 Etheresia Pretorius
Etheresia Pretorius Janette Bester2†
Janette Bester2† Martin J. Page
Martin J. Page Douglas B. Kell
Douglas B. Kell- 1Department of Physiological Sciences, Faculty of Science, Stellenbosch University, Stellenbosch, South Africa
- 2Department of Physiology, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
- 3School of Chemistry, The University of Manchester, Manchester, United Kingdom
- 4The Manchester Institute of Biotechnology, The University of Manchester, Manchester, United Kingdom
Many studies indicate that there is a (mainly dormant) microbial component in the progressive development of Alzheimer-type dementias (ADs); and that in the case of Gram-negative organisms, a chief culprit might be the shedding of the highly inflammagenic lipopolysaccharide (LPS) from their cell walls. We have recently shown that a highly sensitive assay for the presence of free LPS [added to platelet poor plasma (PPP)] lies in its ability (in healthy individuals) to induce blood to clot into an amyloid form. This may be observed in a SEM or in a confocal microscope when suitable amyloid stains (such as thioflavin T) are added. This process could be inhibited by human lipopolysaccharide-binding protein (LBP). In the current paper, we show using scanning electron microscopy and confocal microscopy with amyloid markers, that PPP taken from individuals with AD exhibits considerable amyloid structure when clotting is initiated with thrombin but without added LPS. Furthermore, we could show that this amyloid structure may be reversed by the addition of very small amounts of LBP. This provides further evidence for a role of microbes and their inflammagenic cell wall products and that these products may be involved in pathological clotting in individuals with AD.
Introduction
The progression of AD is accompanied by a great many observable changes, both molecular and physiological, and it is the commonest form of dementia (Takizawa et al., 2015). It is currently estimated that 5.4 million Americans have Alzheimer’s Disease and that by mid-century the number of people living with Alzheimer’s Disease in the United States alone is projected to grow to 13.8 million (Alzheimers Association, 2016). AD is not only recognized as a neuro-inflammatory but also a systemic inflammatory condition, as AD individuals present with abnormal clotting (hypercoagulation), decreased fibrinolysis (hypofibrinolysis), elevated levels of coagulation factors, hyperactivated platelets, and vascular defects that include cerebrovascular dysfunction, decreased cerebral blood flow, and blood–brain barrier (BBB) disruption (Ripollés Piquer et al., 2004; Lee et al., 2008; Cortes-Canteli et al., 2012; Bester et al., 2015; Maiese, 2015; Nielsen et al., 2015; von Bernhardi et al., 2015; Pretorius et al., 2016a).
We have previously shown that AD individuals us hematological abnormalities in terms of fibrin(ogen), platelet, and erythrocyte (RBC) structure, and this is summarized in Figure 1. In brief, AD individuals exhibit pathological levels of circulating cytokines, and “free” iron levels (albeit typically observed as serum ferritin) are also raised (Kell, 2009; Bester et al., 2013; Kell and Pretorius, 2014; Pretorius and Kell, 2014; Pretorius et al., 2016a). These circulating molecules are known to cause both hypercoagulation and hypofibrinolysis (Kell and Pretorius, 2015b). We have also suggested that, at least in part, the upregulation of cytokines and coagulation factors are due to the presence of potent circulating bacterial cell wall products, that include LPSs (Pretorius et al., 2016a). This purposely implies (as reviewed in Kell and Pretorius, 2018) that many of the pathologies seen in AD are due to the presence of the very potent circulating LPS inflammagen molecules (and other such molecules, e.g., lipoteichoic acid from Gram-positive bacteria). The presence of some sort of infection, with the infectious agents typically in a dormant state (Kell and Pretorius, 2015a; Kell et al., 2015; Potgieter et al., 2015), is central to this line of thought. It is supported by a great many papers that suggest that, although various risk factors have been identified and implicated in AD pathogenesis, including family history and genetics, central to the development of AD is in fact the presence of infections (e.g., Ripollés Piquer et al., 2004; Kamer et al., 2008a,b; Miklossy, 2008, 2011a,b; Honjo et al., 2009; Eriksson et al., 2011; Itzhaki and Wozniak, 2012; Amor et al., 2013; de Souza Rolim et al., 2014; Itzhaki, 2014; Karim et al., 2014; Shaik et al., 2014; Singhal et al., 2014; Singhrao et al., 2014; Gaur and Agnihotri, 2015; Itzhaki et al., 2016).
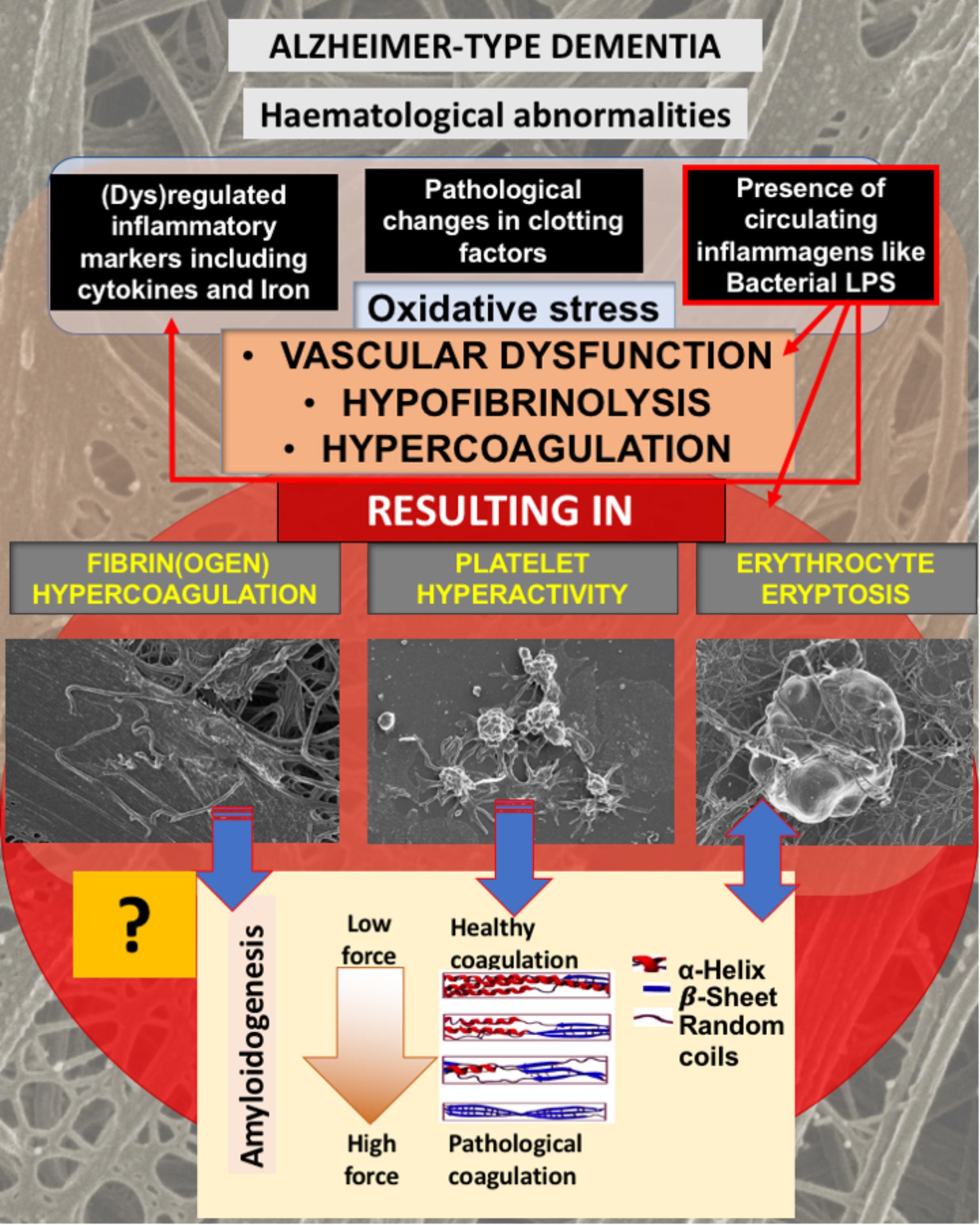
FIGURE 1. Alzheimer-type dementia (AD) is associated with hematological abnormalities that include (dys)regulated cytokines, iron and clotting factors. Increased LPS levels are also known to be present in AD. We have suggested that the presence of LPS not only is one of the causes of (dys)regulated cytokines, clotting factors and oxidative stress, but the cause of fibrin(ogen) and RBC dysfunction. We investigate here if fibrin(ogen) in AD is amyloid in nature, and if LBP can reverse fibrin(ogen) amyloid structure.
We recently reviewed the evidence that dormant, non-growing bacteria are a crucial feature of AD, that their growth in vivo is normally limited by a lack of free iron, and that it is this iron dysregulation that is an important factor in their resuscitation (Potgieter et al., 2015; Pretorius et al., 2016a; Kell and Pretorius, 2018). We have also presented evidence that bacterial cells can be observed by ultrastructural microscopy in the blood of AD patients (Pretorius et al., 2016a). A consequence of this is that these bacterial cells might shed highly inflammatory components such as LPS. LPS is known to be able to induce (apoptotic, ferroptotic, and pyroptotic; Dong et al., 2015) neuronal cell death. LPS is also raised in AD, and it is found inside the brain and closely associated with the amyloid areas in the brains of these individuals (Lee et al., 2008; Deng et al., 2014; Zhao and Lukiw, 2015; Zhao et al., 2015). Recently, Zhan and co-workers also reviewed literature showing that Gram-negative bacteria (E. coli) can induce the formation of extracellular amyloid, and that the degraded myelin basic protein (dMBP) co-localizes with β amyloid (Aβ) and LPS in amyloid plaques in AD brains (Zhan et al., 2018).
We recently also provided evidence that LPS (and LTA from Gram-positive bacteria) could induce amyloid formation in healthy fibrin(ogen), the most abundant plasma protein in blood, after it is added at tiny concentrations to blood from healthy individuals (followed by the clotting agent thrombin) (Pretorius et al., 2016b, 2018a). We then studied the presence of amyloid in these clots (before and after addition of LPS), using confocal microscopy and fluorescent markers for amyloid. In those experiments, we saw that addition of LPS to healthy PPP caused a significant increase of amyloid fluorescent signal, compared to the naïve sample (i.e., samples without added LPS). In these papers, we also showed that LBP can inhibit the formation of such amyloid structures (Pretorius et al., 2016b, 2018a). Furthermore, we showed that (some) of the (naïve) fibrin(ogen) molecules are amyloid in conditions such as type 2 diabetes and Parkinson’s Disease, and that in these conditions, LBP added to PPP of such individuals, could also reduce the extent of amyloid fibrin(ogen) structure (Pretorius et al., 2017a,b, 2018a,b).
Thus, the question now arose as to whether the extent of fibrin-type amyloid in PPP varies between AD individuals and suitably matched controls, and whether the removal of any LPS using the mopping agent, LBP, could remove the amyloid signal present in the (naïve) plasma of AD individuals.
Indeed, Zhang et al. (2009) reported elevated levels of LPS concentrations in plasma from patients with sporadic amyotrophic lateral sclerosis and AD, as compared to healthy controls. The present paper provides further evidence of the presence of LPS in PPP of AD individuals, as we showed that LBP could remove amyloid (fluorescent) signal from AD plasma. Our observation is therefore consistent with the general view set out above that there is a major dormant microbial component to AD.
Materials and Methods
Ethical Statement, Volunteer Details, and Blood Collection
Blood samples were obtained from non-smoking, Alzheimer-type dementia (AD) patients, identified by a Neurologist and under the care of a medical practitioner. Specifically, care was taken to exclude vascular dementia. We also recruited “healthy” age-matched individuals that did not smoke. It should be noted that the term “healthy” is used in this paper to describe an individual who does not have dementia. Ethical clearance was obtained from the Health Sciences Ethical committee from the University of Pretoria, and informed consent was obtained from family members who act as carers of the patients (81/2013, amended 2015). Healthy individuals also filled in consent forms. Blood was collected in two 4 mL citrate tubes and one 4 mL clotting tube for iron level determination. This collection and all handling of samples were performed under very strictly aseptic conditions, to prevent any microbial contamination of samples.
Iron Tests
Serum ferritin, transferrin, and serum iron was tested at a pathology laboratory in South Africa.
LPS-Binding Protein
A final added LBP exposure concentration of 4 ng L-1 LBP was used and LBP was purchased from Sigma (recombinant product SRP6033; >95% pure).
Scanning Electron Microscopy (SEM) of Platelet Poor Plasma (PPP)
At least 30 min after the blood was collected in citrate tubes by venepuncture, PPP were obtained and frozen at -80°C. PPP was prepared by centrifuging citrated whole blood for 15 min at 3,000 g at room temperature. After all samples were collected, PPP were thawed and 10 μL mixed with 5 μL thrombin to create an extensive fibrin network. Thrombin was provided by the South African National Blood Service, and the thrombin solution was at a final exposure concentration of 10 U mL-1 (initial product concentration is 20 U mL-1 made up in PBS containing 0.2% human serum albumin, see footnote 1 for a description of how thrombin units are calculated). A Zeiss ULTRA Plus FEG-SEM with InLens capabilities was used to study the surface morphology of erythrocytes, and micrographs were taken at 1 kV. SEM preparation was done as previously reported (Pretorius et al., 2017c).
Airyscan Confocal Microscopy
PPP was thawed, followed by preparation of clots for analysis using confocal Airyscan methods. We added Thioflavin T (ThT) (a well-established amyloid stain; LeVine, 1999; Biancalana et al., 2009; Biancalana and Koide, 2010; Groenning, 2010; Sulatskaya et al., 2011, 2012; Kuznetsova et al., 2012; Picken and Herrera, 2012; Younan and Viles, 2015; Kuznetsova et al., 2016; Rybicka et al., 2016) at a final concentration of 5 M to 200 μL to either healthy PPP, naïve AD PPP, or after a 10 min exposure of AD PPP to 4 ng L-1 (final concentration) LBP. These PPP samples were incubated (protected from light) for 1 min. This step was followed with the addition of thrombin, added in the ratio 1:2 to create extensive fibrin networks. A coverslip was placed over the prepared clot, and viewed immediately with a Zeiss LSM 510 META confocal microscope with super-resolution (Airyscan) capabilities. The Airyscan detector increases the resolution by a factor of 1.7, achieving super-resolution of 140 nm, and with a Plan-Apochromat 63×/1.4 Oil DIC objective. Excitation was at 488 nm and emitted light was measured at 505–550 nm.
Statistical Analysis and Data-Sharing
Histogram-Based Analysis of SEM and ThT Staining
For each picture, we obtained the histogram of intensities (8-bit scale) using the histogram function of ImageJ. From this we calculated the coefficient of variation (CV; as standard deviation/mean). For details of this analysis method, see (Pretorius et al., 2017b, 2018a). Quantification of fluorescent marker binding (ThT) was done by assessing the variance between (black) background and the presence of fluorescent pixels where ThT fluorescent binding was present in the clots. Increased ThT binding is here reflected as increased fluorescence which shows increased amyloid protein structure in fibrin(ogen) (see Pretorius et al., 2017a, 2018a) for a detailed explanation of the methods. We used the histogram function in ImageJ (FIJI) and calculated the coefficient of variation (CV) (as SD/mean) of the histogram of different pixel intensities as our metric to quantify and discriminate between clots of healthy (age-controlled) naïve PPP and clots from AD with and without LBP.
A healthy clot (i.e., a clot taken from a healthy individual), viewed with SEM looks somewhat like a bowl of spaghetti with elongated fibrin fibers. In AD individuals, this clot structure changes to a dense and matted hypercoagulated clot (Bester et al., 2015). We also used the CV calculation described above to analyze SEM clots. The fibrin fibers of healthy individuals have a greater variation of dark and light areas, due to the elongated fibers, with open areas between the individual fibers. With an increased hypercoagulability and amyloid formation, the clots become matted and dense, resulting in a more uniform grayness. We used this difference in structure as our metric, where increased hypercoagulability is related to an increase in amyloid formation and this is visible as a more uniformly dense morphology with less color gradient.
The statistical analysis of CV data was performed with GraphPad 7, using the one-way ANOVA analysis with Tukey’s multiple comparison’s test comparing the mean of each column with the mean of every other column.
Availability of Data and Material
Raw data, including original micrographs can be accessed at: https://1drv.ms/f/s!AgoCOmY3bkKHiJRrop6cF6uhTnQA1A or https://www.researchgate.net/profile/Etheresia_Pretorius.
Results
As discussed in the introduction, AD is not only known for the presence of neuroinflammation, but also for the presence of hematological abnormalities, including an increased presence of LPS and also (dysregulated) cytokines, iron and clotting factors, which result in oxidative stress and abnormal clotting. Previously we showed that abnormal clotting and the presence of bacterial inflammagens like LPS, result in fibrin(ogen) becoming amyloid in nature, and that we can remove the signal by addition of LBP (Pretorius et al., 2017a,b, 2018b). In Figure 1, we set out our hypothesis: that also in AD, the presence of LPS, together with dysregulated iron levels and oxidative stress, causes fibrin(ogen) to become amyloid and that we can reverse this with LBP. Furthermore, we show this reversal by using both ultrastructure (SEM) and the fluorescent marker ThT using Airyscan (confocal) microscopy. The rationale behind using LBP is that, if the amyloid structure is indeed due to the presence of bacterial inflammagens, LBP would remove it by binding to these inflammagnes, thus preventing it from causing amyloid fibrin(ogen) deposits.
Table 1 shows the demographics of individuals with AD, as well as healthy, age-controlled individuals. Transferrin, iron, % saturation of iron and serum ferritin were measured in these individuals, and these values, particularly serum ferritin, is used as an indication of the level of systemic inflammation (Kell, 2009; Kell and Pretorius, 2014).
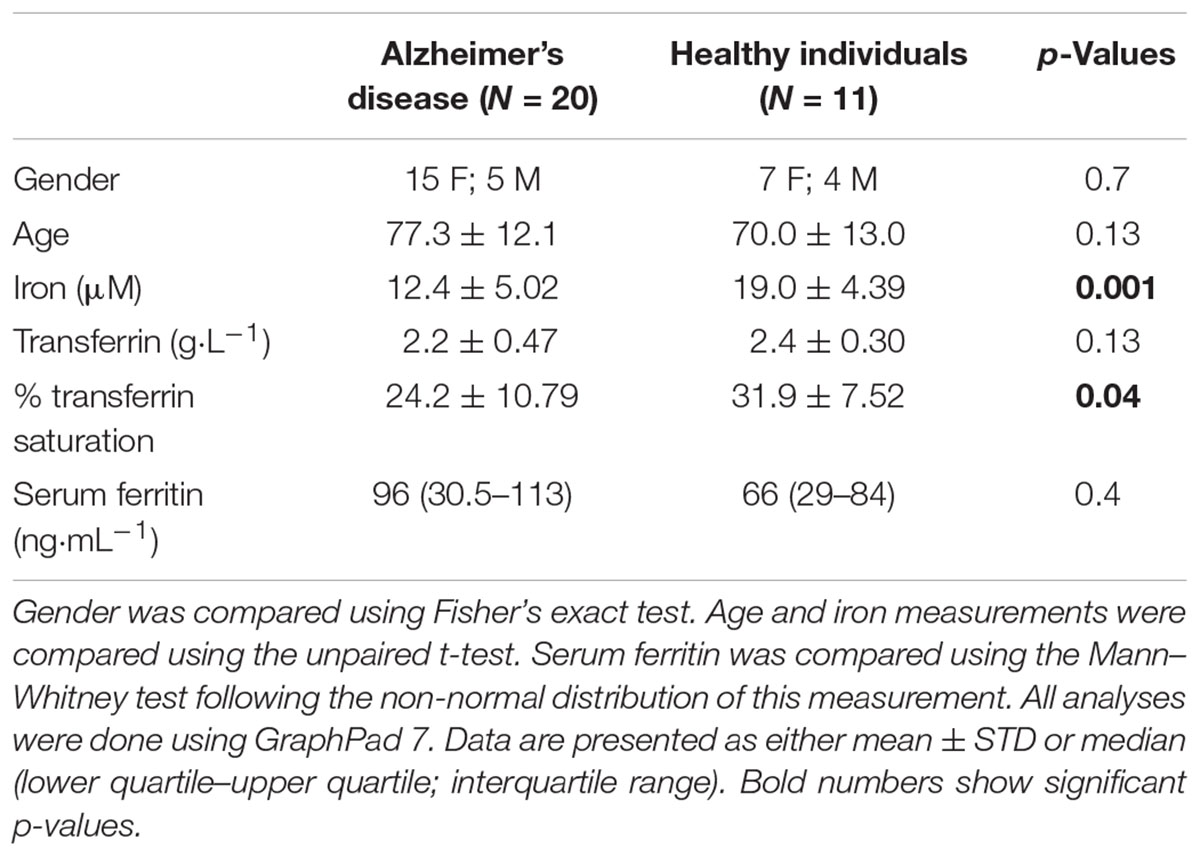
TABLE 1. Demographics for the healthy and the Alzheimer-type dementia individuals used in this study.
In our hypothesis and Figure 1 we argue that there is a link between oxidative stress, increased iron levels and inflammation, and this is directly linked to the presence of bacterial inflammagens like LPS. In our sample, healthy individuals had low mean serum ferritin, where in the AD population it was approximately three times higher. However, despite the large difference in mean serum ferritin values between the two groups, the difference was not statistically significant owing to large variation within the samples.
Table 2 shows results for the analysis of the clots using both SEM and confocal microscopy. Micrographs were analyzed as discussed in Section “Materials and Methods”. Table 2 shows p-values and statistics of CVs calculated from SEM (micrographs showing ultrastructure) and Airyscan (micrographs showing fluorescence). We compared CVs from controls and AD individuals, and that produced the p-values (Table 2).
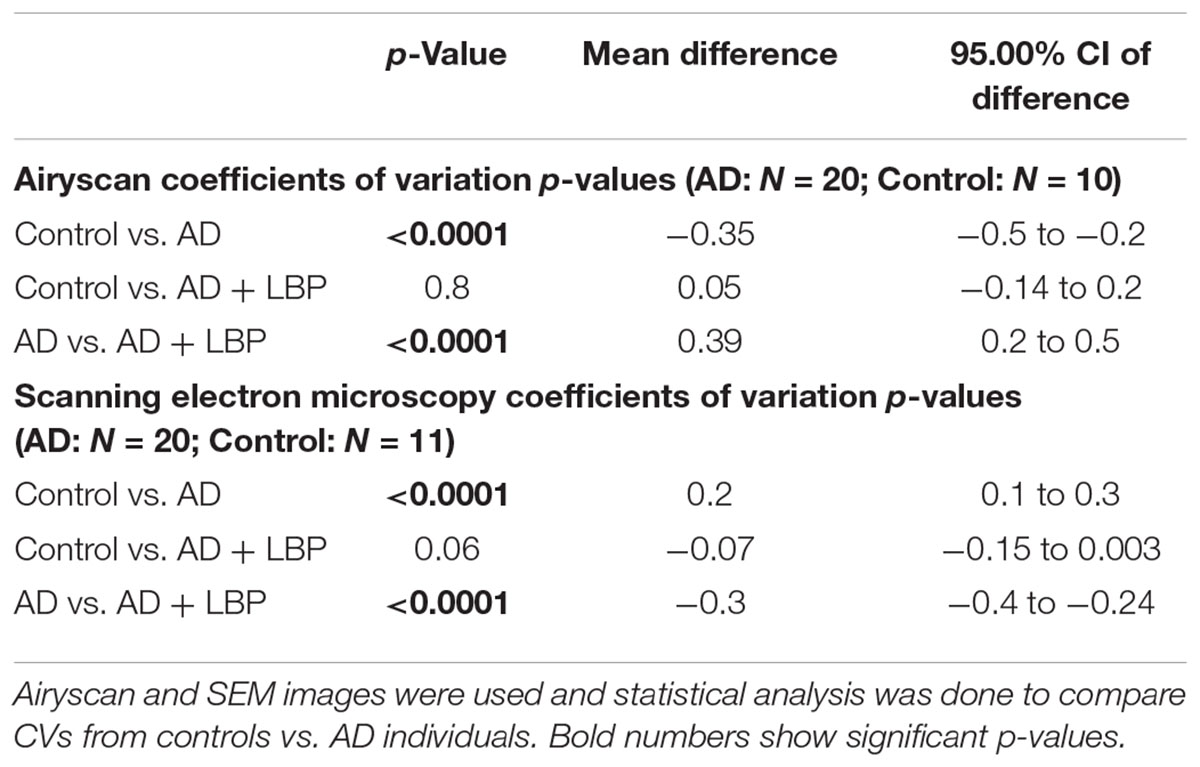
TABLE 2. Data for Alzheimer-type dementia and healthy individuals showing the coefficients of variation (CV) of the intensity of the pixels in the clot images (Tukey’s analysis).
Figure 2A gives an example of the clot structure, as viewed with SEM, from a representative healthy individual. We analyzed each SEM micrograph with ImageJ and produced a histogram that gave us the mean and the standard deviation for each micrograph (see section “Materials and Methods”). Figure 2B shows such a representative histogram of the 8-bit intensity for the SEM micrograph shown in Figure 2A. All micrograph histograms were used to calculate the CVs for each participant (both controls and AD individuals) (statistical analysis shown in Table 2). Figure 3 shows SEM images before and after treatment of a representative examples of three AD PPP clots, with and without LBP.
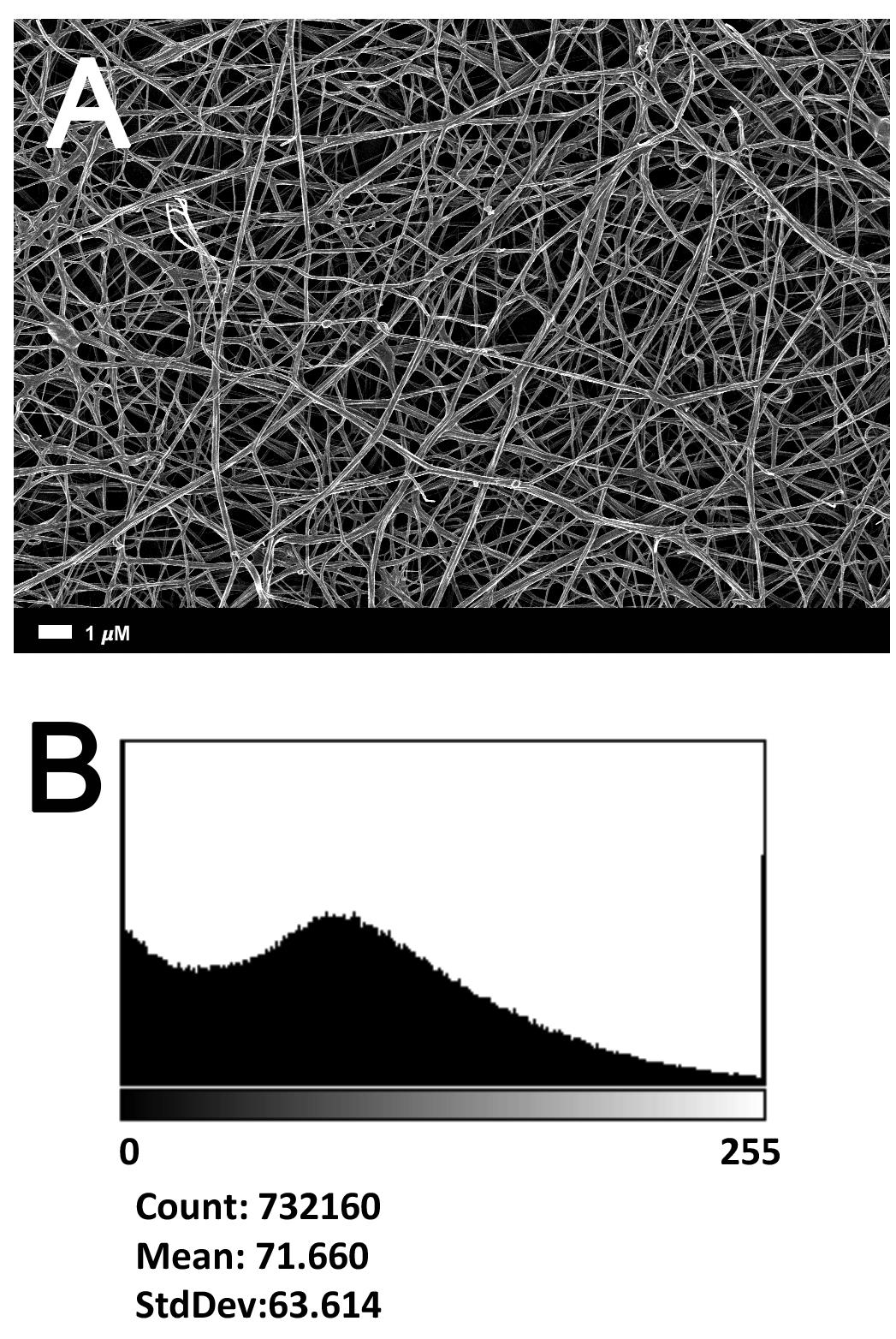
FIGURE 2. (A) Clot structure from a representative healthy individual as seen with SEM. All clots were created by adding thrombin to PPP (prepared after whole blood is centrifuged for 15 min at 3,000 g). (B) Representative histogram of the 8-bit intensity for the SEM clot shown in (A).
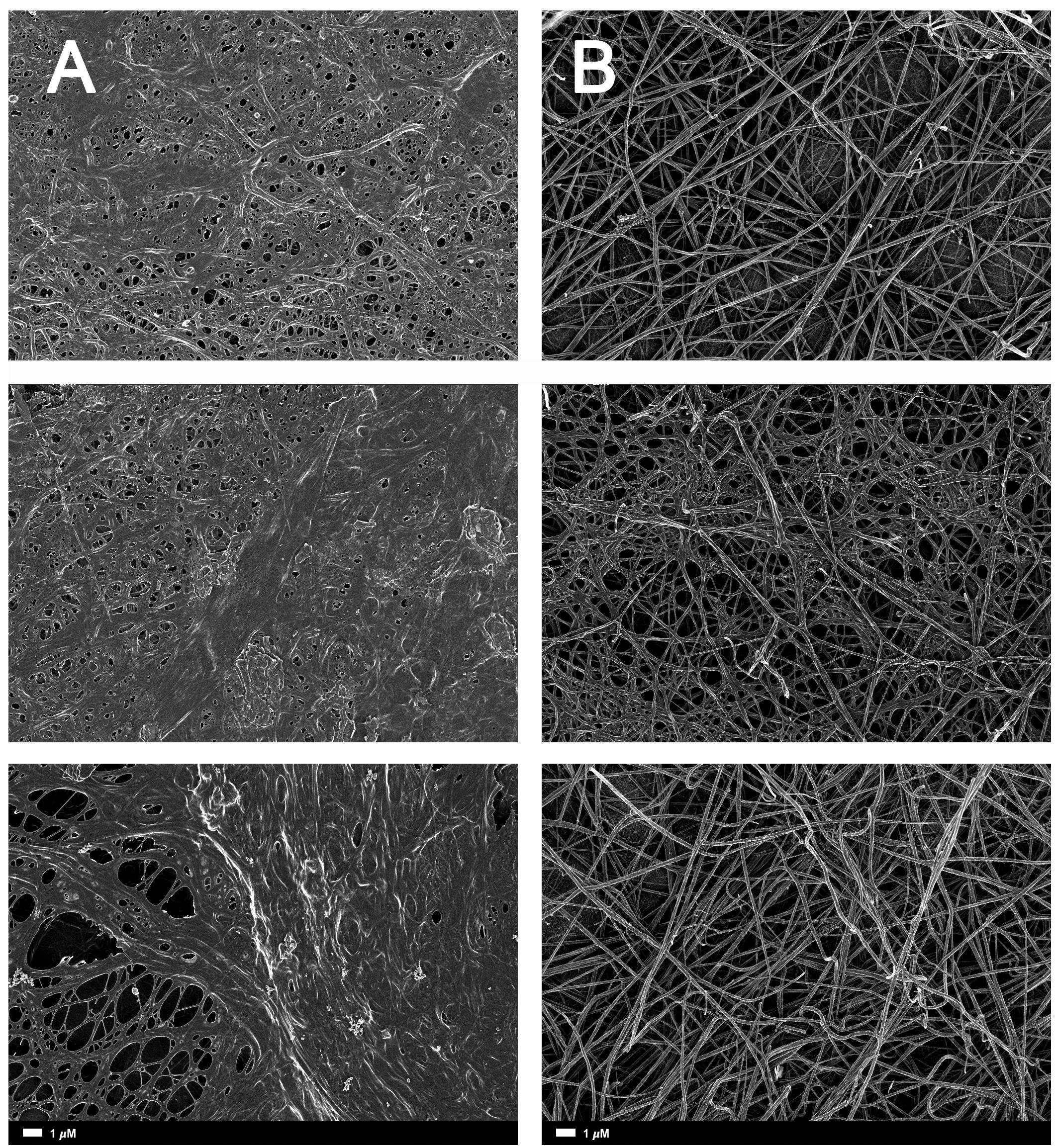
FIGURE 3. (A) Naïve clot structures from representative Alzheimer-type dementia individuals as seen with SEM. (B) The same samples treated with LBP. The size marker is the same for all panels.
Figure 4 show a representative micrograph and its histogram from a healthy individual, using Airyscan confocal microscopy. Figure 5 shows clots from AD individuals before and after LBP treatment. In healthy clots, there is little to no binding of ThT to amyloid fibrin(ogen) proteins. In AD clots, significant ThT binding fluorescence is noted, suggesting increased amyloid formation in fibrin(ogen). When LBP is added to AD PPP, ThT show significantly decreased binding. Figures 6A,B show graphs and boxplots from the CV analysis. LBP added to PPP from AD individuals (with added thrombin to initiate clotting), seems to aid in the removal of amyloid signal so that the fibrin(ogen) structure now looks more like that of the controls (noted by using two techniques: Airyscan and SEM). Furthermore, the p-values between controls vs. AD with added LBP in both the Airyscan and SEM analysis, showed that added LBP makes AD clots not significantly different to the controls (p = 0.8 and 0.06).
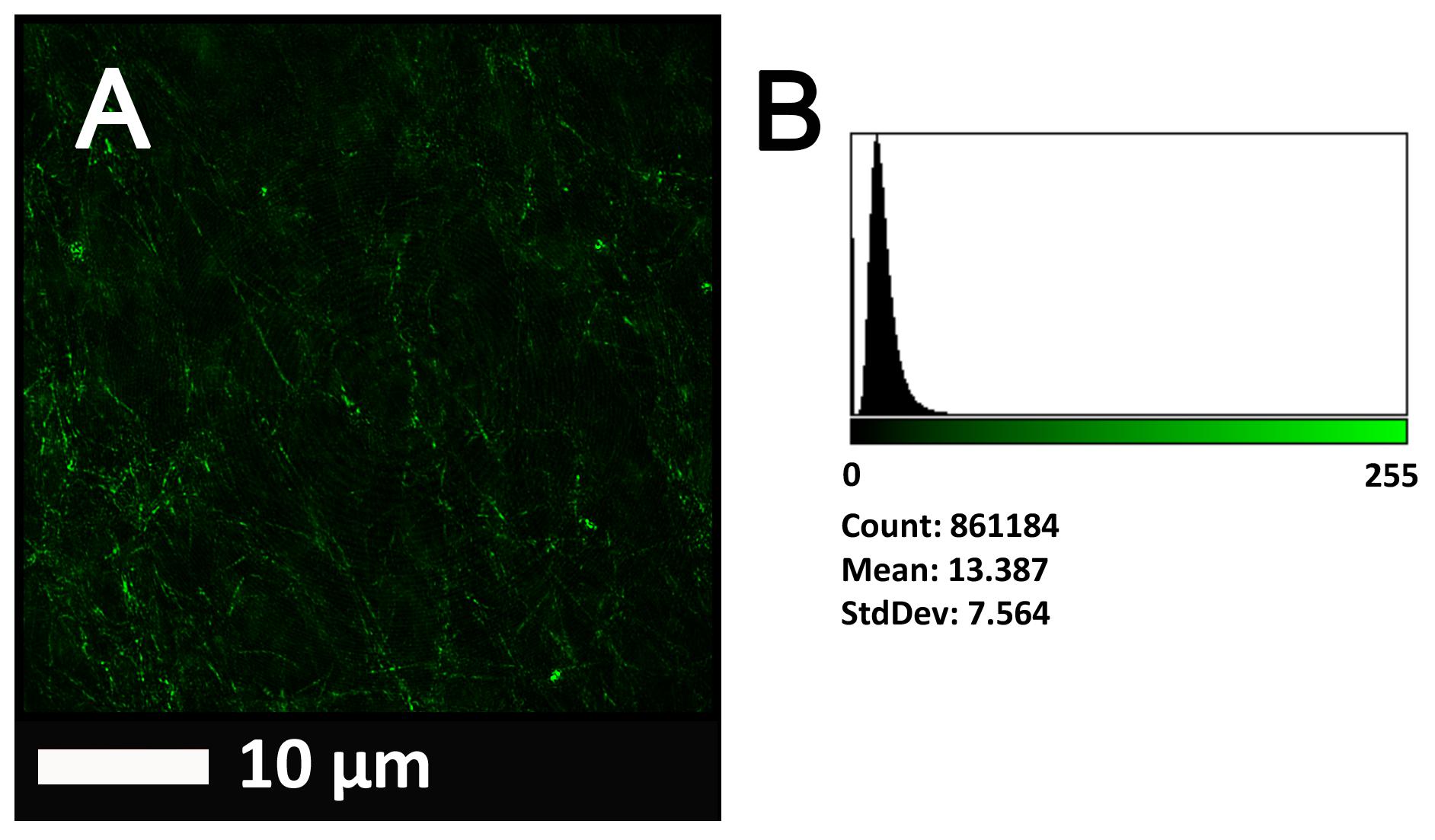
FIGURE 4. (A) Clot structure from a representative healthy individual as seen with Airyscan super-resolution confocal microscopy. PPP from each individual was incubated with the fluorescent marker ThT. PPP were mixed with thrombin to create an extensive fibrin network. (B) Representative histogram of the 8-bit intensity for the Airyscan clot shown in (A).
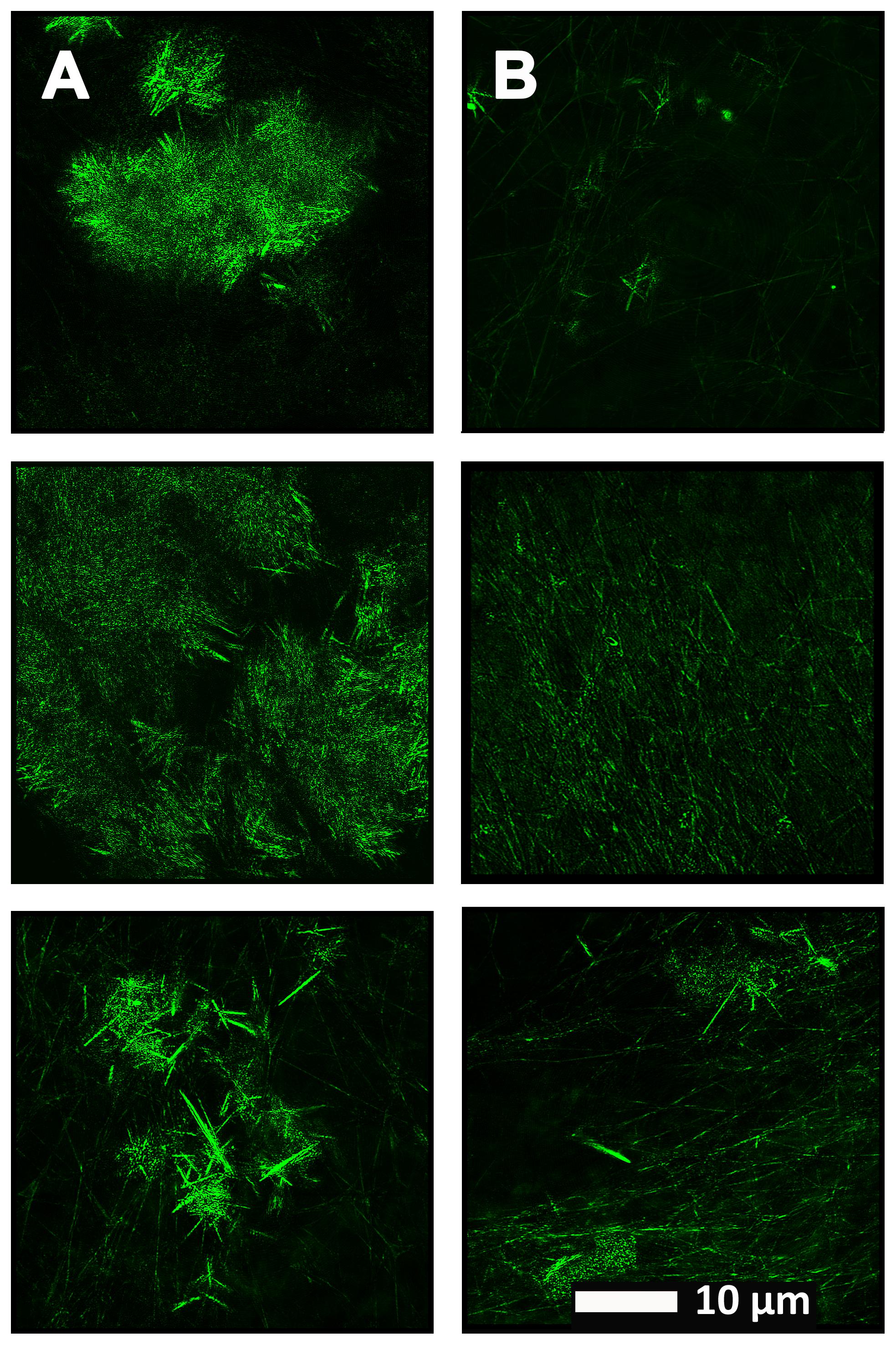
FIGURE 5. (A) Naïve clot structure from representative Alzheimer-type dementia individuals as seen with Airyscan super-resolution confocal microscopy. PPP from each individual was incubated with the fluorescent marker ThT. PPP were mixed with thrombin to create an extensive fibrin network. (B) Micrograph of the PPP clots from the same individual in the opposite column (A), after treatment with LBP, followed by addition of ThT and clot preparation.
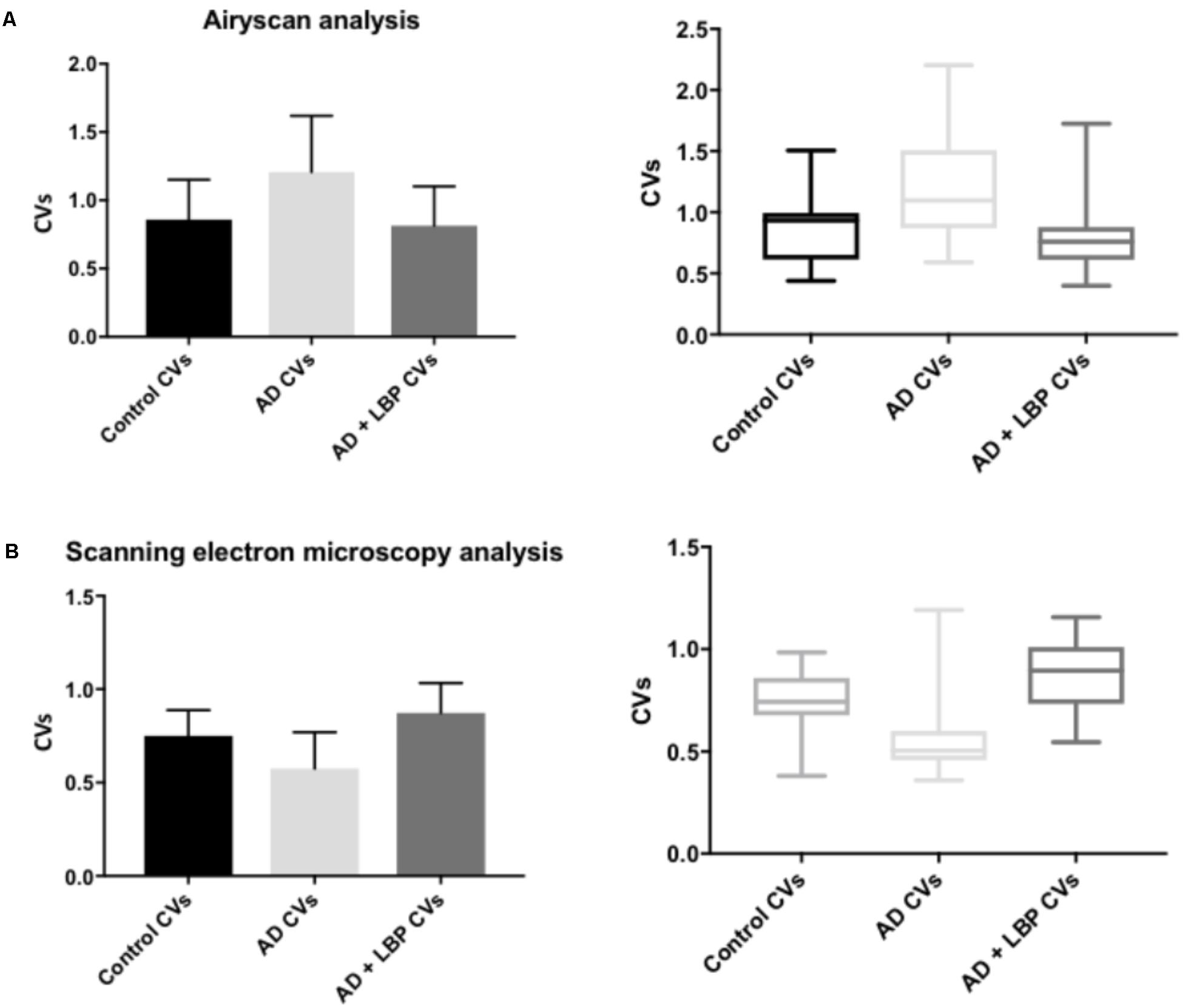
FIGURE 6. Graphs and boxplots from coefficient of variation (CVs) form histogram data of Airyscan analysis (A) and SEM (B). Coefficients of variation (CV) of the intensity of the pixels in the clot images was done using the Tukey’s analysis. Control vs. AD and AD vs. AD + LBP significantly differ by < 0.0001. Control vs. AD + LBP are not significantly different.
Discussion
We have previously determined that in many inflammatory conditions, the “normal” clotting of blood, involving the polymerisation of fibrinogen to fibrin, produces a fibrin fiber structure that becomes amyloid in nature, and that this might be due to the presence (in part) of the potent inflammagen LPS, which comes from the membranes of Gram-negative bacteria (Potgieter et al., 2015, 2016b, 2017b, 2018a; Kell and Pretorius, 2017a,b) and is a potent inflammagen (Walter et al., 2007; Kell and Pretorius, 2015a). This would be consistent with the many studies (reviewed in Miklossy, 2015; Itzhaki et al., 2016; Kell and Pretorius, 2018) that imply that there is a (dormant) microbial component in AD. Previous research (see Poole et al., 2013; Bester et al., 2015; Zhan et al., 2016a,b; Zhao et al., 2017a,b) found LPS inside the brains of Alzheimer’s disease patients, as well as an increase in circulating LPS. LPS is known to cross (and possibly to damage Liu et al., 2001; Xaio et al., 2001; Jaeger et al., 2009; Jangula and Murphy, 2013; Banks et al., 2015) the BBB and lead to β-amyloid depositions (Lee et al., 2008). Furthermore, neurotoxic microbial-derived components from the GI tract microbiome can cross aging GI tracts and BBBs and contribute to progressive proinflammatory neurodegeneration (Zhao and Lukiw, 2018). In a recent review, Zhan and co-workers describe that LPS indeed associates with amyloid plaques, neurons and oligodendrocytes in AD brains (Zhan et al., 2018). These authors also showed that LPS infiltrates the AD nucleus and can induce an inflammatory signaling program in brain cells, including up-regulation of the pro-inflammatory microRNA miRNA-146a via a NF-kB signaling circuit (Zhan et al., 2018).
Here we also show that the hypercoagulable structure of fibrin(ogen) in AD patients is different from healthy individuals, in that they appear to be amyloid (as shown with the fluorescent marker ThT) and that their structure, viewed with SEM, is matted and dense. In healthy clots, fibrin has a typical “spaghetti-like” structure (Kell and Pretorius, 2015b). We could reverse aberrant clotting in AD PPP by the addition of LBP. LBP binds bacterial inflammagens and our results would therefore point to the presence of bacterial inflammagens in AD PPP – that is, LBP could bind to and thus prevent these inflammagens from causing amyloid formation in the AD PPP when clots are formed after addition of thrombin.
When we added LBP to PPP from AD individuals (by incubating their PPP with LBP), we showed that the p-values were not significantly different (p = 0.8 and 0.06) between AD and control donor blood. Therefore, LBP, incorporated in a therapy, might not only prevent aberrant clotting in these individuals, but might also reduce the circulating LPS pool that could eventually cross into their brains via the BBB. Of course, a damaged BBB can admit the transfer (atopobiosis; Potgieter et al., 2015) of the organisms themselves (Miklossy, 2011a, 2015; Bajpai et al., 2014; Tang et al., 2017), where they may be detected ultrastructurally (Mattman, 2001), and that may continue to shed inflammagens. We therefore suggest that LBP might eventually be used as treatment to prevent the damaging effect of LPS on fibrin(ogen) and hypercoagulation, and even to prevent (at least in part) the deposition of amyloid-β (Aβ) plaques in the brain and the loss of cognitive function that accompanies this neurodegenerative disease. However, we note that a control protein, such as human IgG should, in future, be used to present the specific effect of LBP on amyloid formation, to further elucidate the physiological processes discussed in this paper. In future, our hypothesis could also be tested in a transgenic murine model of AD (TgAD) or the 5xFAD (amyloid over-producing) model or equivalent (Vale et al., 2010; Jeong et al., 2018).
Author Contributions
EP study leader, prepared all the figures, and co-wrote the paper. JB prepared and analyzed all the samples. MP statistical analysis and the paper editing. DK study co-leader, and co-wrote and edited the paper. All authors reviewed the manuscript.
Funding
We thank the Biotechnology and Biological Sciences Research Council (Grant No. BB/L025752/1) as well as the National Research Foundation (NRF) of South Africa (91548: Competitive Program) and the Medical Research Council of South Africa (MRC) (Self-Initiated Research Program) for supporting this collaboration.
Conflict of Interest Statement
The authors (DK and EP) declare the following patent application: method for treating Alzheimer’s Disease (P3448ZA00-AS2CA).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This is paper 16 in the series “a dormant blood microbiome in chronic, inflammatory diseases.“ The authors thank Dr. Prashilla Soma: Clinician.
Abbreviations
AD, Alzheimer-type dementia; LBP, lipopolysaccharide-binding protein; LPS, lipopolysaccharide; PPP, platelet poor plasma; SEM, scanning electron microscope.
Footnotes
- ^ https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/1/t6884pis.pdf
References
Alzheimers Association (2016). 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509.
Amor, S., Peferoen, L. A., Vogel, D. Y., Breur, M., van der Valk, P., Baker, D., et al. (2013). Inflammation in neurodegenerative diseases - an update. Immunology 142, 151–166. doi: 10.1111/imm.12233
Bajpai, A., Prasad, K. N., Mishra, P., Singh, A. K., Gupta, R. K., and Ojha, B. K. (2014). Distinct cytokine pattern in response to different bacterial pathogens in human brain abscess. J. Neuroimmunol. 273, 96–102. doi: 10.1016/j.jneuroim.2014.05.009
Banks, W. A., Gray, A. M., Erickson, M. A., Salameh, T. S., Damodarasamy, M., Sheibani, N., et al. (2015). Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflammation 12:223. doi: 10.1186/s12974-015-0434-1
Bester, J., Buys, A. V., Lipinski, B., Kell, D. B., and Pretorius, E. (2013). High ferritin levels have major effects on the morphology of erythrocytes in Alzheimer’s disease. Front. Aging Neurosci. 5:88. doi: 10.3389/fnagi.2013.00088
Bester, J., Soma, P., Kell, D. B., and Pretorius, E. (2015). Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 6, 35284–35303. doi: 10.18632/oncotarget.6074
Biancalana, M., and Koide, S. (2010). Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412. doi: 10.1016/j.bbapap.2010.04.001
Biancalana, M., Makabe, K., Koide, A., and Koide, S. (2009). Molecular mechanism of thioflavin-T binding to the surface of beta-rich peptide self-assemblies. J. Mol. Biol. 385, 1052–1063. doi: 10.1016/j.jmb.2008.11.006
Cortes-Canteli, M., Zamolodchikov, D., Ahn, H. J., Strickland, S., and Norris, E. H. (2012). Fibrinogen and altered hemostasis in Alzheimer’s disease. J. Alzheimers Dis. 32, 599–608. doi: 10.3233/jad-2012-120820
de Souza Rolim, T., Fabri, G. M., Nitrini, R., Anghinah, R., Teixeira, M. J., de Siqueira, J. T., et al. (2014). Oral infections and orofacial pain in Alzheimer’s disease: a case-control study. J. Alzheimers Dis. 38, 823–829. doi: 10.3233/jad-131283
Deng, X., Li, M., Ai, W., He, L., Lu, D., Patrylo, P. R., et al. (2014). Lipolysaccharide-induced neuroinflammation is associated with alzheimer-like amyloidogenic axonal pathology and dendritic degeneration in rats. Adv. Alzheimer Dis. 3, 78–93. doi: 10.4236/aad.2014.32009
Dong, T., Liao, D., Liu, X., and Lei, X. (2015). Using small molecules to dissect non-apoptotic programmed cell death: necroptosis, ferroptosis, and pyroptosis. ChemBioChem 16, 2557–2561. doi: 10.1002/cbic.201500422
Eriksson, I., Gustafson, Y., Fagerstrom, L., and Olofsson, B. (2011). Urinary tract infection in very old women is associated with delirium. Int. Psychogeriatr. 23, 496–502. doi: 10.1017/s1041610210001456
Gaur, S., and Agnihotri, R. (2015). Alzheimer’s disease and chronic periodontitis: is there an association? Geriatr. Gerontol. Int. 15, 391–404. doi: 10.1111/ggi.12425
Groenning, M. (2010). Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J. Chem. Biol. 3, 1–18. doi: 10.1007/s12154-009-0027-5
Honjo, K., van Reekum, R., and Verhoeff, N. P. (2009). Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 5, 348–360. doi: 10.1016/j.jalz.2008.12.001
Itzhaki, R. F. (2014). Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus. Front. Aging Neurosci. 6:202. doi: 10.3389/fnagi.2014.00202
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Braak, H., Bearer, E. L., et al. (2016). Microbes and Alzheimer’s Disease. J. Alzheimers Dis. 51, 979–984. doi: 10.3233/JAD-160152
Itzhaki, R. F., and Wozniak, M. A. (2012). Could antivirals be used to treat Alzheimer’s disease? Future Microbiol. 7, 307–309. doi: 10.2217/fmb.12.10
Jaeger, L. B., Dohgu, S., Sultana, R., Lynch, J. L., Owen, J. B., Erickson, M. A., et al. (2009). Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 23, 507–517. doi: 10.1016/j.bbi.2009.01.017
Jangula, A., and Murphy, E. J. (2013). Lipopolysaccharide-induced blood brain barrier permeability is enhanced by alpha-synuclein expression. Neurosci. Lett. 551, 23–27. doi: 10.1016/j.neulet.2013.06.058
Jeong, Y. O., Shin, S. J., Park, J. Y., Ku, B. K., Song, J. S., Kim, J. J., et al. (2018). MK-0677, a ghrelin agonist, alleviates amyloid beta-related pathology in 5XFAD mice, an animal model of Alzheimer’s disease. Int. J. Mol. Sci. 19:E1800. doi: 10.3390/ijms19061800
Kamer, A. R., Craig, R. G., Dasanayake, A. P., Brys, M., Glodzik-Sobanska, L., and de Leon, M. J. (2008a). Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimers Dement. 4, 242–250. doi: 10.1016/j.jalz.2007.08.004
Kamer, A. R., Dasanayake, A. P., Craig, R. G., Glodzik-Sobanska, L., Bry, M., and de Leon, M. J. (2008b). Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J. Alzheimers Dis. 13, 437–449.
Karim, S., Mirza, Z., Kamal, M. A., Abuzenadah, A. M., Azhar, E. I., Al-Qahtani, M. H., et al. (2014). The role of viruses in neurodegenerative and neurobehavioral diseases. CNS Neurol. Disord. Drug Targets 13, 1213–1223. doi: 10.2174/187152731307141015122638
Kell, D. B. (2009). Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genomics 2:2. doi: 10.1186/1755-8794-2-2
Kell, D. B., Potgieter, M., and Pretorius, E. (2015). Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology. F1000Res. 4:179. doi: 10.12688/f1000research.6709.1
Kell, D. B., and Pretorius, E. (2014). Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 6, 748–773. doi: 10.1039/c3mt00347g
Kell, D. B., and Pretorius, E. (2015a). On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr. Biol. 7, 1339–1377. doi: 10.1039/C5IB00158G
Kell, D. B., and Pretorius, E. (2015b). The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr. Biol. 7, 24–52. doi: 10.1039/C4IB00173G
Kell, D. B., and Pretorius, E. (2017a). Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Prog. Biophys. Mol. Biol. 123, 16–41. doi: 10.1016/j.pbiomolbio.2016.08.006
Kell, D. B., and Pretorius, E. (2017b). To what extent are the terminal stages of sepsis, septic shock, SIRS, and multiple organ dysfunction syndrome actually driven by a toxic prion/amyloid form of fibrin? Semin. Thromb. Hemost. 44, 224–238. doi: 10.1055/s-0037-1604108
Kell, D. B., and Pretorius, E. (2018). No effects without causes. The Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases. Biol. Rev. Camb. Philos. Soc. 93, 1518–1557. doi: 10.1111/brv.12407
Kuznetsova, I. M., Sulatskaya, A. I., Maskevich, A. A., Uversky, V. N., and Turoverov, K. K. (2016). High fluorescence anisotropy of thioflavin T in aqueous solution resulting from its molecular rotor nature. Anal. Chem. 88, 718–724. doi: 10.1021/acs.analchem.5b02747
Kuznetsova, I. M., Sulatskaya, A. I., Uversky, V. N., and Turoverov, K. K. (2012). Analyzing thioflavin T binding to amyloid fibrils by an equilibrium microdialysis-based technique. PLoS One 7:e30724. doi: 10.1371/journal.pone.0030724
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5:37. doi: 10.1186/1742-2094-5-37
LeVine, H. III (1999). Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284. doi: 10.1016/S0076-6879(99)09020-5
Liu, B., Wang, K., Gao, H. M., Mandavilli, B., Wang, J. Y., and Hong, J. S. (2001). Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J. Neurochem. 77, 182–189. doi: 10.1046/j.1471-4159.2001.00216.x
Maiese, K. (2015). A novel prescription for Alzheimer’s disease: targeting hypercoagulable states. Curr. Neurovasc. Res. 12, 1–3. doi: 10.2174/1567202612666150107162329
Mattman, L. (2001). Cell Wall Deficient forms: Stealth Pathogens, 3rd Edn. Boca Raton, FL: CRC Press.
Miklossy, J. (2008). Chronic inflammation and amyloidogenesis in Alzheimer’s disease – role of Spirochetes. J. Alzheimers Dis. 13, 381–391. doi: 10.3233/JAD-2008-13404
Miklossy, J. (2011a). Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflammation 8:90. doi: 10.1186/1742-2094-8-90
Miklossy, J. (2011b). Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med. 13:e30. doi: 10.1017/s1462399411002006
Miklossy, J. (2015). Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front. Aging Neurosci. 7:46. doi: 10.3389/fnagi.2015.00046
Nielsen, V. G., Pretorius, E., Bester, J., Jacobsen, W. K., Boyle, P. K., and Reinhard, J. P. (2015). Carbon monoxide and iron modulate plasmatic coagulation in Alzheimer’s disease. Curr. Neurovasc. Res. 12, 31–39. doi: 10.2174/1567202612666150102150042
Picken, M. M., and Herrera, G. A. (2012). “Thioflavin T stain: an easier and more sensitive method for amyloid detection,” in Amyloid and Related Disorders. Current Clinical Pathology, eds M. M. Picken, G. A. Herrera, and A. Dogan (Switzerland: Humana Press).
Poole, S., Singhrao, S. K., Kesavalu, L., Curtis, M. A., and Crean, S. (2013). Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis. 36, 665–677. doi: 10.3233/jad-121918
Potgieter, M., Bester, J., Kell, D. B., and Pretorius, E. (2015). The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 39, 567–591. doi: 10.1093/femsre/fuv013
Pretorius, E., Bester, J., and Kell, D. B. (2016a). A bacterial component to Alzheimer’s-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J. Alzheimers Dis. 53, 1237–1256. doi: 10.3233/jad-160318
Pretorius, E., Mbotwe, S., Bester, J., Robinson, C. J., and Kell, D. B. (2016b). Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J. R. Soc. Interface 13:20160539. doi: 10.1098/rsif.2016.0539
Pretorius, E., and Kell, D. B. (2014). Diagnostic morphology: biophysical indicators for iron-driven inflammatory diseases. Integr. Biol. 6, 486–510. doi: 10.1039/c4ib00025k
Pretorius, E., Mbotwe, S., and Kell, D. B. (2017a). Lipopolysaccharide-binding protein (LBP) reverses the amyloid state of fibrin seen in plasma of type 2 diabetics with cardiovascular co-morbidities. Sci. Rep. 7:9680. doi: 10.1038/s41598-017-09860-4
Pretorius, E., Page, M. J., Engelbrecht, L., Ellis, G. C., and Kell, D. B. (2017b). Substantial fibrin amyloidogenesis in type 2 diabetes assessed using amyloid-selective fluorescent stains. Cardiovasc. Diabetol. 16:141. doi: 10.1186/s12933-017-0624-5
Pretorius, E., Swanepoel, A. C., DeVilliers, S., and Bester, J. (2017c). Blood clot parameters: thromboelastography and scanning electron microscopy in research and clinical practice. Thromb. Res. 154, 59–63. doi: 10.1016/j.thromres.2017.04.005
Pretorius, E., Page, M. J., Hendricks, L., Nkosi, N. B., Benson, S. R., and Kell, D. B. (2018a). Both lipopolysaccharide and lipoteichoic acids potently induce anomalous fibrin amyloid formation: assessment with novel AmytrackerTM stains. R. Soc. Interface 15:20170941. doi: 10.1098/rsif.2017.0941
Pretorius, E., Page, M. J., Mbotwe, S., and Kell, D. B. (2018b). Lipopolysaccharide-binding protein (LBP) can reverse the amyloid state of fibrin seen or induced in Parkinson’s disease: implications. PLoS One 13:e0192121. doi: 10.1371/journal.pone.0192121
Ripollés Piquer, B., Nazih, H., Neunlist, M., Huvelin, J. M., and Bard, J. M. (2004). Effect of LPS on basal and induced apo E secretion by 25-OH chol and 9cRA in differentiated CaCo-2. J. Cell. Biochem. 91, 786–795. doi: 10.1002/jcb.10786
Rybicka, A., Longhi, G., Castiglioni, E., Abbate, S., Dzwolak, W., Babenko, V., et al. (2016). Thioflavin T: electronic circular dichroism and circularly polarized luminescence induced by amyloid fibrils. Chemphyschem 17, 2931–2937. doi: 10.1002/cphc.201600235
Shaik, M. M., Ahmad, S., Gan, S. H., Abuzenadah, A. M., Ahmad, E., Tabrez, S., et al. (2014). How do periodontal infections affect the onset and progression of Alzheimer’s disease? CNS Neurol. Disord. Drug Targets 13, 460–466. doi: 10.2174/18715273113126660152
Singhal, G., Jaehne, E. J., Corrigan, F., Toben, C., and Baune, B. T. (2014). Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front. Neurosci. 8:315. doi: 10.3389/fnins.2014.00315
Singhrao, S. K., Harding, A., Simmons, T., Robinson, S., Kesavalu, L., and Crean, S. (2014). Oral inflammation, tooth loss, risk factors, and association with progression of Alzheimer’s disease. J. Alzheimers Dis. 42, 723–737. doi: 10.3233/jad-140387
Sulatskaya, A. I., Kuznetsova, I. M., and Turoverov, K. K. (2011). Interaction of thioflavin T with amyloid fibrils: stoichiometry and affinity of dye binding, absorption spectra of bound dye. J. Phys. Chem. B 115, 11519–11524. doi: 10.1021/jp207118x
Sulatskaya, A. I., Kuznetsova, I. M., and Turoverov, K. K. (2012). Interaction of thioflavin T with amyloid fibrils: fluorescence quantum yield of bound dye. J. Phys. Chem. B 116, 2538–2544. doi: 10.1021/jp2083055
Takizawa, C., Thompson, P. L., van Walsem, A., Faure, C., and Maier, W. C. (2015). Epidemiological and economic burden of Alzheimer’s disease: a systematic literature review of data across Europe and the United States of America. J. Alzheimers Dis. 43, 1271–1284. doi: 10.3233/jad-141134
Tang, A. T., Choi, J. P., Kotzin, J. J., Yang, Y., Hong, C. C., Hobson, N., et al. (2017). Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 545, 305–310. doi: 10.1038/nature22075
Vale, C., Alonso, E., Rubiolo, J. A., Vieytes, M. R., LaFerla, F. M., Gimenez-Llort, L., et al. (2010). Profile for amyloid-beta and tau expression in primary cortical cultures from 3xTg-AD mice. Cell Mol. Neurobiol. 30, 577–590. doi: 10.1007/s10571-009-9482-3
von Bernhardi, R., Eugenín-von Bernhardi, L., and Eugenín, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Walter, S., Letiembre, M., Liu, Y., Heine, H., Penke, B., Hao, W., et al. (2007). Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol. Biochem. 20, 947–956. doi: 10.1159/000110455
Xaio, H., Banks, W. A., Niehoff, M. L., and Morley, J. E. (2001). Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 896, 36–42. doi: 10.1016/S0006-8993(00)03247-9
Younan, N. D., and Viles, J. H. (2015). A comparison of three fluorophores for the detection of amyloid fibers and prefibrillar oligomeric assemblies. ThT (Thioflavin T); ANS (1-Anilinonaphthalene-8-sulfonic Acid); and bisANS (4,4’-Dianilino-1,1’-binaphthyl-5,5’-disulfonic Acid). Biochemistry 54, 4297–4306. doi: 10.1021/acs.biochem.5b00309
Zhan, X., Stamova, B., Jin, L. W., DeCarli, C., Phinney, B., and Sharp, F. R. (2016a). Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 87, 2324–2332. doi: 10.1212/WNL.0000000000003391
Zhan, X., Stamova, B. S., Ander, B. P., Liu, D. Z., Jickling, G. C., and Sharp, F. R. (2016b). Lipopolysaccharide (LPS) - ischemia - hypoxia produces amyloid-like plaques in rat brain and LPS co-localizes with amyloid plaques and peri-vascular amyloid in human Alzheimer’s disease brain. Ann. Neurol. 80, S178–S179.
Zhan, X., Stamova, B., and Sharp, F. R. (2018). Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: a review. Front. Aging Neurosci. 10:42. doi: 10.3389/fnagi.2018.00042
Zhang, R., Miller, R. G., Gascon, R., Champion, S., Katz, J., Lancero, M., et al. (2009). Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 206, 121–124. doi: 10.1016/j.jneuroim.2008.09.017
Zhao, Y., Dua, P., and Lukiw, W. J. (2015). Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s disease (AD). J. Alzheimers Dis. Parkinsonism 5:177. doi: 10.4172/2161-0460.1000177
Zhao, Y., and Lukiw, W. J. (2015). Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J. Nat. Sci. 1:e138.
Zhao, Y., and Lukiw, W. J. (2018). Microbiome-mediated upregulation of MicroRNA-146a in sporadic Alzheimer’s disease. Front. Neurol. 9:145. doi: 10.3389/fneur.2018.00145
Zhao, Y. H., Cong, L., Jaber, V., and Lukiw, W. J. (2017a). Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front. Immunol. 8:1064. doi: 10.3389/fimmu.2017.01064
Keywords: Alzheimer-type dementia, amyloid, clotting, dormancy, infection, microbes
Citation: Pretorius E, Bester J, Page MJ and Kell DB (2018) The Potential of LPS-Binding Protein to Reverse Amyloid Formation in Plasma Fibrin of Individuals With Alzheimer-Type Dementia. Front. Aging Neurosci. 10:257. doi: 10.3389/fnagi.2018.00257
Received: 15 January 2018; Accepted: 03 August 2018;
Published: 22 August 2018.
Edited by:
Judith Miklossy, Prevention Alzheimer International Foundation, SwitzerlandReviewed by:
Xinhua Zhan, University of California, Davis, United StatesWalter J. Lukiw, LSU Health Sciences Center New Orleans, United States
Copyright © 2018 Pretorius, Bester, Page and Kell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Etheresia Pretorius, resiap@sun.ac.za Douglas B. Kell, dbk@manchester.ac.uk
†Co-authors