 James W. Murphy1Deepa Rajasekaran1
James W. Murphy1Deepa Rajasekaran1 Janie Merkel2Erin Skeens3Camille Keeler4†Michael E. Hodsdon4,5†George P. Lisi3‡
Janie Merkel2Erin Skeens3Camille Keeler4†Michael E. Hodsdon4,5†George P. Lisi3‡ Elias Lolis1,5*‡
Elias Lolis1,5*‡- 1Department of Pharmacology, Yale School of Medicine, Yale University, New Haven, CT, United States
- 2Yale Center for Molecular Discovery, Yale School of Medicine, Yale University, New Haven, CT, United States
- 3Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI, United States
- 4Department of Laboratory Medicine, Yale School of Medicine, Yale University, New Haven, CT, United States
- 5Yale Cancer Center, New Haven, CT, United States
CXCL12 activates CXCR4 and is involved in embryogenesis, hematopoiesis, and angiogenesis. It has pathological roles in HIV-1, WHIM disease, cancer, and autoimmune diseases. An antagonist, AMD3100, is used for the release of CD34+ hematopoietic stem cells from the bone marrow for autologous transplantation for lymphoma or multiple myeloma patients. Adverse effects are tolerated due to its short-term treatment, but AMD3100 is cardiotoxic in clinical studies for HIV-1. In an effort to determine whether Saccharomyces cerevisiae expressing a functional human CXCR4 could be used as a platform for identifying a ligand from a library of less ∼1,000 compounds, a high-throughput screening was developed. We report that 2-carboxyphenyl phosphate (fosfosal) up-regulates CXCR4 activation only in the presence of CXCL12. This is the first identification of a compound that increases CXCR4 activity by any mechanism. We mapped the fosfosal binding site on CXCL12, described its mechanism of action, and studied its chemical components, salicylate and phosphate, to conclude that they synergize to achieve the functional effect.
Introduction
CXCR4 is a GPCR with physiological roles in cellular homeostasis and a variety of immune response functions, and is activated by the chemokine agonist CXCL12. The agonist exists as a monomer (Crump et al., 1997), dimer (Dealwis et al., 1998), and a decamer (Murphy et al., 2010). Although the monomeric form activates the receptor (Kofuku et al., 2009; Qin et al., 2015), interactions of the various oligomeric states with glycoaminoglycans play a role in establishing a concentration gradient in tissues that allows immune cells expressing CXCR4 to migrate in response to increased CXCL12 concentrations (Fernandez and Lolis, 2002; Handel et al., 2005; Allen et al., 2007). Due to the importance of the monomer in activating the receptor and the oligomers in establishing this concentration gradient, most biophysical studies have investigated this monomer–dimer equilibrium, including factors that alter the equilibrium such as pH, phosphate or sulfate ions, and heparin (Veldkamp et al., 2005; Murphy et al., 2007).
In homeostasis, the monomer and higher order oligomers regulate the release of CD34+ hematopoietic stem cells from the bone marrow (Aiuti et al., 1997). The drug AMD3100 (Plerixafor), a CXCR4 antagonist, was approved as a second-line treatment for patients with multiple myeloma or non-Hodgkin’s lymphoma (De Clercq, 2009); however in a clinical trial as a HIV-1 therapeutic [CXCR4 is one of two co-receptors for HIV-1 (Feng et al., 1996)], AMD3100 was withdrawn from Phase 1 within 30 days due to premature ventricular contractions (Hendrix et al., 2004). Other studies are consistent with CXCL12-CXCR4 cardioprotection during pre- and post-myocardial ischemia, which is prevented by AMD3100 or a CXCR4 inducible knockout in cardiac myocytes (Hu et al., 2007; Huang et al., 2011; Dong et al., 2012; LaRocca et al., 2019). However, the effects of CXCL12-CXCR4 on the cardiovascular system are complicated by varied cell types and cardiac events, indicating more studies are necessary to define mechanisms where activation or antagonism of CXCR4 are beneficial (Bernhagen et al., 2007; Zernecke et al., 2008; Doring et al., 2017, 2019).
AMD3100 antagonism of CXCR4 would be a viable therapeutic approach for other diseases if a strategy to prevent its toxicity is identified. Some of these clinical indications for CXCR4 antagonism include infection by T-tropic strains of HIV which immediately precede AIDS (Feng et al., 1996; Doranz et al., 1997; Donzella et al., 1998), gain-of-function mutations in the orphan disease WHIM syndrome (Walters et al., 2010), and over 20 metastatic cancers (Chatterjee et al., 2014). Short-term treatment of AMD3100 in clinical settings successfully ablated the symptoms of WHIM although it has not been studied long term due to its toxicities (Walters et al., 2010; Dale et al., 2011; McDermott et al., 2011, 2019; Peled et al., 2012). Strategies or compounds that cause biased agonism/antagonism or increased CXCR4 activity may have significant research interest and provide a foundation for possible therapeutics.
Interestingly, AMD3100 functions as a partial agonist for a constitutively active mutant (CAM) of CXCR4, suggesting the feasibility of identifying small molecule agonists for the wild-type form of the receptor (Zhang et al., 2002; Rosenberg et al., 2019). We previously used a genetically manipulated strain of Saccharomyces cerevisiae expressing a functional human CXCR4 to identify two allosteric agonists from a cDNA library coding for ∼160,000 peptides derived from CXCL12 (Sachpatzidis et al., 2003). In the work reported here, we were interested in determining whether this S. cerevisiae strain could be used in a proof-of-principle experiment to identify a small molecule that antagonizes CXCR4 signaling from a small library of 960 bioactive compounds. Surprisingly, we found a compound, 2-carboxyphenyl phosphate (fosfosal), that increased CXCR4 activity but only in the presence of CXCL12. Fosfosal is a pro-drug for salicylate but has better gastric tolerance compared to acetylsalicylic acid and is used in Spain as a nonsteroidal anti-inflammatory drug (NSAID) (Rafanell et al., 1980). The effect of fosfosal in S. cerevisiae expressing CXCR4 in the presence of 200 nM CXCL12 increased signaling at the highest levels of fosfosal. Nuclear magnetic resonance (NMR) was used to determine the affinity of fosfosal for CXCL12, map the site of interaction, and, together with size exclusion chromatography with multi-angle light scattering (SEC-MALS) and molecular docking, determine the mechanism of action. We also evaluated the chemical components of fosfosal, salicylic acid and phosphate, to determine how these chemical groups contribute to fosfosal binding to CXCL12.
Materials and Methods
High-Throughput (HTS) Optimization of S. cerevisiae Expressing a Functional Human CXCR4
All chemicals and reagents were used as received. The Microsource (Microsource Discovery System, Inc., Gaylordsville, CT, United States) small molecule library, consisting of 960 compounds with known biological activity, was used for screening. High-throughput screening (HTS) assays were optimized to detect antagonists of CXCR4 using the agonist CXCL12 and antagonist AMD3100 as controls.
The genetically modified strain of S. cerevisiae (strain CY12946) used in the HTS was transformed with two plasmids (Sachpatzidis et al., 2003), pCXCR4 and pFus1-βgal, constitutively expressed human CXCR4 and the Fus1-dependent promoter for β-galactosidase (β-gal), respectively. The Fus promoter produced β-gal only when CXCR4 was activated by cell signaling. Other genetic changes to this S. cerevisiae strain that make this system appropriate for HTS were: (1) the replacement of the yeast GPCR Ste3 of the pheromone response pathway with CXCR4, thereby eliminating all other GPCRs for selectivity, (2) the replacement of five C-terminal residues of yeast Gα (Gpa1) with those from human Gαi2 to allow coupling with CXCR4, (3) the deletion of Ste14 (coding for a methyltransferase) to dampen background activity of the pheromone response pathway, (4) the deletion of Sst2 that codes for a RGS, a negative regulator of G-protein signaling to facilitate a more robust MAP kinase signaling response (Dohlman et al., 1996), (5) the deletion of Far1, which normally leads to cell cycle arrest, to promote a long-lived signaling response, (6) the addition of the Trp1 gene as a selective marker for lacZ, and (7) the transcription of lacZ upon MAP kinase signaling, allowing β-galactosidase to provide a quantitative measure of CXCR4 activation.
Based on high-throughput optimization, S. cerevisiae strain CY12946 previously transformed with pCXCR4 and pFus1-βgal was grown overnight to saturation (OD600 = 1.5) in nutritional selective dropout media and diluted to 55,000 cells/10 μL. Using three Corning #3574 non-binding white 384-well plates for the screen, 10 μL of complete minimal (CM) dropout media was distributed with a bulk reagent dispenser (Thermo Scientific Multidrop) into all wells. A positive control (100% antagonism) consisted of 10 μL of cells treated with 10 μM AMD3100 with 200 nM CXCL12 added to columns 1 and 2. CXCL12-treated cells were added to columns 3–22 followed by compounds from the library using a pin-tool device. Cells treated with only 200 nM CXCL12 were added to columns 23–24 as the negative control (0% antagonism). Plates were sealed, incubated with shaking at 30°C for 4 h, and treated with 20 μL of Beta-Glo (Promega, converts β-galactosidase activity to luminescence) for 25 min at 25°C. Luminescence measurements were made using an Envision plate reader. Signal to background for the assay was 28:1 and calculated by the mean luminescence of CXCL12 stimulated cells divided by mean luminescence of cells treated with AMD3100 followed by CXCL12. The Z’ value for the assay was >0.8 (Zhang et al., 1999). Wells containing potential antagonists were to be assessed as % inhibition with 0% and 100% representing the mean of columns 23–24 and 1–2, respectively.
Protein Expression and Purification of CXCL12 and 15N-CXCL12
CXC chemokine ligand 12 was expressed and purified as previously described (Murphy et al., 2007). Briefly, a modified human CXCL12α cDNA clone without the secretion sequence, but with a codon for an initiating Met and a deleted codon for the C-terminal Lys68, was cloned into the NdeI and XhoI restriction sites of the pET-22b expression vector and transformed into the Escherichia coli BL21(DE3) strain. Luria–Bertani media (1.5 L) with 100 μg/ml ampicillin was inoculated with BL21(DE3), grown to OD600 of 0.6, and induced with a final concentration of 1.0 mM isopropyl 1-thio-β-d-galactopyranoside. After expressing CXCL12 for an additional 4 h at 37°C, cells were harvested by centrifugation at 5,000 × g for 10 min. Cells were resuspended in phosphate-buffered saline at pH 7.4 and 1% Triton X-100, lysed using a French Press, and centrifuged for 30 min at 30,000 × g. CXCL12 was found in the inclusion bodies, which were washed three times in a buffer containing 100 mM Tris⋅HCl, pH 7.0, 5 mM EDTA, 5 mM DTT, 2 M urea, 2% Triton X-100 and once with wash consisting of 100 mM Tris⋅HCl pH 7.0, 5 mM EDTA, and 5 mM DTT. The inclusion bodies were solubilized with 6 M guanidine HCl, diluted 100-fold into a refolding buffer consisting of 100 mM Tris⋅HCl, pH 8.0, 5 mM EDTA, 0.2 mM oxidized glutathione, and 1 mM reduced glutathione, and stirred at 4°C overnight. Precipitated material was removed, and the refolded protein was applied to an SP Sepharose column and eluted by a NaCl gradient. CXCL12 fractions were combined and further purified by RP-HPLC. Fractions containing CXCL12 were concentrated and lyophilized. Pure CXCL12 was resuspended in sterile H2O (pH 7.0) with 0.1 mM NaN3 and a protease inhibitor cocktail prior to use. For NMR studies, isotopically labeled 15N-CXCL12 was expressed in M9 minimal media containing 1 g/L of 15NH4Cl (98%-15N), IPTG-induced, and purified as described above. CXCL12 concentration was determined by direct amino acid analysis at the W. M. Keck facility (Yale University).
Dose-Response of Fosfosal on CXCR4 Activation
Instead of a decrease in CXCL12 activity with any of the compounds, we observed an increase of activity with fosfosal. A dose-response experiment was used to determine the EC50 of fosfosal with 200 nM CXCL12 for activation of CXCR4 in S. cerevisiae with media described in the high throughput assay. Saturation could not be obtained with fosfosal but a non-linear fit was utilized to determine the EC50 using the Prism software [GraphPad, Inc., Log(agonist) vs. Response, Eq. 1]:
where Bmin/max are the luminescence values at low and high concentrations of fosfosal, and H is the Hill Slope.
Size Exclusion Chromatography With Multi-Angle Light Scattering (SEC-MALS)
An Agilent 960 HPLC system was used for analyzing CXCL12 with a Superdex 75 10/300 GL size exclusion column in tandem with Wyatt Dawn Helios II MALS/Wyatt T-rEx refractive index detectors (Wyatt Technology) under three different conditions: (A) 25 mM HEPES buffer at pH 7.4 with 500 mM NaCl, (B) same as (A) with 100 mM potassium phosphate, and (C) conditions as in (B) with 50 mM fosfosal.
NMR Spectroscopy
Nuclear magnetic resonance experiments were carried out on Varian Inova and Bruker Avance NEO 600 MHz spectrometers. All spectra were collected at 25°C in 50 mM HEPES buffer at pH 7.4 or 50 mM MES buffer at pH 5.6. NMR spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed in SPARKY (Lee et al., 2015). Fosfosal, salicylate, or phosphate was titrated into CXCL12 until no additional shifts were observed in 1H15N NMR resonances. Chemical shift titrations for the determination of fosfosal affinity for CXCL12 were fit with Eq. 2 (adapted from a one-phase association model) and analyzed in Wolfram Alpha (Wolfram Research) and Prism:
where Bunsat and Bsat represent the boundaries (normalized chemical shift values) of free and fosfosal-bound CXCL12, respectively, and Kd is the dissociation constant.
Results
High-Throughput Screening Assay for Antagonists of CXCR4 Activity in S. cerevisiae
We used a genetic strain of S. cerevisiae expressing a functional human CXCR4 to optimize an assay to identify antagonists from a small library of bioactive molecules (Sachpatzidis et al., 2003). In a previous study, the cDNA for a library of ∼160,000 peptides, each with a signal sequence, was co-transfected into this genetically modified strain of S. cerevisiae (Supplementary Figure S1). More recently, we studied the mechanism of a CAM of CXCR4 and small molecules that bound to the CXCR4 CAM in the same strain of S. cerevisiae (Rosenberg et al., 2019). Neither of these previous studies required the addition of recombinant wild-type CXCL12, but in the current study an optimal concentration of CXCL12 was necessary to identify compounds that functionally decrease CXCL12-mediated activity of CXCR4. We first used CXCL12 in a dose-response experiment to determine an EC50 of 269 nM (Figure 1). The EC50 in mammalian cells is 7.9 nM (Murphy et al., 2007). The high EC50 relative to human cells reflects differences between S. cerevisiae and mammalian cells but did not prevent the optimization of the screen using variations of CXCL12 concentration, the number of S. cerevisiae cells, and time of incubation (Supplementary Figure S2). Of the 960 screened compounds, we were surprised to observe one, fosfosal, that increased CXCR4 activity by three standard deviation units above the average for all wells at about 20% above the control of 200 nM CXCL12 alone.
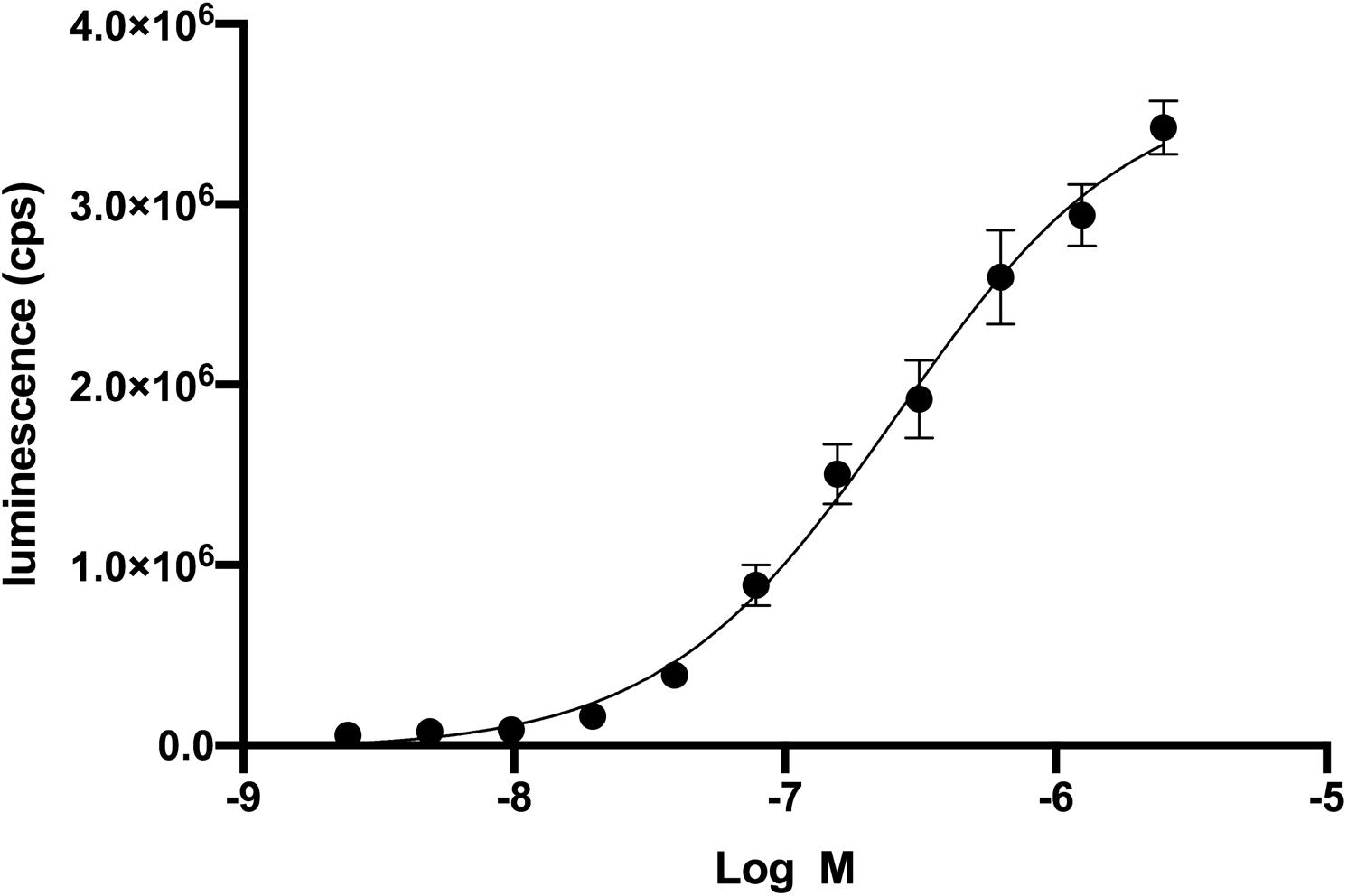
Figure 1. Recombinant CXCL12 activation of CXCR4 in Saccharomyces cerevisiae. Dose-response of CXCL12 activation of CXCR4 results in an EC50 of 269 nM. CXCR4 activation leads to the β-galactosidase enzymatic activity. Beta-glo (Promega) is a coupled enzyme reaction system to quantitate β-galactosidase by using a 6-O-β-galactopyranosyl-luciferin that is cleaved to luciferin and galactose. Luciferin is the substrate for the firefly luciferase and generates luminescence.
Characterization and Mechanism of Fosfosal in CXCL12-Mediated CXCR4 Activation
Fosfosal is a salicylic acid derivative marketed in Spain as a NSAID (Rafanell et al., 1980). We evaluated its dose-response effects in the engineered S. cerevisiae strain and found that fosfosal is inactive when tested on its own (Supplementary Figure S3). Fosfosal only has activity in the presence of CXCL12 with an EC50,app of 217 μM for activation of CXCR4 calculated through a non-linear fit (Equation 1), as fosfosal does not reach saturation due to its solubility (Supplementary Figure S3). We next explored whether there were any specific interactions of fosfosal with CXCL12 in either of its oligomeric states.
Isotopically labeled 15N-CXCL12 was produced as described (Murphy et al., 2007). A global fit of eight CXCL12 NMR resonances with significant chemical shift perturbations upon titration of 16 mM fosfosal into CXCL12 yielded an apparent dissociation constant, Kd,app of 147 μM (Figures 2A,B) after accounting for the concentration of CXCL12 (0.6 mM). At this concentration, apo CXCL12 is expected to be dimeric. We mapped residues with significant chemical shift perturbations or line broadening at saturating fosfosal concentrations onto the CXCL12 structure (Figure 2C). Most notably, His25, Lys27, and Ala40 are in close proximity in the CXCL12 monomer and were proposed to be critical for CXCL12 dimerization in a prior study (Gozansky et al., 2005). Additional chemical shift perturbations localize to the area surrounding these residues spanning the β-strand core of the CXCL12 monomer-monomer interface (Figure 2C). Resonances selected for this binding analysis were in the fast exchange regime, although the addition of fosfosal modulates other sites of slow-to-intermediate exchange, most notably the N-terminus, upon titration into 0.6 mM CXCL12 (Supplementary Figures S4, S5). This suggests the interaction of fosfosal with CXCL12 may propagate a binding signal to the flexible N-terminal region, or alter its architecture.
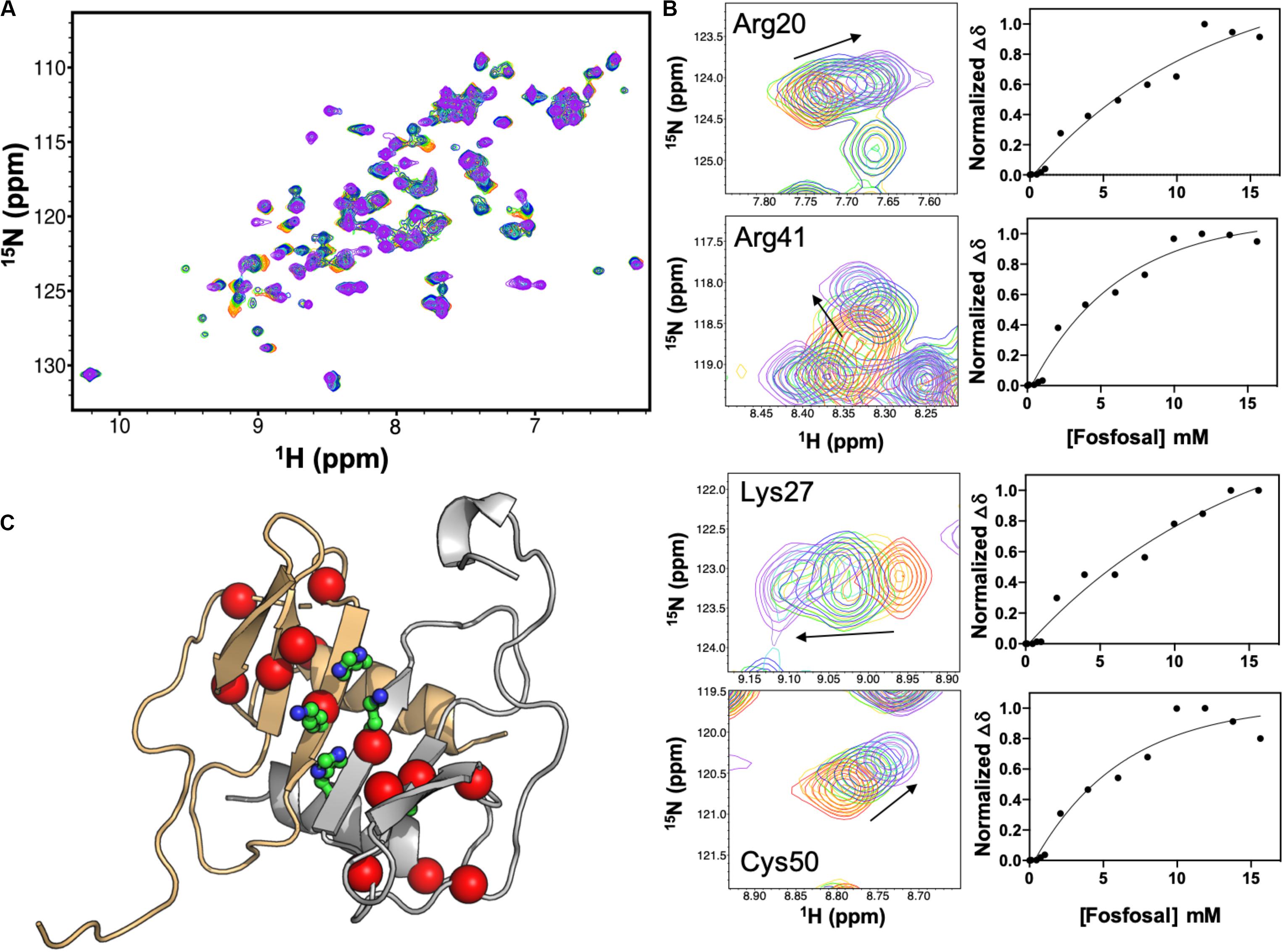
Figure 2. NMR titration of CXCL12 with fosfosal at pH 7.4. (A) Chemical shift perturbations in the 1H-15N HSQC NMR spectrum of 0.6 mM CXCL12 induced by 1–16 mM fosfosal (red → purple). (B) Selected resonances with significant chemical shift perturbations. Global fitting of eight chemical shift profiles gives an apparent binding affinity, Kd,app = 147 μM. (C) Structure of the CXCL12 dimer (PDB 4UAI) showing the monomeric units in gray and orange. Residues His25, Lys27, and Ala40, thought to be critical for dimerization (Gozansky et al., 2005), are shown in green sticks. Residues with significant chemical shift perturbations in fosfosal titrations are mapped onto the structure, clustering around the β-strand core and dimer interface.
We collected an additional 1H-15N NMR titration for fosfosal binding to 0.15 mM CXCL12, under buffer conditions and a concentration that favors CXCL12 monomers in solution. The 1H15N NMR spectra of CXCL12 at each concentration are very similar (Supplementary Figure S6), and Figure 3 shows chemical shift perturbations for fosfosal binding to CXCL12 under conditions favoring dimers and monomers. In the latter case, fosfosal does not induce chemical shift perturbations in 0.15 mM CXCL12 beyond 6 mM. The affected regions of the protein are consistent in both experiments (Figure 3), and the possibility that some contribution to the larger chemical shift perturbations in the dimer titration stem from structural effects propagated by the binding of a second fosfosal molecule to the adjacent CXCL12 subunit must be noted. Molecular docking of fosfosal with the CXCL12 monomer and dimer structures performed with HADDOCK (van Zundert et al., 2016), which incorporates NMR chemical shift data to define binding constraints, also indicates the interaction of fosfosal occurs with the CXCL12 monomer or dimer (Supplementary Figure S7). Interestingly, fosfosal does not modulate slow-to-intermediate exchange in NMR spectra of 0.15 mM CXCL12 (Figure 3 and Supplementary Figures S4, S5), as there are neither “satellite” peaks observed nor N-terminal resonances that reappear upon saturation with the ligand. This may be related to the ability of fosfosal to preferentially modulate dynamics in the CXCL12 dimer that in turn could alter the energetics of non-covalent interactions in the oligomer.
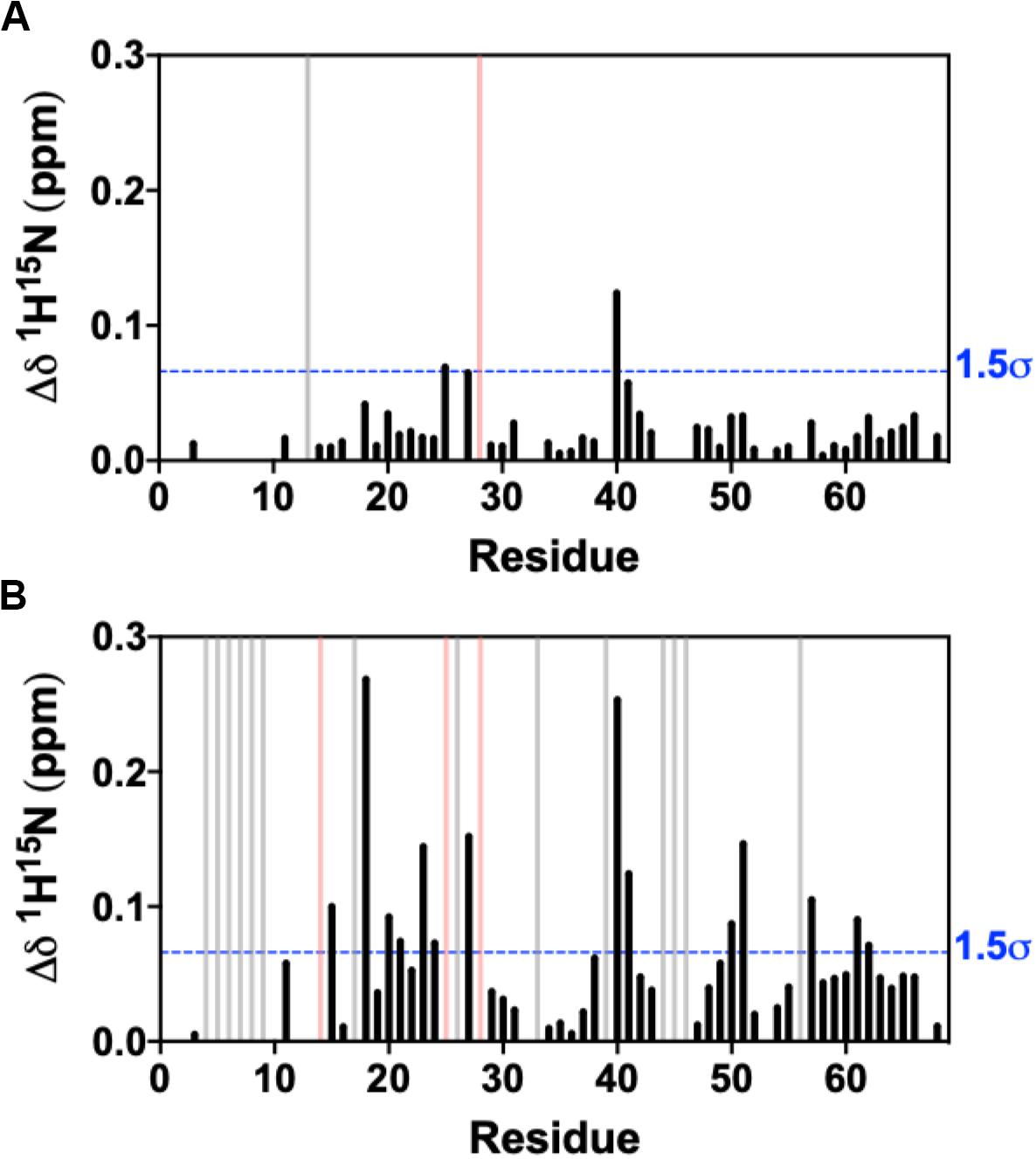
Figure 3. Combined 1H-15N NMR chemical shift perturbations from titration of 30 mM fosfosal stock solution into (A) 0.15 mM CXCL12 and (B) 0.6 mM CXCL12 at pH 7.4. Blue dashed lines indicate 1.5σ above the 10% trimmed mean of all shifts, gray bars denote peaks that became visible during the titration (intermediate → fast exchange), and pink bars represent peaks with observed line broadening.
Size exclusion chromatography with multi-angle light scattering experiments show that the average molecular weight of CXCL12 in solution decreases from 11.6 to 9.4 kDa with 50 mM fosfosal (Supplementary Figure S8) suggesting that the equilibrium between monomer and dimer shifts toward the monomeric state in the presence of fosfosal. Given the SEC-MALS results in conjunction with the NMR data and computational modeling of fosfosal with both forms of CXCL12 using HADDOCK, we conclude fosfosal binds each monomer at surface residues at the dimer interface, affecting the monomer-monomer interactions, and increasing the concentration of the active monomer.
Effects of Chemical Components of Fosfosal on CXCL12
Fosfosal is composed of salicylate and phosphate moieties. In a previous study, negatively charged counter-ions such as phosphate, citrate, sulfate, the glycosaminoglycan heparin, or a non-acidic pH to keep the His25 side chain uncharged stabilized the dimer structure (Veldkamp et al., 2005). The heparin binding site was determined by both NMR (Veldkamp et al., 2005) and X-ray crystallography (Murphy et al., 2007). We studied the relative effects of the salicylate and phosphate moieties on CXCL12 for comparison to fosfosal. In order to mitigate effects from changes in the monomer–dimer equilibrium during these experiments, NMR chemical shifts were monitored upon saturation with fosfosal, sodium phosphate or sodium salicylate at pH 5.6 (Supplementary Figure S9), as acidic pH has been shown to favor monomeric CXCL12 even at high concentrations (van Zundert et al., 2016). The NMR spectrum of CXCL12 was fully assigned at pH 5.6 to account for some notable differences from the pH 7.4 spectrum caused by the change in pH (Supplementary Figure S10). The overall magnitude of the chemical shift perturbations of salicylate and phosphate is diminished relative to those caused by fosfosal, although they map to the same binding region of CXCL12 with residues in regions 24–27 and 39–42 most affected and statistically significant (Figure 4A). Together, the salicylate and phosphate moieties appear to have a predominantly additive effect, where the chemical shift profile of fosfosal is nearly recapitulated when its two components are combined (Figure 4B).
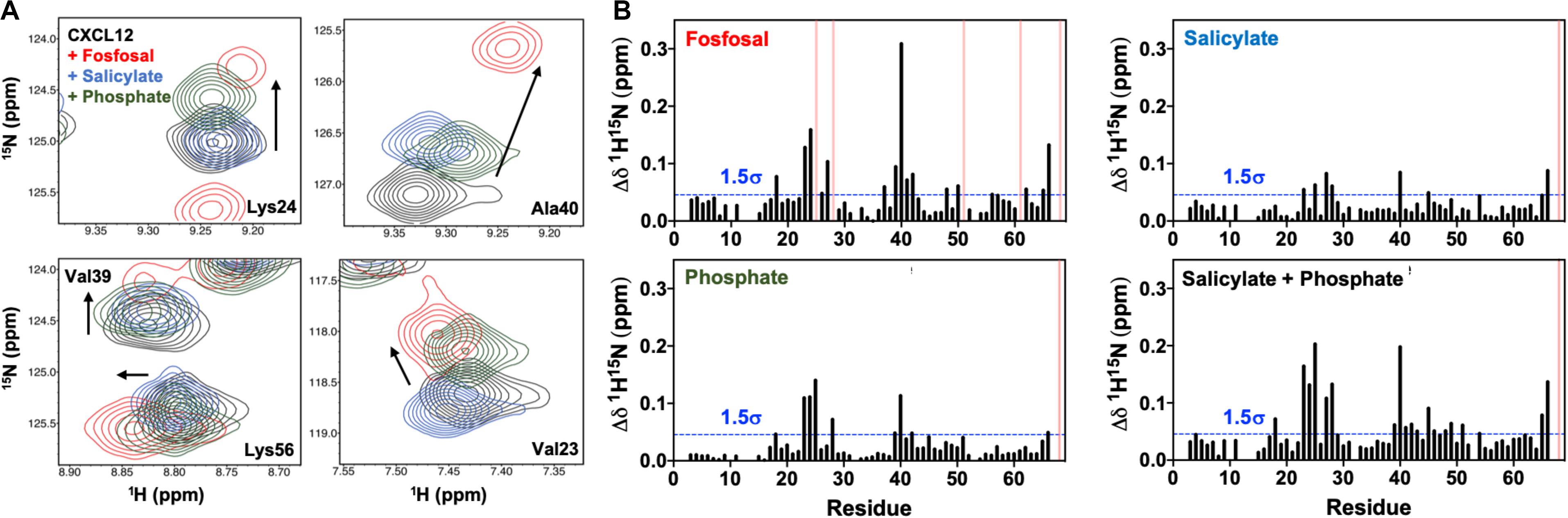
Figure 4. (A) Contribution of salicylate and phosphate moieties to fosfosal binding. Selected chemical shift perturbations induced by saturation of 0.6 mM CXCL12 (black) with fosfosal (red), sodium salicylate (blue), and phosphate (green). NMR spectra were collected in 50 mM MES buffer at pH 5.6, as acidic pH was previously shown to mitigate dimer formation (Veldkamp et al., 2005). (B) Combined 1H-15N NMR chemical shift perturbations observed for 0.6 mM CXCL12 with fosfosal (top left), sodium salicylate (top right), and phosphate (bottom left), and a plot of the perturbations observed from the additive effect of sodium salicylate and phosphate (bottom right). Blue dashed lines indicate 1.5σ above the 10% trimmed mean of all shifts and pink bars represent peaks with observed line broadening.
Discussion
The largest protein family targeted by FDA-approved drugs is GPCRs, which play many physiological roles and are activated by photons, ions, small molecules, and proteins. The drugs that are approved for GPCRs are agonists, antagonists, inverse agonists, and modulators of activity. The dynamic nature of the GPCR protein family makes it amenable to identifying small molecules that bind to different conformational states. Over the past 15 years, technological advances have made the determination of three-dimensional structures of recombinant GPCRs possible for use in structure-guided drug design or virtual screening. Other methods to identify small molecule modulators rely on HTS followed by structure-activity relationships to identify. Optimized assays are capable of identifying small molecule compounds that function as agonists or antagonists. It is far more challenging to identify an agonist, which binds and induces conformational changes to activate a receptor than an antagonist that binds and blocks an agonist from binding to its receptor. Therefore, there are many antagonists for GPCRs but few agonists. For CXCR4, a small molecule agonist with moderate potency for CXCR4 is known (Mishra et al., 2016). Other strategies to modulate receptor activities are focused on chemokines instead of their receptors. A CXCL12 small molecule inhibitor is known to prevent activation of CXCR4 and CXCR7 at low micromolar levels (Veldkamp et al., 2010), but there are no known positive modulators of chemokines that increase the activity of their receptor.
The S. cerevisiae strain expressing a functional CXCR4 used for our HTS has both advantages and disadvantages. The advantages include ease of growth, nutrient selectivity for desired properties, simplicity of genetic modification, and the ability to couple G-protein to intracellular pathways resulting in a quantitative response. The expression of human CXCR4 in a non-mammalian cell also has disadvantages. For example, the EC50 for recombinant CXCL12 is ∼100-fold greater in S. cerevisiae relative to mammalian cells, likely due to a number of factors. Further, the cell wall of S. cerevisiae is a barrier for proteins under a variety of conditions. Another disadvantage is the inability to sulfate tyrosine residues, which contribute to chemokine affinity (Veldkamp et al., 2006). Tyrosine residues at positions 7, 12, and 21 in CXCR4 are not sulfated due to the absence of tyrosylprotein sulfotransferase-1 in the S. cerevisiae genome. There are also differences in the phospholipid bilayers of S. cerevisiae and mammalian cell membranes that may alter the conformational states and physiological function of any human GPCR. The ratio of specific phospholipids is different while other components may be entirely distinct. For example, cholesterol in mammalian cells is replaced by ergosterol in S. cerevisiae. CXCR4 and many other mammalian GPCRs bind cholesterol, which plays a role in stability and function (Liu et al., 2012; Pluhackova et al., 2016). None of these differences, however, prevented the development of a high-throughput assay for CXCR4.
We used a small library with many FDA-approved or bioactive drugs in a proof-of-principle study to determine whether S. cerevisiae can be optimized for an HTS for chemokine receptors. We identified fosfosal as a positive regulator of CXCR4, but only in the presence of CXCL12. The true EC50 of fosfosal could not be determined because it does not reach saturation (Supplementary Figure S3). Due to its weak affinity, we used orthogonal, in vitro techniques with purified recombinant protein to confirm binding and investigate its mechanism. The activity of fosfosal is due to its interactions with residues that map to surface residues of the CXCL12 dimer interface. Although CXCL12 dimers and higher order oligomers (Murphy et al., 2010) play an important role in chemokine function by forming a concentration gradient that is required for migration of cells (Proudfoot et al., 2003), it is the monomer that activates CXCR4 as previously shown by the three-dimensional structures of chemokine-receptor complexes (Burg et al., 2015; Qin et al., 2015; Zheng et al., 2017). The mechanism of how fosfosal increases the CXCL12-mediated CXCR4 activity is based on the following facts: (1) fosfosal increases the activity of CXCL12 in cellular studies with S. cerevisiae, (2) NMR suggests fosfosal interacts with both the dimer and monomer, saturating the monomer over a narrower concentration range of fosfosal, (3) fosfosal promotes a decrease in the average molecular weight of CXCL12 by SEC-MALS, and (4) the CXCL12 monomer is the active species. Molecular docking shows favorable interactions with the CXCL12 dimer, perhaps to modulate the monomer–dimer equilibrium via another conformational step to favor the monomeric active form. Fosfosal-induced conformational changes exclusive to the CXCL12 dimer would be consistent with NMR-detected changes in dynamics found only in this complex; however, we do not have crystallographic data that confirms the docked model. Additionally, the computations with HADDOCK are not precise enough to determine whether the monomer or dimer is favored in vitro or in vivo, as monomeric CXCL12 also strongly docks fosfosal. The majority of these data are consistent with a case where the interaction of fosfosal with monomeric CXCL12 is thermodynamically favored, which would be the expectation based on its activity. We also analyzed the contribution to binding of both the salicylate and phosphate chemical moieties of fosfosal, as both molecules form interactions with CXCL12 in docked complexes and display an additive effect in NMR chemical shift titrations. However, these smaller functional groups do not induce changes in the exchange regimes of amino acids observed with fosfosal (Supplementary Figures S4, S5).
We describe the first molecule that up-regulates CXCR4 activity and affects the monomer-dimer equilibrium of CXCL12 to favor the monomeric form. Since fosfosal is a weak up-regulator that was part of a 960-compound library, much larger compound libraries could be screened to identify more potent regulators of CXCL12. Alternatively, fosfosal analogs can be purchased or synthesized to test for increased the potency and induce CXCL12-mediated CXCR4 signaling. Known ligand databases (i.e., PubChem) have many fosfosal analogs that we will test with structure-based virtual and experimental screening in subsequent studies. This dimeric interaction interface can also be used to investigate molecules that stabilize the CXCL12 dimer and down-regulate CXCR4 activity. Finally, modification of this genetic strain of S. cerevisiae could be used for HTS of other chemokines, receptors, or GPCRs to identify tool compounds for studying these protein–receptor interactions.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
JWM, DR, and JM were involved in the optimization of the high-throughput screening and carried out screening against the library. ES, CK, and JM were involved with expression, purification, and purification of labeled and unlabeled CXCL12, and carried out the NMR experiments. JWM was involved in studying fosfosal in S. cerevisiae and in carrying out SEC-MALS. All co-authors were involved in analyzing the data. GL and EL wrote the manuscript. All authors have given approval to the final version of the manuscript.
Funding
This work was supported, in part, by a Pilot Grant from the Yale Cancer Center (to EL) and start-up funds from Brown University (to GL).
Conflict of Interest
MH is now employed by Eli Lilly and Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the W. M. Keck facility for assistance with amino acid sequencing of CXCL12. The use of HADDOCK 2.4 acknowledges the FP7 WeNMR (project# 261572), H2020 West-Life (project# 675858), and the EOSC-hub (project# 777536) European e-Infrastructure projects for the use of their web portals, which make use of the EGI infrastructure with the dedicated support of CESNET-MetaCloud, INFN-PADOVA, NCG-INGRID-PT, TW-NCHC, SURFsara, and NIKHEF, and the additional support of the national GRID Initiatives of Belgium, France, Italy, Germany, Netherlands, Poland, Portugal, Spain, United Kingdom, Taiwan, and the US Open Science Grid.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00164/full#supplementary-material
Abbreviations
CXCL12, CXC chemokine ligand 12; CXCR4, CXC chemokine receptor 2; fosfosal, 2-(phosphonooxy) benzoic acid; GPCR, G protein-coupled receptor.
References
Aiuti, A., Webb, I. J., Bleul, C., Springer, T., and Gutierrez-Ramos, J. C. (1997). The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J. Exp. Med. 185, 111–120. doi: 10.1084/jem.185.1.111
Allen, S. J., Crown, S. E., and Handel, T. M. (2007). Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820.
Bernhagen, J., Krohn, R., Lue, H., Gregory, J. L., Zernecke, A., Koenen, R. R., et al. (2007). MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596. doi: 10.1038/nm1567
Burg, J. S., Ingram, J. R., Venkatakrishnan, A. J., Jude, K. M., Dukkipati, A., Feinberg, E. N., et al. (2015). Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117. doi: 10.1126/science.aaa5026
Chatterjee, M., Borst, O., Walker, B., Fotinos, A., Vogel, S., Seizer, P., et al. (2014). Macrophage migration inhibitory factor limits activation-induced apoptosis of platelets via CXCR7-dependent Akt signaling. Circ. Res. 115, 939–949. doi: 10.1161/circresaha.115.305171
Crump, M. P., Gong, J. H., Loetscher, P., Rajarathnam, K., Amara, A., Arenzana-Seisdedos, F., et al. (1997). Solution structure and basis for functional activity of stromal cell- derived factor-1, dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16, 6996–7007. doi: 10.1093/emboj/16.23.6996
Dale, D. C., Bolyard, A. A., Kelley, M. L., Westrup, E. C., Makaryan, V., Aprikyan, A., et al. (2011). The CXCR4 antagonist plerixafor is a potential therapy for myelokathexis, WHIM syndrome. Blood 118, 4963–4966. doi: 10.1182/blood-2011-06-360586
De Clercq, E. (2009). The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil). Biochem. Pharmacol. 77, 1655–1664. doi: 10.1016/j.bcp.2008.12.014
Dealwis, C., Fernandez, E. J., Thompson, D. A., Simon, R. J., Siani, M. A., and Lolis, E. (1998). Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1alpha, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. U.S.A. 95, 6941–6946. doi: 10.1073/pnas.95.12.6941
Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293.
Dohlman, H. G., Song, J., Ma, D., Courchesne, W. E., and Thorner, J. (1996). Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein alpha subunit). Mol. Cell. Biol. 16, 5194–5209. doi: 10.1128/mcb.16.9.5194
Dong, F., Harvey, J., Finan, A., Weber, K., Agarwal, U., and Penn, M. S. (2012). Myocardial CXCR4 expression is required for mesenchymal stem cell mediated repair following acute myocardial infarction. Circulation 126, 314–324. doi: 10.1161/circulationaha.111.082453
Donzella, G. A., Schols, D., Lin, S. W., Este, J. A., Nagashima, K. A., Maddon, P. J., et al. (1998). AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co- receptor. Nat. Med. 4, 72–77. doi: 10.1038/nm0198-072
Doranz, B. J., Grovit-Ferbas, K., Sharron, M. P., Mao, S. H., Goetz, M. B., Daar, E. S., et al. (1997). A small-molecule inhibitor directed against the chemokine receptor CXCR4 prevents its use as an HIV-1 coreceptor. J. Exp. Med. 186, 1395–1400. doi: 10.1084/jem.186.8.1395
Doring, Y., Noels, H., van der Vorst, E. P. C., Neideck, C., Egea, V., Drechsler, M., et al. (2017). Vascular CXCR4 limits atherosclerosis by maintaining arterial integrity: evidence from mouse and human studies. Circulation 136, 388–403. doi: 10.1161/circulationaha.117.027646
Doring, Y., van der Vorst, E. P. C., Duchene, J., Jansen, Y., Gencer, S., Bidzhekov, K., et al. (2019). CXCL12 derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation 139, 1338–1340. doi: 10.1161/circulationaha.118.037953
Feng, Y., Broder, C. C., Kennedy, P. E., and Berger, E. A. (1996). HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877. doi: 10.1126/science.272.5263.872
Fernandez, E. J., and Lolis, E. (2002). Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42, 469–499.
Gozansky, E., Louis, J., Caffrey, M., and Clore, G. (2005). Mapping the Binding of the N-terminal Extracellular tail of the CXCR4 receptor to stromal cell-derived factor-1. J. Mol. Biol. 345, 651–658. doi: 10.1016/j.jmb.2004.11.003
Handel, T. M., Johnson, Z., Crown, S. E., Lau, E. K., Sweeney, M., and Proudfoot, A. E. (2005). Regulation of protein function by glycosoaminoglycans–as exemplified by chemokines. Annu. Rev. Biochem. 74, 385–410. doi: 10.1146/annurev.biochem.72.121801.161747
Hendrix, C. W., Collier, A. C., Lederman, M. M., Schols, D., Pollard, R. B., Brown, S., et al. (2004). Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J. Acqu. Immune Def. Syndrom. 37, 1253–1262. doi: 10.1097/01.qai.0000137371.80695.ef
Hu, X., Dai, S., Wu, W. J., Tan, W., Zhu, X., Mu, J., et al. (2007). Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation 116, 654–663. doi: 10.1161/circulationaha.106.672451
Huang, C., Gu, H., Zhang, W., Manukyan, M. C., Shou, W., and Wang, M. (2011). SDF-1/CXCR4 mediates acute protection of cardiac function through myocardial STAT3 signaling following global ischemia/reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 301, H1496–H1505.
Kofuku, Y., Yoshiura, C., Ueda, T., Terasawa, H., Hirai, T., Tominaga, S., et al. (2009). Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J. Biol. Chem. 284, 35240–35250. doi: 10.1074/jbc.m109.024851
LaRocca, T. J., Altman, P., Jarrah, A. A., Gordon, R., Wang, E., Hadri, L., et al. (2019). CXCR4 cardiac specific knockout mice develop a progressive cardiomyopathy. Intern. J. Mol. Sci. 20:2267. doi: 10.3390/ijms20092267
Lee, W., Tonelli, M., and Markley, J. L. (2015). NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. doi: 10.1093/bioinformatics/btu830
Liu, W., Chun, E., Thompson, A. A., Chubukov, P., Xu, F., Katritch, V., et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236. doi: 10.1126/science.1219218
McDermott, D. H., Liu, Q., Ulrick, J., Kwatemaa, N., Anaya-O’Brien, S., Penzak, S. R., et al. (2011). The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood 118, 4957–4962. doi: 10.1182/blood-2011-07-368084
McDermott, D. H., Pastrana, D. V., Calvo, K. R., Pittaluga, S., Velez, D., and Cho, E. (2019). Plerixafor for the treatment of WHIM syndrome. N. Engl. J. Med. 380, 163–170.
Mishra, R. K., Shum, A. K., Platanias, L. C., Miller, R. J., and Schiltz, G. E. (2016). Discovery and characterization of novel small-molecule CXCR4 receptor agonists and antagonists. Sci. Rep. 6:30155.
Murphy, J. W., Cho, Y., Sachpatzidis, A., Fan, C., Hodsdon, M. E., and Lolis, E. (2007). Structural and functional basis of CXCL12 (stromal cell-derived factor-1alpha) binding to heparin. J. Biol. Chem. 282, 10018–10027. doi: 10.1074/jbc.m608796200
Murphy, J. W., Yuan, H., Kong, Y., Xiong, Y., and Lolis, E. J. (2010). Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins 78, 1331–1337. doi: 10.1002/prot.22666
Peled, A., Wald, O., and Burger, J. (2012). Development of novel CXCR4-based therapeutics. Expert Opin. Investig. Drugs 21, 341–353. doi: 10.1517/13543784.2012.656197
Pluhackova, K., Gahbauer, S., Kranz, F., Wassenaar, T. A., and Bockmann, R. A. (2016). Dynamic cholesterol-conditioned dimerization of the G protein coupled chemokine receptor Type 4. PLoS Comput. Biol. 12:e1005169. doi: 10.1371/journal.pcbi.1005169
Proudfoot, A. E., Handel, T. M., Johnson, Z., Lau, E. K., LiWang, P., Clark-Lewis, I., et al. (2003). Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890. doi: 10.1073/pnas.0334864100
Qin, L., Kufareva, I., Holden, L. G., Wang, C., Zheng, Y., Zhao, C., et al. (2015). Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122. doi: 10.1126/science.1261064
Rafanell, J. G., Belles, L., Sanchez, M. S., and Forn, J. (1980). Pharmacological study of 2-phosphonoxybenzoic acid (fosfosal), a new analgesic drug. Arzneimittelforschung 30, 1091–1098.
Rosenberg, E. M., Harrison, R. E. D., Tsou, L. K., Drucker, N., and Humphries, B. (2019). Functional characterization, dynamics, and mechanism of cxcr4 antagonists on a constitutively active mutant. Cell Chem. Biol. 26, 662–673.
Sachpatzidis, A., Benton, B. K., Manfredi, J. P., Wang, H., Hamilton, A., Dohlman, H. G., et al. (2003). Identification of allosteric peptide agonists of CXCR4. J. Biol. Chem. 278, 896–907. doi: 10.1074/jbc.m204667200
van Zundert, G. C. P., Rodrigues, J., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., et al. (2016). The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725. doi: 10.1016/j.jmb.2015.09.014
Veldkamp, C. T., Peterson, F. C., Pelzek, A. J., and Volkman, B. F. (2005). The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 14, 1071–1081. doi: 10.1110/ps.041219505
Veldkamp, C. T., Seibert, C., Peterson, F. C., Sakmar, T. P., and Volkman, B. F. (2006). Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1[alpha] (SDF-1[alpha]/CXCL12). J. Mol. Biol. 359, 1400–1409. doi: 10.1016/j.jmb.2006.04.052
Veldkamp, C. T., Ziarek, J. J., Peterson, F. C., Chen, Y., and Volkman, B. F. (2010). Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J. Am. Chem. Soc. 132, 7242–7243. doi: 10.1021/ja1002263
Walters, K. B., Green, J. M., Surfus, J. C., Yoo, S. K., and Huttenlocher, A. (2010). Live imaging of neutrophil motility in a zebrafish model of WHIM syndrome. Blood 116, 2803–2811. doi: 10.1182/blood-2010-03-276972
Zernecke, A., Bot, I., Djalali-Talab, Y., Shagdarsuren, E., Bidzhekov, K., Meiler, S., et al. (2008). Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 102, 209–217. doi: 10.1161/circresaha.107.160697
Zhang, J.-H., Chung, T. D. Y., and Oldenburg, K. R. (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73. doi: 10.1177/108705719900400206
Zhang, W. B., Navenot, J. M., Haribabu, B., Tamamura, H., Hiramatu, K., Omagari, A., et al. (2002). A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J. Biol. Chem. 277, 24515–24521. doi: 10.1074/jbc.m200889200
Keywords: chemokine, G protein-coupled receptor, CXCL12, CXCR4, high-throughput screening, fosfosal, NMR
Citation: Murphy JW, Rajasekaran D, Merkel J, Skeens E, Keeler C, Hodsdon ME, Lisi GP and Lolis E (2020) High-Throughput Screening of a Functional Human CXCL12-CXCR4 Signaling Axis in a Genetically Modified S. cerevisiae: Discovery of a Novel Up-Regulator of CXCR4 Activity. Front. Mol. Biosci. 7:164. doi: 10.3389/fmolb.2020.00164
Received: 17 March 2020; Accepted: 25 June 2020;
Published: 16 July 2020.
Edited by:
Chiara Giacomelli, University of Pisa, ItalyReviewed by:
Stefano Capaldi, University of Verona, ItalyAndrew Benjamin Herr, Cincinnati Children’s Hospital Medical Center, United States
Copyright © 2020 Murphy, Rajasekaran, Merkel, Skeens, Keeler, Hodsdon, Lisi and Lolis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elias Lolis, ZWxpYXMubG9saXNAeWFsZS5lZHU=
†Present address: Camille Keeler, Department of Chemistry, Wesleyan University, Middletown, CT, United States; Michael E. Hodsdon, Eli Lilly and Company, Lilly Corporate Center, Indianapolis, IN, United States
‡ORCID: George P. Lisi, orcid.org/0000-0001-8878-5655; Elias Lolis, orcid.org/0000-0002-7902-7868