusion chamber of a Zeiss Axiovert 100 TV, perfused continuously with the same pre-warmed medium at 37°C and epi-illuminated alternately at 340 and 380 nm using a filter wheel. Light emitted at 520 nm was recorded every 1–5 s with a Hamamatsu ER camera (Hamamatsu Photonics France). Pixel by pixel ratios of consecutive frames were captured and cytosolic Ca2+ concentration ([Ca2+]cyt) values from regions of interest (ROIs) corresponding to individual neurons were averaged and expressed as the ratio of fluorescence emission following excitation at 340 and 380 nm as reported in detail previously (Sanz-Blasco et al., 2008; Calvo et al., 2015b). For estimation of Ca2+ increase, responses (calculated as area under curve, A.U.C.) were averaged from responsive neurons (selected by their morphology different from glial cells in the brightfield). Fraction was calculated by dividing responsive cells for the total cell number in the field, considering responsive cells the ones showing a change in the slope of the Ca2+ recording after application of the stimulus.
For measurements of SOCE, fura-2-loaded cells were treated with the sarcoplasmic and ER Ca2+ ATPase (SERCA) pump blocker thapsigargin (1 μM) for 15 min in the same SEM devoid of extracellular Ca2+ before imaging as reported elsewhere (Calvo-Rodríguez et al., 2016b). Then cells were subjected to fluorescence imaging and stimulated with 5 mM Ca2+ to monitor the SOCE-dependent rise in [Ca2+]cyt. Recordings were made in the presence of TTX to prevent activation of voltage-gated Ca2+ channels by connected neurons.
For estimation of Ca2+ store content, the rise in [Ca2+]cyt induced by low concentrations of the Ca2+ ionophore ionomycin (400 nM) added in the absence of extracellular Ca2+ was monitored as reported previously (Villalobos and García-Sancho, 1995).
Hippocampal cells cultured for 4–8 DIV (short-term) or 15–21 DIV (long-term) were exposed to either vehicle or 1 μM Aβo for 24 h before the experiment. The day of the experiment, hippocampal cells were washed in SEM and incubated in the same medium containing Rhod-5N AM (1 μM) for 30 min at RT in the dark and washed in SEM for additional 30 min. Then coverslips were placed on the perfusion chamber of a Zeiss Axiovert 100 TV, perfused continuously with the same pre-warmed medium at 37°C and epi-illuminated in the red channel (551 nm). Light emitted at 576 nm was recorded every 5 s with a Hamamatsu ER camera (Hamamatsu Photonics France). Pixel by pixel ratios of consecutive frames were captured and mitochondrial Ca2+ concentration ([Ca2+]mit) values from ROIs corresponding to individual neurons were averaged and expressed as normalized fluorescence emission (F/F0). For estimation of Ca2+ increase, responses (calculated as amplitude of the peak) were averaged from responsive neurons (selected by their morphology different from glial cells in the brightfield). Fraction was calculated by dividing responsive cells for the total cell number in the field, considering responsive cells the ones showing a change in the slope of the Ca2+ recording after application of the stimulus.
Hippocampal cells at different culture periods and exposed to either vehicle or 1 μM Aβo for 24 h were washed with phosphate buffered saline (PBS), fixed with p-formaldehyde 4% and incubated with antibodies against MCU (1:200) and IP3 receptors IP3R1 (1:50), IP3R2 (1:50) and IP3R3 (1:50) at 4°C overnight. Immunopositive cells were revealed using Alexafluor 488-tagged antibodies (1:300). Optical density was measured in selected ROIs corresponding to individual neurons using ImageJ software. Further details have been reported elsewhere (Calvo-Rodríguez et al., 2016b).
Cells in every condition were stained for 10 min with 200 nM MitoTracker green and ERTracker red and imaged directly using a confocal microscope. High resolution images of cells were recorded by using a Leica TCS SP5 confocal microscope (Leica Microsystems, Mannheim, Germany). The Mitotracker and ER-tracker were excited with 488 and 543 laser lines, respectively, and emission was acquired with a charged CCD camera. The images were analyzed in LAS AF Lite software (Leica Microsystems, Mannheim, Germany). Background was subtracted from all images. Colocalization analysis for the two markers was performed on Z-stacks acquired with steps of 0.8 μm. For colocalization analysis, green and red channel images were acquired independently. Analysis were carried out on single-plane images using ImageJ plug-ins. Manders2 and Pearson’s coefficients were calculated applying the ImageJ Colocalization analysis plug-in.
For mitochondrial membrane potential measurements, hippocampal neurons were exposed to either vehicle or Aβo 1 μM for 24 h. Then, cells were washed in SEM and loaded with the mitochondrial potential probe (TMRM, 10 nM) for 30 min at RT in the dark. Then coverslips containing cells were placed on a Zeiss Axiovert 100 TV inverted microscope and subjected to fluorescence imaging. Fluorescence images were captured with the rhodamine filter set with a Hamamatsu ER-Orca fluorescence camera as reported previously (Calvo-Rodríguez et al., 2016b).
For ROS generation measurements, hippocampal neurons were exposed to either vehicle or Aβo 1 μM for 24 h. Then, cells were washed in SEM and loaded with the ROS probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA 2 μM) for 40 min at RT in the dark. Then coverslips containing cells were placed on a Zeiss Axiovert 100 TV inverted microscope and subjected to fluorescence imaging. Fluorescence images were captured with the FITC filter set with a Hamamatsu ER-Orca fluorescence camera.
Changes in fluorescence ratio are expressed as area under the curve (AUC) and maximum increase in ratio (Δratio). Calculation of AUC and Δratios were performed using Origin Lab 7.0. Data are presented as mean ± SEM. When only two means were compared, Student’s t-test was used. For more than two groups, statistical significance of the data was assessed by one-way or multifactorial analysis of variance (ANOVA), depending on the number of factors considered. Differences were considered significant at p < 0.05, where 0.05 is the signification level.
We tested the effects of Aβ1–42 oligomers (Aβo) on the rate of apoptosis in rat hippocampal neurons cultured for 4–8 DIV representing young neurons, or in neurons cultured for 15–21 DIV that represent many characteristics of aged neurons as reported previously (Calvo-Rodríguez et al., 2016a). Short-term (4–8 DIV) and long-term (15–21 DIV) cultured hippocampal neurons were exposed for 24 h to 1 μM Aβo or to vehicle (supplemented MEM, see “Materials and Methods” section). This latter condition was taken as control group. Figure 1 shows that, in resting conditions, only less than 3% of the young neurons display apoptosis. Aβo do not increase the rate of apoptosis in these cultured cells. About 5% of aged neurons display apoptosis in basal conditions. In contrast, Aβo increase the percent of apoptotic cells by nearly four-fold. These results confirm our previous results reported elsewhere (Caballero et al., 2016; Calvo-Rodríguez et al., 2016a) and indicate that Aβo toxicity depends on the time in culture of rat hippocampal neurons.
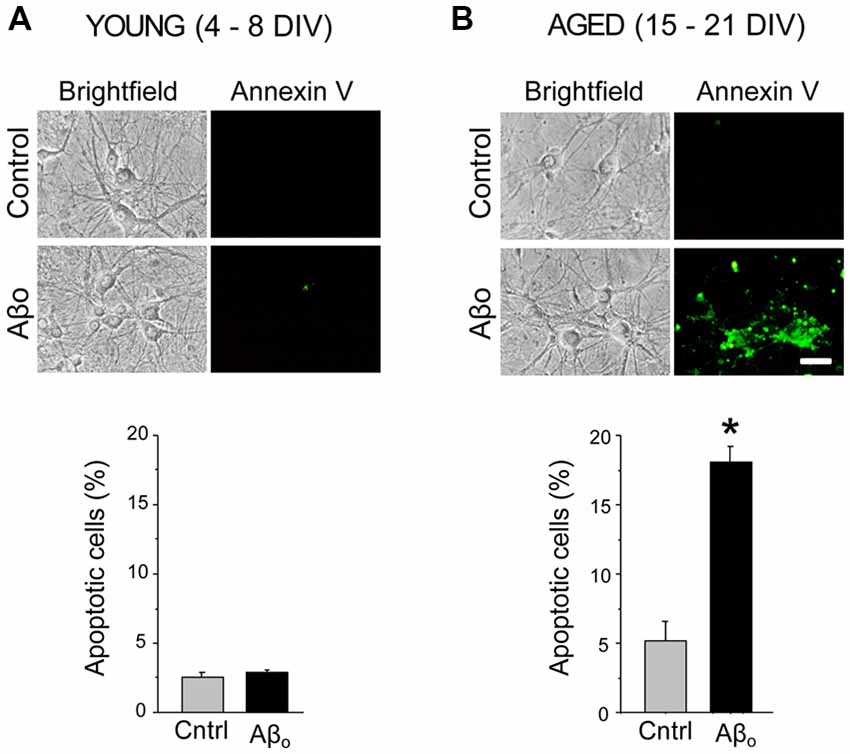
Figure 1. Amyloid β peptide oligomers (Aβo) induce apoptosis only in aged neurons. Short-term (young) and long-term (aged) hippocampal cultures were exposed for 24 h Aβo (1 μM) or vehicle. Then, the relative abundance (percent, %) of apoptotic neurons was assessed by incubation with annexin V. Pictures show representative bright field cultures of short-term (A) and long-term cultures (B) of rat hippocampal neurons. Bars represent mean ± standard external medium (SEM) percentage of apoptotic cells per condition. n = 339 and 330 cells from three independent experiments for young neurons (A) and 452 and 417 cells from four independent experiments, respectively for aged neurons (B). *p < 0.05 compared to control group.
In our hands, cultured rat hippocampal cells display a mix of neurons and glial cells and their relative abundance changes with aging in culture mostly because glial cells proliferate while neuron cells do not. The fraction of neurons amounts 60% at 5 DIV, decreases to 23% at 14 DIV and decreases further to only 15% at 20 DIV. Therefore, for monitoring of effects of Aβo in neurons, it is convenient to carry out measurements at the single cell level in identified neurons. Fortunately, hippocampal neurons can be easily identified and distinguished from astrocytes or glial cells according to morphometric characteristics for the expert eye. Next, we addressed whether treatment of rat hippocampal neurons with Aβo promote differential changes in intracellular Ca2+ homeostasis in young and aged neurons.
We have tested the effects of chronic treatment of Aβo on resting [Ca2+]cyt, a parameter previously reported to vary as hippocampal neurons age in culture (Calvo et al., 2015a). Short-term (4–8 DIV) and long-term (15–21 DIV) cultured hippocampal neurons were exposed for 24 h to 1 μM Aβo or to vehicle (supplemented MEM). The effects of treatments were assessed by calcium imaging of fura-2 loaded neurons. Neurons were clearly distinguished from surrounding glial cells by their morphometric characteristics as previously reported (Calvo-Rodríguez et al., 2016b). Figure 2 shows that short-term cultured, rat hippocampal neurons display spontaneous, synchronous [Ca2+]cyt oscillations. Notice that the average recording runs in parallel to the recordings of individual cells, indicating that oscillations are synchronous. These synchronous oscillations reflect synchronous synaptic activity and the formation of neural networks in vitro (Nunez et al., 1996; Calvo-Rodríguez et al., 2016b). Long-term cultured rat hippocampal neurons exposed to Aβo (1 μM, 24 h) display increased [Ca2+]cyt levels (resting level) when compared to the control neurons (Figure 2C). Moreover, treatment of long-term cultured hippocampal neurons with 1 μM Aβo for 24 h increases significantly the synchronous and spontaneous [Ca2+]cyt oscillations compared to the control group measured as oscillations index (OI), a parameter representing both the frequency and amplitude of [Ca2+] oscillations (Figure 2D; Nunez et al., 1996). In contrast, exposure of short-term cultured neurons to 1 μM Aβo had a little or no effect on both parameters, resting [Ca2+]cyt and OI. These results suggest that prolonged exposure of hippocampal neurons in vitro to Aβo increases the excitability of the neuronal circuit, at the level of basal Ca2+ and spontaneous oscillations. This effect is statistically significant only in aged neurons and Aβo has no effect in this regard in young neurons.
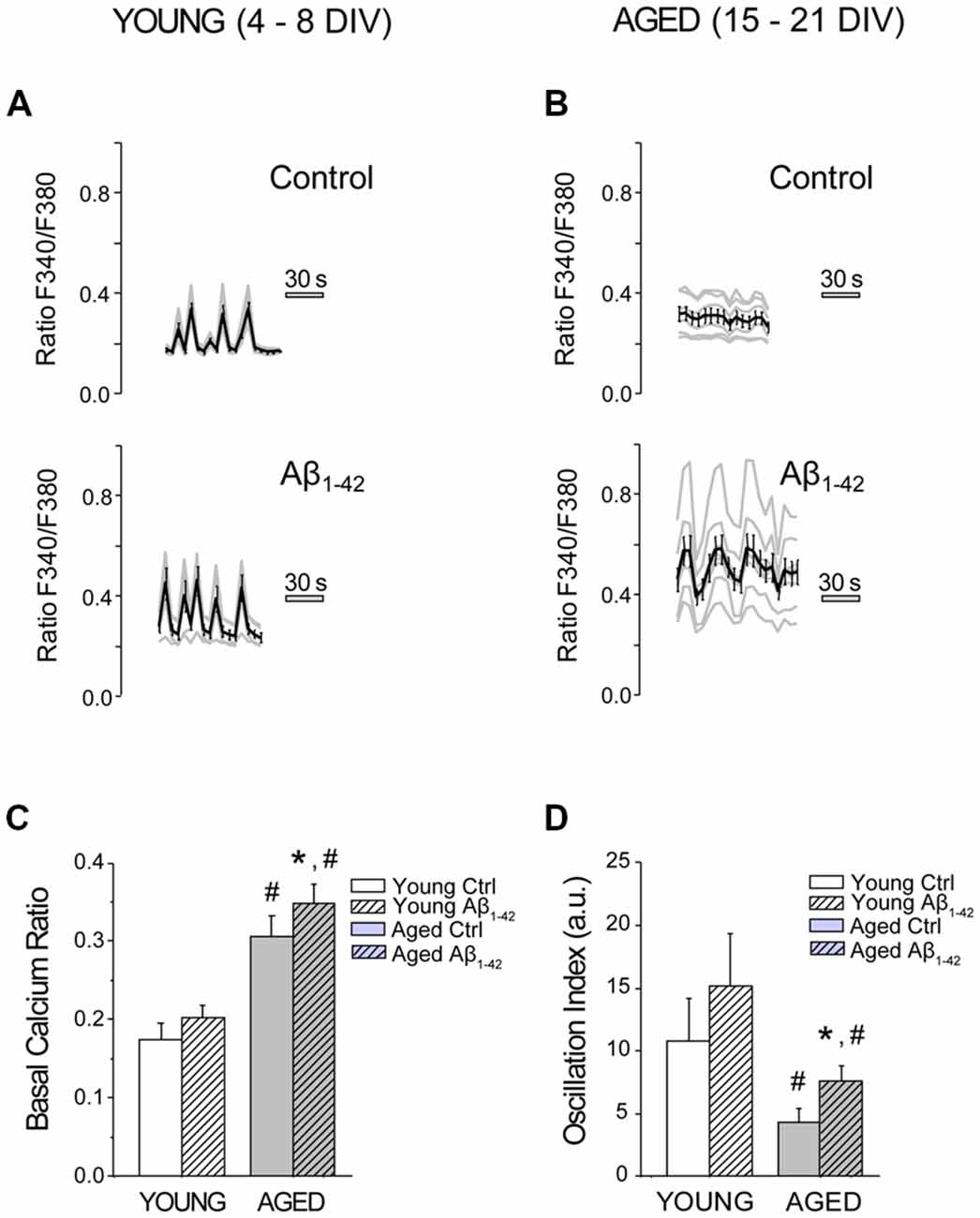
Figure 2. The exposure of aged hippocampal neurons to Aβo increases basal Ca2+ and synchronous and spontaneous Ca2+ oscillations. Hippocampal neurons cultured for different days in vitro (DIV), were exposed to vehicle or Aβo (1 μM, 24 h), and [Ca2+]cyt was monitored by fluorescence imaging using fura-2. (A,B) Traces show individual cells (gray lines) and mean ± SEM (black line) of the increase in [Ca2+]cyt prior to any stimulus in young (A) and aged (B) cultured hippocampal neurons exposed to either Aβo (1 μM) or vehicle. (C,D) Bars represent mean ± SEM of basal Ca2+ (ratio F340/F380; C) and the oscillation index (D) for young and aged neurons, treated in absence (ctrl) of presence of the Aβo (Aβ1–42, hatched bars). n = 113, 97, 131 and 169 neurons from four to six independent experiments, respectively. *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
We have previously shown that SOCE, the Ca2+ entry pathway activated after depletion of intracellular Ca2+ stores, decreases with in vitro aging (Calvo-Rodríguez et al., 2016b). This process has been associated to a downregulation of stromal interaction molecule 1 (Stim1) and Orai1, molecular players involved in SOCE in most cell types. Here, we assessed the effect of Aβo on SOCE in our model of in vitro aging. Short-term and long-term cultured hippocampal neurons were exposed to either vehicle (Control) or Aβo (1 μM, 24 h), and SOCE was monitored by fluorescence imaging of [Ca2+]cyt. For this end, cultured neurons were loaded with fura-2 and treated with Tg (1 μM) for 15 min in medium devoid of extracellular Ca2+ (0 Ca) in order to fully deplete the intracellular Ca2+ stores. Subsequently, medium containing Ca2+ 5 mM and TTX (500 nM) was added for 5 min to activate SOCE. TTX was added to block the electrical connectivity of cultured neurons. La3+ (100 μM) was added later to block this pathway. Pictures in Figure 3 show typical bright field images (transm) and fluorescence images coded in pseudocolor representing [Ca2+]cyt before (basal) and after addition of 5 mM Ca2+ (5 Ca). Representative recordings of fluorescence ratios of individual short-term (young, 4–8 DIV, Figures 3A,C) and long-term (aged, 15–21 DIV, Figures 3B,D) cultured neurons are also shown. Figure 3E shows bars corresponding to the averaged SOCE calculated as ΔRatioF340/F380 (mean ± SEM). Data confirm our previous results showing that SOCE is reduced significantly in in vitro aged neurons relative to young neurons. In addition, they show that AβO decrease SOCE further but this effect is only statistically significant in aged neurons.
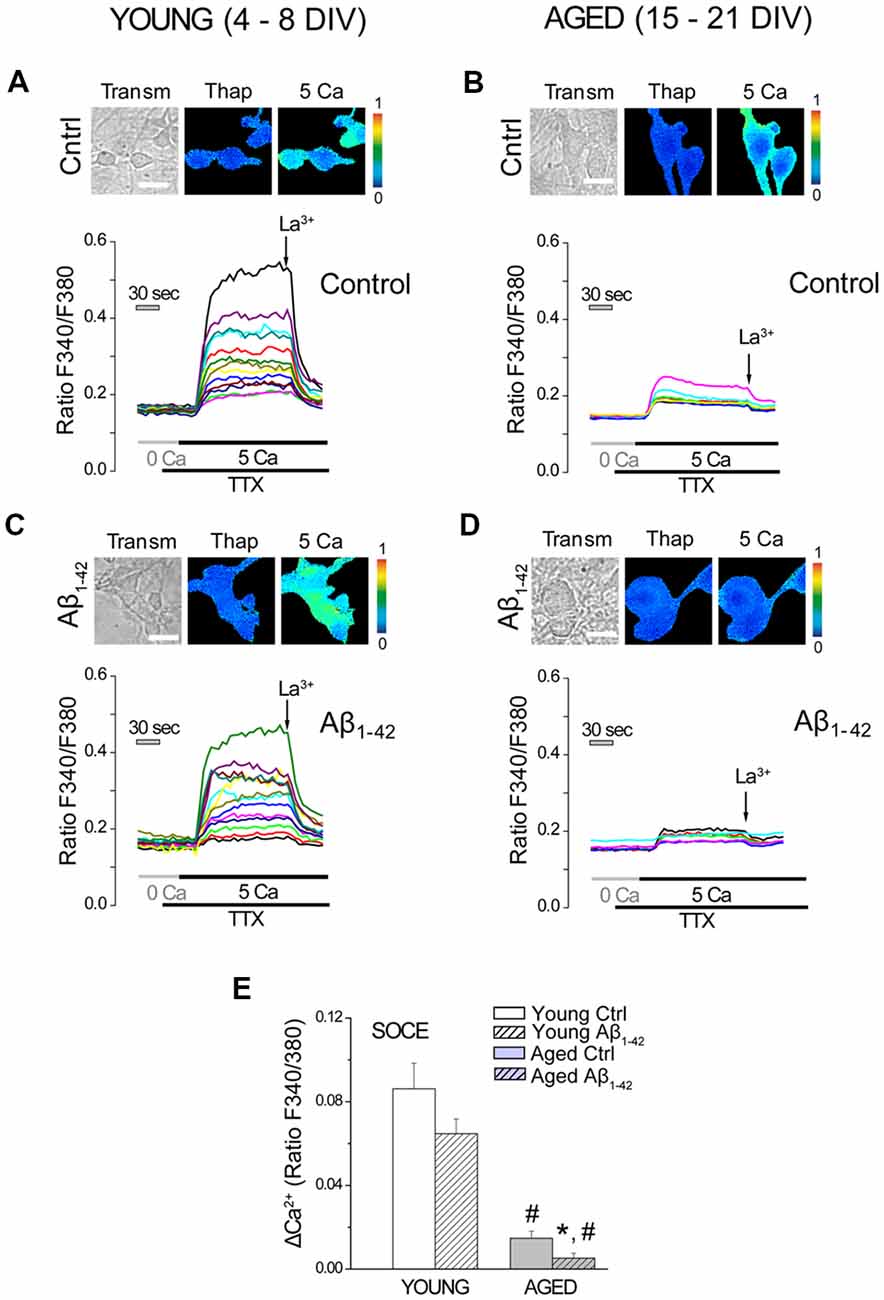
Figure 3. Store operated Ca2+ entry (SOCE) decreases in long-term cultured hippocampal neurons exposed to Aβo. Short- and long-term cultured hippocampal neurons were either exposed to vehicle (ctrl) or Aβo (1 μM, 24 h), and SOCE was monitored by fluorescence imaging of Ca2+. Neurons were incubated with fura-2 and treated with Tg (1 μM) for 15 min in medium devoid of Ca2+ (0 Ca). Subsequently, Ca2+ 5 mM in presence of TTX (500 nM) was added to elicit an increase in the [Ca2+]cyt. La3+ (100 μM) was added to block SOCE. (A–D) Pictures show typical fluorescence images of transmission (transm) and [Ca2+]cyt coded in pseudocolor of thapsigargin (Thap) and after addition of 5 Ca. Pseudocolor scale is shown at right. Bars represent 10 μm. Traces are representative recordings of fluorescence ratios of individual short-term (young, 4–8, A,C) and long-term (aged, 15–21, B,D) cultured neurons. (E) Bars correspond to the averaged SOCE (mean ± SEM). n = 65, 84, 35 and 45 individual cells studied in three and three independent experiments, respectively. *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
We have reported previously that in vitro aging increases Ca2+ store content in rat hippocampal neurons (Calvo-Rodríguez et al., 2016b). Ca2+ store content was assessed after chronic exposure to Aβo in young and aged neurons. For this end, Ca2+ store content was tested using the Ca2+ ionophore ionomycin added in medium devoid of external Ca2+. This procedure has been reported previously to estimate Ca2+ store content in living cells (Villalobos and García-Sancho, 1995; Calvo-Rodríguez et al., 2016b). Ca2+ store content was monitored by fluorescence imaging in short-term and long-term cultured hippocampal neurons, exposed to Aβo (1 μM, 24 h), and compared to the controls (exposed to the vehicle for dilution of Aβo, MEM). These experiments were always carried out in medium devoid of extracellular Ca2+ (0 Ca), in order to avoid the entry of Ca2+ from the external medium. The stimulation of hippocampal neurons with 400 nM ionomycin in Ca2+ free medium (0 Ca; Figure 4) increased [Ca2+]cyt in all neurons studied (measured as AUC). Exposure of the short-term cultured cells to Aβo did not modify the increase in [Ca2+]cyt induced by ionomycin (Figures 4A,C). However, in the long-term cultured cells, chronic exposure to Aβo for 24 h increased significantly the content of the stores, as shown by the enhanced increase in the [Ca2+]cyt induced by ionomycin in 0 Ca medium (Figures 4B,D). These results suggest that the chronic treatment of cultured hippocampal neurons with Aβo increases further Ca2+ store content exclusively in the long-term cultured neurons, an effect that could be related to the increase in the basal Ca2+ levels that were appreciated in the aged neurons exposed to the Aβo, but that does not occur in the young cultured ones.
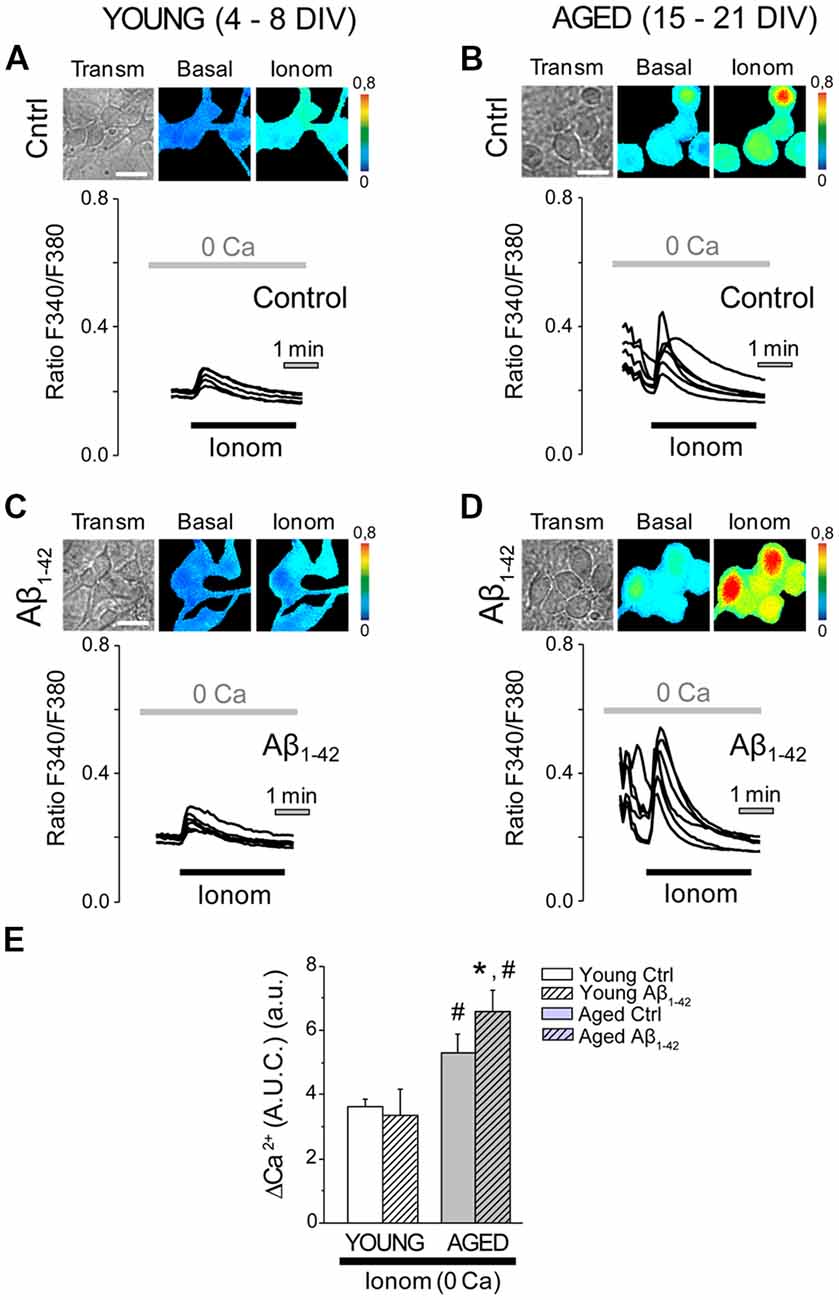
Figure 4. The exposure of aged hippocampal neurons to Aβo increases Ca2+ store content. Hippocampal neurons exposed to either Aβo (1 μM) or vehicle and loaded with fura-2 were stimulated with ionomycin (400 nm) in external medium devoid of Ca2+ (0 Ca) and subjected to fluorescence imaging to estimate the content of Ca2+ deposits. (A–D) Pictures show transmission (transm) and pseudocolor images of fluorescence ratios in basal conditions (basal) and after ionomycin (Ionom) stimulation in short-term (A,C, Young 4–8 DIV) and long-term (B,D, Aged 15–21 DIV) cultured hippocampal neurons not exposed (A,B, control) or exposed (C,D, Aβ1–42) to Aβo (1 μM, 24 h). Pseudocolor bars are shown at right. Bar represent 10 μm. Traces are representative recordings of the Ca2+ release induced by ionomycin (400 nM) in medium devoid of Ca2+ (0 Ca) in individual neurons subjected to fluorescence imaging. (E) Bars represent the average (mean ± SEM) in the increase of [Ca2+]cyt in response to ionomycin (measured as area under the curve, AUC) in short-term (young) and long-term (aged) hippocampal neurons exposed to either Aβo (1 μM, 24 h) or vehicle. n = 55, 42, 81 and 114 individual cells from three to six independent cultures, respectively. *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
In order to understand the functional consequences of the increase in the levels of Ca2+ in the intracellular stores, we then analyzed the release of Ca2+ from the ER that is mobilized by different agonists. The release of Ca2+ mediated by the IP3 receptor was first evaluated using the neurotransmitter acetylcholine (ACh). In order to cancel the contribution of the entry of Ca2+ and just focus on the release from the stores, all the experiments were carried out in the absence of free Ca2+ in the extracellular medium (0 Ca). Application of ACh to cultured hippocampal neurons in 0 Ca produced an increase in the [Ca2+]cyt, as described previously (Calvo-Rodríguez et al., 2016b). Figure 5 shows representative recordings in short-term and long-term cultured hippocampal neurons exposed to either Aβo (1 μM, 24 h) or vehicle, and subjected to fluorescence Ca2+ imaging. It can be observed that the chronic treatment with Aβo does not affect the increase in the [Ca2+]cyt induced by ACh in short-term cultured hippocampal neurons (Figures 5A,C). In contrast, chronic exposure of long-term cultured hippocampal neurons to Aβo promoted a larger [Ca2+]cyt response to ACh when compared to control cells (Figures 5B,D). Figures 5E–G show the quantification of the response. Bars represent the mean value of the [Ca2+]cyt increase, represented as AUC (AUC ± SEM) of the Ca2+ response (Figure 5E). The fraction of cells that presented an increase in the [Ca2+]cyt is also showed (Figure 5F). It can be observed that the fraction of cells that responded to the stimulation with ACh is higher in long-term cultured hippocampal neurons. In order to compare the response between young and aged neurons, a measure of global release of Ca2+ involving both parameters (Ca2+ released and fraction of responding cells) was calculated. For this end we multiplied the AUC of the cells that showed an increase in the [Ca2+]cyt by the fraction of cells that responded to the stimulus. Figure 5G shows the global difference in Ca2+ release from the stores among the four populations. Notice that whereas aging increases the fraction of neurons responsive to ACh, treatment with Aβo enhances instead the release of Ca2+ in each individual cell. Thus, aging increases the fraction of neurons responsive to ACh and Aβo enhances the rise in Ca2+ elicited by ACh in aged neurons. No effect was observed in young neurons.
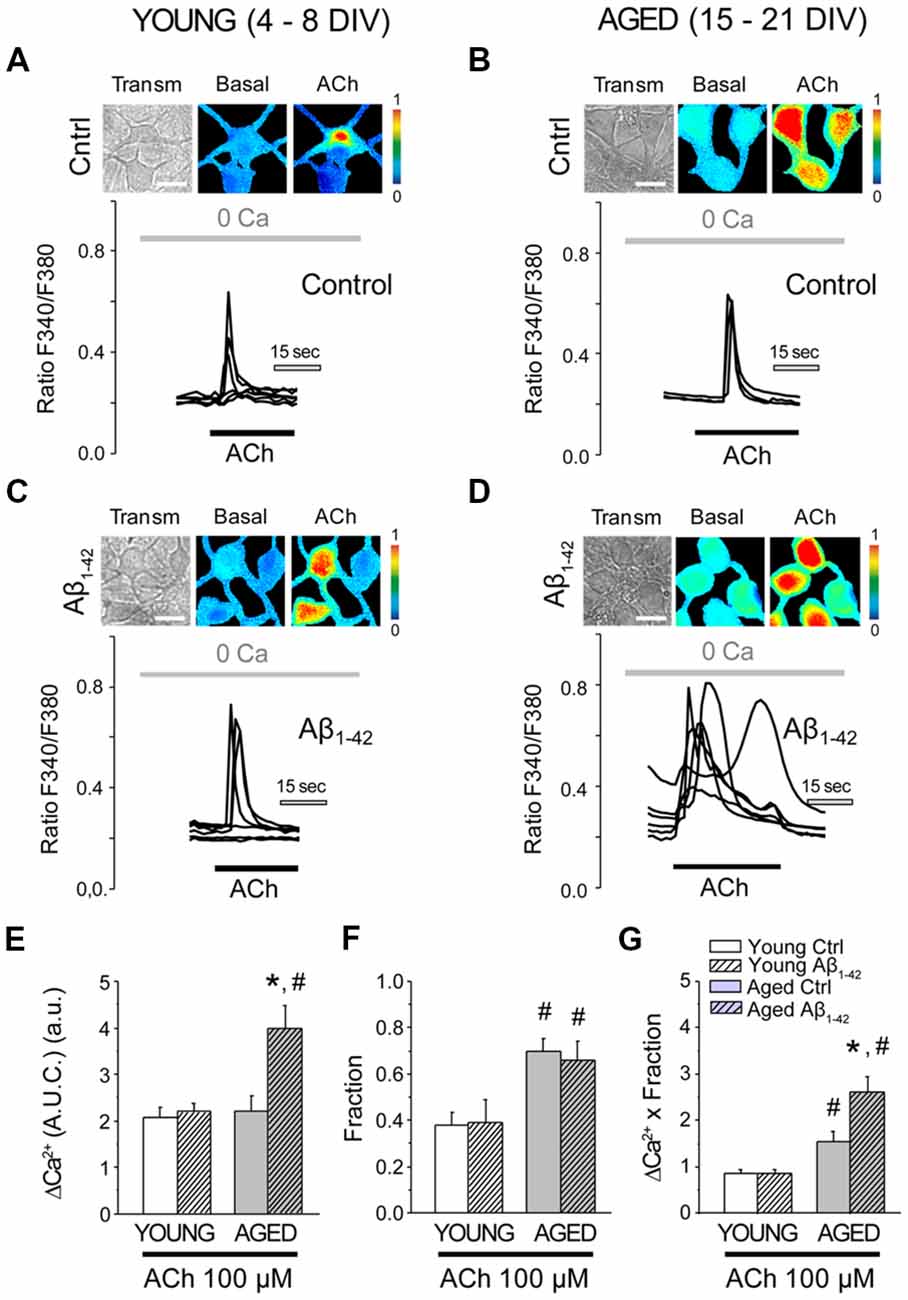
Figure 5. The exposure of aged hippocampal neurons to Aβo increases Ca2+ responses to acetylcholine (ACh) in rat hippocampal neurons. Hippocampal neurons incubated with vehicle or Aβo (1 μM, 24 h) and loaded with fura-2 were subjected to fluorescence imaging for monitoring the Ca2+ released by ACh in short-term and long-term cultured neurons. Cells were stimulated with ACh in Ca2+ free medium. (A–D) Pictures show transmission images (transm) and pseudocolor images of fluorescence ratios taken before (Basal) and after ACh stimulation. Pseudocolor scale is shown at right. Bars correspond to 10 μm. Traces represent significant fluorescence recordings in single neurons of the Ca2+ release induced by ACh (100 μM) in young and aged cultures not exposed (A,B, control) or exposed (C,D, Aβ1–42) to Aβo (1 μM, 24 h). (E–G) Bars represent the summary results for 102, 94, 39 and 59 cells from four independent cultures, respectively (mean ± SEM, AUC) of responsive cells (E), the fraction of responsive cells (F), and the product of both parameters (G). *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
Next, we assessed the effects of Aβo on Ca2+ release induced by activation of ryanodine receptors (RyRs) in short-term and long-term cultured neurons. Caffeine (Caff; 20 mM) was employed as an agonist of the RyRs. Caffeine is an agonist of the RyRs, which are sensitive to Ca2+-induced Ca2+ released (CICR) processes. In this case, nominally free Ca2+ (without adding EGTA) was used as external cellular medium to be able to activate CICR, while minimizing the effect of Ca2+ entry from the extracellular medium into the cytosol. To avoid high osmotic variation following the application of 20 mM Caff, 20 mM glucose was added to the extracellular medium prior to its addition, subsequently replaced by 20 mM Caff (Calvo-Rodríguez et al., 2016b). When hippocampal neurons were stimulated by caffeine in nominally free Ca2+, a rapid and transient increase in the [Ca2+]cyt could be observed (Figure 6). The bars shown in Figure 6E represent the mean ± SEM value of the increase in [Ca2+]cyt (quantified as AUC). ER Ca2+ mobilized by caffeine was similar in the young neurons (exposed or not exposed to 1 μM Aβo for 24 h) and in the non-exposed cultured-aged neurons. However, in cultured-aged neurons previously exposed to Aβo (1 μM, 24 h), the increase in [Ca2+]cyt was significantly higher than in the control group. Figure 6G shows the global increase (AUC Ca2+ increase × fraction) produced by stimulation with caffeine under these conditions. Therefore, Ca2+ release induced by both IP3 receptor and RyR agonists is further enhanced by Aβo treatment in aged neurons but not in young neurons.
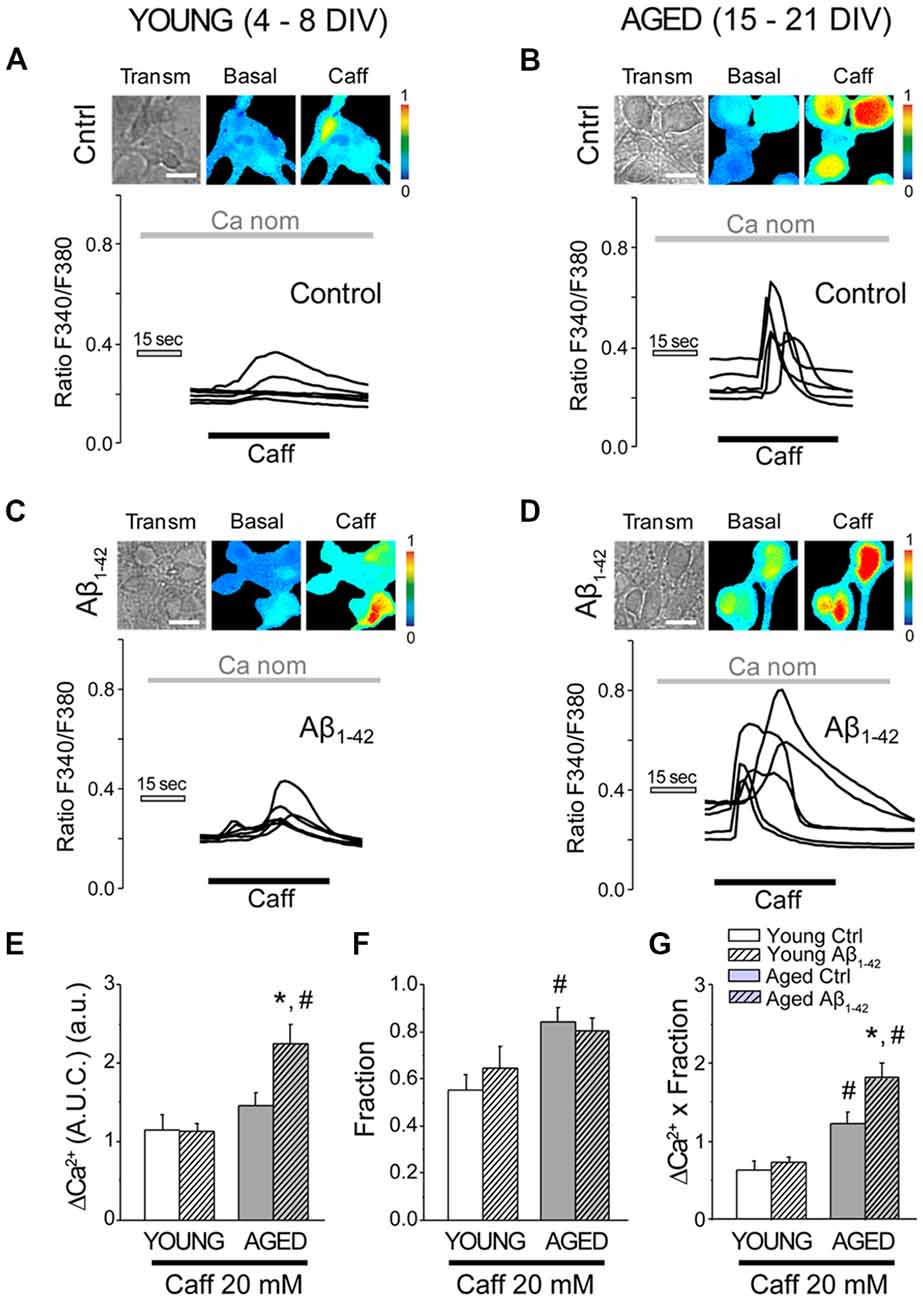
Figure 6. The exposure of aged hippocampal neurons to Aβo increases Ca2+ responses to caffeine in rat hippocampal neurons. Short-term (young, 4–8 DIV) and long-term (aged, 15–21 DIV) cultured hippocampal neurons were either exposed to vehicle or Aβo (1 μM, 24 h) and loaded with fura-2 and subjected to fluorescence imaging. Pictures show representative images of transmission (transm) and pseudocolor coded of basal and the maximum response of Ca2+ to caffeine in young (A,C) and aged cells (B,D). Pseudocolor scale is shown at right. Bars correspond to 10 μm. Traces represent single-cell recordings of young and aged cells non-exposed (ctrl) or exposed (Aβ1–42) to Aβo (1 μM, 24 h) of the Ca2+ response induced by caffeine 20 mM in medium nominal Ca2+. (E–G) Bars represent mean ± SEM of the response of [Ca2+]cyt to caffeine (expressed as AUC) of 56, 78, 94 and 105 cells from three to five independent cultures, respectively (E). Panel (F) shows the fraction of cells that responded. Panel (G) represents the overall result of the multiplication of both factors. *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
Next, we tested the effects of Aβo on ER-mitochondria cross-talk, i.e., Ca2+ transfer from ER to mitochondria. For this end, we measured the increase in mitochondrial [Ca2+] ([Ca2+]mit) induced by Ca2+ release elicited by ACh in medium devoid of Ca2+ (0 Ca), in the same manner as for Figure 5. Short-term and long-term cultured hippocampal neurons were incubated either with vehicle (Control) or Aβo (1 μM, 24 h). Then cells were washed and preincubated with the mitochondrial Ca2+ probe Rhod-5N, a fluorescent calcium probe that targets mitochondria (de la Fuente et al., 2012), and subjected to fluorescence imaging in single neurons. Neurons treated with either vehicle (control) or Aβo were perfused with the agonist ACh in medium devoid of Ca2+ (0 Ca) to induce the release of Ca2+ from the ER. Figure 7 shows bright field and Rhod-5N fluorescence images in basal conditions and after stimulation with ACh (100 μM) in short-term (4–8 DIV) and long-term (15–21 DIV) cultured hippocampal neurons. Representative recordings of [Ca2+]mit obtained in single cells before and after stimulation with ACh are also shown. Bars represent the quantification of the rises in [Ca2+]mit induced by ACh in 0 Ca (left), the fraction of responsive cells (middle) and the global response by taking into account both factors (right). As shown previously using mitochondria-targeted aequorin (Calvo-Rodríguez et al., 2016b), the rise in [Ca2+]mit induced by ACh is larger in aged neurons than in young neurons. Therefore, these results confirm our previous results and validate monitoring of [Ca2+]mit with Rhod-5N. The larger rise in [Ca2+]mit in aged neurons is due to both the increase in mitochondrial Ca2+ uptake in every cell (left panel) and the fraction of responsive cells (middle panel) corresponding to a large global ER-mitochondria cross talk in aged neurons relative to young neurons (right panel; Figure 7C). Interestingly, Aβo increased the rise in [Ca2+]mit induced by ACh in young neurons (left panel) and this effect was not due to changes in the fraction of responsive cells (middle panel). Therefore, Aβo increase ER-cross talk in young neurons (right panel, Figure 7C). It is noteworthy that this effect cannot be due to increased ER Ca2+ content (Figure 4E) and is the only effect induced by Aβo that we have observed in young neurons.
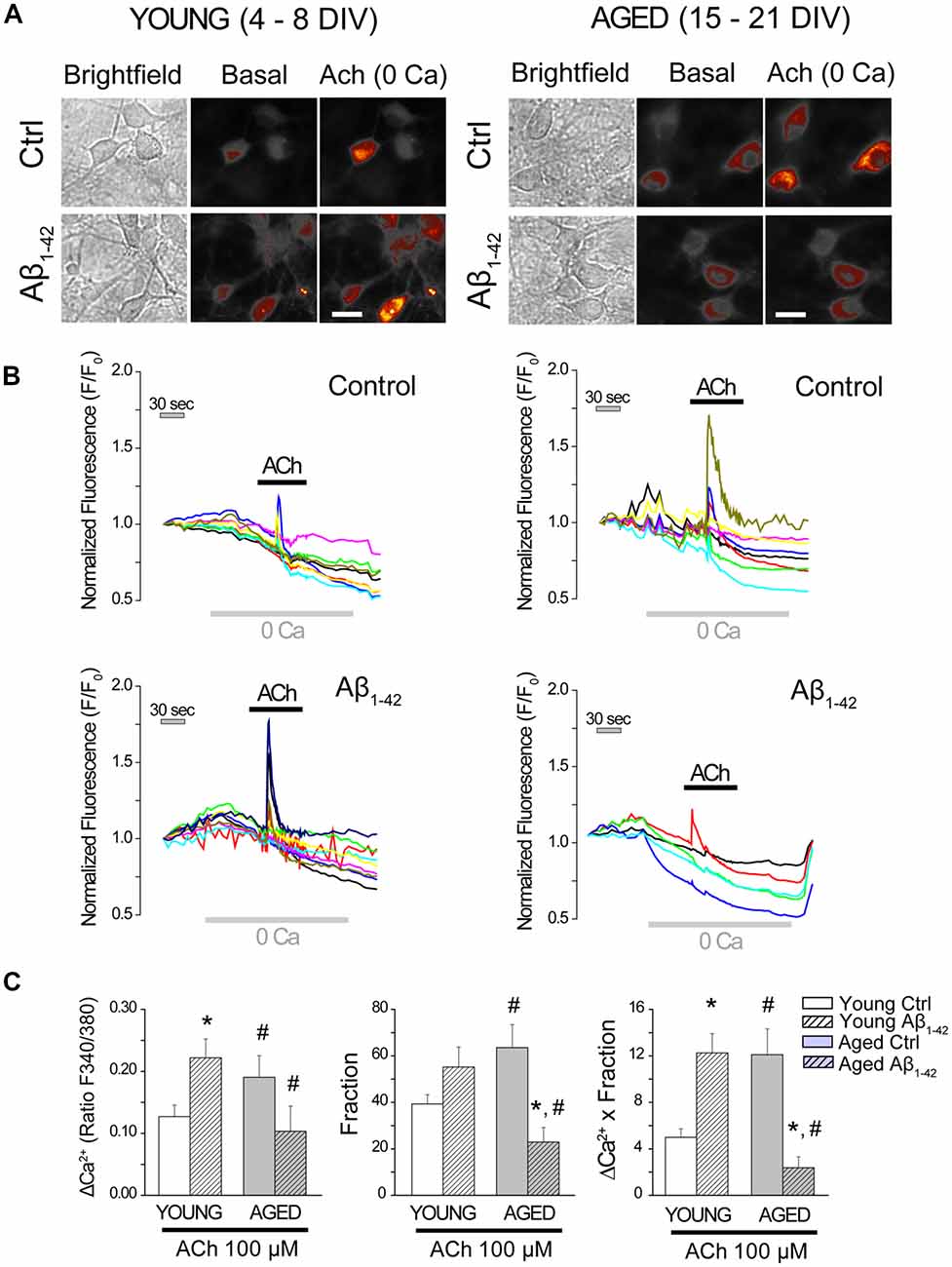
Figure 7. The exposure of hippocampal neurons to Aβo affects the cross-talk endoplasmic reticulum (ER)–mitochondria. Hippocampal neurons cultured for 4–8 (young) and 15–21 (aged) DIV were incubated with Rhod-5N (1 μM) and subjected to fluorescence imaging to measure [Ca2+]mit in single cell. Cells exposed to either vehicle (control) or Aβo (1 μM, 24 h) were stimulated with the agonist ACh in medium devoid of Ca2+ (0 Ca). (A) Pictures show typical fluorescence images of transmission (transm) and Rhod-5N fluorescence in basal conditions of after stimulation with ACh (100 μM) in medium 0 Ca of short and long-term cultured hippocampal neurons. The bars represent 10 μm. (B) Traces represent characteristic response of [Ca2+]mit in single cells by stimulating with ACh (100 μM) in medium 0 Ca. (C) Bars represent mean ± SEM of the increase in [Ca2+]mit induced by ACh in 0 Ca, the fraction of response and the global response by taking into account both factors. The global response to ACh is significantly higher in the young neurons exposed to Aβo (1 μM, 24 h) and significantly smaller in the exposed aged ones. n = 44, 66, 29 and 31 individual cells studied in three and three independent experiments, respectively. *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
It is also remarkable that the effects of Aβo on the rise in [Ca2+]mit induced by ACh in young and aged neurons are opposite. Aβo increases Ca2+ transfer from ER to mitochondria in young neurons and decreases it in aged neurons. In fact, Aβo nearly abolishes the rise in [Ca2+]mit induced by ACh in aged neurons and this effect is due to decreases in both the amount of Ca2+ transferred per neuron and the fraction of neurons responsive to ACh. Taken together, these results show that Aβo significantly increases ER-mitochondria cross-talk in young neurons, but they rather abolish it in aged neurons.
Ca2+ enters the mitochondria through the MCU, located into the inner mitochondrial membrane (Baughman et al., 2011; De Stefani et al., 2011). To address the opposite effects of Aβo on mitochondrial Ca2+ uptake in young and aged neurons, we also tested the effects of Aβo on MCU expression. Short-term and long-term cultured neurons were incubated either with vehicle (ctrl) or Aβo (1 μM, 24 h). Then, immunostaining against MCU was evaluated by immunofluorescence. Figure 8 shows representative images of the fluorescence detection of MCU in short-term (young) and long-term (aged) cultured hippocampal neurons and the quantitative analysis of fluorescence intensity (optical density, arbitrary units) of the MCU. As reported previously (Calvo-Rodríguez et al., 2016b), we found that optical density of MCU immunofluorescence is enhanced in aged neurons relative to young neurons, thus confirming our previous results. In addition, we found that Aβo significantly increased the expression of MCU in the long-term cultured hippocampal neurons but not in young neurons. These results indicate that the opposite effects of Aβo on ER-mitochondria cross talking in young and aged neurons are not due to changes in expression of MCU.
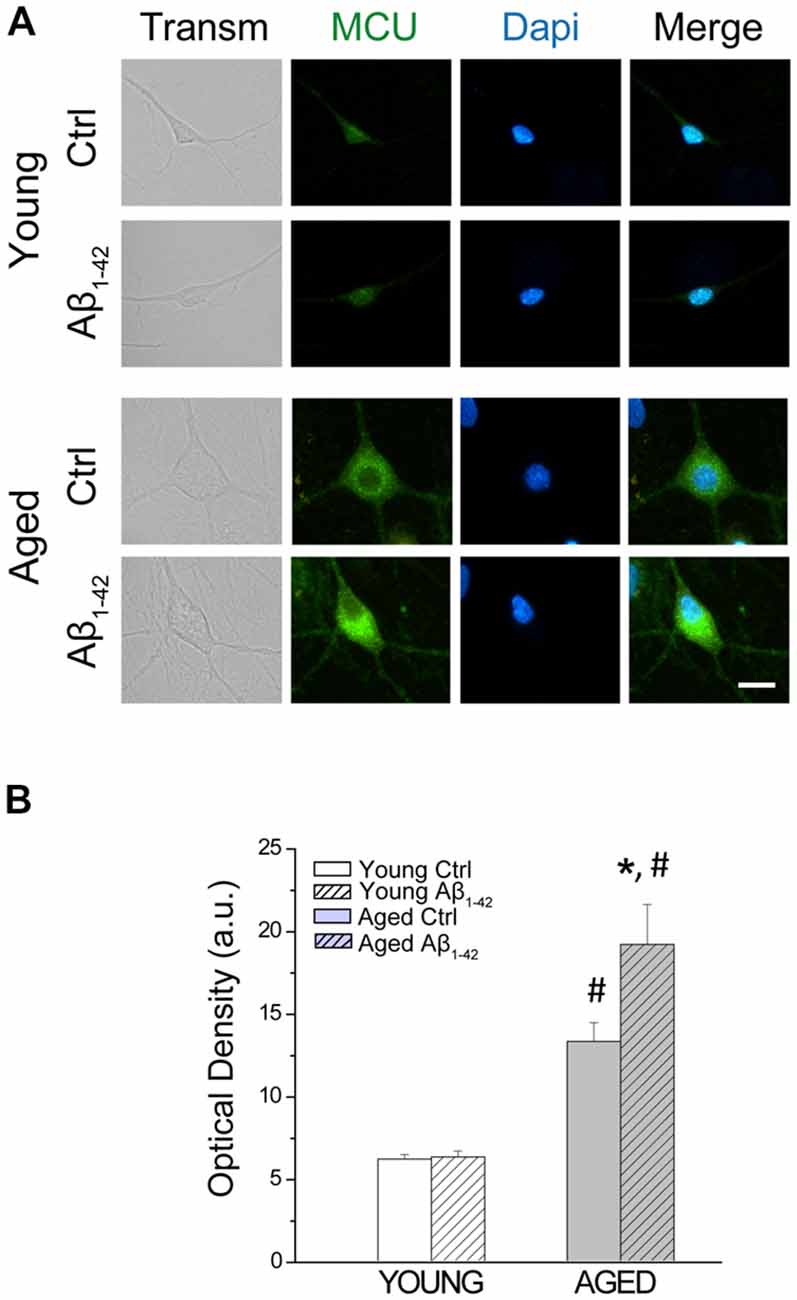
Figure 8. The exposure of aged hippocampal neurons to Aβo increases the expression of the mitochondrial Ca2+ uniporter (MCU). Immunostaining against MCU was assessed in hippocampal neurons incubated either with vehicle (ctrl) or Aβo (1 μM, 24 h). (A) Representative images of the fluorescence detection of MCU in short-term (young) and long-term (aged) cultured hippocampal neurons. Bar represents 10 μm and applies to all images. (B) Quantitative analysis of fluorescence intensity (optical density, arbitrary units) of the MCU. Bars represent average ± SEM. n = 112, 81, 90 and 59 cells respectively from three independent cultures. *p < 0.05 compared to control group. #p < 0.05 compared to the young ones.
Ca2+ release from intracellular stores takes place after activation of IP3 receptors at the ER. In addition, IP3 receptors are an ER component of the MAMs (Hedskog et al., 2013). Therefore, these receptors are critical to the coupling between ER and mitochondria. Moreover, IP3 receptors have been involved in the enhanced cytosolic [Ca2+] induced by oligomers in AD (Ferreiro et al., 2004; Demuro and Parker, 2013). Accordingly, we tested whether expression of the three IP3 receptor isoforms (IP3R1, IP3R2 and IP3R3) changes during in vitro aging. Figure 9A shows representative immunofluorescence images of all three IP3 receptor isoforms obtained in short-term (young) and long-term (aged) cultured hippocampal neurons. Figure 9B shows the quantitative analysis of fluorescence intensity (optical density, arbitrary units) corresponding to expression of all three IP3R isoforms in young and aged neurons. Results show that optical density of all three IP3R isoforms is significantly increased in aged neurons relative to young neurons. These results indicate that changes in expression of IP3 receptors may contribute to enhanced response to Aβo in aged neurons and enhanced ER-mitochondria cross talking in aging.
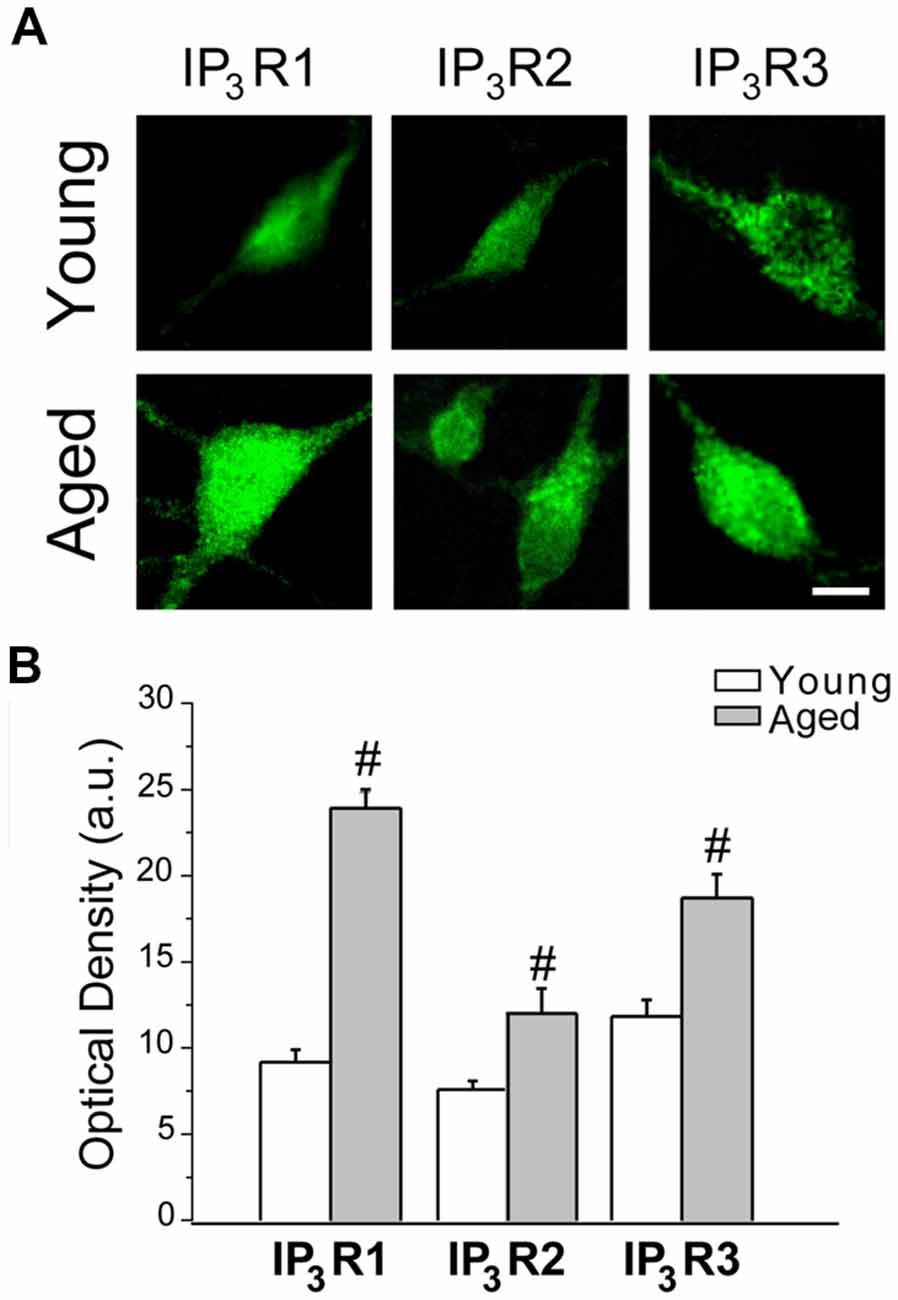
Figure 9. IP3 receptor expression increases in aged rat hippocampal neurons. Immunostaining against IP3R1, IP3R2 and IP3R3 was assessed in short-term and long-term cultured hippocampal neurons representing young and aged neurons. (A) Representative images of the fluorescence detection of the three isoforms in short-term (young) and long-term (aged) cultured hippocampal neurons. Bar represents 10 μm and applies to all images. (B) Quantitative analysis of fluorescence intensity (optical density, arbitrary units) of the three IP3R isoforms. Bars represent average ± SEM. n = 34, 15, 39, 4, 29 and 14 cells, respectively from three independent cultures. #p < 0.05 aged vs. young neurons.
We investigated ER-mitochondria colocalization using confocal fluorescence imaging of young and aged rat hippocampal neurons loaded with both MitoTracker green and ER-Tracker red. Figure 10A shows representative confocal images of signals in the green and the red channels of hippocampal neurons treated with vehicle or Aβo 1 μM for 24 h. For colocalization analysis of ER and mitochondria, we computed the Manders2 and Pearson coefficients. Figure 10B shows Manders2 and Pearson coefficients of colocalization of ER and mitochondria. Results show that colocalization coefficients increase significantly in long-term cultured neurons corresponding to aged neurons relative to short-term cultured neurons representing young neurons. In addition, coefficients are further enhanced in neurons treated with Aβo. These results indicate that both aging and Aβo enhance ER-mitochondria colocalization.
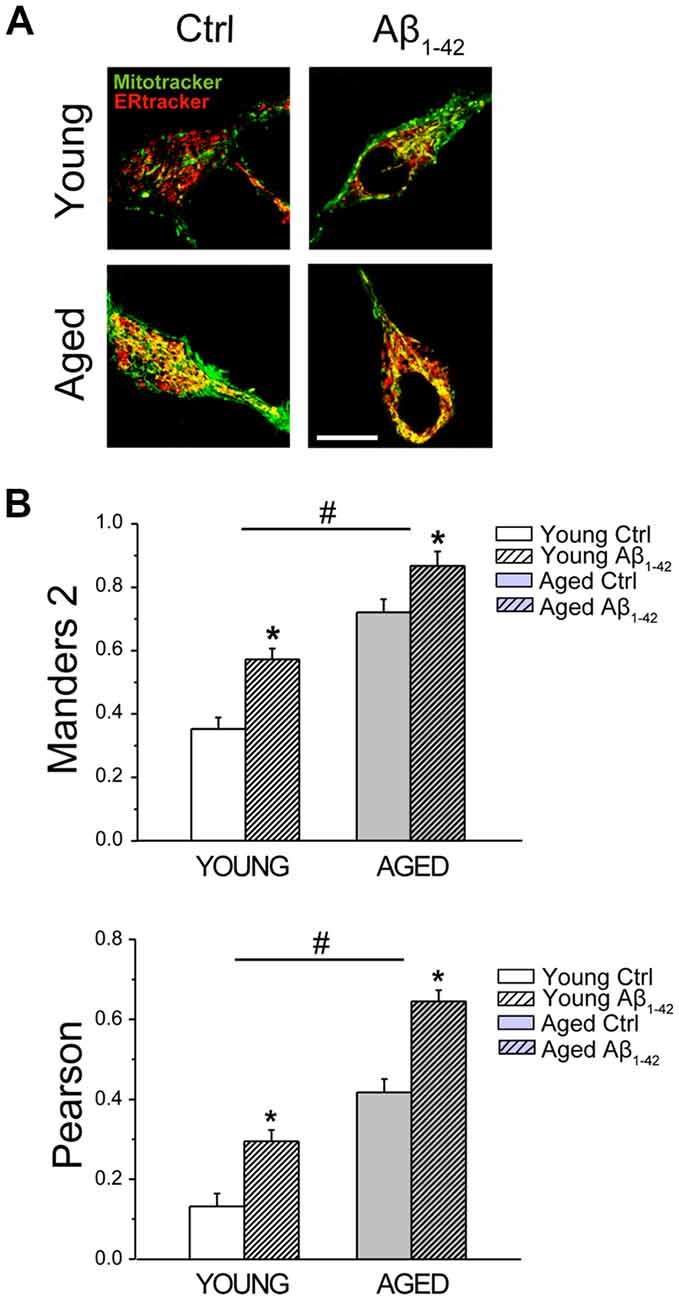
Figure 10. Aging and Aβo increase ER–mitochondria colocalization in rat hippocampal neurons. Short-term (young) and long-term (aged) hippocampal cultures were exposed for 24 h to Aβo (1 μM) or vehicle. Then, cells were incubated with mito-tracker green and ER-tracker red and mitochondrial—ER colocalization was assessed by confocal fluorescence imaging. (A) Pictures show representative confocal fluorescence images of short-term (young) and long-term (aged) cultures of rat hippocampal neurons treated with vehicle (Ctrl) or Aβ1–42 oligomers. Bar represents 10 μm and applies to all images. (B) Bars represent Manders2 and Pearson colocalization coefficients (mean ± SEM) obtained in ROIs corresponding to identified young and aged neurons treated with vehicle (Cntrl) or Aβ1–42. Data corresponds to three experiments. *p < 0.05 vs. control group. #p < 0.05 vs. young neurons.
The above results may contribute to explain how Aβo increase Ca2+ transfer from ER to mitochondria in young neurons (Figure 7). However, they cannot explain why this transfer is rather impaired in aged neurons treated with Aβo. To address this issue, we investigated next the effects of aging and Aβo on mitochondrial membrane potential and ROS generation in rat hippocampal neurons.
We have shown previously that in vitro aging is associated to a loss of mitochondrial membrane potential in rat hippocampal neurons (Calvo-Rodríguez et al., 2016b). Here, we tested whether Aβo influence also mitochondrial membrane potential. For this end, short-term and long-term cultures of rat hippocampal neurons corresponding to young and aged neurons, respectively, were treated with vehicle or Aβo (1 μM, 24 h). Then, cells were loaded with the mitochondrial potential probe TMRM and subjected to fluorescence imaging. Figure 11A shows representative TMRM fluorescence images of young and aged neurons treated with vehicle or Aβo. Bars in Figure 11B show average (mean ± SEM) optical density values of ROIs corresponding to hippocampal neurons. Results confirm that mitochondrial membrane potential decreases in aged neurons relative to young ones. In addition, the results show that Aβo decrease it further in young and aged neurons. The effect of Aβo on mitochondrial membrane potential is relatively larger in aged neurons than in young ones. The loss of mitochondrial membrane potential favors PTPm, a process that is also enhanced by ROS generation. Accordingly, we investigated next the generation of ROS in young and aged neurons either treated or not with Aβo.
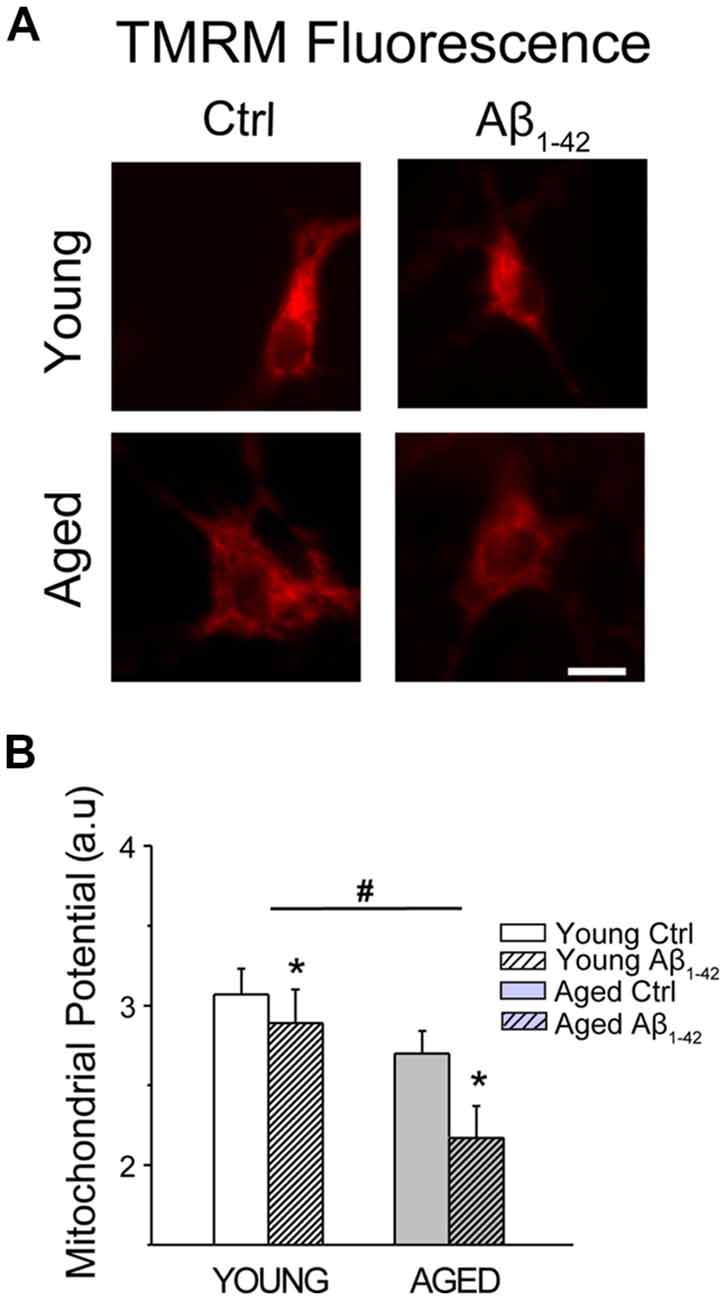
Figure 11. Aging and Aβo decrease mitochondrial membrane potential in rat hippocampal neurons. Short-term (young) and long-term (aged) hippocampal cultures were exposed for 24 h to Aβo (1 μM) or vehicle. Then, cells were incubated with TMRM and mitochondrial membrane potential was assessed by fluorescence imaging. (A) Pictures show representative fluorescence images of short-term (young) and long-term (aged) cultures of rat hippocampal treated with vehicle (Ctrl) or Aβ1–42 oligomers. Scale bar represents 10 μm and applies to all images. (B) Bars represent mean ± SEM values of TMRM fluorescence intensity in selected ROIs corresponding to identified young and aged neurons treated with vehicle (Cntrl) or Aβo. Data correspond to three independent experiments. *p < 0.05 vs. control group. #p < 0.05 vs. young neurons.
We tested whether aging and Aβo influence also ROS generation in rat hippocampal neurons. For this end, short-term and long-term cultures of rat hippocampal neurons representing young and aged neurons, respectively, were treated with vehicle or Aβo (1 μM, 24 h). Then cells were loaded with the ROS probe CM-H2DCFDA and subjected to fluorescence imaging. Figure 12A shows representative fluorescence images of young and aged neurons treated with vehicle or Aβo. Bars in Figure 12B show average (mean ± SEM) optical density values for ROIs corresponding to hippocampal neurons. The results show that aging increases significantly CM-H2DCFDA fluorescence consistently with enhanced ROS generation in aged neurons relative to young ones. In addition, we found that Aβo increase ROS generation further in aged neurons, having no effect in the young ones. These results are consistent with detrimental effects of Aβo in aged neurons but not in young ones.
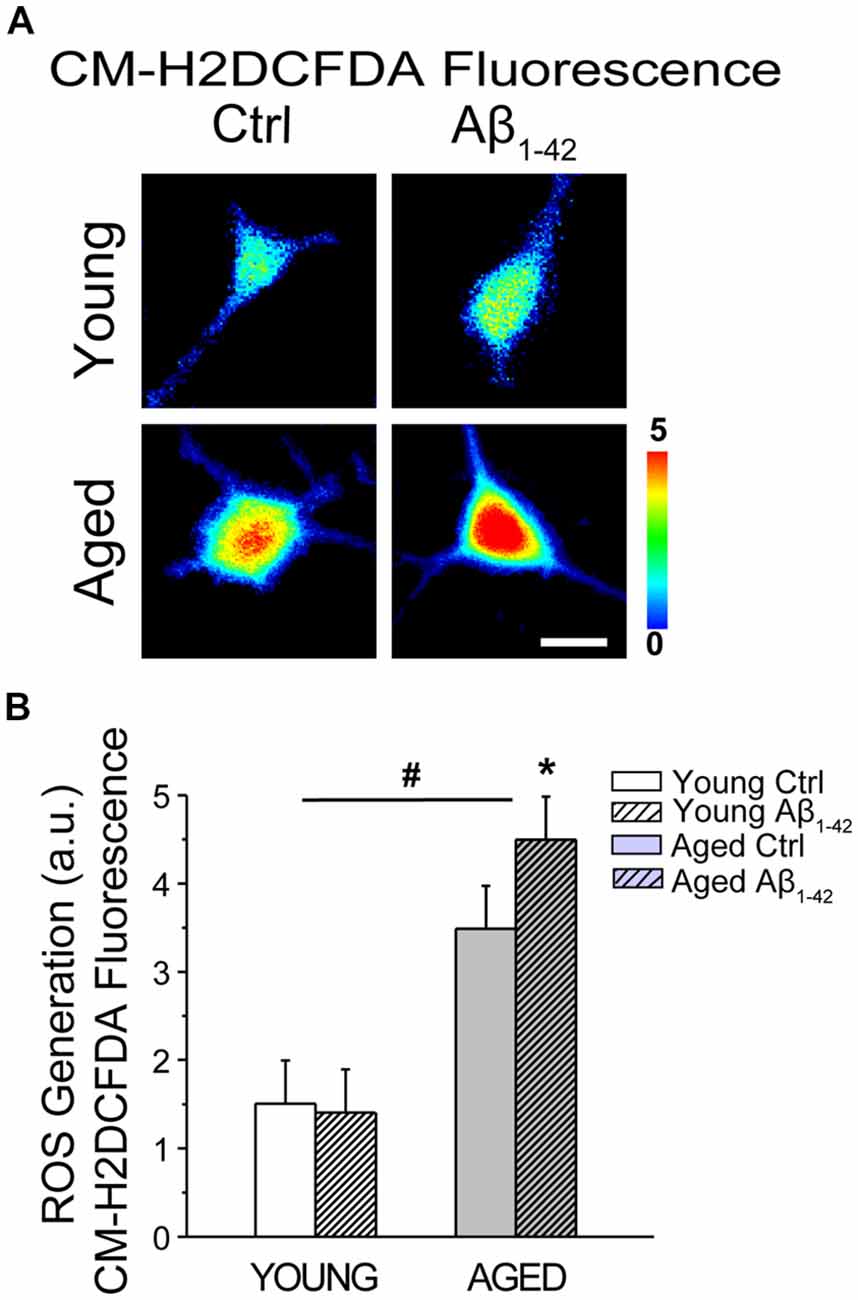
Figure 12. Aging and Aβo enhance reactive oxygen species (ROS) generation in rat hippocampal neurons. Short-term (young) and long-term (aged) hippocampal cultures were exposed for 24 h to Aβo (1 μM) or vehicle. Then, cells were incubated with CM-H2DCFDA and ROS generation was assessed by fluorescence imaging. (A) Pictures show representative CM-H2DCFDA fluorescence images coded in pseudocolor of short-term (young) and long-term (aged) cultures of rat hippocampal treated with vehicle (Ctrl) or Aβ1–42 oligomers. Scale bar represents 10 μm and applies to all images. (B) Bars represent mean ± SEM values of CM-H2DCFDA fluorescence intensity in selected regions of interest (ROIs) corresponding to identified young and aged neurons treated with vehicle (Control) or Aβo. Data corresponds to three experiments. *p < 0.05 vs. control group. #p < 0.05 vs. young neurons.
Accordingly, the present results show that Aβo enhance ER-mitochondrial coupling in short-term cultured hippocampal neurons corresponding to young neurons but this is not associated to a detrimental effect. In contrast, in long-term cultured hippocampal neurons, corresponding to aged neurons, Aβo also increases ER-mitochondrial colocalization, but this effect is associated to loss of ER-mitochondrial coupling and detrimental effect (Figure 13).
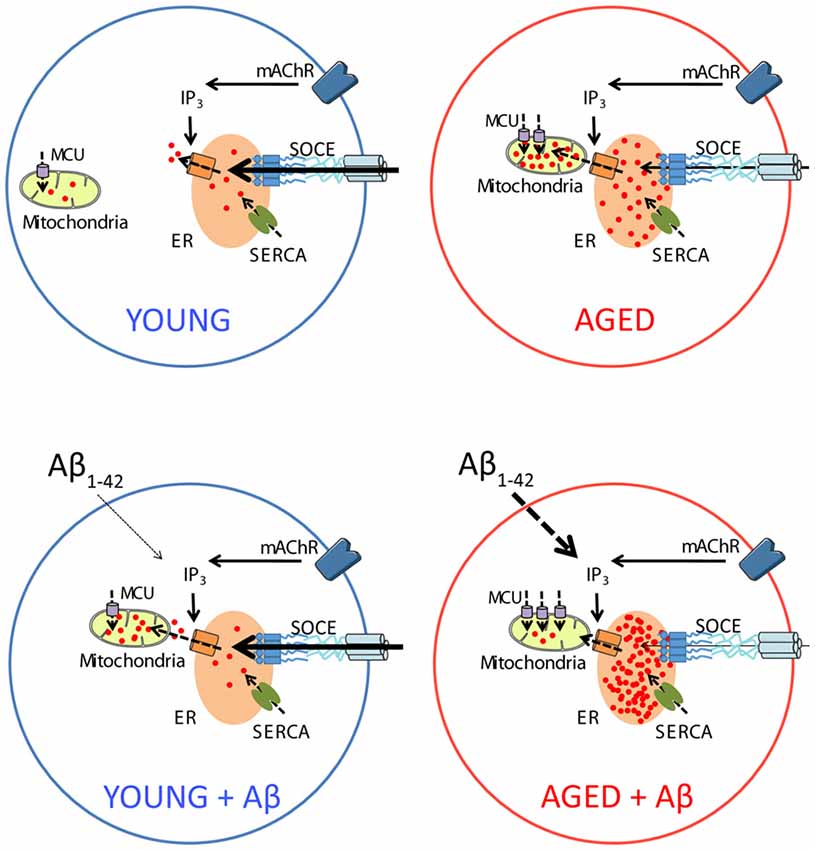
Figure 13. Schematic representing the effects of Aβo on intracellular Ca2+ homeostasis in young and aged neurons in vitro. In young neurons (top left), activation of metabotropic ACh receptors induce IP3 synthesis and release of Ca2+ from intracellular stores in the ER. Depletion of Ca2+ stores activate SOCE that rise intracellular Ca2+ to refill Ca2+ stores after activation of the sarcoplasmic and ER Ca2+ ATPase (SERCA). Ca2+ release is also taken by mitochondria through the MCU for activation of mitochondrial metabolism. Treatment with Aβo increases Ca2+ transfer from ER to mitochondria in young neurons without altering any other parameter or promoting apoptosis (bottom left). Aging increases Ca2+ store content at the ER, MCU expression and Ca2+ transfer from ER to mitochondria but decreases SOCE (top right). Treatment of aged neurons with Aβo exacerbates the increases in Ca2+ store content and MCU expression as well as SOCE loss. However, in contrast to young neurons, treatment with Aβo decreases Ca2+ transfer from ER to mitochondria in aged neurons and promotes apoptosis (bottom right).
Signaling by Ca2+ plays a fundamental role in learning and memory processes, and participates in survival and neuronal death. Intracellular Ca2+ dyshomeostasis has been extensively related to many neurodegenerative diseases, including AD (LaFerla, 2002; Bezprozvanny and Mattson, 2008). Ca2+ pathology in AD involves altered Ca2+ influx processes as well as Ca2+ release from the internal stores and impaired Ca2+ buffering capacity. Furthermore, sustained up-regulation of Ca2+ levels could both initiate and accelerate the features of AD, including plaque deposition and synaptic loss (Stutzmann, 2007), although if Ca2+ dysregulation is a cause or a consequence of AD is still under debate.
Here, we show that chronic exposure to Aβo exacerbates largely the remodeling of subcellular Ca2+ associated to in vitro aging in cultured hippocampal neurons. First, we show that Aβo increase further basal [Ca2+]cyt and spontaneous and synchronous [Ca2+]cyt oscillations. These results are consistent with previous reports in vivo showing that brains of AD patients exhibit increased [Ca2+]cyt levels (Murray et al., 1992). Moreover, recent in vivo studies using intravital Ca2+ imaging of APP/PS1 transgenic mouse, a mouse model of familial AD, show that basal [Ca2+]cyt levels are elevated significantly in approximately 35% of neurites located in areas in close proximity to amyloid plaques (Kuchibhotla et al., 2008). A possible explanation for the increase in the basal [Ca2+]cyt levels is the ability of the Aβo to generate ROS, disturbing the function of the membrane ATPases and, thus leading to an increase in the resting [Ca2+]cyt level (Mark et al., 1995). Previous studies have shown that the increase in the basal [Ca2+]cyt relates to the increase in neuronal excitability and, therefore, with circuit connectivity (Catterall and Few, 2008). Therefore, it could be also speculated that chronic treatment with Aβo increases the excitability of the circuit, as shown by our own in vitro experiments, and previously by other groups that use in vivo imaging in AD mouse models (Busche et al., 2008, 2012; Šišková et al., 2014). Interestingly, the risk of epileptic activity is particularly high in AD patients with early-onset dementia and during early stages of the disease, which a much higher incidence of crises compared to the reference population of the same age (Amatniek et al., 2006).
SOCE, also called capacitative Ca2+ entry, is a Ca2+ influx pathway activated after the release of Ca2+ from the ER in order to replenish intracellular stores that subserves many different cell and physiological functions in a large variety of cell types (Parekh and Putney, 2005). In neurons, SOCE is involved in synaptic processes as well as neuronal excitability (Moccia et al., 2015). In addition, it has been described to be extensively altered in neurodegenerative disorders (Secondo et al., 2018). The components that participate in SOCE are also dysregulated in neurodegenerative disorders. We have previously shown that SOCE decreases with in vitro aging, and this effect is at least partially due to downregulation of Stim1 and Orai1 (Calvo-Rodríguez et al., 2016b). Here, we show that exposure of cultured hippocampal neurons to Aβo has a further detrimental effect on SOCE in the aged neurons without having significant effects in young neurons. Previous work by Tong et al. (2016) using cultured hippocampal neurons expressing mutant PS1 proposed that mutations in PS1 enhance the activity of γ-secretase, thus increasing the cleavage of Stim1 and therefore reducing the activation of Orai1, and SOCE. These effects disrupted dendritic spines arguing that PS1 mutations could contribute to memory loss through SOCE dysregulation. Consistently, treatment of AD mouse models with the SOCE positive modulator NSN21778 stimulates SOCE in the spines and rescues hippocampal long-term potentiation impairment (Zhang et al., 2016). In addition, inasmuch as SOCE contributes to ER Ca2+ refilling, it could be reasoned that SOCE downregulation in the aged neurons, and in aged neurons exposed to Aβo, is a compensatory effect to the enhanced filling state of intracellular stores in aging and Aβo treated neurons. In any case, SOCE is strongly downregulated in aged neurons and this process is further exacerbated by Aβo exposure. Interestingly, this effect of Aβo is only statistically significant in aging neurons suggesting that a permissive mechanism associated to aging is involved in the mechanism linking Aβo to SOCE downregulation.
PSs are integral proteins located in the ER membrane that may form low conductivity channels in the ER, known as leak channels. These channels may release Ca2+ in a passive and constant manner from the ER into the cytoplasm (Tu et al., 2006). Work on transgenic mice has shown that mutations in these proteins, as it occurs in familial AD, result in loss of function of the leak channel activity, causing an increase in the Ca2+ level on the intracellular stores (Nelson et al., 2007). We have previously shown that Ca2+ store content is significantly increased in aged hippocampal neurons (Calvo-Rodríguez et al., 2016b). Our present data confirm these previous results. In addition, our results show that the chronic treatment of hippocampal neurons in vitro with the toxic species of AD, Aβo, increases Ca2+ store content further, thus proving that chronic exposure to Aβo causes a similar effect than mutations in PS. These results are supported by previous experiments in PC12 cells exposed to chronic treatment (24 h) with Aβ1–42, where an excess release of Ca2+ from intracellular stores was shown after stimulation with thapsigargin in free calcium media (Pannaccione et al., 2012). This effect is only observed in long-term cultured hippocampal neurons exposed to Aβo, proving the resistance of the short-term cultured ones to the effects of Aβo. Interestingly, a similar tendency has recently been shown by Lerdkrai et al. (2018). These authors found significant differences in the filling state of the intracellular Ca2+ stores in AD compared with WT mice in vivo, mainly in hyperactive AD cells. In accordance with this line, the chronic exposure of cultured hippocampal neurons to the Aβo notably increases the peak of [Ca2+]cyt induced by both ACh and Caff. This is again pointing to an increased concentration of Ca2+ in the ER after exposure to Aβo. Consistently, previous studies in fibroblast from AD patients (Gibson et al., 1996) or in cells carrying the mutated human PS1 (Stutzmann, 2007), showed an abnormal Ca2+ release through IP3Rs. In addition, aberrant Ca2+ increases mediated by IP3 in fibroblasts of asymptomatic members of families with AD have also been described (Etcheberrigaray et al., 1998). Other studies have also implicated the RyRs as responsible for this increased release of intracellular Ca2+ in animals carrying the PS1 mutation (Chan et al., 2000). Also, Aβ1–42 peptides could be responsible for an increase of the isoform RyR3 in transgenic mouse models of AD (Supnet et al., 2006), influencing the amount of Ca2+ that is released from the stores when they are activated. Therefore, Ca2+ store content is increased in aged neurons and this process is further enhanced by Aβo. Again, Aβo do not influence Ca2+ store content in young neurons.
Mitochondria play also an important role in intracellular Ca2+ homeostasis by shaping neuronal Ca2+ signaling. Mitochondria and ER are structurally and functionally connected in contact points enriched in MAMs (Csordás and Hajnóczky, 2009; Hayashi et al., 2009). MAMs play different roles in cholesterol ester synthesis, phospholipid transport and Ca2+ transfer from ER to mitochondria. It has been previously shown that MAM function is altered in the pathology of AD. Area-Gomez et al. (2012) showed a few years ago that there is an enhanced ER-mitochondria connectivity in AD patients of sporadic and familial AD. Consistently, we reported recently that in vitro aging is associated to enhanced Ca2+ transfer from ER to mitochondria in rat hippocampal neurons (Calvo-Rodríguez et al., 2016b). This view was supported by two findings. First, the increased rise in mitochondrial [Ca2+] induced by ACh in Ca2+ free medium in aged neurons compared to young neurons. Second, the increased rise in [Ca2+]cyt induced by ACh in the presence of the mitochondrial uncoupler FCCP in aged neurons compared to young neurons (Calvo-Rodríguez et al., 2016b). Our present results confirm these previous findings that were carried out using aequorin targeted to mitochondria using an alternative probe for mitochondrial Ca2+, Rhod-5N.
Interestingly, we show now that Aβo enhance Ca2+ transfer from mitochondria to ER in young neurons. This view is supported by the finding that Aβo enhance the rise in [Ca2+]mit elicited by Ca2+ release induced by ACh without increasing either the resting levels of [Ca2+]cyt, the Ca2+ store content or the fraction of cells responsive to ACh. This effect is also independent of changes in the expression of MCU, the calcium channel involved in mitochondrial Ca2+ uptake. Thus, the most likely explanation for our results is that Aβo oligomers reinforce the relative position and/or coupling between ER and mitochondria. Consistently with this view, it has been shown that Aβ peptide increases inositol-1,4,5-triphosphate receptor and voltage-dependent anion channel protein expression and elevates the number of ER-mitochondria contact points and mitochondrial calcium concentrations in cultured hippocampal neurons (Hedskog et al., 2013). Unfortunately, the time in culture of these neurons was not reported. In these lines, we show here that expression of all three IP3 receptor isoforms, including IP3R1, IP3R2, and IP3R3, is increased in aged neurons, which may enhance ER-mitochondrial coupling in aging. In addition, confocal imaging of living neurons and colocalization analysis indicated increased ER-mitochondria colocalization in aged neurons relative to young neurons. Moreover, colocalization was intensified by Aβo in both young and aged neurons. The effects of Aβo on ER—mitochondria coupling in young neurons are not necessarily detrimental since they may favor increased Ca2+ transfer between ER and mitochondria to support increased energy demands and Aβo do not induce apoptosis in young neurons, suggesting a physiological role for Aβo in the modulation of ER—mitochondria coupling.
Surprisingly, and in striking contrast with the results in young neurons, exposure of aged neurons to Aβo decreased rather than increased the rise in [Ca2+]mit induced by ACh. These results cannot be explained by lack of ACh receptors as [Ca2+]cyt responses to ACh are indeed enhanced in aged neurons treated with Aβo. They cannot be explained either by depletion of Ca2+ stores since Ca2+ stores are actually enhanced in aged neurons treated with Aβo. Finally, lack of Ca2+ transfer from ER to mitochondria in aged neurons treated with Aβo cannot be attributed either to MCU downregulation as MCU expression is actually enhanced in aged neurons treated with Aβo. These results suggest that, contrary to our expectations, Aβo nearly abolish ER—mitochondria cross talk in aged neurons and this effect, together with exacerbated remodeling of intracellular Ca2+ induced by aging, is highly detrimental as Aβo induces apoptosis to a large fraction of aged neurons.
It is well established that an adequate flux of Ca2+ transfer from ER to mitochondria is required to maintain the Krebs cycle (Cárdenas et al., 2010). Thus, loss of Ca2+ transfer from ER to mitochondria in aged neurons treated with Aβo may compromise OXPHOS activity leading to mitochondrial damage and collapse of the mitochondrial potential. However, excess of Ca2+ transfer associated to enhanced Ca2+ store content may lead to mitochondrial Ca2+ overload.
Several studies by others have proposed that mitochondrial dysfunction is an early event the pathology of AD. Yao et al. (2009) described mitochondrial bioenergetics deficits before plaque deposition in females in a mouse model of AD. By using Thy-1 APP mice, an AD model that deposits plaques at 6 months of age, Hauptmann et al. (2009) showed mitochondrial dysfunction associated with higher levels of ROS, including mitochondrial membrane potential decrease and reduced ATP levels as soon as 3 months of age, when extracellular deposits are not present. In AD patients, low glucose metabolism at baseline and decline of glucose metabolism is used for monitoring changes in cognition and functionality in AD and mild cognitive impairment (MCI; Landau et al., 2011; Shokouhi et al., 2013). Also, oxidative stress, solidly related to mitochondrial dysfunction, has been proposed before plaque deposition in AD mouse models (Praticò et al., 2001; Rönnbäck et al., 2016) and before clinical symptoms in AD patients (Mosconi et al., 2008). Consistently with previous results (Calvo-Rodríguez et al., 2016b), we show here that in vitro aging is associated to a partial loss of mitochondrial membrane potential and this loss is further decreased by Aβo in both young and aged neurons. The effect is mild in young neurons. However, aged neurons treated with Aβo display a quite large loss of mitochondrial membrane potential, thus reducing rather dramatically the driving force for mitochondrial Ca2+ uptake. In addition, we also found that aged neurons show increased ROS generation compared with young neurons. Moreover, Aβo increase further ROS generation in aged neurons having no effect in young neurons. Accordingly, loss of mitochondrial membrane potential and enhanced ROS generation induced by Aβo in aged neurons may constrain Ca2+ transfer from ER to mitochondria.
In summary, we show that Aβo modulate differentially ER–mitochondria cross talk in young and aged neurons. In young neurons, Aβo increase ER–mitochondria coupling without having a detrimental effect. In contrast, in aged neurons, although Aβo increase also ER-mitochondria colocalization, the loss of mitochondrial membrane potential and enhanced ROS generation impairs Ca2+ transfer from ER to mitochondria despite Ca2+ stores are overloaded and the expression of molecular players involved in Ca2+ transfer increases, thus leading to mitochondrial damage and apoptosis. It has been reported that MAM-associated proteins are up-regulated in AD brain and APP Swe/Lon mouse models (Hedskog et al., 2013), and this effect is mirrored by nanomolar concentrations of Aβ-peptide. Changes in MAM associated proteins in AD could be a rescue response to dysfunctional ER-mitochondria coupling. However, in the face of increased Ca2+ store content associated to age and/or AD-related mutations, the further rise in ER-mitochondria coupling likely induced by Aβo may somehow collapse the own coupling leading to neuron damage. Further research is required to address this critical question. In addition, all the above results have been obtained in identified neurons in an in vitro model of neuronal aging. As stated above, the fraction of glial cells in hippocampal cultures increases with in vitro aging. Whether glial cell may contribute to Ca2+ remodeling in aged neurons treated with Aβo remains to be established.
Taken together, our data show that intraorganellar Ca2+ remodeling is a hallmark of the aging that is exacerbated in the presence of Aβo. Aging decreases SOCE required for spine stability and increases Ca2+ store content and MCU and IP3 receptors expression that favors Ca2+ transfer from ER to mitochondria. Most of these effects are exacerbated by Aβo but only in aged neurons. In contrast, in young neurons Aβo increase ER-mitochondrial colocalization and Ca2+ transfer from ER to mitochondria without having effects on ROS generation or apoptosis and only a minor decrease in mitochondrial membrane potential, probably related to increased use of this potential for ATP synthesis. However, the effect of Aβo may become highly detrimental in the aged neuron and the mechanism for this switch may play a fundamental role in AD and warrants further consideration.
MC-R, LN and CV designed the study and wrote the manuscript. MC-R and EH-P performed research and analyzed data.
This work was supported by grants BFU2012-37146 and BFU2015-70131R from Ministry of Economy and Competitivity (Spain) and grants VA145U13, BIO/VA33/13 and VA294P18 from Regional Government of Castilla y León (Spain). MC-R was supported by a pre-doctoral fellowship from Regional Government of Castilla y León (Spain) and the European Social Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Enrique Pérez-Riesgo for help with statistical analysis and Cristina Sánchez-Vicente for help with colocalization analysis.
Amatniek, J. C., Hauser, W. A., DelCastillo-Castaneda, C., Jacobs, D. M., Marder, K., Bell, K., et al. (2006). Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 47, 867–872. doi: 10.1111/j.1528-1167.2006.00554.x
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Area-Gomez, E., and Schon, E. A. (2017). On the pathogenesis of Alzheimer’s disease: the MAM hypothesis. FASEB J. 31, 864–867. doi: 10.1096/fj.201601309
Atamna, H., and Frey, W. H. II. (2007). Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 7, 297–310. doi: 10.1016/j.mito.2007.06.001
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Berridge, M. J. (2010). Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 459, 441–449. doi: 10.1007/s00424-009-0736-1
Bezprozvanny, I. (2009). Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 15, 89–100. doi: 10.1016/j.molmed.2009.01.001
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 31, 454–463. doi: 10.1016/j.tins.2008.06.005
Borchelt, D. R., Thinakaran, G., Eckman, C. B., Lee, M. K., Davenport, F., Ratovitsky, T., et al. (1996). Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013. doi: 10.1016/s0896-6273(00)80230-5
Brewer, G. J., Torricelli, J. R., Evege, E. K., and Price, P. J. (1993). Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35, 567–576. doi: 10.1002/jnr.490350513
Brewer, L. D., Thibault, O., Staton, J., Thibault, V., Rogers, J. T., Garcia-Ramos, G., et al. (2007). Increased vulnerability of hippocampal neurons with age in culture: temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res. 1151, 20–31. doi: 10.1016/j.brainres.2007.03.020
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Caballero, E., Calvo-Rodríguez, M., Gonzalo-Ruiz, A., Villalobos, C., and Núñez, L. (2016). A new procedure for amyloid β oligomers preparation enables the unambiguous testing of their effects on cytosolic and mitochondrial Ca2+ entry and cell death in primary neurons. Neurosci. Lett. 612, 66–73. doi: 10.1016/j.neulet.2015.11.041
Calvo, M., Sanz-Blasco, S., Caballero, E., Villalobos, C., and Núñez, L. (2015a). Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-inflammatory drugs: role of mitochondrial calcium. J. Neurochem. 132, 403–417. doi: 10.1111/jnc.13004
Calvo, M., Villalobos, C., and Núñez, L. (2015b). Calcium imaging in neuron cell death. Methods Mol. Biol. 1254, 73–85. doi: 10.1007/978-1-4939-2152-2_6
Calvo-Rodríguez, M., de la Fuente, C., García-Durillo, M., García-Rodríguez, C., Villalobos, C., and Núñez, L. (2017). Aging and amyloid β oligomers enhance TLR4 expression, LPS-induced Ca2+ responses, and neuron cell death in cultured rat hippocampal neurons. J. Neuroinflammation 14:24. doi: 10.1186/s12974-017-0802-0
Calvo-Rodríguez, M., García-Durillo, M., Villalobos, C., and Núñez, L. (2016a). Aging enables Ca2+ overload and apoptosis induced by amyloid-β oligomers in rat hippocampal neurons: neuroprotection by non-steroidal anti-inflammatory drugs and R-flurbiprofen in aging neurons. J. Alzheimers Dis. 54, 207–221. doi: 10.3233/jad-151189
Calvo-Rodríguez, M., García-Durillo, M., Villalobos, C., and Núñez, L. (2016b). In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 1863, 2637–2649. doi: 10.1016/j.bbamcr.2016.08.001
Calvo-Rodríguez, M., Villalobos, C., and Nuñez, L. (2015). Fluorescence and bioluminescence imaging of subcellular Ca2+ in aged hippocampal neurons. J. Vis. Exp. 106:e53330. doi: 10.3791/53330
Cárdenas, C., Miller, R. A., Smith, I., Bui, T., Molgó, J., Müller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/j.cell.2010.06.007
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Chan, S. L., Mayne, M., Holden, C. P., Geiger, J. D., and Mattson, M. P. (2000). Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 275, 18195–18200. doi: 10.1074/jbc.m000040200
Csordás, G., and Hajnóczky, G. (2009). SR/ER-mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta 1787, 1352–1362. doi: 10.1016/j.bbabio.2009.06.004
de la Fuente, S., Fonteriz, R. I., Montero, M., and Alvarez, J. (2012). Dynamics of mitochondrial [Ca2+] measured with the low-Ca2+-affinity dye rhod-5N. Cell Calcium 51, 65–71. doi: 10.1016/j.ceca.2011.10.007
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 33, 3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013
DuBoff, B., Feany, M., and Götz, J. (2013). Why size matters—balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci. 36, 325–335. doi: 10.1016/j.tins.2013.03.002
Etcheberrigaray, R., Hirashima, N., Nee, L., Prince, J., Govoni, S., Racchi, M., et al. (1998). Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 5, 37–45. doi: 10.1006/nbdi.1998.0176
Ferreiro, E., Oliveira, C. R., and Pereira, C. (2004). Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-β peptide. J. Neurosci. Res. 76, 872–880. doi: 10.1002/jnr.20135
Gibson, G. E., Zhang, H., Toral-Barza, L., Szolosi, S., and Tofel-Grehl, B. (1996). Calcium stores in cultured fibroblasts and their changes with Alzheimer’s disease. Biochim. Biophys. Acta 1316, 71–77. doi: 10.1016/0925-4439(96)00002-6
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hauptmann, S., Scherping, I., Dröse, S., Brandt, U., Schulz, K. L., Jendrach, M., et al. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005
Hayashi, T., Rizzuto, R., Hajnoczky, G., and Su, T. P. (2009). MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88. doi: 10.1016/j.tcb.2008.12.002
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U S A 110, 7916–7921. doi: 10.1073/pnas.1300677110
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001
Khachaturian, Z. S. (1989). The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging 1, 17–34. doi: 10.1007/bf03323872
Kopeikina, K. J., Carlson, G. A., Pitstick, R., Ludvigson, A. E., Peters, A., Luebke, J. I., et al. (2011). Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 179, 2071–2082. doi: 10.1016/j.ajpath.2011.07.004
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H. Y., Hyman, B. T., and Bacskai, B. J. (2008). Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. doi: 10.1016/j.neuron.2008.06.008
LaFerla, F. M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 3, 862–872. doi: 10.1038/nrn960
Landau, S. M., Harvey, D., Madison, C. M., Koeppe, R. A., Reiman, E. M., Foster, N. L., et al. (2011). Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 32, 1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002
Lemere, C. A., Lopera, F., Kosik, K. S., Lendon, C. L., Ossa, J., Saido, T. C., et al. (1996). The E280A presenilin 1 Alzheimer mutation produces increased A β 42 deposition and severe cerebellar pathology. Nat. Med. 2, 1146–1150. doi: 10.1038/nm1096-1146
Lerdkrai, C., Asavapanumas, N., Brawek, B., Kovalchuk, Y., Mojtahedi, N., Olmedillas Del Moral, M., et al. (2018). Intracellular Ca2+ stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 115, E1279–E1288. doi: 10.1073/pnas.1714409115
Mark, R. J., Hensley, K., Butterfield, D. A., and Mattson, M. P. (1995). Amyloid β-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 15, 6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995
Mattson, M. P. (2007). Calcium and neurodegeneration. Aging Cell 6, 337–350. doi: 10.1111/j.1474-9726.2007.00275.x
Moccia, F., Zuccolo, E., Soda, T., Tanzi, F., Guerra, G., Mapelli, L., et al. (2015). Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 9:153. doi: 10.3389/fncel.2015.00153
Mosconi, L., Pupi, A., and De Leon, M. J. (2008). Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N Y Acad. Sci. 1147, 180–195. doi: 10.1196/annals.1427.007
Müller, M., Ahumada-Castro, U., Sanhueza, M., Gonzalez-Billault, C., Court, F. A., and Cárdenas, C. (2018). Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 12:470. doi: 10.3389/fnins.2018.00470
Murray, F. E., Landsberg, J. P., Williams, R. J., Esiri, M. M., and Watt, F. (1992). Elemental analysis of neurofibrillary tangles in Alzheimer’s disease using proton-induced X-ray analysis. Ciba Found. Symp. 169, 201–210; discussion 210–216. doi: 10.1002/9780470514306.ch12
Nelson, O., Tu, H., Lei, T., Bentahir, M., de Strooper, B., and Bezprozvanny, I. (2007). Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Invest. 117, 1230–1239. doi: 10.1172/jci30447
Nunez, L., Sanchez, A., Fonteriz, R. I., and Garcia-Sancho, J. (1996). Mechanisms for synchronous calcium oscillations in cultured rat cerebellar neurons. Eur. J. Neurosci. 8, 192–201. doi: 10.1111/j.1460-9568.1996.tb01180.x
Pannaccione, A., Secondo, A., Molinaro, P., D’Avanzo, C., Cantile, M., Esposito, A., et al. (2012). A new concept: Aβ1–42 generates a hyperfunctional proteolytic NCX3 fragment that delays caspase-12 activation and neuronal death. J. Neurosci. 32, 10609–10617. doi: 10.1523/JNEUROSCI.6429-11.2012
Parekh, A. B., and Putney, J. W. Jr. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810. doi: 10.1152/physrev.00057.2003
Pérez-Otaño, I., Luján, R., Tavalin, S. J., Plomann, M., Modregger, J., Liu, X. B., et al. (2006). Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat. Neurosci. 9, 611–621. doi: 10.1038/nn1680
Porter, N. M., Thibault, O., Thibault, V., Chen, K. C., and Landfield, P. W. (1997). Calcium channel density and hippocampal cell death with age in long-term culture. J. Neurosci. 17, 5629–5639. doi: 10.1523/JNEUROSCI.17-14-05629.1997
Praticò, D., Uryu, K., Leight, S., Trojanoswki, J. Q., and Lee, V. M. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 21, 4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001
Rönnbäck, A., Pavlov, P. F., Mansory, M., Gonze, P., Marlière, N., Winblad, B., et al. (2016). Mitochondrial dysfunction in a transgenic mouse model expressing human amyloid precursor protein (APP) with the Arctic mutation. J. Neurochem. 136, 497–502. doi: 10.1111/jnc.13410
Santos, R. X., Correia, S. C., Wang, X., Perry, G., Smith, M. A., Moreira, P. I., et al. (2010). A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimers Dis. 20, S401–S412. doi: 10.3233/jad-2010-100666
Sanz-Blasco, S., Valero, R. A., Rodríguez-Crespo, I., Villalobos, C., and Núñez, L. (2008). Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3:e2718. doi: 10.1371/journal.pone.0002718
Secondo, A., Bagetta, G., and Amantea, D. (2018). On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front. Mol. Neurosci. 11:87. doi: 10.3389/fnmol.2018.00087
Shokouhi, S., Claassen, D., Kang, H., Ding, Z., Rogers, B., Mishra, A., et al. (2013). Longitudinal progression of cognitive decline correlates with changes in the spatial pattern of brain 18F-FDG PET. J. Nucl. Med. 54, 1564–1569. doi: 10.2967/jnumed.112.116137
Šišková, Z., Justus, D., Kaneko, H., Friedrichs, D., Henneberg, N., Beutel, T., et al. (2014). Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84, 1023–1033. doi: 10.1016/j.neuron.2014.10.024
Sodero, A. O., Weissmann, C., Ledesma, M. D., and Dotti, C. G. (2011). Cellular stress from excitatory neurotransmission contributes to cholesterol loss in hippocampal neurons aging in vitro. Neurobiol. Aging 32, 1043–1053. doi: 10.1016/j.neurobiolaging.2010.06.001
Stutzmann, G. E. (2007). The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist 13, 546–559. doi: 10.1177/1073858407299730
Supnet, C., Grant, J., Kong, H., Westaway, D., and Mayne, M. (2006). Amyloid-β-(1–42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J. Biol. Chem. 281, 38440–38447. doi: 10.1074/jbc.m606736200
Szabadkai, G., Simoni, A. M., and Rizzuto, R. (2003). Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J. Biol. Chem. 278, 15153–15161. doi: 10.1074/jbc.m300180200
Toescu, E. C., and Verkhratsky, A. (2003). Neuronal ageing from an intraneuronal perspective: roles of endoplasmic reticulum and mitochondria. Cell Calcium 34, 311–323. doi: 10.1016/s0143-4160(03)00142-8
Tong, B. C., Lee, C. S., Cheng, W. H., Lai, K. O., Foskett, J. K., and Cheung, K. H. (2016). Familial Alzheimer’s disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 9:ra89. doi: 10.1126/scisignal.aaf1371
Tu, H., Nelson, O., Bezprozvanny, A., Wang, Z., Lee, S. F., Hao, Y. H., et al. (2006). Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126, 981–993. doi: 10.1016/j.cell.2006.06.059
Villalobos, C., and García-Sancho, J. (1995). Capacitative Ca2+ entry contributes to the Ca2+ influx induced by thyrotropin-releasing hormone (TRH) in GH3 pituitary cells. Pflugers Arch. 430, 923–935. doi: 10.1007/bf01837406
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Xie, H., Guan, J., Borrelli, L. A., Xu, J., Serrano-Pozo, A., and Bacskai, B. J. (2013). Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 33, 17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 14670–14675. doi: 10.1073/pnas.0903563106
Keywords: calcium, Alzheimer’s disease, aging, endoplasmic reticulum, mitochondria, store-operated calcium entry, hippocampal neurons
Citation: Calvo-Rodriguez M, Hernando-Perez E, Nuñez L and Villalobos C (2019) Amyloid β Oligomers Increase ER-Mitochondria Ca2+ Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca2+ Remodeling. Front. Cell. Neurosci. 13:22. doi: 10.3389/fncel.2019.00022
Received: 20 August 2018; Accepted: 17 January 2019;
Published: 08 February 2019.
Edited by:
Francesco Moccia, University of Pavia, ItalyReviewed by:
Cesar Cardenas, Universidad Mayor, ChileCopyright © 2019 Calvo-Rodriguez, Hernando-Perez, Nuñez and Villalobos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Calvo-Rodriguez, bWNhbHZvcm9kcmlndWV6QG1naC5oYXJ2YXJkLmVkdQ==
† Present address: Maria Calvo-Rodriguez, Alzheimer Research Unit, Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Charlestown, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.