
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 18 July 2018
Sec. Alzheimer's Disease and Related Dementias
Volume 10 - 2018 | https://doi.org/10.3389/fnagi.2018.00223
 Yoko Shigemoto1,2
Yoko Shigemoto1,2 Daichi Sone1,3
Daichi Sone1,3 Etsuko Imabayashi1
Etsuko Imabayashi1 Norihide Maikusa1
Norihide Maikusa1 Nobuyuki Okamura4,5Shozo Furumoto6Yukitsuka Kudo5
Nobuyuki Okamura4,5Shozo Furumoto6Yukitsuka Kudo5 Masayo Ogawa1
Masayo Ogawa1 Harumasa Takano1,3Yuma Yokoi3
Harumasa Takano1,3Yuma Yokoi3 Masuhiro Sakata3Tadashi Tsukamoto7Koichi Kato1Noriko Sato2
Masuhiro Sakata3Tadashi Tsukamoto7Koichi Kato1Noriko Sato2 Hiroshi Matsuda1*
Hiroshi Matsuda1*The recent advent of tau-specific positron emission tomography (PET) has enabled in vivo assessment of tau pathology in Alzheimer’s disease (AD). However, because PET scanners have limited spatial resolution, the measured signals of small brain structures or atrophied areas are underestimated by partial volume effects (PVEs). The aim of this study was to determine whether partial volume correction (PVC) improves the precision of measures of tau deposits in early AD. We investigated tau deposits in 18 patients with amyloid-positive early AD and in 36 amyloid-negative healthy controls using 18F-THK5351 PET. For PVC, we applied the SPM toolbox PETPVE12. The PET images were then spatially normalized and subjected to voxel-based group analysis using SPM12 for comparison between the early AD patients and healthy controls. We also compared these two groups in terms of brain atrophy using voxel-based morphometry of MRI. We found widespread neocortical tracer retention predominantly in the posterior cingulate and precuneus areas, but also in the inferior temporal lobes, inferior parietal lobes, frontal lobes, and occipital lobes in the AD patients compared with the controls. The pattern of tracer retention was similar between before and after PVC, suggesting that PVC had little effect on the precision of tau load measures. Gray matter atrophy was detected in the medial/lateral temporal lobes and basal frontal lobes in the AD patients. Interestingly, only a few associations were found between atrophy and tau deposits, even after PVC. In conclusion, PVC did not significantly affect 18F-THK5351 PET measures of tau deposits. This discrepancy between tau deposits and atrophy suggests that tau load precedes atrophy.
Alzheimer’s disease (AD), the most common form of progressive degenerative dementia, is characterized by cognitive deterioration and behavioral impairment. One of the neuropathological hallmarks of AD is the aggregation of hyperphosphorylated tau protein into intracellular neurofibrillary tangles (NFTs) (Braak and Braak, 1991). Tau pathology correlates with cognitive functions better than amyloid-β (Nelson et al., 2012; Murray et al., 2015). The recent advent of tau-specific positron emission tomography (PET) has enabled in vivo assessment of tau pathology in AD, which will aid in early detection, disease staging, and treatment development (Holtzman et al., 2016).
Because PET scanners have limited spatial resolution, the measured signals of small brain structures such as cortical gray matter are underestimated by partial volume effects (PVEs) (Matsuda et al., 2002; Yanase et al., 2005; Gonzalez-Escamilla et al., 2017). AD is characterized by progressive brain atrophy and thus PVEs are estimated to be even more severe in AD patients (Scheltens et al., 1992; Schroeter et al., 2009; Rullmann et al., 2016). Although some previous studies reported that PVC was effective for evaluating tau load in AD (Villemagne et al., 2014; Shidahara et al., 2017), other studies found that PVC produced only small effects (Ossenkoppele et al., 2016; Schöll et al., 2016; Maass et al., 2017). The aim of the present study was to determine whether PVC improves the precision of 18F-THK5351 PET measures of tau deposits in early AD.
In total, 54 participants – 18 patients (13 women, 5 men) with early AD and 36 cognitively normal healthy controls (20 women, 16 men) – underwent both 18F-THK5351 and 11C-PiB PET scans from June 2015 to January 2017. The average interval between 18F-THK5351, 11C-PiB PET, and MRI was 20 ± 20 days (18F-THK5351 to 11C-PiB: 12 ± 13 days; 18F-THK5351 to MRI: 12 ± 14 days; 11C-PiB to MRI: 17 ± 21 days). Diagnosis of probable AD was based on the clinical criteria outlined by the National Institute on Aging-Alzheimer’s Association (NIA-AA) (McKhann et al., 2011). All early AD patients showed visually positive 11C-PiB PET results. None had a history of neurological disorder causing dementia, significant cerebrovascular disease, or major systemic disease. The AD patients were 70.4 ± 7.9 years old [mean ± standard deviation (SD)], their average Mini-Mental State Examination (MMSE) score was 22.0 ± 4.5 (mean ± SD), their global Clinical Dementia Rating (CDR) ranged from 0.5 to 1.0, and CDR sum of boxes was 4.6 ± 3.0.
The cognitively normal controls showed visually negative 11C-PiB PET results. None had a history of neurological or psychiatric disorders and none were taking medications that affect cognition. The controls were 66.0 ± 8.6 years old, with an average MMSE score of 29.2 ± 0.9, a global CDR of 0, and CDR sum of boxes of 0.0 ± 0.3. Participants’ demographic and clinical characteristics are presented in Table 1.
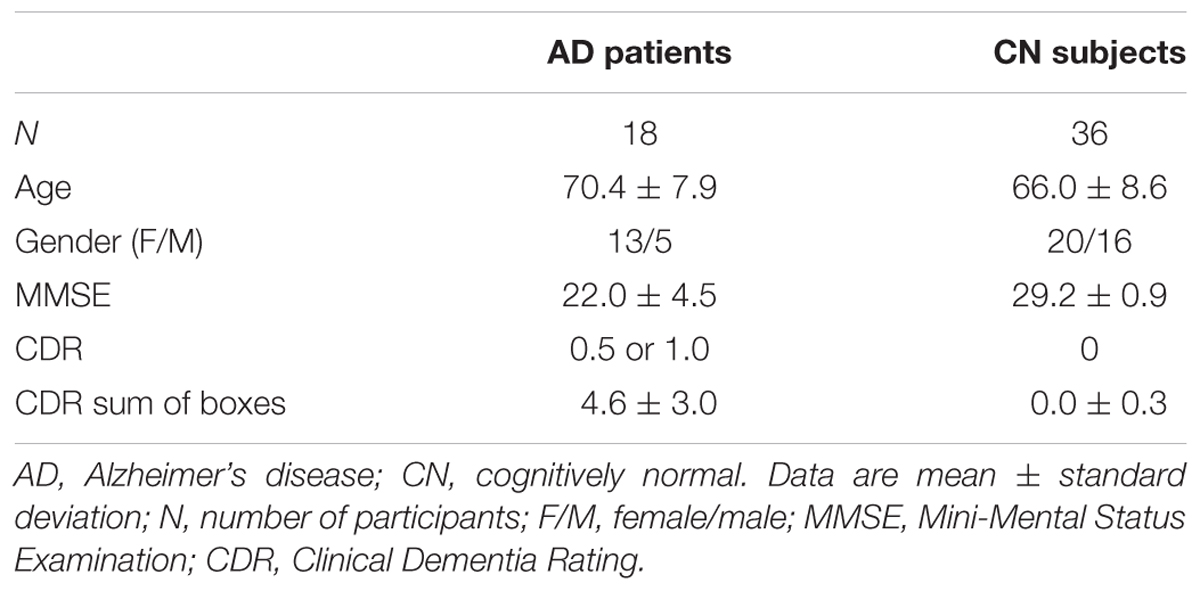
TABLE 1. Demographic characteristics of participants.
All participants gave written informed consent to participate in the study, which was approved by the institutional ethics committee at the National Center of Neurology and Psychiatry.
All participants underwent MRI scans with a 3-T MR imaging system (Verio; Siemens, Erlangen, Germany). Three-dimensional (3D) sagittal T1-weighted magnetization-prepared rapid acquisition with gradient echo (MPRAGE) images were acquired as follows: repetition time (TR)/echo time (TE), 1.900 ms/2.52 ms; flip angle (FA), 9°; in-plane resolution, 1.0 mm × 1.0 mm; 1.0 mm effective slice thickness, gapless; 300 slices; matrix, 256 × 256; field of view (FOV), 25 cm × 25 cm; acquisition time, 4 min 18 s.
Positron emission tomography scans were performed on a combined PET/CT scanner (Biograph 16; Siemens) in 3D acquisition mode. A low-dose CT scan was performed for attenuation correction before all scans. For 18F-THK5351 imaging, 18F-THK5351 at a dose of 185 MBq was injected intravenously 40 min before the PET/CT scan. For 11C-PiB imaging, 11C-PiB at a dose of 555 MBq was injected 50 min before the scan. PET/CT data were reconstructed using a combination of Fourier rebinning and ordered subset expectation maximization.
The 18F-THK5351 PET data were PVE-corrected using the recently developed statistical parametric mapping (SPM) toolbox PETPVE12 (Gonzalez-Escamilla et al., 2017). We used the Müller-Gärtner approach (Müller-Gärtner et al., 1992). For voxel-based analyses, both non-corrected and PVE-corrected PET images were spatially normalized using SPM version 12 (Wellcome Trust Centre for Neuroimaging, London, United Kingdom). PET scans were coregistered to the participants’ T1-weighted images and normalized with diffeomorphic anatomical registration using exponentiated Lie algebra (DARTEL). Each PET image was warped using the deformation fields derived from DARTEL registration of the coregistered T1-weighted image to the reference template. Standardized uptake value ratios (SUVR) for PET images were calculated using the mean activity in the cerebellar gray matter as the reference region. Finally, each PET image was smoothed using an 8 mm full width at half-maximum (FWHM) Gaussian kernel.
The gray matter images automatically segmented through the PVC process were also spatially normalized by DARTEL and smoothed using an 8 mm FWHM Gaussian kernel.
To investigate the direct correlation of atrophy and PVE-corrected 18F-THK5351 retention, 114 cerebral cortical regions of interest (ROIs) were used for calculation of both gray matter volume and 18F-THK5351 SUVR using Neuromorphometrics atlas1.
Differences in tau deposits between the early AD patients and healthy controls, were evaluated by a group comparison using a two-sample t-test in SPM12 with non-corrected and PVE-corrected 18F-THK5351 PET images. We also evaluated morphological group differences on gray matter images using a two-sample t-test in SPM12. Both statistical models included age and sex as covariates. Differences that met the following criteria were deemed significant: a height threshold of p < 0.001 and an extent threshold of p < 0.05 (corrected for multiple comparisons using the family-wise error rate).
We evaluated the correlation of atrophy and 18F-THK5351 retention obtained from the same ROIs using multiple regression models among AD patients and healthy controls respectively. Bonferroni and false discovery rate (FDR) correction were applied to correct multiple comparisons. Data analysis was performed using the SPSS Software, version 25.0 (SPSS, Tokyo, Japan).
Additionally, for the correlation analyses with 18F-THK5351 retention and the cognitive test scores (MMSE and CDR sum of boxes) within AD patients, we used a multiple regression analysis corrected for multiple comparisons in SPM12.
The early AD patients showed significantly increased tau deposits on 18F-THK5351 PET data before and after PVC compared with the healthy controls (Figures 1, 2, 3 and Tables 2, 3). We found widespread neocortical tracer retention predominantly in the posterior cingulate and precuneus areas, as well as in the inferior temporal lobes, inferior parietal lobes, frontal lobes, and occipital lobes; the primary sensory and motor regions were spared. No significant difference of tracer retention was found in the white matter or subcortical structures between the two groups. The pattern of tracer distribution was similar between before and after PVC. PVC marginally reduced the size of clusters throughout the neocortex and no new clusters were produced, suggesting that PVC had little effect.
FIGURE 1. The distribution of voxel-wise group differences before PVC with 18F-THK5351. Widespread neocortical tracer retention was observed predominantly in the posterior cingulate/precuneus areas, as well as in the inferior temporal lobes, parieto-occipital, and frontal lobes, in the early AD patients compared with the healthy controls. AD, Alzheimer’s disease; PVC, partial volume correction.
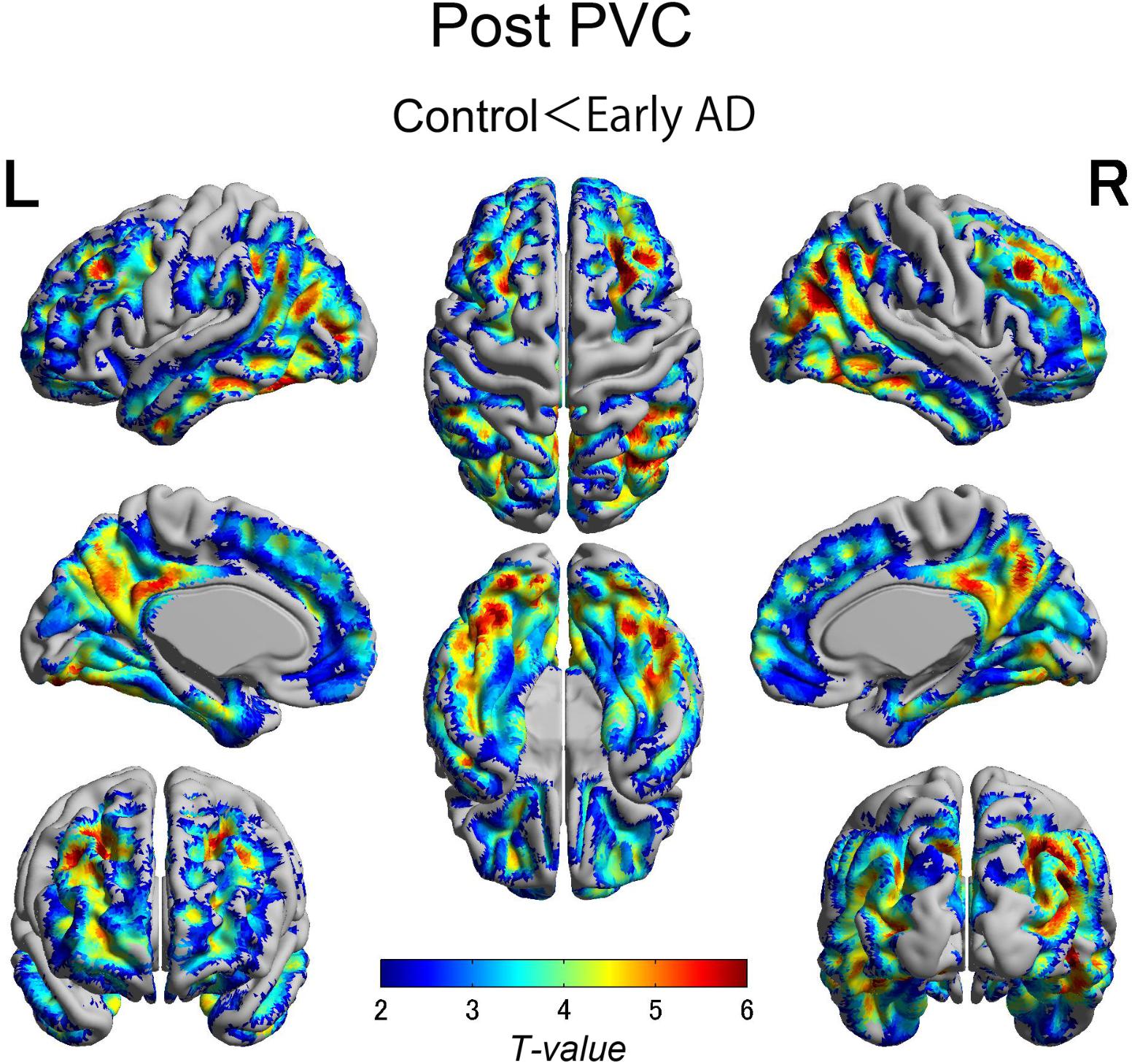
FIGURE 2. The distribution of voxel-wise group differences after PVC with 18F-THK5351. Almost the same tracer retention was observed as that before PVC. PVC, partial volume correction.
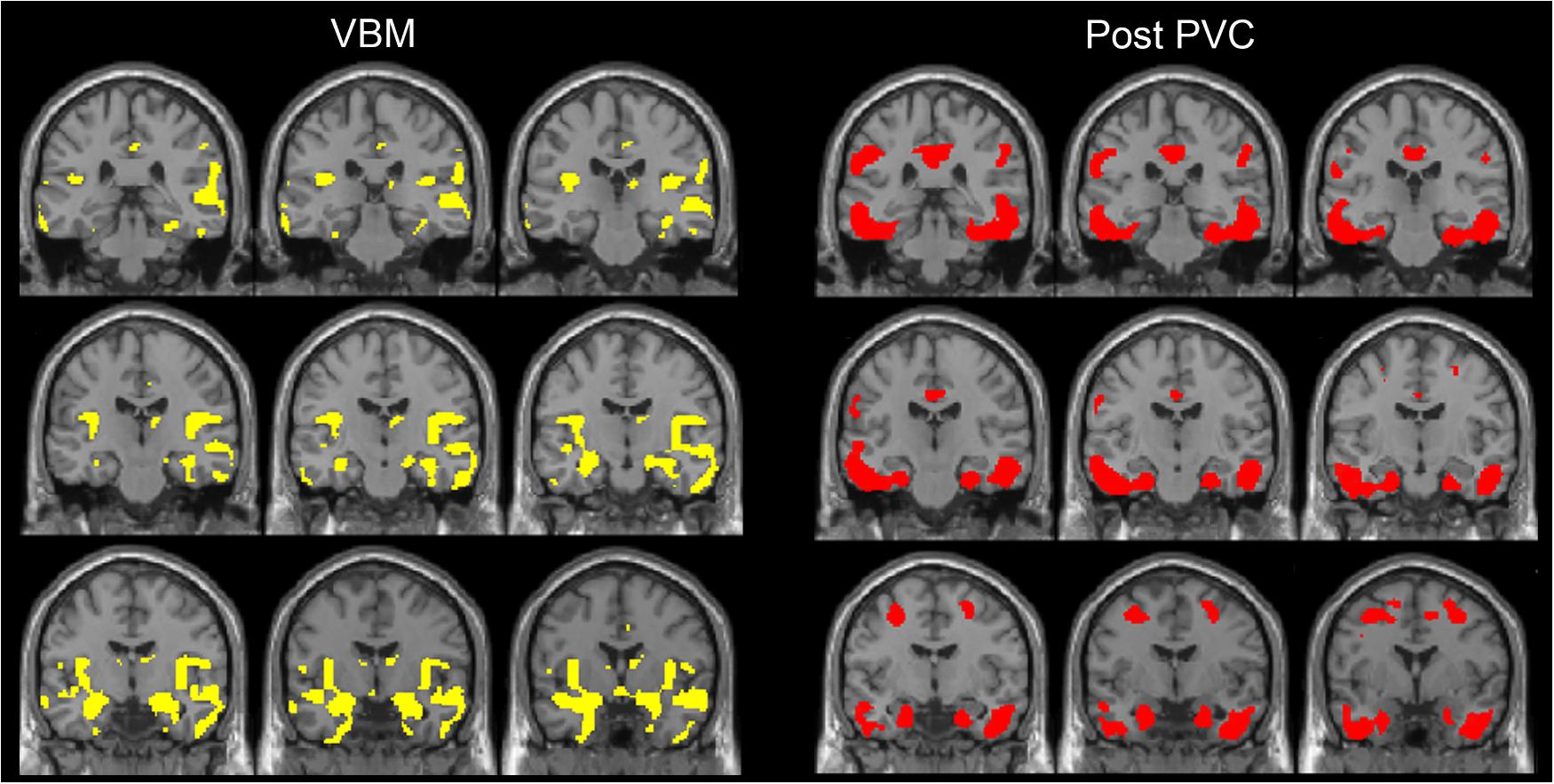
FIGURE 3. The distribution of voxel-wise group differences between early AD patients and controls. A series of coronal sections of VBM (yellow) and Post-PVC with 18F-THK5351 (red) along an anterior–posterior axis through hippocampus are mapped on a standard anatomical space.
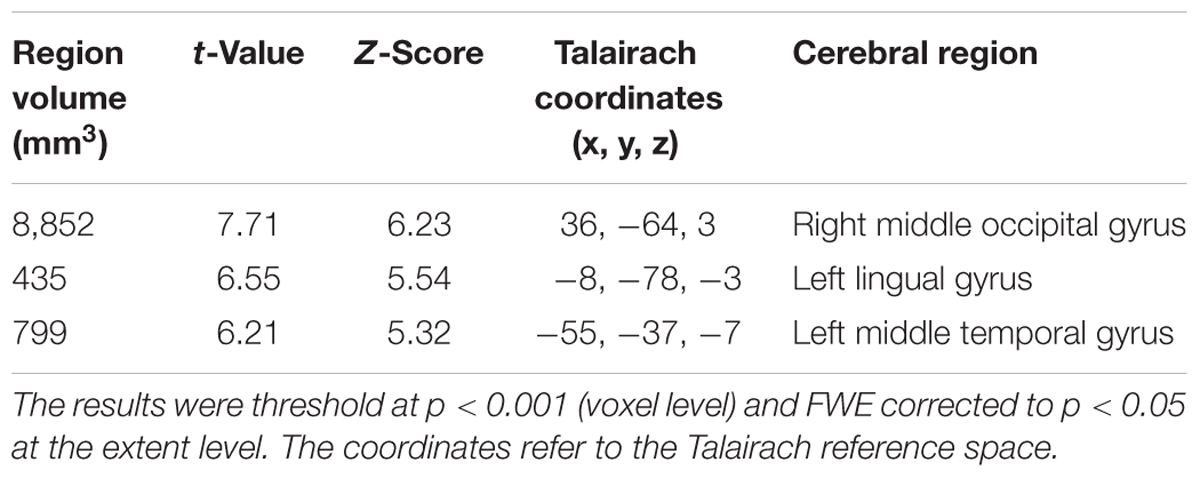
TABLE 2. Clusters of increased tau accumulation with early AD patients compared to healthy controls before PVC.
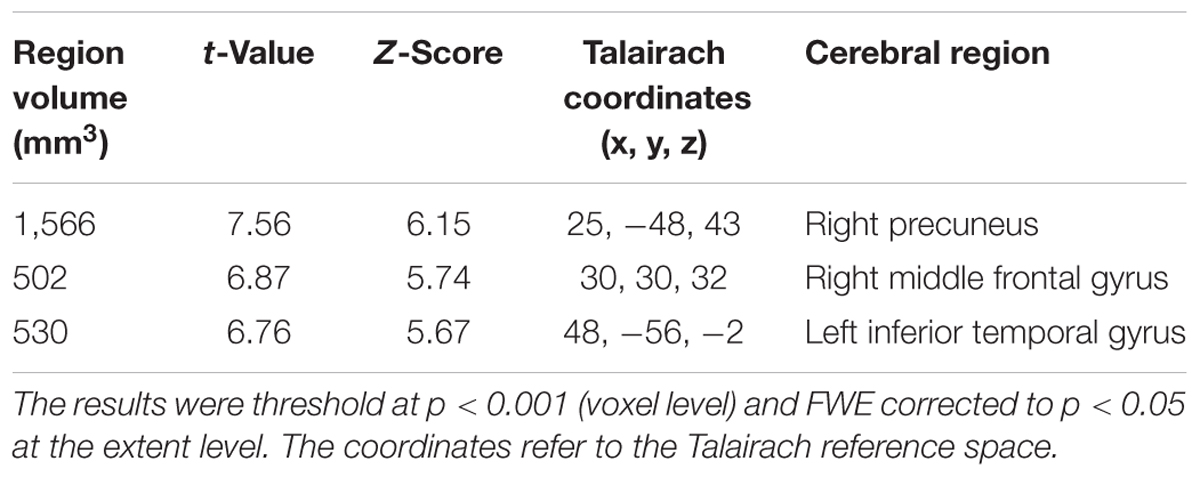
TABLE 3. Clusters of increased tau accumulation with early AD patients compared to healthy controls after PVC.
We also observed gray matter atrophy in the medial temporal lobes including hippocampal head extending into amygdala and lateral temporal lobes, as well as basal frontal lobes in AD patients (Table 4 and Figures 3, 4). No significant difference of atrophy was found in the white matter or subcortical structures between the two groups.
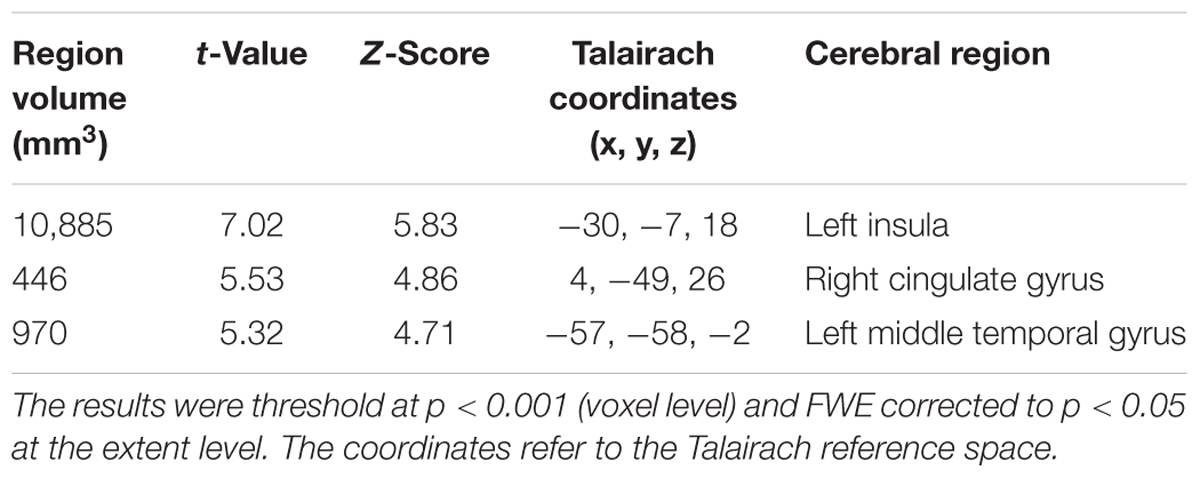
TABLE 4. Clusters of gray matter loss with early AD patients compared to healthy controls.
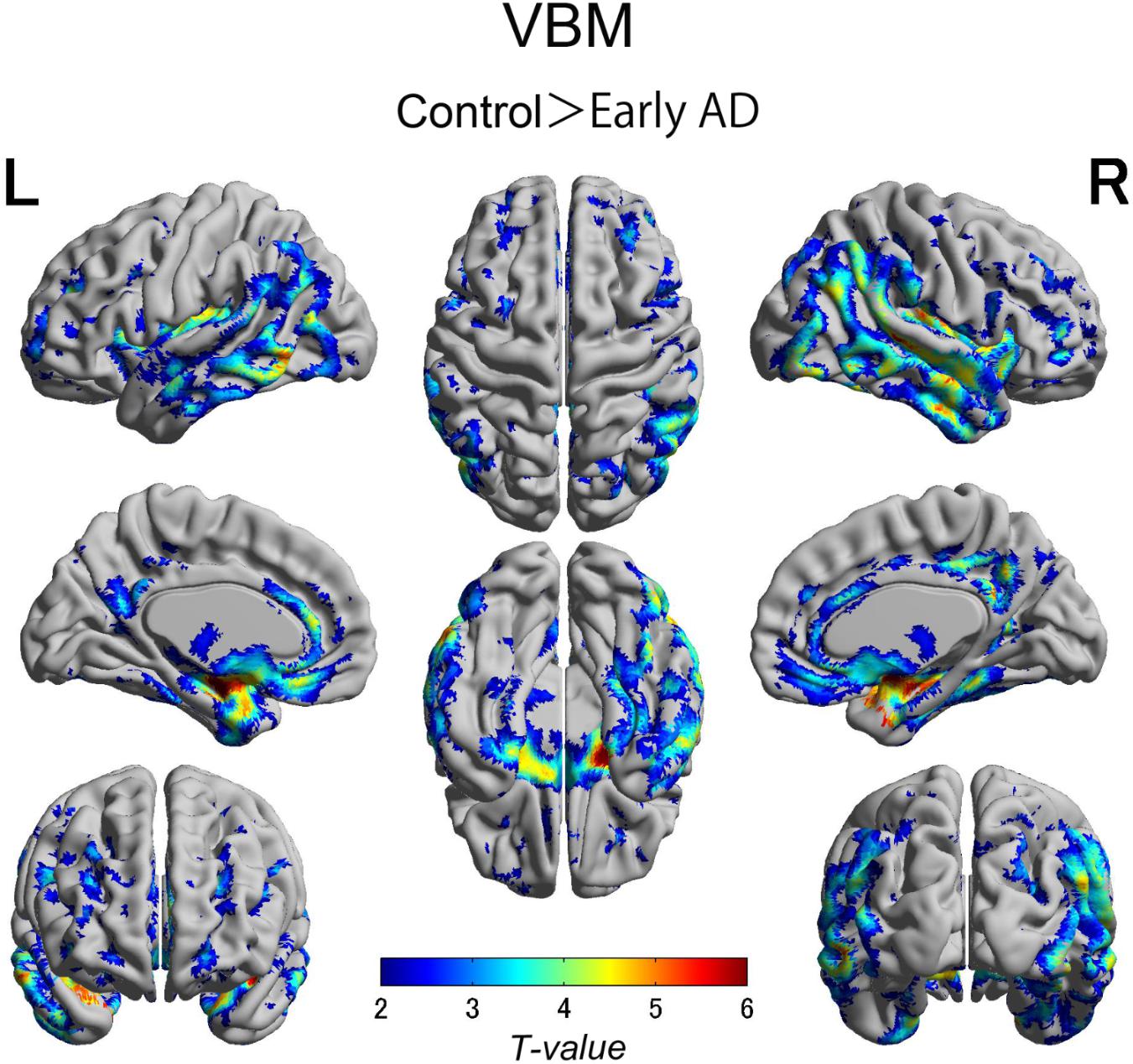
FIGURE 4. Voxel-wise group differences in gray matter volume between the early AD patients and healthy controls. Significant atrophy was observed in the medial temporal lobes including hippocampal head extending into amygdala and lateral temporal lobes as well as basal frontal lobes in the AD patients compared with the controls. AD, Alzheimer’s disease.
Visual evaluation of the atrophied areas and tau deposits measured by 18F-THK5351 PET after PVC in AD patients as compared to healthy controls revealed only slight overlap in the precuneus, posterior cingulate gyrus, lateral parietal cortex, inferior/lateral temporal cortex, and amygdala (Figures 5, 6). Among them, only few areas showed significant correlations in direct comparison of gray matter volume and PVE-corrected 18F-THK5351 retention (Table 5). An FDR correction showed significant negative correlations in the right fusiform gyrus (r = -0.78, p = 0.00012), left fusiform gyrus (r = -0.65, p = 0.0036), left inferior temporal gyrus (r = -0.73, p = 0.00059), right lingual gyrus (r = -0.66, p = 0.0032), left lingual gyrus (r = -0.76, p = 0.00023), right middle temporal gyrus (r = -0.72, p = 0.00075), left middle temporal gyrus (r = -0.75, p = 0.00038), and left parahippocampal gyrus (r = -0.71, p = 0.00089) in AD patients. The right fusiform gyrus, left lingual gyrus, and left middle temporal gyrus correlations survived Bonferroni correction for multiple comparisons. No significant negative correlations were found in healthy controls in this direct comparison.
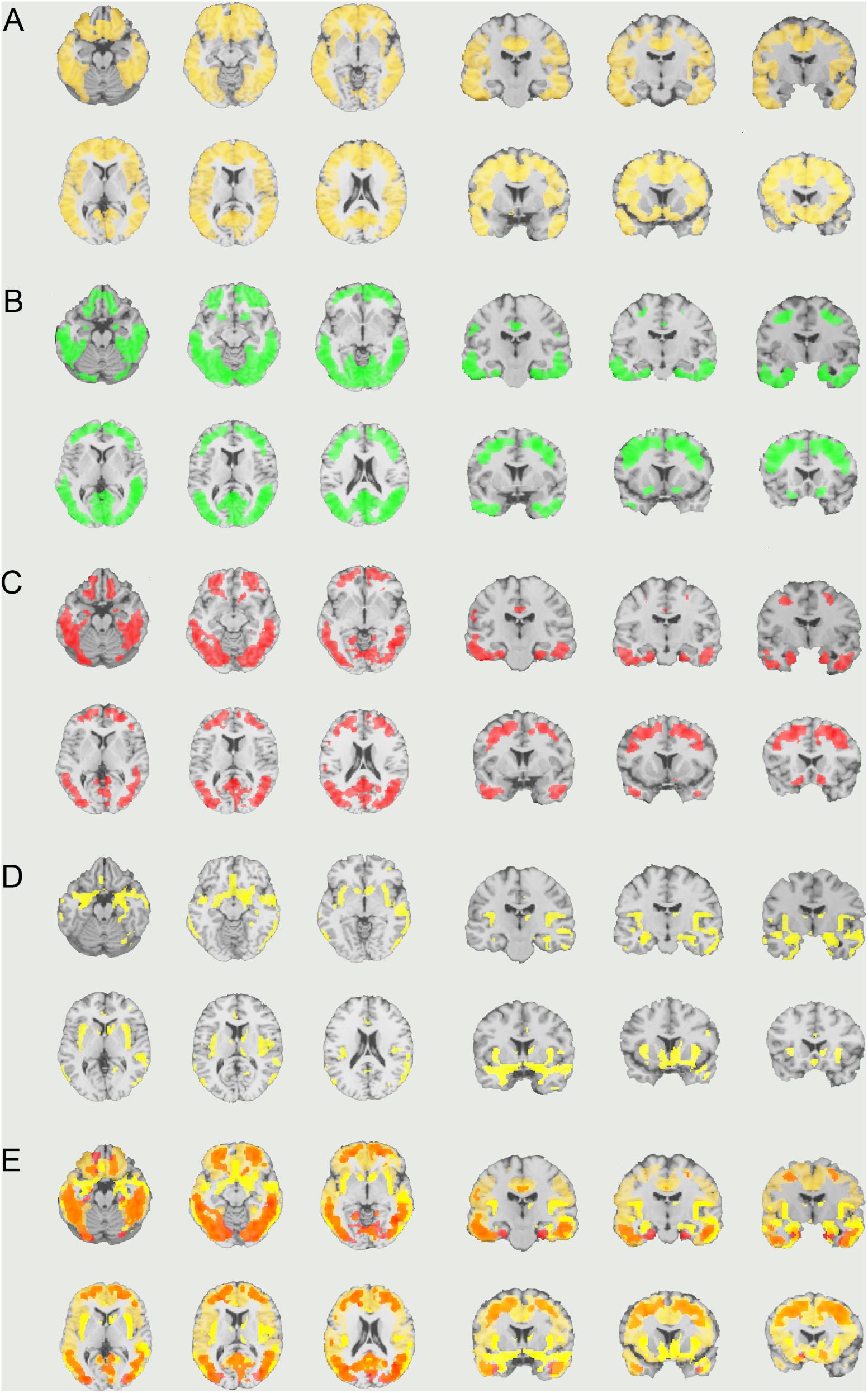
FIGURE 5. The distribution of voxel-wise group differences between early AD patients and controls. (A) PiB (bright yellow), (B) Pre-PVC with 18F-THK5351 (green), (C) Post-PVC with 18F-THK5351 (red), (D) VBM (yellow), and (E) PiB + Post-PVC with 18F-THK5351 + VBM results are mapped on a standard anatomical space. AD, Alzheimer’s disease; PVC, partial volume correction; VBM, voxel-based morphometry.
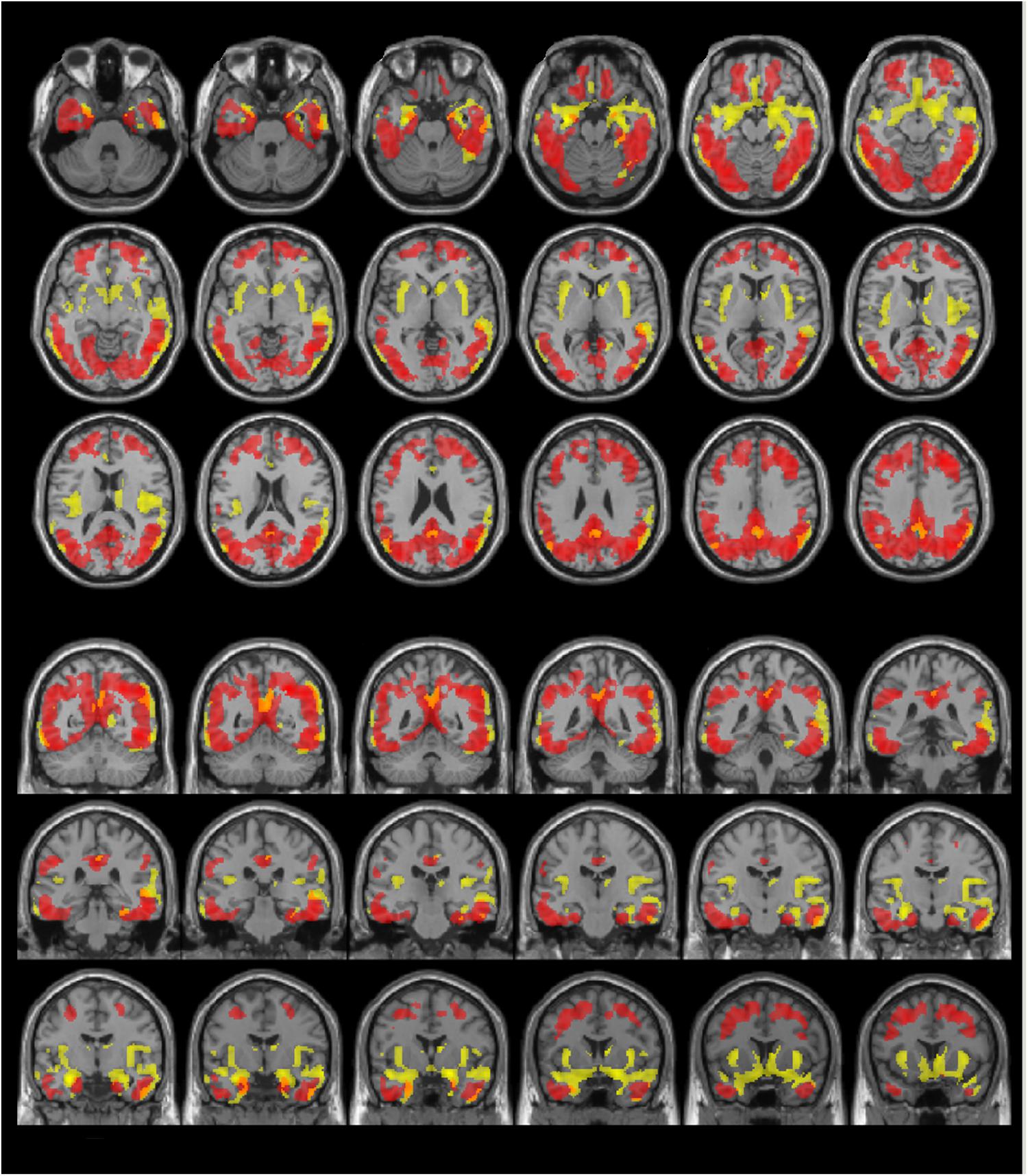
FIGURE 6. The distribution of voxel-wise group differences between early AD patients and controls. Post-PVC with 18F-THK5351 (red) and VBM (yellow) results are mapped on a standard anatomical space. Visual comparison of tau deposits after PVC with 18F-THK5351 and atrophy revealed only slight overlap in the precuneus, posterior cingulate gyrus, lateral parietal cortex, inferior/lateral temporal cortex and amygdala. AD, Alzheimer’s disease; PVC, partial volume correction; VBM, voxel-based morphometry.
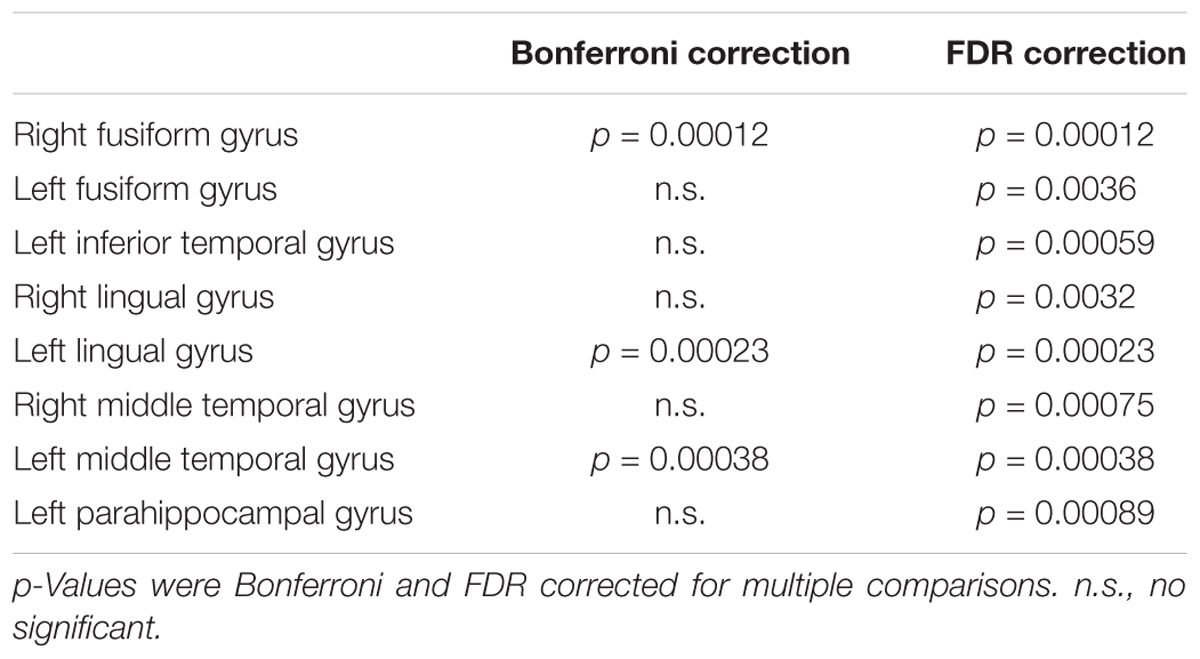
TABLE 5. Significant negative correlations between gray matter volume and 18F-THK5351 retention in early AD patients.
A significant correlation with 18F-THK5351 retention and cognitive test scores (MMSE and CDR sum of boxes) within AD patients was not detected.
In this study, we compared tau deposits as measured by 18F-THK5351 PET before and after PVC along with regional brain atrophy using VBM. PVC was applied to correct for atrophy but had little effect, even in atrophied areas such as the medial/lateral temporal lobes and basal frontal lobes. Somewhat surprisingly, only a few associations were found between atrophy and tau deposits. The finding that tau load seemed to show very few relationships with gray matter atrophy has never been reported in previous tau PET studies.
A pathological hallmark of AD is that NFTs first form in the transentorhinal cortex, followed by the hippocampus, spread from the nearby cortex (predominantly the limbic, insular, and basal frontal areas) to the posterior cingulate cortex, and then spread throughout the neocortex (Braak and Braak, 1991; Braak et al., 2006, 2011). Previous tau PET studies reported tracer retention in the neocortical regions, most prominently throughout the temporal lobes, posterior cingulate/precuneus, and parieto-occipital and frontal lobes, whereas the primary sensory, motor, and visual areas were spared (Villemagne et al., 2014; Johnson et al., 2016; Ossenkoppele et al., 2016; Schöll et al., 2016; Maass et al., 2017). The widespread distribution of tau deposits in neocortical areas detected in our study was consistent with the results of previous studies (Serrano-Pozo et al., 2011; Villemagne et al., 2014; Johnson et al., 2016; Ossenkoppele et al., 2016; Schöll et al., 2016; Maass et al., 2017). Our results also corresponded to neuropathological findings in AD patients (Braak stages V/VI) (Braak et al., 2006, 2011).
We performed PVC to correct for brain atrophy. The pattern of tracer retention was similar between uncorrected and corrected data, suggesting that PVC had little effect, in line with previous studies with 18F-AV-1451 (Ossenkoppele et al., 2016; Schöll et al., 2016; Maass et al., 2017). A potential explanation for ineffectiveness of PVC was that we evaluated early AD patients with mild brain atrophy. However, VBM analysis revealed significant atrophy in the medial/lateral temporal lobes and basal frontal lobes in the AD patients. We could not detect significant tau deposits in these atrophied areas, even after PVC. This significant discrepancy between tau deposits and atrophy revealed in the present study might help to explain the mechanisms of 18F-THK5351 retention.
18F-THK5351 retention was detected in less atrophied areas in the neocortex but not in the most severely atrophied areas; these findings have not previously been reported. According to pathological studies, the spread of NFTs in the neocortex leads to synaptic dysfunction, glial activation, and neuronal loss (Sperling et al., 2014). At the neuronal cytoskeleton level, NFTs begin with the accumulation of soluble phosphorylated tau in cell bodies. As the phosphorylation progresses, this pretangle gradually aggregates and forms insoluble intracellular mature NFTs. After neuronal death, the abnormal hyperphosphorylated tau becomes exposed as extracellular ghost tangles (Braak et al., 1994, 2011). According to previous studies (Harada et al., 2016b; Ono et al., 2017; Saint-Aubert et al., 2017), all three families of radiotracers (11C-PBB3, 18F-THK5117, 18F-THK5351, and 18F-AV-1451) are insensitive to pretangles. However, 11C-PBB3, 18F-THK5117, and 18F-AV-1451 are sensitive to both intracellular mature tangles and extracellular ghost tangles. 18F-THK5351 is sensitive to tangles but discrimination between mature tangles and ghost tangles has not been demonstrated (Harada et al., 2016a). Lowe et al. (2016) recently reported that 18F-AV-1451 has a preference for mature tangles over pretangles and ghost tangles, suggesting that morphological differences might affect the binding intensity. If 18F-THK5351 has a similar preference, this could explain our results because areas with more severe neuronal loss have more ghost tangles (Fukutani et al., 1995), which might be less sensitive to tracer binding.
Interestingly, 18F-THK5351 retention was found in more advanced Braak stages compared with the Braak stage of atrophied areas revealed by VBM. A recent report on retrospective cortical atrophy and tau PET in clinically normal elderly adults revealed a correlation between retrospective parahippocampal atrophy and inferior temporal AV-1451 retention instead of parahippocampal retention, suggesting that tau pathology precedes atrophy (LaPoint et al., 2017). While our results support this hypothesis, longitudinal analysis of the association between tau pathology and atrophy are needed.
Although hippocampal atrophy is a key structural finding in AD (Matsuda, 2016), we did not find significant differences in hippocampal 18F-THK5351 retention between the AD patients and controls even after PVC. Johnson et al. (2016) reported that AV-1451 retention in the hippocampus was relatively low, which is consistent with our findings. They considered that off-target binding in the choroid plexus might affect retention in the hippocampus. However, there must be another reason because the THK series does not have off-target binding in the choroid plexus (Ishiki et al., 2015; Harada et al., 2016a; Saint-Aubert et al., 2017). Schöll et al. (2016) reported that PVC produced only a small increase in tracer retention in the hippocampus, which is in line with our results. Recent imaging and pathological studies revealed that medial temporal atrophy and NFTs that are restricted to the medial temporal lobe in the absence of amyloid plaques are generally observed in cognitively normal healthy elderly adults (primary age-related tauopathy) (Crary et al., 2014; Jack et al., 2016; Shimada et al., 2016). Cognitively impaired individuals have a large proportion of ghost tangles in the medial temporal lobe compared with healthy elderly adults (Santa-Maria et al., 2012; Crary et al., 2014). Thus, we believe that individuals with mature tau deposits and patients with mild cognitive impairment or AD with increased tau deposits that are insensitive to tracer binding show small differences in tracer uptake in the hippocampus.
This study has several limitations. First, this study had small sample sizes of both early AD patients and healthy controls. Second, we did not collect genetic data (apolipoprotein E). Therefore, further longitudinal studies are needed to investigate whether tau deposits predict atrophy. Third, a recent human blocking study using monoamine oxidase B (MAO-B) inhibitor demonstrated significant reduction of 18F-THK5351 SUVs in cortical regions (Ng et al., 2017). Even the reference region such as cerebellar cortex, which is considered the least-affected region, also reduced 18F-THK5351 SUVs by 41.6%. Because 18F-THK5351 retention seems to reflect reactive astrocytes in addition to tau pathology, it is hard to reveal the exact correlation of atrophy and tau pathology. Further studies are needed to clarify 18F-THK5351 binding sensitivity to tau pathology and MAO-B.
We applied PVC to correct for atrophy in early AD. However, the pattern of 18F-THK5351 tracer retention was similar between before and after PVC, suggesting that PVC is not required to estimate tau deposits. Moreover, 18F-THK5351 retention was detected in less atrophied areas but not in the most severely atrophied areas even after PVC. Although this discrepancy between tau deposits and atrophy could be 18F-THK5351 specific findings, our findings suggest that tau load precedes atrophy. Further prospective longitudinal studies of the association between tau pathology and atrophy are needed to confirm our findings.
YS, DS, and HM designed the experiments. HT, YY, MS, and TT collected and sorted the data. YS, DS, EI, NM, MO, and KK carried out the experiments, data analysis, and statistical analysis. YS, DS, and HM wrote the manuscript. SF, YK, NS, and NO revised the manuscript.
This study was supported by the following funding sources: the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) project (Grant No. 16dm0207017h0003), funded by the Japan Agency for Medical Research and Development (AMED), and an Intramural Research (Grant No. 27-9) for Neurological and Psychiatric Disorders from the National Center of Neurology and Psychiatry (Japan).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Braak, E., Braak, H., and Mandelkow, E. M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567. doi: 10.1007/BF00293315
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., and Braak, E. (1991). Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Crary, J. F., Trojanowski, J. Q., Schneider, J. A., Abisambra, J. F., Abner, E. L., Alafuzoff, I., et al. (2014). Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766. doi: 10.1007/s00401-014-1349-0
Fukutani, Y., Kobayashi, K., Nakamura, I., Watanabe, K., Isaki, K., and Cairns, N. J. (1995). Neurons, intracellular and extracellular neurofibrillary tangles in subdivisions of the hippocampal cortex in normal ageing and Alzheimer’s disease. Neurosci. Lett. 200, 57–60. doi: 10.1016/0304-3940(95)12083-G
Gonzalez-Escamilla, G., Lange, C., Teipel, S., Buchert, R., Grothe, M. J., and Alzheimer’s Disease Neuroimaging Initiative (2017). PETPVE12: an SPM toolbox for partial volume effects correction in brain PET – application to amyloid imaging with AV45-PET. Neuroimage 15, 669–677. doi: 10.1016/j.neuroimage.2016.12.077
Harada, R., Okamura, N., Furumoto, S., Furukawa, K., Ishiki, A., Tomita, N., et al. (2016a). 18F-THK5351: a novel pet radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J. Nucl. Med. 57, 208–214. doi: 10.2967/jnumed.115.164848
Harada, R., Okamura, N., Furumoto, S., Tago, T., Yanai, K., Arai, H., et al. (2016b). Characteristics of tau and its ligands in pet imaging. Biomolecules 6:7. doi: 10.3390/biom6010007
Holtzman, D. M., Carrillo, M. C., Hendrix, J. A., Bain, L. J., Catafau, A. M., Gault, L. M., et al. (2016). Tau: from research to clinical development. Alzheimers Dement. 12, 1033–1039. doi: 10.1016/j.jalz.2016.03.018
Ishiki, A., Okamura, N., Furukawa, K., Furumoto, S., Harada, R., Tomita, N., et al. (2015). Longitudinal assessment of tau pathology in patients with Alzheimer’s Disease using [18F]THK-5117 positron emission tomography. PLoS One 10:e0140311. doi: 10.1371/journal.pone.0140311
Jack, C. R. Jr., Knopman, D. S., Chételat, G., Dickson, D., Fagan, A. M., Frisoni, G. B., et al. (2016). Suspected non-Alzheimer disease pathophysiology–concept and controversy. Nat. Rev. Neurol. 12, 117–124. doi: 10.1038/nrneurol.2015.251
Johnson, K. A., Schultz, A., Betensky, R. A., Becker, J. A., Sepulcre, J., Rentz, D., et al. (2016). Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 79, 110–119. doi: 10.1002/ana.24546
LaPoint, M. R., Chhatwal, J. P., Sepulcre, J., Johnson, K. A., Sperling, R. A., and Schultz, A. P. (2017). The association between tau PET and retrospective cortical thinning in clinically normal elderly. Neuroimage 157, 612–622. doi: 10.1016/j.neuroimage.2017.05.049
Lowe, V. J., Curran, G., Fang, P., Liesinger, A. M., Josephs, K. A., Parisi, J. E., et al. (2016). An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol. Commun. 4:58. doi: 10.1186/s40478-016-0315-6
Maass, A., Landau, S., Baker, S. L., Horng, A., Lockhart, S. N., La Joie, R., et al. (2017). Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage 157, 448–463. doi: 10.1016/j.neuroimage.2017.05.058
Matsuda, H. (2016). MRI morphometry in Alzheimer’s disease. Ageing Res. Rev. 30, 17–24. doi: 10.1016/j.arr.2016.01.003
Matsuda, H., Kanetaka, H., Ohnishi, T., Asada, T., Imabayashi, E., Nakano, S., et al. (2002). Brain SPET abnormalities in Alzheimer’s disease before and after atrophy correction. Eur. J. Nucl. Med. Mol. Imaging 29, 1502–1505. doi: 10.1007/s00259-002-0930-2
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Müller-Gärtner, H. W., Links, J. M., Prince, J. L., Bryan, R. N., McVeigh, E., Leal, J. P., et al. (1992). Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. J. Cereb. Blood Flow Metab. 12, 571–583. doi: 10.1038/jcbfm.1992.81
Murray, M. E., Lowe, V. J., Graff-Radford, N. R., Liesinger, A. M., Cannon, A., Przybelski, S. A., et al. (2015). Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 138, 1370–1381. doi: 10.1093/brain/awv050
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Ng, K. P., Pascoal, T. A., Mathotaarachchi, S., Therriault, J., Kang, M. S., Shin, M., et al. (2017). Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res. Ther. 9:25. doi: 10.1186/s13195-017-0253-y
Ono, M., Sahara, N., Kumata, K., Ji, B., Ni, R., Koga, S., et al. (2017). Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 140, 764–780. doi: 10.1093/brain/aww339
Ossenkoppele, R., Schonhaut, D. R., Schöll, M., Lockhart, S. N., Ayakta, N., Baker, S. L., et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139, 1551–1567. doi: 10.1093/brain/aww027
Rullmann, M., Dukart, J., Hoffmann, K. T., Luthardt, J., Tiepolt, S., Patt, M., et al. (2016). Partial-volume effect correction improves quantitative analysis of 18f-florbetaben β-amyloid pet scans. J. Nucl. Med. 57, 198–203. doi: 10.2967/jnumed.115.161893
Saint-Aubert, L., Lemoine, L., Chiotis, K., Leuzy, A., Rodriguez-Vieitez, E., and Nordberg, A. (2017). Tau PET imaging: present and future directions. Mol. Neurodegener. 12:19. doi: 10.1186/s13024-017-0162-3
Santa-Maria, I., Haggiagi, A., Liu, X., Wasserscheid, J., Nelson, P. T., Dewar, K., et al. (2012). The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol. 124, 693–704. doi: 10.1007/s00401-012-1017-1
Scheltens, P., Leys, D., Barkhof, F., Huglo, D., Weinstein, H. C., Vermersch, P., et al. (1992). Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J. Neurol. Neurosurg. Psychiatry 55, 967–972. doi: 10.1136/jnnp.55.10.967
Schöll, M., Lockhart, S. N., Schonhaut, D. R., O’Neil, J. P., Janabi, M., Ossenkoppele, R., et al. (2016). PET imaging of tau deposition in the aging human brain. Neuron 89, 971–982. doi: 10.1016/j.neuron.2016.01.028
Schroeter, M. L., Stein, T., Maslowski, N., and Neumann, J. (2009). Neural correlates of Alzheimer’s disease and mild cognitive impairment: a systematic and quantitative meta-analysis involving 1351 patients. Neuroimage 47, 1196–1206. doi: 10.1016/j.neuroimage.2009.05.037
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Shidahara, M., Thomas, B. A., Okamura, N., Ibaraki, M., Matsubara, K., Oyama, S., et al. (2017). A comparison of five partial volume correction methods for Tau and amyloid PET imaging with [18F]THK5351 and [11C]PIB. Ann. Nucl. Med. 31, 563–569. doi: 10.1007/s12149-017-1185-0
Shimada, H., Kitamura, S., Shinotoh, H., Endo, H., Niwa, F., Hirano, S., et al. (2016). Association between Aβ and tau accumulations and their influence on clinical features in aging and Alzheimer’s disease spectrum brains: a [11C]PBB3-PET study. Alzheimers Dement. 6, 11–20. doi: 10.1016/j.dadm.2016.12.009
Sperling, R., Mormino, E., and Johnson, K. (2014). The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 84, 608–622. doi: 10.1016/j.neuron.2014.10.038
Villemagne, V. L., Furumoto, S., Fodero-Tavoletti, M. T., Mulligan, R. S., Hodges, J., Harada, R., et al. (2014). In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 41, 816–826. doi: 10.1007/s00259-013-2681-7
Keywords: Alzheimer’s disease, tau, positron emission tomography, magnetic resonance imaging, partial volume correction, brain atrophy
Citation: Shigemoto Y, Sone D, Imabayashi E, Maikusa N, Okamura N, Furumoto S, Kudo Y, Ogawa M, Takano H, Yokoi Y, Sakata M, Tsukamoto T, Kato K, Sato N and Matsuda H (2018) Dissociation of Tau Deposits and Brain Atrophy in Early Alzheimer’s Disease: A Combined Positron Emission Tomography/Magnetic Resonance Imaging Study. Front. Aging Neurosci. 10:223. doi: 10.3389/fnagi.2018.00223
Received: 11 September 2017; Accepted: 29 June 2018;
Published: 18 July 2018.
Edited by:
Shin Murakami, Touro University California, United StatesReviewed by:
Rik Vandenberghe, KU Leuven, BelgiumCopyright © 2018 Shigemoto, Sone, Imabayashi, Maikusa, Okamura, Furumoto, Kudo, Ogawa, Takano, Yokoi, Sakata, Tsukamoto, Kato, Sato and Matsuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroshi Matsuda, bWF0c3VkYWhAbmNucC5nby5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.