 Riccardo Cristofani1
Riccardo Cristofani1 Valeria Crippa1
Valeria Crippa1 Maria Elena Cicardi1,2
Maria Elena Cicardi1,2 Barbara Tedesco1
Barbara Tedesco1 Veronica Ferrari1
Veronica Ferrari1 Marta Chierichetti1
Marta Chierichetti1 Elena Casarotto1
Elena Casarotto1 Margherita Piccolella1
Margherita Piccolella1 Elio Messi1
Elio Messi1 Mariarita Galbiati1
Mariarita Galbiati1 Paola Rusmini1
Paola Rusmini1 Angelo Poletti1,3*
Angelo Poletti1,3*- 1Laboratorio di Biologia Applicata, Dipartimento di Scienze Farmacologiche e Biomolecolari, Dipartimento di Eccellenza 2018-2022, Università degli Studi di Milano, Milan, Italy
- 2Department of Neuroscience, Jefferson Weinberg ALS Center, Vickie and Jack Farber Institute for Neuroscience, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, United States
- 3Center of Excellence on Neurodegenerative Diseases (CEND), Università degli Studi di Milano, Milan, Italy
Motor neuron diseases (MNDs) are fatal diseases characterized by loss of motor neurons in the brain cortex, in the bulbar region, and/or in the anterior horns of the spinal cord. While generally sporadic, inherited forms linked to mutant genes encoding altered RNA/protein products have also been described. Several different mechanisms have been found altered or dysfunctional in MNDs, like the protein quality control (PQC) system. In this review, we will discuss how the PQC system is affected in two MNDs—spinal and bulbar muscular atrophy (SBMA) and amyotrophic lateral sclerosis (ALS)—and how this affects the clearance of aberrantly folded proteins, which accumulate in motor neurons, inducing dysfunctions and their death. In addition, we will discuss how the PQC system can be targeted to restore proper cell function, enhancing the survival of affected cells in MNDs.
Introduction
Motor neuron diseases (MNDs) are neurodegenerative diseases (NDs) characterized by the loss of motor neurons in the brain cortex, in the bulbar region, and/or in the anterior horns of the spinal cord; the consequence of motor neuron death is the lack of control on the skeletal muscle fibers. While motor neurons are considered the primary target in MNDs, muscle and glial cells may also be directly involved, and this affects motor neuron survival. MNDs are generally fatal diseases, clinically characterized by severe loss of voluntary movements, muscle weakness, spasticity, and atrophy. MNDs appear as sporadic or inherited forms, which have been extensively studied in the last 30 years. The inherited forms are associated with gene mutations that result in the production of altered RNA or proteins with reduced [loss-of-function (LOF)] or aberrant neurotoxic [gain-of-function (GOF)] functions. Mixed LOF and GOF are also possible. In LOF, the RNA or the protein affected are generally essential for motor neuron viability; thus, their reduced activity often causes motor neuron death [e.g., in spinal muscular atrophy (SMA); Lefebvre et al., 1995]. In these cases, the therapeutic intervention is aimed to restore the proper activity of the missed/altered RNA or protein (Poletti and Fischbeck, 2020), and successful therapies have been recently approved worldwide from regulatory agencies (Finkel et al., 2017; Mendell et al., 2017; Mercuri et al., 2018). In GOF, different neurotoxic mechanisms have been reported to take place in a given mutant RNA or protein. Unfortunately, this makes difficult to identify a common therapeutic target for MNDs. Therefore, these approaches must be specifically designed for each MND’s form. However, it is now clear that many familial MND forms are characterized by alterations of common intracellular pathways, which are often also altered in sporadic MNDs. Thus, these pathways might serve as potential therapeutic targets to reduce motor neuron death. In this review, we will focus on one of the most common pathways affected in MNDs, the protein quality control (PQC) system. In fact, in several MNDs, which include spinal and bulbar muscular atrophy (SBMA) and amyotrophic lateral sclerosis (ALS), the PQC system becomes unable to correctly handle misfolded proteins (mainly produced by the mutant gene), letting them become harmful to motor neurons and/or to glial and skeletal muscle cells.
Misfolded Proteins Associated With Motor Neuron Diseases
Spinal and Bulbar Muscular Atrophy
SBMA is the first MND for which a specific gene mutation has been linked to the disease as the cause of neuronal cell death (La Spada et al., 1991). SBMA, initially defined as a pure MND, is presently also classified as a neuromuscular disease. In fact, in SBMA, the primarily affected cell populations are lower motor neurons localized in the bulbar region of the brain (brain stem containing motor neurons of the lower cranial nerves) or in the anterior horn of the spinal cord (Sobue et al., 1989; La Spada et al., 1991; Brooks and Fischbeck, 1995; Li et al., 1995; Brooks et al., 1997). Dorsal root ganglia (DRG) neurons may also be affected in SBMA (Chua and Lieberman, 2013) and the combination of motor and DRG neurons loss is responsible for the clinical signs which include muscle fasciculations, weakness, and subsequent atrophy, including dysphagia and dysarthria with atrophy of the bulbar, facial, and limb muscles, as well as sensory disturbances at distal extremities (Sobue et al., 1989). So far, there is no evidence for the involvement of other brain cell types (e.g., glial cells or microglia). In addition to neuronal cells, skeletal muscle cells are also directly affected in SBMA (Chua and Lieberman, 2013; Cortes et al., 2014a; Lieberman et al., 2014; Rinaldi et al., 2014; Rusmini et al., 2015; Cicardi et al., 2019). This specific cell susceptibility is because the gene responsible for SBMA encodes for the androgen receptor (AR), and this gene is highly expressed in all the cell types described above (Poletti, 2004; Marron et al., 2005). The same cells express high levels of androgen-activating enzymes (Poletti et al., 1994, 1997, 2001; Pozzi et al., 2003). SBMA patients show mild endocrine alterations, like hypogonadism, possibly due to modification of the gonadal-hypothalamic axis or gynecomastia (Sobue et al., 1989; Kazemi-Esfarjani et al., 1995; Polo et al., 1996; Belsham et al., 1998; Piccioni et al., 2001). These alterations are often associated with reduced AR function.
Since the AR gene locus is on the X-chromosome, SBMA exists only as X-linked inherited form, but only males are affected (La Spada et al., 1991). Notably, the mutated AR protein is inactive in the absence of androgens [testosterone or its derivative 5α-dihydrotestosterone (DHT)], while it acquires toxic properties upon agonist binding (Katsuno et al., 2002, 2003), and the presence of androgens is thus mandatory for symptoms appearance and disease manifestation. This is possible since the AR mutation found in SBMA is radically different from those responsible for partial or complete androgen insensitivity syndrome (PAIS or CAIS) or tumors like prostate cancer (Brinkmann, 2001). In SBMA, the mutant AR gene is characterized by an expansion of a CAG (cytosine, adenine, guanine) tandem repeat (La Spada et al., 1991). The CAG sequence is expressed in exon 1 of the mRNA and then translated into a polyglutamine tract in the AR N-terminus (ARpolyQ). In normal individuals, the polyQ length of AR is highly polymorphic, ranging from 15 to 35 Qs (Edwards et al., 1992; Kuhlenbäumer et al., 2001); in SBMA patients the polyQ size becomes longer than 37 Qs (to a maximum of 72; Fischbeck, 1997; Kuhlenbäumer et al., 2001; Grunseich et al., 2014; Madeira et al., 2018). CAG repeat expansions coding for elongated polyQ tracts have been found in other eight genes, which are unrelated to AR; the mutant protein products of these genes cause other similar NDs (Ross, 2002). The ARpolyQ retains approximately 30% of its transcriptional functions, which explains the endocrine signs present in SBMA, but acquires a novel toxic function that impacts neuronal and muscle cell viability. As mentioned above, this toxic function of ARpolyQ appears after its activation by androgens. These AR ligands (testosterone or DHT) may induce aberrant protein conformations to ARpolyQ (protein misfolding), which becomes highly prone to aggregation (Stenoien et al., 1999; Simeoni et al., 2000; Piccioni et al., 2002). Details of this pathological mechanism are provided below.
Amyotrophic Lateral Sclerosis
ALS is a typical MND characterized by the loss of both the cerebral motor cortex or brainstem (upper) motor neurons and the cranial nerves and ventral horns of the spinal cord (lower) motor neurons. Neurons located in the frontotemporal cortex may be involved in some specific forms of ALS (Robberecht and Philips, 2013), which may clinically manifest in a pure MND form or be associated with a different extension to frontotemporal dementia (ALS-FTD). Differently from SBMA, the surrounding non-neuronal glial cells [astrocytes (Trotti et al., 1999; Boillee et al., 2006; Nagai et al., 2007), oligodendrocytes (Philips et al., 2013), and Schwann cells (Lobsiger et al., 2009; Manjaly et al., 2010)] are indirectly or directly affected in ALS. Reactive microglia are also present in ALS-affected tissues, but not in SBMA (Philips and Robberecht, 2011), proving that neuroinflammation and oxidative stress may play a significant role in ALS (Ferraiuolo et al., 2011). As in SBMA, the striatal skeletal muscle target cells can also be directly affected in ALS (Dobrowolny et al., 2008; Onesto et al., 2011; Cicardi et al., 2018; Meroni et al., 2019). Ninety percent of ALS cases appear as sporadic (sALS) forms, and only 10% of cases are caused by inherited mutations linked to familial (fALS) forms. The two types of ALS are clinically indistinguishable. Up to now, more than 30 genes have been found altered in fALS (Robberecht and Philips, 2013; Cook and Petrucelli, 2019; Mathis et al., 2019; Mejzini et al., 2019), and each of these accounts for disease, which mainly occurs as monogenic disease, even if disease modifier genes might exist. It is noteworthy that several of the gene products that cause a specific fALS have been reported to acquire an aberrant behavior of their wild-type (wt) forms in sALS. This suggests the existence of common pathways that lead to motor neuronal death in both fALS and in sALS (Neumann et al., 2006; Daoud et al., 2009; Bosco and Landers, 2010).
ALS has a very high variability in terms of both age of onset and disease progression, and it seems to occur earlier in males compared to females (Vegeto et al., 2020), with a male/female ratio of 1–3 in the geographic region and population evaluated in the study (Kurtzke, 1982; Haverkamp et al., 1995; Manjaly et al., 2010). The two sexes also show different symptomatology, since in males the disease predominantly begins in the lumbar tract of the spinal cord, while in females ALS mainly begins in the bulbar region (see Blasco et al., 2012 for an extensive review). It is likely that hormonal sex steroids may influence the neurotoxicity of factors involved in the pathogenesis of ALS (see Vegeto et al., 2020 for an extensive review).
Historically, the superoxide dismutase 1 (SOD1) gene is the first gene associated with fALS. However, this mutation only accounts for 15% of all fALS cases. SOD1 encodes a ubiquitously-expressed antioxidant enzyme that acts as a free radical scavenger enzyme (Bendotti et al., 2012). The most frequent fALS form (almost 50% of all fALS) is due to a mutation in the C9orf72 (chromosome 9 open reading frame 72) gene; in particular, the mutation consists of an expansion of a hexanucleotide (G4C2) repeat located in the 5′-untranslated region of the C9orf72 gene. Surprisingly, despite its location in an intronic sequence, the G4C2 expansion (which is transcribed in both directions) is utilized by ribosomes as a starting point for translation; this results in the production of five different dipeptides (DPRs; Ash et al., 2013; Gendron et al., 2013; Lashley et al., 2013; Mori et al., 2013). The process has been identified as an unconventional translation and named “repeat-associated non-ATG (RAN) translation” (Zu et al., 2011). The five DPRs do not have a physiological role, but they only exert toxicity in the expressing cells of affected individuals. Other mutant genes are less frequently represented in fALS: examples are the genes encoding TAR DNA-binding protein 43 (TDP-43), the ALS-linked fused in sarcoma/translocated in liposarcoma (FUS/TLS), the ubiquilin-2, the optineurin, the valosin-containing protein/p97 (VCP/p97), and others. These alterations occur in a few fALS families, but the same proteins (even if in the wt form) can be dysregulated in sALS, suggesting that their functions are crucial to maintain neuronal homeostasis (a list of the most common gene mutations identified so far in fALS is reported in Table 1). In particular, TDP-43 is considered a hallmark for sALS since it mislocalizes from nucleus to cytoplasm, where it aggregates in inclusions. These inclusions are enriched by TDP-43 caspase-3-cleaved fragments containing the C-terminal unstructured domain (Ratti and Buratti, 2016).
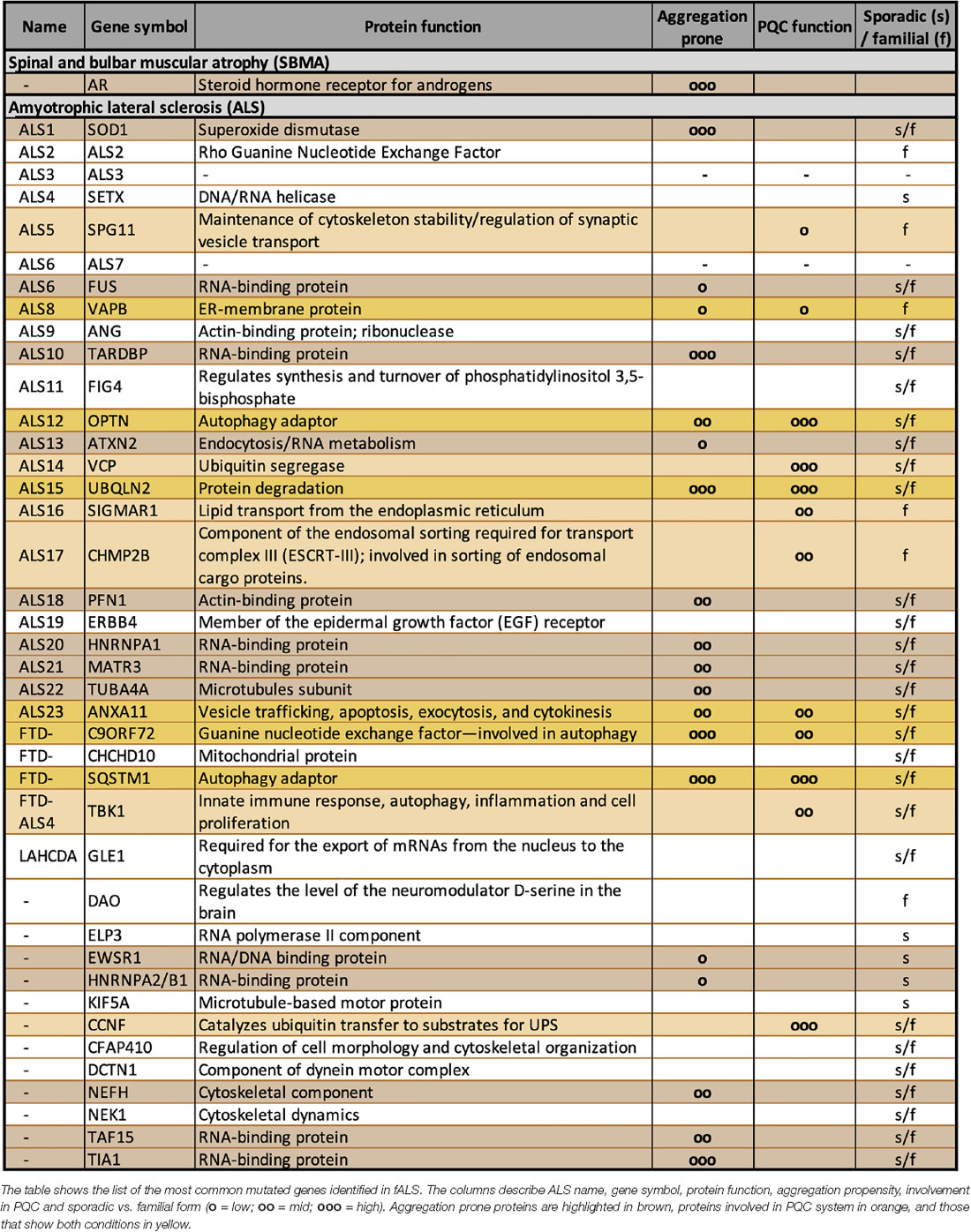
Table 1. Gene mutations reported in familial amyotrophic lateral sclerosis (fALS).
A careful analysis of the gene products identified so far suggests that several of their coded proteins have functions that cluster in specific intracellular processes. One of the most represented pathways is the PQC system (Table 1). In fact, different ALS-associated proteins are directly involved in the PQC system and others indirectly affect the PQC system due to their mutation. Indeed, when mutated, they become unable to properly reach the folded conformation and misfold. Misfolded proteins must be cleared from cells, and with this mechanism they may overwhelm the PQC system capability to handle proteotoxic stresses. As in the case of ARpolyQ and in all other elongated polyQ-containing proteins, which cause adult-onset MNDs, the misfolded ALS proteins tend to segregate from the nuclear or cytoplasmic compartments via a liquid-liquid phase partitioning (Molliex et al., 2015; Patel et al., 2015; Ganassi et al., 2016; Lee et al., 2016; Alberti et al., 2017; Boeynaems et al., 2017; Freibaum and Taylor, 2017; Mackenzie et al., 2017). This leads to an initial seed of aggregates with well-defined physical-chemical properties, which then mature into aggresomes and insoluble inclusions (Davies et al., 1997; DiFiglia et al., 1997; Li et al., 1998; Lieberman et al., 1998; Kopito, 2000; Mediani et al., 2019). The accumulating proteins may thus damage the PQC system by saturating its functional capabilities or by clogging the pathways devoted to protein clearance. For these reasons, by forming aggregates, misfolded ARpolyQ or ALS-associated proteins may perturb not only the PQC system, but also a series of pathways that depend on the proper functioning of the PQC system to maintain the correct cellular homeostasis.
The Protein Quality Control System
Most cell types affected in MNDs are post-mitotic or generally characterized by a poor mitotic index. This means that these cells might accumulate aberrant proteins that cannot be diluted by cell self-renewal or by simple partitioning into duplicated intracellular compartments generated as a result of cell division. Thus, these cells must develop a very sophisticated system to maintain their proper protein homeostasis. Therefore, post-mitotic non-dividing cells like neurons, motor neurons, or skeletal muscle cells, as well as poorly replicating cells, like glial cells, are highly prone to respond to misfolded protein species. Misfolded species may be produced in response to different cell stresses or as a consequence of gene mutations. These cells are able to respond to these stresses in a very powerful way: the overexpression of specific chaperones and co-chaperones, paralleled by the potentiation of the degradative pathways. All these factors are extremely well-coordinated to protect against proteotoxicity, and their synergic activities constitute the PQC system mentioned above. The PQC system thus acts as the first line of defense and because of its protective action, its selective modulation represents a valuable target for therapeutic intervention in all protein misfolding diseases, including MNDs like SBMA and ALS.
The PQC system is composed of a very large number of factors clustered in specific families of proteins that work together to define the fate of every single protein starting from its proper folding after synthesis or denaturation, and it routes proteins to degradation in case the folding fails.
The Chaperones
The family of intracellular chaperones and their co-chaperones is composed of more than 180 different proteins, some of which share a high degree of homology. These chaperones generally act in a specific subcellular compartment: for example, some chaperones localize exclusively in the endoplasmic reticulum, in the mitochondria, in the lysosomes, and/or in the cytoplasm, where they mainly exert their protective activities. Most chaperones are also expressed in a cell- and tissue-specific manner, with some chaperones localized exclusively in one tissue (e.g., in the testis), while others are ubiquitously expressed. In addition, chaperones may be regulated in response to cell stresses. Indeed, chaperones have been discovered as proteins induced by heat shock, and found to protect cells against thermal damages. Because of this, they have been named “heat shock proteins” or HSPs (DiDomenico et al., 1982). This name still stands for many chaperones, even if they have been demonstrated to possess much wider activities against a spectrum of variables capable of damaging intracellular proteins (e.g., oxidative stress, hypoxia, DNA damage, aberrant translation, etc.). Based on their structure and functions, these factors have been classified in subfamilies of chaperones. Originally, chaperones were grouped based on their apparent molecular weight after their biochemical identification in SDS-PAGE (small HSPs, HSP40s, HSP60s, HSP70s, HSP90s, and HSP100), but this classification now reflects their functions in the folding processes. Based on HUGO Gene Nomenclature Committee, a new nomenclature has been adopted for the human HSP families: HSPB (small HSP), DNAJ (HSP40), HSPD (HSP60), HSPA (HSP70), HSPC (HSP90), and HSPH (HSP110; Kampinga et al., 2009; see also Kampinga and Craig, 2010) for an extensive review). Chaperones often require the assistance of co-chaperones, which serve as nucleotide exchange factors (NEFs), like the members of the BCL2-associated athanogene (BAG) protein family (Takayama and Reed, 2001; Figure 1).
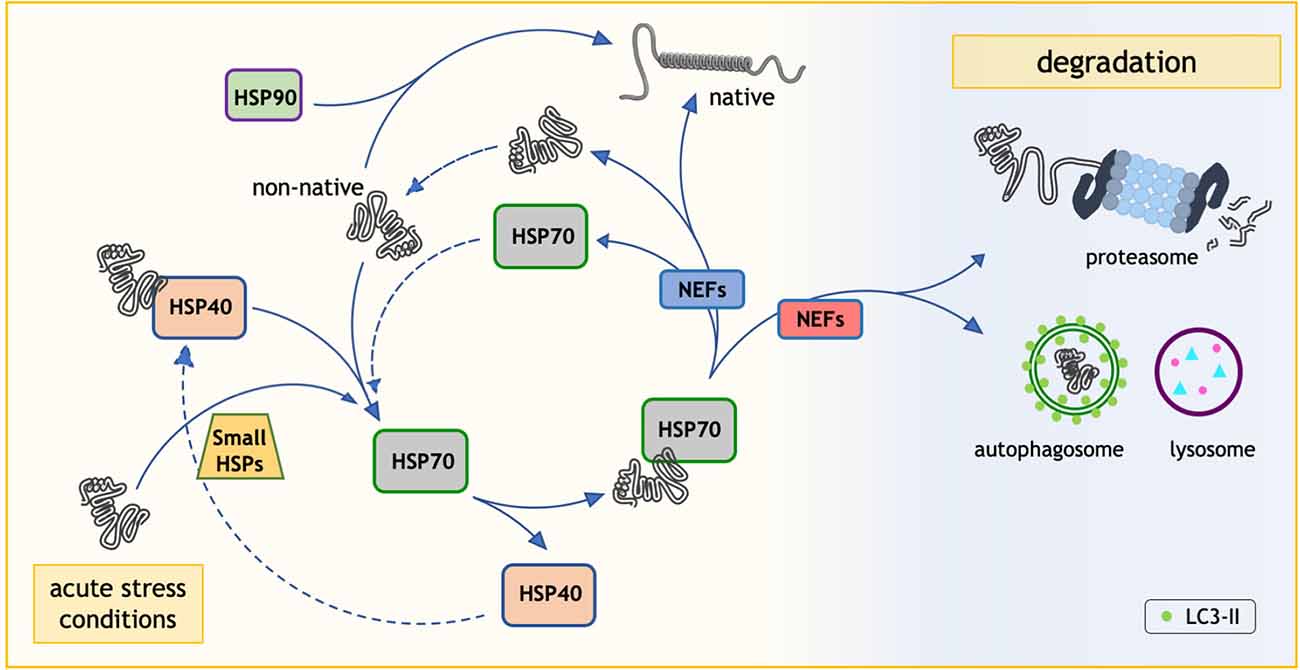
Figure 1. The role of heat shock proteins (HSP70) in the protein quality control (PQC) system. HSP70 plays an essential role in the protein folding process. Through its interaction with HSP40, HSP70 is able to fold the proteins in non-native conformations. HSP70 and HSP40 are not the only HSPs involved. In fact, the HSP90 system can assist protein folding in an independent way and the small HSPs respond to acute stress conditions. Notably, when necessary nucleotide exchange factors (NEFs/BAGs) route HSP70 client proteins to degradative pathways (Ubiquitin-proteasome system and/or autophagy).
The Degradative Systems
Cells, including post-mitotic cells like neurons and skeletal muscle cells, utilize two major degradative systems to enzymatically destroy aberrant proteinaceous materials and recycle their components for other proteins production. This process is assisted by chaperones (and their co-chaperones), which route aberrant proteins to degradative systems.
Proteins undergoing this degradation are damaged proteins or regulatory proteins that ended their functions. The two degradative systems are: (a) the ubiquitin proteasome system (UPS); and (b) the autophago-lysosomal pathway (ALP). Of note, UPS acts both in the cytosolic and nuclear compartment, while ALP acts only in the cell cytoplasm.
(a) The UPS is a highly specific and very selective proteolytic system mainly devoted to the clearance of short-lived proteins. The UPS inactivates proteins controlling cell cycle progression, apoptosis, transcription, and cell differentiation. Moreover, the UPS mediates the immune response and it is responsible for the clearance of damaged monomeric proteins. UPS is based on two subsequent steps: the protein is labeled by a covalent binding to ubiquitin (a small protein of 76 amino acids), which is itself ubiquitinated forming a poly-ubiquitin chain of several molecules of ubiquitin (Pickart, 2001a,b); and this poly-ubiquitinated protein is degraded by the 26S proteasome. The recognition of the protein to be degraded is mediated by different chaperones of the HSP70/HSP40 families in a complex (Figure 1). HSP70 has the ability to interact with specific E3-ubiquitin ligases (such as the C-terminus HSP70 interacting protein, CHIP), which selectively ubiquitinate misfolded proteins (Ciechanover, 1994; Ciechanover and Brundin, 2003; Ciechanover and Kwon, 2015). The ubiquitination cascade is rather complex. Ubiquitination initially requires the activation of E1 enzymes that activate ubiquitin; next, the activated ubiquitin is transferred to E2 enzymes, which in concert with the E3-ubiquitin ligases bind ubiquitin to a lysine residue of the substrate protein. E3-ubiquitin ligases have slightly different functions (Jackson et al., 2000; Joazeiro and Weissman, 2000). In addition, deubiquitinating enzymes (DUBs) are involved in this process (Amerik and Hochstrasser, 2004); DUBs maintain the cellular pool of free ubiquitin by processing ubiquitin precursors and recycling ubiquitin from poly-ubiquitinated substrates. Once polyubiquitinated, the substrate protein is recognized by the SQSTM1/p62, and other proteins of this class (Klionsky et al., 2016) and routed to the proteasome for degradation. The 26S proteasome has a typical barrel shape constituted by a large, multi-subunit protease complex: a 20S core complex with catalytic activity and a 19S regulatory complex, the cap. The cap receives the polyubiquitinated substrate, removes the poly-ubiquitin chain and induces its translocation into the 20S complex. Here, the substrate protein must enter the narrow central 20S cavity for the enzymatic degradation to small peptides. To this aim, folded proteins must be unfolded by the 19S subunit to reach a “linear” conformation. Thus, globular or aggregated proteins are not processed by the proteasome (Ciechanover and Brundin, 2003), and may even clog its catalytic core. Molecular chaperones and co-chaperones cooperating with the proteasomal-mediated degradation of ubiquitinated substrates include the already mentioned HSP70/HSP40 (now identified as HSPAs/DNAJs) and the HSP70/BAG1 complexes (Figure 1; Demand et al., 2001; Alberti et al., 2002; Kampinga and Craig, 2010; Kampinga and Bergink, 2016; Cristofani et al., 2017; Cicardi et al., 2018, 2019). In the latter case, the HSP70/CHIP complex, initially described as required for the substrate ubiquitination, can associate to BAG1, and together with SQSTM1/p62 it drives the ubiquitinated misfolded protein to proteasomal degradation.
(b) The lysosomal-mediated system collects proteins from various origins. The system is typically divided into microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy (normally identified as autophagy). These systems are evolutionarily well-conserved processes required for the degradation of proteins or large cytosolic components via the lysosome (Mizushima et al., 2008). In the case of microautophagy, the cytosolic components are directly engulfed into lysosomes via an invagination of its membrane (Sahu et al., 2011). In CMA, only a specific subset of proteins can be processed: those containing a pentapeptide lysosome-targeting motif KFERQ or related consensus motifs (also generated by specific post-translational modifications; Orenstein et al., 2013; Kaushik and Cuervo, 2018; Kirchner et al., 2019); the sequence allows the direct translocation of cargo into lysosome. CMA requires the docking to the lysosomal receptor lysosome-associated membrane protein 2A (LAMP2A), as well as the protein unfolding by a chaperone complex containing HSC70, BAG1, HSC70-interacting protein (HIP), Hsp-organizing protein (HOP), and HSP40 (DNAJB1; Kampinga et al., 2009; Kampinga and Craig, 2010; Kampinga and Bergink, 2016). Instead, in macroautophagy (which the general term “autophagy” usually refers to), the cytosolic components are engulfed into the autophagosome, a double-membrane vesicle that then fuses with the lysosome, in order to deliver its content to the lysosome for degradation (Xie and Klionsky, 2007). Initially considered as a sort of non-specific degradation for long-lived proteins, organelles, or protein aggregates, it is now clear that autophagy is tightly regulated by several pro-autophagic factors (Mizushima et al., 2008; Sardiello et al., 2009). In this latter form of autophagy, it is also possible to distinguish between “in bulk” autophagy and selective autophagy. While “in bulk” autophagy is characterized by a very high clearance capability but is rather non-specific since it entraps large portion of cytoplasm, selective autophagy is highly specific and involves specific molecular regulators (Kaushik and Cuervo, 2018). Selective autophagy includes chaperone-assisted selective autophagy (CASA; Arndt et al., 2010; Kettern et al., 2011; Sarparanta et al., 2012; Ulbricht et al., 2013, 2015; Ghaoui et al., 2016; Sandell et al., 2016; Cicardi et al., 2019; Cristofani et al., 2019; Rusmini et al., 2019), organelles-specific types of autophagy (mitophagy, lysophagy, ribophagy, granulophagy, etc.), or processes aimed at removing large protein aggregates (aggrephagy; Nivon et al., 2012; Stürner and Behl, 2017; Aparicio et al., 2020).
CASA has attracted large attention in the field of NDs, specifically in MNDs, since this highly selective autophagy is based on the recognition of misfolded substrates by a heteromeric complex composed of a small HSP, the HSPB8, with its co-chaperone BAG3. Once the misfolded protein is bound to HSPB8/BAG3, the HSP70/CHIP dimer (already seen in the UPS pathway) can be recruited. Here, the misfolded protein is rapidly ubiquitinated by CHIP, allowing recognition by the autophagy receptor SQSTM1/p62 (and related proteins) and forming the CASA complex. Some studies include HSP40 or DNAJ proteins in this complex (Sarparanta et al., 2012; Sandell et al., 2016). In this context, the role of SQSTM1/p62 is different from that exerted in association with BAG1/HSP70/CHIP, which allows the use of the UPS pathway. When acting with HSPB8/BAG3/HSP70/CHIP, the SQSTM1/p62 protein interacts with the ubiquitinated misfolded proteins (or other cargoes) and the lipidated form of the microtubule-associated proteins 1A/1B light chain 3B (LC3-II) anchored to the autophagosome membrane. To allow SQSTM1/p62 and LC3-II-action, the CASA complex takes advantage of a dynein binding motif present in the BAG3 sequence. The CASA complex bound to dynein is transported along microtubules to the microtubule organizing center (MTOC). Ubiquitinated and SQSTM1/p62-positive misfolded proteins are concentrated at MTOC to form the aggresomes. Meanwhile, LC3-II decorated-autophagosomes are generated, allowing aggresome insertion into a nascent autophagosome. The autophagosome containing the CASA complex and the misfolded proteins fuses with the lysosome to allow the degradation of the engulfed material following the canonical autophagic pathway.
Selective autophagy is also involved in the degradation of damaged organelles like mitochondria and lysosomes. In mitophagy, the damaged mitochondria stabilize PINK1 on its outer membrane. PINK1 recruits E3-ubiquitin ligases, like Parkin, which amplify the ubiquitination of proteins in the outer membrane mediating recruitment of the autophagic receptors that interact with LC3-II present on the forming autophagosome membrane (Youle and Narendra, 2011). Some of the mitochondrial membrane proteins, like mitofusin, are polyubiquinated with K48 ubiquitin chains. These proteins are substrates of VCP/p97, an AAA+ ATPase, that segregates these proteins from the mitochondria membrane and promotes their degradation via UPS. The removal of these proteins is necessary for mitochondria degradation (Tanaka et al., 2010; Tanaka, 2010; Kimura et al., 2013). In lysophagy, ruptured lysosomes expose galectins (Gal-3, Gal-8) as damage signals. Gal-8 is directly recognized by autophagy receptors, while Gal-3 recruits and binds TRIM16. Gal-3/TRIM16 complex promotes ubiquitination of lysosomal proteins and recruits autophagy initiation factors to trigger local phagophore formation (Thurston et al., 2012; Chauhan et al., 2016). Moreover, K63-ubiquitinated proteins recruit autophagy receptors, while K48-ubiquitinated proteins are targeted by VCP/p97 to UPS degradation. VCP/p97 recruitment to lysosome membranes and functioning are mediated by its cofactors and adaptors YOD1, UBXD1, and PLAA. The removal of K48 polyubiquitinated proteins is a critical step to promote lysosome degradation (Fujita et al., 2013; Akutsu et al., 2016; Papadopoulos et al., 2017).
The Unfolded Protein Response (UPR) and the Endoplasmic Reticulum-Associated Degradation (ERAD)
UPR and ERAD are two other key pathways devoted to the PQC in cells. UPR is typically activated in the presence of an abnormal excess of misfolded proteins, while ERAD mediates their degradation by taking advantage of the cytosolic proteasome mentioned above. In fact, the accumulation of misfolded proteins in the endoplasmic reticulum (ER) activates the UPR. This action is mediated by three different “sensors”—inositol requiring enzyme 1 (IRE1α), PKR-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6; Hetz, 2012)—that signal to dedicated pathways to stimulate either protein folding or protein degradation. During this process, ribosomes are forced to attenuate protein translation. ERAD has a specific function in PQC system since the ER is a major site for protein folding. When aberrant ER-resident proteins are processed by ERAD, they are released into the cytosol for proteasomal (when these are still soluble) or for autophagic clearance (when they are in an aggregated form; Hetz, 2012). Even in the case of ERAD, the proteins are ubiquitinated by specific E3-ubiquitin ligases, like HRD1 in the SEL1L-HRD1 protein complex (where SEL1L acts as a cofactor). Ubiquitinated misfolded proteins can be “retro-translocated” or “dislocated” (extracted) from the ER membrane and transported to the cytosol mainly by the activity of VCP/p97. VCP/p97 in complex with UFD1-NPL4 first binds HRD1 and the ubiquitinated proteins, then addresses substrates to the proteasome via shuttle cofactors (Ye et al., 2005; Senft and Ronai, 2015). Even in the case of the UPR-ERAD, a central role is played by an HSP70, BiP (or HSPA5 or GRP-78), which has low intrinsic ATPase activity, enhanced by co-chaperones of the DNAJ-proteins (the same class of the HSP40, like ERdj4 or DNAJB9). In addition to protein folding, the ER controls the Ca2+ homeostasis, being the major intracellular Ca2+ reservoir (Hetz and Mollereau, 2014). When misfolded proteins accumulate in the ER, the depletion of ER Ca2+ impacts on cell activity and enhances stress. Store-operated Ca2+ influx is activated in these conditions to assure the replenishment of Ca2+ levels (Szegezdi et al., 2006). If ER stress is prolonged, the ability of the UPR to restore ER homeostasis is reduced and this may cause ER stress-induced apoptosis by activation of caspase 12 (Yoneda et al., 2001). Once activated, the UPR-ERAD converges on the proteasome or to autophagy; therefore, in this review we will only focus on the proper degradative pathways. Details on UPR-ERAD can be found elsewhere (for an extensive review see Hwang and Qi, 2018).
Release Mediated by Extracellular Vesicles
Emerging data strongly suggest that the extracellular secretion may also play an important role in the maintenance of intracellular protein homeostasis by cooperating with or even being a part of the PQC system (Desdín-Micó and Mittelbrunn, 2017; Xu et al., 2018; Guix, 2020). In fact, it has been found that several NDs-related proteins are secreted in double membrane spherical particles known as extracellular vesicles. This is the case for the amyloid-beta peptide and tau/phosphorylated tau for Alzheimer’s disease (Pérez et al., 2019), alpha-synuclein for Parkinson’s disease (Longoni et al., 2019), misfolded/mutant SOD1, TDP-43 and its pathological-related C-terminal fragments (of 35 kDa and 25 kDa) and FUS for ALS (Basso and Bonetto, 2016; Iguchi et al., 2016; Hanspal et al., 2017; Sproviero et al., 2018), and progranulin, TDP-43, and C9orf72 DPRs for FTD and ALS-FTD (Benussi et al., 2016; Iguchi et al., 2016; Westergard et al., 2016). The extracellular vesicles are heterogeneous in size and are mainly classified into three different types: exosomes, microvesicles, and apoptotic bodies. These vesicles differ for size, proteins, and lipids composition and intracellular origin. In fact, exosomes are secreted membrane vesicles (approximately 30–120 nm in diameter) formed intracellularly and released from exocytosis of multivesicular bodies, whereas apoptotic bodies (approximately 1,000–4,000 nm in diameter) are released by dying/apoptotic cells. Microvesicles (approximately 200–1,000 nm in diameter) are shed from cells by an outward protrusion (or budding) of the plasma membrane followed by fission of their membrane stalk (for a detailed review see Akers et al., 2013; Colombo et al., 2014; van Niel et al., 2018). A tight connection between PQC and extracellular vesicles is particularly true for exosomes (Xu et al., 2018). As stated above, exosomes are intraluminal vesicles of the endosomal compartment that maturate into a structure called the multivesicular body after a very dynamic process. The multivesicular body may release its content into the lysosome for degradation or, under certain conditions, it may fuse with the plasma membrane and secrete its intraluminal vesicles, the exosomes. Interestingly, components of the CASA complex may also affect/take part in extracellular vesicles pathway: for example, STUB1/CHIP deficiency resulted in an increased secretion of small extracellular vesicles that are enriched in ubiquitinated and/or undegraded proteins and protein oligomers (Ferreira et al., 2019), and BAG3 is found to be involved in the exosome secretion of mutant Huntingtin upon proteasome blockade (Diaz-Hidalgo et al., 2016). These evidences suggest that extracellular vesicles have to be considered as new actors in the proteostasis scenario, together with chaperones and the degradative systems.
How the Protein Quality Control System Protects Against Misfolded Protein Toxicity in SBMA and ALS
Data collected over the last 30 years suggest that ARpolyQ and several ALS-associated proteins (listed in Table 1) may lead to PQC system alterations (Kabashi and Durham, 2006; Voisine et al., 2010; Rusmini et al., 2016, 2017; Cristofani et al., 2018, 2019). At the same time, the boost of key proteins involved in PQC system regulation is protective in SBMA and ALS (Waza et al., 2006; Yu et al., 2011; Giorgetti et al., 2015; Crippa et al., 2016b; Rusmini et al., 2016, 2017, 2019, 2020; Cristofani et al., 2018, 2019; Mandrioli et al., 2019).
Figure 2 summarizes how all the PQC system components work synergistically to prevent misfolded protein accumulation in these diseases.
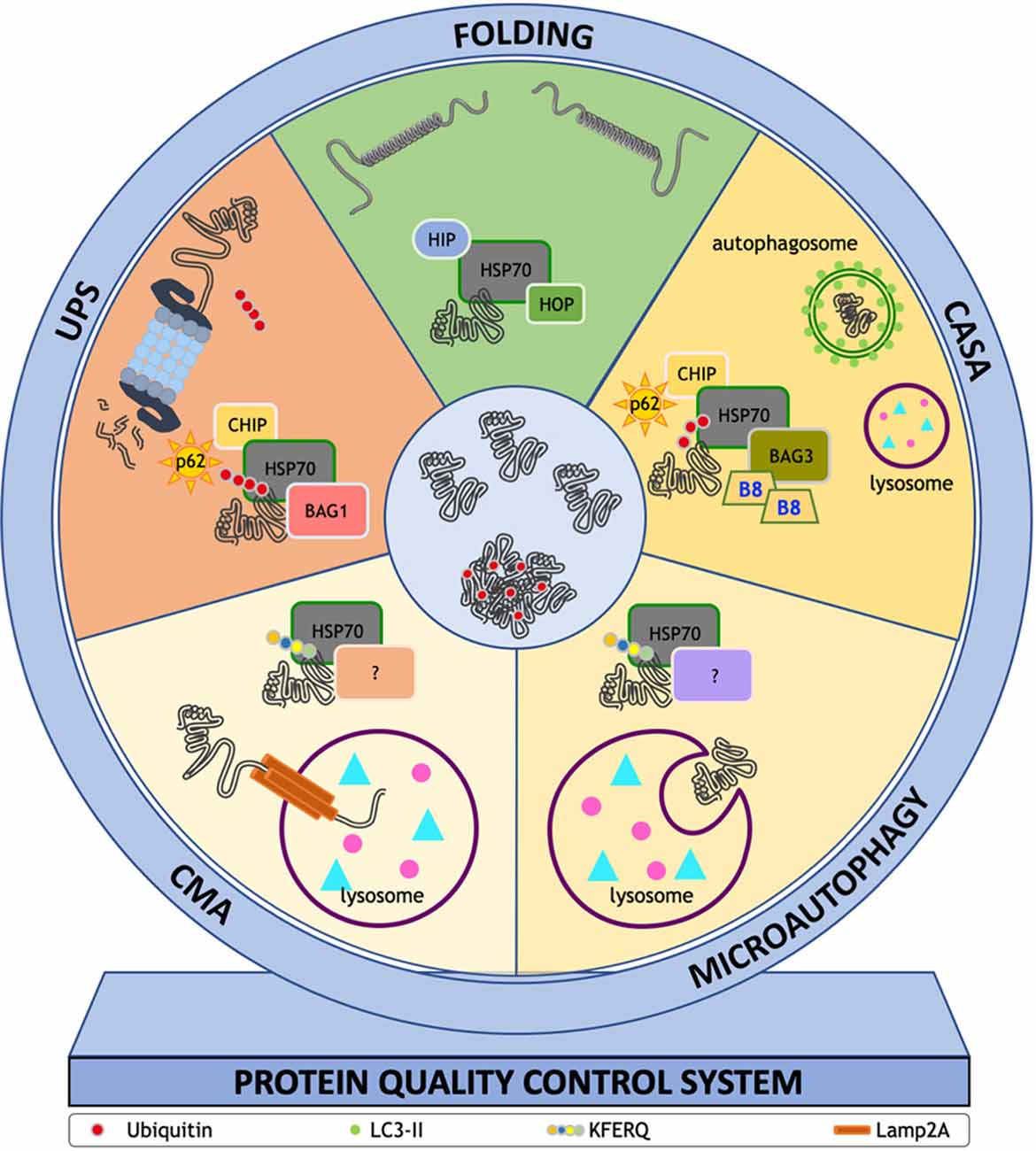
Figure 2. ThePQC system. The fate of misfolded proteins is finely tuned by the PQC system. This system is centered on a group of chaperones and co-chaperones assisting proteins to reach their correct conformation or directing proteins to degradative systems. Each pathway needs specific proteins that assist the action of HSP70: (i) HSC70 interacting protein 1 (HIP) and HSP70-HSP90 organizing protein (HOP) in the folding process; (ii) sequestosome 1 (SQSTM1/p62), E3-ubiquitin ligase C-terminus HSP70 interacting protein (CHIP), BAG family molecular chaperone regulator 3 (BAG3) and heat shock protein B8 (HSPB8, B8 in figure) in chaperone assisted selective autophagy (CASA); (iii) Lysosome-associated membrane glycoprotein 2 (Lamp2A) in chaperone mediated autophagy (CMA); and (iv) SQSTM1/p62, CHIP, BAG family molecular chaperone regulator 1 (BAG1) in ubiquitin proteasome system (UPS). The HSP70 interactors at lysosome membrane remain to be determined [indicated in figure as (?)] even if coimmunoprecipitation and colocalization studies identified HSP90, HSP40, HOP, HIP, and BAG1. Their role in CMA and microautophagy remains to be determined.
Folding Process
The first line of PQC system intervention on the misfolded protein is an attempt to restore the proper protein folding. Even if the folding process is well-understood, many questions still remain open in the case of disease-associated misfolded proteins; in particular, to what extent the refolding of a protein may occur after the first aggregation steps. The main actors in the folding process are the HSP70s (also named HSPAs), which are similar to nanomachines capable of switching conformation using hydrolysis of ATP (Figure 1). This allows HSP70 to change conformation in order to assist protein folding, disaggregation, and degradation (see Kampinga and Craig, 2010; Kampinga and Bergink, 2016 for an extensive review). HSP70 is a hub that requires the assistance of HSP40s (or DNAJ proteins) in order to recognize the protein to be folded, and of NEFs, like the BAGs, which exchange ADP/ATP during the hydrolytic process (Sondermann et al., 2001; Rauch and Gestwicki, 2014).
Misfolded proteins responsible for SBMA and ALS are able to alter this finely-tuned process. These misfolded proteins escape the correct folding and expose unstructured domains highly prone to aggregate. Such domains are present in the ARpolyQ in its poorly structured N-terminus containing the polyQ stretch, in the prion-like domains of TDP-43 and FUS proteins, and maybe also in the five DPRs derived from the C9orf72 mRNA, which do not possess tertiary structures. These unstructured domains may clamp together in a liquid-liquid partitioning of phases, forming membraneless organelles attracting other compatible molecules (e.g., RNAs or proteins which normally interact with these unstructured proteins). The mutations in these proteins greatly enhance their capability to generate liquid-liquid intracellular compartments, which soon after their formation may mature into aggresomes, stable aggregates, and even insoluble inclusions trapping specific intracellular factors (Molliex et al., 2015; Patel et al., 2015; Ganassi et al., 2016; Alberti et al., 2017; Boeynaems et al., 2017; Mackenzie et al., 2017; Mateju et al., 2017; Marrone et al., 2018). Specific proteins are known to accelerate the conversion of the aggregates formed after phase separation into stable insoluble aggregates. Conversely, chaperones and co-chaperones may prevent this conversion, delaying the maturation into stable structures and facilitating the disassembling of the newly formed membraneless organelles. This activity of chaperones and co-chaperones should permit the refolding process of a misfolded protein even after its entrapping in the aggresomes when they are still dynamic (Jaru-Ampornpan et al., 2013; Mattoo and Goloubinoff, 2014; O’Driscoll et al., 2015; Zaarur et al., 2015; Mathew and Stirling, 2017; Kitamura et al., 2018; Alexandrov et al., 2019).
Alterations of UPS in SBMA and ALS
Evidence suggests that ARpolyQ, SOD1, TDP-43, and its ALS associated fragments, as well as other ALS-proteins, including at least one out of five DPRs of C9orf72, are processed via the proteasome (Rusmini et al., 2007, 2010, 2011, 2013, 2019; Sau et al., 2007; Crippa et al., 2010b, 2016a; Onesto et al., 2011; Cristofani et al., 2017, 2018; Cicardi et al., 2018, 2019). However, the large amounts of misfolded proteins formed when specific gene mutations occur may overwhelm the UPS capability to degrade them efficiently. This process is accentuated in aged cells in which the chaperone and UPS activities are reduced (Ciechanover and Brundin, 2003; Terry et al., 2004, 2006; Wang K. et al., 2018; Hegde et al., 2019). It is also possible, as in the case of the elongated polyQ tract of the AR, that the proteasome proteolytic capability is unable to digest the long polyQ sequence since no consensus cleavage sites for its enzymatic activity are present between the Qs; thus, long uninterrupted polyQ size might block the narrow catalytic site, where only a single protein can enter and be degraded. We showed that while the wtAR with a 23Q stretch can be cleared via the proteasome even in presence of androgens (Rusmini et al., 2007), the mutant ARpolyQ may impair the UPS function; in fact, by expressing the ARpolyQ in basal condition (absence of androgens), we noted an accumulation of the proteasome activity reporter GFP-CL1 (GFPu) as an indication that the elongated polyQ is poorly processed by the UPS and interferes with the activity of this degradative pathway. Interestingly, the inactivated ARpolyQ does not play toxicity in all cell models tested. Surprisingly, when the ARpolyQ is activated by androgens (which bound at the AR C-terminus), the protein is thought to acquire toxic conformations (Stenoien et al., 1999; Simeoni et al., 2000; Piccioni et al., 2002; Poletti, 2004), but the UPS is fully functional (Rusmini et al., 2007) since the GFPu reporter is fully degraded by the UPS. An explanation for this unexpected phenomenon is that by inducing the ARpolyQ toxic conformation, androgens also induce its misfolding (possibly via a phase partitioning phenomenon; Eftekharzadeh et al., 2019; Escobedo et al., 2019) and sequestration into subcellular compartments (the aggregates), protecting the cell from this dangerous protein conformation (Rusmini et al., 2007, 2010). The formation of aggregates acts as a sink that permits UPS desaturation from the excess of “free” polyQ to be processed. Meanwhile, aggregates might stimulate autophagy for ARpolyQ clearance (see below). It is thus expected that also autophagy alterations might contribute to the accumulation of stable insoluble ARpolyQ aggregates in cells (Rusmini et al., 2013; Giorgetti et al., 2015; Cristofani et al., 2017; Cicardi et al., 2019). As will be discussed below, the potentiation of CASA restores normal ARpolyQ clearance. A similar UPS role has been found involved in the clearance of ALS-misfolded proteins. Mutant SOD1 is mainly cleared by the UPS, and its pharmacological inhibition with MG132 results in an accumulation of ubiquitin-positive SOD1 aggregates in cells (Crippa et al., 2010a,b, 2016a; Cicardi et al., 2018). This aggregated SOD1 is poorly removed by autophagy but, as seen for ARpolyQ, the induction of CASA restores complete clearance of aggregating mutant SOD1 (Crippa et al., 2010a,b, see below). TDP-43 and its 35 kDa and 25 kDa TDP-43 fragments follow the same route of degradation identified for inactive ARpolyQ and mutant SOD1 (Crippa et al., 2016a; Cicardi et al., 2018, 2019). Even in this case, UPS inhibition results in an accumulation and mislocalization of TDP-43 and fragments, aside from the 25 kDa TDP-43 fragment. CASA induction reverts also this phenotype (Crippa et al., 2016a; Cicardi et al., 2018). It is unclear whether autophagy defects play a major role in the accumulation of these TDP-43-related aberrant species and this may underline differences in the type of toxicity exerted by these MNDs proteins. Recent works have shown that TDP-43 inclusions and TDP-43 hyperphosphorylation (typical hallmarks of ALS-motor neurons) are also present in muscles in sALS patients. This discovery raised a question: whether TDP-43 misfolded species could accumulate and exert toxicity in muscle cells. We found that the insoluble TDP-43 fragments also accumulate in muscle C2C12 cells, but their aggregation is reverted by tuning the expression of key components of the CASA complex. Whether the accumulation of these fragments in muscle tissue is causative of muscle atrophy is yet to be elucidated (Cicardi et al., 2018).
Also, the C9orf72 DPRs degradation is mediated by UPS and autophagy, even with different behavior of the five DPRs, since only the polyGP is efficiently degraded by the UPS (Cristofani et al., 2018). PolyGP is also degraded via autophagy that is able to efficiently remove polyPA, polyGR, and polyGA (Cristofani et al., 2018). Conversely, only the polyPR seems to be resistant to both degradative systems in basal condition. The reasons for these differences are still unclear, but CASA activation prevents the accumulation of all five DPRs (Cristofani et al., 2018), as will be described below.
Collectively, these data suggest that several MND-associated misfolded proteins can be cleared by the UPS system, possibly in a monomeric state. It is expected that UPS overwhelming will result in an accumulation of these species that, once concentrated in specific subcellular compartments (liquid-liquid, aggresomes, etc.), may reversibly aggregate. This mechanism might protect from misfolded protein toxicity since these species are sequestered, limiting their potential toxicity. If the accumulation persists, the aggregates may mature to more stable and potentially toxic species, and thus must be removed using alternative strategies by the cells.
Alteration of Autophagy and CASA in SBMA and ALS
Alteration of autophagy has been reported in animal and cell models of SBMA (Montie and Merry, 2009; Yu et al., 2011; Doi et al., 2013; Rusmini et al., 2013, 2015, 2019; Chua et al., 2014; Cortes et al., 2014b; Thellung et al., 2018; Cicardi et al., 2019) and ALS (Kabuta et al., 2006; Morimoto et al., 2007; Li et al., 2008; Wang et al., 2012; Crippa et al., 2013; Xiao et al., 2015; Evans and Holzbaur, 2019; Nguyen et al., 2019). Despite this, the complexity of the autophagic pathway makes it difficult to fully understand which level of this multistep process is affected by the presence of misfolded proteins. It is evident from experimental data that autophagy activation has a beneficial role in disease since pharmacological or genetic induction of autophagy ameliorates disease phenotype (e.g., delaying disease onset, slowing down its progression or ameliorating motor behavior; Montie et al., 2009; Wang et al., 2012; Castillo et al., 2013; Kim et al., 2013; Tohnai et al., 2014; Zhang et al., 2014; Giorgetti et al., 2015; Li et al., 2015; Perera et al., 2018; Wang Y. et al., 2018; Rusmini et al., 2019). Unfortunately, not all studies agree with these observations (Zhang et al., 2011). By focusing on CASA, which has already been mentioned above, it must be noted that HSP70 chaperones and others require the assistance of co-chaperones, like the member of the NEF family (Kampinga and Craig, 2010), which includes the BAGs (Takayama and Reed, 2001). Cells utilize different BAGs to route misfolded proteins either to the UPS or to autophagy (Figure 1). BAG1 associates to HSP70 and CHIP to route misfolded proteins to the UPS, while BAG3, in association with HSPB8, interacts with HSP70/CHIP to route misfolded proteins to autophagy (Figure 2). This allows to select which pathway has to be followed by misfolded proteins to be efficiently cleared from cells; the perturbation of this equilibrium may result in misfolded proteins accumulation (Cristofani et al., 2017, 2019; Rusmini et al., 2017). The importance of the CASA complex in cell protection against proteotoxicity is underlined by the fact that mutations in the genes coding for almost all components of the CASA complex have been associated with human diseases. Indeed, mutations in HSPB8 cause diseases of motoneurons and/or muscle cells [Charcot-Marie-Tooth (CMT) type 2L disease, hereditary distal motor neuropathy type II (dHMN-II), or distal myopathy; Fontaine et al., 2006; Irobi et al., 2010; Ghaoui et al., 2016; Al-Tahan et al., 2019]. Mutations in BAG3 are causative of dilated cardiomyopathy (Arimura et al., 2011), muscular dystrophy (Selcen et al., 2009), giant axonal neuropathy, and late-onset axonal CMT neuropathy (Jaffer et al., 2012; Shy et al., 2018). Interestingly, three BAG3 mutations involve the Pro209 residue (Pro209Leu, Pro209Ser, Pro209Glu), which falls in one of the two HSPB8-interacting Ile-Pro-Val (IPV) motifs. These Pro209 mutants still retain the ability to bind to all CASA members but they impair HSP70 client processing, and they accumulate at the aggresome preventing target protein degradation and sequestering CASA members (Meister-Broekema et al., 2018; Adriaenssens et al., 2020). Mutation in STUB1/CHIP have been found in Gordon Holmes syndrome (multisystemic neurodegeneration; Hayer et al., 2017) and more recently in SCA48 (Genis et al., 2018), and a destabilized CHIP (linked to six different variants) is present in SCA16 (Pakdaman et al., 2017; Kanack et al., 2018); also, a missense mutation in the CHIP-ubiquitin ligase domain was reported as the cause of a form of spinocerebellar autosomal recessive 16 (SCAR16; Shi et al., 2013, 2018). Mutations of the SQSTM1/p62, which recognizes the CHIP-ubiquitinated cargo inside the CASA complex (some authors include it as a member of this complex), are responsible for fALS (Fecto et al., 2011; Teyssou et al., 2013). Of note, it has been suggested that in skeletal muscle, DNAJB6 of the DNAJ/Hsp40 family (HSP70 co-chaperones) suppresses aggregation of misfolded proteins involved in NDs (Hageman et al., 2010) and participates to the formation of the CASA complex (Sarparanta et al., 2012). Interestingly, a mutation in DNAJB6 causes Limb-girdle muscular dystrophies (LGMDs), characterized by aggregates of DNAJB6 sequestering CASA complex proteins (Sandell et al., 2016).
The CASA complex is involved in mutant SOD1-associated fALS (Crippa et al., 2013). Indeed, mutant SOD1 induces a robust autophagic response both in the spinal cord and in muscle. BAG1, BAG3, HSPB8, LC3, and SQSTM1/p62 are significantly upregulated in mutant SOD1 transgenic ALS mice at the symptomatic stage (16 weeks). Notably, the autophagic response is much higher in muscle than in spinal cord, supporting the absence of high molecular weight insoluble species of mutant SOD1 in muscle; this also suggests that the toxicity exerted by mutant SOD1 in muscle cells is probably not related to the classical mechanism of intracellular protein aggregation (Galbiati et al., 2012, 2014). Interestingly, an analysis performed in SBMA knock-in mouse model revealed that the CASA complex is highly upregulated in skeletal muscle after disease onset, while no variations were observed in the spinal cord. In fact, HSPB8 and BAG3 mRNA and protein levels are increased in SBMA mice at the symptomatic stage compared to control, as well as the co-chaperone BAG1, involved in routing misfolded proteins to UPS. The increased BAG3 to BAG1 ratio suggested that autophagy is the main proteolytic pathway activated in muscle tissue during SBMA progression and CASA complex is involved in reducing ARpolyQ toxicity in skeletal muscle, which is a primary site of SBMA pathogenesis (Rusmini et al., 2015). HSPB8 seems to be a limiting factor for the CASA complex (Crippa et al., 2010a,b). HSPB8 overexpression rescues from protein accumulation and aggregation of mutant SOD1 and TDP-43 in cell models of ALS (Crippa et al., 2010a,b), while its silencing has opposite effects favoring misfolded proteins accumulation in motor neurons (Crippa et al., 2010a,b). Overlapping data were obtained with other misfolded proteins implicated in Alzheimer’s disease, Parkinson’s disease, a form of spinal cerebellar ataxia, (SCA3), SBMA, fALS, and FTD. In fact, HSPB8 enhances the autophagy clearance of beta-amyloid, alpha-synuclein (α-syn), the polyQ proteins huntingtin, ataxin-3, and ARpolyQ, as well as all five DPRs from the C9orf72 mRNA (Chávez Zobel et al., 2003; Wilhelmus et al., 2006; Carra et al., 2008a,b; Crippa et al., 2010b, 2016a; Bruinsma et al., 2011; Seidel et al., 2012; Rusmini et al., 2013, 2016; Giorgetti et al., 2015; Cicardi et al., 2018), while HSPB8 downregulation has the opposite effects (Crippa et al., 2010b, 2016a,b; Rusmini et al., 2013; Cristofani et al., 2017).
Since HSPB8 may be a limiting factor of CASA complex activity and its overexpression is sufficient to restore autophagy, it is clear that this protein represents a valid therapeutic target for these NDs. It has been demonstrated that HSPB8 expression is induced by estrogens and other selective estrogen receptor modulators (SERMs; Sun et al., 2007; Piccolella et al., 2014, 2017; Meister-Broekema et al., 2018), and this could help to explain why gender differences occur in the appearance of several NDs (Vegeto et al., 2020). Recently, we set up a high throughput screening (HTS) using a reporter luciferase gene under the transcriptional control of the human HSPB8 promoter. With this system, we found that colchicine [a Food and Drug Administration (FDA)- and European Medicine Agency (EMA)-approved drug] stimulates HSPB8 expression and enhances the autophagy clearance of the insoluble TDP-43 species (Crippa et al., 2016a) in models of ALS. The drug is presently in phase II clinical trial for ALS (Mandrioli et al., 2019). Other HSPB8 inducers are some disaccharides, like trehalose, melibiose, or lactulose (Rusmini et al., 2013, 2019; Giorgetti et al., 2015). Trehalose has been tested in mouse models of Huntington’s disease, ALS, Parkinson’s disease, Alzheimer’s disease, succinate semialdehyde dehydrogenase deficiency, and oculopharyngeal muscular dystrophy, and found to be capable of ameliorating disease course and symptomatology (Tanaka et al., 2004; Davies et al., 2006; Rodriguez-Navarro et al., 2010; Perucho et al., 2012; Schaeffer and Goedert, 2012; Castillo et al., 2013; Du et al., 2013; Sarkar et al., 2014; Zhang et al., 2014; He et al., 2016). The mechanism of action of trehalose and its analogs, melibiose and lactulose, was recently uncovered. These disaccharides induce transient lysosomal permeabilization and possibly calcium release from lysosomes. These events trigger the Transcription Factor EB (TFEB) pathway, mediated by the calcium-dependent phosphatase PPP3/calcineurin, which dephosphorylates TFEB. Trehalose-activated TFEB migrates into the nucleus where it acts on CLEAR responsive elements to enhance the expression of genes controlling autophagy and lysosomal biogenesis. With this mechanism trehalose/TFEB-mediated activation of autophagy promotes the clearance of damaged lysosomes through lysophagy, but in parallel exerts neuroprotection by promoting the degradation of mutant and misfolded proteins from neurons (Rusmini et al., 2019).
Both colchicine and trehalose also induce BAG3 expression (Lei et al., 2015; Crippa et al., 2016b), indicating that these compounds may act via a general potentiation of CASA. Other drugs have been found able to stimulate BAG3 expression (e.g., proteasome inhibitors, TNF-related apoptosis-inducing ligand, fludarabine, cytosine arabinoside, and etoposide) but unfortunately, these drugs are used in chemotherapy with relevant side effects, and are thus not suitable for NDs (Romano et al., 2003; Chiappetta et al., 2007; Rapino et al., 2014). However, they might serve as molecule templates for the development of safer and better tolerated derivatives.
Alteration of CMA in SBMA and ALS
Nothing is known so far about the involvement of CMA in the degradation of ARpolyQ in SBMA. Instead, recent data suggest that CMA may play a role in NDs, including ALS (Ormeño et al., 2020). Indeed, CMA is essential in Parkinson’s disease where its dysregulation modifies the onset or progression of the disease (Arias and Cuervo, 2011; Cuervo, 2011; Alfaro et al., 2018; Kaushik and Cuervo, 2018). Alpha-synuclein protein, leucine-rich repeat kinase 2 (LRRK2), Parkinson disease protein 7 (PARK7), and DJ-1, as well as myocyte-specific enhancer factor 2D protein (MEF2D), which are dysregulated or mutated in Parkinson’s disease, are CMA substrates (Vogiatzi et al., 2008; Yang et al., 2009; Arias and Cuervo, 2011; Cuervo, 2011; Orenstein et al., 2013; Murphy et al., 2015; Alfaro et al., 2018; Kaushik and Cuervo, 2018). Alzheimer’s disease is also associated with CMA since the beta-amyloid peptide (Aβ), the microtubule-associated protein Tau or the Regulator of calcineurin 1 (RCAN1) are involved in Alzheimer’s disease and are dysregulated when CMA is altered (Liu et al., 2009; Wang et al., 2009, 2010; Park et al., 2016). CMA also plays a role in Huntington’s disease (Koga et al., 2011; Qi et al., 2012), and mutant huntingtin can sequester LAMP2A and HSC70, two major players of CMA (Alfaro et al., 2018).
In ALS, CMA has been involved in TDP-43 metabolism (Huang et al., 2014). These data were recently corroborated by a study of the group of Budini et al., who pointed out that also TDP-43 can be a CMA substrate (Ormeño et al., 2020). This study started from the observation that TDP-43 contains a KFERQ-like domain, the consensus sequence that allows the interaction with HSC70 (Huang et al., 2014); mutation in this domain blocks the ubiquitin-dependent binding of TDP-43 with HSC70. Other authors have shown that LAMP2A downregulation induces the intracellular accumulation of the ALS-associated TDP-43 fragments of 35 and 25 kDa (Huang et al., 2014), and TDP-43 can also be forced to be degraded via CMA (Tamaki et al., 2018). Ormeño et al. (2020) showed in vitro that a recombinant form of TDP-43 is processed by isolated rat liver lysosomes, a process that can be reduced by competition with the GAPDH protein, a typical CMA substrate. Endogenous TDP-43 accumulates in CMA+ lysosomes of the brain (Ormeño et al., 2020). By using an artificial TDP-43 aggregate-prone protein, Ormeño et al. (2020) demonstrated its interaction with HSC70 and LAMP2A, which causes an upregulation of CMA activity and lysosomal damage. These data open up the question of how CMA is involved not only in the few fALS forms associated with mutations of TDP-43, but also in the vast majority of sALS forms characterized by an intense mislocalization and accumulation of TDP-43 in affected neuronal and motor neuronal cells of ALS patients. By analyzing the two CMA regulators (LAMP2A and HSC70) in peripheral blood mononuclear cells (PBMCs) of ALS patients, it was found that the levels of the lysosome receptor LAMP2A were similar in control and ALS PBMCs, while the expression of the cytosolic chaperone HSC70 was found reduced, but the total amount of insoluble TDP-43 protein was found increased and accompanied by aberrant intracellular localization (Arosio et al., 2020). In parallel, HSC70 downregulation in human neuroblastoma cells correlates with the increased accumulation of the TDP-43 protein (Arosio et al., 2020). These data are in line with experimental observation showing that HSC70 is reduced in motor neurons of TDP-43-based ALS fly models, as well as in iPSCs C9orf72 models differentiated to motor neurons (Coyne et al., 2017). In addition to these observations, in ALS-PBMCs, the ratio of the expression levels and protein of BAG1 and BAG3, which determines the equilibrium between proteasome and autophagy (including CASA), was also found altered (Arosio et al., 2020). Thus, even if CMA is not directly affected in ALS-PBMCs, the reduction of the CMA regulator HSC70 may be involved in ALS pathogenesis.
Alteration of UPR-ERAD in SBMA and ALS
As mentioned above, proteasome and autophagy work together in response to proteotoxic stimuli. Both pathways are also involved downstream in the UPR occurring in the ER. The UPR, activated in the ER lumen, generates a transient translational inhibition along with the induction of chaperones and the stimulation of the degradative pathways. Misfolding proteins here are identified by BiP/GRP78 (an HSP70), which assists the ERAD, also activating PERK and IRE1. The PERK receptor attenuates translation in response to UPR involving oligomerization and autophosphorylation of PERK with eIF2alpha phosphorylation. In parallel, the transcription factor XBP1 activated by alternative splicing induces UPR stress genes, while cleaved activated ATF6 exits the ER and moves to the nucleus to stimulate other UPR genes. Collectively, this restores ER activities: in SBMA, an ARpolyQ N-terminal fragment activates ER stress-inducible promoter via ATF6, IRE1, and PERK. Indeed, ARpolyQ toxicity is enhanced by ATF6 blockage and reverted by ATF6 overexpression. Also, stimulation of PERK increases ARpolyQ toxicity (Thomas et al., 2005). Thus, ARpolyQ induces UPR, while UPR stimulation is protective in SBMA (Rusmini et al., 2016). In a SBMA knock-in mouse model, the downregulation of transcription factor C/EBP homologous protein (CHOP), involved in UPR-ERAD, worsened muscle atrophy (Yu et al., 2011). In parallel, in mouse embryonic stem cells (ESCs), ARpolyQ inclusions sequester CHIP and BiP/GRP78, inducing ER stress and apoptosis. UPR was found with the induction of the ER chaperones BiP/GRP78 and GRP94 and the stress markers ATF6, phosphorylated PERK, GADD153/CHOP, and spliced XBP-1. Notably, BiP/GRP78 overexpression reverted this phenotype, while BiP/GRP78 downregulation had the opposite effect (Yang et al., 2013). As mentioned above, ER stress and Ca2+ homeostasis are tightly connected. In mouse model of SBMA (Sopher et al., 2004; Malik et al., 2011; Montague et al., 2014) alteration of Ca2+ homeostasis has been reported in embryonic motor neurons in response to ER stress causing ER-stress-induced apoptosis (Montague et al., 2014). ARpolyQ specifically depleted ER Ca2+ levels and the store-operated Ca2+ influx (Hetz and Mollereau, 2014; Tadic et al., 2014), possibly via the reduction of the sarcoendoplasmic reticulum Ca2+ ATPases (SERCA) 2b pump activity. This pump allows ER Ca2+ re-uptake (Foradori and Handa, 2008), and its dysregulation activates caspase 12 (Montague et al., 2014). Thus, ER stress is also involved in SBMA pathogenesis and may represent an additional therapeutic target for this disease.
ER morphology alterations occur both in ALS patients and ALS mouse models (Dal Canto and Gurney, 1995; Dal Canto, 1995; Oyanagi et al., 2008; Lautenschlaeger et al., 2012), possibly because of protein accumulation in ER causing ER stress (Sasaki, 2010). Also, the Golgi apparatus is affected in ALS (Fujita et al., 2000; Stieber et al., 2000). Mutant SOD1 inclusions in ER are positive for BiP/GRP78 and calnexin (Wate et al., 2005; Kikuchi et al., 2006), while some ER chaperones are upregulated in ALS patients and mice (Atkin et al., 2006). Notably, mutant SOD1 specifically binds Derlin-1, which controls the ERAD machinery, and triggers ER stress-induced apoptosis (Nishitoh et al., 2008). ER stress in ALS may also result from altered ER calcium homeostasis (Grosskreutz et al., 2010) or by ER-mitochondria calcium cycle unbalance (Damiano et al., 2006; Grosskreutz et al., 2010; Jaronen et al., 2014). In addition, ATF6, phospho-PERK, and phospho-eIF2α are elevated in ALS mice and cell models (Atkin et al., 2006, 2008; Saxena et al., 2009). In the spinal cord of ALS patients and mice, IRE1 is increased (Atkin et al., 2006, 2008) and its phosphorylated form correlated with spliced XBP1 in ALS mice (Kikuchi et al., 2006). Notably, autophagy is induced in double knockout/transgenic mice with mutant G86R-SOD1 and XBP1 blockage (Hetz et al., 2008; Hetz, 2012; Hetz and Mollereau, 2014), suggesting that autophagy may serve to protect when UPR/ERAD fails. A recent study performed by the group of de Belleroche suggests that at least 40 different target genes, associated with ERAD and regulated by XBP1 or ATF6, are altered in spinal cord specimens from ALS patients; this is paralleled by severe alterations and activation of the IRE1α-XBP1 and ATF6 pathways (Montibeller and de Belleroche, 2018). Among these genes, co-chaperones of the DNAJ family (DNAJB9 and DNAJC10) modulating HSPA5 (BiP/Grp78, which is the only HSP70 in the ER; Kampinga and Bergink, 2016) were increased in this dataset. Both DNAJB9 and DNAJC10 are involved in ERAD (Behnke et al., 2015) and may suppress cell death induced by ER stress (Kurisu et al., 2003). As occurs in SBMA, misfolded proteins also impact ERAD-UPR in ALS, suggesting that similar strategies based on the reinforcement of this pathway can contribute to restore protein homeostasis in affected cells.
Conclusions
In conclusion, data accumulated over the past 30 years have suggested that specific proteins cause MNDs by triggering aberrant responses in neurons and other cells involved in this group of diseases. The alteration of the PQC system is presently thought to be one of the major factors responsible for both the onset and progression rate of the disease. PQC systems failure could be directly associated with a mutant protein involved in one of the PQC pathways, or indirectly associated with effects caused by the overproduction of misfolded proteins that saturate or impair the PQC system activity. This leads to a reduced PQC potential to maintain the proper cellular homeostasis, especially during cell stresses. Notably, this system is presently considered a potential druggable target, since it provides huge numbers of players with activity that can be pharmacologically or genetically enhanced or modulated. Indeed, several of the cooperative factors playing a role in the PQC system can be specifically induced or downregulated, allowing the potentiation of a single arm of this defense mechanism. In many cases, the restoration of the proper function of one PQC arm has positive effects on the other arms of the system; they together provide a redundant mechanism capable of efficiently clearing most of the aberrant aggregating proteins, thus reducing cell death. Different approaches aimed to potentiate one or more arms of the PQC system have already been preclinically tested and are under investigation in clinical trials. Hopefully, these approaches will identify new treatments to counteract neurodegeneration in MNDs.
Author Contributions
RC, MG, VC, PR, and AP designed and wrote the manuscript and critically discussed all sections of the minireview. In addition, RC prepared the figures. MEC, VF, BT, EC, MC, EM, and MP critically revised the manuscript and the figures. All authors have provided final approval of the version to be published.
Funding
The following grants are gratefully acknowledged: Fondazione Telethon, Italy (n. GGP14039 to AP, GGP19128 to AP); Kennedy’s disease association (2018 grant to RC); Fondazione Cariplo, Italy (n. 2014-0686 to AP; n. 2017_0747 to VC); Fondazione AriSLA, Italy (n. ALS_HSPB8 to AP; ALS_Granulopathy to AP; MLOpathy to AP; Target-RAN to AP); Association Francaise contre les Myopathies, France (AFM Telethon n. 16406 to AP); Università degli Studi di Milano e piano di sviluppo UNIMI—linea B (to VC and PR) and Bando Straordinario per Progetti Interdipartimentali (Bando SEED 2019: #TDP-43-iPSC to VC); Italian Ministry of University and Research (MIUR), PRIN—Progetti di ricerca di interesse nazionale (n. 2015LFPNMN to AP; n. 2017F2A2C5 to AP); Fondo per il Finanziamento delle Attività Base di Ricerca (FFABR; MIUR, to MG, EM, and to PR); Agenzia Italiana del Farmaco (AIFA; Co_ALS to AP); Italian Ministry of Health (n. GR-2011-02347198 to VC); Fondazione Regionale per la Ricerca Biomedica (FRRB; Regione Lombardia, TRANS_ALS, project nr. 2015-0023, to AP); and EU Joint Programme—Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND (www.jpnd.eu). This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement N° 643417 (Grant ID: 01ED1601A, CureALS; to AP); Italian Ministry of University and Research (Progetto Dipartimenti di Eccellenza).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adriaenssens, E., Tedesco, B., Mediani, L., Asselbergh, B., Crippa, V., Antoniani, F., et al. (2020). BAG3 Pro209 mutants associated with myopathy and neuropathy relocate chaperones of the CASA-complex to aggresomes. Sci. Rep. 10:8755. doi: 10.1038/s41598-020-65664-z
Akers, J. C., Gonda, D., Kim, R., Carter, B. S., and Chen, C. C. (2013). Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 113, 1–11. doi: 10.1007/s11060-013-1084-8
Akutsu, M., Dikic, I., and Bremm, A. (2016). Ubiquitin chain diversity at a glance. J. Cell Sci. 129, 875–880. doi: 10.1242/jcs.183954
Alberti, S., Demand, J., Esser, C., Emmerich, N., Schild, H., and Hohfeld, J. (2002). Ubiquitylation of BAG-1 suggests a novel regulatory mechanism during the sorting of chaperone substrates to the proteasome. J. Biol. Chem. 277, 45920–45927. doi: 10.1074/jbc.m204196200
Alberti, S., Mateju, D., Mediani, L., and Carra, S. (2017). Granulostasis: protein quality control of RNP granules. Front. Mol. Neurosci. 10:84. doi: 10.3389/fnmol.2017.00084
Alexandrov, A. I., Grosfeld, E. V., Dergalev, A. A., Kushnirov, V. V., Chuprov-Netochin, R. N., Tyurin-Kuzmin, P. A., et al. (2019). Analysis of novel hyperosmotic shock response suggests ‘beads in liquid’ cytosol structure. Biol. Open 8:bio044529. doi: 10.1242/bio.044529
Alfaro, I. E., Albornoz, A., Molina, A., Moreno, J., Cordero, K., Criollo, A., et al. (2018). Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front. Endocrinol. 9:778. doi: 10.3389/fendo.2018.00778
Al-Tahan, S., Weiss, L., Yu, H., Tang, S., Saporta, M., Vihola, A., et al. (2019). New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 5:e349. doi: 10.1212/nxg.0000000000000349
Amerik, A. Y., and Hochstrasser, M. (2004). Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 1695, 189–207. doi: 10.1016/j.bbamcr.2004.10.003
Aparicio, R., Hansen, M., Walker, D. W., and Kumsta, C. (2020). The selective autophagy receptor SQSTM1/p62 improves lifespan and proteostasis in an evolutionarily conserved manner. Autophagy 16, 772–774. doi: 10.1080/15548627.2020.1725404
Arias, E., and Cuervo, A. M. (2011). Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 23, 184–189. doi: 10.1016/j.ceb.2010.10.009
Arimura, T., Ishikawa, T., Nunoda, S., Kawai, S., and Kimura, A. (2011). Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum. Mutat. 32, 1481–1491. doi: 10.1002/humu.21603
Arndt, V., Dick, N., Tawo, R., Dreiseidler, M., Wenzel, D., Hesse, M., et al. (2010). Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 20, 143–148. doi: 10.1016/j.cub.2009.11.022
Arosio, A., Cristofani, R., Pansarasa, O., Crippa, V., Riva, C., Sirtori, R., et al. (2020). HSC70 expression is reduced in lymphomonocytes of sporadic ALS patients and contributes to TDP-43 accumulation. Amyotroph. Lateral Scler. Frontotemporal. Degener. 21, 51–62. doi: 10.1080/21678421.2019.1672749
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atkin, J. D., Farg, M. A., Turner, B. J., Tomas, D., Lysaght, J. A., Nunan, J., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Basso, M., and Bonetto, V. (2016). Extracellular vesicles and a novel form of communication in the brain. Front. Neurosci. 10:127. doi: 10.3389/fnins.2016.00127
Behnke, J., Feige, M. J., and Hendershot, L. M. (2015). BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J. Mol. Biol. 427, 1589–1608. doi: 10.1016/j.jmb.2015.02.011
Belsham, D. D., Evangelou, A., Roy, D., Duc, V. L., and Brown, T. J. (1998). Regulation of gonadotropin-releasing hormone (GnRH) gene expression by 5α-dihydrotestosterone in GnRH-secreting GT1–7 hypothalamic neurons. Endocrinology 139, 1108–1114. doi: 10.1210/endo.139.3.5846
Bendotti, C., Marino, M., Cheroni, C., Fontana, E., Crippa, V., Poletti, A., et al. (2012). Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog. Neurobiol. 97, 101–126. doi: 10.1016/j.pneurobio.2011.10.001
Benussi, L., Ciani, M., Tonoli, E., Morbin, M., Palamara, L., Albani, D., et al. (2016). Loss of exosomes in progranulin-associated frontotemporal dementia. Neurobiol. Aging 40, 41–49. doi: 10.1016/j.neurobiolaging.2016.01.001
Blasco, H., Guennoc, A. M., Veyrat-Durebex, C., Gordon, P. H., Andres, C. R., Camu, W., et al. (2012). Amyotrophic lateral sclerosis: a hormonal condition? Amyotroph. Lateral Scler. 13, 585–588. doi: 10.3109/17482968.2012.706303
Boeynaems, S., Bogaert, E., Kovacs, D., Konijnenberg, A., Timmerman, E., Volkov, A., et al. (2017). Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 65, 1044.e5–1055.e5. doi: 10.1016/j.molcel.2017.02.013
Boillee, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Bosco, D. A., and Landers, J. E. (2010). Genetic determinants of amyotrophic lateral sclerosis as therapeutic targets. CNS Neurol. Disord. Drug Targets 9, 779–790. doi: 10.2174/187152710793237494
Brinkmann, A. O. (2001). Molecular basis of androgen insensitivity. Mol. Cell Endocrinol. 179, 105–109. doi: 10.1016/s0303-7207(01)00466-x
Brooks, B. P., and Fischbeck, K. H. (1995). Spinal and bulbar muscular atrophy: a trinucleotide-repeat expansion neurodegenerative disease. Trends Neurosci. 18, 459–461. doi: 10.1016/0166-2236(95)94497-s
Brooks, B. P., Paulson, H. L., Merry, D. E., Salazar-Grueso, E. F., Brinkmann, A. O., Wilson, E. M., et al. (1997). Characterization of an expanded glutamine repeat androgen receptor in a neuronal cell culture system. Neurobiol. Dis. 3, 313–323. doi: 10.1006/nbdi.1997.0126
Bruinsma, I. B., Bruggink, K. A., Kinast, K., Versleijen, A. A., Segers-Nolten, I. M., Subramaniam, V., et al. (2011). Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 79, 2956–2967. doi: 10.1002/prot.23152
Carra, S., Seguin, S. J., Lambert, H., and Landry, J. (2008a). HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 283, 1437–1444. doi: 10.1074/jbc.m706304200
Carra, S., Seguin, S. J., and Landry, J. (2008b). HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4, 237–239. doi: 10.4161/auto.5407
Castillo, K., Nassif, M., Valenzuela, V., Rojas, F., Matus, S., Mercado, G., et al. (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. doi: 10.4161/auto.25188
Chauhan, S., Kumar, S., Jain, A., Ponpuak, M., Mudd, M. H., Kimura, T., et al. (2016). TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev. Cell 39, 13–27. doi: 10.1016/j.devcel.2016.08.003
Chávez Zobel, A. T., Loranger, A., Marceau, N., Theriault, J. R., Lambert, H., and Landry, J. (2003). Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G αB-crystallin mutant. Hum. Mol. Genet. 12, 1609–1620. doi: 10.1093/hmg/ddg173
Chiappetta, G., Ammirante, M., Basile, A., Rosati, A., Festa, M., Monaco, M., et al. (2007). The antiapoptotic protein BAG3 is expressed in thyroid carcinomas and modulates apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J. Clin. Endocrinol. Metab. 92, 1159–1163. doi: 10.1210/jc.2006-1712
Chua, J. P., and Lieberman, A. P. (2013). Pathogenic mechanisms and therapeutic strategies in spinobulbar muscular atrophy. CNS Neurol. Disord. Drug Targets 12, 1146–1156. doi: 10.2174/187152731131200124
Chua, J. P., Reddy, S. L., Merry, D. E., Adachi, H., Katsuno, M., Sobue, G., et al. (2014). Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum. Mol. Genet. 23, 1376–1386. doi: 10.1093/hmg/ddt527
Cicardi, M. E., Cristofani, R., Crippa, V., Ferrari, V., Tedesco, B., Casarotto, E., et al. (2019). Autophagic and proteasomal mediated removal of mutant androgen receptor in muscle models of spinal and bulbar muscular atrophy. Front. Endocrinol. 10:569. doi: 10.3389/fendo.2019.00569
Cicardi, M. E., Cristofani, R., Rusmini, P., Meroni, M., Ferrari, V., Vezzoli, G., et al. (2018). Tdp-25 routing to autophagy and proteasome ameliorates its aggregation in amyotrophic lateral sclerosis target cells. Sci. Rep. 8:12390. doi: 10.1038/s41598-018-29658-2
Ciechanover, A. (1994). The ubiquitin-proteasome proteolytic pathway. Cell 79, 13–21. doi: 10.1016/0092-8674(94)90396-4
Ciechanover, A., and Brundin, P. (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. doi: 10.1016/s0896-6273(03)00606-8
Ciechanover, A., and Kwon, Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47:e147. doi: 10.1038/emm.2014.117
Colombo, M., Raposo, G., and Thery, C. (2014). Biogenesis, secretion and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289. doi: 10.1146/annurev-cellbio-101512-122326
Cook, C., and Petrucelli, L. (2019). Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101, 1057–1069. doi: 10.1016/j.neuron.2019.02.032
Cortes, C. J., Ling, S. C., Guo, L. T., Hung, G., Tsunemi, T., Ly, L., et al. (2014a). Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 82, 295–307. doi: 10.1016/j.neuron.2014.03.001
Cortes, C. J., Miranda, H. C., Frankowski, H., Batlevi, Y., Young, J. E., Le, A., et al. (2014b). Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 17, 1180–1189. doi: 10.1038/nn.3787
Coyne, A. N., Lorenzini, I., Chou, C. C., Torvund, M., Rogers, R. S., Starr, A., et al. (2017). Post-transcriptional inhibition of Hsc70–4/HSPA8 expression leads to synaptic vesicle cycling defects in multiple models of ALS. Cell Rep. 21, 110–125. doi: 10.1016/j.celrep.2017.09.028
Crippa, V., Boncoraglio, A., Galbiati, M., Aggarwal, T., Rusmini, P., Giorgetti, E., et al. (2013). Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front. Cell. Neurosci. 7:234. doi: 10.3389/fncel.2013.00234
Crippa, V., Carra, S., Rusmini, P., Sau, D., Bolzoni, E., Bendotti, C., et al. (2010a). A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy 6, 958–960. doi: 10.4161/auto.6.7.13042
Crippa, V., Sau, D., Rusmini, P., Boncoraglio, A., Onesto, E., Bolzoni, E., et al. (2010b). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. doi: 10.1093/hmg/ddq257
Crippa, V., Cicardi, M. E., Ramesh, N., Seguin, S. J., Ganassi, M., Bigi, I., et al. (2016a). The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 25, 3908–3924. doi: 10.1093/hmg/ddw232
Crippa, V., D’Agostino, V. G., Cristofani, R., Rusmini, P., Cicardi, M. E., Messi, E., et al. (2016b). Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 6:22827. doi: 10.1038/srep22827
Cristofani, R., Crippa, V., Rusmini, P., Cicardi, M. E., Meroni, M., Licata, N. V., et al. (2017). Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 13, 1280–1303. doi: 10.1080/15548627.2017.1308985
Cristofani, R., Crippa, V., Vezzoli, G., Rusmini, P., Galbiati, M., Cicardi, M. E., et al. (2018). The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 23, 1–12. doi: 10.1007/s12192-017-0806-9
Cristofani, R., Rusmini, P., Galbiati, M., Cicardi, M. E., Ferrari, V., Tedesco, B., et al. (2019). The regulation of the small heat shock protein b8 in misfolding protein diseases causing motoneuronal and muscle cell death. Front. Neurosci. 13:796. doi: 10.3389/fnins.2019.00796
Cuervo, A. M. (2011). Chaperone-mediated autophagy: dice’s ‘wild’ idea about lysosomal selectivity. Nat. Rev. Mol. Cell Biol. 12, 535–541. doi: 10.1038/nrm3150
Dal Canto, M. C. (1995). Comparison of pathological alterations in ALS and a murine transgenic model: pathogenetic implications. Clin. Neurosci. 3, 332–337.
Dal Canto, M. C., and Gurney, M. E. (1995). Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 676, 25–40. doi: 10.1016/0006-8993(95)00063-v
Damiano, M., Starkov, A. A., Petri, S., Kipiani, K., Kiaei, M., Mattiazzi, M., et al. (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 96, 1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x
Daoud, H., Valdmanis, P. N., Kabashi, E., Dion, P., Dupre, N., Camu, W., et al. (2009). Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 46, 112–114. doi: 10.1136/jmg.2008.062463
Davies, J. E., Sarkar, S., and Rubinsztein, D. C. (2006). Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 15, 23–31. doi: 10.1093/hmg/ddi422
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/s0092-8674(00)80513-9
Demand, J., Alberti, S., Patterson, C., and Höhfeld, J. (2001). Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 11, 1569–1577. doi: 10.1016/s0960-9822(01)00487-0
Desdín-Micó, G., and Mittelbrunn, M. (2017). Role of exosomes in the protection of cellular homeostasis. Cell Adh. Migr. 11, 127–134. doi: 10.1080/19336918.2016.1251000
Diaz-Hidalgo, L., Altuntas, S., Rossin, F., D’Eletto, M., Marsella, C., Farrace, M. G., et al. (2016). Transglutaminase type 2-dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim. Biophys. Acta 1863, 2084–2092. doi: 10.1016/j.bbamcr.2016.05.005
DiDomenico, B. J., Bugaisky, G. E., and Lindquist, S. (1982). Heat shock and recovery are mediated by different translational mechanisms. Proc. Natl. Acad. Sci. U S A 79, 6181–6185. doi: 10.1073/pnas.79.20.6181
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dobrowolny, G., Aucello, M., Molinaro, M., and Musaro, A. (2008). Local expression of mIgf-1 modulates ubiquitin, caspase and CDK5 expression in skeletal muscle of an ALS mouse model. Neurol. Res. 30, 131–136. doi: 10.1179/174313208x281235
Doi, H., Adachi, H., Katsuno, M., Minamiyama, M., Matsumoto, S., Kondo, N., et al. (2013). p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J. Neurosci. 33, 7710–7727. doi: 10.1523/jneurosci.3021-12.2013
Du, J., Liang, Y., Xu, F., Sun, B., and Wang, Z. (2013). Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J. Pharm. Pharmacol. 65, 1753–1756. doi: 10.1111/jphp.12108
Edwards, A., Hammond, H. A., Jin, L., Caskey, C. T., and Chakraborty, R. (1992). Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics 12, 241–253. doi: 10.1016/0888-7543(92)90371-x
Eftekharzadeh, B., Banduseela, V. C., Chiesa, G., Martinez-Cristobal, P., Rauch, J. N., Nath, S. R., et al. (2019). Hsp70 and Hsp40 inhibit an inter-domain interaction necessary for transcriptional activity in the androgen receptor. Nat. Commun. 10:3562. doi: 10.1038/s41467-019-11594-y
Escobedo, A., Topal, B., Kunze, M. B. A., Aranda, J., Chiesa, G., Mungianu, D., et al. (2019). Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 10:2034. doi: 10.1038/s41467-019-09923-2
Evans, C. S., and Holzbaur, E. L. F. (2019). Autophagy and mitophagy in ALS. Neurobiol. Dis. 122, 35–40. doi: 10.1016/j.nbd.2018.07.005
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Ferreira, J. V., Rosa Soares, A., Ramalho, J. S., Ribeiro-Rodrigues, T., Maximo, C., Zuzarte, M., et al. (2019). Exosomes and STUB1/CHIP cooperate to maintain intracellular proteostasis. PLoS One 14:e0223790. doi: 10.1371/journal.pone.0223790
Finkel, R. S., Mercuri, E., Darras, B. T., Connolly, A. M., Kuntz, N. L., Kirschner, J., et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732. doi: 10.1056/NEJMoa1702752
Fischbeck, K. H. (1997). Kennedy disease. J. Inherit. Metab. Dis. 20, 152–158. doi: 10.1023/a:1005344403603
Fontaine, J. M., Sun, X., Hoppe, A. D., Simon, S., Vicart, P., Welsh, M. J., et al. (2006). Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 20, 2168–2170. doi: 10.1096/fj.06-5911fje
Foradori, C. D., and Handa, R. J. (2008). Living or dying in three quarter time: neonatal orchestration of hippocampal cell death pathways by androgens and excitatory GABA. Exp. Neurol. 213, 1–6. doi: 10.1016/j.expneurol.2008.04.035
Freibaum, B. D., and Taylor, J. P. (2017). The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 10:35. doi: 10.3389/fnmol.2017.00035
Fujita, N., Morita, E., Itoh, T., Tanaka, A., Nakaoka, M., Osada, Y., et al. (2013). Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J. Cell Biol. 203, 115–128. doi: 10.1083/jcb.201304188
Fujita, Y., Okamoto, K., Sakurai, A., Gonatas, N. K., and Hirano, A. (2000). Fragmentation of the Golgi apparatus of the anterior horn cells in patients with familial amyotrophic lateral sclerosis with SOD1 mutations and posterior column involvement. J. Neurol. Sci. 174, 137–140. doi: 10.1016/s0022-510x(00)00265-3
Galbiati, M., Crippa, V., Rusmini, P., Cristofani, R., Cicardi, M. E., Giorgetti, E., et al. (2014). ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 79, 70–78. doi: 10.1016/j.neuint.2014.10.007
Galbiati, M., Onesto, E., Zito, A., Crippa, V., Rusmini, P., Mariotti, R., et al. (2012). The anabolic/androgenic steroid nandrolone exacerbates gene expression modifications induced by mutant SOD1 in muscles of mice models of amyotrophic lateral sclerosis. Pharmacol. Res. 65, 221–230. doi: 10.1016/j.phrs.2011.12.001
Ganassi, M., Mateju, D., Bigi, I., Mediani, L., Poser, I., Lee, H. O., et al. (2016). A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 63, 796–810. doi: 10.1016/j.molcel.2016.07.021
Gendron, T. F., Bieniek, K. F., Zhang, Y. J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
Genis, D., Ortega-Cubero, S., San Nicolas, H., Corral, J., Gardenyes, J., de Jorge, L., et al. (2018). Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91, e1988–e1998. doi: 10.1212/wnl.0000000000006550
Ghaoui, R., Palmio, J., Brewer, J., Lek, M., Needham, M., Evila, A., et al. (2016). Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 86, 391–398. doi: 10.1212/WNL.0000000000002324
Giorgetti, E., Rusmini, P., Crippa, V., Cristofani, R., Boncoraglio, A., Cicardi, M. E., et al. (2015). Synergic prodegradative activity of Bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum. Mol. Genet. 24, 64–75. doi: 10.1093/hmg/ddu419
Grosskreutz, J., Van Den Bosch, L., and Keller, B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. doi: 10.1016/j.ceca.2009.12.002
Grunseich, C., Kats, I. R., Bott, L. C., Rinaldi, C., Kokkinis, A., Fox, D., et al. (2014). Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscul. Disord. 24, 978–981. doi: 10.1016/j.nmd.2014.06.441
Guix, F. X. (2020). The interplay between aging-associated loss of protein homeostasis and extracellular vesicles in neurodegeneration. J. Neurosci. Res. 98, 262–283. doi: 10.1002/jnr.24526
Hageman, J., Rujano, M. A., van Waarde, M. A., Kakkar, V., Dirks, R. P., Govorukhina, N., et al. (2010). A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369. doi: 10.1016/j.molcel.2010.01.001
Hanspal, M. A., Dobson, C. M., Yerbury, J. J., and Kumita, J. R. (2017). The relevance of contact-independent cell-to-cell transfer of TDP-43 and SOD1 in amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2762–2771. doi: 10.1016/j.bbadis.2017.07.007
Haverkamp, L. J., Appel, V., and Appel, S. H. (1995). Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 118, 707–719. doi: 10.1093/brain/118.3.707
Hayer, S. N., Deconinck, T., Bender, B., Smets, K., Zuchner, S., Reich, S., et al. (2017). STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations. Orphanet J. Rare Dis. 12:31. doi: 10.1186/s13023-017-0580-x
He, Q., Koprich, J. B., Wang, Y., Yu, W. B., Xiao, B. G., Brotchie, J. M., et al. (2016). Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV α-synuclein rat model of Parkinson’s disease. Mol. Neurobiol. 53, 2258–2268. doi: 10.1007/s12035-015-9173-7
Hegde, A. N., Smith, S. G., Duke, L. M., Pourquoi, A., and Vaz, S. (2019). Perturbations of ubiquitin-proteasome-mediated proteolysis in aging and Alzheimer’s disease. Front. Aging Neurosci. 11:324. doi: 10.3389/fnagi.2019.00324
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Lee, A. H., Gonzalez-Romero, D., Thielen, P., Castilla, J., Soto, C., et al. (2008). Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. U S A 105, 757–762. doi: 10.1073/pnas.0711094105
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Huang, C. C., Bose, J. K., Majumder, P., Lee, K. H., Huang, J. T., Huang, J. K., et al. (2014). Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 127, 3024–3038. doi: 10.1242/jcs.136150
Hwang, J., and Qi, L. (2018). Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605. doi: 10.1016/j.tibs.2018.06.005
Iguchi, Y., Eid, L., Parent, M., Soucy, G., Bareil, C., Riku, Y., et al. (2016). Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139, 3187–3201. doi: 10.1093/brain/aww237
Irobi, J., Almeida-Souza, L., Asselbergh, B., De Winter, V., Goethals, S., Dierick, I., et al. (2010). Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 19, 3254–3265. doi: 10.1093/hmg/ddq234
Jackson, P. K., Eldridge, A. G., Freed, E., Furstenthal, L., Hsu, J. Y., Kaiser, B. K., et al. (2000). The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 10, 429–439. doi: 10.1016/s0962-8924(00)01834-1
Jaffer, F., Murphy, S. M., Scoto, M., Healy, E., Rossor, A. M., Brandner, S., et al. (2012). BAG3 mutations: another cause of giant axonal neuropathy. J. Peripher. Nerv. Syst. 17, 210–216. doi: 10.1111/j.1529-8027.2012.00409.x
Jaronen, M., Goldsteins, G., and Koistinaho, J. (2014). ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Front. Cell. Neurosci. 8:402. doi: 10.3389/fncel.2014.00402
Jaru-Ampornpan, P., Liang, F. C., Nisthal, A., Nguyen, T. X., Wang, P., Shen, K., et al. (2013). Mechanism of an ATP-independent protein disaggregase: II. Distinct molecular interactions drive multiple steps during aggregate disassembly. J. Biol. Chem. 288, 13431–13445. doi: 10.1074/jbc.m113.462861
Joazeiro, C. A., and Weissman, A. M. (2000). RING finger proteins: mediators of ubiquitin ligase activity. Cell 102, 549–552. doi: 10.1016/s0092-8674(00)00077-5
Kabashi, E., and Durham, H. D. (2006). Failure of protein quality control in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1038–1050. doi: 10.1016/j.bbadis.2006.06.006
Kabuta, T., Suzuki, Y., and Wada, K. (2006). Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J. Biol. Chem. 281, 30524–30533. doi: 10.1074/jbc.m603337200
Kampinga, H. H., and Bergink, S. (2016). Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 15, 748–759. doi: 10.1016/s1474-4422(16)00099-5
Kampinga, H. H., and Craig, E. A. (2010). The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592. doi: 10.1038/nrm2941
Kampinga, H. H., Hageman, J., Vos, M. J., Kubota, H., Tanguay, R. M., Bruford, E. A., et al. (2009). Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111. doi: 10.1007/s12192-008-0068-7
Kanack, A. J., Newsom, O. J., and Scaglione, K. M. (2018). Most mutations that cause spinocerebellar ataxia autosomal recessive type 16 (SCAR16) destabilize the protein quality-control E3 ligase CHIP. J. Biol. Chem. 293, 2735–2743. doi: 10.1074/jbc.ra117.000477
Katsuno, M., Adachi, H., Doyu, M., Minamiyama, M., Sang, C., Kobayashi, Y., et al. (2003). Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med. 9, 768–773. doi: 10.1038/nm878
Katsuno, M., Adachi, H., Kume, A., Li, M., Nakagomi, Y., Niwa, H., et al. (2002). Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–854. doi: 10.1016/s0896-6273(02)00834-6
Kaushik, S., and Cuervo, A. M. (2018). The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381. doi: 10.1038/s41580-018-0001-6
Kazemi-Esfarjani, P., Trifiro, M. A., and Pinsky, L. (1995). Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum. Mol. Genet. 4, 523–527. doi: 10.1093/hmg/4.4.523
Kettern, N., Rogon, C., Limmer, A., Schild, H., and Hohfeld, J. (2011). The Hsc/Hsp70 co-chaperone network controls antigen aggregation and presentation during maturation of professional antigen presenting cells. PLoS One 6:e16398. doi: 10.1371/journal.pone.0016398
Kikuchi, H., Almer, G., Yamashita, S., Guegan, C., Nagai, M., Xu, Z., et al. (2006). Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. U S A 103, 6025–6030. doi: 10.1073/pnas.0509227103
Kim, J., Kim, T. Y., Cho, K. S., Kim, H. N., and Koh, J. Y. (2013). Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 59, 80–85. doi: 10.1016/j.nbd.2013.07.011
Kimura, Y., Fukushi, J., Hori, S., Matsuda, N., Okatsu, K., Kakiyama, Y., et al. (2013). Different dynamic movements of wild-type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin-mediated mitochondrial quality control system. Genes Cells 18, 1131–1143. doi: 10.1111/gtc.12103
Kirchner, P., Bourdenx, M., Madrigal-Matute, J., Tiano, S., Diaz, A., Bartholdy, B. A., et al. (2019). Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 17:e3000301. doi: 10.1371/journal.pbio.3000301
Kitamura, A., Iwasaki, N., and Kinjo, M. (2018). Molecular chaperone HSP70 prevents formation of inclusion bodies of the 25-kDa C-terminal fragment of TDP-43 by preventing aggregate accumulation. Cell Stress Chaperones 23, 1177–1183. doi: 10.1007/s12192-018-0930-1
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Koga, H., Martinez-Vicente, M., Arias, E., Kaushik, S., Sulzer, D., and Cuervo, A. M. (2011). Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 31, 18492–18505. doi: 10.1523/JNEUROSCI.3219-11.2011
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. doi: 10.1016/s0962-8924(00)01852-3
Kuhlenbäumer, G., Kress, W., Ringelstein, E. B., and Stögbauer, F. (2001). Thirty-seven CAG repeats in the androgen receptor gene in two healthy individuals. J. Neurol. 248, 23–26. doi: 10.1007/s004150170265
Kurisu, J., Honma, A., Miyajima, H., Kondo, S., Okumura, M., and Imaizumi, K. (2003). MDG1/ERdj4, an ER-resident DnaJ family member, suppresses cell death induced by ER stress. Genes Cells 8, 189–202. doi: 10.1046/j.1365-2443.2003.00625.x
Lashley, T., Hardy, J., and Isaacs, A. M. (2013). RANTing about C9orf72. Neuron 77, 597–598. doi: 10.1016/j.neuron.2013.02.009
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E., and Fischbeck, K. H. (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. doi: 10.1038/352077a0
Lautenschlaeger, J., Prell, T., and Grosskreutz, J. (2012). Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 166–177. doi: 10.3109/17482968.2011.641569
Lee, K. H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics and function of membrane-less organelles. Cell 167, 774.e17–788.e17. doi: 10.1016/j.cell.2016.10.002
Lefebvre, S., Burglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lei, Z., Brizzee, C., and Johnson, G. V. (2015). BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol. Aging 36, 241–248. doi: 10.1016/j.neurobiolaging.2014.08.012
Li, Y., Guo, Y., Wang, X., Yu, X., Duan, W., Hong, K., et al. (2015). Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1–G93A mouse model. Neuroscience 298, 12–25. doi: 10.1016/j.neuroscience.2015.03.061
Li, M., Miwa, S., Kobayashi, Y., Merry, D., Yamamoto, M., Tanaka, F., et al. (1998). Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann. Neurol. 44, 249–254. doi: 10.1002/ana.410440216
Li, M., Sobue, G., Doyu, M., Mukai, E., Hashizume, Y., and Mitsuma, T. (1995). Primary sensory neurons in X-linked recessive bulbospinal neuropathy: histopathology and androgen receptor gene expression. Muscle Nerve 18, 301–308. doi: 10.1002/mus.880180306
Li, L., Zhang, X., and Le, W. (2008). Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4, 290–293. doi: 10.4161/auto.5524
Lieberman, A. P., Robitaille, Y., Trojanowski, J. Q., Dickson, D. W., and Fischbeck, K. H. (1998). Polyglutamine-containing aggregates in neuronal intranuclear inclusion disease. Lancet 351:884. doi: 10.1016/s0140-6736(05)70296-8
Lieberman, A. P., Yu, Z., Murray, S., Peralta, R., Low, A., Guo, S., et al. (2014). Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 7, 774–784. doi: 10.1016/j.celrep.2014.02.008
Liu, H., Wang, P., Song, W., and Sun, X. (2009). Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 23, 3383–3392. doi: 10.1096/fj.09-134296
Lobsiger, C. S., Boillee, S., McAlonis-Downes, M., Khan, A. M., Feltri, M. L., Yamanaka, K., et al. (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U S A 106, 4465–4470. doi: 10.1073/pnas.0813339106
Longoni, B., Fasciani, I., Kolachalam, S., Pietrantoni, I., Marampon, F., Petragnano, F., et al. (2019). Neurotoxic and neuroprotective role of exosomes in Parkinson’s disease. Curr. Pharm. Des. 25, 4510–4522. doi: 10.2174/1381612825666191113103537
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808.e9–816.e9. doi: 10.1016/j.neuron.2017.07.025
Madeira, J. L. O., Souza, A. B. C., Cunha, F. S., Batista, R. L., Gomes, N. L., Rodrigues, A. S., et al. (2018). A severe phenotype of Kennedy disease associated with a very large CAG repeat expansion. Muscle Nerve 57, E95–E97. doi: 10.1002/mus.25952
Malik, B., Nirmalananthan, N., Bilsland, L. G., La Spada, A. R., Hanna, M. G., Schiavo, G., et al. (2011). Absence of disturbed axonal transport in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 20, 1776–1786. doi: 10.1093/hmg/ddr061
Mandrioli, J., Crippa, V., Cereda, C., Bonetto, V., Zucchi, E., Gessani, A., et al. (2019). Proteostasis and ALS: protocol for a phase II, randomized, double blind, placebo controlled, multicenter clinical trial for Colchicine in ALS (Co-ALS). BMJ Open 9:e028486. doi: 10.1136/bmjopen-2018-028486
Manjaly, Z. R., Scott, K. M., Abhinav, K., Wijesekera, L., Ganesalingam, J., Goldstein, L. H., et al. (2010). The sex ratio in amyotrophic lateral sclerosis: a population based study. Amyotroph. Lateral Scler. 11, 439–442. doi: 10.3109/17482961003610853
Marron, T. U., Guerini, V., Rusmini, P., Sau, D., Brevini, T. A., Martini, L., et al. (2005). Androgen-induced neurite outgrowth is mediated by neuritin in motor neurones. J. Neurochem. 92, 10–20. doi: 10.1111/j.1471-4159.2004.02836.x
Marrone, L., Poser, I., Casci, I., Japtok, J., Reinhardt, P., Janosch, A., et al. (2018). Isogenic FUS-eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy. Stem Cell Reports 10, 375–389. doi: 10.1016/j.stemcr.2017.12.018
Mateju, D., Franzmann, T. M., Patel, A., Kopach, A., Boczek, E. E., Maharana, S., et al. (2017). An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 36, 1669–1687. doi: 10.15252/embj.201695957
Mathew, V., and Stirling, P. C. (2017). Protein quality control meets transcriptome remodeling under stress. Cell Stress 1, 134–135. doi: 10.15698/cst2017.12.115
Mathis, S., Goizet, C., Soulages, A., Vallat, J. M., and Masson, G. L. (2019). Genetics of amyotrophic lateral sclerosis: a review. J. Neurol. Sci. 399, 217–226. doi: 10.1016/j.jns.2019.02.030
Mattoo, R. U., and Goloubinoff, P. (2014). Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell. Mol. Life Sci. 71, 3311–3325. doi: 10.1007/s00018-014-1627-y
Mediani, L., Guillen-Boixet, J., Vinet, J., Franzmann, T. M., Bigi, I., Mateju, D., et al. (2019). Defective ribosomal products challenge nuclear function by impairing nuclear condensate dynamics and immobilizing ubiquitin. EMBO J. 38:e101341. doi: 10.15252/embj.2018101341
Meister-Broekema, M., Freilich, R., Jagadeesan, C., Rauch, J. N., Bengoechea, R., Motley, W. W., et al. (2018). Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat. Commun. 9:5342. doi: 10.1038/s41467-018-07718-5
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms and therapeutics: where are we now? Front. Neurosci. 13:1310. doi: 10.3389/fnins.2019.01310
Mendell, J. R., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., et al. (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722. doi: 10.1056/NEJMoa1706198
Mercuri, E., Darras, B. T., Chiriboga, C. A., Day, J. W., Campbell, C., Connolly, A. M., et al. (2018). Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635. doi: 10.1056/NEJMoa1710504
Meroni, M., Crippa, V., Cristofani, R., Rusmini, P., Cicardi, M. E., Messi, E., et al. (2019). Transforming growth factor β 1 signaling is altered in the spinal cord and muscle of amyotrophic lateral sclerosis mice and patients. Neurobiol. Aging 82, 48–59. doi: 10.1016/j.neurobiolaging.2019.07.001
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. doi: 10.1016/j.cell.2015.09.015
Montague, K., Malik, B., Gray, A. L., La Spada, A. R., Hanna, M. G., Szabadkai, G., et al. (2014). Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy. Brain 137, 1894–1906. doi: 10.1093/brain/awu114
Montibeller, L., and de Belleroche, J. (2018). Amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD) are characterised by differential activation of ER stress pathways: focus on UPR target genes. Cell Stress Chaperones 23, 897–912. doi: 10.1007/s12192-018-0897-y
Montie, H. L., Cho, M. S., Holder, L., Liu, Y., Tsvetkov, A. S., Finkbeiner, S., et al. (2009). Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 18, 1937–1950. doi: 10.1093/hmg/ddp115
Montie, H. L., and Merry, D. E. (2009). Autophagy and access: understanding the role of androgen receptor subcellular localization in SBMA. Autophagy 5, 1194–1197. doi: 10.4161/auto.5.8.9726
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morimoto, N., Nagai, M., Ohta, Y., Miyazaki, K., Kurata, T., Morimoto, M., et al. (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1167, 112–117. doi: 10.1016/j.brainres.2007.06.045
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Spiro, A. S., Furuta, A., Cooper, A., et al. (2015). Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 30, 1639–1647. doi: 10.1002/mds.26141
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, D. K. H., Thombre, R., and Wang, J. (2019). Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 697, 34–48. doi: 10.1016/j.neulet.2018.04.006
Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., et al. (2008). ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464. doi: 10.1101/gad.1640108
Nivon, M., Abou-Samra, M., Richet, E., Guyot, B., Arrigo, A. P., and Kretz-Remy, C. (2012). NF-kappaB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex. J. Cell Sci. 125, 1141–1151. doi: 10.1242/jcs.091041
O’Driscoll, J., Clare, D., and Saibil, H. (2015). Prion aggregate structure in yeast cells is determined by the Hsp104-Hsp110 disaggregase machinery. J. Cell Biol. 211, 145–158. doi: 10.1083/jcb.201505104
Onesto, E., Rusmini, P., Crippa, V., Ferri, N., Zito, A., Galbiati, M., et al. (2011). Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 118, 266–280. doi: 10.1111/j.1471-4159.2011.07298.x
Orenstein, S. J., Kuo, S. H., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi: 10.1038/nn.3350
Ormeño, F., Hormazabal, J., Moreno, J., Riquelme, F., Rios, J., Criollo, A., et al. (2020). Chaperone mediated autophagy degrades TDP-43 protein and is affected by TDP-43 aggregation. Front. Mol. Neurosci. 13:19. doi: 10.3389/fnmol.2020.00019
Oyanagi, K., Yamazaki, M., Takahashi, H., Watabe, K., Wada, M., Komori, T., et al. (2008). Spinal anterior horn cells in sporadic amyotrophic lateral sclerosis show ribosomal detachment from and cisternal distention of the rough endoplasmic reticulum. Neuropathol. Appl. Neurobiol. 34, 650–658. doi: 10.1111/j.1365-2990.2008.00941.x
Pakdaman, Y., Sanchez-Guixe, M., Kleppe, R., Erdal, S., Bustad, H. J., Bjorkhaug, L., et al. (2017). in vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins. Biosci. Rep. 37:BSR20170251. doi: 10.1042/bsr20170251
Papadopoulos, C., Kirchner, P., Bug, M., Grum, D., Koerver, L., Schulze, N., et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150. doi: 10.15252/embj.201695148
Park, J. S., Kim, D. H., and Yoon, S. Y. (2016). Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 49, 337–342. doi: 10.5483/bmbrep.2016.49.6.212
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Perera, N. D., Sheean, R. K., Lau, C. L., Shin, Y. S., Beart, P. M., Horne, M. K., et al. (2018). Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 14, 534–551. doi: 10.1080/15548627.2017.1385674
Pérez, M., Avila, J., and Hernández, F. (2019). Propagation of tau via extracellular vesicles. Front. Neurosci. 13:698. doi: 10.3389/fnins.2019.00698
Perucho, J., Casarejos, M. J., Gomez, A., Solano, R. M., de Yebenes, J. G., and Mena, M. A. (2012). Trehalose protects from aggravation of amyloid pathology induced by isoflurane anesthesia in APP(swe) mutant mice. Curr. Alzheimer Res. 9, 334–343. doi: 10.2174/156720512800107573
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482. doi: 10.1093/brain/aws339
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/s1474-4422(11)70015-1
Piccioni, F., Pinton, P., Simeoni, S., Pozzi, P., Fascio, U., Vismara, G., et al. (2002). Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J. 16, 1418–1420. doi: 10.1096/fj.01-1035fje
Piccioni, F., Simeoni, S., Andriola, I., Armatura, E., Bassanini, S., Pozzi, P., et al. (2001). Polyglutamine tract expansion of the androgen receptor in a motoneuronal model of spinal and bulbar muscular atrophy. Brain Res. Bull. 56, 215–220. doi: 10.1016/s0361-9230(01)00652-9
Piccolella, M., Crippa, V., Cristofani, R., Rusmini, P., Galbiati, M., Cicardi, M. E., et al. (2017). The small heat shock protein B8 (HSPB8) modulates proliferation and migration of breast cancer cells. Oncotarget 8, 10400–10415. doi: 10.18632/oncotarget.14422
Piccolella, M., Crippa, V., Messi, E., Tetel, M. J., and Poletti, A. (2014). Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells. Pharmacol. Res. 79, 13–20. doi: 10.1016/j.phrs.2013.10.002
Pickart, C. M. (2001a). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Pickart, C. M. (2001b). Ubiquitin enters the new millennium. Mol. Cell 8, 499–504. doi: 10.1016/s1097-2765(01)00347-1
Poletti, A. (2004). The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front. Neuroendocrinol. 25, 1–26. doi: 10.1016/j.yfrne.2004.03.001
Poletti, A., and Fischbeck, K. H. (2020). Combinatorial treatment for spinal muscular atrophy: an editorial for ‘combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in spinal muscular atrophy cells’ on. J. Neurochem. 153, 146–149. doi: 10.1111/jnc.14974
Poletti, A., Melcangi, R. C., Negri-Cesi, P., Maggi, R., and Martini, L. (1994). Steroid binding and metabolism in the luteinizing hormone-releasing hormone-producing neuronal cell line GT1–1. Endocrinology 135, 2623–2628. doi: 10.1210/endo.135.6.7988451
Poletti, A., Negri-Cesi, P., Melcangi, R. C., Colciago, A., Martini, L., and Celotti, F. (1997). Expression of androgen-activating enzymes in cultured cells of developing rat brain. J. Neurochem. 68, 1298–1303. doi: 10.1046/j.1471-4159.1997.68031298.x
Poletti, A., Rampoldi, A., Piccioni, F., Volpi, S., Simeoni, S., Zanisi, M., et al. (2001). 5α-reductase type 2 and androgen receptor expression in gonadotropin releasing hormone GT1–1 cells. J. Neuroendocrinol. 13, 353–357. doi: 10.1046/j.1365-2826.2001.00635.x
Polo, A., Teatini, F., D’Anna, S., Manganotti, P., Salviati, A., Dallapiccola, B., et al. (1996). Sensory involvement in X-linked spino-bulbar muscular atrophy (Kennedy’s syndrome): an electrophysiological study. J. Neurol. 243, 388–392. doi: 10.1007/bf00868997
Pozzi, P., Bendotti, C., Simeoni, S., Piccioni, F., Guerini, V., Marron, T. U., et al. (2003). Androgen 5-α-reductase type 2 is highly expressed and active in rat spinal cord motor neurones. J. Neuroendocrinol. 15, 882–887. doi: 10.1046/j.1365-2826.2003.01074.x
Qi, L., Zhang, X. D., Wu, J. C., Lin, F., Wang, J., DiFiglia, M., et al. (2012). The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One 7:e46834. doi: 10.1371/journal.pone.0046834
Rapino, F., Jung, M., and Fulda, S. (2014). BAG3 induction is required to mitigate proteotoxicity via selective autophagy following inhibition of constitutive protein degradation pathways. Oncogene 33, 1713–1724. doi: 10.1038/onc.2013.110
Ratti, A., and Buratti, E. (2016). Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 138, 95–111. doi: 10.1111/jnc.13625
Rauch, J. N., and Gestwicki, J. E. (2014). Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J. Biol. Chem. 289, 1402–1414. doi: 10.1074/jbc.m113.521997
Rinaldi, C., Bott, L. C., and Fischbeck, K. H. (2014). Muscle matters in Kennedy’s disease. Neuron 82, 251–253. doi: 10.1016/j.neuron.2014.04.005
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14, 248–264. doi: 10.1038/nrn3430
Rodriguez-Navarro, J. A., Rodriguez, L., Casarejos, M. J., Solano, R. M., Gomez, A., Perucho, J., et al. (2010). Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 39, 423–438. doi: 10.1016/j.nbd.2010.05.014
Romano, M. F., Festa, M., Petrella, A., Rosati, A., Pascale, M., Bisogni, R., et al. (2003). BAG3 protein regulates cell survival in childhood acute lymphoblastic leukemia cells. Cancer Biol. Ther. 2, 508–510. doi: 10.4161/cbt.2.5.524
Ross, C. A. (2002). Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 35, 819–822. doi: 10.1016/s0896-6273(02)00872-3
Rusmini, P., Bolzoni, E., Crippa, V., Onesto, E., Sau, D., Galbiati, M., et al. (2010). Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol. Dis. 40, 361–369. doi: 10.1016/j.nbd.2010.06.016
Rusmini, P., Cortese, K., Crippa, V., Cristofani, R., Cicardi, M. E., Ferrari, V., et al. (2019). Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651. doi: 10.1080/15548627.2018.1535292
Rusmini, P., Crippa, V., Cristofani, R., Rinaldi, C., Cicardi, M. E., Galbiati, M., et al. (2016). The role of the protein quality control system in SBMA. J. Mol. Neurosci. 58, 348–364. doi: 10.1007/s12031-015-0675-6
Rusmini, P., Crippa, V., Giorgetti, E., Boncoraglio, A., Cristofani, R., Carra, S., et al. (2013). Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol. Aging 34, 2585–2603. doi: 10.1016/j.neurobiolaging.2013.05.026
Rusmini, P., Cristofani, R., Galbiati, M., Cicardi, M. E., Meroni, M., Ferrari, V., et al. (2017). The role of the heat shock protein B8 (HSPB8) in motoneuron diseases. Front. Mol. Neurosci. 10:176. doi: 10.3389/fnmol.2017.00176
Rusmini, P., Cristofani, R., Tedesco, B., Ferrari, V., Messi, E., Piccolella, M., et al. (2020). Enhanced clearance of neurotoxic misfolded proteins by the natural compound berberine and its derivatives. Int. J. Mol. Sci. 21:3443. doi: 10.3390/ijms21103443
Rusmini, P., Polanco, M. J., Cristofani, R., Cicardi, M. E., Meroni, M., Galbiati, M., et al. (2015). Aberrant autophagic response in the muscle of a knock-in mouse model of spinal and bulbar muscular atrophy. Sci. Rep. 5:15174. doi: 10.1038/srep15174
Rusmini, P., Sau, D., Crippa, V., Palazzolo, I., Simonini, F., Onesto, E., et al. (2007). Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol. Aging 28, 1099–1111. doi: 10.1016/j.neurobiolaging.2006.05.015
Rusmini, P., Simonini, F., Crippa, V., Bolzoni, E., Onesto, E., Cagnin, M., et al. (2011). 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol. Dis. 41, 83–95. doi: 10.1016/j.nbd.2010.08.023
Sahu, R., Kaushik, S., Clement, C. C., Cannizzo, E. S., Scharf, B., Follenzi, A., et al. (2011). Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139. doi: 10.1016/j.devcel.2010.12.003
Sandell, S., Huovinen, S., Palmio, J., Raheem, O., Lindfors, M., Zhao, F., et al. (2016). Diagnostically important muscle pathology in DNAJB6 mutated LGMD1D. Acta Neuropathol. Commun. 4:9. doi: 10.1186/s40478-016-0276-9
Sardiello, M., Palmieri, M., di Ronza, A., Medina, D. L., Valenza, M., Gennarino, V. A., et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. doi: 10.1126/science.1174447
Sarkar, S., Chigurupati, S., Raymick, J., Mann, D., Bowyer, J. F., Schmitt, T., et al. (2014). Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicology 44, 250–262. doi: 10.1016/j.neuro.2014.07.006
Sarparanta, J., Jonson, P. H., Golzio, C., Sandell, S., Luque, H., Screen, M., et al. (2012). Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450–455, S451–S452. doi: 10.1038/ng.1103
Sasaki, S. (2010). Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol 69, 346–355. doi: 10.1097/nen.0b013e3181d44992
Sau, D., De Biasi, S., Vitellaro-Zuccarello, L., Riso, P., Guarnieri, S., Porrini, M., et al. (2007). Mutation of SOD1 in ALS: a gain of a loss of function. Hum. Mol. Genet. 16, 1604–1618. doi: 10.1093/hmg/ddm110
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schaeffer, V., and Goedert, M. (2012). Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy 8, 1686–1687. doi: 10.4161/auto.21488
Seidel, K., Vinet, J., Dunnen, W. F., Brunt, E. R., Meister, M., Boncoraglio, A., et al. (2012). The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 38, 39–53. doi: 10.1111/j.1365-2990.2011.01198.x
Selcen, D., Muntoni, F., Burton, B. K., Pegoraro, E., Sewry, C., Bite, A. V., et al. (2009). Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 65, 83–89. doi: 10.1002/ana.21553
Senft, D., and Ronai, Z. A. (2015). UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141–148. doi: 10.1016/j.tibs.2015.01.002
Shi, C. H., Rubel, C., Soss, S. E., Sanchez-Hodge, R., Zhang, S., Madrigal, S. C., et al. (2018). Disrupted structure and aberrant function of CHIP mediates the loss of motor and cognitive function in preclinical models of SCAR16. PLoS Genet. 14:e1007664. doi: 10.1371/journal.pgen.1007664
Shi, Y., Wang, J., Li, J. D., Ren, H., Guan, W., He, M., et al. (2013). Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One 8:e81884. doi: 10.1371/journal.pone.0081884
Shy, M., Rebelo, A. P., Feely, S. M., Abreu, L. A., Tao, F., Swenson, A., et al. (2018). Mutations in BAG3 cause adult-onset Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 89, 313–315. doi: 10.1136/jnnp-2017-315929
Simeoni, S., Mancini, M. A., Stenoien, D. L., Marcelli, M., Weigel, N. L., Zanisi, M., et al. (2000). Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 9, 133–144. doi: 10.1093/hmg/9.1.133
Sobue, G., Hashizume, Y., Mukai, E., Hirayama, M., Mitsuma, T., and Takahashi, A. (1989). X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain 112, 209–232. doi: 10.1093/brain/112.1.209
Sondermann, H., Scheufler, C., Schneider, C., Hohfeld, J., Hartl, F. U., and Moarefi, I. (2001). Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science 291, 1553–1557. doi: 10.1126/science.1057268
Sopher, B. L., Thomas, P. S. Jr., LaFevre-Bernt, M. A., Holm, I. E., Wilke, S. A., Ware, C. B., et al. (2004). Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron 41, 687–699. doi: 10.1016/s0896-6273(04)00082-0
Sproviero, D., La Salvia, S., Giannini, M., Crippa, V., Gagliardi, S., Bernuzzi, S., et al. (2018). Pathological proteins are transported by extracellular vesicles of sporadic amyotrophic lateral sclerosis Patients. Front. Neurosci. 12:487. doi: 10.3389/fnins.2018.00487
Stenoien, D. L., Cummings, C. J., Adams, H. P., Mancini, M. G., Patel, K., DeMartino, G. N., et al. (1999). Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1 and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 8, 731–741. doi: 10.1093/hmg/8.5.731
Stieber, A., Gonatas, J. O., Collard, J., Meier, J., Julien, J., Schweitzer, P., et al. (2000). The neuronal Golgi apparatus is fragmented in transgenic mice expressing a mutant human SOD1, but not in mice expressing the human NF-H gene. J. Neurol. Sci. 173, 63–72. doi: 10.1016/s0022-510x(99)00301-9
Stürner, E., and Behl, C. (2017). The role of the multifunctional BAG3 protein in cellular protein quality control and in disease. Front. Mol. Neurosci. 10:177. doi: 10.3389/fnmol.2017.00177
Sun, X., Fontaine, J. M., Bartl, I., Behnam, B., Welsh, M. J., and Benndorf, R. (2007). Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones 12, 307–319. doi: 10.1379/csc-276.1
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Tadic, V., Prell, T., Lautenschlaeger, J., and Grosskreutz, J. (2014). The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8:147. doi: 10.3389/fncel.2014.00147
Takayama, S., and Reed, J. C. (2001). Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell Biol. 3, E237–E241. doi: 10.1038/ncb1001-e237
Tamaki, Y., Shodai, A., Morimura, T., Hikiami, R., Minamiyama, S., Ayaki, T., et al. (2018). Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci. Rep. 8:6030. doi: 10.1038/s41598-018-24463-3
Tanaka, A. (2010). Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Lett. 584, 1386–1392. doi: 10.1016/j.febslet.2010.02.060
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N. R., Doi, H., et al. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 10, 148–154. doi: 10.1038/nm985
Terry, D. F., McCormick, M., Andersen, S., Pennington, J., Schoenhofen, E., Palaima, E., et al. (2004). Cardiovascular disease delay in centenarian offspring: role of heat shock proteins. Ann. N Y Acad. Sci. 1019, 502–505. doi: 10.1196/annals.1297.092
Terry, D. F., Wyszynski, D. F., Nolan, V. G., Atzmon, G., Schoenhofen, E. A., Pennington, J. Y., et al. (2006). Serum heat shock protein 70 level as a biomarker of exceptional longevity. Mech. Ageing Dev. 127, 862–868. doi: 10.1016/j.mad.2006.08.007
Teyssou, E., Takeda, T., Lebon, V., Boillee, S., Doukoure, B., Bataillon, G., et al. (2013). Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 125, 511–522. doi: 10.1007/s00401-013-1090-0
Thellung, S., Scoti, B., Corsaro, A., Villa, V., Nizzari, M., Gagliani, M. C., et al. (2018). Pharmacological activation of autophagy favors the clearing of intracellular aggregates of misfolded prion protein peptide to prevent neuronal death. Cell Death Dis. 9:166. doi: 10.1038/s41419-017-0252-8
Thomas, M., Yu, Z., Dadgar, N., Varambally, S., Yu, J., Chinnaiyan, A. M., et al. (2005). The unfolded protein response modulates toxicity of the expanded glutamine androgen receptor. J. Biol. Chem. 280, 21264–21271. doi: 10.1074/jbc.m500144200
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. doi: 10.1038/nature10744
Tohnai, G., Adachi, H., Katsuno, M., Doi, H., Matsumoto, S., Kondo, N., et al. (2014). Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 23, 3552–3565. doi: 10.1093/hmg/ddu066
Trotti, D., Rolfs, A., Danbolt, N. C., Brown, R. H. Jr., and Hediger, M. A. (1999). SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 2:848. doi: 10.1038/12227
Ulbricht, A., Arndt, V., and Hohfeld, J. (2013). Chaperone-assisted proteostasis is essential for mechanotransduction in mammalian cells. Commun. Integr. Biol. 6:e24925. doi: 10.4161/cib.24925
Ulbricht, A., Gehlert, S., Leciejewski, B., Schiffer, T., Bloch, W., and Hohfeld, J. (2015). Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 11, 538–546. doi: 10.1080/15548627.2015.1017186
van Niel, G., D’Angelo, G., and Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228. doi: 10.1038/nrm.2017.125
Vegeto, E., Villa, A., Della Torre, S., Crippa, V., Rusmini, P., Cristofani, R., et al. (2020). The role of sex and sex hormones in neurodegenerative diseases. Endocr. Rev. 41, 273–319. doi: 10.1210/endrev/bnz005
Vogiatzi, T., Xilouri, M., Vekrellis, K., and Stefanis, L. (2008). Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556. doi: 10.1074/jbc.m801992200
Voisine, C., Pedersen, J. S., and Morimoto, R. I. (2010). Chaperone networks: tipping the balance in protein folding diseases. Neurobiol. Dis. 40, 12–20. doi: 10.1016/j.nbd.2010.05.007
Wang, I. F., Guo, B. S., Liu, Y. C., Wu, C. C., Yang, C. H., Tsai, K. J., et al. (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U S A 109, 15024–15029. doi: 10.1073/pnas.1206362109
Wang, Y., Liu, F. T., Wang, Y. X., Guan, R. Y., Chen, C., Li, D. K., et al. (2018). Autophagic modulation by trehalose reduces accumulation of TDP-43 in a cell model of amyotrophic lateral sclerosis via TFEB activation. Neurotox. Res. 34, 109–120. doi: 10.1007/s12640-018-9865-7
Wang, Y., Martinez-Vicente, M., Kruger, U., Kaushik, S., Wong, E., Mandelkow, E. M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. doi: 10.1093/hmg/ddp367
Wang, Y., Martinez-Vicente, M., Kruger, U., Kaushik, S., Wong, E., Mandelkow, E. M., et al. (2010). Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 6, 182–183. doi: 10.4161/auto.6.1.10815
Wang, K., Shang, Y., and Dou, F. (2018). Brain aging: Hsp90 and neurodegenerative diseases. Adv. Exp. Med. Biol. 1086, 93–103. doi: 10.1007/978-981-13-1117-8_6
Wate, R., Ito, H., Zhang, J. H., Ohnishi, S., Nakano, S., and Kusaka, H. (2005). Expression of an endoplasmic reticulum-resident chaperone, glucose-regulated stress protein 78, in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 110, 557–562. doi: 10.1007/s00401-005-1080-y
Waza, M., Adachi, H., Katsuno, M., Minamiyama, M., Tanaka, F., Doyu, M., et al. (2006). Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J. Mol. Med. 84, 635–646. doi: 10.1007/s00109-006-0066-0
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17, 645–652. doi: 10.1016/j.celrep.2016.09.032
Wilhelmus, M. M., Boelens, W. C., Otte-Holler, I., Kamps, B., Kusters, B., Maat-Schieman, M. L., et al. (2006). Small heat shock protein HspB8: its distribution in Alzheimer’s disease brains and its inhibition of amyloid-β protein aggregation and cerebrovascular amyloid-β toxicity. Acta Neuropathol. 111, 139–149. doi: 10.1007/s00401-005-0030-z
Xiao, Y., Ma, C., Yi, J., Wu, S., Luo, G., Xu, X., et al. (2015). Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol. Rep. 3:e12271. doi: 10.14814/phy2.12271
Xie, Z., and Klionsky, D. J. (2007). Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109. doi: 10.1038/ncb1007-1102
Xu, J., Camfield, R., and Gorski, S. M. (2018). The interplay between exosomes and autophagy—partners in crime. J. Cell Sci. 131:jcs215210. doi: 10.1242/jcs.215210
Yang, Y. C., Fu, H. C., Hsiao, B. L., Sobue, G., Adachi, H., Huang, F. J., et al. (2013). Androgen receptor inclusions acquire GRP78/BiP to ameliorate androgen-induced protein misfolding stress in embryonic stem cells. Cell Death Dis. 4:e607. doi: 10.1038/cddis.2013.122
Yang, Q., She, H., Gearing, M., Colla, E., Lee, M., Shacka, J. J., et al. (2009). Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323, 124–127. doi: 10.1126/science.1166088
Ye, Y., Shibata, Y., Kikkert, M., van Voorden, S., Wiertz, E., and Rapoport, T. A. (2005). Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. U S A 102, 14132–14138. doi: 10.1073/pnas.0505006102
Yoneda, T., Imaizumi, K., Oono, K., Yui, D., Gomi, F., Katayama, T., et al. (2001). Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940. doi: 10.1074/jbc.m010677200
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yu, Z., Wang, A. M., Adachi, H., Katsuno, M., Sobue, G., Yue, Z., et al. (2011). Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 7:e1002321. doi: 10.1371/journal.pgen.1002321
Zaarur, N., Xu, X., Lestienne, P., Meriin, A. B., McComb, M., Costello, C. E., et al. (2015). RuvbL1 and RuvbL2 enhance aggresome formation and disaggregate amyloid fibrils. EMBO J. 34, 2363–2382. doi: 10.15252/embj.201591245
Zhang, X., Chen, S., Song, L., Tang, Y., Shen, Y., Jia, L., et al. (2014). MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10, 588–602. doi: 10.4161/auto.27710
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., et al. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412–425. doi: 10.4161/auto.7.4.14541
Keywords: motor neuron, protein quality control, CASA complex, HSPB8, BAG3, BAG1
Citation: Cristofani R, Crippa V, Cicardi ME, Tedesco B, Ferrari V, Chierichetti M, Casarotto E, Piccolella M, Messi E, Galbiati M, Rusmini P and Poletti A (2020) A Crucial Role for the Protein Quality Control System in Motor Neuron Diseases. Front. Aging Neurosci. 12:191. doi: 10.3389/fnagi.2020.00191
Received: 24 March 2020; Accepted: 02 June 2020;
Published: 21 July 2020.
Edited by:
Ines Moreno-Gonzalez, University of Malaga, SpainReviewed by:
Anna Maria Colangelo, University of Milano-Bicocca, ItalySarat C. Vatsavayai, University of California, San Francisco, United States
Copyright © 2020 Cristofani, Crippa, Cicardi, Tedesco, Ferrari, Chierichetti, Casarotto, Piccolella, Messi, Galbiati, Rusmini and Poletti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelo Poletti, YW5nZWxvLnBvbGV0dGlAdW5pbWkuaXQ=