 Yulong Xu*
Yulong Xu* Yanli Wang
Yanli Wang Changning Wang
Changning Wang- Athinoula A. Martinos Center for Biomedical Imaging, Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA, United States
α1-Adrenoceptor is implicated in numerous neuronal diseases. The development of new modulators targeting this receptor as well as the investigation of the role of α1-adrenoceptor in healthy and disease conditions, however, is hindered by the lack of specific positron emission tomography (PET) radiotracers. Iloperidone shows a high binding affinity to α1-adrenoceptor and moderate selectivity over other brain receptors. We report herein the synthesis and characterization of carbon-11 labeled iloperidone for imaging of α1-adrenoceptor in brain. The radiolabeling of [11C]iloperidone was carried out conveniently in one step by treating precursor with [11C]CH3I in DMF in the presence of K2CO3. Then, [11C]iloperidone was purified by semi-preparative HPLC, and characterized in C57BL/6 mice using PET/CT scanning. The desired product [11C]iloperidone was obtained in an average decay corrected radiochemical of 12% (n = 3) and over 99% radiochemical purity. The average molar radioactivity was 357 GBq/μmol with total synthetic time of 35–40 min. PET/CT scanning in C57BL/6 mice showed favorable pharmacokinetic properties and high brain exposure of [11C]iloperidone. Blocking experiments by pretreatment with the unlabeled iloperidone showed the significant blocking effects with about 25% reduction in brain uptake. These results suggested that [11C]iloperidone can serve as a lead compound for the further development of specific radiotracers for PET imaging of α1-adrenoceptor in brain clinically.
Introduction
The α1-adrenoceptor is G protein-coupled receptor, widely distributed in both the peripheral and the central nervous system (CNS; Gross et al., 1989; Zhong and Minneman, 1999). In human CNS, α1-adrenoceptor is mainly localized in the cerebral cortex, thalamus and hippocampus, while moderately in striatum, and cerebellum (Palacios et al., 1987; Kalaria, 1989). Over the past decade, α1-adrenoceptor has gained interest due to its biological functions in regulating both positively motivated behaviors and stress reactions (Selken and Nichols, 2007; Stone et al., 2007). The α1-adrenoceptor has been implicated in numerous neuronal diseases, such as Alzheimer’s disease, Parkinson’s disease, schizophrenia, substance abuse, and affective disorders (Maletic et al., 2017; Wu et al., 2017; Akinaga et al., 2019; Datta et al., 2019). Consequently, a great deal of interests lay in the development of α1-adrenoceptor modulators, which has opened a new avenue for identifying novel CNS disease modifying therapeutics.
Positron emission tomography (PET) is a fully translational imaging technique, allowing the detection of picomolar sensitivity (Bergstroöm et al., 2003). An α1-adrenoceptor specific PET radiotracer would be highly beneficial in the development of novel α1-adrenoceptor modulators as well as in understanding the role of α1-adrenoceptor in progression of CNS disease. Accordingly, substantial research effort has been devoted to the development of efficient PET radiotracer for probing α1-adrenoceptor (Figure 1; Airaksinen et al., 2013; Risgaard et al., 2013). Among these PET ligands, [11C]LuAA27122 exhibited favorable blood-brain barrier (BBB) penetration, binding at α1-adrenoceptor rich regions in cynomolgus monkey brain while lacking specificity on α1-adrenoceptor, which has been confirmed through autoradiography studies in vitro. No specific α1-adrenoceptor PET radiotracer with sufficient brain exposure in clinic has hampered investigation of the role of α1-adrenoceptor in healthy and disease conditions as well as the development of new modulators targeting this receptor.
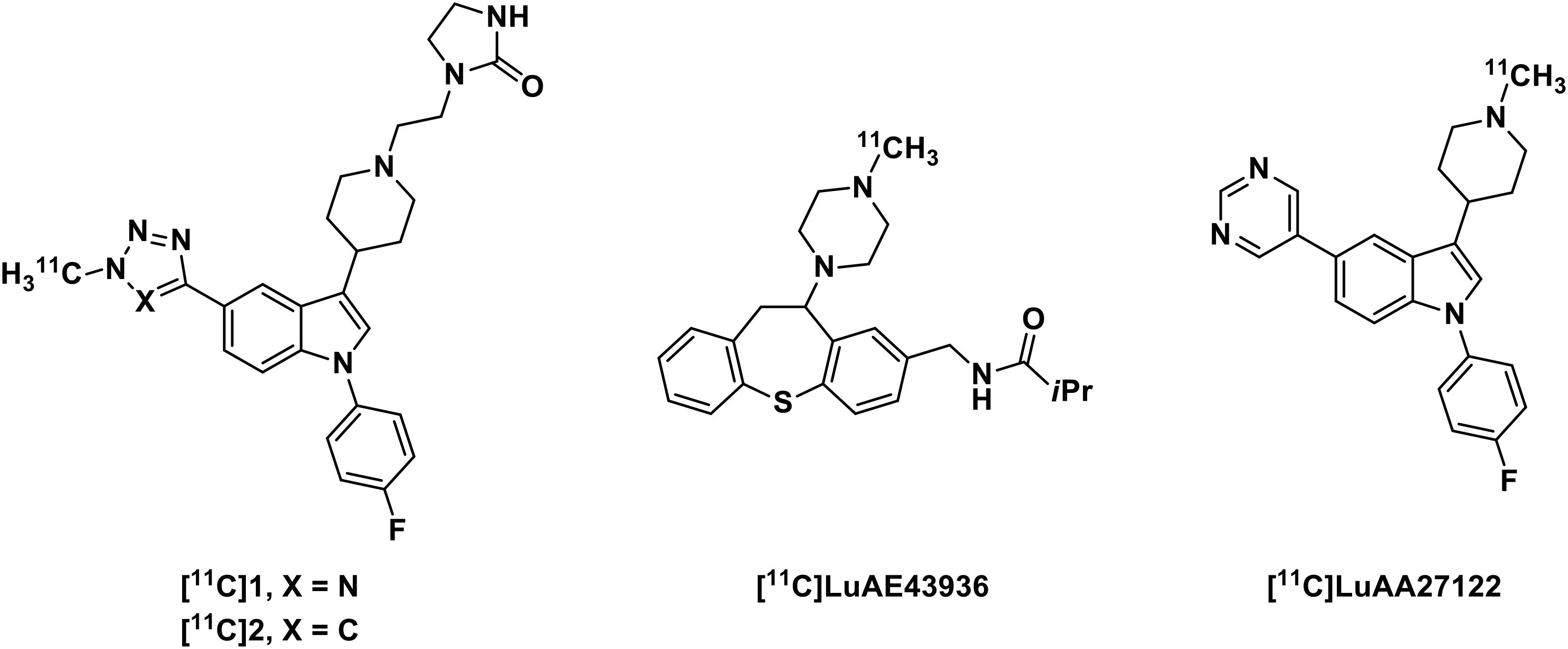
Figure 1. Structures of some representative α1-adrenoceptor PET radiotracers.
As our continuing interest in PET radiotracers for CNS (Wang et al., 2015, 2017; Chen et al., 2019), we decided to pursue an efficient α1-adrenoceptor PET ligand with desirable binding affinity and selectivity, as well as suitable metabolic stability. Iloperidone, an effective antipsychotic drug in clinic, demonstrates a high binding affinity to α1-adrenoceptor (IC50 = 0.4 nM) and sufficient selectivity over other brain receptors, such as serotonin 5-HT2 (IC50 = 9.0 nM), 5-HT1A (IC50 = 210 nM), dopamine D1 (IC50 = 750 nM) and D2 (IC50 = 110 nM) receptors (Strupczewski et al., 1995). Functionally, iloperidone shows antagonistic properties at dopamine D2 and serotonin 5-HT2 receptors, presumably acting as an antagonist at α1-adrenoceptor, which may contribute to its antipsychotic effects (Stahl, 2013). Recently, Joshi’s group demonstrated the repurposing of iloperidone on anti-hypertensive and ocular hypotensive activity in animals, which was related to its effect on peripheral α1-adrenoceptor (Joshi et al., 2020). As a result, the biological properties of iloperidone support its carbon-11 labeled “hot” compound as a promising PET radiotracer for α1-adrenoceptor in the human brain. Herein, we describe our results on the radiosynthesis of [11C]iloperidone as well as PET imaging evaluation in mice for α1-adrenoceptor.
Materials and Methods
General
All commercially available reagents were used without further purification unless otherwise stated. Iloperidone was purchased from Fisher Scientific. Analytical thin layer chromatography (TLC) was performed using Silica Gel GF254 plates (Merck Millipore co,.ltd, 0.2 mm thick). Compounds were purified using CombiFlash Rf 150 (Teledyne ISCO co,.ltd). 1H spectra was recorded on Bruker 500 MHz. Chemical shifts in 1H NMR spectra was reported in parts per million (ppm) on the δ scale from an internal standard of CDCl3 (7.26 ppm). Data were reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad), coupling constant in hertz (Hz), and integration. MS data was recorded on Agilent Technologies 6310 quadrupole mass spectrometer.
All mouse studies were carried out at Massachusetts General Hospital (PHS Assurance of Compliance No. A3596-01). The Subcommittee on Research Animal Care (SRAC) serves as the Institutional Animal Care and Use Committee (IACUC) for the Massachusetts General Hospital. SRAC reviewed and approved all procedures detailed in this paper. All mice were socially housed in cages appropriate for the physical and behavioral health of the individual animal and were given unlimited access to food and water, with additional nutritional supplements provided as prescribed by the attending veterinary staff.
Preparation of Precursor
A mixture of iloperidone (10 mg, 0.02 mmol) and concentrated H2SO4 (1 mL) was stirred at 65°C for 23 h. The cooled reaction was poured into 1 g of ice and was stirred vigorously for 10 min. The aqueous mixture was extracted with CH2Cl2 and the resultant CH2Cl2 extract was washed with 5% sodium hydroxide. The basic phases were combined and washed with CH2Cl2. The aqueous mixture was cooled in an ice bath and concentrated hydrochloric acid was added until a precipitate formed. The crude product was purified via flash column chromatography on silica gel to provide the precursor in 85% yield (7 mg). 1H NMR (500 MHz, CDCl3) δ 7.98 (s, 1H), 7.57 (d, J = 2.1 Hz, 1H), 7.42 (dd, J = 8.3, 2.1 Hz, 1H), 7.23 (dd, J = 8.5, 2.1 Hz, 1H), 7.08 (td, J = 8.9, 2.2 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 4.09 (t, J = 5.2 Hz, 2H), 3.29 (s, 3H), 2.74 (s, 2H), 2.56 (s, 3H), 2.36 (s, 2H), 2.05 (s, 4H), 1.30 (s, 1H), 1.25 (s, 2H), 1.24 (s, 1H). MS (ESI) m/z 413.3 [M + H]+.
Radiosynthesis of [11C]Iloperidone
The radiosynthesis of [11C]iloperidone was carried out according to the procedure described previously by our group (Chen et al., 2019). Briefly, [11C]CO2 was obtained via the 14N (p, α) 11C reaction on nitrogen with 2.5% oxygen, with 11 MeV protons (Siemens Eclipse cyclotron), and trapped on molecular sieves in a TRACERlab FX-MeI synthesizer (General Electric). [11C]CH4 was obtained by the reduction of [11C]CO2 in the presence of Ni/hydrogen at 350°C, which re-circulated through an oven containing I2 to produce [11C]CH3I via a radical reaction.
The prepared [11C]CH3I was trapped in anhydrous DMF (300 μL) containing precursor (1.0 mg) and K2CO3 (5.0 mg). The reaction vessel was heated at 80°C and kept there for 3 min. The radioactive mixture containing [11C]iloperidone was quenched by addition of an HPLC mobile phase (0.7 mL) and then applied to a reverse phase semipreparative HPLC (Phenomenex Gemini-NX 5u C18 110A, 250 × 10 mm, 5.0 mL/min, 65% H2O + 0.1% TFA/35% CH3CN). A radioactive fraction having a retention time of 10 min was collected in a flask, and diluted in water (30 mL). The final product was reformulated by loading onto a solid-phase exchange (SPE) C-18 cartridge (Waters WAT020515 Sep-Pak Plus Short C18), rinsing with water (4 × 5 mL), eluting with EtOH (0.3 mL), and diluting with saline (2.7 mL). The chemical and radiochemical purity of the final product was tested by analytical HPLC (VARIAN Puruit XRs 5 C18, 150 × 4.6 mm), eluting with a gradient of 10–90% CH3CN in H2O of 0.1% TFA, at a flow rate of 1.5 mL/min. Confirmation of the identity of [11C]iloperidone was achieved by co-injection with iloperidone as reference standard. For the determination of molar activity, mass (μmol) of [11C]iloperidone with a known radioactivity was determined by HPLC comparison of UV absorbance at 254 nm with those of known concentrations of non-radioactive iloperidone.
PET/CT Acquisition and Post Processing
Positron emission tomography/CT imaging was performed in anesthetized (isoflurane) C57BL/6 mice (20–25 g, female; n = 2 for baseline, and n = 2 for blocking) to minimize discomfort. Highly trained animal technicians monitored animal safety throughout all procedures. The mice were fixed on the bed of a Triumph Trimodality PET/CT scanner (Gamma Medica, Northridge, CA, United States) in the prone position, and injected with [11C]iloperidone (150–200 μL, ∼7.4 MBq) via a lateral tail vein catheterization at the start of PET acquisition. For blocking studies, iloperidone (5 mg/kg, iv) was injected at 10 min prior to [11C]iloperidone injection. Dynamic PET acquisition lasted for 60 min and was followed by CT for anatomic co-registration. PET data were reconstructed using a 3D-MLEM method resulting in a full width at half-maximum resolution of 1 mm. Reconstructed images were exported from the scanner in DICOM format along with an anatomic CT. These files were imported to AMIDE software (version 1.0.4).
PET/CT Image Analysis
Positron emission tomography images were analyzed using the freely available AMIDE software (version 1.0.4). Volumes of interests (VOIs) were drawn manually as spheres guided by high resolution CT structural images and summed PET data, with a radius no less than 1 mm to minimize partial volume effects. Time-activity curves (TACs) were exported in terms of decay corrected activity at specified time points with gradually increasing intervals. The TACs were expressed as the percentage of injected dose per gram (% ID/g).
Results and Discussion
As shown in Scheme 1, the radiolabeling of [11C]iloperidone was carried out conveniently in one step. The precursor was prepared by treating iloperidone in concentrated H2SO4 at 65°C for 23 h in the yield of 85%. The [11C]CH3I reacted with the precursor in DMF at 80°C for 3 min in the presence of K2CO3, and the radioactive mixture was purified by semi-preparative HPLC, eluting with 0.1% trifluoroacetic acid solution of water and acetonitrile (65:35) at a flow rate of 5 mL/min. The fraction containing the product was collected at around 10 min and passed through a C18 Sep-Pak column, then eluted with 20 mL water. The desired product [11C]iloperidone was obtained by eluting 0.3 mL of ethanol through the C18 Sep-Pak column in 2.7 mL of sterile saline, resulting the final formulation with 12% radiochemical yield (n = 3, decay corrected) and over 99% radiochemical purity. The total time of [11C]iloperidone synthesis was 35–40 min with a molar activity of 357 GBq/μmol.
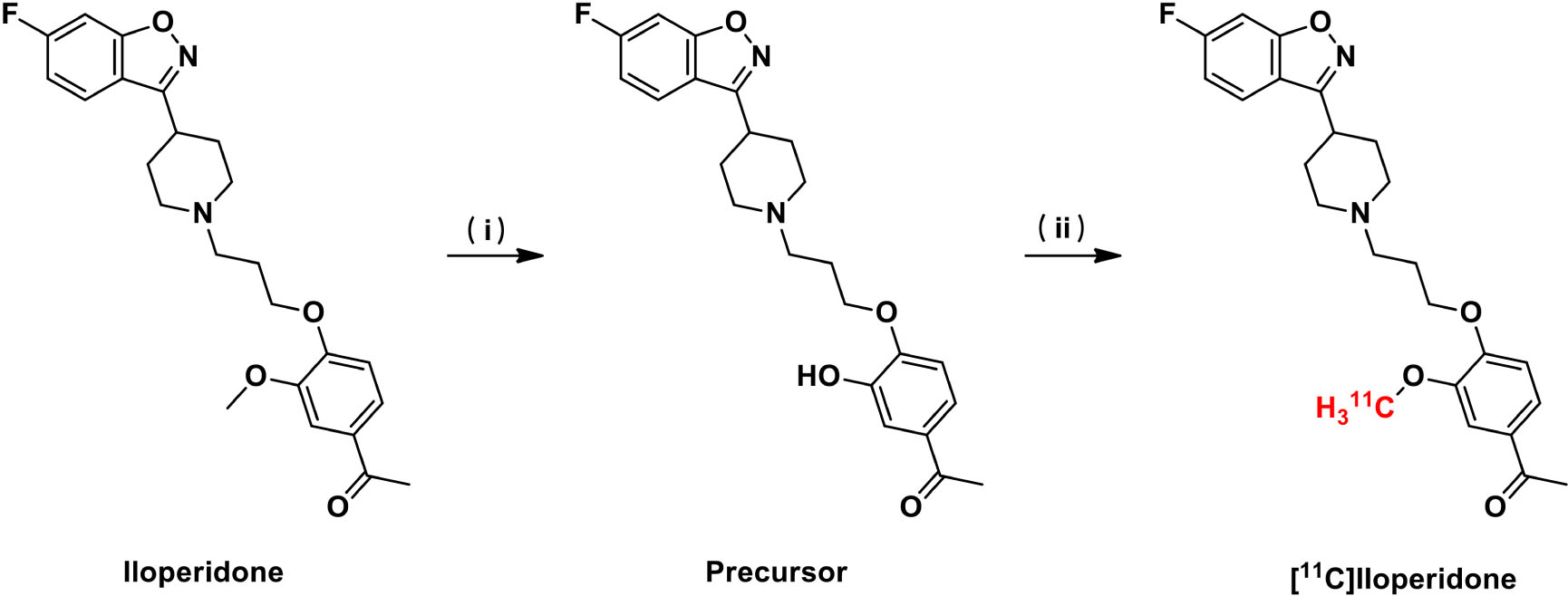
Scheme 1. Radiosynthesis of [11C]iloperidone. Reagents and conditions: (i) H2SO4, 65°C, yield 85%. (ii) [11C]CH3I, K2CO3, DMF, 80°C, yield 12% (decay corrected).
After reliable and successful radiosynthesis of [11C]iloperidone, PET/CT scanning was performed in C57BL/6 mice (20–25 g, female, n = 2) to evaluate its biodistribution, which can quantify accumulation and determine metabolic pathways of [11C]iloperidone in different organs. As shown in Figure 2, [11C]iloperidone showed high initial uptakes in liver and kidney (11.49% ID/g and 9.70% ID/g at 5 min post injection, respectively), and subsequent slow clearance (10.48% ID/g and 9.42% ID/g at 55 min post injection, respectively), demonstrating that [11C]iloperidone was mainly excreted through bile and urine. However, very low uptake in muscle were observed for [11C]iloperidone (0.75% ID/g at 5 min post injection). The blood displayed the highest uptake with 9.96% ID/g at 5 min, followed by lung with 6.18% ID/g at 5 min, brain with 5.79% ID/g at 5 min, and heart with 5.48% ID/g at 5 min. Notably, [11C]iloperidone had a relatively slow clearance rate in blood (7.15% ID/g at 55 min post injection), indicating a long-time blood level.
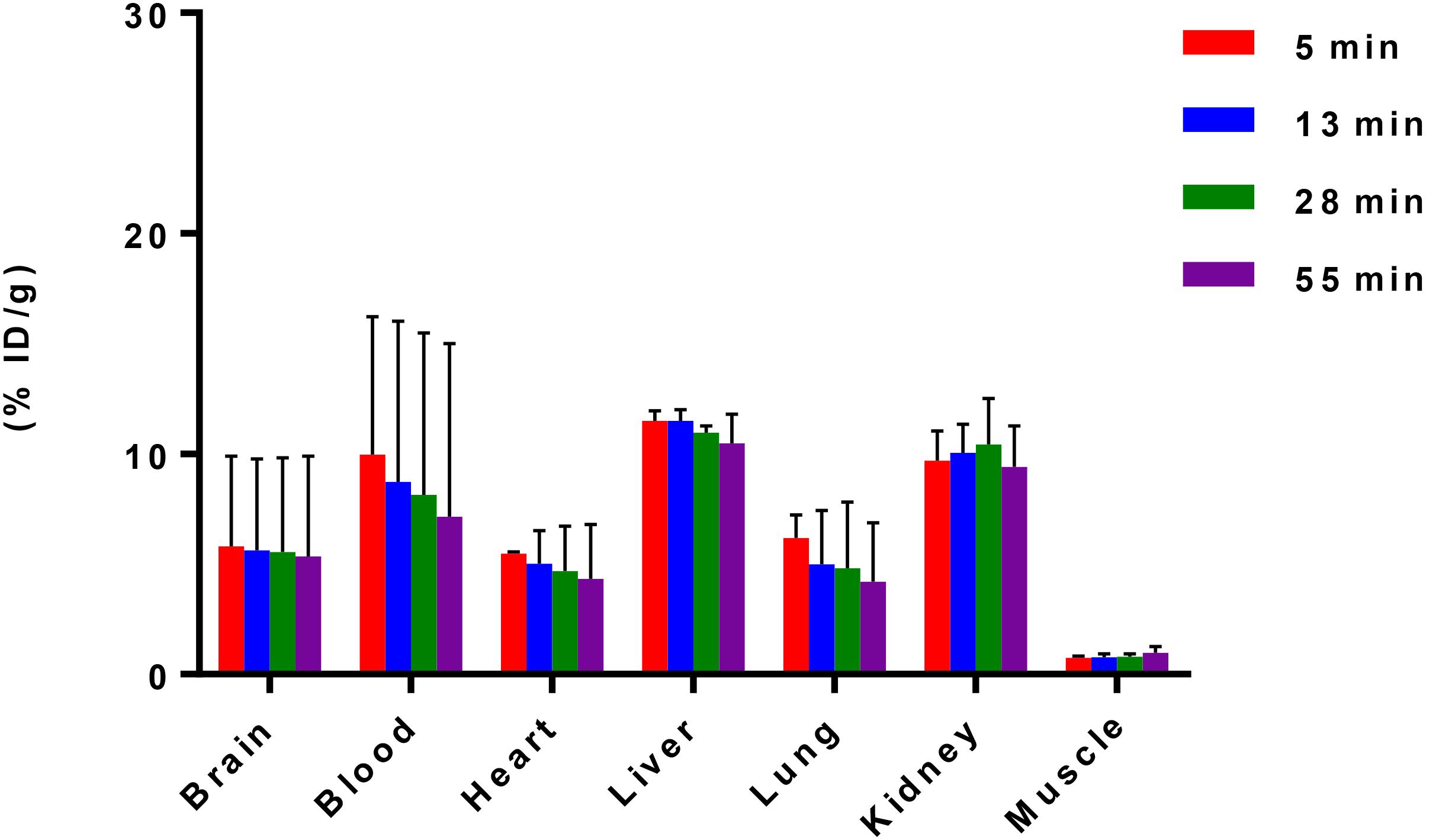
Figure 2. Biodistribution of [11C]iloperidone in selected organs of C57BL/6 mice (20–25 g, female, and n = 2). Data are expressed as the percentage of injected dose per gram (% ID/g).
According to TAC in mouse brain, [11C]iloperidone exhibited high brain exposure with uptake reaching a plateau at 5 min post injection, followed by a very slow washout (Figure 3). These results demonstrated that [11C]iloperidone might perform irreversible kinetics toward α1-adrenoceptor in brain. Additionally, the regional distribution of [11C]iloperidone in brain was homogenous (Figure 4), demonstrating no evident regional binding difference, which might attribute to the moderate selectivity of iloperidone on α1-adrenoceptor. To test the specificity of [11C]iloperidone toward α1-adrenoceptor in brain, we carried out blocking experiments by pretreatment of “cold” iloperidone (5 mg/kg) in C57BL/6 mice (20–25 g, female, and n = 2). The blocking TAC showed that the binding of [11C]iloperidone was blocked in mouse brain (Figures 3, 4), and the brain uptake was about 25% reduced based on the area under curve (AUC, 13 min to 55 min) comparing with baseline.
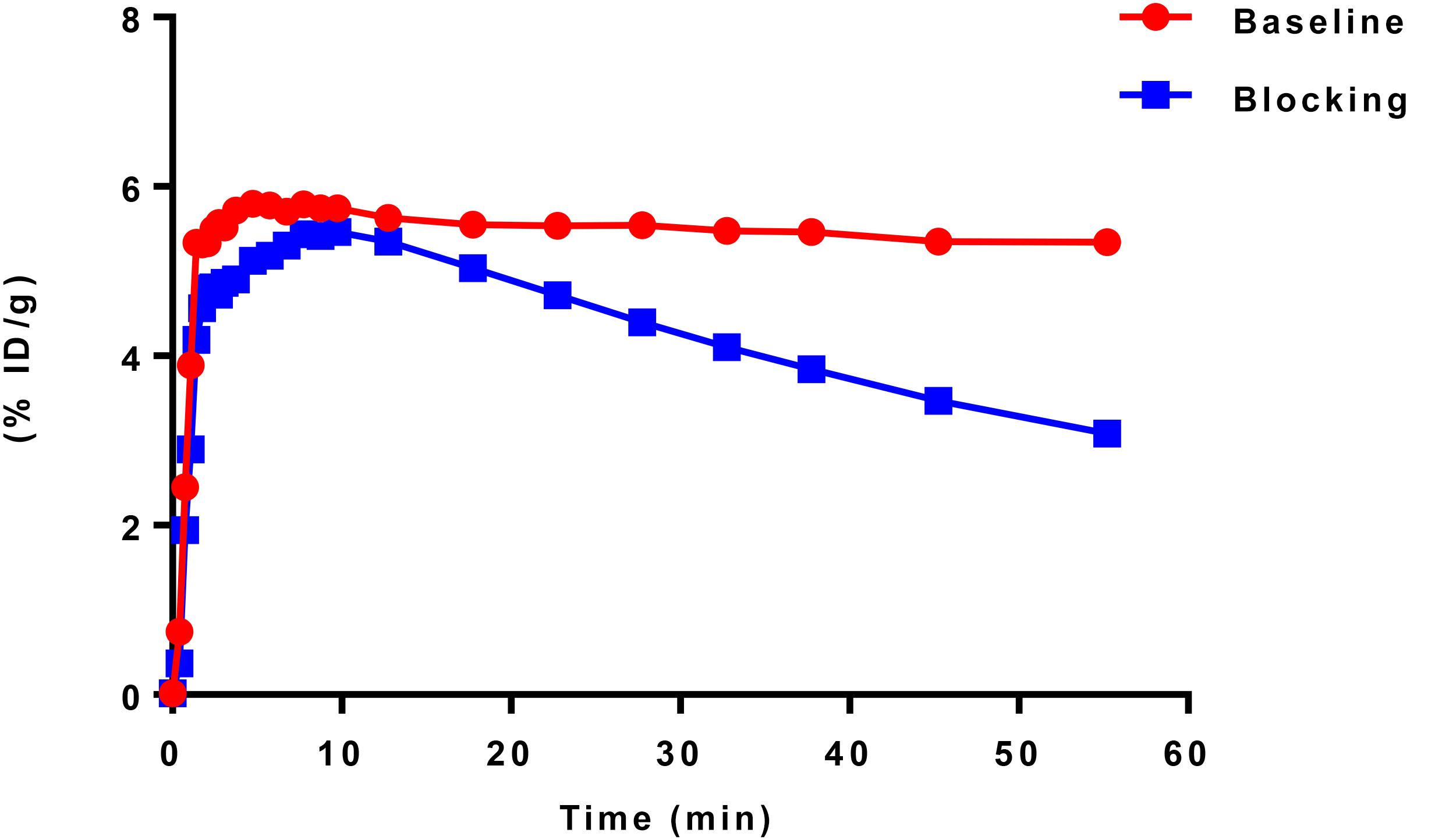
Figure 3. TACs of baseline (20–25 g, female, and n = 2) and blocking (20–25 g, female, and n = 2) experiments in C57BL/6 mouse brains. Data are expressed as the percentage of injected dose per gram (% ID/g).
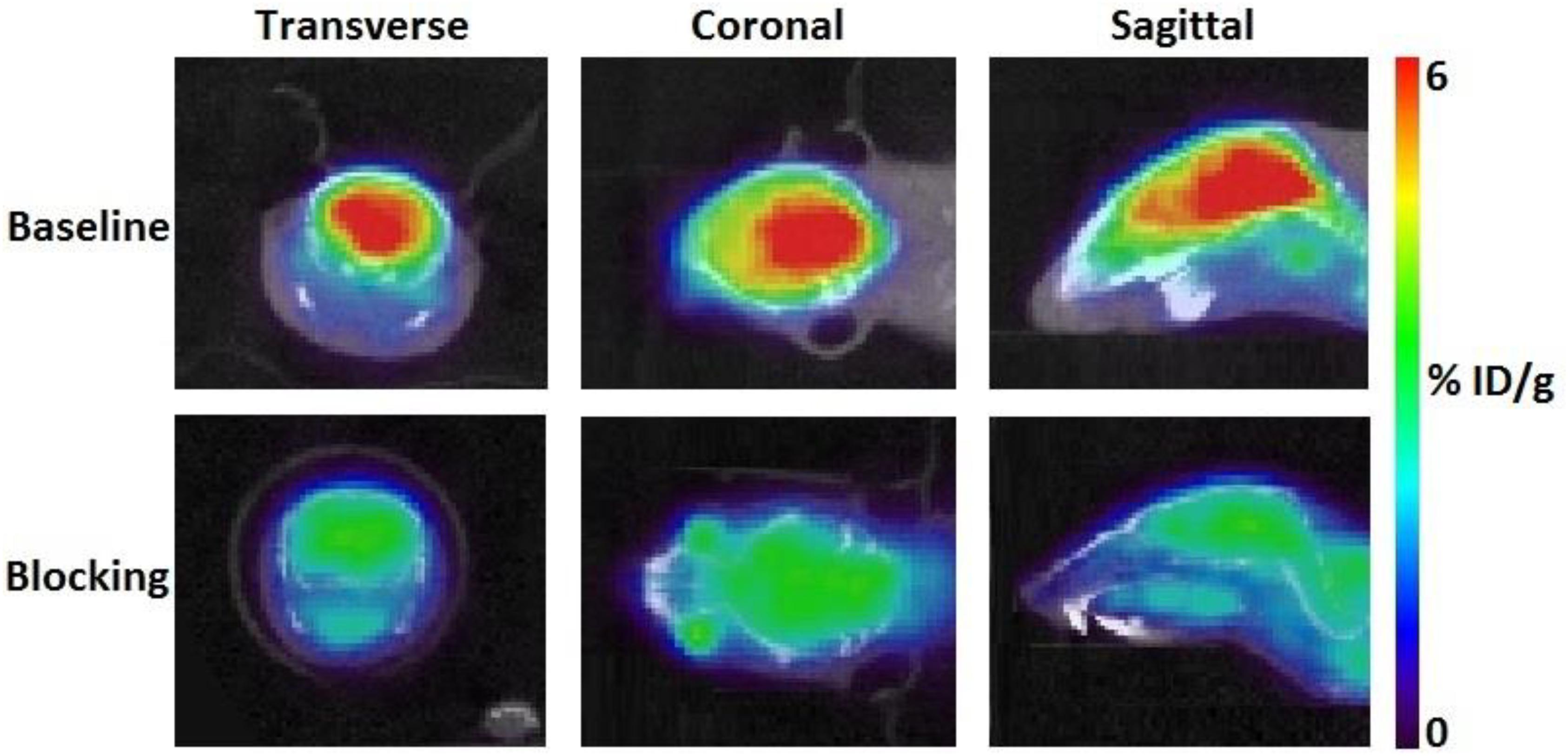
Figure 4. Transverse, coronal and sagittal PET/CT images of [11C]iloperidone in baseline (above) and blocking (below) experiments.
Conclusion
In summary, we have synthesized and characterized carbon-11 labeled iloperidone for imaging α1-adrenoceptor in brain. The radiolabeling of [11C]iloperidone was carried out conveniently in one step with good radiochemical yield and high radiochemical purity. The PET imaging studies in C57BL/6 mice demonstrated that [11C]iloperidone displayed favorable pharmacokinetic properties and exhibited high brain exposure. Blocking experiments by pretreatment with the unlabeled iloperidone showed the significant blocking effects with about 25% reduction in brain uptake. Despite of the promising preclinical profile in C57BL/6 mice, the clinical use of [11C]iloperidone might be hindered by its moderate specificity on α1-adrenoceptor. The efficient brain penetration of [11C]iloperidone, however, suggested that it can serve as a lead compound for the further structure activity relationship (SAR) exploration and optimization studies. We are continuing in this vein and will report our findings in due course.
Data Availability Statement
All datasets presented in this study are included in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by Subcommittee on Research Animal Care at Massachusetts General Hospital.
Author Contributions
YX and CW initiated this research project. YX, HW, and YW processed the data and performed the data analysis. YX and CW designed, wrote, and reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by a pilot funding from the Athinoula A. Martinos Centerfor Biomedical Imaging at the Massachusetts General Hospital (CW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors.
Acknowledgments
The authors are grateful to the Athinoula A. Martinos Center Radiopharmacy Lab staff for the assistance in radiochemistry.
References
Airaksinen, A. J., Finnema, S. J., Balle, T., Varnäs, K., Bang-Andersen, B., Gulyás, B., et al. (2013). Radiosynthesis and evaluation of new α1-adrenoceptor antagonists as PET radioligands for brain imaging. Nucl. Med. Biol. 40, 747–754. doi: 10.1016/j.nucmedbio.2013.05.007
Akinaga, J., García-Sáinz, J. A., and Pupo, A. S. (2019). Updates in the function and regulation of α1-Adrenoceptors. Br. J. Pharmacol. 176, 2343–2357.
Bergstroöm, M., Grahnen, A., and Långstroöm, B. (2003). Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur. J. Clin. Pharmacol. 59, 357–366. doi: 10.1007/s00228-003-0643-x
Chen, Z., Wang, X., Xu, Y., and Wang, C. (2019). Synthesis of 11C-labeled DNA polymerase-β inhibitor 5-methoxyflavone and PET/CT imaging thereof. Nucl. Med. Biol. 78, 17–22. doi: 10.1016/j.nucmedbio.2019.10.005
Datta, D., Yang, S.-T., Galvin, V. C., Solder, J., Luo, F., Morozov, Y. M., et al. (2019). Noradrenergic α1-adrenoceptor actions in the primate dorsolateral prefrontal cortex. J. Neurosci. 39, 2722–2734. doi: 10.1523/jneurosci.2472-18.2019
Gross, G., Hanft, G., and Mehdorn, H. M. (1989). Demonstration of alpha 1A- and alpha 1B-adrenoceptor binding sites in human brain tissue. Eur. J. Pharmacol. 169, 325–328. doi: 10.1016/0014-2999(89)90032-0
Joshi, S. V., Patel, E. P., Vyas, B. A., Lodha, S. R., and Kalyankar, G. G. (2020). Repurposing of iloperidone: antihypertensive and ocular hypotensive activity in animals. Eur. J. Pharm. Sci. 143:105173. doi: 10.1016/j.ejps.2019.105173
Kalaria, R. N. (1989). Characterization of [125I]HEAT binding to alpha 1-receptors in human brain: assessment in aging and alzheimer’s disease. Brain Res. 501, 287–294. doi: 10.1016/0006-8993(89)90645-8
Maletic, V., Eramo, A., Gwin, K., Offord, S. J., and Duffy, R. A. (2017). The role of norepinephrine and its α-adrenergic receptors in the pathophysiology and treatment of major depressive disorder and schizophrenia: a systematic review. Front Psychiatry 8:42. doi: 10.3389/fpsyt.2017.00042
Palacios, J. M., Hoyer, D., and Cortés, R. (1987). Alpha 1-adrenoceptors in the mammalian brain: similar pharmacology but different distribution in rodents and primates. Brain Res. 419, 65–75. doi: 10.1016/0006-8993(87)90569-5
Risgaard, R., Ettrup, A., Balle, T., Dyssegaard, A., Hansen, H. D., Lehel, S., et al. (2013). Radiolabelling and PET brain imaging of the α1-adrenoceptor antagonist Lu AE43936. Nucl. Med. Biol. 40, 135–140. doi: 10.1016/j.nucmedbio.2012.09.010
Selken, J., and Nichols, D. E. (2007). Alpha1-adrenergic receptors mediate the locomotor response to systemic administration of (+/-)-3,4-methylenedioxymethamphetamine (MDMA) in rats. Pharmacol. Biochem. Behav. 86, 622–630. doi: 10.1016/j.pbb.2007.02.006
Stahl, S. M. (2013). Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 18, 285–288. doi: 10.1017/s1092852913000850
Stone, E. A., Quartermain, D., Lin, Y., and Lehmann, M. L. (2007). Central alpha1-adrenergic system in behavioral activity and depression. Biochem. Pharmacol. 73, 1063–1075. doi: 10.1016/j.bcp.2006.10.001
Strupczewski, J. T., Bordeau, K. J., Chiang, Y., Glamkowski, E. J., Conway, P. G., Corbett, R., et al. (1995). 3-[[(Aryloxy)alkyl]piperidinyl]-1,2-benzisoxazoles as D2/5-HT2 antagonists with potential atypical antipsychotic activity: antipsychotic profile of iloperidone (HP 873). J. Med. Chem. 38, 1119–1131. doi: 10.1021/jm00007a009
Wang, C., Placzek, M. S., Bittner, G. C. V., Schroeder, F. A., and Hooker, J. M. (2015). A novel radiotracer for imaging monoacylglycerol lipase in the brain using positron emission tomography. ACS Chem. Neurosci. 7, 484–489. doi: 10.1021/acschemneuro.5b00293
Wang, C., Schroeder, F. A., and Hooker, J. M. (2017). Development of new positron emission tomography radiotracer for BET imaging. ACS Chem. Neurosci. 8, 17–21. doi: 10.1021/acschemneuro.6b00288
Wu, Z. H., Zhang, Q. J., Du, C. X., Xi, Y., Li, W. J., Guo, F. Y., et al. (2017). Prelimbic α1-adrenoceptors are involved in the regulation of depressive-like behaviors in the hemiparkinsonian rats. Brain Res. Bul. 134, 99–108. doi: 10.1016/j.brainresbull.2017.07.011
Keywords: α1-adrenoceptor, neuronal diseases, PET/CT imaging, [11C]iloperidone, brain exposure
Citation: Xu Y, Wang Y, Wang H and Wang C (2020) Synthesis and Characterization of Carbon-11 Labeled Iloperidone for Imaging of α1-Adrenoceptor in Brain. Front. Mol. Biosci. 7:586327. doi: 10.3389/fmolb.2020.586327
Received: 23 July 2020; Accepted: 01 September 2020;
Published: 24 September 2020.
Edited by:
Steven Hou, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Hongmei Jia, Beijing Normal University, ChinaLisheng Cai, National Institute of Mental Health, National Institutes of Health (NIH), United States
Copyright © 2020 Xu, Wang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changning Wang, Y3dhbmcxNUBtZ2guaGFydmFyZC5lZHU=; Yulong Xu, WXVsb25nLlh1QG1naC5oYXJ2YXJkLmVkdQ==