
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci., 04 December 2019
Sec. Alzheimer's Disease and Related Dementias
Volume 11 - 2019 | https://doi.org/10.3389/fnagi.2019.00327
This article is part of the Research TopicUntangling the Role of Tau in Physiology and PathologyView all 23 articles
 Pascual Sánchez-Juan1,2*
Pascual Sánchez-Juan1,2* Sonia Moreno1,3
Sonia Moreno1,3 Itziar de Rojas1,3
Itziar de Rojas1,3 Isabel Hernández1,3
Isabel Hernández1,3 Sergi Valero1,3
Sergi Valero1,3 Montse Alegret1,3Laura Montrreal1Pablo García González1,3Carmen Lage1,2Sara López-García1,2Eloy Rodrííguez-Rodríguez1,2
Montse Alegret1,3Laura Montrreal1Pablo García González1,3Carmen Lage1,2Sara López-García1,2Eloy Rodrííguez-Rodríguez1,2 Adelina Orellana1
Adelina Orellana1 Lluís Tárraga1,3
Lluís Tárraga1,3 Mercè Boada1,3
Mercè Boada1,3 Agustín Ruiz1,3 on behalf of GR@ACE Study Group and DEGESCO Consortium
Agustín Ruiz1,3 on behalf of GR@ACE Study Group and DEGESCO ConsortiumAn ancestral inversion of 900 kb on chromosome 17q21, which includes the microtubule-associated protein tau (MAPT) gene, defines two haplotype clades in Caucasians (H1 and H2). The H1 haplotype has been linked inconsistently with AD. In a previous study, we showed that an SNP tagging this haplotype (rs1800547) was associated with AD risk in a large population from the Dementia Genetics Spanish Consortium (DEGESCO) including 4435 cases and 6147 controls. The association was mainly driven by individuals that were non-carriers of the APOE ε4 allele. Our aim was to replicate our previous findings in an independent sample of 4124 AD cases and 3290 controls from Spain (GR@ACE project) and to analyze the effect of the H1 sub-haplotype structure on the risk of AD. The H1 haplotype was associated with AD risk (OR = 1.12; p = 0.0025). Stratification analysis showed that this association was mainly driven by the APOE ε4 non-carriers (OR = 1.15; p = 0.0022). Pooled analysis of both Spanish datasets (n = 17,996) showed that the highest AD risk related to the MAPT H1/H2 haplotype was in those individuals that were the oldest [third tertile (>77 years)] and did not carry APOE ε4 allele (p = 0.001). We did not find a significant association between H1 sub-haplotypes and AD. H1c was nominally associated but lost statistical significance after adjusting by population sub-structure. Our results are consistent with the hypothesis that genetic variants linked to the MAPT H1/H2 are tracking a genuine risk allele for AD. The fact that this association is stronger in APOE ε4 non-carriers partially explains previous controversial results and might be related to a slower alternative causal pathway less dependent on brain amyloid load.
Dementia is related to many underlying pathologies, with Alzheimer’s disease (AD) being the most common. AD is pathologically defined by the deposits of two proteins: tau which accumulates intracellularly and β-amyloid that accumulates extracellularly and within the walls of the blood vessels of the central nervous system. Dementia of AD-type is a complex entity with a common clinical syndrome that is likely to be reached by different routes influenced by genetic and environmental factors. This complexity is likely to have contributed to the constant failures of AD clinical trials. In particular, therapies based on the amyloid cascade hypothesis have not demonstrated any disease-modifying effect, despite some of these attempts have been proved to be effective in permanently removing brain β-amyloid plaques (Nicoll et al., 2019). This has led the pharmaceutical industry to focus on other therapeutic targets such as the tau protein (Corvol and Buée, 2019).
Neurofibrillary tangles composed of truncated and hyperphosphorylated tau proteins are hallmarks of AD pathology (Goedert, 2004). Tau protein plays an essential role in the central nervous system by promoting microtubule assembly and stability in neuronal cells. Emerging evidence supports that tau function is essential for normal synaptic mechanisms and it may be dysregulated in AD potentially through interaction with genetic risk factors in an Aβ-dependent or Aβ-independent manner (Dourlen et al., 2019). Additionally, tau has been proposed to spread through the brain from neuron to neuron by a “prion-like” mechanism (Clavaguera et al., 2015).
Tau protein is encoded by the MAPT gene (MAPT: OMIM: ∗157140), located at chromosome 17q21-22. There are two common MAPT extended haplotypes in Caucasians resulting from an ancestral inversion: H1 and H2. The H1 haplotype has been linked with familial and sporadic neurodegenerative disorders like progressive supranuclear palsy (PSP) (Conrad et al., 1997; Baker et al., 1999; Higgins et al., 2000; Pastor et al., 2004, 2016), corticobasal degeneration (CBD) (Houlden et al., 2001), Frontotemporal Dementia (FTD) (Verpillat et al., 2002), and Parkinson’s disease (PD) (Martin et al., 2001; Simón-Sánchez et al., 2009; Pastor et al., 2016). Genome-wide association studies (GWAS) have shown that the MAPT H1 haplotype is associated with CBD, PSP, and FTD (Yokoyama et al., 2017).
The H1 haplotype is further divided into sub-haplotypes, of those H1c has been associated with several neurodegenerative diseases (Myers et al., 2005; Heckman et al., 2019). H1c has been associated with higher levels of tau in plasma and CSF (Myers et al., 2007; Chen et al., 2017) and inconsistently with AD (Myers et al., 2005).
Although recent genetic studies show that several AD GWAS-associated genes, especially BIN1, are potentially involved in tau pathways (Dourlen et al., 2019), MAPT itself has not emerged until recently as a locus associated with AD. The IGAP consortium found a significant association between an SNP tagging MAPT H1 haplotype (rs2732703) and AD in subjects not carrying APOE ε4, however, the authors concluded that their conditional analysis pointed out that MAPT was probably not the causal gene (Jun et al., 2016).
Microtubule-associated protein tau H1/H2 haplotype frequency varies according to populations, with H2 frequency being maximum in the Mediterranean region and decreasing gradually as we move away from that area (Donnelly et al., 2010). These differences might contribute to explain controversial results among studies, as genetic stratification in genetically heterogeneous populations might constitute an important confounder. It is therefore essential to study these variants in large genetically homogeneous populations. In a previous study, we showed that H1 MAPT haplotype was strongly associated with risk of PSP, PD, and AD in 4435 cases and 6147 controls from Spain (Pastor et al., 2016). Therefore, it seems that MAPT H1/H2 haplotypes might play a relevant role within the genetic architecture of several neurodegenerative pathologies in our country. It is worth mention that the prevalence of the haplotype H2 in our control sample was one of the highest reported worldwide (29%) (Pastor et al., 2016).
In the present study, we aimed to replicate our previous findings in an independent sample. To do that, we assessed the association between the AD risk and the MAPT H1/H2 haplotype and H1 sub-haplotypes in 4,124 AD cases and 3,290 controls from Spain (GR@ACE/DEGESCO project).
A detailed description of the methods and population of the GR@ACE study has been published elsewhere (Moreno-Grau et al., 2019)1.
The GR@ACE study comprises 4,120 AD cases and 3,289 control individuals. Cases were recruited from Fundació ACE, Institut Català de Neurociències Aplicades (Barcelona, Spain). Diagnoses were established by a neurology working-group according to the DSM-IV criteria for dementia and to the National Institute on Aging and Alzheimer’s Association’s (NIA-AA) 2011 guidelines for defining AD. In the present study, we considered AD cases, dementia individuals diagnosed with probable or possible AD at any moment of their clinical course.
Control individuals were recruited from three centers: Fundació ACE (Barcelona, Spain), Valme University Hospital (Seville, Spain) and the Spanish National DNA Bank Carlos III (University of Salamanca, Spain)2. Written informed consent was obtained from all participants. The Ethics and Scientific Committees have approved this research protocol (Acta 25/2016, Ethics Committee, Hospital Clinic I Provincial de Barcelona, Barcelona, Spain).
Participants were genotyped using the Axiom 815K Spanish Biobank array (Thermo Fisher). Genotyping was performed in the Spanish National Center for Genotyping (CeGEN, Santiago de Compostela, Spain).
We removed samples with genotype call rates below 97%, excess heterozygosity, duplicates, samples genetically related to other individuals in the cohort or sample mix-up (PIHAT > 0.1875). If a sex discrepancy was detected, the sample was removed unless the discrepancy was safely resolved. To detect population outliers of non-European ancestry (>6 SD from European population mean), principal component analysis (PCA) was conducted using SMARTPCA from EIGENSOFT 6.1.4.
We removed variants with a call rate < 95% or that grossly deviated from Hardy–Weinberg equilibrium in controls (P-value ≤ 1 × 10–4), markers with a different missing rate between case and control (P-value < 5 × 10–4 for the difference) or minor allele frequency (MAF) below 0.01. Imputation was carried out using Haplotype reference consortium (HRC) panel in Michigan Imputation servers3. Only common markers (MAF > 0.01) with a high imputation quality (R2 > 0.30) were selected to conduct downstream association analyses.
Allelic and genotypic frequencies were compared using χ2 statistics. Adjusted analyses were performed using multiple logistic regression. We used rs1800547 to tag MAPT H2 haplotype. Additionally, we used six tagging variants (rs1467967, rs242557, rs3785883, rs2471738, rs8070723, rs7521), to construct most common MAPT H1 sub-haplotypes as previously described (Pittman et al., 2005; Allen et al., 2014). To control for population sub-structure, results were co-variated by the main four principal components detected in this population (Moreno-Grau et al., 2019). All analyses were performed in PLINK 1.7.
We included 3290 controls with a mean age of 54.3 ± 14.4 years, and 48.9% of females, and 4124 AD cases with a mean age of 79.0 ± 7.5 years, 69.6% of females. No gross deviation from Hardy Weinberg equilibrium was found in controls for any of the MAPT studied variants (Table 1).
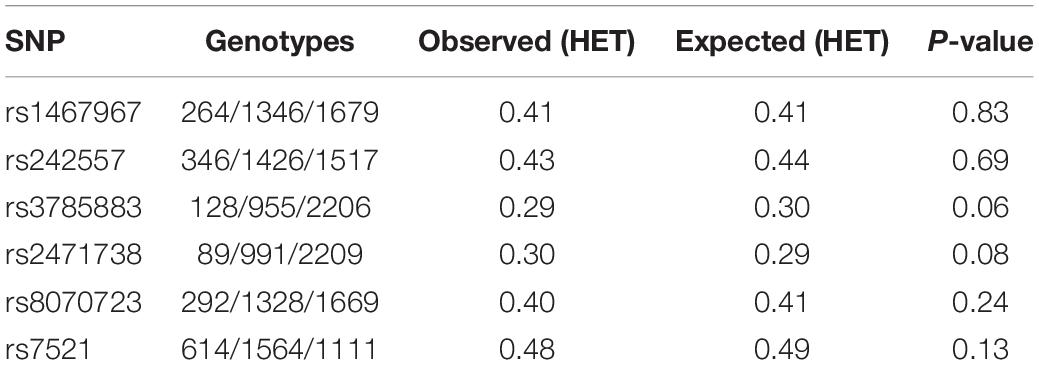
Table 1. Hardy–Weinberg equilibrium test in controls.
Table 2 shows the allelic and genotypic frequency distribution of the SNPrs1800547 tagging the H1/H2 haplotype. We found a statistically significant overrepresentation of the MAPT H1 haplotype, present in 73.3% of AD compared to 71.1% of controls (p = 0.00025). When we stratified the sample by APOE ε4 status, the association of the H1 haplotype was driven by non-carriers of APOE ε4 (p = 0.0022) (Table 2). The association followed exactly the same pattern as our previous study (Pastor et al., 2016). Pooling both Spanish population confirmed that MAPT H1 was significantly more common in AD compared to controls (73.5 versus 70.7% respectively; p = 1.0 × 10–5), and this association was predominantly due to the APOE ε4 non-carriers (p = 8.0 × 10–5) (Table 2).
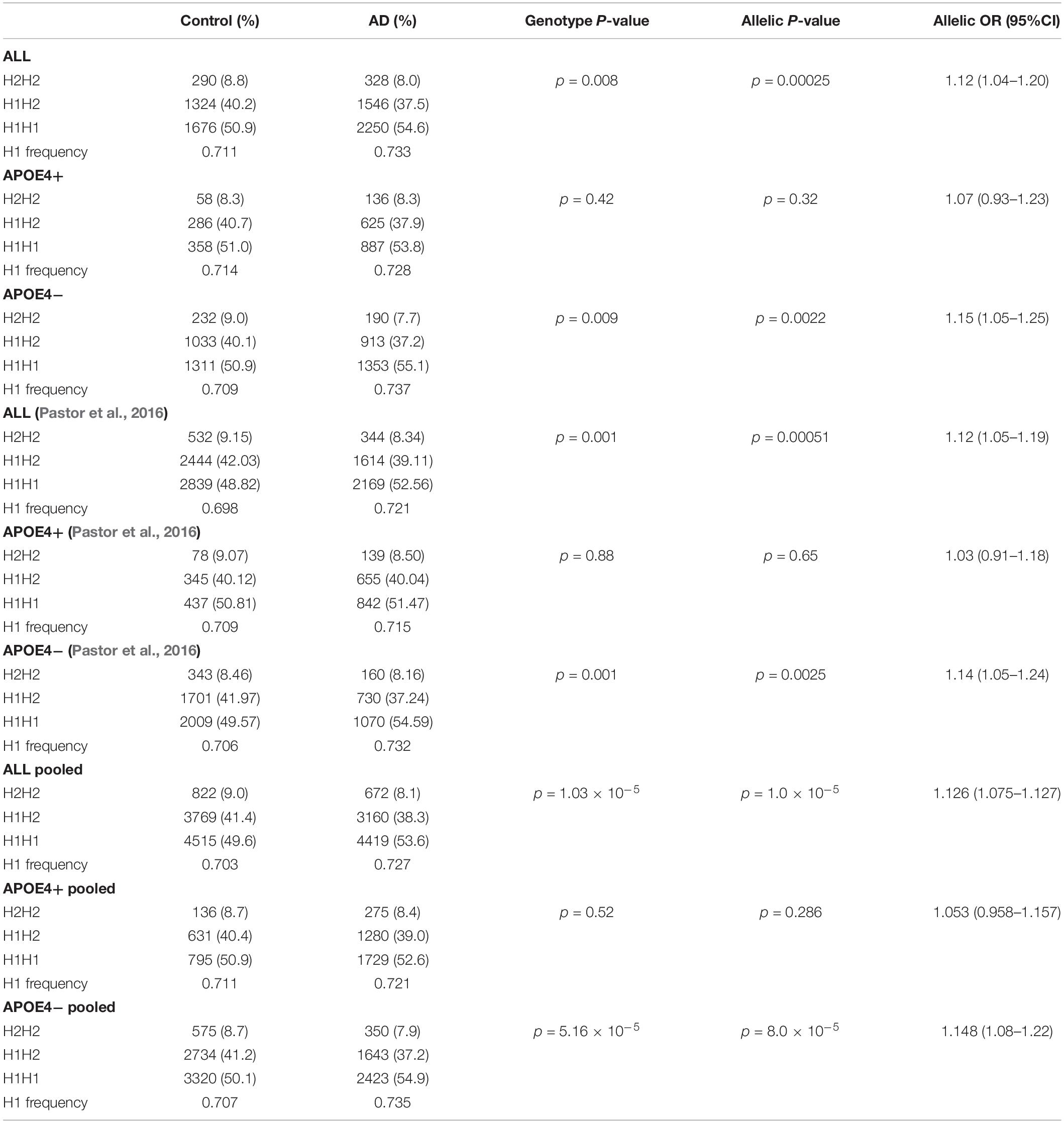
Table 2. Microtubule-associated protein tau H1/H2 haplotypes and AD risk.
Table 3 shows the sub-haplotypes of MAPT in cases and controls. In addition to the protective effect of H2, only H1c was statistically significantly associated with AD. However, when we adjusted by the four main genetic components H1c was not statistically significant. After stratifying by APOE ε4 these results did not change substantially, and in addition to H2, only two rare sub-haplotypes (H1u and H1v) were nominally associated with AD. After adjustment, none of the associations survived multiple comparisons correction (Table 4).
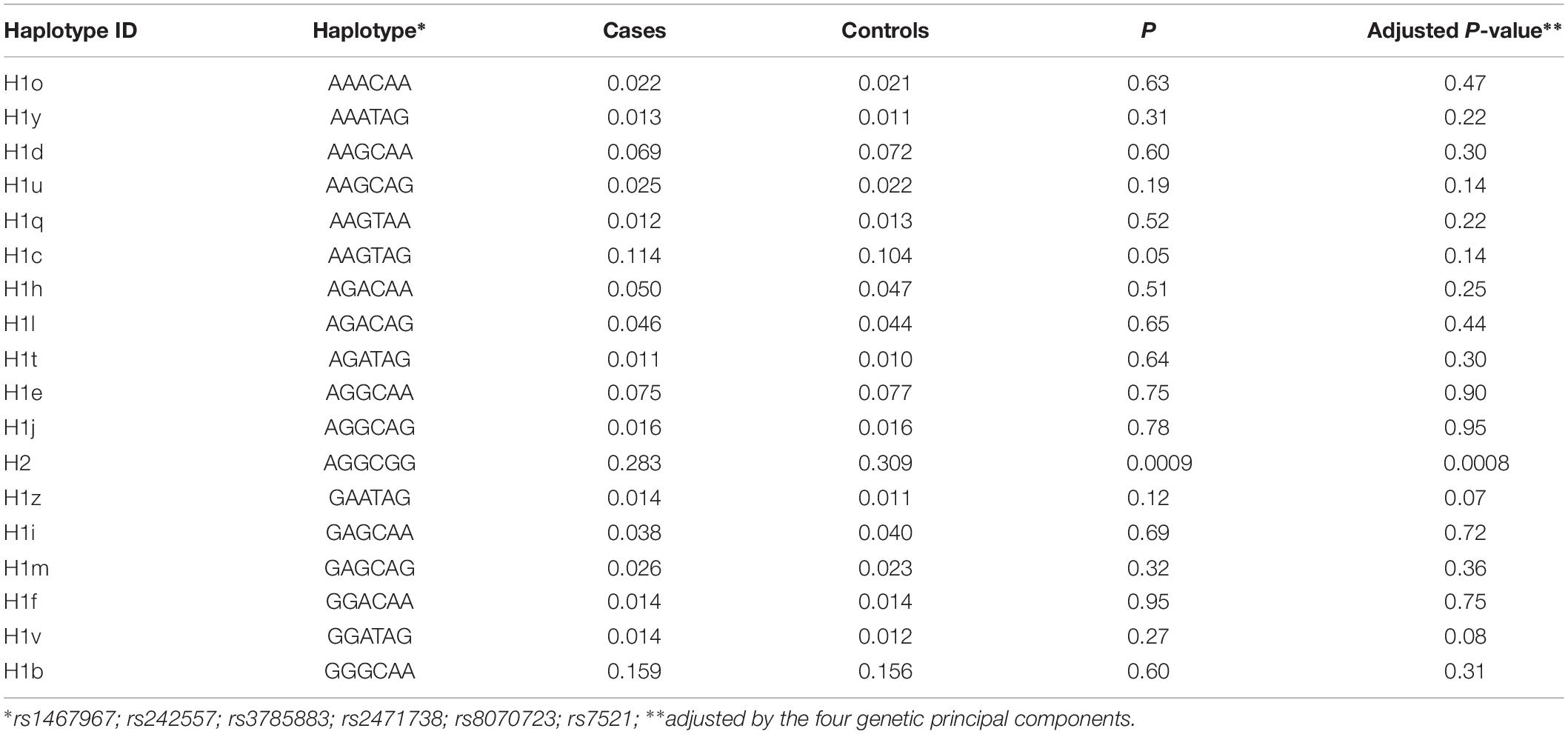
Table 3. Microtubule-associated protein tau sub-haplotypes.
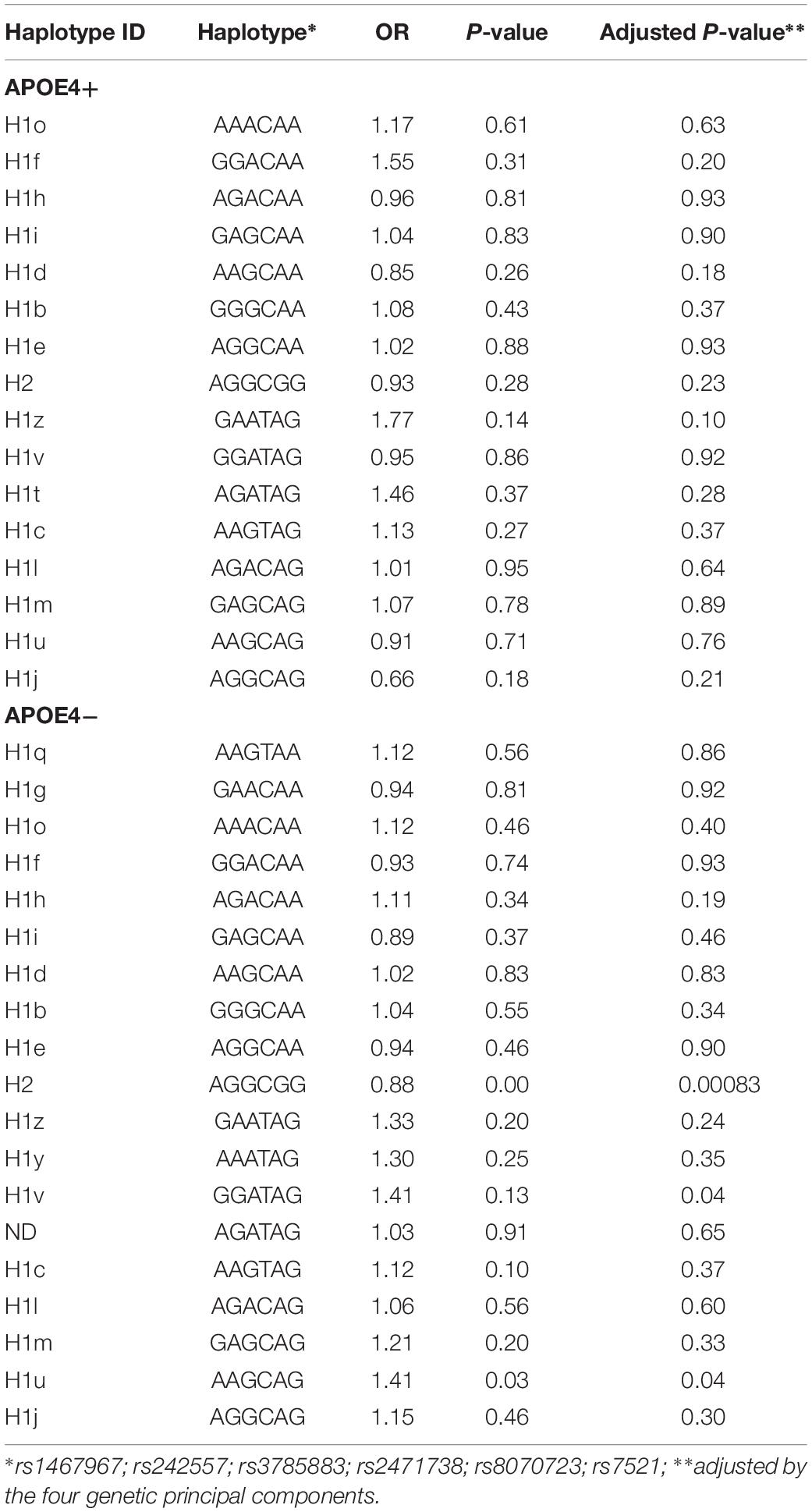
Table 4. Microtubule-associated protein tau sub-haplotypes stratified by APOE ε4 status.
Figure 1 shows the genotypic distribution of the SNP rs1800547 (MAPT H1/H2) stratified by APOE ε4 across age tertiles for the pooled population. In the available entire sample of 15,522 individuals, we appreciated that within the APOE ε4 non-carriers the association between MAPT H1/H2 and AD increased with age, and it was stronger in the oldest individuals.
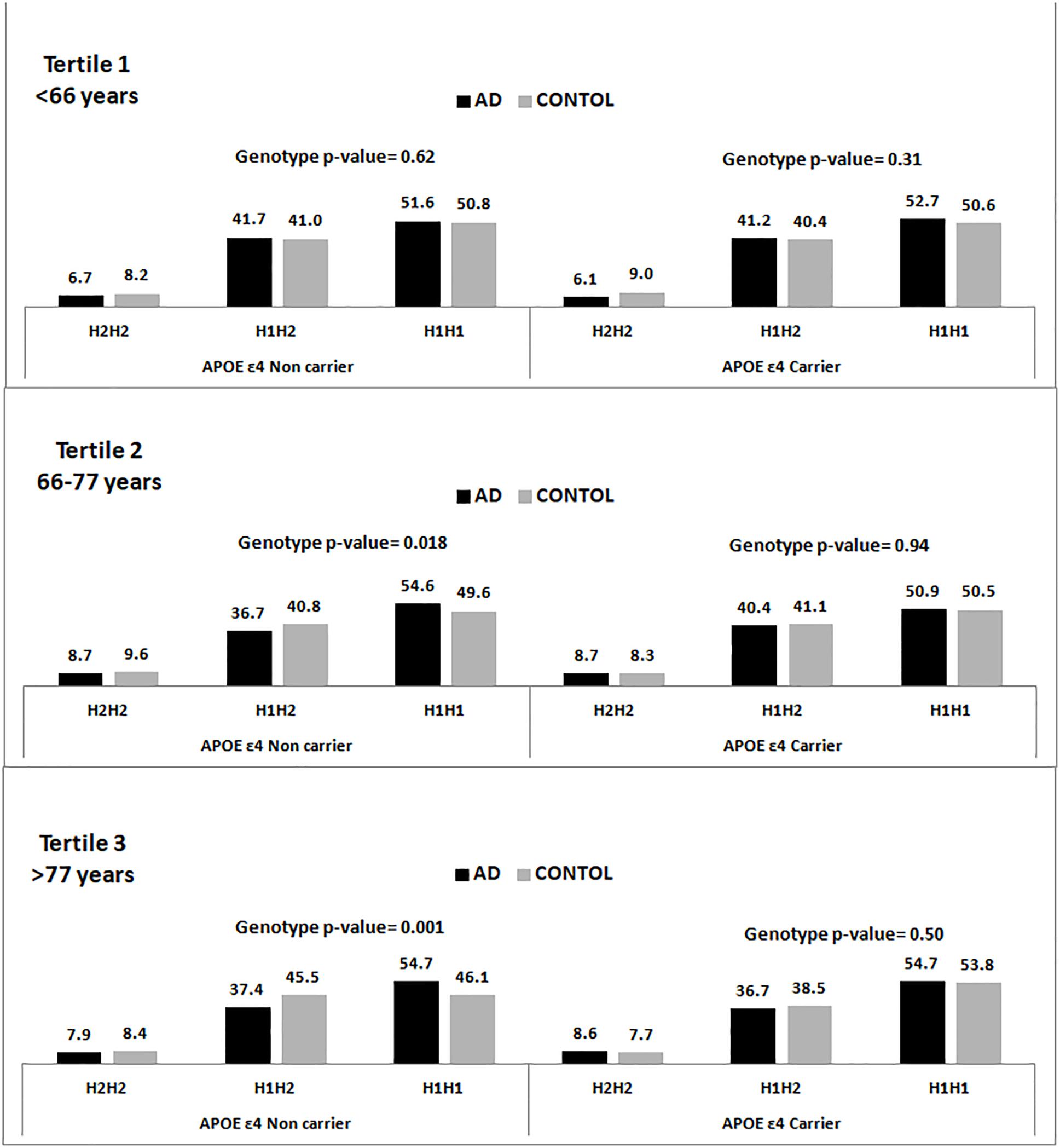
Figure 1. Genotype frequency distribution of the rs1800547 SNP tagging MAPT H1/H2 haplotype stratified by APOE ε4 status and age tertiles.
Our data, from a large and homogeneous single country population, shows that individuals carrying the H1 MAPT haplotype are at higher risk to develop AD dementia. The association is predominantly present in the APOE ε4 non-carriers and it is stronger in the eldest. These results replicate our previous findings (Pastor et al., 2016) and are concordant with the IGAP study (Jun et al., 2016) showing an association of another SNP tagging H1 (rs2732703) with AD only in the APOE ε4 non-carriers.
Our pooled analysis, including 15522 individuals, strongly support an etiological role of the MAPT region in AD. This association has been difficult to replicate, and results of previous studies assessing MAPT H1/H2 haplotype as a risk factor for AD have been controversial, although some of them were considerably under-powered (Russ et al., 2001; Mukherjee et al., 2007; Babić Leko et al., 2018). A robust statistical association only emerged in large sample studies and meta-GWAS, after stratifying by APOE ε4 (Jun et al., 2016; Pastor et al., 2016). An alternative interpretation of our results would be that the causal variant could be in linkage disequilibrium with the MAPT H1 haplotype but outside the MAPT gene, as other authors have suggested (Jun et al., 2016). A less likely explanation might be contamination of non-AD tauopathies in APOE ε4 non-carriers.
There are several factors that might be related to our findings. Taken together, our two studies, comprise one of the largest single-country population assessing MAPT H1/H2 and AD risk to date. This is of special value, as the inversion haplotype frequency has been shown to differ significantly across populations, and it is estimated to be 20% in Europeans, 6% in Africans, and less than 1% in East Asians (Holzer et al., 2004). This ethnic variability might cause population sub-structure biasing the results in countries with a high degree of population admixture. This is likely to be less problematic in our study as our sample come from a single country, and we have tested population sub-structure in Spain which does not represent a substantial problem for common genetic variants analyses (Moreno-Grau et al., 2019). Additionally, in our sub-haplotype analysis, we controlled for population sub-structure by adjusting for genetic principal components. It is worth mentioning that, despite it is commonly reported that the inversion is found at a frequency of around 20% throughout Europe, it shows a great range of frequencies within Europe (from 5 to 37.5%). The H2 haplotype, which identifies the inversion, is most frequent around the Mediterranean decreasing outward in all directions (Donnelly et al., 2010). Our controls presented the inversion in nearly 30% of individuals, this high frequency of the H2 haplotype has increased our power to detect the association compared to other populations in which this variant is less prevalent.
A potential limitation of our study is the age difference between the cases and the controls, which are significantly younger. However, this could only jeopardize the validity of our findings in the case of a survival bias. But to our knowledge, the MAPT H1/H2 haplotype has not been associated with mortality. The most likely consequence of our age unbalance is a decrease in our power to detect the association, as some of the controls carrying the H1 haplotype may still develop AD in the future. It is therefore likely that we are underestimating the true association between the H1/H2 haplotype and AD.
Microtubule-associated protein tau H1 has been associated with many neurodegenerative diseases: PSP, PD, CBD, FTD, and AD. It has been reported that the MAPT H1 is more efficient at driving gene expression than the H2 haplotype (Kwok et al., 2004). This has been shown to be particularly true with the H1c sub-haplotype (Myers et al., 2007). However, the H1 sub-haplotype association with AD is controversial, and H1c findings have been difficult to replicate (Myers et al., 2005; Abraham et al., 2009; Allen et al., 2014). It is likely that population stratification might have played a role. In our data H1c was nominally associated with AD, however, the statistical significance was lost after adjusting by the principal components supporting the notion that no H1sub-haplotype is specifically increasing AD-risk, and stratification by APOE ε4 did not change substantially these results.
Our results add further evidence for an etiological role of MAPT gene variants in clinical AD, supporting the role of the APOE ε4 allele as a modulator of this association. As seen in our previous study in population from Spain (Pastor et al., 2016) and by the international consortium IGAP (Jun et al., 2016), this association is significantly stronger in APOE ε4 non-carriers. Recent studies with tau and amyloid PET supports a view of AD as a tauopathy driven by amyloid, suggesting that tau pathology would appear in middle temporal lobe earlier than amyloid deposits, but the co-occurrence of both would be needed for tau pathology to expand beyond the temporal lobes (Schöll et al., 2016). This is also coherent with the new findings of AD meta-GWAS which shows the involvement of both amyloid and tau pathways (Dourlen et al., 2019; Kunkle et al., 2019). Therefore, it seems that tau and amyloid deposits might follow, at least initially, independent trajectories up to the point when they reach a threshold in which β-amyloid might accelerate tau pathology. A recent single case publication showing that a patient with a presenilin 1 mutation was resistant to cognitive impairment, likely due to a homozygous mutation in APOE, supports the hypothesis that APOE might play an important role in this tau pathology acceleration (Arboleda-Velasquez et al., 2019). On the other hand, APOE status is associated to prevalence of brain amyloid pathology, as shown by a large PET and CSF study in non-demented population that found that APOE ε4 carriers had two to three times higher prevalence than non-carriers (Jansen et al., 2015).
We hypothesize that the MAPT H1 variant might increase the risk of tau pathology which might be related to different amyloid thresholds to disparate tau pathology. We speculate that the association is only significant in the APOE ε4 non-carriers because in the carriers the amyloid would mask the H1 MAPT effect, while in the non-carriers the “tau etiologic pathway” would play a more relevant role and might be able to increase AD risk with a lower amyloid involvement. It is likely that this phenomenon might take longer to develop which might explain why this association is not significant in the youngest patients and stronger in the third tertile. This hypothesis could be tested studying the trajectory of individuals classified according to APOE ε4 status and MAPT H1/H2 haplotype in prospective cohorts with sequential amyloid and tau PET assessments.
Our study highlights the complexity of AD and suggests the existence of different pathogenic routes influenced by the genetic background. To look for successful therapeutic strategies it will be very important to take into account this mechanistic diversity and to combat specifically the pathology demonstrated on each afflicted individual.
Research Center and Memory Clinic, Fundació ACE, Institut Català de Neurociències Aplicades, Universitat Internacional de Catalunya, Barcelona, Spain: C. Abdelnour, N. Aguilera, E. Alarcon, M. Alegret, M. Boada, M. Buendia, P. Cañabate, I. de Rojas, S. Diego, A. Espinosa, A. Gailhajenet, P. García González, S. Gil, M. Guitart, I. Hernández, M. Ibarria, A. Lafuente, E. Martín, A. Mauleón, G. Monté-Rubio, L. Montrreal, S. Moreno-Grau, M. Moreno, A. Orellana, G. Ortega, A. Pancho, E. Pelejà, A. Pérez-Cordon, S. Preckler, O. Rodríguez-Gómez, M. Rosende-Roca, A. Ruiz, S. Ruiz, A. Sanabria, M. A. Santos-Santos, O. Sotolongo-Grau, L. Tárraga, S. Valero, and L. Vargas. Center for Networked Biomedical Research on Neurodegenerative Diseases, National Institute of Health Carlos III, Ministry of Economy and Competitiveness, Madrid, Spain: C. Abdelnour, M. Alegret, M. Boada, P. Cañabate, A. Espinosa, I. Hernández, S. Moreno-Grau, G. Ortega, O. Rodríguez-Gómez, A. Ruiz, S. Ruiz, A. Sanabria, L. Tárraga, and S. Valero. Department of Surgery, Biochemistry and Molecular Biology, School of Medicine, University of Málaga, Málaga, Spain: E. Alarcon, I. Quintela, and L. M. Real. Grupo de Medicina Xenómica, Centro Nacional de Genotipado (CEGEN-PRB3-ISCIII), Universidade de Santiago de Compostela, Santiago, Spain: A. Carracedo and O. Maroñas. Fundación Pública Galega de Medicina Xenómica, CIBERER, IDIS, Santiago, Spain: A. Carracedo. Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas, Hospital Clínico San Carlos, Madrid, Spain: A. Corbatón, M. T. Martínez, and M. Serrano-Rios. Centro Andaluz de Estudios Bioinformáticos, Seville, Spain: A. González Pérez and M. E. Sáez. Unidad Clínica de Enfermedades Infecciosas y Microbiología, Hospital Universitario de Valme, Seville, Spain: J. Macias, J. A. Pineda, and L. M. Real.
Unidad de Trastornos del Movimiento, Servicio de Neurología y Neurofisiología, Instituto de Biomedicina de Sevilla, Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville, Spain: A. D. Adarmes-Gómez, D. Buiza-Rueda, F. Carrillo, M. Carrión-Claro, P. Gómez-Garre, S. Jesús, M. A. Labrador Espinosa, D. Macias, P. Mir, and T. Periñán-Tocino. Network Center for Biomedical Research in Neurodegenerative Diseases, National Institute of Health Carlos III, Madrid, Spain: A. D. Adarmes-Gómez, R. Blesa, M. Boada, D. Buiza-Rueda, M. J. Bullido, M. Calero, F. Carrillo, M. Carrión-Claro, J. Clarimón, J. Fortea, A. Frank-García, P. Gómez-Garre, I. Hernández, S. Jesús, M. A. Labrador Espinosa, C. Lage, A. Lleó, S. López-García, D. Macias, A. Martín Montes, M. Medina, P. Mir, S. Moreno-Grau, J. Pérez Tur, T. Periñán-Tocino, G. Piñol Ripoll, A. Rábano, E. Rodríguez-Rodríguez, A. Ruiz, P. Sánchez-Juan, I. Sastre, L. Tárraga, and S. Valero. Research Center and Memory Clinic, Fundació ACE, Institut Català de Neurociències Aplicades, Universitat Internacional de Catalunya, Barcelona, Spain: E. Alarcón-Martín, M. Boada, I. de Rojas, I. Hernández, M. Marquié, G. Monté-Rubio, L. Montrreal, S. Moreno-Grau, A. Orellana, A. Ruiz, O. Sotolongo-Grau, L. Tárraga, and S. Valero. Department of Surgery, Biochemistry and Molecular Biology, School of Medicine, University of Málaga, Málaga, Spain: E. Alarcón-Martín, J. M. Cruz-Gamero, and J. L. Royo. Fundació per la Recerca Biomèdica i Social Mútua Terrassa, and Memory Disorders Unit, Department of Neurology, Hospital Universitari Mutua de Terrassa, School of Medicine, University of Barcelona, Barcelona, Spain: I. Álvarez, M. Diez-Fairen, and P. Pastor. Laboratorio de Genética, Hospital Universitario Central de Asturias, Oviedo, Spain: V. Álvarez. Instituto de Investigación Biosanitaria del Principado de Asturias, Oviedo, Spain: V. Álvarez, C. Martínez, and M. Menéndez-González. Department of Neurology, Hospital Universitario Son Espases, Palma, Spain: G. Amer-Ferrer. Unidad de Demencias, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain: M. Antequera, C. Antúnez, A. Legaz, S. Manzanares, J. Marín-Muñoz, B. Martínez, V. Martínez, M. P. Vicente, and L. Vivancos. Servei de Neurologia, Hospital Universitari i Politècnic La Fe, Valencia, Spain: M. Baquero and J. A. Burguera. Unidad de Demencias, Servicio de Neurología y Neurofisiología, Instituto de Biomedicina de Sevilla, Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville, Spain: M. Bernal, E. Franco, M. Marín, and S. Rodrigo. Sant Pau Memory Unit, Neurology Department, Sant Pau Biomedical Research Institute, Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain: R. Blesa, J. Clarimón, J. Fortea, and A. Lleó. Centro de Biologia Molecular Severo Ochoa (CSIC), Universidad Autonoma de Madrid, Madrid, Spain: M. J. Bullido, A. Martín Montes, and I. Sastre. Instituto de Investigacion Sanitaria “Hospital la Paz”, Madrid, Spain: M. J. Bullido, T. del Ser, A. Frank-García, and M. Medina. CIEN Foundation, Queen Sofia Foundation Alzheimer Center, Madrid, Spain: M. Calero, A. B. Pastor, and A. Rábano. Instituto de Salud Carlos III, Madrid, Spain: M. Calero, S. Garcia Madrona, and G. Garcia-Ribas. Hospital Universitario Ramón y Cajal, Madrid, Spain: M. J. Casajeros. BIOMICs, Centro de Investigación Lascaray, Universidad del País Vasco UPV/EHU, Leioa, Spain: M. M. de Pancorbo. Neurology Service, Hospital Universitario La Paz (UAM), Madrid, Spain: A. Frank-García and A. Martín Montes. Alzheimer Research Center and Memory Clinic, Andalusian Institute for Neuroscience, Málaga, Spain: J. M. García-Alberca, S. Hevilla, and T. Marín. Neurology Service, Marqués de Valdecilla University Hospital, IDIVAL, University of Cantabria, Santander, Spain: C. Lage, S. López-García, E. Rodríguez-Rodríguez, and P. Sánchez-Juan. Hospital Donostia de San Sebastían, Donostia, Spain: A. López de Munáin. Fundación para la Formación e Investigación Sanitarias de la Región de Murcia, Murcia, Spain: S. Manzanares. Servicio de Neurología, Hospital de Cabueñes, Gijón, Spain: C. Martínez. Centro de Investigación y Terapias Avanzadas, Fundación CITA-Alzheimer, Donostia, Spain: P. Martínez-Lage Álvarez. Navarrabiomed, Pamplona, Spain: M. Mendioroz Iriarte. Servicio de Neurología, Hospital Universitario Central de Asturias, Oviedo, Spain: M. Menéndez-González. Barcelonaβeta Brain Research Center, Fundació Pasqual Maragall, Barcelona, Spain: J. L. Molinuevo. Unitat de Genètica Molecular, Institut de Biomedicina de València, CSIC, Valencia, Spain: J. Pérez Tur. Unidad Mixta de Neurologia y Genètica. Instituto de Investigación Sanitaria La Fe, Valencia, Spain: J. Pérez Tur. Unitat Trastorns Cognitius, Hospital Universitari Santa Maria de Lleida, Institut de Recerca Biomédica de Lleida, Lleida, Spain: G: Piñol Ripoll. BT-CIEN, Madrid, Spain: A. Rábano. Hospital Universitario La Princesa, Madrid, Spain: D. Real de Asúa. Hospital Clínic de Barcelona, Barcelona, Spain: R. Sanchez del Valle Díaz.
The datasets generated for this study can be accessed from the https://ega-archive.org/studies/EGAS00001003424.
The studies involving human participants were reviewed and approved by Acta 25/2016, Ethics Committee, Hospital Clinic I Provincial de Barcelona, Barcelona, Spain. The patients/participants provided their written informed consent to participate in this study.
PS-J and AR: data collection, data analysis, study design, manuscript drafting, and manuscript critical review. SM, IR, IH, SV, MA, LM, PG, CL, SL-G, ER-R, and AO: data collection, data analysis, and manuscript critical review. LT and MB: data collection, data analysis, manuscript critical review, and study design.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Genome Research at Fundacio ACE/Dementia Genetics Spanish Consortium (GR@ACE/DEGESCO) would like to thank patients and controls who participated in this project. GR@ACE/DEGESCO GWAS program was funded by Grifols SA, Fundacion Bancaria “La Caixa,” and Fundació ACE, Institut Català de Neurociències Aplicades. PS-J and AR have also received support by grant PI16/01861. Accion Estrategica en Salud integrated in the Spanish National I+D+i Plan and financed by Instituto de Salud Carlos III (ISCIII) – Subdireccion General de Evaluacion and the Fondo Europeo de Desarrollo Regional (FEDER – “Una Manera de Hacer Europa”). PS-J was supported by IDIVAL, Instituto de Salud Carlos III [Fondo de Investigacion Sanitario, PI08/0139, PI12/02288, PI16/01652, JPND (DEMTEST PI11/03028)], and the CIBERNED program. We thank Biobanco Valdecilla for their support. LM was supported by Consejería de Salud de la Junta de Andalucía (Grant PI-0001/2017). DEGESCO was also sponsored by the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED, Spain). Control samples and data from patients included in this study were provided in part by the National DNA Bank Carlos III (www.bancoadn.org, University of Salamanca, Spain) and Hospital Universitario Virgen de Valme (Sevilla, Spain) and they were processed following standard operating procedures with the appropriate approval of the Ethical and Scientific Committee. The genotyping service to generate GR@ACE/DEGESCO GWAS data was carried out at CEGEN-PRB3-ISCIII; it was supported by grant PT17/0019, of the PE I+D+i 2013–2016, funded by ISCIII and ERDF. GR@ACE/DEGESCO consortia would also like to thank to all researchers contributing to this project. A complete list of collaborators involved in the GR@ACE/DEGESCO GWAS can be found at https://ciberned.es/proyectos/ degesco.html.
Abraham, R., Sims, R., Carroll, L., Hollingworth, P., O’Donovan, M. C., Williams, J., et al. (2009). An association study of common variation at the MAPT locus with late-onset Alzheimer’s disease. Am. J. Med. Genet.. Part B Neuropsychiatr. Genet. 150B, 1152–1155. doi: 10.1002/ajmg.b.30951
Allen, M., Kachadoorian, M., Quicksall, Z., Zou, F., Chai, H. S., Younkin, C., et al. (2014). Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimer’s Res. Ther. 6:39. doi: 10.1186/alzrt268
Arboleda-Velasquez, J. F., Lopera, F., O’Hare, M., Delgado-Tirado, S., Marino, C., Chmielewska, N., et al. (2019). Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. doi: 10.1038/s41591-019-0611-3 [Epub ahead of print].
Babić Leko, M., Willumsen, N., Nikolac Perković, M., Klepac, N., Borovečki, F., Hof, P. R., et al. (2018). Association of MAPT haplotype-tagging polymorphisms with cerebrospinal fluid biomarkers of Alzheimer’s disease: a preliminary study in a croatian cohort. Brain Behav. 8:e01128. doi: 10.1002/brb3.1128
Baker, M., Litvan, I., Houlden, H., Adamson, J., Dickson, D., Perez-Tur, J., et al. (1999). Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 8, 711–715. doi: 10.1093/hmg/8.4.711
Chen, J., Yu, J.-T., Wojta, K., Wang, H.-F., Zetterberg, H., Blennow, K., et al. (2017). Genome-wide association study identifies MAPT locus influencing human plasma tau levels. Neurology 88, 669–676. doi: 10.1212/WNL.0000000000003615
Clavaguera, F., Hench, J., Goedert, M., and Tolnay, M. (2015). Invited review: prion-like transmission and spreading of tau pathology. Neuropathol. Appl. Neurobiol. 41, 47–58. doi: 10.1111/nan.12197
Conrad, C., Andreadis, A., Trojanowski, J. Q., Dickson, D. W., Kang, D., Chen, X., et al. (1997). Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann. Neurol. 41, 277–281. doi: 10.1002/ana.410410222
Corvol, J.-C., and Buée, L. (2019). A new step towards targeting tau. Lancet. Neurol. 18, 517–518. doi: 10.1016/S1474-4422(19)30161-9
Donnelly, M. P., Paschou, P., Grigorenko, E., Gurwitz, D., Mehdi, S. Q., Kajuna, S. L. B., et al. (2010). The distribution and most recent common ancestor of the 17q21 inversion in humans. Am. J. Hum. Genet. 86, 161–171. doi: 10.1016/j.ajhg.2010.01.007
Dourlen, P., Kilinc, D., Malmanche, N., Chapuis, J., and Lambert, J.-C. (2019). The new genetic landscape of Alzheimer’s disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 138, 221–236. doi: 10.1007/s00401-019-02004-0
Goedert, M. (2004). Tau protein and neurodegeneration. Sem. Cell Dev. Biol. 15, 45–49. doi: 10.1016/j.semcdb.2003.12.015
Heckman, M. G., Brennan, R. R., Labbé, C., Soto, A. I., Koga, S., DeTure, M. A., et al. (2019). Association of MAPT Subhaplotypes with risk of progressive supranuclear palsy and severity of tau pathology. JAMA Neurol. 76, 710–717. doi: 10.1001/jamaneurol.2019.0250
Higgins, J. J., Golbe, L. I., De Biase, A., Jankovic, J., Factor, S. A., and Adler, R. L. (2000). An extended 5’-tau susceptibility haplotype in progressive supranuclear palsy. Neurology 55, 1364–1367. doi: 10.1212/wnl.55.9.1364
Holzer, M., Craxton, M., Jakes, R., Arendt, T., and Goedert, M. (2004). Tau gene (MAPT) sequence variation among primates. Gene 341, 313–322. doi: 10.1016/j.gene.2004.07.013
Houlden, H., Baker, M., Morris, H. R., MacDonald, N., Pickering-Brown, S., Adamson, J., et al. (2001). Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56, 1702–1706. doi: 10.1212/wnl.56.12.1702
Jansen, W. J., Ossenkoppele, R., Knol, D. L., Tijms, B. M., Scheltens, P., Verhey, F. R. J., et al. (2015). Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313, 1924–1938. doi: 10.1001/jama.2015.4668
Jun, G., Ibrahim-Verbaas, C. A., Vronskaya, M., Lambert, J.-C., Chung, J., Naj, A. C., et al. (2016). A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117. doi: 10.1038/mp.2015.23
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Kwok, J. B. J., Teber, E. T., Loy, C., Hallupp, M., Nicholson, G., Mellick, G. D., et al. (2004). Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann. Neurol. 55, 329–334. doi: 10.1002/ana.10826
Martin, E. R., Scott, W. K., Nance, M. A., Watts, R. L., Hubble, J. P., Koller, W. C., et al. (2001). Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA 286, 2245–2250.
Moreno-Grau, S., Rojas, I., de, Hernandez, I., Quintela, I., Montrreal, L., Alegret, M., et al. (2019). Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer disease and three causality networks of AD: the GR@ACE project. bioRxiv. [preprint]. doi: 10.1101/528901
Mukherjee, O., Kauwe, J. S. K., Mayo, K., Morris, J. C., and Goate, A. M. (2007). Haplotype-based association analysis of the MAPT locus in late onset Alzheimer’s disease. BMC Genet. 8:3. doi: 10.1186/1471-2156-8-3
Myers, A. J., Kaleem, M., Marlowe, L., Pittman, A. M., Lees, A. J., Fung, H. C., et al. (2005). The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum. Mol. Genet. 14, 2399–2404. doi: 10.1093/hmg/ddi241
Myers, A. J., Pittman, A. M., Zhao, A. S., Rohrer, K., Kaleem, M., Marlowe, L., et al. (2007). The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis. 25, 561–570. doi: 10.1016/j.nbd.2006.10.018
Nicoll, J. A. R., Buckland, G. R., Harrison, C. H., Page, A., Harris, S., Love, S., et al. (2019). Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain 142, 2113–2126. doi: 10.1093/brain/awz142
Pastor, P., Ezquerra, M., Perez, J. C., Chakraverty, S., Norton, J., Racette, B. A., et al. (2004). Novel haplotypes in 17q21 are associated with progressive supranuclear palsy. Ann. Neurol. 56, 249–258. doi: 10.1002/ana.20178
Pastor, P., Moreno, F., Clarimón, J., Ruiz, A., Combarros, O., Calero, M., et al. (2016). (MAPT)H1 haplotype is associated with late-onset Alzheimer’s Disease risk in APOE ε4 noncarriers: results from the dementia genetics spanish consortium. J. Alzheimer’s Dis. 49, 343–352. doi: 10.3233/JAD-150555
Pittman, A. M., Myers, A. J., Abou-Sleiman, P., Fung, H. C., Kaleem, M., Marlowe, L., et al. (2005). Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J. Med. Genet. 42, 837–846. doi: 10.1136/jmg.2005.031377
Russ, C., Powell, J. F., Zhao, J., Baker, M., Hutton, M., Crawford, F., et al. (2001). The microtubule associated protein Tau gene and Alzheimer’s disease–an association study and meta-analysis. Neurosci. Lett. 314, 92–96. doi: 10.1016/s0304-3940(01)02289-3
Schöll, M., Lockhart, S. N., Schonhaut, D. R., O’Neil, J. P., Janabi, M., Ossenkoppele, R., et al. (2016). PET imaging of tau deposition in the aging human brain. Neuron 89, 971–982. doi: 10.1016/j.neuron.2016.01.028
Simón-Sánchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Verpillat, P., Camuzat, A., Hannequin, D., Thomas-Anterion, C., Puel, M., Belliard, S., et al. (2002). Association between the extended tau haplotype and frontotemporal dementia. Arch. Neurol. 59, 935–939.
Keywords: Alzheimer’s disease, MAPT, H1H2, APOE, genetic association
Citation: Sánchez-Juan P, Moreno S, de Rojas I, Hernández I, Valero S, Alegret M, Montrreal L, García González P, Lage C, López-García S, Rodríguez-Rodríguez E, Orellana A, Tárraga L, Boada M and Ruiz A (2019) The MAPT H1 Haplotype Is a Risk Factor for Alzheimer’s Disease in APOE ε4 Non-carriers. Front. Aging Neurosci. 11:327. doi: 10.3389/fnagi.2019.00327
Received: 27 June 2019; Accepted: 12 November 2019;
Published: 04 December 2019.
Edited by:
Fernanda Laezza, The University of Texas Medical Branch at Galveston, United StatesReviewed by:
Rohan de Silva, University College London, United KingdomCopyright © 2019 Sánchez-Juan, Moreno, de Rojas, Hernández, Valero, Alegret, Montrreal, García González, Lage, López-García, Rodríguez-Rodríguez, Orellana, Tárraga, Boada and Ruiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascual Sánchez-Juan, cGFzY3VhbGplc3VzLnNhbmNoZXpAc2NzYWx1ZC5lcw==; cHNhbmp1YUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.