 Mor Savyon
Mor Savyon Simone Engelender*
Simone Engelender*- Department of Biochemistry, The B. Rappaport Faculty of Medicine and Institute of Medical Research, Technion – Israel Institute of Technology, Haifa, Israel
The accumulation and aggregation of α-synuclein are central to Parkinson’s disease (PD), yet the molecular mechanisms responsible for these events are not fully understood. Post-translational modifications of α-synuclein regulate several of its properties, including degradation, interaction with proteins and membranes, aggregation and toxicity. SUMOylation is a post-translational modification involved in various nuclear and extranuclear processes, such as subcellular protein targeting, mitochondrial fission and synaptic plasticity. Protein SUMOylation increases in response to several stressful situations, from viral infections to trauma. In this framework, an increasing amount of evidence has implicated SUMOylation in several neurodegenerative diseases, including PD. This review will discuss recent findings in the role of SUMOylation as a regulator of α-synuclein accumulation, aggregation and toxicity, and its possible implication in neurodegeneration that underlies PD.
Introduction
Parkinson’s disease (PD) is an incurable neurological disorder that affects over six million individuals worldwide and is predicted to increase as the population ages (Dorsey et al., 2018). Brains of PD patients exhibit extensive loss of dopaminergic neurons, responsible for the cardinal motor symptoms of the disease- resting tremor, bradykinesia, muscle rigidity and postural instability. Dysfunction of lower brain regions and peripheral nervous system underlies the non-motor features of the disease, including constipation, sleep disorders, and depression (Engelender and Isacson, 2017). Despite the availability of symptomatic treatments, there is still no intervention that halts the progression of the disease (Oertel and Schulz, 2016).
While the molecular mechanisms involved in the neuronal loss in PD remain unknown, the misfolding and accumulation of α-synuclein in PD brains seem to play a pathological role in the disease. In accordance, brains of patients with sporadic and familial forms of the disease contain proteinaceous inclusion bodies, known as Lewy bodies, which are predominantly composed of aggregated α-synuclein (Spillantini et al., 1997, 1998). Nevertheless, although Lewy bodies are a pathological hallmark of the disease, its role in neuronal toxicity remains controversial. Several studies have shown that oligomeric forms of α-synuclein are toxic to neurons (Outeiro et al., 2008; Karpinar et al., 2009; Kalia et al., 2013), and in vitro studies found that oligomerization and not fibrillation is common to different α-synuclein disease mutations (Conway et al., 2000). Furthermore, in vivo studies reported that mice expressing oligomer-prone α-synuclein mutants have a more severe dopaminergic loss compared to those expressing α-synuclein mutants that are more prone to form fibrils (Winner et al., 2011).
On the other hand, several studies pointed at the possibility that more organized α-synuclein fibrils, similar to those found in Lewy bodies, may also be toxic (Alam et al., 2019). In agreement, injection of pre-formed α-synuclein fibrils to brains is toxic to neurons promoting aggregation of endogenous α-synuclein and neurodegeneration (Luk et al., 2012). Based on these studies, both oligomeric and fibrillar α-synuclein species may contribute to neuronal toxicity. Regardless of the exact α-synuclein species that are more prone to cause toxicity, understanding how it accumulates in PD is essential to disclose the mechanisms that lead to disease.
Exposure of different cell lines to stressors, such as oxidative stress (Ostrerova-Golts et al., 2000), proteotoxic stress (Tanaka et al., 2001), nitrosative stress (Paxinou et al., 2001) and improper calcium homeostasis (Hettiarachchi et al., 2009), promote the accumulation and aggregation of α-synuclein. Exposure to stressors change post-translational modifications (PTMs) of proteins (Appella and Anderson, 2001; Kramer et al., 2015); therefore, it is conceivable that different types of cellular stresses may change α-synuclein PTMs, including phosphorylation (Fujiwara et al., 2002), truncation (Baba et al., 1998), nitration (Paxinou et al., 2001), glycation (Vicente Miranda et al., 2017), ubiquitination (Hasegawa et al., 2002), SUMOylation (Dorval and Fraser, 2006; Rott et al., 2017) and acetylation (Anderson et al., 2006). Thus, changes in α-synuclein PTMs may represent a missing link in the pathogenesis of PD.
The Biology of α-Synuclein
α-synuclein is a 140 amino acid protein predominantly expressed in neurons. It is located at the presynaptic nerve terminals in close association with synaptic vesicles (Maroteaux et al., 1988; Withers et al., 1997; Kahle et al., 2000; Ribeiro et al., 2002). α-synuclein is an intrinsically disordered protein that lacks a stable three-dimensional structure (Weinreb et al., 1996; Conway et al., 1998; Davidson et al., 1998). Still, it is divided into three main domains: i) an N-terminal region (amino acids 1-60) containing several conserved KTKEGV repeats that render a more alpha-helical structure and enables interaction with membranes (Davidson et al., 1998; Jo et al., 2000; McLean et al., 2000); ii) a central portion containing a hydrophobic amino acid stretch known as the non-amyloid component of the amyloid plaques (NAC; amino acids 61-95) and that is involved in the formation of beta-sheet structure and aggregation (Bodles et al., 2001; Murray et al., 2003); and iii) a C-terminal region (amino acids 96-140), which is abundant in negatively charged amino acid residues (Park et al., 2002; Murray et al., 2003). Despite its relatively disordered structure, α-synuclein can shift into a more compact conformation, which conceals the NAC region and prevents its exposure to the cytoplasm, withholding its tendency to aggregate spontaneously (Theillet et al., 2016). In agreement with this possibility, the cleavage of α-synuclein C-terminal region promotes its aggregation and toxicity in cell lines and mice (Tofaris et al., 2006), suggesting that the C-terminal region may play a critical role in concealing α-synuclein NAC region.
The exact function of α-synuclein is still unclear. Nevertheless, studies have connected it with the regulation of synaptic vesicle pool (Murphy et al., 2000; Cabin et al., 2002), the release of neurotransmitters (Abeliovich et al., 2000), SNARE-complex assembly (Burre et al., 2010) and dilation of exocytotic fusion pore (Logan et al., 2017). α-synuclein was also reported to interact with additional sub-cellular organelles and compartments, including the mitochondria (Devi et al., 2008) and the nucleus (Goers et al., 2003; Pinho et al., 2019).
Mutations (A53T/E, A30P, E46K, H50Q, and G51D) and multiplications of the α-synuclein gene (SNCA) cause autosomal dominant PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003; Chartier-Harlin et al., 2004; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Lesage et al., 2013; Pasanen et al., 2014), yet the exact contribution of each mutation to the disease is not clear. Nevertheless, since the known mutations are located in the N-terminal region of the protein, changes in α-synuclein binding with membranes could play a role in the neuronal dysfunction caused by the PD mutants (Davidson et al., 1998; McLean et al., 2000). Another possibility, perhaps not mutually exclusive, is that increased oligomerization and aggregation (but not fibrillation) may be responsible for the toxicity of α-synuclein PD mutants (Conway et al., 2000). Supporting this idea, families with SNCA multiplications have increased levels of α-synuclein, increased α-synuclein aggregation and prominent neurodegeneration (Singleton et al., 2003; Book et al., 2018).
Overview of α-Synuclein Post-Translational Modifications
Post-translational modifications are major cellular events that occur in response to endogenous and exogenous stimuli, allowing the cell to function in normal and stressful conditions. The development of mass spectrometry technology allowed gathering information regarding PTMs and how they regulate protein activity, localization, and stability (Nørregaard Jensen, 2004; Ren et al., 2014).
Numerous α-synuclein PTMs have been identified over the years, including phosphorylation, ubiquitination, truncation, nitration, glycation, acetylation, and SUMOylation. The most comprehensively studied PTM is phosphorylation. α-synuclein is extensively phosphorylated at Ser129, as identified by mass spectrometry in purified Lewy bodies (Fujiwara et al., 2002; Anderson et al., 2006). Different protein kinases were shown to phosphorylate α-synuclein at Ser129, including casein kinases, G protein-coupled receptor kinases and polo-like kinases (Okochi et al., 2000; Pronin et al., 2000; Mbefo et al., 2010). Accordingly, phosphorylation at Ser129 leads to increased α-synuclein fibrillation in vitro (Fujiwara et al., 2002) as well as aggregation and toxicity in different cell lines and transgenic models (Kahle et al., 2002; Smith et al., 2005; Chau et al., 2009; Chen et al., 2009; Zhou et al., 2011) (for more details see Table 1). Moreover, phosphorylation at Ser129 also changes α-synuclein localization and properties. For instance, it was shown to increase the nuclear localization of α-synuclein in H4 cells (Pinho et al., 2019), to modulate the binding of α-synuclein to membranes (Visanji et al., 2011; Kuwahara et al., 2012) and to increase its binding affinity to metal-ions (Lu et al., 2011), suggesting that phosphorylation at Ser129 may regulate both α-synuclein normal and pathological functions.
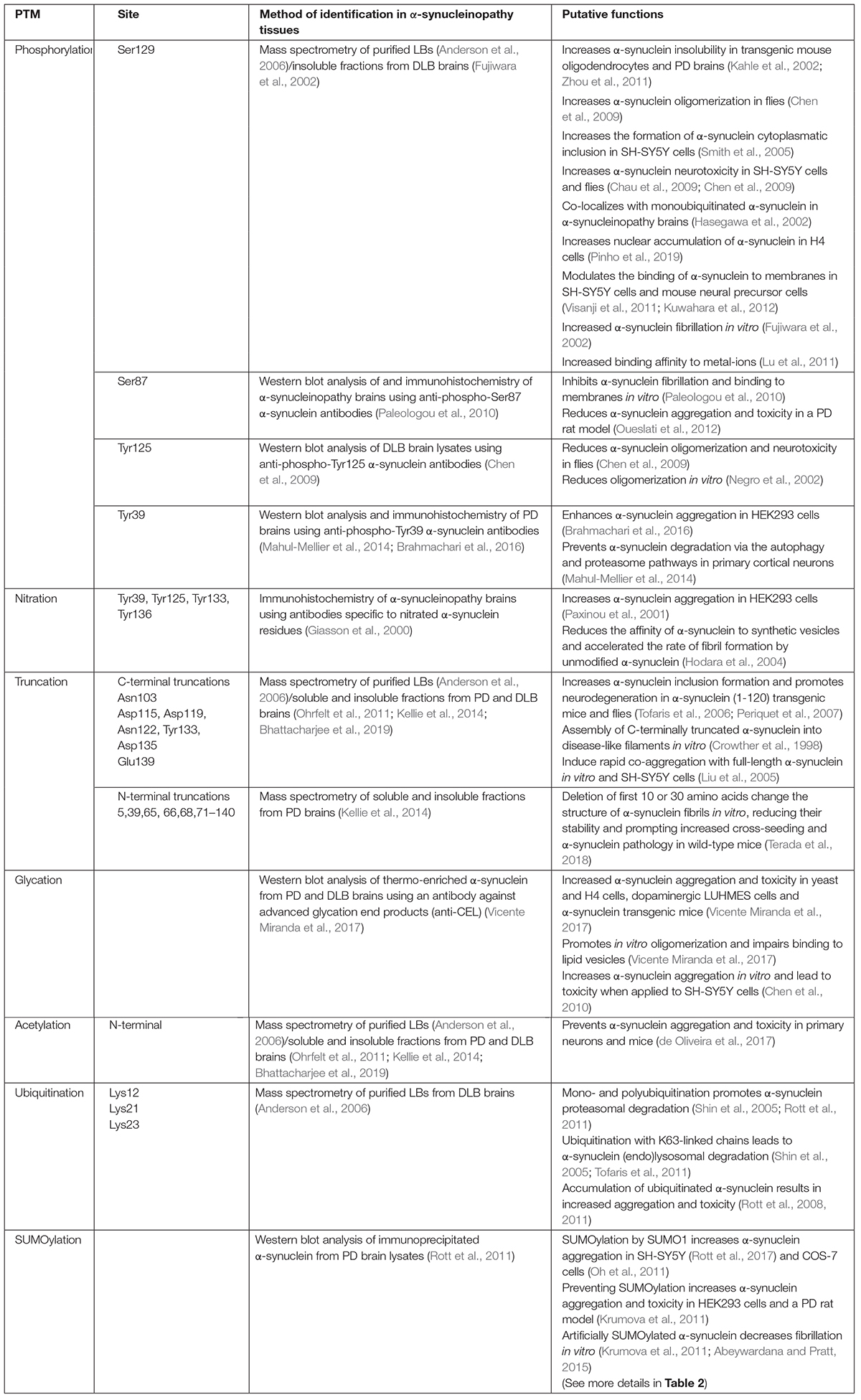
Table 1. α-synuclein post-translational modifications.
α-synuclein is also phosphorylated at Ser87, which was first detected in HEK293 and PC12 cells by phosphoamino-acid analysis and site-directed mutations (Okochi et al., 2000). Its relevance to the disease was confirmed by immunostaining of PD tissues using an anti-phospho-Ser87 α-synuclein antibody (Paleologou et al., 2010). In contrast to Ser129, phosphorylation at Ser87 inhibits α-synuclein fibrillation and binding to membranes in vitro (Paleologou et al., 2010), and also reduces α-synuclein aggregation and toxicity in a PD rat model (Oueslati et al., 2012; Table 1).
Phosphorylation of α-synuclein also occurs at tyrosine residues. Mutation analysis studies revealed that tyrosine kinases, such as Fyn and Syk, phosphorylate α-synuclein at tyrosine residues 125, 133 and 136 (Nakamura et al., 2001; Negro et al., 2002). Among these sites, phosphorylation at Tyr125 was detected in brain lysates by Western blot analysis using antibodies against phospho-Tyr125 α-synuclein and was found to be reduced in patients with Dementia with Lewy bodies (DLB) (Chen et al., 2009; Table 1), suggesting that the increased P-Ser129/P-Tyr125 ratio could contribute to the pathogenicity of α-synuclein.
More recently, the tyrosine kinase c-Abl was shown to interact with and to phosphorylate α-synuclein at tyrosine residue 39 (Mahul-Mellier et al., 2014; Brahmachari et al., 2016). Using antibodies against phospho-Tyr39 α-synuclein, this phosphorylation was found to be increased in lysates from substantia nigra of PD brains and to accumulate in Lewy bodies (Brahmachari et al., 2016; Table 1). Different from Tyr125, phosphorylation at Tyr39 enhances α-synuclein aggregation in HEK293 cells (Brahmachari et al., 2016), and prevents the proteasomal and autophagic degradation of α-synuclein in primary neurons (Mahul-Mellier et al., 2014). These findings are promising as a c-Abl inhibitor that crosses the blood-brain barrier, nilotinib, promotes α-synuclein autophagic degradation, and prevents dopaminergic degeneration in PD mouse models overexpressing α-synuclein (Hebron et al., 2013). Nevertheless, nilotinib failed to prevent α-synuclein aggregation and neuronal loss in a mouse model of MSA (Lopez-Cuina et al., 2020), suggesting that c-Abl inhibitors may be more effective to treat α-synucleinopathies with predominant α-synuclein neuronal accumulation.
Acetylation is also an outstanding α-synuclein PTM as it is found in Lewy bodies and brain regions affected in the disease (Anderson et al., 2006; Ohrfelt et al., 2011; Kellie et al., 2014; Bhattacharjee et al., 2019). Obliteration of α-synuclein acetylation sites in vivo leads to its enhanced aggregation and toxicity in primary neurons and mice (de Oliveira et al., 2017), implying that acetylation may prevent α-synuclein aggregation. Other PTMs, such as nitration and glycation, which are respectively related to nitro-oxidative stress and increased levels of blood glucose, are, on the other hand, associated with increased aggregation and toxicity of α-synuclein (Giasson et al., 2000; Paxinou et al., 2001; Vicente Miranda et al., 2017) (for more details see Table 1). Thus, the roles of PTMs are diverse, and their combined contribution to the pathogenesis of the disease is still far to be understood.
α-synuclein is also conjugated to polypeptides, such as ubiquitin and SUMO (small ubiquitin-related modifier). Biochemical separation and mass spectrometry analysis of purified Lewy bodies consistently identified the conjugation of α-synuclein by one, two, or three ubiquitin molecules, specifically at lysines 12, 21 and 23 (Hasegawa et al., 2002; Tofaris et al., 2003; Anderson et al., 2006). α-synuclein is degraded by the proteasome in cell lines and mice brain tissues (Bennett et al., 1999; Webb et al., 2003; Ebrahimi-Fakhari et al., 2011; Rott et al., 2011). Furthermore, mono- and polyubiquitination by the E3 ubiquitin-ligases SIAH and CHIP promote the proteasomal degradation of α-synuclein (Shin et al., 2005; Rott et al., 2011; Table 1). In contrast, conjugation of ubiquitin chains formed at lysine 63 by the E3 ubiquitin-ligase Nedd4 leads to endo-lysosomal degradation (Tofaris et al., 2011). Most importantly, impairment of degradation pathways leads to the accumulation of ubiquitinated α-synuclein, rendering increased α-synuclein aggregation and toxicity in cell lines (Rott et al., 2008, 2011).
SUMOylation is another type of polypeptide conjugation that is essential for nuclear and extranuclear functions (Henley et al., 2018) and is responsive to cellular stressors (Enserink, 2015). SUMOylation also covalently modifies α-synuclein. We argue in this review that increased levels of SUMOylation may compete with ubiquitin and prevent α-synuclein degradation (Rott et al., 2017; Rousseaux et al., 2018). Further details on the different roles of SUMOylation on α-synuclein functions will be discussed below in detail.
Truncation of α-synuclein was initially described by biochemical means in purified Lewy bodies (Baba et al., 1998). Later, it was identified by mass spectrometry analysis in both purified Lewy bodies (Anderson et al., 2006; Bhattacharjee et al., 2019) and lysates of PD tissues (Ohrfelt et al., 2011; Kellie et al., 2014), and shown to occur at different α-synuclein residues at the C- and N-termini (for more details see Table 1). Although not all reported α-synuclein truncations were found enriched in Lewy bodies or PD tissues (Ohrfelt et al., 2011; Kellie et al., 2014; Bhattacharjee et al., 2019), the importance of truncations is implied by their increased tendency to aggregate in vitro, in cell lines and in vivo models (Crowther et al., 1998; Liu et al., 2005; Tofaris et al., 2006; Periquet et al., 2007). Also, the C-terminally truncated α-synuclein species assemble into filaments that resemble those found in diseased brains (Crowther et al., 1998) and leads to the formation of pathological inclusions and neurotoxicity in transgenic mice (Tofaris et al., 2006).
Although the development of protocols for Lewy body purification and solubilization from brain tissues has allowed an in-depth characterization of α-synuclein PTMs in PD and other α-synucleinopathies (Iwatsubo et al., 1996; Anderson et al., 2006), some PTMs, such as SUMOylation, nitration and glycation, have not been detected in α-synucleinopathy brains using conventional mass spectrometry techniques (Anderson et al., 2006; Bhattacharjee et al., 2019). Nevertheless, they have been identified in Lewy bodies of PD and DLB tissues by immunohistochemistry, and confirmed by biochemical means to directly modify α-synuclein (Giasson et al., 2000; Danielson et al., 2009; Kim et al., 2011; Rott et al., 2017; Vicente Miranda et al., 2017). The reason for failing to identify these PTMs by mass spectrometry in α-synucleinopathy brains is still not clear. One possibility is that some PTMs are more labile than others and are more easily deconjugated before analysis (Dapic et al., 2019). It is also possible that solubilization may not be complete before mass spectrometry analysis. For instance, even when brain lysates were incubated with stringent buffers containing detergents, such as sarkosyl, insoluble material from Lewy bodies was not entirely solubilized (Iwatsubo et al., 1996). Besides, technical limitations in the mass spectrometry technique per se could be responsible for this constraint. For instance, most protocols employed in traditional mass spectrometry analysis use proteolytic digestion before the electrospray ionization, rendering fragments that can be challenging in terms of size and charge to be detected by the mass spectrometer equipment (Dapic et al., 2019). Based on the diversity of PTMs in mammalian cells and the caveats described above, it is conceivable that several other α-synuclein PTMs still need to be uncovered by improved mass spectrometry techniques, such as top-down analysis (Breuker, 2018). We dedicated this review to discuss recent findings on α-synuclein SUMOylation and its possible implications in PD.
The SUMOylation Pathway
Small Ubiquitin-like Modifier (SUMO) is a family of proteins that are covalently conjugated to lysine residues on a ΨKX(D/E) consensus motif of target proteins, where Ψ represents a hydrophobic residue, K is a target lysine, X is any residue, and D/E represent acidic residues (Matunis et al., 1996; Rodriguez et al., 2001; Zhao et al., 2014). SUMOylation regulates intra-nuclear cellular events, including cell division, transcription, DNA repair and nuclear transport (Zhao, 2018). Nevertheless, several studies have shown that extranuclear proteins can also be SUMOylated, implicating SUMOylation in several extranuclear roles, such as endocytosis, synaptic activity and signaling transduction (Henley et al., 2018). At the protein level, SUMOylation can affect the structure, stability, localization and protein-protein interaction. Moreover, several SUMOylation targets are proteins related to neurodegeneration, including tau and α-synuclein (Dorval and Fraser, 2006).
Protein SUMOylation involves sequential events that rely on the activation of three enzymes, known as E1 SUMO-activating, E2 SUMO-conjugating and E3 SUMO-ligase, each one uniquely contributes to the completion of this cascade and the final SUMOylation of the substrate (Muller et al., 2001). The SUMO peptide is first matured for conjugation by SUMO cysteine proteases that belong to the ubiquitin-like-specific protease 1 (Ulp)/sentrin-specific protease (SENP) family, which expose a glycine residue at its C-terminus (Hickey et al., 2012). The E1 SUMO-activating enzyme, a SAE1/2 heterodimer, then forms a thioester bond between its active-site cysteine and the mature SUMO peptide, in an ATP-dependent matter. The attached SUMO is subsequently transferred to a cysteine at the active-site of Ubc9, the only known E2 SUMO-conjugating enzyme of the pathway (Wilkinson and Henley, 2010). Finally, the E3 SUMO-ligase plays a facilitating role in the transfer of SUMO from Ubc9 to the substrate, allowing the completion of the substrate SUMOylation (Kahyo et al., 2001). It is important to note that different from the ubiquitin system, the E2 SUMO-conjugating enzyme Ubc9 can, to a certain extent, directly transfer SUMO to the substrate (Knipscheer et al., 2007). Recycling of SUMO peptides occurs as a result of deSUMOylation of SUMOylated proteins also by the SUMO proteases from the Ulp/SENP family.
Mechanistically, ubiquitination and SUMOylation share several common features. As ubiquitination, SUMOylation is a dynamic multistep conjugation and de-conjugation cascade, directed by ATP-dependent enzymes. Also, similar to ubiquitin, SUMO can be covalently attached to a single (monoSUMOylation) or multiple (multi-monoSUMOylation) lysine residues, or form SUMO chains (polySUMOylation) on the substrate protein (Wilkinson and Henley, 2010; Hickey et al., 2012). On the other hand, some features of the SUMO pathway are different from those of ubiquitination. For instance, while the ubiquitin system relies on a single ubiquitin isoform, the SUMOylation pathway exhibits several distinct SUMO isoforms, including the extensively studied SUMO1 to SUMO3, and the more recently described SUMO4 and SUMO5 isoforms (Bohren et al., 2004; Wilkinson and Henley, 2010). SUMO2 and SUMO3 share a high degree of similarity and are therefore regarded as SUMO2/3. Further distinguishing SUMO isoforms, SUMO2/3, but not SUMO1, support polySUMOylation with chain formation (Tatham et al., 2001). The additional SUMO isoforms, SUMO4 and SUMO5, were described to support polySUMOylation, but their roles await further investigation (Liang et al., 2016; Baczyk et al., 2017). Another critical difference is that ubiquitination usually serves as a signal for proteasomal protein degradation, while SUMOylation either affects lysine ubiquitination or regulates cellular activities, such as protein shuttling between compartments and activity of proteins (Matunis et al., 1996; Hershko and Ciechanover, 1998; Wilson, 2017).
SUMOylation machinery and protein SUMOylation dramatically increase in response to cellular stresses, including heat shock, DNA damage, viral infections, and nutrient and hypoxic stress, suggesting that SUMOylation could regulate the ability of cells to cope with stressors (Zhou et al., 2004; Enserink, 2015). It was suggested that SUMO2/3 was responsive to the different types of cellular stressors (Saitoh and Hinchey, 2000), while SUMO1 was regarded as a housekeeping conjugation involved in the regulation of protein subcellular localization and ubiquitination (Matunis et al., 1996). However, several studies support a broader scenario, where SUMO1 may also mediate the response to several cellular stressors, including heat-shock as well as genotoxic and hypoxic stress (Hong et al., 2001; Huang et al., 2003; Agbor et al., 2011). In agreement, proteasome inhibition and the proteotoxic stress it causes leads to the increased SUMOylation of proteins by SUMO2/3 or SUMO1, which accumulate in the promyelocytic leukemia protein (PML) nuclear bodies (Sha et al., 2019). Supporting an even broader role of SUMO isoforms in response to stress, polymorphism in the SUMO4 gene was linked to type 1 diabetes mellitus and increased SUMOylation by SUMO4 was detected in pre-eclamptic placentas (Bohren et al., 2004; Baczyk et al., 2017).
SUMOylation in PD
The occurrence of PD is associated with exposure to several types of stressors, including toxins and viral infections, as well as inflammation. Populations exposed to insecticides, such as paraquat and rotenone, have increased incidence of PD (Tanner et al., 2011). Also, parkinsonism can occur after influenza viral infections followed by encephalitis lethargica (Maurizi, 1987), and studies injecting the influenza H5N1 strain in mice caused overt neuroinflammation, accumulation of α-synuclein and nigral degeneration (Jang et al., 2009). Brain trauma and the neuroinflammation it produces, also lead to the accumulation of α-synuclein and increase the risk of developing PD (Newell et al., 1999). Because these insults activate the SUMO machinery (Enserink, 2015), SUMOylation may mediate the toxic outcomes of the different stressors in PD and other α-synucleinopathies. In agreement, injection of rotenone in mice brain leads to a concomitant increase of higher molecular weight species of SUMO1 and α-synuclein (Weetman et al., 2013). Moreover, the treatment of oligodendroglial cells with polyunsaturated fatty acids followed by oxidative stress lead to the formation of α-synuclein aggregates positive for ubiquitin and SUMO1 (Riedel et al., 2011).
SUMOylation participates in several pathways connected to PD, including changes in α-synuclein biology and pathology (discussed in detail in the sections below), regulation of DJ-1 activity (Ariga et al., 2013), modulation of transcription factors involved in mitochondrial and lysosomal biogenesis (Harder et al., 2004), and regulation of mitochondrial fission machinery (Harder et al., 2004; Guo et al., 2013).
Mutations in the DJ-1 gene lead to autosomal recessive early-onset parkinsonism (Bonifati et al., 2003). DJ-1 is present in different cellular compartments and has several attributed functions, among them transcriptional regulation, antioxidative properties, chaperone and mitochondrial regulation (Ariga et al., 2013). In particular, the DJ-1 redox-dependent molecular chaperone activity was suggested to inhibit α-synuclein aggregate formation (Shendelman et al., 2004). DJ-1 was also shown to directly bind the mitochondrial F0F1-ATP synthase b subunit enabling proper mitochondrial function, a process that was prevented by DJ-1 PD mutations (Chen et al., 2019), highlighting the importance of DJ-1 to maintain mitochondrial and α-synuclein homeostasis. SUMOylation of DJ-1 was shown to be essential to enable DJ-1 transcriptional activity, and the consequent neuroprotection it exerts, a function that is prevented by PD mutations (Shinbo et al., 2006). Future investigation of the levels of DJ-1 SUMOylation in PD may further shed light on the role of SUMOylation in the disease.
Accumulation of dysfunctional mitochondria is observed in affected PD brain regions, in particular in the proximity or even inside Lewy bodies (Shahmoradian et al., 2019). Also, mitochondrial complex I activity is consistently decreased in dopaminergic neurons of the substantia nigra (Schapira et al., 1989). Moreover, genome-wide expression studies identified that bioenergetics-related nuclear genes controlled by the mitochondrial biogenesis master regulator, PGC-1a, are underexpressed in PD patients (Zheng et al., 2010), suggesting that PGC-1a activity may be decreased in PD and may contribute to the mitochondrial dysfunction observed in the disease. In this framework, SUMOylation of PGC-1a was shown to reduce its transcriptional activity in HeLa cells (Rytinki and Palvimo, 2009), raising the possibility that increased SUMOylation could play a role in the accumulation of dysfunctional mitochondria in the disease.
Another aspect critical for the proper mitochondrial function is the dynamics of the organelle, dictated by fusion and fission processes (Cao et al., 2017). PINK1 and parkin, are genes mutated in autosomal recessive PD that regulate mitochondrial dynamics. More specifically, the PINK1/parkin pathway regulates mitochondrial dynamics by promoting mitochondrial fission as their PD mutants reduce mitochondrial fission (Poole et al., 2008). The dynamin-related protein 1 (DRP-1) is a critical GTPase that promotes mitochondrial fission by encircling constricted mitochondria and assisting the formation of two separated mitochondria (Youle and Karbowski, 2005). DRP-1 was shown to be SUMOylated by SUMO1 and SUMO2/3 isoforms (Harder et al., 2004; Figueroa-Romero et al., 2009), and a surprising number of SUMO1 conjugates were found in mitochondria at fission sites of COS-7 cells, implying that SUMOylated DRP-1 could facilitate the fragmentation of the organelle (Harder et al., 2004). On the other hand, DRP-1 conjugation to SUMO2/3 was shown to prevent mitochondrial fragmentation and to protect against neuronal damage after oxygen/glucose deprivation (Guo et al., 2013). Although the definitive role of SUMOylation on DRP-1 ability to promote mitochondrial fission requires further clarification, the differences in the experimental models (COS-7 cells vs. primary neurons) and the different SUMO isoforms analyzed (SUMO1 vs. SUMO2/3) may help explain the differences. Additional studies addressing the role of DRP-1 SUMOylation in PD may help understand the mechanisms involved in the accumulation of dysfunctional mitochondria in the disease and their connection to the increased aggregation of α-synuclein (Lautenschager and Kaminski Schierle, 2019).
Other proteins involved on α-synuclein homeostasis have also been connected to the SUMO pathway. Lysosomal function is suggested to be dysfunctional in PD (Dehay et al., 2013), and SUMOylation of the Transcription Factor EB (TFEB), a master regulator of lysosomal biogenesis, decreases its transcriptional activity (Miller et al., 2005). Thus, increased TFEB SUMOylation could hamper the renewal of lysosomes to properly carry out α-synuclein degradation through the lysosomal-autophagy pathway.
Another protein implicated in PD is HSP90, which interacts with α-synuclein and is also present in Lewy bodies (Uryu et al., 2006). The role of HSP90 in α-synuclein aggregation is still a matter of debate. Some studies suggest that HSP90 inhibits α-synuclein aggregation (Daturpalli et al., 2013), while inhibition of HSP90 was also shown to prevent α-synuclein oligomer formation and to rescue α-synuclein-induced toxicity (Putcha et al., 2010). Although HSP90 actions in PD are not fully understood, chemical SUMOylation of HSP90 was recently shown to modulate co-chaperone binding in vitro (Wolmarans et al., 2019). Therefore, SUMOylation may play a role in the ability of HSP90 to modulate α-synuclein aggregation into Lewy bodies.
SUMO1 and one of α-synuclein SUMO-ligase, PIAS2, were detected in Lewy bodies of PD brains (Kim et al., 2011; Rott et al., 2017). Most importantly, the levels of SUMOylated α-synuclein isolated by immunoprecipitation are increased in the cerebral cortex of PD patients with dementia (Rott et al., 2017). Moreover, SUMO1 is also associated with lysosomal remnants in glial cytoplasmic inclusions of Multiple System Atrophy (MSA) brains (Pountney et al., 2005; Wong et al., 2013; Radford et al., 2014), and SUMO1 is found by confocal microscopy to co-localize with both α-synuclein and HSP90 in the disease inclusions (Wong et al., 2013). While data of immunoprecipitation experiments suggest that HSP90 may be conjugated to SUMO1 in glial cytoplasmic inclusions (Wong et al., 2013), the validation of SUMO1 conjugation to α-synuclein in MSA brains still need further investigation. Overall, although SUMO1 covalent conjugation to α-synuclein in α-synucleinopathy lesions need to be confirmed by improved mass spectrometry techniques, the current findings suggest that different proteins present in α-synucleinopathy lesions, including α-synuclein (Rott et al., 2017) and HSP90 (Uryu et al., 2006), could be SUMOylated and could also play a role in neurodegeneration. Altogether, understanding the regulation of α-synuclein SUMOylation, and possibly other proteins (such as HSP90), may help shed light on the mechanisms that lead to PD and other α-synucleinopathies.
α-Synuclein SUMOylation
α-synuclein contains two SUMOylation consensus lysines (Lys96 and Lys102), which favor conjugation by SUMO1 over SUMO2/3 (Dorval and Fraser, 2006; Table 2). Different SUMO-ligases SUMOylate α-synuclein, including human Polycomb protein 2 (hPc2), PIAS2 and TRIM28 (Krumova et al., 2011; Oh et al., 2011; Rott et al., 2017; Rousseaux et al., 2018), and they tend to lead to predominant monoSUMOylation or multi-monoSUMOylation (Krumova et al., 2011; Rott et al., 2017). Specific α-synuclein SUMOylation by the SUMO-ligases was confirmed using different experimental approaches, including in vitro conjugation assays and immunoprecipitations using cell lines, primary neuronal cultures as well as brain tissues (Krumova et al., 2011; Rott et al., 2017; Rousseaux et al., 2018; Zhu et al., 2018).
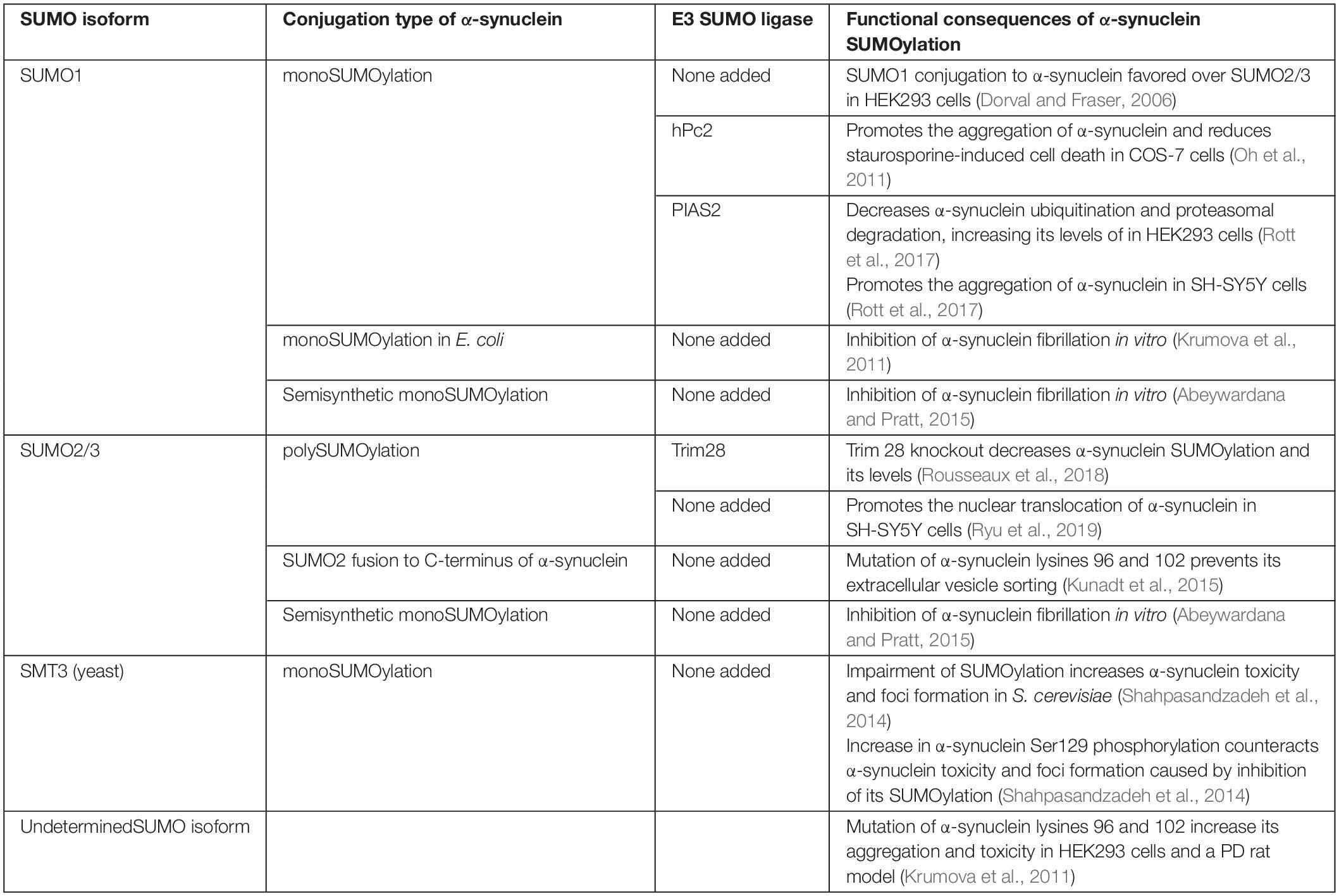
Table 2. α-synuclein SUMOylation.
According to the consensus motif (Ψ-K-x-D/E), α-synuclein lysines 96 and 102 have a very high SUMOylation score (Zhao et al., 2014). Nevertheless, mass spectrometry analysis of purified α-synuclein from SUMO2 transgenic mice identified that several other α-synuclein lysines (11 out of the total 15) are SUMOylated, implying that α-synuclein is also SUMOylated in non-consensus lysines in vivo (Krumova et al., 2011). Accordingly, mutations in Lys96 and Lys102 promoted only a partial decrease in α-synuclein SUMOylation (Krumova et al., 2011; Rott et al., 2017). Altogether, the variety of α-synuclein lysine residues that can be conjugated by SUMO, together with the redundancy of SUMO-ligases and SUMO isoforms that can contribute to α-synuclein SUMOylation, support the idea that differences in SUMOylation patterns could lead to a complex regulation of α-synuclein normal and pathological functions, including degradation, intracellular distribution, protein interactions and aggregation.
SUMOylation Affecting the Subcellular Localization of α-Synuclein
SUMOylation is involved in targeting proteins to different subcellular compartments, in particular to the nucleus (Pichler and Melchior, 2002). Thus, it is conceivable that SUMOylation could play a critical role in the intracellular targeting of α-synuclein. Even though α-synuclein is primarily located at nerve terminals; recent biochemical analysis has consistently identified α-synuclein in the nucleus of neurons (Pinho et al., 2019). Aggregated and Ser129 phosphorylated α-synuclein accumulate in the nucleus of neurons of DLB brains (Pinho et al., 2019). Additional studies have reported that α-synuclein is present in the nucleus of nigral neurons of mice upon injection of paraquat (Goers et al., 2003). Besides, α-synuclein interacts and aggregates with histones (Goers et al., 2003; Kontopoulos et al., 2006; Jiang et al., 2017). Supporting the possible role of SUMOylation in the regulation of α-synuclein translocation to the nucleus, the nuclear SUMO-ligase TRIM28 promotes the accumulation of α-synuclein in the nucleus of neurons (Rousseaux et al., 2016; Figure 1). Another study showed increased nuclear translocation of SUMOylated α-synuclein upon interaction with karyopherin (Ryu et al., 2019).
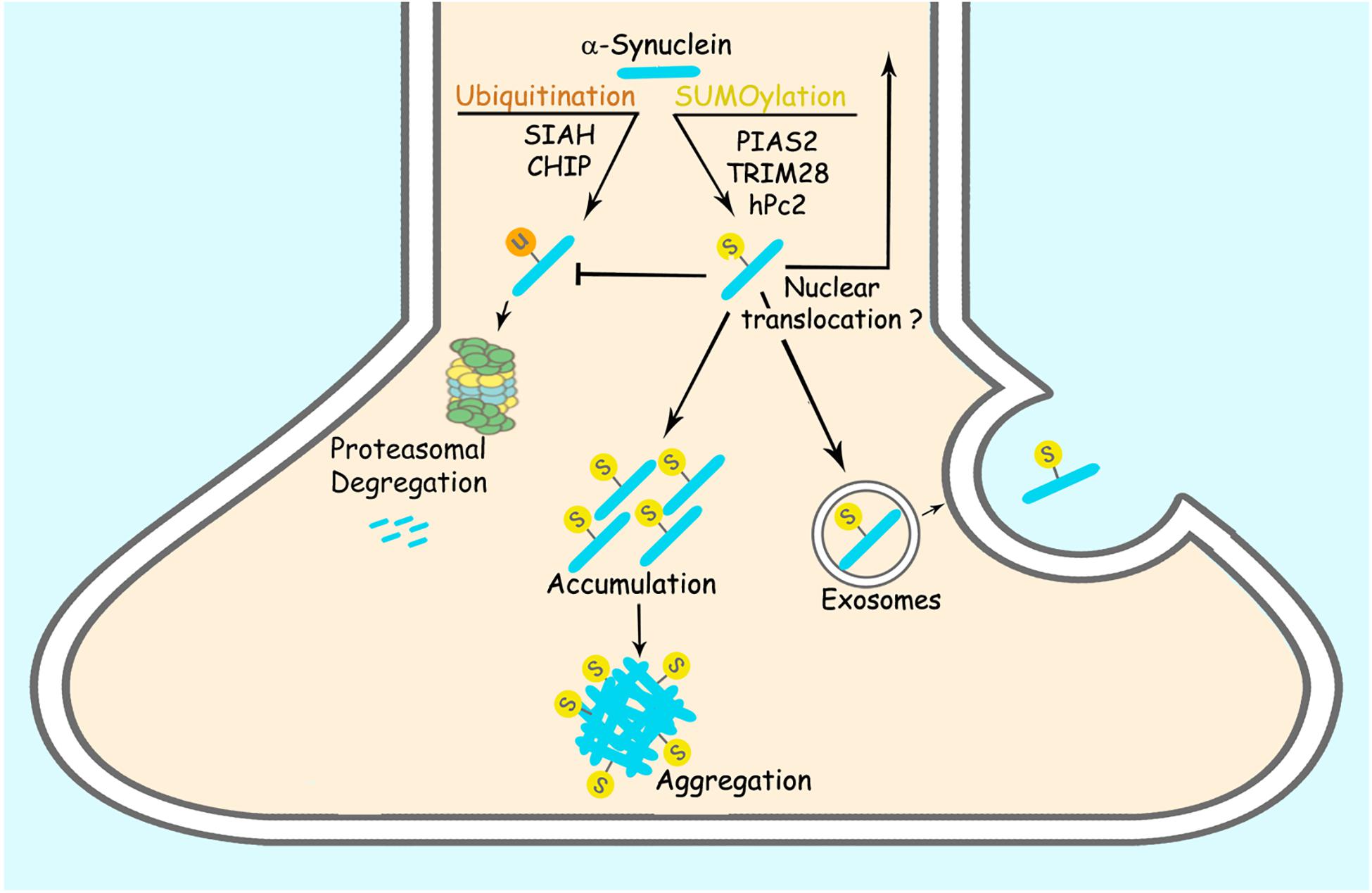
Figure 1. SUMOylation regulates α-synuclein accumulation and aggregation. PIAS2, TRIM28 and hPc2 SUMOylate α-synuclein. α-synuclein SUMOylation counteracts its ubiquitination and proteasomal degradation, leading to increased α-synuclein steady-state levels. SUMOylation of α-synuclein also leads to its increased aggregation per se. SUMOylation of α-synuclein was also suggested to increase its exosomal release and nuclear translocation.
A recent report failed to observe significant SUMOylation of extranuclear proteins using a His6-HA-SUMO1 knock-in mouse model (Daniel et al., 2017). Nevertheless, because this knock-in mouse model has an approximate 30% decreased of Flag-HA-SUMO1 expression (Daniel et al., 2017), it may hinder the detection of protein SUMOylation that occurs outside the nucleus, such as synaptic and mitochondrial proteins (Henley et al., 2018). In this framework, SUMOylation of α-synuclein was also suggested to influence its release from neurons. SUMOylation-deficient α-synuclein was shown to have decreased extracellular release, implying that SUMOylation of α-synuclein may increase its binding to membranes, facilitating, therefore, its release from cells within extracellular vesicles (Kunadt et al., 2015; Figure 1 and Table 2). In addition, the study showed that SUMO2 per se is also targeted to extracellular vesicles, in a process requiring the Endosomal Sorting Complex Required for Transport (ESCRT) complex (Kunadt et al., 2015), suggesting that ESCRT machinery could also be a SUMOylation target and as a consequence, could indirectly regulate cell-to-cell propagation of α-synuclein aggregates or seeds.
Moreover, further supporting a more widespread role of SUMOylation, a recent super-resolution microscopy study found that, although SUMO1, SUMO2/3 and Ubc9 are mainly nuclear in neurons, they also partially co-localize with pre- and post-synaptic markers, such as synaptophysin and PSD95 (Colnaghi et al., 2019). Since α-synuclein is a pre-synaptic protein, the finding of SUMOylation machinery at the pre-synapse further support the importance of α-synuclein SUMOylation. In this framework, it is possible that SUMOylation may also modulate the role of α-synuclein at the presynapse.
The Effect of SUMOylation on α-Synuclein Levels
SUMOylation regulates the stability of a plethora of proteins (Galisson et al., 2011), implying that the degree of SUMOylation could also modulate the levels of α-synuclein in the brain. While SUMOylation can promote the degradation of proteins by priming SUMO-targeted ubiquitin ligases (STUbLs) (Uzunova et al., 2007), SUMOylation can also outcompete and prevent the ubiquitination and proteasomal degradation of different proteins (Desterro et al., 1998; Rott et al., 2017; Table 2). SUMOylation of α-synuclein by PIAS2 and TRIM28 promotes its accumulation (Rott et al., 2017; Rousseaux et al., 2018) while hPc2 causes no change in α-synuclein levels (Oh et al., 2011), indicating that different SUMO-ligases may promote distinct forms of α-synuclein SUMOylation, where some of them would prevent α-synuclein degradation while others would not. In this framework, knockdown of PIAS2 and TRIM28 in HEK293 cells and neurons resulted in decreased levels of α-synuclein that were not ascribed to changes in expression (Rousseaux et al., 2016; Rott et al., 2017). Accordingly, SUMOylation of α-synuclein by PIAS2 competes with and decreases the α-synuclein ubiquitination promoted by both the ubiquitin-ligases SIAH-2 and Nedd4 (Rott et al., 2017; Figure 1). In agreement, PIAS2 knockdown increases SIAH-2-dependent α-synuclein monoubiquitination and proteasomal degradation (Rott et al., 2017). Although TRIM28 knockout mice show decreased levels of α-synuclein, the mechanism by which TRIM28 leads to the accumulation of α-synuclein is still not clear (Rousseaux et al., 2018). Further supporting the role of SUMOylation in counteracting α-synuclein ubiquitination and degradation, pharmacological treatment with the SUMO E1 inhibitor, ginkgolic acid, decreases the steady-state levels of α-synuclein in both HEK293 cells and primary neuronal cultures by promoting its degradation through the proteasome (Rott et al., 2017). In an experimental condition where α-synuclein aggregates were formed by depolarizing SH-SY5Y cells and neurons with KCl, inhibition of SUMOylation by ginkgolic acid stimulated the clearance of α-synuclein aggregates by promoting macroautophagy (Vijayakumaran et al., 2019). Therefore, ginkgolic acid could represent a viable therapeutic strategy to increase proteasomal degradation of ubiquitinated α-synuclein as well as α-synuclein aggregates by macroautophagy stimulation.
SUMOylation also increases the levels of tau, huntingtin, ataxin-3 and SOD1 (Fei et al., 2006; O’Rourke et al., 2013; Zhou et al., 2013; Luo et al., 2014; Ochaba et al., 2016), supporting a broader role of SUMOylation in neurodegenerative diseases. Therefore, dysregulation of SUMOylation may lead to the accumulation of critical proteins involved not only in PD and other α-synucleinopathies but also in additional neurodegenerative diseases, such as Alzheimer’s disease, Huntington’s disease, Spinocerebellar Ataxia-3 and amyotrophic lateral sclerosis.
SUMOylation and α-Synuclein Aggregation
The synthetic fusion of yeast SUMO, encoded by the SMT3 gene, as well as proteins such as maltose-binding protein and NusA, to the N- or C-terminus of recombinant proteins, increase the folding and solubilization of proteins in vitro (Zuo et al., 2005; Marblestone, 2006). Nevertheless, studies using approaches to increase SUMOylation of internal lysine residues of proteins in cell lines and in in vivo models have shown the opposite effect. For instance, SUMOylation of neurodegenerative-related proteins, including tau, huntingtin, SOD1 and α-synuclein, have shown to increase their aggregation (Fei et al., 2006; O’Rourke et al., 2013; Luo et al., 2014; Ochaba et al., 2016; Rott et al., 2017). In addition to differences in the attachment of SUMO isoforms to target proteins (whether to N- or C-terminus or to internal lysines), variations in the SUMO isoforms themselves may play a role in changing the solubility of proteins. For instance, yeast SUMO may have different aggregation properties compared to human SUMO1 and SUMO2/3. Along with this possibility, yeast SUMO shares 47% homology with SUMO1, which in turn is 50% homologous to SUMO2/3 (Melchior, 2000; Marblestone, 2006). In this framework, α-synuclein can be conjugated to either SUMO1 or SUMO2/3, which give rise to mono- or polySUMOylation, respectively (Table 2).
How could we explain the ability of conjugated SUMO to increase the aggregation of disease-causing proteins? One possibility is that proteins involved in neurodegenerative diseases are intrinsically prone to aggregation and that different PTMs with some hydrophobic character would more easily tilt the equilibrium toward their aggregation (O’Rourke et al., 2013; Morimoto et al., 2016). Another possibility is that excessive SUMOylation at multiple lysine residues, due to enhanced SUMOylation machinery upon aging and stress (Tempe et al., 2008; Scurr et al., 2017; Henley et al., 2018), could trigger the aggregation of crucial proteins in different neurodegenerative diseases. Alternatively, non-covalent interaction between SUMOylated disease-causing proteins and adjacent SUMOylated proteins, could work as a molecular “glue” (Matunis et al., 2006), facilitating the formation of aggregates in neurodegenerative diseases.
Regarding α-synuclein, SUMOylation by PIAS2 promotes its aggregation both in vitro and in cell lines (Rott et al., 2017). This aggregation was observed without any additional treatment to the in vitro reactions and cells (Rott et al., 2017), supporting the notion that SUMOylation by PIAS2 per se is enough to promote α-synuclein aggregation (Figure 1). In agreement, α-synuclein PD mutants (A53T, A30P, and E46K) are significantly more SUMOylated than the wild type protein, leading to their robust aggregation (Rott et al., 2017). Accordingly, substantia nigra of PD patients contains increased formic acid- and proteinase K-resistant SUMOylated proteins, as well as SUMOylated α-synuclein (Rott et al., 2017). Stress caused by the accumulation of α-synuclein inclusions may further contribute to the increase of α-synuclein SUMOylation in a positive-feedback loop, as suggested by the formation of SUMO positive aggresome-like α-synuclein inclusions in different cell lines treated with proteasome inhibitors (Kim et al., 2011; Oh et al., 2011; Wong et al., 2013).
On the other hand, in a paradigm where a PDZ binding domain-tagged α-synuclein was co-expressed with PDZ domain-EGFP in HEK293 cells, the aggregation of EGFP (used as a marker for α-synuclein aggregation) increased when lysines 96 and 102 were mutated to arginines (Krumova et al., 2011), suggesting that SUMOylation would decrease the aggregation of α-synuclein. The reason for the discrepancies in the possible role of SUMOylation on α-synuclein aggregation in cell lines is not clear. However, they could be related to differences in the α-synuclein constructs used. For instance, the interaction of α-synuclein with EGFP could facilitate their aggregation, making them more noticeable even in the absence of any method to remove non-aggregated α-synuclein, such as detergent or proteinase K (Rott et al., 2008, 2017). Alternatively, differences of SUMO isoforms and SUMO-ligases could also contribute to the apparent discrepancies of α-synuclein aggregation seen in the different studies. Accordingly, in yeast, impaired SUMOylation by SMT3 results in increased aggregation of α-synuclein (Shahpasandzadeh et al., 2014; Table 2), suggesting that not only the SUMO isoform but also the protein context may also play a role in the aggregation of SUMOylated α-synuclein.
The role of SUMOylation in the formation of α-synuclein fibrils was also investigated. In vitro SUMOylation, either by conjugation or by a semi-synthetic approach, reduce the ability of α-synuclein to form fibrils (Krumova et al., 2011; Abeywardana and Pratt, 2015). The reduction in fibrillation was more pronounced when SUMO was attached to Lys102 compared to when it was attached to Lys96 (Abeywardana and Pratt, 2015). Moreover, SUMO1 was more efficient in reducing fibrillation than SUMO3 (Abeywardana and Pratt, 2015), further supporting the possibility of SUMO isoform-dependent α-synuclein aggregation and fibrillation. Although these findings suggest that SUMOylation may hinder the formation of α-synuclein fibrils, SUMOylation still supports the formation of oligomeric α-synuclein aggregates (Rott et al., 2017), suggesting that SUMOylation could inhibit the conversion of α-synuclein oligomeric into fibrils forms. Because oligomeric α-synuclein has been suggested to be toxic to neurons and non-fibrillar proteinaceous material are prominent at the core of Lewy bodies (Winner et al., 2011; Fusco et al., 2017), inhibition of the transition from oligomeric to fibril forms by SUMOylation could help explain the contribution of SUMO to α-synuclein toxicity in PD.
SUMOylation of α-Synuclein and Toxicity
The aggregation of α-synuclein increases upon conjugation with SUMO1 and α-synuclein PD mutants have increased tendency to be SUMOylated, triggering their prompt and robust aggregation (Rott et al., 2017), supporting the idea that SUMOylation may mediate the accumulation of toxic α-synuclein species. Also, knockdown of TRIM28 increased dopaminergic neuron survival in a transgenic mice model overexpressing α-synuclein (Rousseaux et al., 2016).
On the other hand, α-synuclein SUMOylation by hPc2 increases COS-7 cell survival after exposure to the apoptosis inducer staurosporine (Oh et al., 2011). Also, expression of α-synuclein K96R, K102R mutants in cultured neurons, and rat substantia nigra displayed increased toxicity (Krumova et al., 2011), suggesting that SUMOylation of α-synuclein could also play a protective role depending on the cellular context and SUMO-ligase employed. Nevertheless, because lysine residues are conjugated to several PTMs in addition to SUMOylation, including ubiquitination and glycation (Zee and Garcia, 2012), the use of α-synuclein constructs with mutated lysines are challenging to interpret due to the possible confounding effects with different PTMs. Overall, a better understanding of how SUMOylation may contribute to α-synuclein toxicity will provide essential clues to the mechanisms of neuronal toxicity observed in the disease.
The Interplay Between α-Synuclein SUMOylation and Phosphorylation
In a recent proteomic study, almost ten percent of protein SUMOylation occurred proximal to phosphorylation sites, and several SUMOylation sites were dependent on prior phosphorylation events, implying a significant cross-talk between SUMOylation and phosphorylation (Hendriks et al., 2017). Since phosphorylation at Ser129 is an abundant PTM in Lewy bodies (Fujiwara et al., 2002; Anderson et al., 2006), an interplay between SUMOylation and phosphorylation may also occur in α-synuclein. Supporting this possibility, SUMOylation of α-synuclein in yeast down-regulates Ser129 phosphorylation promoted by CDK5 and PLK2 and increases the clearance of α-synuclein aggregates (Shahpasandzadeh et al., 2014). There are still no studies investigating whether α-synuclein SUMOylation may also influence Ser129 phosphorylation of α-synuclein in mammalian cells. Nevertheless, in an example of their interplay in mammalian cell models relevant to neurodegenerative diseases, SUMOylation of tau induces its hyperphosphorylation at multiple AD-related sites (Luo et al., 2014). In a reciprocating manner, phosphorylation of tau increases its SUMOylation, leading to tau accumulation and aggregation (Luo et al., 2014), suggesting that in a more complex milieu, such as mammalian cells, SUMOylation and phosphorylation could lead to a feed-forward mechanism to increase protein aggregation and pathology. Future studies need to be carried out to investigate if and how α-synuclein SUMOylation and phosphorylation influence each in neuronal models.
Concluding Remarks
Based on previous findings that proteins SUMOylation increases upon stress and that several disease-causing proteins aggregate and become toxic upon SUMOylation, we hypothesize that, among the repertoire of α-synuclein modifications, SUMOylation may represent a PTM that can lead to α-synuclein pathology. Supporting this possibility, several studies have shown that α-synuclein is SUMOylated in different cell models and brain tissues, and that SUMOylation by certain SUMO-ligases and SUMO isoforms leads to α-synuclein accumulation, aggregation and toxicity. Most importantly, SUMOylated α-synuclein is increased in α-synucleinopathy brains. In conclusion, even though studies using improved mass spectrometry techniques are required to allow a more detailed characterization of SUMOylated α-synuclein in brain tissues, the biochemical and cell biological findings indicate that α-synuclein SUMOylation may represent a novel target for better therapeutic intervention.
Author Contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Israel Academy of Sciences, The Allen and Jewel Prince Center for Neurodegenerative Disorders of the Brain, Dears Foundation for Neurodegenerative Diseases, Isidore C. and Penny W. Myers Fund for Parkinson’s Research, and the Technion Research Funds to SE.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
PD, Parkinson’s disease; SIAH, Seven in Absentia homolog.
References
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Abeywardana, T., and Pratt, M. R. (2015). Extent of inhibition of alpha-synuclein aggregation in vitro by SUMOylation is conjugation site-and SUMO isoform-selective. Biochemistry 54, 959–961. doi: 10.1021/bi501512m
Agbor, T. A., Cheong, A., Comerford, K. M., Scholz, C. C., Bruning, U., Clarke, A., et al. (2011). Small ubiquitin-related modifier (SUMO)-1 promotes glycolysis in hypoxia. J. Biol. Chem. 286, 4718–4726. doi: 10.1074/jbc.M110.115931
Alam, P., Bousset, L., Melki, R., and Otzen, D. E. (2019). alpha-synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J. Neurochem. 150, 522–534. doi: 10.1111/jnc.14808
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Appella, E., and Anderson, C. W. (2001). Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268, 2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x
Ariga, H., Takahashi-Niki, K., Kato, I., Maita, H., Niki, T., and Iguchi-Ariga, S. M. (2013). Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell. Longev. 2013:683920. doi: 10.1155/2013/683920
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Baczyk, D., Audette, M. C., Drewlo, S., Levytska, K., and Kingdom, J. C. (2017). SUMO-4: a novel functional candidate in the human placental protein SUMOylation machinery. PLoS One 12:e0178056. doi: 10.1371/journal.pone.0178056
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Bhattacharjee, P., Ohrfelt, A., Lashley, T., Blennow, K., Brinkmalm, A., and Zetterberg, H. (2019). Mass spectrometric analysis of Lewy body-enriched alpha-synuclein in Parkinson’s disease. J. Prot. Res. 18, 2109–2120. doi: 10.1021/acs.jproteome.8b00982
Bodles, A. M., Guthrie, D. J., Greer, B., and Irvine, G. B. (2001). Identification of the region of non-Abeta component (NAC) of Alzheimer’s disease amyloid responsible for its aggregation and toxicity. J. Neurochem. 78, 384–395. doi: 10.1046/j.1471-4159.2001.00408.x
Bohren, K. M., Nadkarni, V., Song, J. H., Gabbay, K. H., and Owerbach, D. (2004). A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 279, 27233–27238. doi: 10.1074/jbc.M402273200
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Book, A., Guella, I., Candido, T., Brice, A., Hattori, N., Jeon, B., et al. (2018). A meta-analysis of alpha-synuclein multiplication in familial Parkinsonism. Front. Neurol. 9:1021. doi: 10.3389/fneur.2018.01021
Brahmachari, S., Ge, P., Lee, S. H., Kim, D., Karuppagounder, S. S., Kumar, M., et al. (2016). Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J. Clin. Invest. 126, 2970–2988. doi: 10.1172/JCI85456
Breuker, K. (2018). Mass spectrometry: raw protein from the top down. Nat. Chem. 10, 114–116. doi: 10.1038/nchem.2936
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Cabin, D. E., Shimazu, K., Murphy, D., Cole, N. B., Gottschalk, W., McIlwain, K. L., et al. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 22, 8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002
Cao, X., Wang, H., Wang, Z., Wang, Q., Zhang, S., Deng, Y., et al. (2017). In vivo imaging reveals mitophagy independence in the maintenance of axonal mitochondria during normal aging. Aging Cell 16, 1180–1190. doi: 10.1111/acel.12654
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. doi: 10.1016/S0140-6736(04)17103-1
Chau, K. Y., Ching, H. L., Schapira, A. H., and Cooper, J. M. (2009). Relationship between alpha synuclein phosphorylation, proteasomal inhibition and cell death: relevance to Parkinson’s disease pathogenesis. J. Neurochem. 110, 1005–1013. doi: 10.1111/j.1471-4159.2009.06191.x
Chen, L., Periquet, M., Wang, X., Negro, A., McLean, P. J., Hyman, B. T., et al. (2009). Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest. 119, 3257–3265. doi: 10.1172/JCI39088
Chen, L., Wei, Y., Wang, X., and He, R. (2010). Ribosylation rapidly induces alpha-synuclein to form highly cytotoxic molten globules of advanced glycation end products. PLoS One 5:e9052. doi: 10.1371/journal.pone.0009052
Chen, R., Park, H. A., Mnatsakanyan, N., Niu, Y., Licznerski, P., Wu, J., et al. (2019). Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 10:469. doi: 10.1038/s41419-019-1679-x
Colnaghi, L., Russo, L., Natale, C., Restelli, E., Cagnotto, A., Salmona, M., et al. (2019). Super resolution microscopy of SUMO proteins in neurons. Front. Cell. Neurosci. 13:486. doi: 10.3389/fncel.2019.00486
Conway, K. A., Harper, J. D., and Lansbury, P. T. (1998). Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320. doi: 10.1038/3311
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., and Lansbury, P. T. Jr. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Acad. Natl. Sci. U.S.A. 97, 571–576. doi: 10.1073/pnas.97.2.571
Crowther, R. A., Jakes, R., Spillantini, M. G., and Goedert, M. (1998). Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 436, 309–312. doi: 10.1016/s0014-5793(98)01146-6
Daniel, J. A., Cooper, B. H., Palvimo, J. J., Zhang, F. P., Brose, N., and Tirard, M. (2017). Analysis of SUMO1-conjugation at synapses. eLife 6:e26338. doi: 10.7554/eLife.26338
Danielson, S. R., Held, J. M., Schilling, B., Oo, M., Gibson, B. W., and Andersen, J. K. (2009). Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 81, 7823–7828. doi: 10.1021/ac901176t
Dapic, I., Baljeu-Neuman, L., Uwugiaren, N., Kers, J., Goodlett, D. R., and Corthals, G. L. (2019). Proteome analysis of tissues by mass spectrometry. Mass Spectrom. Rev. 38, 403–441. doi: 10.1002/mas.21598
Daturpalli, S., Waudby, C. A., Meehan, S., and Jackson, S. E. (2013). Hsp90 inhibits alpha-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 425, 4614–4628. doi: 10.1016/j.jmb.2013.08.006
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
de Oliveira, R. M., Vicente Miranda, H., Francelle, L., Pinho, R., Szego, E. M., Martinho, R., et al. (2017). The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 15:e2000374. doi: 10.1371/journal.pbio.2000374
Dehay, B., Martinez-Vicente, M., Caldwell, G. A., Caldwell, K. A., Yue, Z., Cookson, M. R., et al. (2013). Lysosomal impairment in Parkinson’s disease. Mov. Disord. 28, 725–732. doi: 10.1002/mds.25462
Desterro, J. M., Rodriguez, M. S., and Hay, R. T. (1998). SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 2, 233–239. doi: 10.1016/s1097-2765(00)80133-1
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Dorsey, E. R., Sherer, T., Okun, M. S., and Bloem, B. R. (2018). The emerging evidence of the Parkinson pandemic. J. Parkinsons Dis. 8, S3–S8. doi: 10.3233/JPD-181474
Dorval, V., and Fraser, P. E. (2006). Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 281, 9919–9924. doi: 10.1074/jbc.M510127200
Ebrahimi-Fakhari, D., Cantuti-Castelvetri, I., Fan, Z., Rockenstein, E., Masliah, E., Hyman, B. T., et al. (2011). Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 31, 14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011
Engelender, S., and Isacson, O. (2017). The threshold theory for Parkinson’s disease. Trends Neurosci. 40, 4–14. doi: 10.1016/j.tins.2016.10.008
Enserink, J. M. (2015). Sumo and the cellular stress response. Cell Div. 10:4. doi: 10.1186/s13008-015-0010-1
Fei, E., Jia, N., Yan, M., Ying, Z., Sun, Q., Wang, H., et al. (2006). SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 347, 406–412. doi: 10.1016/j.bbrc.2006.06.092
Figueroa-Romero, C., Iniguez-Lluhi, J. A., Stadler, J., Chang, C. R., Arnoult, D., Keller, P. J., et al. (2009). SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 23, 3917–3927. doi: 10.1096/fj.09-136630
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Fusco, G., Chen, S. W., Williamson, P. T. F., Cascella, R., Perni, M., Jarvis, J. A., et al. (2017). Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443. doi: 10.1126/science.aan6160
Galisson, F., Mahrouche, L., Courcelles, M., Bonneil, E., Meloche, S., Chelbi-Alix, M. K., et al. (2011). A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol. Cell. Proteomics 10:M110004796. doi: 10.1074/mcp.M110.004796
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Goers, J., Manning-Bog, A. B., McCormack, A. L., Millett, I. S., Doniach, S., Di Monte, D. A., et al. (2003). Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 42, 8465–8471. doi: 10.1021/bi0341152
Guo, C., Hildick, K. L., Luo, J., Dearden, L., Wilkinson, K. A., and Henley, J. M. (2013). SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 32, 1514–1528. doi: 10.1038/emboj.2013.65
Harder, Z., Zunino, R., and McBride, H. (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 14, 340–345. doi: 10.1016/j.cub.2004.02.004
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M., et al. (2002). Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Hebron, M. L., Lonskaya, I., and Moussa, C. E. (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 22, 3315–3328. doi: 10.1093/hmg/ddt192
Hendriks, I. A., Lyon, D., Young, C., Jensen, L. J., Vertegaal, A. C., and Nielsen, M. L. (2017). Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 24, 325–336. doi: 10.1038/nsmb.3366
Henley, J. M., Carmichael, R. E., and Wilkinson, K. A. (2018). Extranuclear SUMOylation in Neurons. Trends Neurosci. 41, 198–210. doi: 10.1016/j.tins.2018.02.004
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hettiarachchi, N. T., Parker, A., Dallas, M. L., Pennington, K., Hung, C. C., Pearson, H. A., et al. (2009). alpha-synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 111, 1192–1201. doi: 10.1111/j.1471-4159.2009.06411.x
Hickey, C. M., Wilson, N. R., and Hochstrasser, M. (2012). Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 13, 755–766. doi: 10.1038/nrm3478
Hodara, R., Norris, E. H., Giasson, B. I., Mishizen-Eberz, A. J., Lynch, D. R., Lee, V. M., et al. (2004). Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 279, 47746–47753. doi: 10.1074/jbc.M408906200
Hong, Y., Rogers, R., Matunis, M. J., Mayhew, C. N., Goodson, M. L., Park-Sarge, O. K., et al. (2001). Regulation of heat shock transcription factor 1 by stress-induced SUMO-1 modification. J. Biol. Chem. 276, 40263–40267. doi: 10.1074/jbc.M104714200
Huang, T. T., Wuerzberger-Davis, S. M., Wu, Z. H., and Miyamoto, S. (2003). Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 115, 565–576. doi: 10.1016/s0092-8674(03)00895-x
Iwatsubo, T., Yamaguchi, H., Fujimuro, M., Yokosawa, H., Ihara, Y., Trojanowski, J. Q., et al. (1996). Purification and characterization of Lewy bodies from the brains of patients with diffuse Lewy body disease. Am. J. Pathol. 148, 1517–1529.
Jang, H., Boltz, D., Sturm-Ramirez, K., Shepherd, K. R., Jiao, Y., Webster, R., et al. (2009). Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 14063–14068. doi: 10.1073/pnas.0900096106
Jiang, P., Gan, M., Yen, S. H., McLean, P. J., and Dickson, D. W. (2017). Histones facilitate alpha-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 133, 547–558. doi: 10.1007/s00401-016-1660-z
Jo, E., McLaurin, J., Yip, C. M., St George-Hyslop, P., and Fraser, P. E. (2000). alpha-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334. doi: 10.1074/jbc.M004345200
Kahle, P. J., Neumann, M., Ozmen, L., Muller, V., Jacobsen, H., Schindzielorz, A., et al. (2000). Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000
Kahle, P. J., Neumann, M., Ozmen, L., Muller, V., Jacobsen, H., Spooren, W., et al. (2002). Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 3, 583–588. doi: 10.1093/embo-reports/kvf109
Kahyo, T., Nishida, T., and Yasuda, H. (2001). Involvement of PIAS1 in the SUMOylation of tumor suppressor p53. Mol. Cell 8, 713–718. doi: 10.1016/s1097-2765(01)00349-5
Kalia, L. V., Kalia, S. K., McLean, P. J., Lozano, A. M., and Lang, A. E. (2013). alpha-synuclein oligomers and clinical implications for Parkinson disease. Ann. Neurol. 73, 155–169. doi: 10.1002/ana.23746
Karpinar, D. P., Balija, M. B., Kugler, S., Opazo, F., Rezaei-Ghaleh, N., Wender, N., et al. (2009). Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 28, 3256–3268. doi: 10.1038/emboj.2009.257
Kellie, J. F., Higgs, R. E., Ryder, J. W., Major, A., Beach, T. G., Adler, C. H., et al. (2014). Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 4:5797. doi: 10.1038/srep05797
Kim, Y. M., Jang, W. H., Quezado, M. M., Oh, Y., Chung, K. C., Junn, E., et al. (2011). Proteasome inhibition induces alpha-synuclein SUMOylation and aggregate formation. J. Neurol. Sci. 307, 157–161. doi: 10.1016/j.jns.2011.04.015
Knipscheer, P., van Dijk, W. J., Olsen, J. V., Mann, M., and Sixma, T. K. (2007). Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 26, 2797–2807. doi: 10.1038/sj.emboj.7601711
Kontopoulos, E., Parvin, J. D., and Feany, M. B. (2006). alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. doi: 10.1093/hmg/ddl243
Kramer, P. A., Duan, J., Qian, W. J., and Marcinek, D. J. (2015). The measurement of reversible redox dependent post-translational modifications and their regulation of mitochondrial and skeletal muscle function. Front. Physiol. 6:347. doi: 10.3389/fphys.2015.00347
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). SUMOylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. doi: 10.1083/jcb.201010117
Kunadt, M., Eckermann, K., Stuendl, A., Gong, J., Russo, B., Strauss, K., et al. (2015). Extracellular vesicle sorting of alpha-synuclein is regulated by SUMOylation. Acta Neuropathol. 129, 695–713. doi: 10.1007/s00401-015-1408-1
Kuwahara, T., Tonegawa, R., Ito, G., Mitani, S., and Iwatsubo, T. (2012). Phosphorylation of alpha-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 287, 7098–7109. doi: 10.1074/jbc.M111.237131
Lautenschager, J., and Kaminski Schierle, G. S. (2019). Mitochondrial degradation of amyloidogenic proteins - A new perspective for neurodegenerative diseases. Prog. Neurobiol. 181:101660. doi: 10.1016/j.pneurobio.2019.101660
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honore, A., Rozas, N., et al. (2013). G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Liang, Y. C., Lee, C. C., Yao, Y. L., Lai, C. C., Schmitz, M. L., and Yang, W. M. (2016). SUMO5, a Novel Poly-SUMO Isoform. Regulates PML NUCLEAR Bodies. Sci. Rep. 6:26509. doi: 10.1038/srep26509
Liu, C. W., Giasson, B. I., Lewis, K. A., Lee, V. M., Demartino, G. N., and Thomas, P. J. (2005). A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J. Biol. Chem. 280, 22670–22678. doi: 10.1074/jbc.M501508200
Logan, T., Bendor, J., Toupin, C., Thorn, K., and Edwards, R. H. (2017). alpha-synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 20, 681–689. doi: 10.1038/nn.4529
Lopez-Cuina, M., Guerin, P. A., Canron, M. H., Delamarre, A., Dehay, B., Bezard, E., et al. (2020). Nilotinib fails to prevent synucleinopathy and cell loss in a mouse model of multiple system atrophy. Mov. Disord. doi: 10.1002/mds.28034 [Epub ahead of print].
Lu, Y., Prudent, M., Fauvet, B., Lashuel, H. A., and Girault, H. H. (2011). Phosphorylation of alpha-synuclein at Y125 and S129 alters its metal binding properties: implications for understanding the role of alpha-synuclein in the pathogenesis of Parkinson’s disease and related disorders. ACS Chem. Neurosci. 2, 667–675. doi: 10.1021/cn200074d
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luo, H. B., Xia, Y. Y., Shu, X. J., Liu, Z. C., Feng, Y., Liu, X. H., et al. (2014). SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 111, 16586–16591. doi: 10.1073/pnas.1417548111
Mahul-Mellier, A. L., Fauvet, B., Gysbers, A., Dikiy, I., Oueslati, A., Georgeon, S., et al. (2014). c-Abl phosphorylates alpha-synuclein and regulates its degradation: implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 23, 2858–2879. doi: 10.1093/hmg/ddt674
Marblestone, J. G. (2006). Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 15, 182–189. doi: 10.1110/ps.051812706
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
Matunis, M. J., Coutavas, E., and Blobel, G. (1996). A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 135, 1457–1470. doi: 10.1083/jcb.135.6.1457
Matunis, M. J., Zhang, X. D., and Ellis, N. A. (2006). SUMO: the glue that binds. Dev. Cell 11, 596–597. doi: 10.1016/j.devcel.2006.10.011
Maurizi, C. P. (1987). Was a neurovirulent influenza virus the cause of amyotrophic lateral sclerosis and parkinsonism-dementia on Guam? Med. Hypotheses 23, 325–326. doi: 10.1016/0306-9877(87)90024-7
Mbefo, M. K., Paleologou, K. E., Boucharaba, A., Oueslati, A., Schell, H., Fournier, M., et al. (2010). Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822. doi: 10.1074/jbc.M109.081950
McLean, P. J., Kawamata, H., Ribich, S., and Hyman, B. T. (2000). Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 275, 8812–8816. doi: 10.1074/jbc.275.12.8812
Melchior, F. (2000). SUMO–nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol. 16, 591–626. doi: 10.1146/annurev.cellbio.16.1.591
Miller, A. J., Levy, C., Davis, I. J., Razin, E., and Fisher, D. E. (2005). SUMOylation of MITF and its related family members TFE3 and TFEB. J. Biol. Chem. 280, 146–155. doi: 10.1074/jbc.M411757200
Morimoto, D., Walinda, E., Fukada, H., Sugase, K., and Shirakawa, M. (2016). Ubiquitylation directly induces fold destabilization of proteins. Sci. Rep. 6:39453. doi: 10.1038/srep39453
Muller, S., Hoege, C., Pyrowolakis, G., and Jentsch, S. (2001). SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2, 202–210. doi: 10.1038/35056591
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000
Murray, I. V., Giasson, B. I., Quinn, S. M., Koppaka, V., Axelsen, P. H., Ischiropoulos, H., et al. (2003). Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42, 8530–8540. doi: 10.1021/bi027363r
Nakamura, T., Yamashita, H., Takahashi, T., and Nakamura, S. (2001). Activated Fyn phosphorylates alpha-synuclein at tyrosine residue 125. Biochem. Biophys. Res. Commun. 280, 1085–1092. doi: 10.1006/bbrc.2000.4253
Negro, A., Brunati, A. M., Donella-Deana, A., Massimino, M. L., and Pinna, L. A. (2002). Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J. 16, 210–212. doi: 10.1096/fj.01-0517fje
Newell, K. L., Boyer, P., Gomez-Tortosa, E., Hobbs, W., Hedley-Whyte, E. T., Vonsattel, J. P., et al. (1999). alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J. Neuropathol. Exp. Neurol. 58, 1263–1268. doi: 10.1097/00005072-199912000-00007
Nørregaard Jensen, O. (2004). Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 8, 33–41. doi: 10.1016/j.cbpa.2003.12.009
Ochaba, J., Monteys, A. M., O’Rourke, J. G., Reidling, J. C., Steffan, J. S., Davidson, B. L., et al. (2016). PIAS1 regulates mutant huntingtin accumulation and Huntington’s disease-associated phenotypes in vivo. Neuron 90, 507–520. doi: 10.1016/j.neuron.2016.03.016
Oertel, W., and Schulz, J. B. (2016). Current and experimental treatments of Parkinson disease: a guide for neuroscientists. J. Neurochem. 139(Suppl. 1), 325–337. doi: 10.1111/jnc.13750
Oh, Y., Kim, Y. M., Mouradian, M. M., and Chung, K. C. (2011). Human Polycomb protein 2 promotes alpha-synuclein aggregate formation through covalent SUMOylation. Brain Res. 1381, 78–89. doi: 10.1016/j.brainres.2011.01.039
Ohrfelt, A., Zetterberg, H., Andersson, K., Persson, R., Secic, D., Brinkmalm, G., et al. (2011). Identification of novel alpha-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem. Res. 36, 2029–2042. doi: 10.1007/s11064-011-0527-x
Okochi, M., Walter, J., Koyama, A., Nakajo, S., Baba, M., Iwatsubo, T., et al. (2000). Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 275, 390–397. doi: 10.1074/jbc.275.1.390
O’Rourke, J. G., Gareau, J. R., Ochaba, J., Song, W., Raskó, T., Reverter, D., et al. (2013). SUMO-2 and PIAS1 modulate insoluble mutant Huntingtin protein accumulation. Cell Rep. 4, 362–375. doi: 10.1016/j.celrep.2013.06.034
Ostrerova-Golts, N., Petrucelli, L., Hardy, J., Lee, J. M., Farer, M., and Wolozin, B. (2000). The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 20, 6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000
Oueslati, A., Paleologou, K. E., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2012). Mimicking phosphorylation at serine 87 inhibits the aggregation of human alpha-synuclein and protects against its toxicity in a rat model of Parkinson’s disease. J. Neurosci. 32, 1536–1544. doi: 10.1523/JNEUROSCI.3784-11.2012
Outeiro, T. F., Putcha, P., Tetzlaff, J. E., Spoelgen, R., Koker, M., Carvalho, F., et al. (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 3:e1867. doi: 10.1371/journal.pone.0001867
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30, 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
Park, S. M., Jung, H. Y., Chung, K. C., Rhim, H., Park, J. H., and Kim, J. (2002). Stress-induced aggregation profiles of GST-alpha-synuclein fusion proteins: role of the C-terminal acidic tail of alpha-synuclein in protein thermosolubility and stability. Biochemistry 41, 4137–4146. doi: 10.1021/bi015961k
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging. 35, e2181–e2185. doi: 10.1016/j.neurobiolaging.2014.03.024
Paxinou, E., Chen, Q., Weisse, M., Giasson, B. I., Norris, E. H., Rueter, S. M., et al. (2001). Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 21, 8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007
Pichler, A., and Melchior, F. (2002). Ubiquitin-related modifier SUMO1 and nucleocytoplasmic transport. Traffic 3, 381–387. doi: 10.1034/j.1600-0854.2002.30601.x
Pinho, R., Paiva, I., Jercic, K. G., Fonseca-Ornelas, L., Gerhardt, E., Fahlbusch, C., et al. (2019). Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 28, 31–50. doi: 10.1093/hmg/ddy326
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Poole, A. C., Thomas, R. E., Andrews, L. A., McBride, H. M., Whitworth, A. J., and Pallanck, L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 105, 1638–1643. doi: 10.1073/pnas.0709336105
Pountney, D. L., Chegini, F., Shen, X., Blumbergs, P. C., and Gai, W. P. (2005). SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci. Lett. 381, 74–79. doi: 10.1016/j.neulet.2005.02.013
Pronin, A. N., Morris, A. J., Surguchov, A., and Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522. doi: 10.1074/jbc.M003542200
Putcha, P., Danzer, K. M., Kranich, L. R., Scott, A., Silinski, M., Mabbett, S., et al. (2010). Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J. Pharmacol Exp. Ther. 332, 849–857. doi: 10.1124/jpet.109.158436
Radford, R., Wong, M., and Pountney, D. L. (2014). “Neurodegenerative aspects of Multiple System Atrophy,” in Handbook of Neurotoxicity, ed. R. Kostrzewa (New York, NY: Springer).
Ren, R. J., Dammer, E. B., Wang, G., Seyfried, N. T., and Levey, A. I. (2014). Proteomics of protein post-translational modifications implicated in neurodegeneration. Transl. Neurodegener. 3:23. doi: 10.1186/2047-9158-3-23
Ribeiro, C. S., Carneiro, K., Ross, C. A., Menezes, J. R., and Engelender, S. (2002). Synphilin-1 is developmentally localized to synaptic terminals, and its association with synaptic vesicles is modulated by alpha-synuclein. J. Biol. Chem. 277, 23927–23933. doi: 10.1074/jbc.M201115200
Riedel, M., Goldbaum, O., Wille, M., and Richter-Landsberg, C. (2011). Membrane lipid modification by docosahexaenoic acid (DHA) promotes the formation of alpha-synuclein inclusion bodies immunopositive for SUMO-1 in oligodendroglial cells after oxidative stress. J. Mol. Neurosci. 43, 290–302. doi: 10.1007/s12031-010-9439-5
Rodriguez, M. S., Dargemont, C., and Hay, R. T. (2001). SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659. doi: 10.1074/jbc.M009476200
Rott, R., Szargel, R., Haskin, J., Bandopadhyay, R., Lees, A. J., Shani, V., et al. (2011). alpha-synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 18666–18671. doi: 10.1073/pnas.1105725108
Rott, R., Szargel, R., Haskin, J., Shani, V., Shainskaya, A., Manov, I., et al. (2008). Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328. doi: 10.1074/jbc.M704809200
Rott, R., Szargel, R., Shani, V., Hamza, H., Savyon, M., Abd Elghani, F., et al. (2017). SUMOylation and ubiquitination reciprocally regulate alpha-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 13176–13181. doi: 10.1073/pnas.1704351114
Rousseaux, M. W., de Haro, M., Lasagna-Reeves, C. A., De Maio, A., Park, J., Jafar-Nejad, P., et al. (2016). TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. eLife 5:e19809. doi: 10.7554/eLife.19809
Rousseaux, M. W., Revelli, J. P., Vazquez-Velez, G. E., Kim, J. Y., Craigen, E., Gonzales, K., et al. (2018). Depleting Trim28 in adult mice is well tolerated and reduces levels of alpha-synuclein and tau. eLife 7:e36768. doi: 10.7554/eLife.36768
Rytinki, M. M., and Palvimo, J. J. (2009). SUMOylation attenuates the function of PGC-1alpha. J. Biol. Chem. 284, 26184–26193. doi: 10.1074/jbc.M109.038943
Ryu, S., Baek, I., and Liew, H. (2019). Sumoylated α-synuclein translocates into the nucleus by karyopherin α6. Mol. Cell. Toxicol. 15, 103–109. doi: 10.1007/s13273-019-0012-1
Saitoh, H., and Hinchey, J. (2000). Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275, 6252–6258. doi: 10.1074/jbc.275.9.6252
Schapira, A. H., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1:1269. doi: 10.1016/s0140-6736(89)92366-0
Scurr, L. L., Haferkamp, S., and Rizos, H. (2017). The role of SUMOylation in senescence. Adv. Exp. Med. Biol. 963, 215–226. doi: 10.1007/978-3-319-50044-7_13
Sha, Z., Blyszcz, T., Gonzalez-Prieto, R., Vertegaal, A. C. O., and Goldberg, A. L. (2019). Inhibiting ubiquitination causes an accumulation of SUMOylated newly synthesized nuclear proteins at PML bodies. J. Biol. Chem. 294, 15218–15234. doi: 10.1074/jbc.RA119.009147
Shahmoradian, S. H., Lewis, A. J., Genoud, C., Hench, J., Moors, T. E., Navarro, P. P., et al. (2019). Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. doi: 10.1038/s41593-019-0423-2
Shahpasandzadeh, H., Popova, B., Kleinknecht, A., Fraser, P. E., Outeiro, T. F., and Braus, G. H. (2014). Interplay between SUMOylation and phosphorylation for protection against alpha-synuclein inclusions. J. Biol. Chem. 289, 31224–31240. doi: 10.1074/jbc.M114.559237
Shendelman, S., Jonason, A., Martinat, C., Leete, T., and Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2:e362. doi: 10.1371/journal.pbio.0020362
Shin, Y., Klucken, J., Patterson, C., Hyman, B. T., and McLean, P. J. (2005). The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 280, 23727–23734. doi: 10.1074/jbc.M503326200
Shinbo, Y., Niki, T., Taira, T., Ooe, H., Takahashi-Niki, K., Maita, C., et al. (2006). Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 13, 96–108. doi: 10.1038/sj.cdd.4401704
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Smith, W. W., Margolis, R. L., Li, X., Troncoso, J. C., Lee, M. K., Dawson, V. L., et al. (2005). alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 25, 5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R. E., et al. (2001). Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 10, 919–926. doi: 10.1093/hmg/10.9.919
Tanner, C. M., Kamel, F., Ross, G. W., Hoppin, J. A., Goldman, S. M., Korell, M., et al. (2011). Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 119, 866–872. doi: 10.1289/ehp.1002839
Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., et al. (2001). Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276, 35368–35374. doi: 10.1074/jbc.M104214200
Tempe, D., Piechaczyk, M., and Bossis, G. (2008). SUMO under stress. Biochem. Soc. Trans. 36, 874–878. doi: 10.1042/BST0360874
Terada, M., Suzuki, G., Nonaka, T., Kametani, F., Tamaoka, A., and Hasegawa, M. (2018). The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 293, 13910–13920. doi: 10.1074/jbc.RA118.001862
Theillet, F. X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., et al. (2016). Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Tofaris, G. K., Garcia Reitbock, P., Humby, T., Lambourne, S. L., O’Connell, M., Ghetti, B., et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006
Tofaris, G. K., Kim, H. T., Hourez, R., Jung, J. W., Kim, K. P., and Goldberg, A. L. (2011). Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 17004–17009. doi: 10.1073/pnas.1109356108
Tofaris, G. K., Razzaq, A., Ghetti, B., Lilley, K. S., and Spillantini, M. G. (2003). Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411. doi: 10.1074/jbc.M308041200
Uryu, K., Richter-Landsberg, C., Welch, W., Sun, E., Goldbaum, O., Norris, E. H., et al. (2006). Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am. J. Pathol. 168, 947–961. doi: 10.2353/ajpath.2006.050770
Uzunova, K., Gottsche, K., Miteva, M., Weisshaar, S. R., Glanemann, C., Schnellhardt, M., et al. (2007). Ubiquitin-dependent Proteolytic Control of SUMO Conjugates. J. Biol. Chem. 282, 34167–34175. doi: 10.1074/jbc.M706505200
Vicente Miranda, H., Szego, E. M., Oliveira, L. M. A., Breda, C., Darendelioglu, E., de Oliveira, R. M., et al. (2017). Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 140, 1399–1419. doi: 10.1093/brain/awx056
Vijayakumaran, S., Nakamura, Y., Henley, J. M., and Pountney, D. L. (2019). Ginkgolic acid promotes autophagy-dependent clearance of intracellular alpha-synuclein aggregates. Mol. Cell. Neurosci. 101:103416. doi: 10.1016/j.mcn.2019.103416
Visanji, N. P., Wislet-Gendebien, S., Oschipok, L. W., Zhang, G., Aubert, I., Fraser, P. E., et al. (2011). Effect of Ser-129 phosphorylation on interaction of alpha-synuclein with synaptic and cellular membranes. J. Biol. Chem. 286, 35863–35873. doi: 10.1074/jbc.M111.253450
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Weetman, J., Wong, M. B., Sharry, S., Rcom-H’cheo-Gauthier, A., Gai, W. P., Meedeniya, A., et al. (2013). Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson’s disease. Neurosci. Lett. 544, 119–124. doi: 10.1016/j.neulet.2013.03.057
Weinreb, P. H., Zhen, W., Poon, A. W., Conway, K. A., and Lansbury, P. T. Jr. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. doi: 10.1021/bi961799n
Wilkinson, K. A., and Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145. doi: 10.1042/BJ20100158
Wilson, V. G. (2017). Introduction to SUMOylation. Adv. Exp. Med. Biol. 963, 1–12. doi: 10.1007/978-3-319-50044-7_1
Winner, B., Jappelli, R., Maji, S. K., Desplats, P. A., Boyer, L., Aigner, S., et al. (2011). In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199. doi: 10.1073/pnas.1100976108
Withers, G. S., George, J. M., Banker, G. A., and Clayton, D. F. (1997). Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 99, 87–94. doi: 10.1016/s0165-3806(96)00210-6
Wolmarans, A., Kwantes, A., and LaPointe, P. (2019). A novel method for site-specific chemical SUMOylation: SUMOylation of Hsp90 modulates co-chaperone binding in vitro. Biol. Chem. 400, 487–500. doi: 10.1515/hsz-2018-0251
Wong, M. B., Goodwin, J., Norazit, A., Meedeniya, A. C., Richter-Landsberg, C., Gai, W. P., et al. (2013). SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox. Res. 23, 1–21. doi: 10.1007/s12640-012-9358-z
Youle, R. J., and Karbowski, M. (2005). Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 6, 657–663. doi: 10.1038/nrm1697
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zee, B. M., and Garcia, B. A. (2012). Discovery of lysine post-translational modifications through mass spectrometric detection. Essays Biochem. 52, 147–163. doi: 10.1042/bse0520147
Zhao, Q., Xie, Y., Zheng, Y., Jiang, S., Liu, W., Mu, W., et al. (2014). GPS-SUMO: a tool for the prediction of SUMOylation sites and SUMO-interaction motifs. Nucleic Acids Res. 42, W325–W330. doi: 10.1093/nar/gku383
Zhao, X. (2018). SUMO-mediated regulation of nuclear functions and signaling processes. Mol. Cell 71, 409–418. doi: 10.1016/j.molcel.2018.07.027
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., et al. (2010). PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Science Transl. Med. 2:52ra73. doi: 10.1126/scitranslmed.3001059
Zhou, J., Broe, M., Huang, Y., Anderson, J. P., Gai, W. P., Milward, E. A., et al. (2011). Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol. 121, 695–704. doi: 10.1007/s00401-011-0815-1
Zhou, W., Ryan, J. J., and Zhou, H. (2004). Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein SUMOylation by cellular stresses. J. Biol. Chem. 279, 32262–32268. doi: 10.1074/jbc.M404173200
Zhou, Y. F., Liao, S. S., Luo, Y. Y., Tang, J. G., Wang, J. L., Lei, L. F., et al. (2013). SUMO-1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity. PLoS One 8:e54214. doi: 10.1371/journal.pone.0054214
Zhu, L. N., Qiao, H. H., Chen, L., Sun, L. P., Hui, J. L., Lian, Y. L., et al. (2018). SUMOylation of alpha-synuclein influences on alpha-synuclein aggregation induced by methamphetamine. Front. Cell. Neurosci. 12:262. doi: 10.3389/fncel.2018.00262
Keywords: Parkinson’s disease, α-synuclein, post-translational modifications, SUMOylation, ubiquitination, protein aggregation
Citation: Savyon M and Engelender S (2020) SUMOylation in α-Synuclein Homeostasis and Pathology. Front. Aging Neurosci. 12:167. doi: 10.3389/fnagi.2020.00167
Received: 20 January 2020; Accepted: 14 May 2020;
Published: 25 June 2020.
Edited by:
Ralf J. Braun, Danube Private University, AustriaReviewed by:
Paul Fraser, University of Toronto, CanadaDean Louis Pountney, Griffith University, Australia
Kevin Wilkinson, University of Bristol, United Kingdom
Copyright © 2020 Savyon and Engelender. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Engelender, c2ltb25lQHRlY2huaW9uLmFjLmls