 Joshua M. Lorenz-Guertin
Joshua M. Lorenz-Guertin Matthew J. Bambino
Matthew J. Bambino Tija C. Jacob
Tija C. Jacob
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 23 August 2018
Sec. Cellular Neurophysiology
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00265
This article is part of the Research Topic Postsynaptic Exocytosis, Endocytosis and Recycling: Mechanisms, Regulation and Physiological Relevance in Synaptic Function and Plasticity View all 11 articles
GABA type A receptors (GABAARs) mediate the majority of fast inhibitory neurotransmission in the central nervous system (CNS). Most prevalent as heteropentamers composed of two α, two β, and a γ2 subunit, these ligand-gated ionotropic chloride channels are capable of extensive genetic diversity (α1-6, β1-3, γ1-3, δ, 𝜀, 𝜃, π, ρ1-3). Part of this selective GABAAR assembly arises from the critical role for γ2 in maintaining synaptic receptor localization and function. Accordingly, mutations in this subunit account for over half of the known epilepsy-associated genetic anomalies identified in GABAARs. Fundamental structure–function studies and cellular pathology investigations have revealed dynamic GABAAR trafficking and synaptic scaffolding as critical regulators of GABAergic inhibition. Here, we introduce in vitro and in vivo findings regarding the specific role of the γ2 subunit in receptor trafficking. We then examine γ2 subunit human genetic variation and assess disease related phenotypes and the potential role of altered GABAAR trafficking. Finally, we discuss new-age imaging techniques and their potential to provide novel insight into critical regulatory mechanisms of GABAAR function.
The adult central nervous system (CNS) is critically dependent on fast inhibitory neurotransmission evoked by GABAA receptors (GABAARs). GABAARs are ligand-gated ionotropic chloride (Cl-) channels ubiquitously expressed throughout the CNS that play a fundamental role in restraining and sculpting neuronal activity. Disruptions in GABAAR dependent neurotransmission leads to insufficient inhibitory effects throughout the brain, contributing to the pathogenesis of epilepsy, neurodevelopmental disorders, depression, schizophrenia and stroke (Hines et al., 2012). Activation of GABAARs by the neurotransmitter GABA induces ion channel opening, Cl- influx, and subsequent membrane hyperpolarization. These heteropentameric structures are predominantly composed of two α (α1-6), two β (β1-3), and either a γ (γ1-3) or a δ subunit (Olsen and Sieghart, 2009) (Figures 1A,B). GABAARs belong to the Cys-loop superfamily of pentameric ligand-gated ion channels (pLGICs) including strychnine-sensitive glycine receptors, nicotinic acetylcholine (nACh) receptors, and 5-hydroxytryptamine type-3 (5-HT3) receptors. Individual subunits have a common structure consisting of a large N-terminus extracellular domain (ECD) that participates in endogenous ligand binding, a transmembrane domain (TM) comprised of four α-helical regions (M1-4) and a barely extruding extracellular C-terminus. The M2 region of the subunits forms the ion channel pore. The hydrophobic M regions are connected by a small intracellular loop between M1-M2 and a much larger intracellular domain (ICD; previously termed intracellular loop) between M3 and M4 (Sigel and Steinmann, 2012) that mediates interactions with intracellular proteins critical for receptor trafficking and synaptic clustering (Figure 1C). Recently, GABAAR structures for the human β3 homopentamer bound to benzamidine (Miller and Aricescu, 2014), chimeric α5TM/β3ECD bound to the neurosteroid allopregnanolone (Miller et al., 2017), and human α1β2γ2 heteropentamer bound to GABA and the benzodiazepine site antagonist Flumazenil (Zhu et al., 2018) were resolved, advancing our growing understanding of GABAAR molecular architecture. Importantly, nearly all pLGIC family structural data lacks the large ICD (Nemecz et al., 2016) (exception 5-HT3 receptor; Hassaine et al., 2014), leaving functionally relevant information about this region left undiscovered.
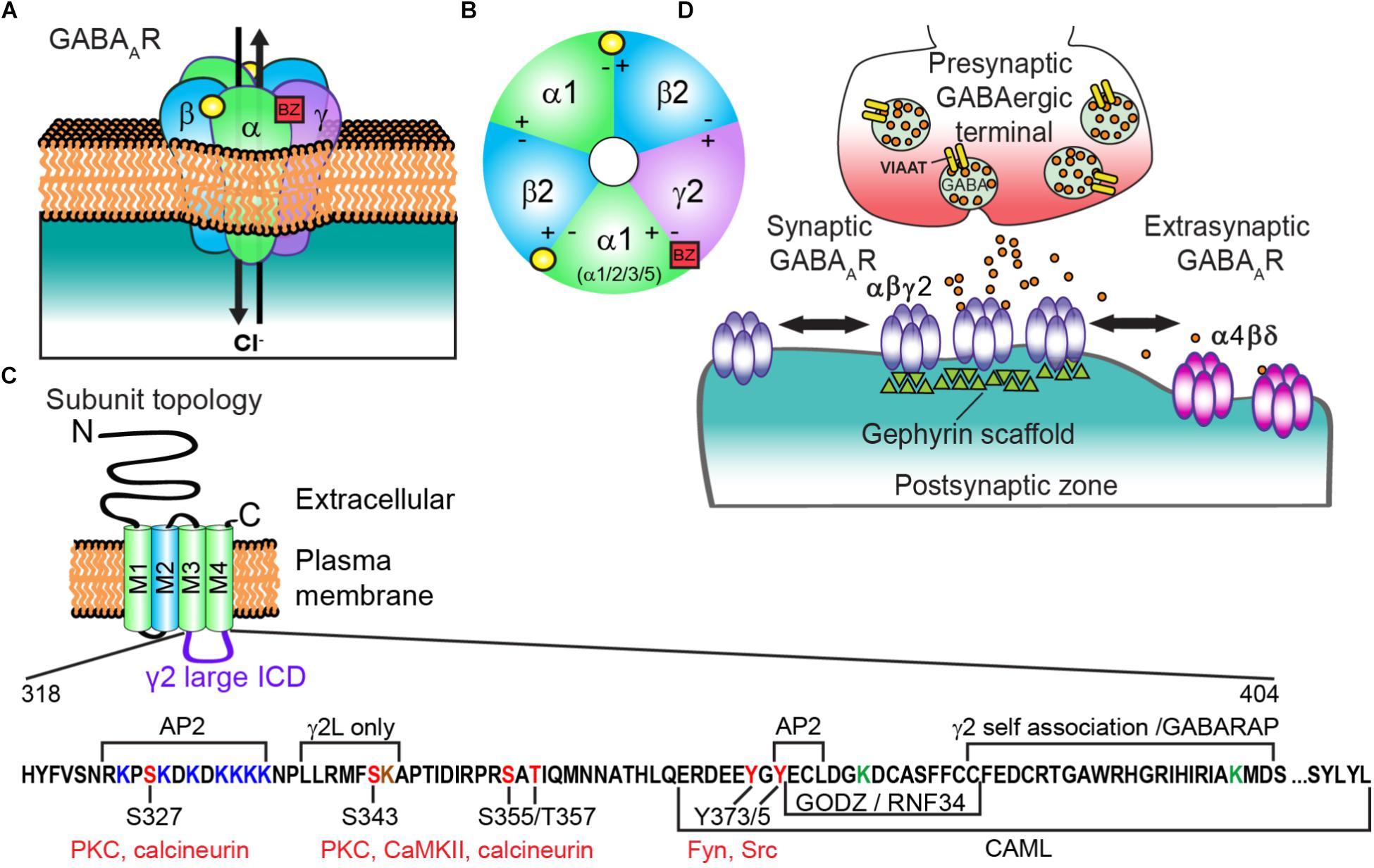
FIGURE 1. Generic GABAAR structure and subunit topology; and regulatory sites of the γ2 intracellular domain (ICD). (A) GABAAR heteropentamer composed of αβγ subunits. Binding of the neurotransmitter GABA (yellow circle) at the αβ interface triggers ion channel opening and allows the rapid influx of Cl- and membrane hyperpolarization in the mature nervous system. (B) Extracellular representation of the most prevalent cortical receptor subtype composed of α1β2γ2 subunits showing all five subunits contributing to the central ion pore and the general binding sites of GABA (yellow circle) and benzodiazepines (BZs) (red square). BZs bind at the interface of an α1/2/3/5 and γ subunit. (C) All subunits have a common topology including an extracellular N-terminal domain (ECD), short C-terminal tail, and four transmembrane regions (M1-4) which compose the transmembrane domain (TM). M2 (blue) contributes to formation of the receptor ion channel pore, while the ICD between M3 and M4 contains sites of phosphorylation and protein interactions that modulate channel function and/or trafficking. The γ2 L isoform intracellular domain (ICD = AA 318-404, residue numbering does not include signal peptide) is shown here with identified regulatory sites and regions of protein interaction. Seven lysine residues (red) contribute to γ2-containing GABAAR ubiquitination and endo-lysosomal targeting in HEK cells, with mutation of three additional lysine residues needed to block receptor downregulation by E3 ligase RNF34 overexpression (ICD green Ks and K259 in smaller M1-M2 loop not shown in diagram). Note the γ2L specific K344 residue (brown) has not been tested in ubiquitination studies (D) GABAARs composed of α(1-3)βγ subunits are largely synaptically localized via gephyrin interactions and contribute to phasic currents, whereas α(4 or 6)βδ receptors are extrasynaptic and generate tonic current.
Presynaptic terminal release of GABA onto postsynaptically clustered GABAARs initiates fast, transient receptor activation. In contrast, activation of extrasynaptic GABAARs by ambient “spill over” GABA generates a persistent tonic current (Figure 1D). Most GABAARs evoking fast synaptic inhibition in the mature cortex contain α1β2γ2 subunits, although α/β content can vary widely (Olsen and Sieghart, 2009), prompting a unifying role of γ2 in synaptic function. Importantly, the benzodiazepine drug class selectively binds between the interface of a γ2 subunit and either an α1/2/3/5 subunit to potentiate GABAAR function and elicit behavioral effects including sedative/hypnotic, anti-convulsant, myorelaxant, and/or anti-anxiety effects (Vinkers and Olivier, 2012) (Figures 1A,B). Here we summarize (1) known molecular interactors and mechanisms regulating γ2 trafficking (2) the importance of this subunit physiologically and human γ2 genetic variants compromising structure and function in vitro and in vivo and (3) application of modern imaging techniques to discover novel insight into synaptic GABAAR modulation.
During biosynthesis, GABAAR subunits are first assembled in the endoplasmic reticulum (ER) and then transported to the Golgi apparatus (Golgi) for further maturation (Figure 2). Forward trafficking of γ2-GABAARs from the ER is negatively regulated by Cleft lip and palate transmembrane protein (CLPTM1) in vitro and in vivo (Figure 2) (Ge et al., 2018). Overexpressing CLPTM1 reduces surface and synaptic levels of γ2, resulting in reduced amplitude and frequency of inhibitory postsynaptic current (IPSC), where the opposite effect is seen by CLPTM1 knockdown (KD). Importantly, CLPTM1 also regulates tonic inhibition and interacts with the extrasynaptic subunits α4 and δ, suggesting this protein non-selectively binds many GABAAR subtypes. Upon entry into the Golgi, the γ2 subunit undergoes palmitoylation via the Golgi-specific DHHC zinc finger enzyme (GODZ; also known as ZDHHC3) (Keller et al., 2004; Fang et al., 2006). This process is key for receptor clustering, innervation, and inhibitory strength in vitro and in vivo (Keller et al., 2004; Fang et al., 2006; Kilpatrick et al., 2016). GABAAR forward trafficking to the cell surface depends on the microtubule-dependent molecular motor kinesins (KIFs) (Figure 2). The KIF21B protein co-precipitates with the GABAAR γ2 subunit (Labonte et al., 2014). RNA KD of KIF21B reduces receptor surface levels and the intensity of extrasynaptic γ2 clusters, but does not affect synaptic GABAARs levels. Additionally, the KIF5 family plays a critical role in trans-Golgi to surface GABAAR trafficking (Twelvetrees et al., 2010). Conditional knockout (KO) of KIF5A in mice results in deficits of GABAAR plasma membrane levels, epilepsy phenotypes, and high lethality rate within 21 days postnatal (Nakajima et al., 2012).
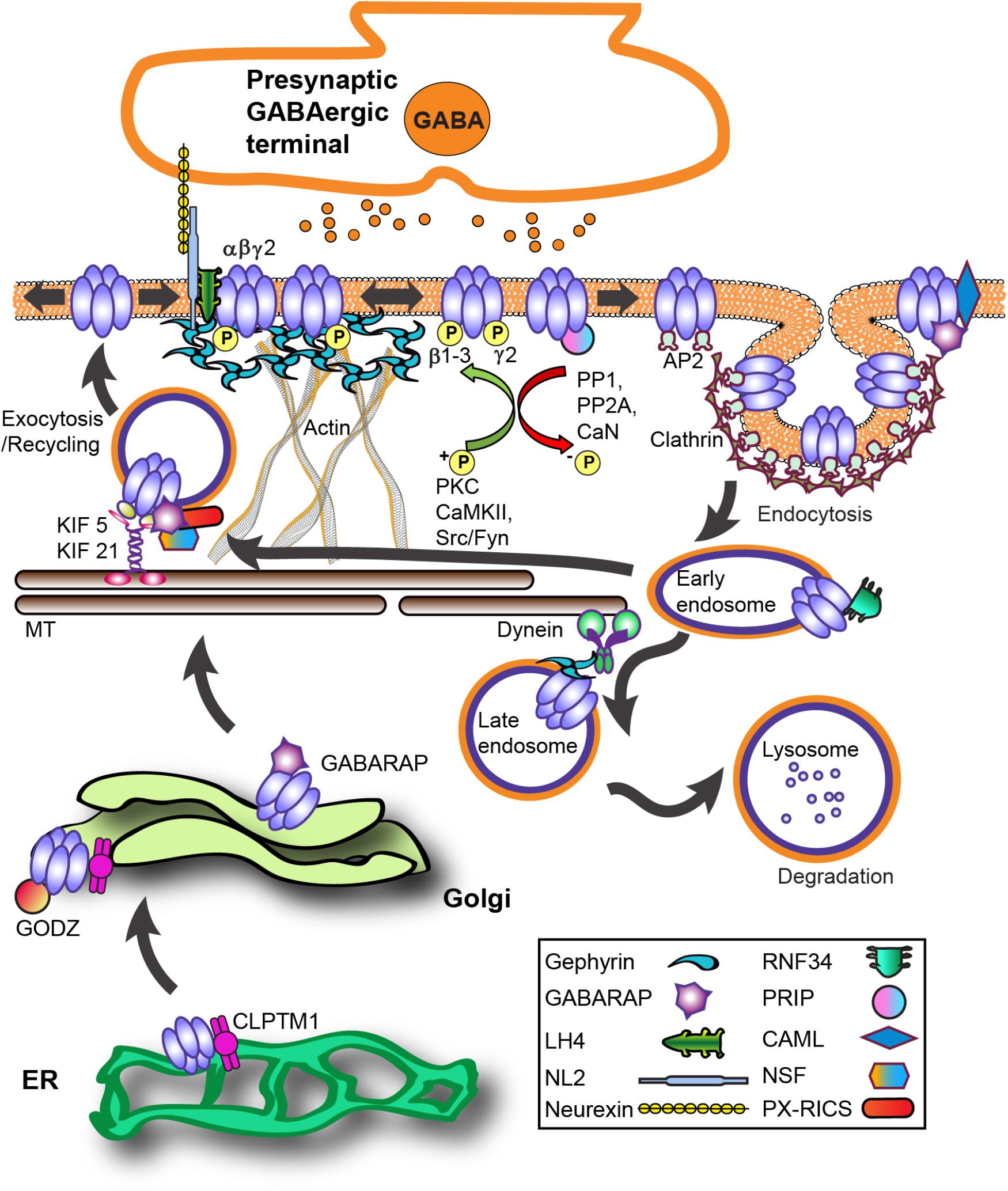
FIGURE 2. GABAAR trafficking and key interacting proteins at GABAergic synapses. The process of GABAAR synthesis, assembly and forward trafficking is highly regulated. Forward trafficking of γ2-GABAARs from the ER is negatively regulated by CLPTM1. Subunits are assembled into pentameric receptors in the endoplasmic reticulum (ER) where proper folding allows receptors to avoid proteosomal degradation and exit to the Golgi. In the Golgi, palmitoylation of γ subunits by the palmitoyltransferase GODZ is a key step in promoting forward trafficking to the synapse. GABARAP interacts with γ subunits and microtubules and overexpression augments receptor plasma membrane levels. PX-RICS forms an adaptor complex with GABARAP to facilitate γ2-GABAARs forward trafficking. PRIP1/2 and NSF interact with GABAARs both indirectly via GABARAP and directly with β subunits. The kinesin KIF5 is the main microtubule (MT)-dependent motor transporting inhibitory synapse components although recent work shows KIF21 contributes to extrasynaptic receptor delivery. LH4 forms a complex between γ2 and NL2. NL2 is central in GABAAR synapse development via its trans-synaptic association with axonal neurexins and also binds gephyrin. GABAARs primarily undergo clathrin-dependent endocytosis via β and γ subunit interactions with the clathrin-adaptor protein 2 (AP2) complex. Phosphorylation of AP2-interaction motifs within receptor subunits increases cell-surface receptor levels and enhances GABAAR neurotransmission by reducing AP2 binding to receptors. After internalization, clathrin-coated vesicles fuse with early endosomes, allowing for subsequent receptor recycling or targeting for degradation in lysosomes. CAML interaction with the γ2 subunit promotes forward trafficking and recycling. Ubiquitination of GABAAR contributes to lysosomal targeting, with the ubiquitin E3 ligase RNF34 directly interacting with the γ2 subunit. Protein abbreviations: CAML (calcium-modulating cyclophilin ligand), CLPTM1 (Cleft lip and palate transmembrane protein), GABARAP (GABAAR - associated protein), GODZ (Golgi-specific DHHC zinc finger enzyme), KIF 5/21 (microtubule-dependent molecular motor kinesins), LH4 (lipoma HMGIC fusion partner-like protein 4), NL2 (neuroligin 2), NSF (N-ethylmaleimide-sensitive factor ATPase), PRIP (phospholipase C-related catalytically inactive proteins), PX-RICS [Rho GTPase Activating Protein 32 (ARHGAP32) isoform 1], RNF34 (ring finger protein 34 E3 ligase).
Notably, KIF5A (not KIF5B, KIF5C) selectively interacts with the GABAAR-associated protein (GABARAP) in vivo (Nakajima et al., 2012). The well-characterized GABARAP (Figure 2) is part of the ubiquitin-like protein (UBL) family implicated in numerous cellular processes (van der Veen and Ploegh, 2012). GABARAP interacts with GABAAR γ subunits and microtubules, is heavily localized at the Golgi apparatus and cell surface (Wang et al., 1999), and overexpression augments GABAAR plasma membrane levels (Leil et al., 2004). However, GABARAP KO mice have unhindered distribution of γ2-GABAARs and gephyrin, suggesting functional redundancy with other trafficking proteins (O’Sullivan et al., 2005). Some evidence suggests GABARAP preferentially associates with serine phosphorylated γ2-GABAARs, while dephosphorylation by protein phosphatase 1 (PP1) decreases this interaction (Qian et al., 2011).
A number of GABARAP interacting proteins mediate GABAAR trafficking or localization (Figure 2). For instance, increased association with the PDZ domain-containing protein GRIP is seemingly involved in NMDA receptor-dependent GABAAR synaptic plasticity (Marsden et al., 2007). The phospholipase C-related catalytically inactive proteins 1 and 2 (PRIP1/2) and the N-ethylmaleimide-sensitive factor ATPase (NSF) interact with GABAARs both indirectly via GABARAP and directly with β subunits (Figure 2) (Kanematsu et al., 2002; Terunuma et al., 2004; Goto et al., 2005; Mizokami et al., 2007). NSF is a key component of SNARE-mediated fusion and is involved in receptor cell surface transit (Chou et al., 2010). Notably, the γ2 subunit and PRIP share an overlapping binding site on GABARAP (Kanematsu et al., 2002). PRIP1/2 KO mice demonstrate diminished benzodiazepine sensitivity and Zn2+ modulation concurrent with lower plasma membrane GABAAR expression, consistent with impaired γ2 subunit trafficking. KO of PRIP-1, the primary brain subtype, leads to mice displaying an epileptic phenotype that can be successfully suppressed by diazepam (DZP), but interictal discharges persist (Zhu et al., 2012). Interestingly, DZP potentiation of miniature inhibitory postsynaptic currents (mIPSC) remains unchanged, but baseline and DZP potentiated tonic GABA current amplitude in PRIP-1 KO neurons was reduced. PRIP-1 KO and PRIP1/2 double KO mice show anxiety-related behaviors and abnormal locomotion related to GABAAR dysfunction and reduced benzodiazepine sensitivity. Recently the Rho GTPase Activating Protein 32 (ARHGAP32) isoform 1 (PX-RICS) was shown to form an adaptor complex with GABARAP and the scaffold proteins 14-3-3ζ/𝜃 to facilitate γ2-GABAARs forward trafficking via dynein/dynactin and promote surface expression (Nakamura T. et al., 2016). KO of PX-RICS in mice generates an Autism Spectrum Disorder (ASD) phenotype with increased susceptibility to kainate-induced epileptic seizures, decreased GABAAR plasma membrane levels, and lowered mIPSC amplitude. Transgenic overexpression of 14-3-3ζ in mice protects against neuronal death caused by prolonged seizures (Brennan et al., 2013). In contrast, 14-3-3ζ mutations or deletions have been identified in patients with pathology associated with GABAAR deficits including schizophrenia, autism and generalized epilepsy (Tenney et al., 2011; Fromer et al., 2014; Toma et al., 2014).
Following insertion at the plasma membrane, γ2-GABAARs undergo Brownian diffusion until interaction with the inhibitory postsynaptic scaffolding protein gephyrin causes constraint and accumulation (Figures 1D, 2). Specifically, GABAAR α1/2/3/5 and β2/3 subunits (at lower affinity) mediate gephyrin-receptor binding (Tretter et al., 2008, 2011; Mukherjee et al., 2011; Kowalczyk et al., 2013; Brady and Jacob, 2015). While no direct interaction between γ2 and gephyrin has been identified, the synaptic levels of these proteins are intimately tied, shown by KO studies of gephyrin (Kneussel et al., 1999) and γ2 (Schweizer et al., 2003). Interestingly, chimeric studies indicate the γ2 M4 is sufficient to cause GABAAR accumulation opposite GABAergic terminals, while the large ICD of γ2 is necessary for gephyrin recruitment and rescue of synaptic function in γ2 KO cultured neurons (Alldred et al., 2005). It is likely that an indirect interaction occurs between γ2 and gephyrin across a bridge of other key synaptic proteins. Recently, six unrelated patients were identified with microdeletions in the gephyrin gene resulting in a range of neurodevelopmental deficits including ASD, schizophrenia or epilepsy (Lionel et al., 2013). The recently discovered GABAAR regulatory Lhfpl (GARLH) family proteins lipoma HMGIC fusion partner-like 3 and 4 (LH3 and LH4) forms a native complex between γ2 and the transsynaptic protein neuroligin 2 (NL2) (Figure 2) (Yamasaki et al., 2017). NL2 is central in GABAAR synapse development via its trans-synaptic association with axonal neurexins (Sudhof, 2008). Diminishing LH4 levels in culture and in vivo dramatically reduced γ2-GABAAR and gephyrin synaptic clustering and inhibitory strength (Davenport et al., 2017; Yamasaki et al., 2017). Curiously, despite the dramatic reduction in synaptic inhibition, epilepsy susceptibility or overt behavioral phenotypes in these mice have yet to be reported in the constitutive LH4 KO mouse. Importantly, gephyrin is known to directly bind the intracellular domain of NL2 (Poulopoulos et al., 2009). Thus γ2 subunit-LH4-NL2-gephyrin interactions could provide a molecular framework to support γ2’s role in GABAAR synaptic recruitment and maintenance.
Synaptic plasticity, or the dynamic modulation of synaptic output, is heavily influenced by receptor phosphorylation via altering channel function or receptor trafficking. Phosphoregulation of γ2 S327 is an important mediator of GABAAR retention at synapses. Detailed electrophysiology and in vivo studies have identified the PKC𝜀 isoform specifically phosphorylates the γ2 S327 residue (Figures 1C, 2), ultimately fine-tuning responsiveness to ethanol and benzodiazepines (Qi et al., 2007). Additionally, protocols that induce calcium-entry via glutamate application, strong NMDA receptor activation, or robust neuronal activity enhance receptor lateral mobility, decrease synaptic cluster size, and reduce mIPSC amplitude via the phosphatase calcineurin (CaN) (Bannai et al., 2009) and dephosphorylation of the γ2 subunit S327 residue (Figures 1C, 2) (Muir et al., 2010). More broadly, activation of all PKC isoforms by 1 h PMA (PKC activator; 30 nM) treatment decreases surface γ2-GABAAR levels that can be reversed by specific inhibition of PKC𝜀 catalytic activity in HEK cells and PKC𝜀 specific activation reduces GABAAR current amplitude (Chou et al., 2010). This effect was in part attributed to changes in GABAAR trafficking occurring though PKC𝜀 association and phosphorylation of NSF. The scaffolding protein 14-3-3-𝜃 acts as a bridge for the PKCγ isoform to interact with γ2 in cerebellar Purkinje neurons and N2a cells (Qian et al., 2012). 14-3-3-𝜃 KD in mice by siRNA microinjection reduces γ2-GABAAR overall serine phosphorylation, while KD of 14-3-3-𝜃 or PKCγ reverses the PMA (200 nM, 30 min) induced upregulation of C cell surface expression in N2a cells. These apparently conflicting reports on PKC kinase family modulation highlights the complexity of this signaling pathway in γ2-GABAAR regulation, with varied effects dependent on the pharmacological agents used, treatment times, model, and PKC isoforms.
An important consideration for γ2 subunit regulation is its presence in a short (γ2S) or long (γ2L) isoform; the γ2L isoform has 8 additional amino acids (LLRMFSFK) in the large ICD with the serine site (S343) capable of being phosphorylated by Protein kinase C (PKC) and Calcium/calmodulin-dependent protein kinase type II (CaMKII) (Figure 1C) (Whiting et al., 1990; Moss et al., 1992; McDonald and Moss, 1994). Expression levels of γ2S remain constant throughout development, while γ2L levels increase during neuronal maturation (Wang and Burt, 1991). Early in vitro expression studies found that the additional amino acids in the γ2L subunit may play a role in the response to diazepam and be critical for ethanol enhancement of GABA current (Wafford et al., 1991). Both mutation of S343 to a phosphomimetic aspartate or to non-phosphorylatable valine resulted in cell surface trafficking of γ2L when expressed alone, similar to γ2S (Boileau et al., 2010). This work also proposed an accessory protein role for γ2S as an external modulator of GABAAR function to confer zinc blockade protection for receptors. When comparing synaptic clustering of γ2L vs. γ2S subunit large ICD (partial subunit chimeras) in spinal cord neurons, postsynaptic γ2L ICD chimera accumulation is higher, and can be enhanced by PKC activation by phorbol ester phorbol-12,13-dibutyrate (PDBu) and reversed by mutating the S343 residue of γ2L (Meier and Grantyn, 2004). The physiological role of CaMKII direct phosphorylation on γ2 has not yet been described, although CaMKII is required for a type of inhibitory long term potentiation (iLTP) in Purkinje neurons known as rebound potentiation (Kano et al., 1996) and increased association between the γ2 subunit and GABARAP (Kawaguchi and Hirano, 2007). CaMKII plays other critical roles in GABAergic plasticity including promoting receptor surface levels (Wang et al., 1995; Marsden et al., 2007, 2010; Saliba et al., 2012) and recruitment of the synaptic scaffold protein gephyrin, while reducing GABAAR lateral diffusion (Petrini et al., 2014).
Non-synaptic GABAARs on the cell surface are capable of undergoing internalization (Bogdanov et al., 2006), a fundamental cellular process that regulates receptor signaling and function (Figure 2). GABAAR internalization is primarily clathrin-mediated in concert with GTPase dynamin activity and the adaptor protein AP2 complex (Kittler et al., 2000), although clathrin-independent GABAAR endocytosis has been described (Cinar and Barnes, 2001; Rowland et al., 2006). AP2 interacts with the ICD of GABAAR β subunits and the extrasynaptic δ subunit in a phospho-dependent manner (McDonald et al., 1998; Brandon et al., 2002, 2003; Herring et al., 2005; Kittler et al., 2005; Smith et al., 2008; Gonzalez et al., 2012; Smith et al., 2012). The γ2 subunit also contains two AP2 interaction domains on its ICD, a 12 basic amino acid region and a classical YGYECL motif (Smith et al., 2008) (Figure 1C). Phosphorylation at Y365/367 residues within the YGYECL motif by the non-receptor tyrosine-protein kinases Fyn and Src family kinases (Moss et al., 1995; Brandon et al., 2001; Jurd et al., 2010) reduces AP2 binding, as does mutation of Y365/7 to phenylalanine (Kittler et al., 2008; Tretter et al., 2009). Homozygous tyrosine to phenylalanine (Y365/7F) knock-in mice are developmentally lethal, suggesting phosphoregulation of these residues is critical for GABAAR function or trafficking in vivo. Heterozygous Y365/7F knock-in mutant mice show inhibition of AP2 binding to the γ2 subunit, surface and synaptic accumulation of receptors and ultimately spatial memory deficits (Tretter et al., 2009). Further investigation revealed that brain-derived neurotrophic factor (BDNF) enhances Y365/7 phosphorylation and stabilizes γ2-containing GABAAR, consistent with heterozygous Y365/7F mice showing an anti-depressant phenotype in the forced swim task and tail-suspension test and increased neurogenesis effects that are resistant to further enhancement by BDNF (Vithlani et al., 2013).
GABAAR endocytosis can be increased by stimuli of opposite polarities, either excitotoxic protocols such as in vitro seizure (Goodkin et al., 2005, 2008; Naylor et al., 2005; Lorenz-Guertin et al., 2017) and oxygen-glucose deprivation (OGD) (Arancibia-Carcamo et al., 2009), or by prolonged inhibition with agonist exposure (Chaumont et al., 2013; Gutierrez et al., 2014). Internalization is in part regulated by phosphatase activity under these conditions. For example, inhibition of CaN or the serine/threonine protein phosphatase 1 (PP1) and 2A (PP2A) reverses a status epilepticus induced decrease in surface γ2-GABAARs and mIPSC amplitude (Joshi et al., 2015). Importantly, genetic GABAAR mutants also affect intracellular trafficking. For instance, the γ2 R82Q (numbering without signal peptide R43Q) mutation linked to childhood absence epilepsy and febrile seizures (FS) showed increased basal receptor endocytosis rates relative to wild-type (Chaumont et al., 2013). In summary, endogenous signaling pathways, pharmacological treatments, and pathological stimuli or genetic variation can modulate GABAAR endocytosis networks [kinase and phosphatase regulation reviewed in Lorenz-Guertin and Jacob (2017)].
Internalized GABAARs can either be recycled back to the cell surface or targeted for degradation at lysosomes (Figure 2) (Kittler et al., 2004; Arancibia-Carcamo et al., 2009). Interaction of the integral membrane protein calcium-modulating cyclophilin ligand (CAML) with the γ2 subunit cytoplasmic and fourth transmembrane domain regions promotes forward trafficking and recycling (Yuan et al., 2008). Neurons lacking CAML demonstrate diminished recycling of endocytosed GABAARs and decreased inhibitory strength. Broad PKC activity is implicated as a negative regulator of GABAAR recycling activity following internalization (Connolly et al., 1999). 5-HT2 serotonergic negative modulation of GABAAR currents is also thought to occur through a PKC-RACK1 (receptor for activated C kinase) mechanism (Feng et al., 2001).
Synaptic receptors destined for degradation undergo ubiquitination of 7 lysine residues within the ICD of the γ2 subunit (Figure 1C) (Arancibia-Carcamo et al., 2009). Lysine to arginine (K7R) mutation at these ubiquitination sites diminishes late endosome targeting of receptors in heterologous cells, and reverses loss of surface receptor clusters following OGD treatment (Arancibia-Carcamo et al., 2009). The ring finger protein 34 (RNF34) E3 ligase directly binds the γ2 ICD, co-immunoprecipitates with γ2 in vivo and can be identified at inhibitory synapses (Figure 2) (Jin et al., 2014). Interestingly, the short 14 amino acid motif in the γ2 ICD sufficient for RNF34 binding is identical to the GODZ binding region (Figure 1C), and is also highly conserved among the γ subunits. γ2-GABAAR degradation is accelerated upon overexpression of RNF34 resulting in smaller GABAAR synaptic clusters and diminished inhibitory current strength. Proteosomal and lysosomal inhibitor experiments suggest RNF34 ubiquitination of γ2 contributes to degradation by both of these pathways in HEK cells. Notably, co-expression of RNF34 with the γ2 ubiquitin resistant K7R mutant did not inhibit degradation of this subunit. On the contrary, additional lysine mutations (K8R, K9R, K10R) were able to prevent downregulation of γ2 by RNF34, suggesting these residues may be important for ubiquitination-degradation.
Only a handful of stimuli clearly induce lysosomal degradation of GABAARs, likely due to the receptor’s crucial role in maintaining neuronal inhibition and the tight regulation of receptor surface levels that must therefore occur. Our lab previously found 24 h benzodiazepine treatment in cultured hippocampal neurons enhances lysosomal-mediated degradation of α2-containing receptors (Jacob et al., 2012). More recently, we identified that a GABAAR antagonist bicuculline acute seizure model also induces lysosomal targeting of surface GABAARs in cultured cortical neurons (Lorenz-Guertin et al., 2017). It is likely that stimulus specific subunit ubiquitination patterns ultimately dictate receptor fate. This remains a highly understudied area of research in GABAAR trafficking.
The network of proteins governing inhibitory synapse clustering, trafficking, and plasticity are unresolved, as evidenced by three recent in vivo inhibitory synapse proteomic screenings utilizing either knock-in mice expressing GFP-tagged α2 subunit (Nakamura Y. et al., 2016), adeno-associated viral (AAV) expression of fusion proteins including gephyrin (Uezu et al., 2016), or mice expressing a Thy1-His6-Flag-YFP-γ2 subunit transgene (Ge et al., 2018). Initial analysis from these experiments has revealed novel inhibitory protein constituents including the metabotropic glutamate receptor subunit mGluR5, the Dbl family GEF Ephexin, metabotropic GABA B receptor (GABABR) auxiliary subunit KCTD12, and inhibitory synaptic regulator protein 1 (InSyn1) (Nakamura Y. et al., 2016; Uezu et al., 2016). Most recently, tandem affinity purification proteomics revealed the critical GABAAR forward trafficking component CLPTM1, and two novel interactors including integral membrane protein 2C (ITM2C) and Golgi glycoprotein 1 (GLG1) (Ge et al., 2018). Considering new candidate interactor proteins are identified with slight derivations in methodology (140 in Uezu et al., 2016; 149 in Nakamura Y. et al., 2016; 39 additional in Ge et al., 2018), future investigations will need to both confirm the validity and importance of these observed proteins in GABAAR function and modulation.
Due to the fundamental importance of γ2 GABAAR inhibition in the CNS, embryonic KO animals die within days of birth (Gunther et al., 1995). Developmentally delayed KO of γ2 using a CaMKIICre transgene expression system results in mice who are phenotypically normal 3 weeks post-natal, but by week 4 exhibit a rapid decline in health including epileptic episodes and eventually death (Schweizer et al., 2003). A large drop in gephyrin immunoreactivity also occurs coincident with loss of γ2 expression without changing GABAergic presynaptic innervation as measured by vesicular inhibitory amino acid transporter (VIAAT) levels.
Partial KD of brain wide γ2 levels results in impaired behavior including an enhanced anxious-depressive phenotype (Crestani et al., 1999; Chandra et al., 2005; Earnheart et al., 2007; Shen et al., 2010). In addition, heterozygous γ2+/- mice show defective spine maturation and synaptogenesis (Ren et al., 2015). Ablating forebrain γ2 expression in embryonic glutamatergic neurons using homozygous EMX1Cre-induced inactivation also recapitulated the depressive-anxiety phenotype and reduced hippocampal neurogenesis similar to total heterozygous γ2 KO mice (Earnheart et al., 2007). In contrast, KD of γ2 in neurons at post-natal day 13/14 did not affect hippocampal neurogenesis, but anxiety- and depressive-like behavior still formed (Shen et al., 2012). Numerous studies have examined brain-region or cell-type specific γ2 KD or KO describing circuit specific roles that will not be discussed here (Buhr et al., 1997; Wingrove et al., 1997; Wulff et al., 2007, 2009; Lee et al., 2010; Leppa et al., 2011, 2016; Zecharia et al., 2012; Stojakovic et al., 2018).
Homozygous deletion of γ2L in mice results in near complete replacement with γ2S subunit (Homanics et al., 1999). When examining γ2 isoform specific ablation, in vitro findings (refer to earlier discussion in Synaptic Accumulation and Functional Regulation) would suggest GABAAR incorporating γ2L vs. γ2S would incur distinct changes in functional and pharmacological properties of GABAAR. Yet, this isoform switch did not result in changed responsiveness to ethanol in behavioral or electrophysiology experiments, although a mild increase in anxiety was observed (Homanics et al., 1999). Interestingly, the γ2L -/- mice did show a modest increase in behavioral sensitivity and GABAAR affinity for benzodiazepine agonists (Quinlan et al., 2000). Isoform switching of γ2 in vivo has been described to occur in response to certain cues such as chronic intermittent ethanol administration in rats (Petrie et al., 2001; Cagetti et al., 2003) and in schizophrenic brains of humans (Huntsman et al., 1998). The relevance of γ2 isoform switching and predominance to pathophysiology in vivo remains poorly understood.
Amongst all the subunit genes, mutations in GABRG2 encoding the γ2 subunit are most commonly linked to epileptogenesis (Macdonald et al., 2012). Indeed, heterozygous γ2 R82Q mutant mice were one of the first in vivo models for childhood absence epilepsy, recapitulating a familial mutation phenotype including onset, behavior, and treatment responsiveness (Tan et al., 2007). GABRG2 genetic anomalies including missense, nonsense, frameshift, splice-site, insertion and deletion mutations are associated with epilepsy phenotypes ranging from mild FS to moderate generalized tonic-clonic seizures or more severe disorders such as Dravet syndrome (DS) or epileptic encephalopathies (further information found in Kang and Macdonald, 2016). In order to bridge the gap between known γ2 trafficking mechanisms, identified protein interaction sites and human pathology, we examined γ2 subunit genetic variation using the Genome Aggregation Database (gnomAD) (Lek et al., 2016), a dataset of exome sequence data from 123,136 individuals and whole genome sequencing from 15,496 unrelated individuals without any severe pediatric disease and their first-degree relatives. We focused specifically on synonymous (codon substitutions result in no amino acid sequence change) and non-synonymous (alter amino acid sequence) mutations. Although synonymous codon changes were previously labeled as “silent” mutations and thought to have limited consequences, recent data indicates these may also impact function and contribute to disease through effects on cis-regulatory elements, mRNA structure, and protein expression. Non-synonymous mutations that result in a stop codon are referred to as nonsense mutations whereas missense mutations result in the exchange of one amino acid for another. Non-synonymous mutations may affect structural and functional properties and be associated with a disease condition; however, others may be functionally neutral and not related to a disease phenotype. Protein domains which show significant diversity in mutations identify regions of genetic flexibility, while regions with low allele frequency events (standard threshold of 0.1%) identify potentially pathogenic mutations that are not evolutionarily favored (Dudley et al., 2012). In the γ2S isoform, we identified and plotted the distribution of 104 synonymous and 122 non-synonymous missense variants (Figure 3A) (Jay and Brouwer, 2016). Five additional non-synonymous variants were found in the γ2L specific sequence (LLRMFSFK: L377R, R379W, R379Q, F381L, S382C), while no synonymous variants were identified (Figure 3A). Of note, there is a third putative γ2 isoform which appears conserved in humans and primates including the great apes and old world monkeys but absent in rodents that was not evaluated here for human genetic variation (ENST00000414552, Y211 is substituted by W, followed by 40 additional amino acids in the N-terminal extracellular domain). Overall, the latter half of the ECD, TM and linker regions showed low levels of missense variation when compared to synonymous variation (Figure 3A).
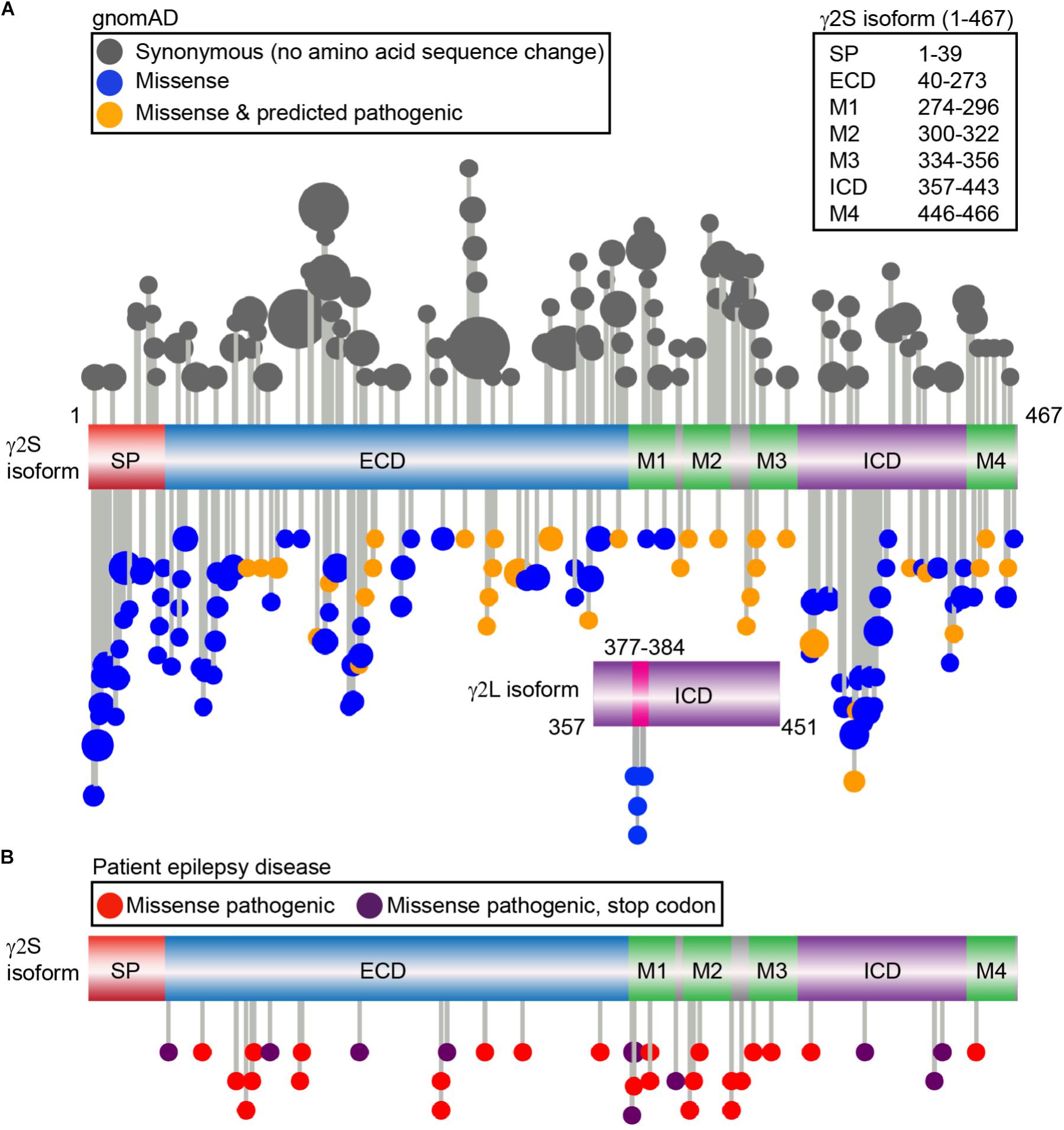
FIGURE 3. Genetic variation of the γ2 GABAAR subunit in gnomAD vs. genetic epilepsies. (A) The gnomAD dataset (individuals without any severe pediatric disease or their first-degree relatives) was used to identify a total of 104 control synonymous variants (gray) and 122 missense non-synonymous variants and plotted; each variant is represented by a lollipop marker that scales with allele frequency. Missense variants were categorized as neutral (blue) or deleterious (orange) through bioinformatics analysis using PROVEAN and SIFT predictions. Linear representation of the γ2 GABAAR subunit with domains: signal peptide (SP; red); extracellular N-terminal region (ECD, blue), transmembrane domain including the four transmembrane helical regions (M1-M4, green); small loops between transmembrane regions (gray); and large intracellular domain between M3-M4 (ICD, purple). The residue numbers correspond to the γ2S sequence (UniProt P18507). The independent ICD below shows the additional residues present in the γ2L isoform (UniProt P18507-2), and 5 distinct missense variants identified, predicted as neutral by PROVEAN: L377R, R379W, R379Q, F381L, S382C. (B) Patient epilepsy disease related missense (red) and nonsense variants leading to early stop codons (dark purple) were compiled as described in Material and Methods and plotted on the linear protein structure. All genetic variant AA residue numbering includes signal peptide.
We next turned to the patient epilepsy disease case variants to determine if these are over-represented in similar regions. Disease case variants were gathered from National Center for Biotechnical Information (NCBI), ClinVar, and Human Gene Mutation Databases (HGMD), yielding a total of 49 pathogenic or likely pathogenic mutations including 25 missense, 11 nonsense, 9 frameshift, and 4 intron splice variants. The distribution of the 36 epilepsy-related missense and nonsense mutations was mapped across the γ2 subunit protein domains (Figure 3B). The 11 γ2 nonsense variants resulted in early stop codons (X) throughout the following domains: (1) ECD = Q40X, L91X, R136X, Y180X, G273X; (2) M1 = Y274X (2 unique stop codon mutant variants), W295X; (3) ICD = Q390X, R425X, W429X. The 25 γ2 subunit missense mutations showed wider distribution throughout the ECD, M1-4, M2-M3 linker and ICD regions. Comparison of the disease-associated and gnomAD missense variants identified significantly greater percentages of epilepsy related variants in the M2 and M2-M3 linker regions (Table 1). In contrast, signal peptide missense mutations were not found and ICD missense mutations were less prevalent in epilepsy patients (Table 1).
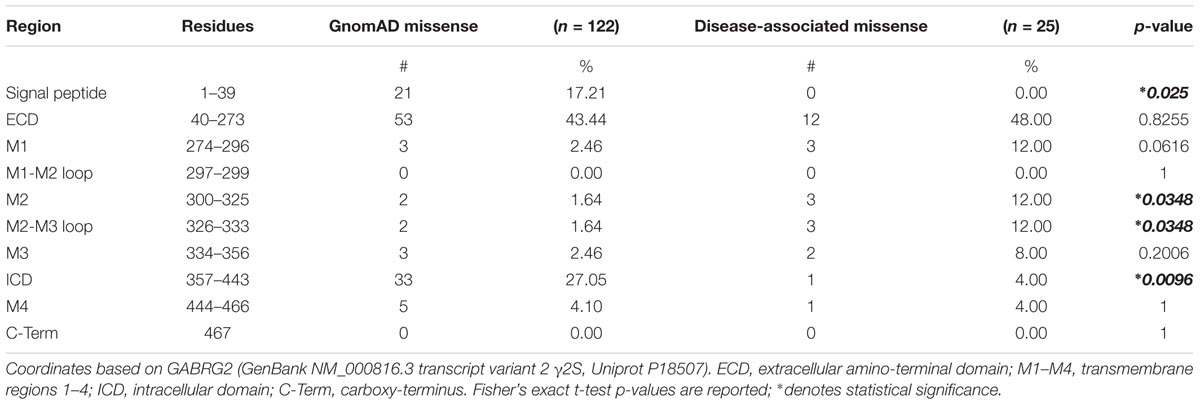
TABLE 1. Genetic variation across GABRG2 domains.
In the field of medical genomics, identification of potentially pathological mutations is a significant challenge, prompting the development of multiple bioinformatics methods to assess non-synonymous variants. We used the sequence homology-based genetic analysis bioinformatics programs PROVEAN (Protein Variation Effect Analyzer) and SIFT (Sorting Intolerant from Tolerant) to assess non-synonymous variants in the gnomAD population and predict the effects on γ2 subunit biological function. Interestingly, 35 of the 122 non-synonymous gnomAD variants were also predicted to be putatively damaging/deleterious by both of the two bioinformatics tools (scoring agreement at 81.9%, Figure 3A, orange colored variants). Neutral scored non-synonymous variants included S386P and T388A (aka S355 and T357 phosphorylation sites, Figure 1C). None of the γ2L isoform missense variants were predicted by PROVEAN as damaging, although S382C (aka S343, the PKC/CaMKII phosphorylation site, see earlier Synaptic Accumulation and Functional Regulation, Figure 1C) was predicted as possibly damaging by SIFT. Among the gnomAD population six variants were identified that overlapped the epilepsy patient missense group (L57F, N79S, M199V, R177Q, A334T, R363Q): three were predicted as deleterious (N79S, M199V, A334T) and 3 as neutral (L57F, R177Q, R363Q). PROVEAN and SIFT bioinformatics analysis of the 25 epilepsy patient missense variants showed four as neutral (L57F, A106T, L307V and R363Q), two had conflicting predictions (L74V, R304K), and all others were scored as damaging. As the gnomAD population is relatively free from significant clinical disorders, this implies masking by epistatic genetic interactions, consistent with phenotypic variability seen in epilepsy patients and animal epilepsy models. In addition, although in silico prediction tools show overall robust performance, particularly when software are used in combination (Leong et al., 2015; Masica and Karchin, 2016), this suggests pathological variants can be missed. Improving clinically admissible predictions from these in silico tools is a current high priority focus in medical bioinformatics (Masica and Karchin, 2016; Ernst et al., 2018). To expand our insight into the cellular pathology underlying the thirty-six patient cases, we next cross-examined database information (NCBI, ClinVar, HGMD) and the current literature for disease phenotypic and cellular study based analysis.
The most common patient phenotypes associated with nonsense and missense mutations ranged in severity and included FS, generalized tonic-clonic seizures (GTCS), GTCS with FS, genetic epilepsy with FS (GEFS), genetic epilepsy (GE), DS, and epileptic encephalopathy with severe global developmental delays (EEDD). FS are a relatively mild pathology which occur in the presence of fevers and display tonic-clonic seizure activity in individuals between 6 months and 5 years of age (Boillot et al., 2015). FS which have prolonged episode duration and occur past 6 years of age are termed FS+ and are generally associated with increased risk for developing epilepsy later in life. Moderate forms of epilepsy include GTCS and GE both with and without FS, where FS can co-occur with persistent seizure episodes past childhood and can present intense seizure activity more commonly known as a “grand mal” seizure as in the case of GTCS (Johnston et al., 2014; Wang et al., 2016; Fisher et al., 2017). The most severe phenotypes reported are DS and EEDD. In particular, DS is subset of epileptic encephalopathy and is characterized by a wide range of seizure type activity as well as psychomotor development delays, ataxia and hyperkinesis emerging between the ages of 1–4 (Ishii et al., 2014; Fisher et al., 2017). In contrast, EEDD have broader phenotypic manifestations and deficits as a result of global neurodevelopmental impairments with treatment-resistant seizures (Shen et al., 2017). Less common reported patient phenotypes included myoclonic epilepsy, absence seizures, complex partial seizures, tonic infantile spasms, tonic seizures, Rolandic epilepsy, and ASD with learning difficulties. In vitro studies have been invaluable in gaining in depth understanding of etiology, cellular pathology, and functional effects of these epilepsy patient variants.
In vitro studies on 17 of the γ2 pathogenic variants have revealed reduced surface expression in 15 cases, in part resulting from ER retention and trafficking defects (Table 2). The severe disease DS epilepsy phenotype is associated with three nonsense mutations (Q40X, R136X, Q390X) and one missense (P302L) mutation (Table 2). The early occurrence of Q40X and R136X within the ECD resulted in premature termination codons (PTCs) and mRNA degradation via nonsense mediated mRNA decay (NMD) with decreased γ2 protein levels. The introduction of upstream PTCs limited the availability of trafficable γ2, diminished overall receptor surface expression and synaptic localization and resulted in significant GABAergic deficits (Ishii et al., 2014). Conversely, the Q390X (previously known as Q351X) mutation occurs in the ICD and escapes NMD but is instead subject to ubiquitin-proteasome degradation (Kang et al., 2013). In vitro experiments found Q390X to have comparable mRNA levels to other late sequence nonsense mutations but dissimilar protein expression due to different degradation rates. Q390X displayed a substantially longer half-life as compared to wild-type γ2 and other nonsense mutant subunits in addition to an increased ability to oligomerize with and sequester wild-type α and β subunits. This alternative disruption in receptor trafficking provides evidence that expressed non-functional truncated subunits may be modifiers of epilepsy phenotype severity. Interestingly, P302L was the only missense mutation reported in a patient with DS (Hernandez et al., 2017). Of note, this mutation resides in M2 and contributes to the formation of the ion channel pore which likely explains its severe phenotype. This is supported by P302L mutant electrophysiological studies and structural modeling which suggests a shift in pore activity resulting in slow activation, low conductance states, and fast desensitization of GABAAR (Hernandez et al., 2017). In contrast, all six cases of EEDD were found in patients with missense mutations (A106T, I107T, P282S, R323W, R323Q, F343L) dispersed throughout structural domains (ECD, M1 and M2) and exhibited additional epileptic phenotypes such as GTCS, GEFS, and tonic seizures (Shen et al., 2017). In fact, the I107T mutation is located in the ECD which typically tolerates missense mutations as evidenced by relatively mild phenotypes; however, this mutation was found to exhibit the most severe cellular pathologies as compared to other disease variants emphasizing the need to further investigate these mutations and their ramifications on cellular processes.
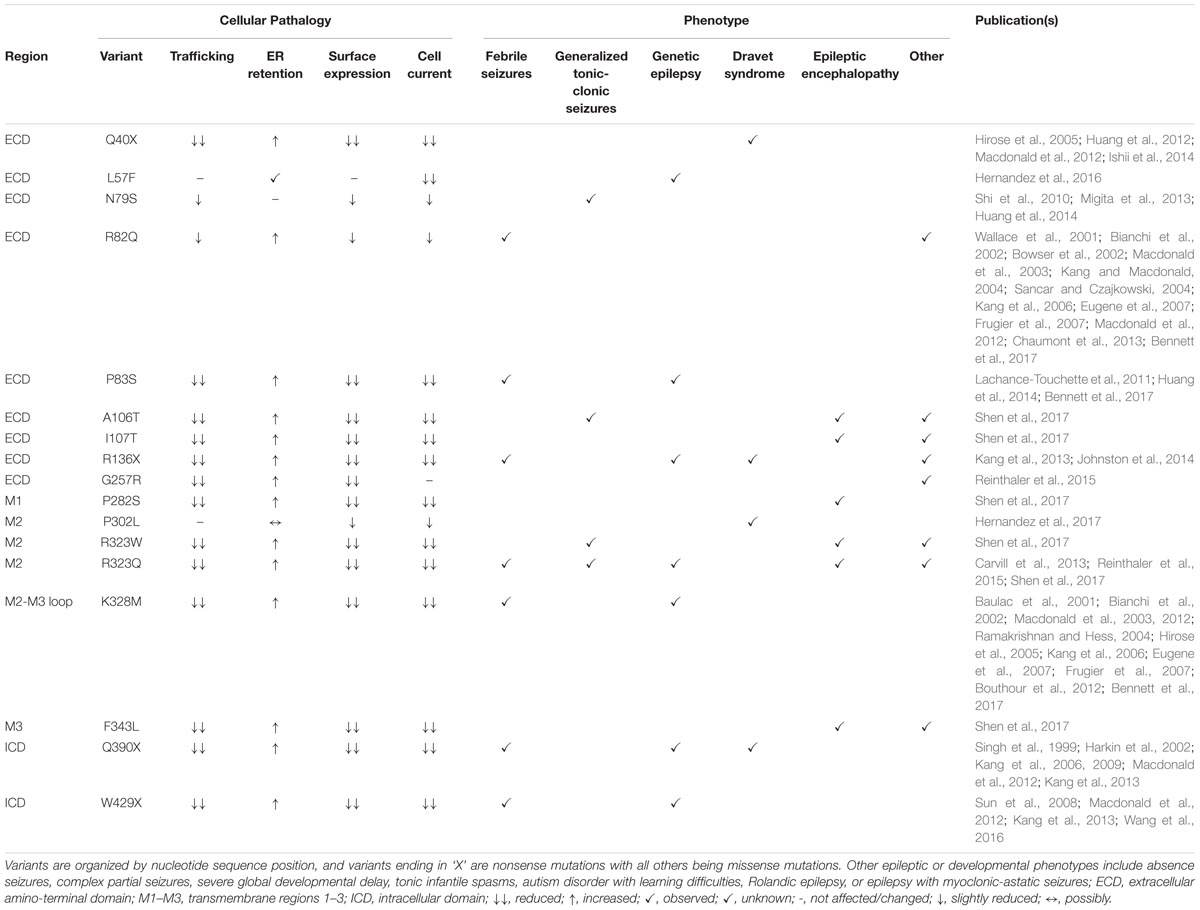
TABLE 2. GABRG2 missense and nonsense patient mutations with associated cellular pathologies and reported clinical phenotypes.
The moderate epileptic phenotype GEFS without co-occurring conditions was observed in three cases with two missense (P83S and K328M) and one nonsense (W429X) variants reported with structural locations in the ECD, M2-M3 linker, and ICD, respectively (Table 2). P83S was found to reduce GABA-evoked whole cell currents mainly through a plasma membrane and trafficking-dependent manner (Lachance-Touchette et al., 2011; Huang et al., 2014; Bennett et al., 2017). In contrast, K328M (previously known as K289M) is found in the short extracellular loop between the M2-M3 regions and was found to increase receptor deactivation, implicating this region in receptor kinetic properties (Macdonald et al., 2012). Conversely, W429X displayed less drastic protein degradation and subunit oligomerization pathologies compared to the previously discussed DS variant Q390X (Wang et al., 2016). The later downstream incidence of W429X combined with slightly higher surface expression compared to Q390X may explain the milder epilepsy phenotype (Sun et al., 2008; Macdonald et al., 2012; Kang et al., 2013; Wang et al., 2016).
Throughout the reviewed mutations, only two variants (L57F and N79S) deviated from a pathology associated with reduced γ2 containing GABAAR plasma membrane levels and were located in the ECD. L57F was present in an individual with GE and found to have normal surface and trafficking characteristics compared to wild-type γ2 receptors; however, altered current density properties and function were observed possibly due to minor structural perturbations in the α1-helix of the ECD (Hernandez et al., 2016). Comparatively, the N79S mutation was the sole occurrence of GTCS without co-occurring phenotypes and presented slight but significant impairments in plasma membrane levels and peak current amplitude (Huang et al., 2014) suggesting it is more of a susceptibility variant as opposed to an epilepsy mutation (Shi et al., 2010; Migita et al., 2013; Huang et al., 2014). Moreover, the resilience of the ECD is further supported by R82Q (previously known as R43Q), a well characterized missense mutation associated with mild phenotypic manifestations like FS and absence seizures with trafficking deficient pathologies (Macdonald et al., 2012). Overall, the 13 frameshift and intron splice variant mutations analyzed were associated with mild phenotypes, though further studies are needed to elucidate their pathological mechanisms (Table 3). However, frameshift mutations within the ICD (E402Dfs∗3 generating a stop codon at Y404X critical Src/Fyn phospho site discussed earlier; and S443delC resulting in an altered and elongated carboxy terminus with +50 novel AA) were associated with more moderate-severe phenotypes like GTCS and GEFS+ underscoring the importance for intracellular regulation via the ICD (Macdonald et al., 2012).
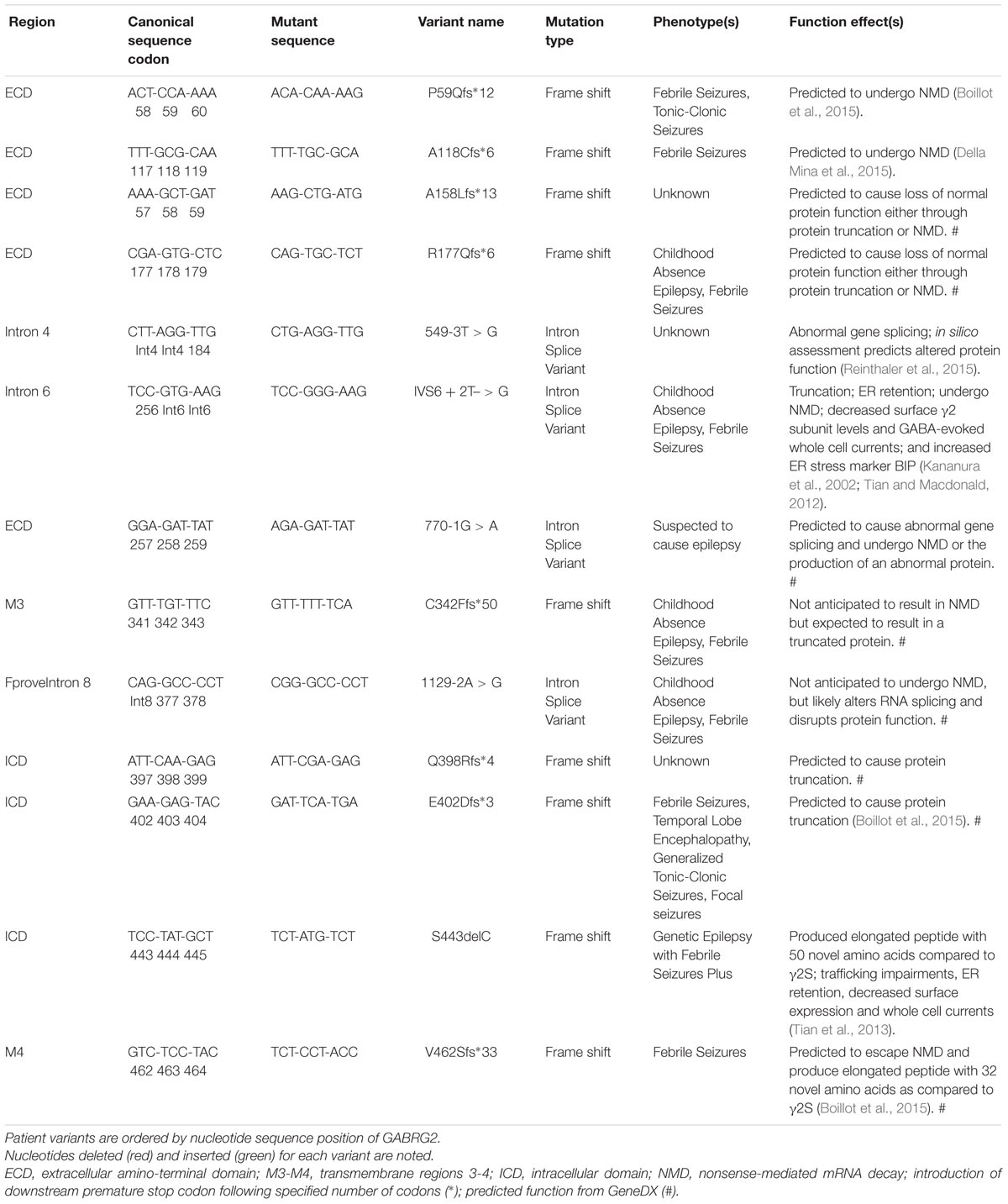
TABLE 3. Patient frameshift mutations and intron splice variants associated or likely associated with various epilepsy phenotypes.
In summary, both deficits in GABAAR surface trafficking and the functional role of specific γ2 subunit regions are critical factors modulating phenotypic outcome, with some missense mutations resulting in phenotypes as severe as nonsense mutations. Furthermore, expressed non-functional truncated subunits may be correlated with more severe manifestations and be modifiers of disease phenotypes. Disease case variants in the pore lining M2 region showed particularly severe phenotypes, consistent with the reduced genetic variation in this region in gnomAD non-synonymous variants. Clearly, in vitro studies of recombinant receptor trafficking, electrophysiology and assembly have provided important insight into the underlying cellular pathology and functional effects of these epilepsy patient variants. Greater understanding of the consequences of γ2 genetic variation, both for revealing disease mechanisms and for GABAAR synaptic plasticity will be gained through application of innovative imaging methods in the neuronal context.
Advancing imaging techniques are providing critical insight into GABAAR trafficking extending beyond basic endo/exocytic trafficking of receptors. Live-cell imaging using pH-sensitive GFP (pHluorin) tagged GABAARs subunits and fluorescence recovery after photobleaching (FRAP) experiments first identified GABAAR synaptic retention, limiting diffusion at synaptic release sites, and the crucial role of gephyrin in this process (Jacob et al., 2005). Receptor subunits with pHluorin tags have further described GABAAR surface levels and lysosomal degradation (Jacob et al., 2012; Lorenz-Guertin et al., 2017) and novel exocytic machinery and insertion sites of receptors (Gu et al., 2016). The pHluorin-FRAP technique is often performed in addition to the newer workhorse of diffusion studies, quantum dot (QD) single-particle tracking. QD studies have revealed precise quantitative properties of synaptic and extrasynaptic GABAAR diffusion during baseline conditions (Renner et al., 2012), excitatory stimulation (including iLTP) (Bannai et al., 2009, 2015; Muir et al., 2010; Niwa et al., 2012; Muir and Kittler, 2014; Petrini et al., 2014), GABAAR agonist and/or drug treatment (Gouzer et al., 2014; Levi et al., 2015; de Luca et al., 2017), GABAB receptor activation (Gerrow and Triller, 2014), purinergic (P2x2 receptor) activation (Shrivastava et al., 2011), and changes in gephyrin or radixin phosphorylation (Hausrat et al., 2015; Battaglia et al., 2018). Receptor functional regulation by changes in surface diffusion, perhaps completely independent of changes in surface levels, represents a paradigm shift in our basic understanding of synaptic plasticity. Indeed current studies of human genetic variants in recombinant systems are unlikely to detect these fundamentally important properties due to lack of a neuronal context, the appropriate GABAAR subunit complement, interacting proteins, and general overexpression problems. For example, QD neuronal studies of the γ2 K328M disease variant revealed an additional phenotype of enhanced temperature sensitive receptor diffusion, likely contributing to the FS pathology in patients (Bouthour et al., 2012).
To address multiple trafficking questions within a single assay, our group recently engineered a GABAAR γ2 subunit dual fluorescent sensor encoding a pHluorin tag and a fluorogen-activating peptide (FAP) (γ2pHFAP) (Lorenz-Guertin et al., 2017). FAPs are antibody single chain variable fragments characterized to selectively bind inorganic dyes with high specificity and affinity (Szent-Gyorgyi et al., 2008). The dyes are non-fluorescent until bound by a FAP and individual dyes have unique characteristics including cell permeability, pH-sensitivity, fluorescent properties, and in vivo administration capability (Fisher et al., 2010; Grover et al., 2012; Saunders et al., 2012; Zhang et al., 2015; He et al., 2016). We have used the FAP-dye system in neurons to selectively examine cell surface GABAARs undergoing internalization, early endosomal accumulation and targeting to late endosomes/lysosomes via confocal live-imaging (Lorenz-Guertin et al., 2017). Pulse-labeling γ2pHFAP with cell impermeable dye allows for detection of surface receptor turnover rates independent of a change in total GABAAR surface levels, as we demonstrated using a mild seizure protocol. As more GABAARs subunits are engineered to express the FAP tag, and additional unique dyes are synthesized to address specific experimental questions, the utility of this imaging approach continues to grow.
Other innovative imaging approaches advancing our ability to detect changes in GABAAR synaptic plasticity include optogenetic toolkits for controlling GABAAR activity (Lin et al., 2014, 2015), spatially regulated GABA activation using two-photon photolysis (Oh et al., 2016), proximity ligation assays to measure endogenous protein interaction (Smith et al., 2014; Tseng et al., 2015; Ghosh et al., 2016), and super-resolution imaging and other fluorescent tools to examine inhibitory gephyrin scaffolding (Gross et al., 2013, 2016; Sigal et al., 2015; Maric et al., 2017; Pennacchietti et al., 2017). Fluorescence resonance energy transfer (FRET) techniques have been limitedly applied to studying GABAAR trafficking or receptor subunit composition (Ding et al., 2010; Shrivastava et al., 2011), collectively suggesting imaging techniques will be a rich resource of novel GABAAR knowledge.
In summary, we live in an unprecedented time for understanding human disease pathology and neurodevelopment through integration of “big data” on human genetic variation and protein interaction networks/interactomes, in combination with high resolution live-imaging approaches. Future efforts to resolve GABAAR pathologies will benefit from connecting genetic variants to their cellular mechanisms of pathology within the complexity of neuronal signaling. Importantly, increased understanding of surface and intracellular pool regulated trafficking of GABAAR will provide mechanisms to treat overall reduced receptor levels in various disease states. Future treatment of genetic epilepsy syndromes are likely to involve CRISPR-Cas9 gene editing (Ma et al., 2017), RNA focused REPAIR editing approaches, or application of improved drugs that act as chaperones to promote receptor trafficking. The new imaging based methods described here are particularly likely to show high utility in both identifying cellular pathology of human GABAAR genetic variants and for drug screening efforts in a neuronal context.
The prevalence of γ2 subunit non-synonymous and synonymous variations in gnomAD1, currently a dataset of exome sequence data from 123,136 individuals and whole genome sequencing from 15,496 unrelated individuals, was assessed and restricted to those meeting the “PASS” quality threshold (Lek et al., 2016). Individuals known to be affected by severe pediatric disease are not contained in this data set, or their first-degree relatives. Next “pathogenic” and “likely pathogenic” patient case variants not present in the gnomAD dataset were investigated in National Center for Biotechnical Information variation viewer (NCBIvv)1, ClinVar, and Human Gene Mutation Databases (HGMD) utilizing the following search parameters: GRCh37.p13 annotation release 105 assembly and NM_000816.3 (transcript variant 2, γ2S). The search in NCBIvv identified 17 variants (accessed January 2018). The ClinVar search (accessed February 2018) confirmed 16/17 candidate variants with the outlier (R323W) having been newly identified in the literature (Shen et al., 2017)2. In addition to those confirmed, the ClinVar investigation produced 10 additional mutations. Some variants identified in ClinVar had associated predicted functions (submitted by GeneDX genetics company)3. Finally, HGMD (hgmd2018.1; accessed March 2018) interrogation uncovered 22 disease-causing mutations that were absent from NCBIvv and ClinVar inquiries4. Using these candidate case variants and their associated database information, the current literature was evaluated for disease phenotypic and cellular study based implications yielding a total of 49 pathogenic or likely pathogenic mutations including 25 missense, 11 nonsense, 9 frameshift, and 4 intron splice variants. We used lollipops-v.1.3.1 software (Jay and Brouwer, 2016) to plot the distribution of synonymous, non-synonymous and disease case mutations in GABRG2 along a linear γ2S assembly (P18507, ENST00000361925) and a linear segment representation of the additional eight encoded amino acids within the ICD in the γ2L isoform (P18507-2, ENST00000356592). The missense and nonsense disease case variants studied at the cellular trafficking level were included in Table 2. The frameshift and intron splice variants were annotated in Table 3.
PROVEAN (Protein Variation Effect Analyzer5 (Choi et al., 2012) and SIFT (Sorting Intolerant from Tolerant) algorithms (Hu and Ng, 2013) are bioinformatics tools which predict whether an amino acid substitution or indel (insertion or deletion) has an impact on a protein’s biological function using homology based genetic analysis. Currently PROVEAN provides scoring via both PROVEAN and SIFT algorithms. PROVEAN utilizes pairwise sequence alignment scores to generate pre-computed predictions at every amino acid position in all human and mouse protein sequences. Mutations are predicted to be deleterious or tolerant based on the prediction cutoff value of -2.5: scores smaller than -2.5 are considered deleterious. Similarly, SIFT predicts whether the amino acid substitution alter the protein function based on sequence homology and the physical properties of amino acids. The intolerant range of SIFT is ≤0.05 for predicted damaging/deleterious mutations and a score of >0.05 predicts the tolerant range.
• gnomAD, http://gnomad.broadinstitute.org/
• ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
• Human Gene Mutation Database, http://www.hgmd.org/
• Lollipops v.1.3.1, https://github.com/pbnjay/lollipops/releases
• UniProt, http://www.uniprot.org/
JL-G and TJ wrote and edited the sections “γ2 Subunit Trafficking and Interactors” and “Looking Forward: Imaging Advances.” MB and TJ analyzed, wrote, and prepared the section “Human Genetic Variation of γ2 and Pathological Implications” and associated tables. TJ prepared all the figures. MB prepared all the tables.
This work was generously funded by T32GM008424 and R56MH114908-01, the William C. deGroat Neuropharmacology Departmental Fellowship, and Pharmacology and Chemical Biology Startup Funds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MG and handling Editor declared their shared affiliation at the time of review.
Alldred, M. J., Mulder-Rosi, J., Lingenfelter, S. E., Chen, G., and Luscher, B. (2005). Distinct gamma2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J. Neurosci. 25, 594–603. doi: 10.1523/jneurosci.4011-04.2005
Arancibia-Carcamo, I. L., Yuen, E. Y., Muir, J., Lumb, M. J., Michels, G., Saliba, R. S., et al. (2009). Ubiquitin-dependent lysosomal targeting of GABA(A) receptors regulates neuronal inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 17552–17557. doi: 10.1073/pnas.0905502106
Bannai, H., Levi, S., Schweizer, C., Inoue, T., Launey, T., Racine, V., et al. (2009). Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron 62, 670–682. doi: 10.1016/j.neuron.2009.04.023
Bannai, H., Niwa, F., Sherwood, M. W., Shrivastava, A. N., Arizono, M., Miyamoto, A., et al. (2015). Bidirectional control of synaptic GABAAR clustering by glutamate and calcium. Cell Rep. 13, 2768–2780. doi: 10.1016/j.celrep.2015.12.002
Battaglia, S., Renner, M., Russeau, M., and Come, E. (2018). Activity-dependent inhibitory synapse scaling is determined by gephyrin phosphorylation and subsequent regulation of GABAA receptor diffusion. eNeuro 5:ENEURO.0203-17.2017. doi: 10.1523/eneuro.0203-17.2017
Baulac, S., Huberfeld, G., Gourfinkel-An, I., Mitropoulou, G., Beranger, A., Prud’homme, J. F., et al. (2001). First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet. 28, 46–48. doi: 10.1038/88254
Bennett, C. A., Petrovski, S., Oliver, K. L., and Berkovic, S. F. (2017). ExACtly zero or once: a clinically helpful guide to assessing genetic variants in mild epilepsies. Neurol. Genet. 3:e163. doi: 10.1212/NXG.0000000000000163
Bianchi, M. T., Song, L., Zhang, H., and Macdonald, R. L. (2002). Two different mechanisms of disinhibition produced by GABA(A) receptor mutations linked to epilepsy in humans. J. Neurosci. 22, 5321–5327. doi: 10.1523/JNEUROSCI.22-13-05321.2002
Bogdanov, Y., Michels, G., Armstrong-Gold, C., Haydon, P. G., Lindstrom, J., Pangalos, M., et al. (2006). Synaptic GABAA receptors are directly recruited from their extrasynaptic counterparts. EMBO J. 25, 4381–4389. doi: 10.1038/sj.emboj.7601309
Boileau, A. J., Pearce, R. A., and Czajkowski, C. (2010). The short splice variant of the gamma 2 subunit acts as an external modulator of GABA(A) receptor function. J. Neurosci. 30, 4895–4903. doi: 10.1523/jneurosci.5039-09.2010
Boillot, M., Morin-Brureau, M., Picard, F., Weckhuysen, S., Lambrecq, V., Minetti, C., et al. (2015). Novel GABRG2 mutations cause familial febrile seizures. Neurol. Genet. 1:e35. doi: 10.1212/NXG.0000000000000035
Bouthour, W., Leroy, F., Emmanuelli, C., Carnaud, M., Dahan, M., Poncer, J. C., et al. (2012). A human mutation in Gabrg2 associated with generalized epilepsy alters the membrane dynamics of GABAA receptors. Cereb. Cortex 22, 1542–1553. doi: 10.1093/cercor/bhr225
Bowser, D. N., Wagner, D. A., Czajkowski, C., Cromer, B. A., Parker, M. W., Wallace, R. H., et al. (2002). Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [gamma 2(R43Q)] found in human epilepsy. Proc. Natl. Acad. Sci. U.S.A. 99, 15170–15175. doi: 10.1073/pnas.212320199
Brady, M. L., and Jacob, T. C. (2015). Synaptic localization of alpha5 GABA (A) receptors via gephyrin interaction regulates dendritic outgrowth and spine maturation. Dev. Neurobiol. 75, 1241–1251. doi: 10.1002/dneu.22280
Brandon, N., Jovanovic, J., and Moss, S. (2002). Multiple roles of protein kinases in the modulation of gamma-aminobutyric acid(A) receptor function and cell surface expression. Pharmacol. Ther. 94, 113–122. doi: 10.1016/S0163-7258(02)00175-4
Brandon, N. J., Delmas, P., Hill, J., Smart, T. G., and Moss, S. J. (2001). Constitutive tyrosine phosphorylation of the GABA(A) receptor gamma 2 subunit in rat brain. Neuropharmacology 41, 745–752. doi: 10.1016/S0028-3908(01)00121-6
Brandon, N. J., Jovanovic, J. N., Colledge, M., Kittler, J. T., Brandon, J. M., Scott, J. D., et al. (2003). A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABA(A) receptors by cAMP-dependent protein kinase via selective interaction with receptor beta subunits. Mol. Cell. Neurosci. 22, 87–97. doi: 10.1016/S1044-7431(02)00017-9
Brennan, G. P., Jimenez-Mateos, E. M., McKiernan, R. C., Engel, T., Tzivion, G., and Henshall, D. C. (2013). Transgenic overexpression of 14-3-3 zeta protects hippocampus against endoplasmic reticulum stress and status epilepticus in vivo. PLoS One 8:e54491. doi: 10.1371/journal.pone.0054491
Buhr, A., Baur, R., and Sigel, E. (1997). Subtle changes in residue 77 of the gamma subunit of alpha1beta2gamma2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J. Biol. Chem. 272, 11799–11804. doi: 10.1074/jbc.272.18.11799
Cagetti, E., Liang, J., Spigelman, I., and Olsen, R. W. (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol. Pharmacol. 63, 53–64. doi: 10.1124/mol.63.1.53
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O’Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi: 10.1038/ng.2646
Chandra, D., Korpi, E. R., Miralles, C. P., De Blas, A. L., and Homanics, G. E. (2005). GABAA receptor gamma 2 subunit knockdown mice have enhanced anxiety-like behavior but unaltered hypnotic response to benzodiazepines. BMC Neurosci. 6:30. doi: 10.1186/1471-2202-6-30
Chaumont, S., Andre, C., Perrais, D., Boue-Grabot, E., Taly, A., and Garret, M. (2013). Agonist-dependent endocytosis of gamma-aminobutyric acid type A (GABAA) receptors revealed by a gamma2(R43Q) epilepsy mutation. J. Biol. Chem. 288, 28254–28265. doi: 10.1074/jbc.M113.470807
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688. doi: 10.1371/journal.pone.0046688
Chou, W. H., Wang, D., McMahon, T., Qi, Z. H., Song, M., Zhang, C., et al. (2010). GABAA receptor trafficking is regulated by protein kinase C(epsilon) and the N-ethylmaleimide-sensitive factor. J. Neurosci. 30, 13955–13965. doi: 10.1523/jneurosci.0270-10.2010
Cinar, H., and Barnes, E. M. Jr. (2001). Clathrin-independent endocytosis of GABA(A) receptors in HEK 293 cells. Biochemistry 40, 14030–14036. doi: 10.1021/bi011025t
Connolly, C. N., Kittler, J. T., Thomas, P., Uren, J. M., Brandon, N. J., Smart, T. G., et al. (1999). Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. J. Biol. Chem. 274, 36565–36572. doi: 10.1074/jbc.274.51.36565
Crestani, F., Lorez, M., Baer, K., Essrich, C., Benke, D., Laurent, J. P., et al. (1999). Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat. Neurosci. 2, 833–839. doi: 10.1038/12207
Davenport, E. C., Pendolino, V., Kontou, G., McGee, T. P., Sheehan, D. F., Lopez-Domenech, G., et al. (2017). An essential role for the tetraspanin LHFPL4 in the cell-type-specific targeting and clustering of synaptic GABAA receptors. Cell Rep. 21, 70–83. doi: 10.1016/j.celrep.2017.09.025
de Luca, E., Ravasenga, T., Petrini, E. M., Polenghi, A., Nieus, T., Guazzi, S., et al. (2017). Inter-synaptic lateral diffusion of GABAA receptors shapes inhibitory synaptic currents. Neuron 95, 63–69.e5. doi: 10.1016/j.neuron.2017.06.022
Della Mina, E., Ciccone, R., Brustia, F., Bayindir, B., Limongelli, I., Vetro, A., et al. (2015). Improving molecular diagnosis in epilepsy by a dedicated high-throughput sequencing platform. Eur. J. Hum. Genet. 23, 354–362. doi: 10.1038/ejhg.2014.92
Ding, L., Feng, H. J., Macdonald, R. L., Botzolakis, E. J., Hu, N., and Gallagher, M. J. (2010). GABA(A) receptor alpha1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABA(A) receptors. J. Biol. Chem. 285, 26390–26405. doi: 10.1074/jbc.M110.142299
Dudley, J. T., Kim, Y., Liu, L., Markov, G. J., Gerold, K., Chen, R., et al. (2012). Human genomic disease variants: a neutral evolutionary explanation. Genome Res. 22, 1383–1394. doi: 10.1101/gr.133702.111
Earnheart, J. C., Schweizer, C., Crestani, F., Iwasato, T., Itohara, S., Mohler, H., et al. (2007). GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. J. Neurosci. 27, 3845–3854. doi: 10.1523/jneurosci.3609-06.2007
Ernst, C., Hahnen, E., Engel, C., Nothnagel, M., Weber, J., Schmutzler, R. K., et al. (2018). Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med. Genomics 11:35. doi: 10.1186/s12920-018-0353-y
Eugene, E., Depienne, C., Baulac, S., Baulac, M., Fritschy, J. M., Le Guern, E., et al. (2007). GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J. Neurosci. 27, 14108–14116. doi: 10.1523/JNEUROSCI.2618-07.2007
Fang, C., Deng, L., Keller, C. A., Fukata, M., Fukata, Y., Chen, G., et al. (2006). GODZ-mediated palmitoylation of GABA(A) receptors is required for normal assembly and function of GABAergic inhibitory synapses. J. Neurosci. 26, 12758–12768. doi: 10.1523/jneurosci.4214-06.2006
Feng, J., Cai, X., Zhao, J., and Yan, Z. (2001). Serotonin receptors modulate GABA(A) receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. J. Neurosci. 21, 6502–6511. doi: 10.1523/JNEUROSCI.21-17-06502.2001
Fisher, G. W., Adler, S. A., Fuhrman, M. H., Waggoner, A. S., Bruchez, M. P., and Jarvik, J. W. (2010). Detection and quantification of beta2AR internalization in living cells using FAP-based biosensor technology. J. Biomol. Screen. 15, 703–709. doi: 10.1177/1087057110370892
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., et al. (2017). Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia 58, 522–530. doi: 10.1111/epi.13670
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Frugier, G., Coussen, F., Giraud, M. F., Odessa, M. F., Emerit, M. B., Boue-Grabot, E., et al. (2007). A gamma 2(R43Q) mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell surface. J. Biol. Chem. 282, 3819–3828. doi: 10.1074/jbc.M608910200
Ge, Y., Kang, Y., Cassidy, R. M., Moon, K. M., Lewis, R., Wong, R. O. L., et al. (2018). Clptm1 limits forward trafficking of GABAA receptors to scale inhibitory synaptic strength. Neuron 97, 596–610.e8. doi: 10.1016/j.neuron.2017.12.038
Gerrow, K., and Triller, A. (2014). GABAA receptor subunit composition and competition at synapses are tuned by GABAB receptor activity. Mol. Cell. Neurosci. 60, 97–107. doi: 10.1016/j.mcn.2014.04.001
Ghosh, H., Auguadri, L., Battaglia, S., Simone Thirouin, Z., Zemoura, K., Messner, S., et al. (2016). Several posttranslational modifications act in concert to regulate gephyrin scaffolding and GABAergic transmission. Nat. Commun. 7:13365. doi: 10.1038/ncomms13365
Gonzalez, C., Moss, S. J., and Olsen, R. W. (2012). Ethanol promotes clathrin adaptor-mediated endocytosis via the intracellular domain of delta-containing GABAA receptors. J. Neurosci. 32, 17874–17881. doi: 10.1523/jneurosci.2535-12.2012
Goodkin, H. P., Joshi, S., Mtchedlishvili, Z., Brar, J., and Kapur, J. (2008). Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 28, 2527–2538. doi: 10.1523/jneurosci.3426-07.2008
Goodkin, H. P., Yeh, J. L., and Kapur, J. (2005). Status epilepticus increases the intracellular accumulation of GABAA receptors. J. Neurosci. 25, 5511–5520. doi: 10.1523/jneurosci.0900-05.2005
Goto, H., Terunuma, M., Kanematsu, T., Misumi, Y., Moss, S. J., and Hirata, M. (2005). Direct interaction of N-ethylmaleimide-sensitive factor with GABA(A) receptor beta subunits. Mol. Cell. Neurosci. 30, 197–206. doi: 10.1016/j.mcn.2005.07.006
Gouzer, G., Specht, C. G., Allain, L., Shinoe, T., and Triller, A. (2014). Benzodiazepine-dependent stabilization of GABA(A) receptors at synapses. Mol. Cell. Neurosci. 63, 101–113. doi: 10.1016/j.mcn.2014.10.004
Gross, G. G., Junge, J. A., Mora, R. J., Kwon, H. B., Olson, C. A., Takahashi, T. T., et al. (2013). Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78, 971–985. doi: 10.1016/j.neuron.2013.04.017
Gross, G. G., Straub, C., Perez-Sanchez, J., Dempsey, W. P., Junge, J. A., Roberts, R. W., et al. (2016). An E3-ligase-based method for ablating inhibitory synapses. Nat Methods 13, 673–678. doi: 10.1038/nmeth.3894
Grover, A., Schmidt, B. F., Salter, R. D., Watkins, S. C., Waggoner, A. S., and Bruchez, M. P. (2012). Genetically encoded pH sensor for tracking surface proteins through endocytosis. Angew. Chem. Int. Ed. Engl. 51, 4838–4842. doi: 10.1002/anie.201108107
Gu, Y., Chiu, S. L., Liu, B., Wu, P. H., Delannoy, M., Lin, D. T., et al. (2016). Differential vesicular sorting of AMPA and GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 113, E922–E931. doi: 10.1073/pnas.1525726113
Gunther, U., Benson, J., Benke, D., Fritschy, J. M., Reyes, G., Knoflach, F., et al. (1995). Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 92, 7749–7753. doi: 10.1073/pnas.92.17.7749
Gutierrez, M. L., Ferreri, M. C., and Gravielle, M. C. (2014). GABA-induced uncoupling of GABA/benzodiazepine site interactions is mediated by increased GABAA receptor internalization and associated with a change in subunit composition. Neuroscience 257, 119–129. doi: 10.1016/j.neuroscience.2013.10.077
Harkin, L. A., Bowser, D. N., Dibbens, L. M., Singh, R., Phillips, F., Wallace, R. H., et al. (2002). Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 70, 530–536. doi: 10.1086/338710
Hassaine, G., Deluz, C., Grasso, L., Wyss, R., Tol, M. B., Hovius, R., et al. (2014). X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 512, 276–281. doi: 10.1038/nature13552
Hausrat, T. J., Muhia, M., Gerrow, K., Thomas, P., Hirdes, W., Tsukita, S., et al. (2015). Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory. Nat. Commun. 6:6872. doi: 10.1038/ncomms7872
He, J., Wang, Y., Missinato, M. A., Onuoha, E., Perkins, L. A., Watkins, S. C., et al. (2016). A genetically targetable near-infrared photosensitizer. Nat. Methods 13, 263–268. doi: 10.1038/nmeth.3735
Hernandez, C. C., Klassen, T. L., Jackson, L. G., Gurba, K., Hu, N., Noebels, J. L., et al. (2016). Deleterious rare variants reveal risk for loss of GABAA receptor function in patients with genetic epilepsy and in the general population. PLoS One 11:e0162883. doi: 10.1371/journal.pone.0162883
Hernandez, C. C., Kong, W., Hu, N., Zhang, Y., Shen, W., Jackson, L., et al. (2017). Altered channel conductance states and gating of GABAA receptors by a pore mutation linked to Dravet syndrome. eNeuro 4:ENEURO.0251-16.2017. doi: 10.1523/ENEURO.0251-16.2017
Herring, D., Huang, R., Singh, M., Dillon, G. H., and Leidenheimer, N. J. (2005). PKC modulation of GABAA receptor endocytosis and function is inhibited by mutation of a dileucine motif within the receptor beta 2 subunit. Neuropharmacology 48, 181–194. doi: 10.1016/j.neuropharm.2004.09.015
Hines, R. M., Davies, P. A., Moss, S. J., and Maguire, J. (2012). Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 22, 552–558. doi: 10.1016/j.conb.2011.10.007
Hirose, S., Mitsudome, A., Okada, M., and Kaneko, S. (2005). Genetics of idiopathic epilepsies. Epilepsia 46, 38–43. doi: 10.1111/j.0013-9580.2005.461011.x
Homanics, G. E., Harrison, N. L., Quinlan, J. J., Krasowski, M. D., Rick, C. E., de Blas, A. L., et al. (1999). Normal electrophysiological and behavioral responses to ethanol in mice lacking the long splice variant of the gamma2 subunit of the gamma-aminobutyrate type A receptor. Neuropharmacology 38, 253–265. doi: 10.1016/S0028-3908(98)00177-4
Hu, J., and Ng, P. C. (2013). SIFT Indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS One 8:e77940. doi: 10.1371/journal.pone.0077940
Huang, X., Hernandez, C. C., Hu, N., and Macdonald, R. L. (2014). Three epilepsy-associated GABRG2 missense mutations at the gamma+/beta- interface disrupt GABAA receptor assembly and trafficking by similar mechanisms but to different extents. Neurobiol. Dis. 68, 167–179. doi: 10.1016/j.nbd.2014.04.015
Huang, X., Tian, M., Hernandez, C. C., Hu, N., and Macdonald, R. L. (2012). The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol. Dis. 48, 115–123. doi: 10.1016/j.nbd.2012.06.013
Huntsman, M. M., Tran, B. V., Potkin, S. G., Bunney, W. E., and Jones, E. G. (1998). Altered ratios of alternatively spliced long and short gamma2 subunit mRNAs of the gamma-amino butyrate type A receptor in prefrontal cortex of schizophrenics. Proc. Natl. Acad. Sci. U.S.A. 95, 15066–15071. doi: 10.1073/pnas.95.25.15066
Ishii, A., Kanaumi, T., Sohda, M., Misumi, Y., Zhang, B., Kakinuma, N., et al. (2014). Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. 108, 420–432. doi: 10.1016/j.eplepsyres.2013.12.005
Jacob, T. C., Bogdanov, Y. D., Magnus, C., Saliba, R. S., Kittler, J. T., Haydon, P. G., et al. (2005). Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J. Neurosci. 25, 10469–10478. doi: 10.1523/jneurosci.2267-05.2005
Jacob, T. C., Michels, G., Silayeva, L., Haydon, J., Succol, F., and Moss, S. J. (2012). Benzodiazepine treatment induces subtype-specific changes in GABA(A) receptor trafficking and decreases synaptic inhibition. Proc. Natl. Acad. Sci. U.S.A. 109, 18595–18600. doi: 10.1073/pnas.1204994109
Jay, J. J., and Brouwer, C. (2016). Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS One 11:e0160519. doi: 10.1371/journal.pone.0160519
Jin, H., Chiou, T. T., Serwanski, D. R., Miralles, C. P., Pinal, N., and De Blas, A. L. (2014). Ring finger protein 34 (RNF34) interacts with and promotes gamma-aminobutyric acid type-A receptor degradation via ubiquitination of the gamma2 subunit. J. Biol. Chem. 289, 29420–29436. doi: 10.1074/jbc.M114.603068
Johnston, A. J., Kang, J. Q., Shen, W., Pickrell, W. O., Cushion, T. D., Davies, J. S., et al. (2014). A novel GABRG2 mutation, p.R136∗, in a family with GEFS+ and extended phenotypes. Neurobiol. Dis. 64, 131–141. doi: 10.1016/j.nbd.2013.12.013
Joshi, S., Rajasekaran, K., Hawk, K. M., Brar, J., Ross, B. M., Tran, C. A., et al. (2015). Phosphatase inhibition prevents the activity-dependent trafficking of GABAA receptors during status epilepticus in the young animal. Epilepsia 56, 1355–1365. doi: 10.1111/epi.13098
Jurd, R., Tretter, V., Walker, J., Brandon, N. J., and Moss, S. J. (2010). Fyn kinase contributes to tyrosine phosphorylation of the GABA receptor gamma2 subunit. Mol. Cell. Neurosci. 44, 129–134. doi: 10.1016/j.mcn.2010.03.002
Kananura, C., Haug, K., Sander, T., Runge, U., Gu, W., Hallmann, K., et al. (2002). A splice-site mutations in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch. Neurol. 59, 1137–1141. doi: 10.1001/archneur.59.7.1137
Kanematsu, T., Jang, I. S., Yamaguchi, T., Nagahama, H., Yoshimura, K., Hidaka, K., et al. (2002). Role of the PLC-related, catalytically inactive protein p130 in GABA(A) receptor function. EMBO J. 21, 1004–1011. doi: 10.1093/emboj/21.5.1004
Kang, J. Q., and Macdonald, R. L. (2004). The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J. Neurosci. 24, 8672–8677. doi: 10.1523/jneurosci.2717-04.2004
Kang, J. Q., and Macdonald, R. L. (2016). Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 73, 1009–1016. doi: 10.1001/jamaneurol.2016.0449
Kang, J. Q., Shen, W., and Macdonald, R. L. (2006). Why does fever trigger febrile seizures? GABAA receptor gamma2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J. Neurosci. 26, 2590–2597. doi: 10.1523/jneurosci.4243-05.2006
Kang, J. Q., Shen, W., and Macdonald, R. L. (2009). The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J. Neurosci. 29, 2845–2856. doi: 10.1523/JNEUROSCI.4772-08.2009
Kang, J. Q., Shen, W., and Macdonald, R. L. (2013). Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann. Neurol. 74, 547–559. doi: 10.1002/ana.23947
Kano, M., Kano, M., Fukunaga, K., and Konnerth, A. (1996). Ca(2+)-induced rebound potentiation of gamma-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc. Natl. Acad. Sci. U.S.A. 93, 13351–13356. doi: 10.1073/pnas.93.23.13351
Kawaguchi, S. Y., and Hirano, T. (2007). Sustained structural change of GABA(A) receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J. Neurosci. 27, 6788–6799. doi: 10.1523/jneurosci.1981-07.2007
Keller, C. A., Yuan, X., Panzanelli, P., Martin, M. L., Alldred, M., Sassoe-Pognetto, M., et al. (2004). The gamma2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ. J. Neurosci. 24, 5881–5891. doi: 10.1523/jneurosci.1037-04.2004
Kilpatrick, C. L., Murakami, S., Feng, M., Wu, X., Lal, R., Chen, G., et al. (2016). Dissociation of Golgi-associated DHHC-type zinc finger protein (GODZ)- and sertoli cell gene with a zinc finger domain-β (SERZ-β)-mediated palmitoylation by loss of function analyses in knock-out mice. J. Biol. Chem. 291, 27371–27386. doi: 10.1074/jbc.M116.732768
Kittler, J. T., Chen, G., Honing, S., Bogdanov, Y., McAinsh, K., Arancibia-Carcamo, I. L., et al. (2005). Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc. Natl. Acad. Sci. U.S.A. 102, 14871–14876. doi: 10.1073/pnas.0506653102
Kittler, J. T., Chen, G., Kukhtina, V., Vahedi-Faridi, A., Gu, Z., Tretter, V., et al. (2008). Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma2 subunit. Proc. Natl. Acad. Sci. U.S.A. 105, 3616–3621. doi: 10.1073/pnas.0707920105
Kittler, J. T., Delmas, P., Jovanovic, J. N., Brown, D. A., Smart, T. G., and Moss, S. J. (2000). Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J. Neurosci. 20, 7972–7977. doi: 10.1523/JNEUROSCI.20-21-07972.2000
Kittler, J. T., Thomas, P., Tretter, V., Bogdanov, Y. D., Haucke, V., Smart, T. G., et al. (2004). Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc. Natl. Acad. Sci. U.S.A. 101, 12736–12741. doi: 10.1073/pnas.0401860101
Kneussel, M., Brandstatter, J. H., Laube, B., Stahl, S., Muller, U., and Betz, H. (1999). Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J. Neurosci. 19, 9289–9297. doi: 10.1523/JNEUROSCI.19-21-09289.1999
Kowalczyk, S., Winkelmann, A., Smolinsky, B., Forstera, B., Neundorf, I., Schwarz, G., et al. (2013). Direct binding of GABAA receptor beta2 and beta3 subunits to gephyrin. Eur. J. Neurosci. 37, 544–554. doi: 10.1111/ejn.12078
Labonte, D., Thies, E., and Kneussel, M. (2014). The kinesin KIF21B participates in the cell surface delivery of gamma2 subunit-containing GABAA receptors. Eur. J. Cell Biol. 93, 338–346. doi: 10.1016/j.ejcb.2014.07.007
Lachance-Touchette, P., Brown, P., Meloche, C., Kinirons, P., Lapointe, L., Lacasse, H., et al. (2011). Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur. J. Neurosci. 34, 237–249. doi: 10.1111/j.1460-9568.2011.07767.x
Lee, K., Porteous, R., Campbell, R. E., Luscher, B., and Herbison, A. E. (2010). Knockdown of GABA(A) receptor signaling in GnRH neurons has minimal effects upon fertility. Endocrinology 151, 4428–4436. doi: 10.1210/en.2010-0314
Leil, T. A., Chen, Z. W., Chang, C. S., and Olsen, R. W. (2004). GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J. Neurosci. 24, 11429–11438. doi: 10.1523/JNEUROSCI.3355-04.2004
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Leong, I. U., Stuckey, A., Lai, D., Skinner, J. R., and Love, D. R. (2015). Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med. Genet. 16:34. doi: 10.1186/s12881-015-0176-z
Leppa, E., Linden, A. M., Aller, M. I., Wulff, P., Vekovischeva, O., Luscher, B., et al. (2016). Increased motor-impairing effects of the neuroactive steroid pregnanolone in mice with targeted inactivation of the GABAA receptor gamma2 subunit in the cerebellum. Front. Pharmacol. 7:403. doi: 10.3389/fphar.2016.00403
Leppa, E., Linden, A. M., Vekovischeva, O. Y., Swinny, J. D., Rantanen, V., Toppila, E., et al. (2011). Removal of GABA(A) receptor gamma2 subunits from parvalbumin neurons causes wide-ranging behavioral alterations. PLoS One 6:e24159. doi: 10.1371/journal.pone.0024159
Levi, S., Le Roux, N., Eugene, E., and Poncer, J. C. (2015). Benzodiazepine ligands rapidly influence GABAA receptor diffusion and clustering at hippocampal inhibitory synapses. Neuropharmacology 88, 199–208. doi: 10.1016/j.neuropharm.2014.06.002
Lin, W. C., Davenport, C. M., Mourot, A., Vytla, D., Smith, C. M., Medeiros, K. A., et al. (2014). Engineering a light-regulated GABAA receptor for optical control of neural inhibition. ACS Chem. Biol. 9, 1414–1419. doi: 10.1021/cb500167u
Lin, W.C., Tsai, M.C., Davenport, C.M., Smith, C.M., Veit, J., Wilson, N.M., et al. (2015). A comprehensive optogenetic pharmacology toolkit for in vivo control of GABA(A) receptors and synaptic inhibition. Neuron 88, 879–891. doi: 10.1016/j.neuron.2015.10.026
Lionel, A. C., Vaags, A. K., Sato, D., Gazzellone, M. J., Mitchell, E. B., Chen, H. Y., et al. (2013). Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 22, 2055–2066. doi: 10.1093/hmg/ddt056
Lorenz-Guertin, J. M., and Jacob, T. C. (2017). GABA type A receptor trafficking and the architecture of synaptic inhibition. Dev. Neurobiol. 78, 238–270. doi: 10.1002/dneu.22536
Lorenz-Guertin, J. M., Wilcox, M. R., Zhang, M., Larsen, M. B., Pilli, J., Schmidt, B. F., et al. (2017). A versatile optical tool for studying synaptic GABAA receptor trafficking. J. Cell Sci. 130, 3933–3945. doi: 10.1242/jcs.205286
Ma, H., Marti-Gutierrez, N., Park, S. W., Wu, J., Lee, Y., Suzuki, K., et al. (2017). Correction of a pathogenic gene mutation in human embryos. Nature 548, 413–419. doi: 10.1038/nature23305
Macdonald, R. L., Bianch, M. T., and Feng, H. (2003). Mutations linked to generalized epilepsy in humans reduce GABA(A) receptor current. Exp. Neurol. 184, S58–S67. doi: 10.1016/S0014-4886(03)00405-9
Macdonald, R. L., Kang, J. -Q., and Gallagher, M. J. (2012). GABA(A) Receptor Subunit Mutations and Genetic Epilepsies. Bethesda, MD: National Center for Biotechnology Information.
Maric, H. M., Hausrat, T. J., Neubert, F., Dalby, N. O., Doose, S., Sauer, M., et al. (2017). Gephyrin-binding peptides visualize postsynaptic sites and modulate neurotransmission. Nat. Chem. Biol. 13, 153–160. doi: 10.1038/nchembio.2246
Marsden, K. C., Beattie, J. B., Friedenthal, J., and Carroll, R. C. (2007). NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. J. Neurosci. 27, 14326–14337. doi: 10.1523/jneurosci.4433-07.2007
Marsden, K. C., Shemesh, A., Bayer, K. U., and Carroll, R. C. (2010). Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc. Natl. Acad. Sci. U.S.A. 107, 20559–20564. doi: 10.1073/pnas.1010346107
Masica, D.L., and Karchin, R. (2016). Towards increasing the clinical relevance of in silico methods to predict pathogenic missense variants. PLoS Comput. Biol. 12:e1004725. doi: 10.1371/journal.pcbi.1004725
McDonald, B. J., Amato, A., Connolly, C. N., Benke, D., Moss, S. J., and Smart, T. G. (1998). Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat. Neurosci. 1, 23–28. doi: 10.1038/223
McDonald, B. J., and Moss, S. J. (1994). Differential phosphorylation of intracellular domains of gamma-aminobutyric acid type A receptor subunits by calcium/calmodulin type 2-dependent protein kinase and cGMP-dependent protein kinase. J. Biol. Chem. 269, 18111–18117.
Meier, J., and Grantyn, R. (2004). Preferential accumulation of GABAA receptor gamma 2L, not gamma 2S, cytoplasmic loops at rat spinal cord inhibitory synapses. J. Physiol. 559(Pt 2), 355–365. doi: 10.1113/jphysiol.2004.066233
Migita, K., Yamada, J., Nikaido, Y., Shi, X., Kaneko, S., Hirose, S., et al. (2013). Properties of a Novel GABAA receptor gamma2 subunit mutation associated with seizures. J. Pharmacol. Sci. 121, 84–87. doi: 10.1254/jphs.12222SC
Miller, P. S., and Aricescu, A. R. (2014). Crystal structure of a human GABAA receptor. Nature 512, 270–275. doi: 10.1038/nature13293
Miller, P. S., Scott, S., Masiulis, S., De Colibus, L., and Pardon, E. (2017). Structural basis for GABAA receptor potentiation by neurosteroids. Nat. Struct. Mol. Biol. 24, 986–992. doi: 10.1038/nsmb.3484
Mizokami, A., Kanematsu, T., Ishibashi, H., Yamaguchi, T., Tanida, I., Takenaka, K., et al. (2007). Phospholipase C-related inactive protein is involved in trafficking of gamma2 subunit-containing GABA(A) receptors to the cell surface. J. Neurosci. 27, 1692–1701. doi: 10.1523/jneurosci.3155-06.2007
Moss, S. J., Doherty, C. A., and Huganir, R. L. (1992). Identification of the cAMP-dependent protein kinase and protein kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma 2L subunits of the gamma-aminobutyric acid type A receptor. J. Biol. Chem. 267, 14470–14476.
Moss, S. J., Gorrie, G. H., Amato, A., and Smart, T. G. (1995). Modulation of GABAA receptors by tyrosine phosphorylation. Nature 377, 344–348. doi: 10.1038/377344a0
Muir, J., Arancibia-Carcamo, I. L., MacAskill, A. F., Smith, K. R., Griffin, L. D., and Kittler, J. T. (2010). NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma2 subunit. Proc. Natl. Acad. Sci. U.S.A. 107, 16679–16684. doi: 10.1073/pnas.1000589107
Muir, J., and Kittler, J. T. (2014). Plasticity of GABAA receptor diffusion dynamics at the axon initial segment. Front. Cell. Neurosci. 8:151. doi: 10.3389/fncel.2014.00151
Mukherjee, J., Kretschmannova, K., Gouzer, G., Maric, H. M., Ramsden, S., Tretter, V., et al. (2011). The residence time of GABA(A)Rs at inhibitory synapses is determined by direct binding of the receptor alpha1 subunit to gephyrin. J. Neurosci. 31, 14677–14687. doi: 10.1523/jneurosci.2001-11.2011
Nakajima, K., Yin, X., Takei, Y., Seog, D. H., Homma, N., and Hirokawa, N. (2012). Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron 76, 945–961. doi: 10.1016/j.neuron.2012.10.012
Nakamura, T., Arima-Yoshida, F., Sakaue, F., Nasu-Nishimura, Y., Takeda, Y., Matsuura, K., et al. (2016). PX-RICS-deficient mice mimic autism spectrum disorder in Jacobsen syndrome through impaired GABAA receptor trafficking. Nat. Commun. 7:10861. doi: 10.1038/ncomms10861
Nakamura, Y., Morrow, D. H., Modgil, A., Huyghe, D., Deeb, T. Z., Lumb, M. J., et al. (2016). Proteomic characterization of inhibitory synapses using a novel pHluorin-tagged gamma-aminobutyric acid receptor, type A (GABAA), alpha2 subunit knock-in mouse. J. Biol. Chem. 291, 12394–12407. doi: 10.1074/jbc.M116.724443
Naylor, D. E., Liu, H., and Wasterlain, C. G. (2005). Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 25, 7724–7733. doi: 10.1523/jneurosci.4944-04.2005
Nemecz, A., Prevost, M. S., Menny, A., and Corringer, P. J. (2016). Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron 90, 452–470. doi: 10.1016/j.neuron.2016.03.032
Niwa, F., Bannai, H., Arizono, M., Fukatsu, K., Triller, A., and Mikoshiba, K. (2012). Gephyrin-independent GABA(A)R mobility and clustering during plasticity. PLoS One 7:e36148. doi: 10.1371/journal.pone.0036148
Oh, W. C., Lutzu, S., Castillo, P. E., and Kwon, H. B. (2016). De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353, 1037–1040. doi: 10.1126/science.aaf5206
Olsen, R. W., and Sieghart, W. (2009). GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56, 141–148. doi: 10.1016/j.neuropharm.2008.07.045
O’Sullivan, G. A., Kneussel, M., Elazar, Z., and Betz, H. (2005). GABARAP is not essential for GABA receptor targeting to the synapse. Eur. J. Neurosci. 22, 2644–2648. doi: 10.1111/j.1460-9568.2005.04448.x
Pennacchietti, F., Vascon, S., Nieus, T., Rosillo, C., Das, S., Tyagarajan, S. K., et al. (2017). Nanoscale molecular reorganization of the inhibitory postsynaptic density is a determinant of GABAergic synaptic potentiation. J. Neurosci. 37, 1747–1756. doi: 10.1523/jneurosci.0514-16.2016
Petrie, J., Sapp, D. W., Tyndale, R. F., Park, M. K., Fanselow, M., and Olsen, R. W. (2001). Altered gabaa receptor subunit and splice variant expression in rats treated with chronic intermittent ethanol. Alcohol. Clin. Exp. Res. 25, 819–828. doi: 10.1111/j.1530-0277.2001.tb02285.x
Petrini, E. M., Ravasenga, T., Hausrat, T. J., Iurilli, G., Olcese, U., Racine, V., et al. (2014). Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP. Nat. Commun. 5:3921. doi: 10.1038/ncomms4921
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Papadopoulos, T., et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. doi: 10.1016/j.neuron.2009.08.023
Qi, Z. H., Song, M., Wallace, M. J., Wang, D., Newton, P. M., McMahon, T., et al. (2007). Protein kinase C epsilon regulates gamma-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of gamma2 subunits. J. Biol. Chem. 282, 33052–33063. doi: 10.1074/jbc.M707233200
Qian, Z., Micorescu, M., Yakhnitsa, V., and Barmack, N. H. (2012). Climbing fiber activity reduces 14-3-3-theta regulated GABA(A) receptor phosphorylation in cerebellar Purkinje cells. Neuroscience 201, 34–45. doi: 10.1016/j.neuroscience.2011.11.021
Qian, Z., Yakhnitsa, V., and Barmack, N. H. (2011). Climbing fiber-evoked Purkinje cell discharge reduces expression of GABA(A) receptor-associated protein and decreases its interaction with GABA(A) receptors. J. Neurochem. 117, 197–208. doi: 10.1111/j.1471-4159.2010.07119.x
Quinlan, J. J., Firestone, L. L., and Homanics, G. E. (2000). Mice lacking the long splice variant of the gamma 2 subunit of the GABA(A) receptor are more sensitive to benzodiazepines. Pharmacol. Biochem. Behav. 66, 371–374. doi: 10.1016/S0091-3057(00)00225-2
Ramakrishnan, L., and Hess, G. P. (2004). On the mechanism of a mutated and abnormally functioning γ-aminobutyric acid (A) receptor linked to epilepsy. Biochemistry 43, 7534–7540. doi: 10.1021/bi036181+
Reinthaler, E. M., Dejanovic, B., Lal, D., Semtner, M., Merkler, Y., Reinhold, A., et al. (2015). Rare variants in gamma-aminobutyric acid type A receptor genes in rolandic epilepsy and related syndromes. Ann. Neurol. 77, 972–986. doi: 10.1002/ana.24395
Ren, Z., Sahir, N., Murakami, S., Luellen, B. A., Earnheart, J. C., Lal, R., et al. (2015). Defects in dendrite and spine maturation and synaptogenesis associated with an anxious-depressive-like phenotype of GABAA receptor-deficient mice. Neuropharmacology 88, 171–179. doi: 10.1016/j.neuropharm.2014.07.019
Renner, M., Schweizer, C., Bannai, H., Triller, A., and Levi, S. (2012). Diffusion barriers constrain receptors at synapses. PLoS One 7:e43032. doi: 10.1371/journal.pone.0043032
Rowland, A. M., Richmond, J. E., Olsen, J. G., Hall, D. H., and Bamber, B. A. (2006). Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 26, 1711–1720. doi: 10.1523/jneurosci.2279-05.2006
Saliba, R. S., Kretschmannova, K., and Moss, S. J. (2012). Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 31, 2937–2951. doi: 10.1038/emboj.2012.109
Sancar, F., and Czajkowski, C. (2004). A GABAA receptor mutation linked to human epilepsy (gamma2R43Q) impairs cell surface expression of alpha beta gamma receptors. J. Biol. Chem. 279, 47034–47039. doi: 10.1074/jbc.M403388200
Saunders, M. J., Szent-Gyorgyi, C., Fisher, G. W., Jarvik, J. W., Bruchez, M. P., and Waggoner, A. S. (2012). Fluorogen activating proteins in flow cytometry for the study of surface molecules and receptors. Methods 57, 308–317. doi: 10.1016/j.ymeth.2012.02.003
Schweizer, C., Balsiger, S., Bluethmann, H., Mansuy, I. M., Fritschy, J. M., Mohler, H., et al. (2003). The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 24, 442–450. doi: 10.1016/S1044-7431(03)00202-1
Shen, D., Hernandez, C. C., Shen, W., Hu, N., Poduri, A., Shiedley, B., et al. (2017). De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 140, 49–67. doi: 10.1093/brain/aww272
Shen, Q., Fuchs, T., Sahir, N., and Luscher, B. (2012). GABAergic control of critical developmental periods for anxiety- and depression-related behavior in mice. PLoS One 7:e47441. doi: 10.1371/journal.pone.0047441
Shen, Q., Lal, R., Luellen, B. A., Earnheart, J. C., Andrews, A. M., and Luscher, B. (2010). gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol. Psychiatry 68, 512–520. doi: 10.1016/j.biopsych.2010.04.024
Shi, X., Huang, M. C., Ishii, A., Yoshida, S., Okada, M., Morita, K., et al. (2010). Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. J. Hum. Genet. 55, 375–378. doi: 10.1038/jhg.2010.47
Shrivastava, A. N., Triller, A., Sieghart, W., and Sarto-Jackson, I. (2011). Regulation of GABA(A) receptor dynamics by interaction with purinergic P2X(2) receptors. J. Biol. Chem. 286, 14455–14468. doi: 10.1074/jbc.M110.165282
Sigal, Y. M., Speer, C. M., Babcock, H. P., and Zhuang, X. (2015). Mapping synaptic input fields of neurons with super-resolution imaging. Cell 163, 493–505. doi: 10.1016/j.cell.2015.08.033
Sigel, E., and Steinmann, M. E. (2012). Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 287, 40224–40231. doi: 10.1074/jbc.R112.386664
Singh, R., Scheffer, I. E., Crossland, K., and Berkovic, S. F. (1999). Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Annu. Neurol. 45, 75–81. doi: 10.1002/1531-8249(199901)45:1<75::AID-ART13>3.0.CO;2-W
Smith, K. R., Davenport, E. C., Wei, J., Li, X., Pathania, M., Vaccaro, V., et al. (2014). GIT1 and betaPIX are essential for GABA(A) receptor synaptic stability and inhibitory neurotransmission. Cell Rep. 9, 298–310. doi: 10.1016/j.celrep.2014.08.061
Smith, K. R., McAinsh, K., Chen, G., Arancibia-Carcamo, I. L., Haucke, V., Yan, Z., et al. (2008). Regulation of inhibitory synaptic transmission by a conserved atypical interaction of GABA(A) receptor beta- and gamma-subunits with the clathrin AP2 adaptor. Neuropharmacology 55, 844–850. doi: 10.1016/j.neuropharm.2008.06.072
Smith, K. R., Muir, J., Rao, Y., Browarski, M., Gruenig, M. C., Sheehan, D. F., et al. (2012). Stabilization of GABA(A) receptors at endocytic zones is mediated by an AP2 binding motif within the GABA(A) receptor beta3 subunit. J. Neurosci. 32, 2485–2498. doi: 10.1523/jneurosci.1622-11.2011
Stojakovic, A., Walczak, M., and Cieslak, P. E. (2018). Several behavioral traits relevant for alcoholism are controlled by 2 subunit containing GABAA receptors on dopamine neurons in mice. Neuropsychopharmacology 43, 1548–1556. doi: 10.1038/s41386-018-0022-z
Sudhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Sun, H., Zhang, Y., Liang, J., Liu, X., Ma, X., Wu, H., et al. (2008). SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J. Hum. Genet. 53, 769–774. doi: 10.1007/s10038-008-0306-y
Szent-Gyorgyi, C., Schmidt, B. F., Creeger, Y., Fisher, G. W., Zakel, K. L., Adler, S., et al. (2008). Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26, 235–240. doi: 10.1038/nbt1368
Tan, H. O., Reid, C. A., Single, F. N., Davies, P. J., Chiu, C., Murphy, S., et al. (2007). Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc. Natl. Acad. Sci. U.S.A. 104, 17536–17541. doi: 10.1073/pnas.0708440104
Tenney, J. R., Hopkin, R. J., and Schapiro, M. B. (2011). Deletion of 14-3-3{varepsilon} and CRK: a clinical syndrome with macrocephaly, developmental delay, and generalized epilepsy. J. Child Neurol. 26, 223–227. doi: 10.1177/0883073810379638
Terunuma, M., Jang, I. S., Ha, S. H., Kittler, J. T., Kanematsu, T., Jovanovic, J. N., et al. (2004). GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J. Neurosci. 24, 7074–7084. doi: 10.1523/JNEUROSCI.1323-04.2004
Tian, M., and Macdonald, R. L. (2012). The intronic GABRG2 mutation, IVS6+2T->G, associated with childhood absence epilepsy altered subunit mRNA intron splicing, activated nonsense-mediated decay, and produced a stable truncated gamma2 subunit. J. Neurosci. 32, 5937–5952. doi: 10.1523/JNEUROSCI.5332-11.2012
Tian, M., Mei, D., Freri, E., Hernandez, C. C., Granata, T., Shen, W., et al. (2013). Impaired surface alpha beta gamma GABA(A) receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol. Dis. 50, 135–141. doi: 10.1016/j.nbd.2012.10.008
Toma, C., Torrico, B., Hervas, A., Valdes-Mas, R., Tristan-Noguero, A., Padillo, V., et al. (2014). Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 19, 784–790. doi: 10.1038/mp.2013.106
Tretter, V., Jacob, T. C., Mukherjee, J., Fritschy, J. M., Pangalos, M. N., and Moss, S. J. (2008). The clustering of GABA(A) receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J. Neurosci. 28, 1356–1365. doi: 10.1523/jneurosci.5050-07.2008
Tretter, V., Kerschner, B., Milenkovic, I., Ramsden, S. L., Ramerstorfer, J., Saiepour, L., et al. (2011). Molecular basis of the gamma-aminobutyric acid A receptor alpha3 subunit interaction with the clustering protein gephyrin. J. Biol. Chem. 286, 37702–37711. doi: 10.1074/jbc.M111.291336
Tretter, V., Revilla-Sanchez, R., Houston, C., Terunuma, M., Havekes, R., Florian, C., et al. (2009). Deficits in spatial memory correlate with modified {gamma}-aminobutyric acid type A receptor tyrosine phosphorylation in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 106, 20039–20044. doi: 10.1073/pnas.0908840106
Tseng, W. C., Jenkins, P. M., Tanaka, M., Mooney, R., and Bennett, V. (2015). Giant ankyrin-G stabilizes somatodendritic GABAergic synapses through opposing endocytosis of GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 112, 1214–1219. doi: 10.1073/pnas.1417989112
Twelvetrees, A. E., Yuen, E. Y., Arancibia-Carcamo, I. L., MacAskill, A. F., Rostaing, P., Lumb, M. J., et al. (2010). Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 65, 53–65. doi: 10.1016/j.neuron.2009.12.007
Uezu, A., Kanak, D. J., Bradshaw, T. W., Soderblom, E. J., Catavero, C. M., Burette, A. C., et al. (2016). Identification of an elaborate complex mediating postsynaptic inhibition. Science 353, 1123–1129. doi: 10.1126/science.aag0821
van der Veen, A. G., and Ploegh, H. L. (2012). Ubiquitin-like proteins. Annu. Rev. Biochem. 81, 323–357. doi: 10.1146/annurev-biochem-093010-153308
Vinkers, C. H., and Olivier, B. (2012). Mechanisms underlying tolerance after long-term benzodiazepine use: a future for subtype-selective GABA(A) receptor modulators? Adv. Pharmacol. Sci. 2012:416864. doi: 10.1155/2012/416864
Vithlani, M., Hines, R. M., Zhong, P., Terunuma, M., Hines, D. J., Revilla-Sanchez, R., et al. (2013). The ability of BDNF to modify neurogenesis and depressive-like behaviors is dependent upon phosphorylation of tyrosine residues 365/367 in the GABA(A)-receptor gamma2 subunit. J. Neurosci. 33, 15567–15577. doi: 10.1523/jneurosci.1845-13.2013
Wafford, K. A., Burnett, D. M., Leidenheimer, N. J., Burt, D. R., Wang, J. B., Kofuji, P., et al. (1991). Ethanol sensitivity of the GABAA receptor expressed in Xenopus oocytes requires 8 amino acids contained in the gamma 2L subunit. Neuron 7, 27–33. doi: 10.1016/0896-6273(91)90071-7
Wallace, R. H., Marini, C., Petrou, S., Harkin, L. A., Bowser, D. N., Panchal, R. G., et al. (2001). Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 28, 49–52. doi: 10.1038/88259
Wang, H., Bedford, F. K., Brandon, N. J., Moss, S. J., and Olsen, R. W. (1999). GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature 397, 69–72. doi: 10.1038/16264
Wang, J., Shen, D., Xia, G., Shen, W., Macdonald, R. L., Xu, D., et al. (2016). Differential protein structural disturbances and suppression of assembly partners produced by nonsense GABRG2 epilepsy mutations: implications for disease phenotypic heterogeneity. Sci. Rep. 6:35294. doi: 10.1038/srep35294
Wang, J. B., and Burt, D. R. (1991). Differential expression of two forms of GABAA receptor gamma 2-subunit in mice. Brain Res. Bull. 27, 731–735. doi: 10.1016/0361-9230(91)90054-N
Wang, R. A., Cheng, G., Kolaj, M., and Randic, M. (1995). Alpha-subunit of calcium/calmodulin-dependent protein kinase II enhances gamma-aminobutyric acid and inhibitory synaptic responses of rat neurons in vitro. J. Neurophysiol. 73, 2099–2106. doi: 10.1152/jn.1995.73.5.2099
Whiting, P., McKernan, R. M., and Iversen, L. L. (1990). Another mechanism for creating diversity in gamma-aminobutyrate type A receptors: RNA splicing directs expression of two forms of gamma 2 phosphorylation site. Proc. Natl. Acad. Sci. U.S.A. 87, 9966–9970. doi: 10.1073/pnas.87.24.9966
Wingrove, P. B., Thompson, S. A., Wafford, K. A., and Whiting, P. J. (1997). Key amino acids in the gamma subunit of the gamma-aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine site. Mol. Pharmacol. 52, 874–881. doi: 10.1124/mol.52.5.874
Wulff, P., Goetz, T., Leppa, E., Linden, A. M., Renzi, M., Swinny, J. D., et al. (2007). From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat. Neurosci. 10, 923–929. doi: 10.1038/nn1927
Wulff, P., Ponomarenko, A. A., Bartos, M., Korotkova, T. M., Fuchs, E. C., Bahner, F., et al. (2009). Hippocampal theta rhythm and its coupling with gamma oscillations require fast inhibition onto parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. U.S.A. 106, 3561–3566. doi: 10.1073/pnas.0813176106
Yamasaki, T., Hoyos-Ramirez, E., Martenson, J. S., Morimoto-Tomita, M., and Tomita, S. (2017). GARLH family proteins stabilize GABAA receptors at synapses. Neuron 93, 1138–1152.e6. doi: 10.1016/j.neuron.2017.02.023
Yuan, X., Yao, J., Norris, D., Tran, D. D., Bram, R.J., Chen, G., et al. (2008). Calcium-modulating cyclophilin ligand regulates membrane trafficking of postsynaptic GABAA receptors. Mol. Cell. Neurosci. 38, 277–289. doi: 10.1016/j.mcn.2008.03.002
Zecharia, A. Y., Yu, X., Gotz, T., Ye, Z., Carr, D. R., Wulff, P., et al. (2012). GABAergic inhibition of histaminergic neurons regulates active waking but not the sleep-wake switch or propofol-induced loss of consciousness. J. Neurosci. 32, 13062–13075. doi: 10.1523/jneurosci.2931-12.2012
Zhang, M., Chakraborty, S. K., Sampath, P., Rojas, J. J., Hou, W., Saurabh, S., et al. (2015). Fluoromodule-based reporter/probes designed for in vivo fluorescence imaging. J. Clin. Invest. 125, 3915–3927. doi: 10.1172/JCI81086
Zhu, G., Yoshida, S., Migita, K., Yamada, J., Mori, F., Tomiyama, M., et al. (2012). Dysfunction of extrasynaptic GABAergic transmission in phospholipase C-related, but catalytically inactive protein 1 knockout mice is associated with an epilepsy phenotype. J. Pharmacol. Exp. Ther. 340, 520–528. doi: 10.1124/jpet.111.182386
Keywords: GABAA receptor, trafficking, genetic variation, human, epilepsy, imaging
Citation: Lorenz-Guertin JM, Bambino MJ and Jacob TC (2018) γ2 GABAAR Trafficking and the Consequences of Human Genetic Variation. Front. Cell. Neurosci. 12:265. doi: 10.3389/fncel.2018.00265
Received: 17 May 2018; Accepted: 02 August 2018;
Published: 23 August 2018.
Edited by:
David Perrais, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Christine Laura Dixon, University College London, United KingdomCopyright © 2018 Lorenz-Guertin, Bambino and Jacob. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tija C. Jacob, dGNqMTFAcGl0dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.