 Honghua Zheng
Honghua Zheng Baoying Cheng
Baoying Cheng Yanfang Li
Yanfang Li Xin Li1
Xin Li1 Xiaofen Chen
Xiaofen Chen Yun-wu Zhang
Yun-wu Zhang- 1Fujian Provincial Key Laboratory of Neurodegenerative Disease and Aging Research, Institute of Neuroscience, Medical College, Xiamen University, Xiamen, China
- 2Shenzhen Research Institute, Xiamen University, Shenzhen, China
Alzheimer’s disease (AD) is the leading cause of age-related dementia among the elderly population. Recent genetic studies have identified rare variants of the gene encoding the triggering receptor expressed on myeloid cells-2 (TREM2) as significant genetic risk factors in late-onset AD (LOAD). TREM2 is specifically expressed in brain microglia and modulates microglial functions in response to key AD pathologies such as amyloid-β (Aβ) plaques and tau tangles. In this review article, we discuss recent research progress in our understanding on the role of TREM2 in microglia and its relevance to AD pathologies. In addition, we discuss evidence describing new TREM2 ligands and the role of TREM2 signaling in microglial survival and energy metabolism. A comprehensive understanding of TREM2 function in the pathogenesis of AD offers a unique opportunity to explore the potential of this microglial receptor as an alternative target in AD therapy.
Alzheimer’s disease (AD), the most common form of dementia, is a neurodegenerative disease clinically characterized by progressive memory loss and cognitive deficits that are accompanied by pathological deposition of senile plaques and neurofibrillary tangles (NFTs) in the brain (Duyckaerts et al., 2009; Jankowsky and Zheng, 2017). AD is an age-related disease with a higher incidence in people over 65 years; after which AD incidence rate doubles every 5 years. By 2050, one new case of AD is expected to develop every 33 s, resulting in nearly 1 million new cases per year and a total of 115 million cases (Alzheimer’s Association, 2016). Given the heavy economic and social burdens associated with the care of AD patients, significant efforts, both public and private, have been undertaken to understand the pathogenesis of AD and develop early diagnostic methods and effective intervention therapies.
Microglia, known as resident immune cells in the brain, are classified based on morphology and functional state into resting (ramified) and activated/phagocytic (ameboid) microglia (Sarlus and Heneka, 2017). Fate-mapping studies have identified the immature yolk sac as the predominant source of microglia progenitors (Ginhoux et al., 2010; Schafer and Stevens, 2015). Recent advances in genomic research have uncovered significant contributions of microglia to inheritable disease risks for AD (Malik et al., 2015; Efthymiou and Goate, 2017; Salter and Stevens, 2017; Sarlus and Heneka, 2017). Genetic variants of the triggering receptor expressed on myeloid cells-2 (TREM2), which is dominantly expressed in microglia in central nervous system (CNS), result in increased risk of developing late-onset AD (LOAD) and other neurodegenerative disorders (Guerreiro et al., 2013; Jonsson et al., 2013; Painter et al., 2015; Ulrich and Holtzman, 2016). These genetic findings have attracted attention to the role of microglia in neurodegenerative diseases and TREM2 in regulating microglial function (Calcagno et al., 2018). Besides its neuroinflammatory and phagocytic role in pathogenesis, microglia plays important roles in clustering or limiting the diffusion or growth of amyloid-β (Aβ) plaques (Wang et al., 2016; Zhao R. et al., 2017) and tau tangles (Kitazawa et al., 2004; Bloom, 2014; Hung et al., 2016). Given our rapidly expanding understanding of the microglia function and dysfunction in CNS disorders, as well as the important roles of TREM2 in AD, microglia is generally accepted as playing a pivotal role in the maintenance of brain homeostasis (Salter and Stevens, 2017). In this review article, we discuss recent progress in our understanding of the role of TREM2 in microglia functions related to AD pathology, as well as signaling pathways activated by TREM2 ligands and their role in microglial survival and metabolism. Comprehensive understanding of TREM2 function in the pathogenesis of AD offers a unique opportunity to explore this microglial receptor as a target alternative to Aβ and tau in AD therapy.
Neuropathological Hallmarks of AD
AD is pathologically characterized by a buildup of extracellular senile plaques composed of Aβ peptide, intracellular NFTs composed of hyperphosphorylated tau (p-tau) protein and reactive gliosis, including microgliosis (Efthymiou and Goate, 2017; Leyns and Holtzman, 2017). Currently, soluble β-amyloid precursor protein (APP), Aβ, tau and p-tau in cerebrospinal fluid and blood are candidate biomarkers for AD (Hampel et al., 2010; Shekhar et al., 2016; Lucey et al., 2017; Tatebe et al., 2017).
Aβ Plaques
Aβ peptides, the main component of extracellular senile plaques, are sequentially cleaved from APP by distinct β- and γ-secretases (Zhang et al., 2014). In contrast, α-secretase cleavage of APP within the Aβ domain precludes the formation of toxic Aβ peptides (Nathalie and Jean-Noël, 2008). Aβ clearance involves phagocytosis and endocytosis via microglial scavenger receptors and extracellular degradation by Aβ-degrading enzymes (Hickman et al., 2018). Aβ can form oligomers, which are widely regarded as the most toxic and pathogenic forms of Aβ (Tu et al., 2014; Arbel-Ornath et al., 2017; Cline et al., 2018). It has been proposed that the over-production and/or decreased degradation of Aβ and subsequent formation of Aβ oligomers instigates the activation of microglia and astrocytes, dystrophy of neurites, and ultimately the symptoms of dementia (Viola and Klein, 2015; Brody et al., 2017; Cline et al., 2018; Doig, 2018). Microglia surround and limit Aβ plaques in AD brains and recent studies have reported reduced microglia clearance of Aβ in the absence of microglial scavenger receptors (Salter and Stevens, 2017; Ulland and Colonna, 2018). These findings support a direct link between abnormal microglial function and Aβ plaques in AD.
Pathological Tau
Hyperphosphorylation of microtubule-associated protein tau (MAPT) is another key pathological hallmark of AD (Spillantini and Goedert, 1998; Lee et al., 2001; Wu et al., 2017). Tau is an axon-enriched protein that binds to and stabilizes microtubules, playing a crucial role in neuronal function. Studies have also revealed profound tau pathology in the visual system, leading to early retinal ganglion cell (RGC) damage in a mouse model of AD (Chiasseu et al., 2017). Recent studies propose inhibition or reduction of tau hyperphosphorylation as a potential therapeutic strategy for AD (Brunden et al., 2009; Li and Götz, 2017). Given that pathological tau correlates better with the degree of dementia than Aβ deposition, therapeutic strategies targeting pathological tau may provide a more promising approach for the treatment of AD. Because microglia can engulf, degrade and clear tau, it has recently been proposed microglia may play a significant role in the spreading of tau pathology (Leyns and Holtzman, 2017; Wang et al., 2017; Hickman et al., 2018). Nevertheless, the underlying pathways and the potential significance of microglia-mediated tau pathology require further investigation.
Microgliopathy
Reactive gliosis, microglia dysfunction and neuroinflammation, sometimes described under the term “microgliopathy,” are pathological hallmarks of AD (Zhang et al., 2013; Sasaki, 2017) and other neurodegenerative diseases, including Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and frontal temporal dementia (FTD; Salter and Stevens, 2017). Molecules associated with these “microgliopathies” mainly include the 12 kDa DNAX activating protein (DAP12) and TREM2 in Nasu-Hakola disease (NHD; Paloneva et al., 2000, 2002); and the colony stimulating factor 1 receptor (CSF1R) in hereditary diffuse leukoencephalopathy with spheroids (HDSL; Rademakers et al., 2011). Interestingly, variants of these genes are associated with increased risk for AD and cognitive decline (Efthymiou and Goate, 2017), implying a pivotal role for microglia in AD pathogenesis.
Genetics of AD
Mutations in the APP, presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes can cause Familial AD (FAD), which is typically associated with early-onset (<65 years) AD (Selkoe, 2001; De Strooper and Annaert, 2010; Tcw and Goate, 2017). In contrast, sporadic or LOAD, which accounts for the majority of the cases (90%–95%), results from a complex genetic architecture. Apolipoprotein E epsilon 4 (APOE ε4) is the most common genetic risk factor for AD; APOE4 carriers have a 3–8-fold higher risk of developing AD than non-carriers (Corder et al., 1993). In addition to APOE, recent genome-wide association studies (GWAS) have identified more than 20 additional susceptibility loci associated with AD, including rare coding variants of TREM2 that confer AD risk comparable to that of APOE4 carriers (Benitez and Cruchaga, 2013; Guerreiro and Hardy, 2013; Guerreiro et al., 2013). Findings from gene network analyses also revealed higher AD risk associated with several rare variants of genes that are highly expressed in microglia (Dos Santos et al., 2017; Efthymiou and Goate, 2017; Hickman et al., 2018; Ulland and Colonna, 2018), including complement receptor 1 (CR1; Corneveaux et al., 2010), the membrane-spanning 4-domain subfamily A (MS4A) gene cluster, ATP-binding cassette transporter A7 (ABCA7; Steinberg et al., 2015), the cluster of differentiation 33 (CD33; Bradshaw et al., 2013), Phospholipase C Gamma 2 (PLCG2) and B3 domain-containing transcription factor ABI3 (ABI3; Sims et al., 2017). These genetic findings provide strong evidence that microglia-mediated immune response contributes directly to the development of AD.
Genetic Investigation of TREM2 in AD and Other Neurodegenerative Diseases
The importance of TREM2 in neuronal health was first demonstrated by genetic studies that identified TREM2 variants in families with NHD (also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy or PLOSL), a fatal disease characterized by presenile dementia and bone cysts (Paloneva et al., 2001, 2002; Klünemann et al., 2005; Bianchin et al., 2006). Heterozygous rare variants R47H, R62H and H157Y of TREM2 are associated with an increased risk of developing AD in European, African American and Asian populations (Guerreiro et al., 2013; Jonsson et al., 2013; Jin et al., 2014; Jiang et al., 2016). Besides AD, TREM2 variants have been linked to other neurodegenerative diseases, including ALS (Cady et al., 2014), PD (Rayaprolu et al., 2013) and FTD (Borroni et al., 2014; Lill et al., 2015). A recent study showed that AD mice heterozygous for the Trem2 R47H allele were comparable to AD mice lacking one copy of Trem2 (Cheng-Hathaway et al., 2018). Moreover, in 5XFAD mouse models in which the mouse endogenous Trem2 was replaced by human normal TREM2 or human TREM2 R47H, Aβ-induced microglial response was found significantly reduced in TREM2 R47H mice (Song et al., 2018), further supporting that R47H variant impairs TREM2 function in vivo. Interestingly, two other independent mouse models with mouse Trem2 R47H knock-in showed reduced Trem2 mRNA and protein levels, indicating that mouse Trem2 R47H variant activates a cryptic splice site and leads to Trem2 haploinsufficiency only in mice but not in humans (Xiang et al., 2018). Thus, humanized TREM2 R47H knock-in mice should be used to study the cellular consequences caused by human TREM2 R47H coding variant and it will be interesting to study the genetic variability and the extended mouse phenotype in the context of AD.
TREM2 is highly expressed in white matter in normal human brain, with transcripts detected in all brain regions (Forabosco et al., 2013). It is also highly expressed in plaque-associated microglia in human AD brain (Frank et al., 2008; Yuan et al., 2016; Yin et al., 2017). Studies in animal models have consistently found increased TREM2 expression during aging and disease progression (Guerreiro et al., 2013; Jay et al., 2015; Wang et al., 2015). A growing body of evidence provides new insight into the multifaceted roles of TREM2 in regulating extracellular Aβ pathology (Wang et al., 2016; Lee et al., 2018), hyperphosphorylation and aggregation of tau (Bemiller et al., 2017; Leyns and Holtzman, 2017; Sayed et al., 2018), microgliosis and inflammation in AD (Zhong et al., 2015, 2017b; Zheng et al., 2016; Jay et al., 2017a; Ulrich et al., 2017). Interestingly, it has been reported that TREM2 can be cleaved by the γ-secretase, which is required for the generation of Aβ peptides (Wunderlich et al., 2013). TREM2 has also been found to interact with PSEN1, the key component of the γ-secretase complex, in a manner independent of γ-secretase activity to affect TREM2-mediated phagocytic capacity in microglia (Zhao Y. et al., 2017).
Additionally, the ectodomain of TREM2, namely soluble TREM2 (sTREM2), can be generated by proteolytic processing within the protein stalk (Wunderlich et al., 2013) or by alternative splicing (Jin et al., 2014). sTREM2 has been identified in the CSF of multiple sclerosis (MS) patients (Piccio et al., 2008) and a number of AD cohorts (Gispert et al., 2016; Heslegrave et al., 2016). A recent study found that sTREM2 can trigger microglial activation, induce inflammatory responses and promote microglial survival (Zhong et al., 2017a), which suggests the possibility of targeting sTREM2 as potential AD therapy. These results also provide a paradigm in which sTREM2 may act independently of, and perhaps oppose to, the role of full-length TREM2 in regulating inflammatory responses. Additional work is needed to explore other functions of sTREM2 in microglia and to define the precise molecular pathways downstream of sTREM2 actions.
Ligands of TREM2 Associated With AD
Many potential TREM2 ligands have been proposed (Figure 1), including Aβ, apoE, anionic lipids, etc.
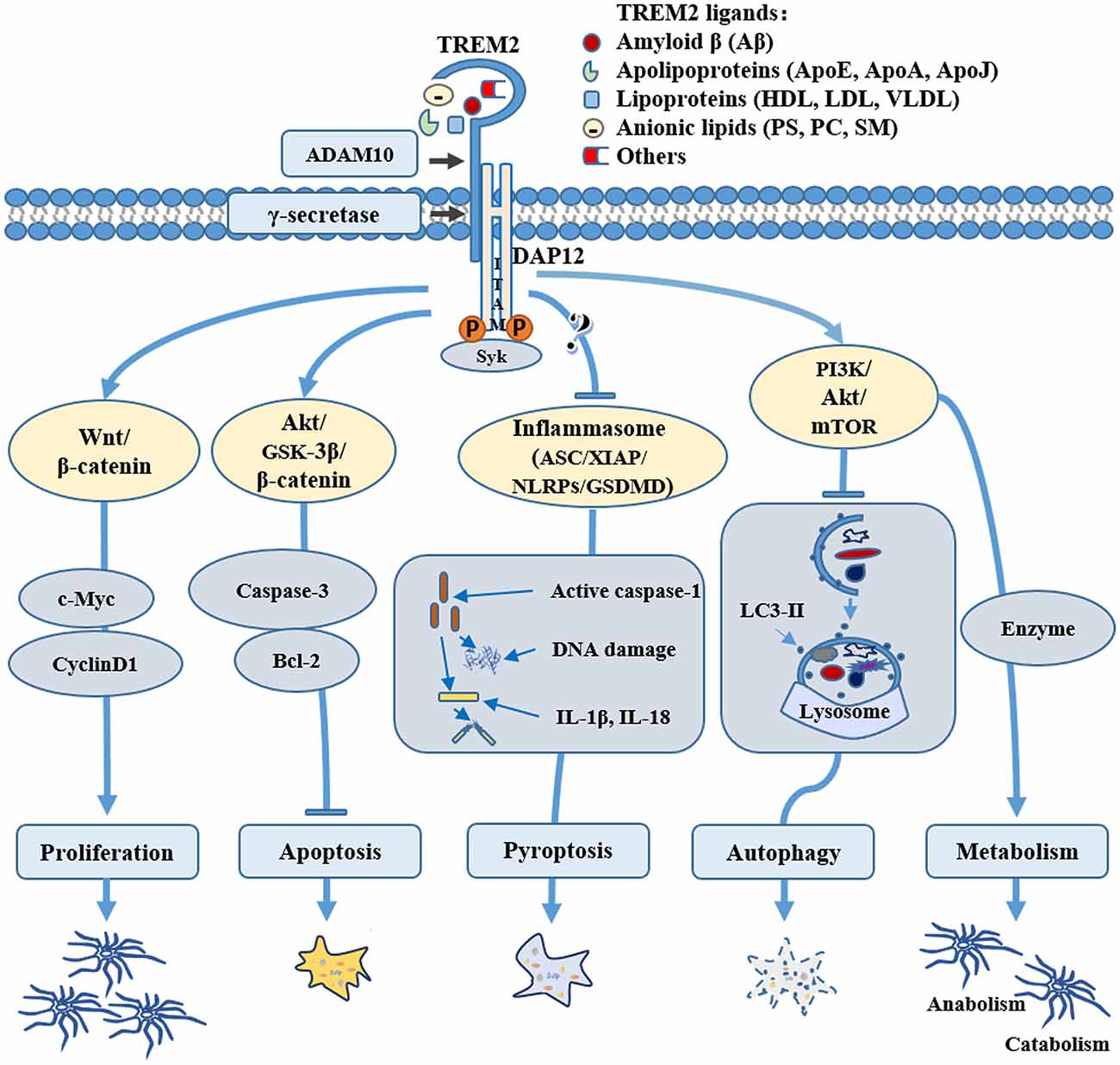
Figure 1. Role of triggering receptor expressed on myeloid cells-2 (TREM2) in microglial survival and metabolism. TREM2 may mediate microglial cell survival through various pathways: TREM2 can promote microglial proliferation by activating the Wnt/β-catenin signaling pathway and inhibit microglial apoptosis through the Akt/GSK3β pathway; TREM2 can regulate the key components of inflammasome, such as NLR family pyrin domain containing 3 (NLRP3) and/or gasdermin D (GSDMD) to inhibit microglial pyroptosis; TREM2 can also activate the PI3K/AKT/mTOR pathway, regulating microglial autophagy and sustaining cellular energetic and biosynthetic metabolism, therefore promoting the survival of microglial cells. Through binding to various ligands, such as amyloid-β (Aβ), apolipoproteins, lipoprotein or anionic lipids, TREM2 can be stimulated physiologically or pathologically, directing microglia to different fates during normal conditions or the pathogenesis of Alzheimer’s disease (AD) and other neurological diseases.
Aβ
Although the role of TREM2 in regulating Aβ plaques has been controversial, all Trem2−/– studies have shown decreased clustering of myeloid or microglial cells around plaques, suggesting a defect in microglia activation (Ulrich et al., 2014; Jay et al., 2015, 2017a; Wang et al., 2015, 2016; Konishi and Kiyama, 2018). Importantly, samples from human R47H carriers also showed fewer microglia clustering around Aβ plaques with diffuse morphology, which are more toxic to neurons (Yuan et al., 2016).
However, how TREM2 impacts microglia-plaque interaction and Aβ-associated cognitive deficits remains unclear. Recent studies have shed new light onto this issue by showing evidence of direct binding of TREM2 to Aβ oligomers with nanomolar affinity, whereas AD-associated TREM2 mutations show reduced Aβ binding (Zhao et al., 2018; Zhong et al., 2018). Very recently, TREM2 was found to interact directly with various forms of Aβ, with the highest affinity interactions with soluble Aβ42 oligomers (Lessard et al., 2018). These findings have offered key insight into the role of TREM2 in Aβ pathologies, in which TREM2 strongly affects the ability of microglia to respond to plaques (Colonna and Wang, 2016; Udeochu et al., 2018). Indeed, increasing TREM2 gene dosage reduces plaque area and appears to promote the formation of plaques that are more compact and associated with fewer dystrophic neurites (Lee et al., 2018).
ApoE
In 2015, two groups independently identified a direct interaction between TREM2 and apoE (Atagi et al., 2015; Bailey et al., 2015). Both groups found that TREM2 binds all apoE isoforms, lipidated and non-lipidated, and that the R47H variant reduced its binding affinity to apoE. Separate groups identified a set of lipoprotein particles, including very low density lipoprotein (VLDL), LDL and high-density lipoprotein (HDL), as well as apolipoproteins, including CLU/apoJ and apoE, as TREM2 ligands (Yeh et al., 2016; Jay et al., 2017b; Song et al., 2017). Binding of these ligands was abolished or reduced in disease-associated TREM2 mutations (Song et al., 2017).
Given that apoE genotype is by far the strongest genetic factor modulating Aβ deposition, and the risk of suffering AD associated with TREM2 variants is comparable to that of the apoE4 allele, an interaction between apoE and TREM2 connects these two key AD risk factors, which could have pivotal roles in the pathogenesis of AD (Engstrom et al., 2017; Jendresen et al., 2017; Pankiewicz et al., 2017). Indeed, the TREM2-apoE pathway has been identified as a major regulator of microglia phenotypic change in neurodegenerative diseases (Krasemann et al., 2017). TREM2 activation of the apoE signaling pathway can restore the homeostatic signature of microglia in ALS and AD mouse models, and prevent neuronal loss in an acute model of neurodegeneration (Krasemann et al., 2017). Interestingly, recent work demonstrated that apoE is required for microglial association to Aβ plaques where it performs a plaque-trimming function similar to that of TREM2; thus supporting an apoE-TREM2 axis in mediating microglial function (Ulrich et al., 2018). These studies indicate that apoE and TREM2 may share similar mechanisms in regulating microglial cell responses in Aβ pathology (Shi and Holtzman, 2018).
Anionic Lipids and Other Ligands
Bacteria and polyanionic molecules, including dextran sulfate, lipopolysaccharide (LPS) and lipoteichoic acid (LTA), were the first potential ligands identified for TREM2 (Daws et al., 2003; Charles et al., 2008; Quan et al., 2008; N’Diaye et al., 2009). TREM2 has also been shown to bind a number of anionic molecules from mammalian cells. Studies using solid-state ELISA or TREM2 reporter cells support the concept of phospholipids as TREM2 ligands, including phosphatidylethanolamine (PE), phosphatidylserine (PS), and cardiolipin, phosphatidylcholine (PC) or sphingomyelin (SM; Cannon et al., 2012; Poliani et al., 2015; Wang et al., 2015). In addition to phospholipids, nucleic acids released by damaged cells can also act as TREM2 ligands. Both cellular fractions containing nuclei and purified DNA can activate TREM2 signaling (Kawabori et al., 2015). Furthermore, it has been shown that TREM2 can also bind proteoglycans (such as heparan sulfates or HS), which are a large group of cell-surface proteins. Interestingly, proteoglycan binding to TREM2 is impacted by AD risk variants (Kober et al., 2016). The potential roles of these ligands in TREM2 signaling pathway in physiological and pathological conditions is currently the target of intense research as they may represent potential new sources of therapeutic and diagnostic tools.
TREM2 and Microglial Survival
TREM2-mediated microglia activation can display a number of forms (Figure 1), including promotion of cell survival and proliferation, regulation of cytokine production, and/or phagocytosis (Schafer and Stevens, 2015; Leyns and Holtzman, 2017). TREM2 stimulation by these ligands is also important for microglial function in response to pathogens and other causes of cell damage. For example, Aβ-induced microglial depolarization, K+ inward current induction, cytokine expression and secretion, migration, proliferation, apoptosis and morphological changes are all dependent on TREM2 activation (Zhao et al., 2018). Moreover, recent evidence revealed the crucial role of TREM2 in maintaining microglial cell survival (Wang et al., 2015) and optimal metabolic fitness (Ulland et al., 2017).
TREM2 and Microglial Proliferation
Studies have found that mice lacking TREM2 (Trem2−/–) have fewer microglia and increased microglia apoptosis (Ulland et al., 2015; Jay et al., 2017b; Yeh et al., 2017). Cell proliferation was also reduced in microglia derived from Trem2−/– mice compared with those from wild type (WT) mice both in vivo and in vitro (Zheng et al., 2017). In Trem2−/– mouse brain, TREM2 deficiency reduced the viability and proliferation of microglia, reduced microgliosis, induced cell cycle arrest at the G1/S checkpoint, and decreased the stability of β-catenin, a key component of the canonical Wnt signaling pathway responsible for maintaining key biological processes, including cell survival (Zheng et al., 2017). TREM2 is also involved in macrophage CSF (M-CSF) mediated survival and proliferation of macrophages and osteoclast precursors. M-CSF, also known as CSF-1, acting via the CSF-1R can induce calmodulin-dependent kinase-mediated phosphorylation of β-catenin in a TREM2-dependent mechanism (Otero et al., 2009, 2012). However, whether there is a synergy between the TREM2 and CSF1R remains elusive and further studies on the cell- and tissue-specific functions of the TREM2/CSF1R signaling pathway in healthy and diseased brains may provide insight into this (Kober and Brett, 2017).
TREM2 and Microglial Apoptosis
TREM2 deficient microglia show high rates of apoptotic cell death (Wang et al., 2015). Our recent work also demonstrated that TREM2 depletion accelerates microglial apoptosis by showing significantly increased levels of cleaved caspase 3 and decreased levels of pro-caspase 3 and Bcl-2 in Trem2−/– microglia compared to WT microglia (Zheng et al., 2017). Furthermore, sTREM2, purified from conditioned media of transfected HEK293T cells, was found to attenuate apoptotic cell death caused by withdrawal of granulocyte-M-CSF (GM-CSF) in primary microglia cultures from both WT and Trem2−/– mice (Zhong et al., 2017a). Indeed, previous studies have shown that TREM2 suppresses apoptosis in lung macrophages during Sendai virus infection (Wu et al., 2015). Together these results imply a vital role for TREM2-mediated signaling in programmed cell death.
TREM2 and Microglial Autophagy
The term autophagy (derived from the Greek words auto: self and phagos: eating) indicates a self-sacrificing mechanism of cell elimination and removal (Eisenberg-Lerner et al., 2009; Zhu and Zhang, 2018). At the basal level, autophagy plays a vital role in keeping cell homeostasis through digestion of dysfunctional organelles and proteins (Metaxakis et al., 2018; Mizushima, 2018). Defective autophagy pathways or alterations in autophagy-related genes have been revealed in various human pathologies including neurodegenerative disorders and microglia dysfunction (Mizushima et al., 2008; Jiang and Mizushima, 2014; Kim et al., 2016; Moors et al., 2017). Autophagy involves the activation of three major autophagic pathways: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA); and the final destination of all three autophagic pathways is the lysosome (Ghavami et al., 2014; Deng et al., 2017).
Studies have provided evidence on how autophagy may affect microglial function during progression of age-associated neurodegenerative diseases (Plaza-Zabala et al., 2017). Recycling of TREM2 in microglia is regulated by Beclin-1, a protein involved in autophagy (Salminen et al., 2013) and levels of TREM2 are reduced in AD brain microglia (Lucin et al., 2013), implying a potential link between TREM2 and autophagy. A recent study by Colonna and Wang (2016) provided evidence in support of this prediction by showing deficient TREM2-mediated autophagy in human AD brain and mouse AD model (Ulland et al., 2017). The lipidation of microtubule-associated light chain 3 (LC3) is an important marker and effector of autophagy. The ratio of lipidated LC3II to non-lipidated LC3I was markedly higher in microglia from 5XFAD mice lacking TREM2 (Trem2−/–) than in WT 5XFAD mice. This is consistent with the increased number of autophagic vesicles observed in TREM2-deficient microglia (Ulland et al., 2017). These results indicate that TREM2 plays an important role in microglial autophagy; however, identification of the mechanism and/or signaling pathways need to be verified.
TREM2 and Microglial Pyroptosis
Inflammasome is a type of tissue-damage sensor that is necessary for the conversion of IL-1β immature pro-form to the mature, active form and it is implicated in a form of cell death called pyroptosis (Keller et al., 2008; Latz et al., 2013; Sharma and Kanneganti, 2016). Increasing evidence shows aberrant expression of inflammasome-related proteins in AD brain (Tan et al., 2014; Olsen and Singhrao, 2016; Saresella et al., 2016). Although one study demonstrated that TREM2 promoted host resistance against P. aeruginosa by inhibiting caspase-1-dependent pyroptosis (Qu et al., 2018), it is still unclear whether and how TREM2 may be involved in inflammasomes and pyroptosis in microglia. Due to the complex relationship between TREM2 and inflammatory pathways (Jay et al., 2017b), further research is needed to elucidate whether and how TREM2 plays a biological role in microglial pyroptosis in AD (Figure 1). For example, it would be interesting to investigate whether TREM2 can regulate key components of inflammasome, such as NLR family pyrin domain containing 3 (NLRP3) and/or gasdermin D (GSDMD) in AD.
TREM2 and Metabolism
Similar to other cell types, microglia express the full complement of gene products required for both glycolytic and oxidative metabolism (Ma et al., 2017). Evidence suggests that microglia metabolize glucose, acetoacetate (AcAc) and β-hydroxybutyrate (BHB), increase aerobic glycolysis and decrease respiration when activated by various stimuli (Ghosh et al., 2018; Zhou et al., 2018). Mutations in TREM2 were found to disrupt microglial energy state and function, thus sabotaging microglia’s ability to protect the brain against toxic amyloid plaques (Hong and Stevens, 2017; Ulland et al., 2017). Ulland et al. (2017) also found that microglia in AD patients carrying TREM2 risk variants and in TREM2-deficient mice with AD-like pathology were defective in rapamycin (mTOR) signaling, which affects ATP levels and biosynthetic pathways. Metabolic derailment and autophagy were offset in vitro through Dectin-1, a receptor that elicits TREM2-like intracellular signals, and cyclocreatine, a creatine analog that acts as a source of ATP (Ulland et al., 2017). This study indicates that TREM2 enables microglial responses during AD by sustaining cellular energetic and biosynthetic metabolism. TREM2 has also been linked to lipid metabolism. TREM2 promotes adipogenesis and diet-induced obesity by upregulating adipogenic regulators in association with inhibiting the Wnt10b/β-catenin signaling pathway (Park et al., 2015). The connection between TREM2 and lipid metabolic pathways has also been confirmed by expression analysis (Piccio et al., 2008; Poliani et al., 2015). Studies consistently demonstrate that a TREM2 loss-of-function mutation T66M can cause a significant reduction in brain glucose metabolism (Kleinberger et al., 2017). These studies indicate that TREM2 play critical roles in maintaining the anabolism and catabolism of nutrients.
Conclusion and Prospects
Substantial genetic findings point to a central role of microglia in neurodegenerative diseases. TREM2 can support microglial cell survival by promoting microglial proliferation and inhibiting apoptosis, autophagy and perhaps pyroptosis. TREM2 also supports a higher metabolic rate in microglia, while dysfunction of TREM2 impairs brain metabolism. Besides anionic lipids, several new ligands of TREM2, such as Aβ and apoE, have been identified. It remains unclear what exact signaling pathways these ligands activate and how they are involved in microglia survival and metabolism. Therefore, further research is needed to elucidate the biological role of these ligands in TREM2 signaling pathway and in microglia survival in AD pathogenesis. Given that clinical trials targeting the accumulation of Aβ and hyperphosphorylated tau in the brain have nearly all ended in failure, deciphering the role of TREM2 in microglia will advance our understanding and provide novel therapeutic strategies for the treatment of AD and other neurodegenerative diseases.
Author Contributions
HZ wrote and reviewed the manuscript. BC drew the figures. XL arranged the references. YL, XC and YZ reviewed the manuscript.
Funding
This work was supported in part by grants from the Natural Science Key Foundation of Fujian Educational Department of China (JZ160403), the National Natural Science Foundation of China (81771164 and 81870845), the Natural Science Foundation of Guangdong Province of China (2016A030313821, 2017A030313604 and 2018A030313158), the Open Research Fund of State Key Laboratory of Cellular Stress Biology, Xiamen University (SKLCSB2018KF017), the National Key Research and Development Program of China (2016YFC1305903) and Fundamental Research Funds for the Central Universities (20720180049).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alzheimer’s Association. (2016). 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509. doi: 10.1016/j.jalz.2016.03.001
Arbel-Ornath, M., Hudry, E., Boivin, J. R., Hashimoto, T., Takeda, S., Kuchibhotla, K. V., et al. (2017). Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 12:27. doi: 10.1186/s13024-017-0169-9
Atagi, Y., Liu, C.-C., Painter, M. M., Chen, X.-F., Verbeeck, C., Zheng, H., et al. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050. doi: 10.1074/jbc.M115.679043
Bailey, C. C., DeVaux, L. B., and Farzan, M. (2015). The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 290, 26033–26042. doi: 10.1074/jbc.M115.677286
Bemiller, S. M., Mccray, T. J., Allan, K., Formica, S. V., Xu, G., Wilson, G., et al. (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 12:74. doi: 10.1186/s13024-017-0216-6
Benitez, B. A., Cruchaga, C., and United States-Spain Parkinson’s Disease Research Group. (2013). TREM2 and neurodegenerative disease. N. Engl. J. Med. 369, 1567–1568. doi: 10.1056/NEJMc1306509
Bianchin, M. M., Lima, J. E., Natel, J., and Sakamoto, A. C. (2006). The genetic causes of basal ganglia calcification, dementia and bone cysts: DAP12 and TREM2. Neurology 66, 615–616; author reply 615–616. doi: 10.1212/01.wnl.0000216105.11788.0f
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Borroni, B., Ferrari, F., Galimberti, D., Nacmias, B., Barone, C., Bagnoli, S., et al. (2014). Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 35, 934.e7–934.e10. doi: 10.1016/j.neurobiolaging.2013.09.017
Bradshaw, E. M., Chibnik, L. B., Keenan, B. T., Ottoboni, L., Raj, T., Tang, A., et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. doi: 10.1038/nn.3435
Brody, D. L., Jiang, H., Wildburger, N., and Esparza, T. J. (2017). Non-canonical soluble amyloid-β aggregates and plaque buffering: controversies and future directions for target discovery in Alzheimer’s disease. Alzheimers Res. Ther. 9:62. doi: 10.1186/s13195-017-0293-3
Brunden, K. R., Trojanowski, J. Q., and Lee, V. M. (2009). Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discov. 8, 783–793. doi: 10.1038/nrd2959
Cady, J., Koval, E. D., Benitez, B. A., Zaidman, C., Jockel-Balsarotti, J., Allred, P., et al. (2014). TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453. doi: 10.1001/jamaneurol.2013.6237
Calcagno, N., Baufeld, C., Madore, C., and Butovsky, O. (2018). TREMendous 2 be social. Immunity 48, 842–843. doi: 10.1016/j.immuni.2018.04.034
Cannon, J. P., O’Driscoll, M., and Litman, G. W. (2012). Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 64, 39–47. doi: 10.1007/s00251-011-0562-4
Charles, J. F., Humphrey, M. B., Zhao, X., Quarles, E., Nakamura, M. C., Aderem, A., et al. (2008). The innate immune response to Salmonella enterica serovar Typhimurium by macrophages is dependent on TREM2-DAP12. Infect. Immun. 76, 2439–2447. doi: 10.1128/iai.00115-08
Cheng-Hathaway, P. J., Reed-Geaghan, E. G., Jay, T. R., Casali, B. T., Bemiller, S. M., Puntambekar, S. S., et al. (2018). The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 13:29. doi: 10.1186/s13024-018-0262-8
Chiasseu, M., Alarcon-Martinez, L., Belforte, N., Quintero, H., Dotigny, F., Destroismaisons, L., et al. (2017). Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:58. doi: 10.1186/s13024-017-0199-3
Cline, E. N., Bicca, M. A., Viola, K. L., and Klein, W. L. (2018). The amyloid-β oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 64, S567–S610. doi: 10.3233/jad-179941
Colonna, M., and Wang, Y. (2016). TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207. doi: 10.1038/nrn.2016.7
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Corneveaux, J. J., Myers, A. J., Allen, A. N., Pruzin, J. J., Ramirez, M., Engel, A., et al. (2010). Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum. Mol. Genet. 19, 3295–3301. doi: 10.1093/hmg/ddq221
Daws, M. R., Sullam, P. M., Niemi, E. C., Chen, T. T., Tchao, N. K., and Seaman, W. E. (2003). Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 171, 594–599. doi: 10.4049/jimmunol.171.2.594
De Strooper, B., and Annaert, W. (2010). Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu. Rev. Cell Dev. Biol. 26, 235–260. doi: 10.1146/annurev-cellbio-100109-104117
Deng, Z., Sheehan, P., Chen, S., and Yue, Z. (2017). Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Mol. Neurodegener. 12:90. doi: 10.1186/s13024-017-0232-6
Doig, A. J. (2018). Positive feedback loops in Alzheimer’s disease: the Alzheimer’s feedback hypothesis. J. Alzheimers Dis. 66, 25–36. doi: 10.3233/jad-180583
Dos Santos, L. R., Pimassoni, L. H. S., Sena, G. G. S., Camporez, D., Belcavello, L., Trancozo, M., et al. (2017). Validating GWAS variants from microglial genes implicated in Alzheimer’s disease. J. Mol. Neurosci. 62, 215–221. doi: 10.1007/s12031-017-0928-7
Duyckaerts, C., Delatour, B., and Potier, M. C. (2009). Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36. doi: 10.1007/s00401-009-0532-1
Efthymiou, A. G., and Goate, A. M. (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Eisenberg-Lerner, A., Bialik, S., Simon, H. U., and Kimchi, A. (2009). Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 16, 966–975. doi: 10.1038/cdd.2009.33
Engstrom, A. K., Snyder, J. M., Maeda, N., and Xia, Z. (2017). Gene-environment interaction between lead and Apolipoprotein E4 causes cognitive behavior deficits in mice. Mol. Neurodegener. 12:14. doi: 10.1186/s13024-017-0155-2
Forabosco, P., Ramasamy, A., Trabzuni, D., Walker, R., Smith, C., Bras, J., et al. (2013). Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging 34, 2699–2714. doi: 10.1016/j.neurobiolaging.2013.05.001
Frank, S., Burbach, G. J., Bonin, M., Walter, M., Streit, W., Bechmann, I., et al. (2008). TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. doi: 10.1002/glia.20710
Ghavami, S., Shojaei, S., Yeganeh, B., Ande, S. R., Jangamreddy, J. R., Mehrpour, M., et al. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49. doi: 10.1016/j.pneurobio.2013.10.004
Ghosh, S., Castillo, E., Frias, E. S., and Swanson, R. A. (2018). Bioenergetic regulation of microglia. Glia 66, 1200–1212. doi: 10.1002/glia.23271
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gispert, J. D., Suarez-Calvet, M., Monte, G. C., Tucholka, A., Falcon, C., Rojas, S., et al. (2016). Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement. 12, 1259–1272. doi: 10.1016/j.jalz.2016.06.005
Guerreiro, R., and Hardy, J. (2013). TREM2 and neurodegenerative disease. N. Engl. J. Med. 369, 1569–1570. doi: 10.1056/NEJMc1306509
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Hampel, H., Blennow, K., Shaw, L. M., Hoessler, Y. C., Zetterberg, H., and Trojanowski, J. Q. (2010). Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 45, 30–40. doi: 10.1016/j.exger.2009.10.010
Heslegrave, A., Heywood, W., Paterson, R., Magdalinou, N., Svensson, J., Johansson, P., et al. (2016). Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 11:3. doi: 10.1186/s13024-016-0071-x
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hong, S., and Stevens, B. (2017). TREM2: keeping microglia fit during good times and bad. Cell Metab. 26, 590–591. doi: 10.1016/j.cmet.2017.09.010
Hung, A. S., Liang, Y., Chow, T. C., Tang, H. C., Wu, S. L., Wai, M. S., et al. (2016). Mutated tau, amyloid and neuroinflammation in Alzheimer disease-A brief review. Prog. Histochem. Cytochem. 51, 1–8. doi: 10.1016/j.proghi.2016.01.001
Jankowsky, J. L., and Zheng, H. (2017). Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:89. doi: 10.1186/s13024-017-0231-7
Jay, T. R., Hirsch, A. M., Broihier, M. L., Miller, C. M., Neilson, L. E., Ransohoff, R. M., et al. (2017a). Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37, 637–647. doi: 10.1523/JNEUROSCI.2110-16.2016
Jay, T. R., Von Saucken, V. E., and Landreth, G. E. (2017b). TREM2 in neurodegenerative diseases. Mol. Neurodegener. 12:56. doi: 10.1186/s13024-017-0197-5
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jendresen, C., Arskog, V., Daws, M. R., and Nilsson, L. N. (2017). The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflammation 14:59. doi: 10.1186/s12974-017-0835-4
Jiang, P., and Mizushima, N. (2014). Autophagy and human diseases. Cell Res. 24, 69–79. doi: 10.1038/cr.2013.161
Jiang, T., Tan, L., Chen, Q., Tan, M. S., Zhou, J. S., Zhu, X. C., et al. (2016). A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol. Aging 42, 217.e1–217.e3. doi: 10.1016/j.neurobiolaging.2016.02.023
Jin, S. C., Benitez, B. A., Karch, C. M., Cooper, B., Skorupa, T., Carrell, D., et al. (2014). Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum. Mol. Genet. 23, 5838–5846. doi: 10.1093/hmg/ddu277
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kawabori, M., Kacimi, R., Kauppinen, T., Calosing, C., Kim, J. Y., Hsieh, C. L., et al. (2015). Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 35, 3384–3396. doi: 10.1523/JNEUROSCI.2620-14.2015
Keller, M., Ruegg, A., Werner, S., and Beer, H. D. (2008). Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831. doi: 10.1016/j.cell.2007.12.040
Kim, Y., Kim, C., Son, S. M., Song, H., Hong, H. S., Han, S. H., et al. (2016). The novel RAGE interactor PRAK is associated with autophagy signaling in Alzheimer’s disease pathogenesis. Mol. Neurodegener. 11:4. doi: 10.1186/s13024-016-0068-5
Kitazawa, M., Yamasaki, T. R., and LaFerla, F. M. (2004). Microglia as a potential bridge between the amyloid β-peptide and tau. Ann. N Y Acad. Sci. 1035, 85–103. doi: 10.1196/annals.1332.006
Kleinberger, G., Brendel, M., Mracsko, E., Wefers, B., Groeneweg, L., Xiang, X., et al. (2017). The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 36, 1837–1853. doi: 10.15252/embj.201796516
Klünemann, H. H., Ridha, B. H., Magy, L., Wherrett, J. R., Hemelsoet, D. M., Keen, R. W., et al. (2005). The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64, 1502–1507. doi: 10.1212/01.wnl.0000160304.00003.ca
Kober, D. L., Alexander-Brett, J. M., Karch, C. M., Cruchaga, C., Colonna, M., Holtzman, M. J., et al. (2016). Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 5:e20391. doi: 10.7554/eLife.20391
Kober, D. L., and Brett, T. J. (2017). TREM2-ligand interactions in health and disease. J. Mol. Biol. 429, 1607–1629. doi: 10.1016/j.jmb.2017.04.004
Konishi, H., and Kiyama, H. (2018). Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front. Cell. Neurosci. 12:206. doi: 10.3389/fncel.2018.00206
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566.e9–581.e9. doi: 10.1016/j.immuni.2017.08.008
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L. L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032.e5–1048.e5. doi: 10.1016/j.neuron.2018.02.002
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Lessard, C. B., Malnik, S. L., Zhou, Y., Ladd, T. B., Cruz, P. E., Ran, Y., et al. (2018). High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol. Med. 10:e9027. doi: 10.15252/emmm.201809027
Leyns, C. E. G., and Holtzman, D. M. (2017). Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 12:50. doi: 10.1186/s13024-017-0192-x
Li, C., and Götz, J. (2017). Tau-based therapies in neurodegeneration: opportunities and challenges. Nat. Rev. Drug Discov. 16, 863–883. doi: 10.1038/nrd.2017.155
Lill, C. M., Rengmark, A., Pihlstrøm, L., Fogh, I., Shatunov, A., Sleiman, P. M., et al. (2015). The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 11, 1407–1416. doi: 10.1016/j.jalz.2014.12.009
Lucey, B. P., Fagan, A. M., Holtzman, D. M., Morris, J. C., and Bateman, R. J. (2017). Diurnal oscillation of CSF Aβ and other AD biomarkers. Mol. Neurodegener. 12:36. doi: 10.1186/s13024-017-0161-4
Lucin, K. M., O’Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., et al. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886. doi: 10.1016/j.neuron.2013.06.046
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., et al. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 12:7. doi: 10.1186/s13024-017-0150-7
Malik, M., Parikh, I., Vasquez, J. B., Smith, C., Tai, L., Bu, G., et al. (2015). Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 10:52. doi: 10.1186/s13024-015-0048-1
Metaxakis, A., Ploumi, C., and Tavernarakis, N. (2018). Autophagy in age-associated neurodegeneration. Cells 7:E37. doi: 10.3390/cells7050037
Mizushima, N. (2018). A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527. doi: 10.1038/s41556-018-0092-5
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Moors, T. E., Hoozemans, J. J., Ingrassia, A., Beccari, T., Parnetti, L., Chartier-Harlin, M. C., et al. (2017). Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 12:11. doi: 10.1186/s13024-017-0154-3
Nathalie, P., and Jean-Noël, O. (2008). Processing of amyloid precursor protein and amyloid peptide neurotoxicity. Curr. Alzheimer Res. 5, 92–99. doi: 10.2174/156720508783954721
N’Diaye, E. N., Branda, C. S., Branda, S. S., Nevarez, L., Colonna, M., Lowell, C., et al. (2009). TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J. Cell Biol. 184, 215–223. doi: 10.1083/jcb.200808080
Olsen, I., and Singhrao, S. K. (2016). Inflammasome involvement in Alzheimer’s disease. J. Alzheimers Dis. 54, 45–53. doi: 10.3233/jad-160197
Otero, K., Shinohara, M., Zhao, H., Cella, M., Gilfillan, S., Colucci, A., et al. (2012). TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J. Immunol. 188, 2612–2621. doi: 10.4049/jimmunol.1102836
Otero, K., Turnbull, I. R., Poliani, P. L., Vermi, W., Cerutti, E., Aoshi, T., et al. (2009). Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and β-catenin. Nat. Immunol. 10, 734–743. doi: 10.1038/ni.1744
Painter, M. M., Atagi, Y., Liu, C. C., Rademakers, R., Xu, H., Fryer, J. D., et al. (2015). TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 10:43. doi: 10.1186/s13024-015-0040-9
Paloneva, J., Autti, T., Raininko, R., Partanen, J., Salonen, O., Puranen, M., et al. (2001). CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology 56, 1552–1558. doi: 10.1212/wnl.56.11.1552
Paloneva, J., Kestilä, M., Wu, J., Salminen, A., Böhling, T., Ruotsalainen, V., et al. (2000). Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25, 357–361. doi: 10.1038/77153
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. doi: 10.1086/342259
Pankiewicz, J. E., Baquero-Buitrago, J., Sanchez, S., Lopez-Contreras, J., Kim, J., Sullivan, P. M., et al. (2017). APOE genotype differentially modulates effects of anti-aβ, passive immunization in APP transgenic mice. Mol. Neurodegener. 12:12. doi: 10.1186/s13024-017-0156-1
Park, M., Yi, J. W., Kim, E. M., Yoon, I. J., Lee, E. H., Lee, H. Y., et al. (2015). Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes 64, 117–127. doi: 10.2337/db13-1869
Piccio, L., Buonsanti, C., Cella, M., Tassi, I., Schmidt, R. E., Fenoglio, C., et al. (2008). Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131, 3081–3091. doi: 10.1093/brain/awn217
Plaza-Zabala, A., Sierra-Torre, V., and Sierra, A. (2017). Autophagy and microglia: novel partners in neurodegeneration and aging. Int. J. Mol. Sci. 18:E598. doi: 10.3390/ijms18030598
Poliani, P. L., Wang, Y., Fontana, E., Robinette, M. L., Yamanishi, Y., Gilfillan, S., et al. (2015). TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161–2170. doi: 10.1172/jci77983
Qu, W., Wang, Y., Wu, Y., Liu, Y., Chen, K., Liu, X., et al. (2018). Triggering receptors expressed on myeloid cells 2 promotes corneal resistance against pseudomonas aeruginosa by inhibiting caspase-1-dependent pyroptosis. Front. Immunol. 9:1121. doi: 10.3389/fimmu.2018.01121
Quan, D. N., Cooper, M. D., Potter, J. L., Roberts, M. H., Cheng, H., and Jarvis, G. A. (2008). TREM-2 binds to lipooligosaccharides of Neisseria gonorrhoeae and is expressed on reproductive tract epithelial cells. Mucosal Immunol. 1, 229–238. doi: 10.1038/mi.2008.1
Rademakers, R., Baker, M., Nicholson, A. M., Rutherford, N. J., Finch, N., Soto-Ortolaza, A., et al. (2011). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44, 200–205. doi: 10.1038/ng.1027
Rayaprolu, S., Mullen, B., Baker, M., Lynch, T., Finger, E., Seeley, W. W., et al. (2013). TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 8:19. doi: 10.1186/1750-1326-8-19
Salminen, A., Kaarniranta, K., Kauppinen, A., Ojala, J., Haapasalo, A., Soininen, H., et al. (2013). Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 106–107, 33–54. doi: 10.1016/j.pneurobio.2013.06.002
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi: 10.1038/nm.4397
Saresella, M., La Rosa, F., Piancone, F., Zoppis, M., Marventano, I., Calabrese, E., et al. (2016). The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 11:23. doi: 10.1186/s13024-016-0088-1
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249. doi: 10.1172/JCI90606
Sasaki, A. (2017). Microglia and brain macrophages: an update. Neuropathology 37, 452–464. doi: 10.1111/neup.12354
Sayed, F. A., Telpoukhovskaia, M., Kodama, L., Li, Y., Zhou, Y., Le, D., et al. (2018). Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. U S A 115, 10172–10177. doi: 10.1073/pnas.1811411115
Schafer, D. P., and Stevens, B. (2015). Microglia function in central nervous system development and plasticity. Cold Spring Harb. Perspect. Biol. 7:a020545. doi: 10.1101/cshperspect.a020545
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Sharma, D., and Kanneganti, T. D. (2016). The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J. Cell Biol. 213, 617–629. doi: 10.1083/jcb.201602089
Shekhar, S., Kumar, R., Rai, N., Kumar, V., Singh, K., Upadhyay, A. D., et al. (2016). Estimation of tau and phosphorylated Tau181 in serum of Alzheimer’s disease and mild cognitive impairment patients. PLoS One 11:e0159099. doi: 10.1371/journal.pone.0159099
Shi, Y., and Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. doi: 10.1038/s41577-018-0051-1 [Epub ahead of print].
Sims, R., van der Lee, S. J., Naj, A. C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384. doi: 10.1038/ng.3916
Song, W., Hooli, B., Mullin, K., Jin, S. C., Cella, M., Ulland, T. K., et al. (2017). Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Mol. Metab. 13, 381–387. doi: 10.1016/j.jalz.2016.07.004
Song, W. M., Joshita, S., Zhou, Y., Ulland, T. K., Gilfillan, S., and Colonna, M. (2018). Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 215, 745–760. doi: 10.1084/jem.20171529
Spillantini, M. G., and Goedert, M. (1998). Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 21, 428–433. doi: 10.1016/s0166-2236(98)01337-x
Steinberg, S., Stefansson, H., Jonsson, T., Johannsdottir, H., Ingason, A., Helgason, H., et al. (2015). Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat. Genet. 47, 445–447. doi: 10.1038/ng.3246
Tan, M. S., Tan, L., Jiang, T., Zhu, X. C., Wang, H. F., Jia, C. D., et al. (2014). Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 5:e1382. doi: 10.1038/cddis.2014.348
Tatebe, H., Kasai, T., Ohmichi, T., Kishi, Y., Kakeya, T., Waragai, M., et al. (2017). Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 12:63. doi: 10.1186/s13024-017-0206-8
Tcw, J., and Goate, A. M. (2017). Genetics of β-amyloid precursor protein in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 7:a024539. doi: 10.1101/cshperspect.a024539
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Udeochu, J., Sayed, F. A., and Gan, L. (2018). TREM2 and amyloid β: a love-hate relationship. Neuron 97, 991–993. doi: 10.1016/j.neuron.2018.02.018
Ulland, T. K., and Colonna, M. (2018). TREM2—a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675. doi: 10.1038/s41582-018-0072-1
Ulland, T. K., Song, W. M., Huang, S. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649.e13–663.e13. doi: 10.1016/j.cell.2017.07.023
Ulland, T. K., Wang, Y., and Colonna, M. (2015). Regulation of microglial survival and proliferation in health and diseases. Semin. Immunol. 27, 410–415. doi: 10.1016/j.smim.2016.03.011
Ulrich, J. D., Finn, M. B., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener. 9:20. doi: 10.1186/1750-1326-9-20
Ulrich, J. D., and Holtzman, D. M. (2016). TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem. Neurosci. 7, 420–427. doi: 10.1021/acschemneuro.5b00313
Ulrich, J. D., Ulland, T. K., Colonna, M., and Holtzman, D. M. (2017). Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94, 237–248. doi: 10.1016/j.neuron.2017.02.042
Ulrich, J. D., Ulland, T. K., Mahan, T. E., Nyström, S., Nilsson, K. P., Song, W. M., et al. (2018). ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 215, 1047–1058. doi: 10.1084/jem.20171265
Viola, K. L., and Klein, W. L. (2015). Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 129, 183–206. doi: 10.1007/s00401-015-1386-3
Wang, Y., Balaji, V., Kaniyappan, S., Krüger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12:5. doi: 10.1186/s13024-016-0143-y
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wu, K., Byers, D. E., Jin, X., Agapov, E., Alexander-Brett, J., Patel, A. C., et al. (2015). TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 212, 681–697. doi: 10.1084/jem.20141732
Wu, X. L., Piña-Crespo, J., Zhang, Y. W., Chen, X. C., and Xu, H. X. (2017). Tau-mediated neurodegeneration and potential implications in diagnosis and treatment of Alzheimer’s disease. Chin. Med. J. 130, 2978–2990. doi: 10.4103/0366-6999.220313
Wunderlich, P., Glebov, K., Kemmerling, N., Tien, N. T., Neumann, H., and Walter, J. (2013). Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036. doi: 10.1074/jbc.m113.517540
Xiang, X., Piers, T. M., Wefers, B., Zhu, K., Mallach, A., Brunner, B., et al. (2018). The Trem2 R47H Alzheimer’s risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Mol. Neurodegener. 13:49. doi: 10.1186/s13024-018-0280-6
Yeh, F. L., Hansen, D. V., and Sheng, M. (2017). TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med. 23, 512–533. doi: 10.1016/j.molmed.2017.03.008
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ and thereby facilitates uptake of amyloid-β by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yin, Z., Raj, D., Saiepour, N., Van Dam, D., Brouwer, N., Holtman, I. R., et al. (2017). Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 55, 115–122. doi: 10.1016/j.neurobiolaging.2017.03.021
Yuan, P., Condello, C., Keene, C. D., Wang, Y. M., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. doi: 10.1016/j.cell.2013.03.030
Zhang, X., Li, Y., Xu, H., and Zhang, Y. W. (2014). The γ-secretase complex: from structure to function. Front. Cell. Neurosci. 8:427. doi: 10.3389/fncel.2014.00427
Zhao, R., Hu, W., Tsai, J., Li, W., and Gan, W. B. (2017). Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:47. doi: 10.1186/s13024-017-0188-6
Zhao, Y., Li, X., Huang, T., Jiang, L. L., Tan, Z., Zhang, M., et al. (2017). Intracellular trafficking of TREM2 is regulated by presenilin 1. Exp. Mol. Med. 49:e405. doi: 10.1038/emm.2017.200
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023.e7–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zheng, H., Jia, L., Liu, C. C., Rong, Z., Zhong, L., Yang, L., et al. (2017). TREM2 promotes microglial survival by activating wnt/β-catenin pathway. J. Neurosci. 37, 1772–1784. doi: 10.1523/JNEUROSCI.2459-16.2017
Zheng, H., Liu, C. C., Atagi, Y., Chen, X. F., Jia, L., Yang, L., et al. (2016). Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 42, 132–141. doi: 10.1016/j.neurobiolaging.2016.03.004
Zhong, L., Chen, X. F., Wang, T., Wang, Z., Liao, C., Wang, Z., et al. (2017a). Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 214, 597–607. doi: 10.1084/jem.20160844
Zhong, L., Zhang, Z. L., Li, X., Liao, C., Mou, P., Wang, T., et al. (2017b). TREM2/DAP12 complex regulates inflammatory responses in microglia via the JNK signaling pathway. Front. Aging Neurosci. 9:204. doi: 10.3389/fnagi.2017.00204
Zhong, L., Chen, X. F., Zhang, Z. L., Wang, Z., Shi, X. Z., Xu, K., et al. (2015). DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877. doi: 10.1074/jbc.m115.645986
Zhong, L., Wang, Z., Wang, D., Wang, Z., Martens, Y. A., Wu, L., et al. (2018). Amyloid-β modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 13:15. doi: 10.1186/s13024-018-0247-7
Zhou, Y., Ulland, T. K., and Colonna, M. (2018). TREM2-dependent effects on microglia in Alzheimer’s disease. Front. Aging Neurosci. 10:202. doi: 10.3389/fnagi.2018.00202
Keywords: Alzheimer’s disease, TREM2, microglia, survival, metabolism
Citation: Zheng H, Cheng B, Li Y, Li X, Chen X and Zhang Y (2018) TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 10:395. doi: 10.3389/fnagi.2018.00395
Received: 15 October 2018; Accepted: 13 November 2018;
Published: 23 November 2018.
Edited by:
Xiongwei Zhu, Case Western Reserve University, United StatesReviewed by:
Md. Golam Sharoar, University of Connecticut Health Center, United StatesJian-Zhi Wang, Huazhong University of Science and Technology, China
Copyright © 2018 Zheng, Cheng, Li, Li, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honghua Zheng, aG9uZ2h1YUB4bXUuZWR1LmNu