
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Mol. Biosci. , 19 May 2020
Sec. Molecular Diagnostics and Therapeutics
Volume 7 - 2020 | https://doi.org/10.3389/fmolb.2020.00081
This article is part of the Research Topic Reversible and Irreversible Covalent Inhibitors View all 4 articles
 Deepak Gurbani1†
Deepak Gurbani1† Guangyan Du2,3†
Guangyan Du2,3† Nathaniel J. Henning2,3†Suman Rao2,3,4Asim K. Bera1Tinghu Zhang2,3*Nathanael S. Gray2,3*
Nathaniel J. Henning2,3†Suman Rao2,3,4Asim K. Bera1Tinghu Zhang2,3*Nathanael S. Gray2,3* Kenneth D. Westover1*‡
Kenneth D. Westover1*‡Unregulated Src activity promotes malignant processes in cancer, but no Src-directed targeted therapies are used clinically, possibly because early Src inhibitors produce off-target effects leading to toxicity. Improved selective Src inhibitors may enable Src-directed therapies. Previously, we reported an irreversible Src inhibitor, DGY-06-116, based on the hybridization of dasatinib and a promiscuous covalent kinase probe SM1-71. Here, we report biochemical and biophysical characterization of this compound. An x-ray co-crystal structure of DGY-06-116: Src shows a covalent interaction with the kinase p-loop and occupancy of the back hydrophobic kinase pocket, explaining its high potency, and selectivity. However, a reversible analog also shows similar potency. Kinetic analysis shows a slow inactivation rate compared to other clinically approved covalent kinase inhibitors, consistent with a need for p-loop movement prior to covalent bond formation. Overall, these results suggest that a strong reversible interaction is required to allow sufficient time for the covalent reaction to occur. Further optimization of the covalent linker may improve the kinetics of covalent bond formation.
SRC was among the first oncogenes to be discovered (Stehelin et al., 1976) and encodes a non-receptor protein tyrosine kinase that regulates many cancer-related cellular processes including mitogenesis, angiogenesis, adhesion, invasion, migration, and survival (Sen and Johnson, 2011). Src activity drives malignant phenotypes in hematologic and solid cancers including breast, prostate, lung, colorectal, and pancreatic cancer (Araujo and Logothetis, 2010; de Felice et al., 2016; Appel et al., 2017). Genetic ablation of Src in animal models reverses cancer phenotypes without systemic toxicity (Trevino et al., 2006; Ammer et al., 2009; Marcotte et al., 2012), suggesting that Src inhibition may be effective in treating certain cancers (Araujo and Logothetis, 2009; Zhang et al., 2009; Chen et al., 2014; Anderson et al., 2017; Appel et al., 2017). Src has also been implicated in cancer drug resistance (Carretero et al., 2010; Sen et al., 2011). Nevertheless, selective Src inhibition has not been demonstrated as a driver of efficacy for any of the clinically used multi-targeted Src drugs.
Dasatinib and bosutinib inhibit multiple kinases including Src, but are approved as anti-Bcr-Abl therapies to treat chronic myelogenous leukemia and acute lymphoblastic leukemia (Shah et al., 2010; Keskin et al., 2016; Cortes et al., 2019). Src-directed trials using dasatinib failed in part due to dose-limiting toxicity (Araujo and Logothetis, 2010; Algazi et al., 2012; Araujo et al., 2012; Secord et al., 2012; Sharma et al., 2012; Schott et al., 2016) including grade 3 to 4 diarrhea, thrombocytopenia, neutropenia, and anemia (Buglio et al., 2012; Daud et al., 2012). These toxicities may be due to the multi-targeted nature of these compounds that also inhibit members of the Src family of kinases (SFKs), Bcr-Abl, c-Kit, PDGFR, c-Fms, and EphA2. Improved Src inhibitors with better selectivity may enable Src-directed cancer therapies.
Engineering selectivity into Src inhibitors is challenging because of the high degree of sequence homology between Src family members and other receptor tyrosine kinases (Duan et al., 2014; Elias and Ditzel, 2015). One strategy for achieving selectivity in kinases is to utilize covalent chemistry, targeting non-conserved cysteines near the inhibitor binding site. Prior work showed that Src, in particular, is amenable to this approach by targeting non-conserved cysteines in the p-loop (Kwarcinski et al., 2012). In that work, promiscuous scaffolds, including the dasatinib scaffold, were derivatized to include reactive warheads that could react with p-loop cysteines, resulting in enhanced selectivity for kinases that include a p-loop cysteine. The work generated hypotheses regarding the importance of p-loop dynamics for this class of inhibitor, but structural data were not reported. Recently, we found an opportunity to build upon this strategy when we found that SM1-71, a 2, 4-disubstituted pyrimidine that includes a cysteine-reactive warhead, can covalently modify 23 different kinases including Src. Our Src-SM1-71 crystal structure (PDB: 6ATE) revealed the Src p-loop in a kinked conformation (Rao et al., 2019). We subsequently showed that SM-1-71 could be optimized for Src inhibition by hybridization with dasatinib (Figures 1A–D, Figure S1; Du et al., 2020). Here, we present formal biochemical, biophysical, and structural characterization of Src inhibition by DGY-06-116.
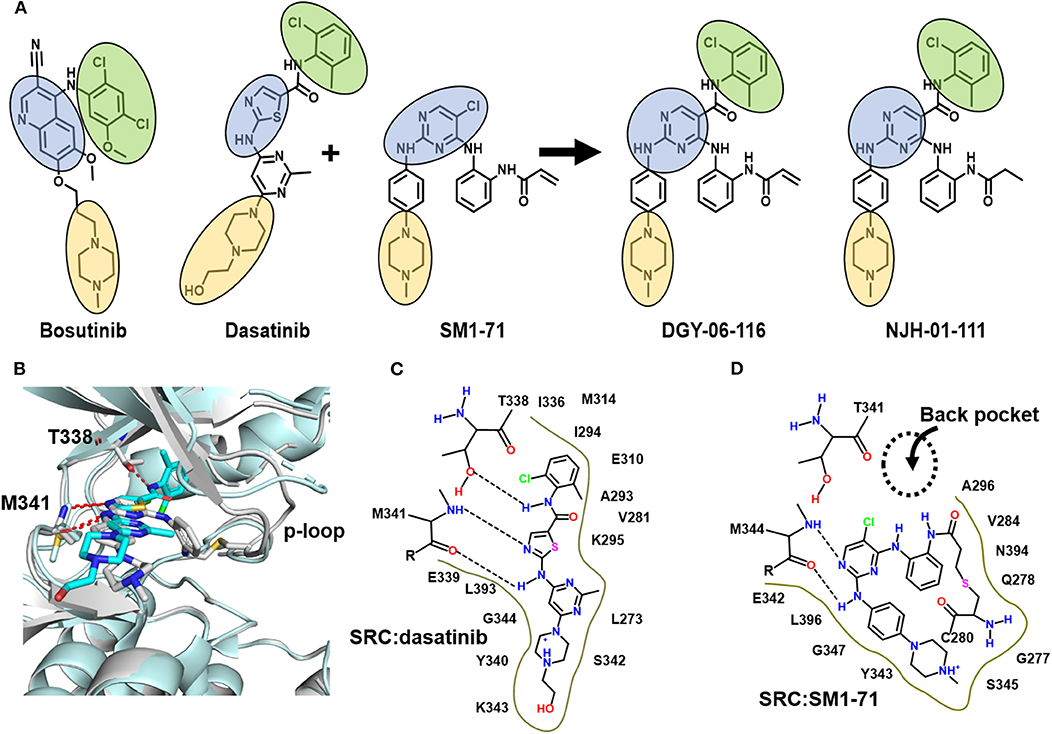
Figure 1. DGY-06-116 is a hybrid of dasatinib and SM171. (A) Src inhibitors are composed of a kinase hinge-binding component (blue), back pocket-binding component (green), and solvent-exposed component (yellow). DGY-06-116 (second from right) resulted from hybridizing the SM1-71 core (third from left) with the back-pocket component of dasatinib (second from left). NJH-01-111 (first from right) is a non-covalent analog. (B) Superposition of Src-dasatinib (PDB: 3G5D; cyan) and Src-SM1-71 (PDB: 6ATE; gray) structures suggested that the substitution would be tolerated. Schematic representation of interactions between Src and (C) dasatinib or (D) SM1-71. Dotted lines are hydrogen bonds.
DGY-06-116 was previously characterized for the ability to bind Src (Du et al., 2020). Here, we evaluated the relative potency of inhibitors on Src enzymatic activity using a mobility shift assay (MSA), which measures phosphorylation of a peptide substrate of Src. Given that covalent inhibitors can show time-dependent effects on IC50 values, we use a 1-h time point for all samples so relative potencies are comparable. At 1-h incubation, DGY-06-116 showed an IC50, 1 h of 2.6 nM. This was substantially better than SM1-71 (IC50, 1 h of 26.6 nM), bosutinib (IC50 of 9.5 nM), and its non-covalent analog NJH-01-111 (IC50 of 5.3 nM) (Figure 2). To estimate the contribution of covalent binding to the overall potency of DGY-06-116, we also tested SrcC280S, which cannot form a covalent bond because of the cysteine-to-serine mutation. SrcC280S showed excellent kinase activity, although specific activity was less (~50%) than that of wild-type protein (Figure S2B). DGY-06-116 showed comparable IC50, 1 h values for mutant and wild-type Src (Figure 2), suggesting that reversible binding substantially contributes to the potency of this compound. To confirm this, we also tested a non-covalent analog of DGY-06-116, NJH-01-111, in which the acrylamide warhead is replaced with propionamide (Figure 1A). The IC50 of NJH-01-111 could not be distinguished from DGY-06-116, confirming that the core scaffold supports the potency of this compound class (Figure 2).
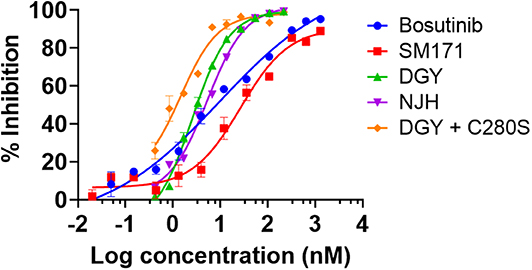
Figure 2. DGY-06-116 inhibits Src enzymatic activity. DGY-06-116 potency at 1 h exceeds SM1-71 and bosutinib. However, a non-covalent analog NJH-01-111 also shows potent activity. DGY-06-116 also potently inhibits SrcC280S. Error bars are the standard deviation of three replicates.
To further understand the nature of the interactions between DGY-06-116 and Src, we determined a co-crystal structure of DGY-06-116 with Src (PDB code: 6E6E) at 2.15 Å resolution. Data collection and refinement statistics are shown in Table 1. This structure includes eight molecules in the asymmetric unit. The main difference between individual protomers was variation in an N-terminal lobe loop conformation, but there were no differences in the conformations of the ATP binding sites (Figure S3). DGY-06-116 was easily modeled into the predicted binding site for all, with the warhead forming continuous electron density with Cys-280 (Figure 3A). As seen with SM171, the p-loop of the Src kinase is bent and thus allows a covalent bond to form, a phenomenon that is not observed with other Src structures (Figure 3B). As expected, the nitrogen of DGY-06-116′s carboxamide linker hydrogen bonds to gatekeeper Thr-338. The nitrogen is also part of a coordinated hydrogen bonding network that includes an active-site water (HOH432), catalytic Lys-298, the backbone of Cys-280, and the carbonyl DGY-06-116 (Figure 3C). The chloro-methyl phenyl substituent is situated in the back pocket of the Src kinase, creating hydrophobic interactions with Ile-297 and Leu-396. The anilinopyrimidine forms two hydrogen bonds with the backbone of Met-341 in the hinge region and the methyl piperazinyl tail extends to the solvent channel forming hydrophobic contacts with Val-284 and Gly-347 (Figure 3C). These results confirm that DGY-06-116 forms a covalent bond with Cys-280, but also suggest that hydrophobic interactions with the back pocket significantly contribute to affinity, possibly explaining the high potency of the non-covalent analog NJH-01-111.
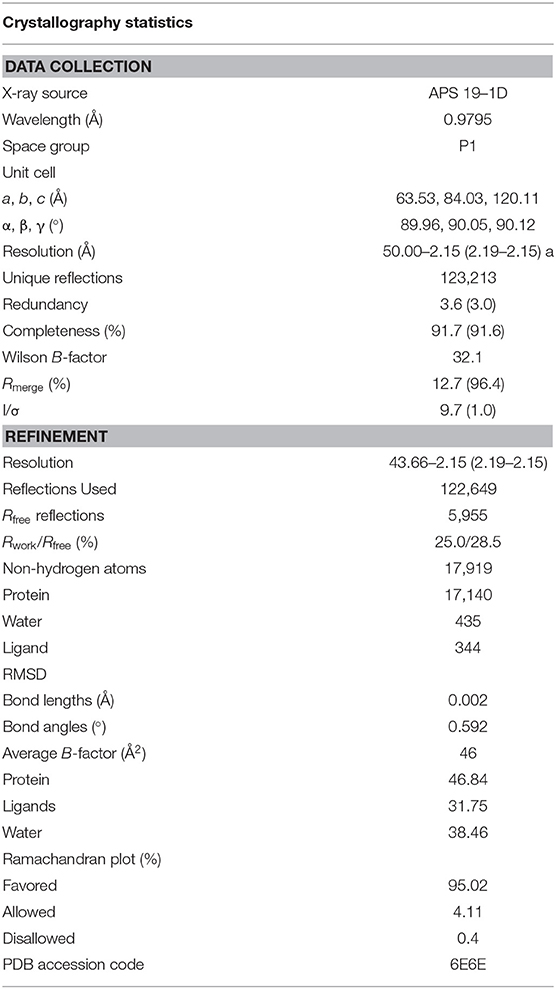
Table 1. Data collection and refinement statistics.
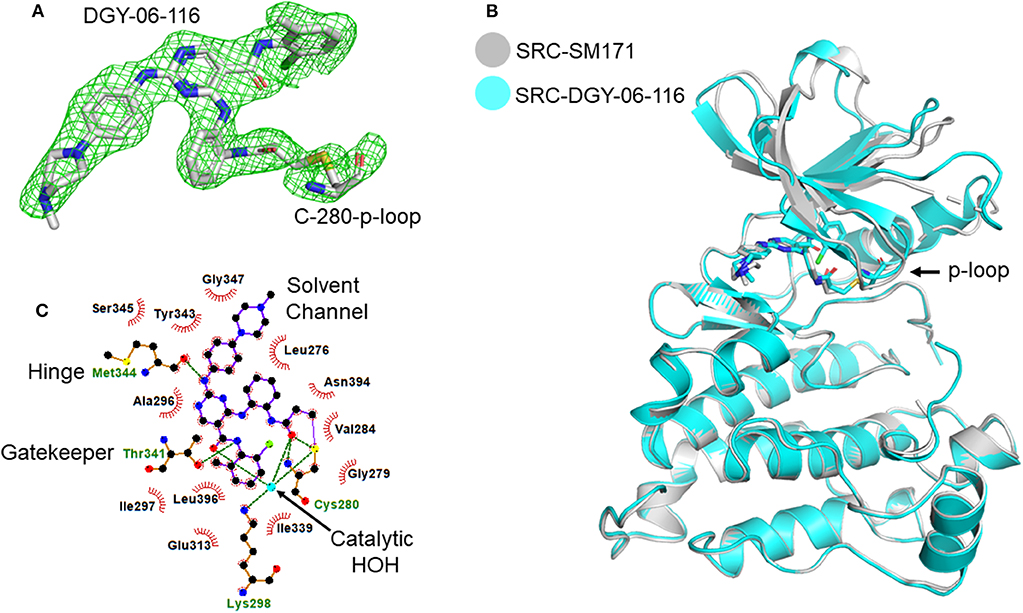
Figure 3. Structural characterization of the Src-DGY-06-116 complex. (A) A Fo-Fc map at 2.5σ shows continuous electron density observed for DGY-06-116 forming a covalent linkage to Cys-280 of the p-loop. (B) DGY-06-116 binding to Src kinase leads to bending of p-loop Cys-280 consistent with design. (C) Two-dimensional representations of interactions between DGY and Src kinase domain residues. Hydrogen bonds are shown as green dashes. Hydrophobic interactions are shown as red spikes.
While the crystal structure clearly indicates covalent binding to Src, the enzymatic assay could not distinguish between DGY-06-116 and NJH-01-111. Nevertheless, clear differences are seen in the selectivity of these compounds (Du et al., 2020). Previously, we used the MSA to measure the inactivation rate of other covalent inhibitors (Tan et al., 2017). However, in this case, we could not because the IC50, 1 h of for DGY-06-116 was near the enzyme concentration used in the assay and therefore close to the theoretical sensitivity limit. To evaluate the inactivation rate, we used a surface plasmon resonance assay to estimate kinact/KI (Copeland, 2013; Miyahisa et al., 2015). In our setup, biotinylated Src kinase was immobilized on the biosensor. Binding kinetics for DGY-06-116 and NJH-01-111 were found to be similar, indicating a similar initial binding event (Figures 4A,B). Although the decay appeared similar on visual inspection, we were unable to fit the curve for DGY using a one-state model. We considered that the p-loop must move into position to allow a covalent bond to form. We therefore used a two-state model to calculate kinact/KI. For the non-covalent inhibitor NJH-01-111, the value was only 1.7 M−1 s−1, indicating that a covalent bond did not form. In contrast, kinact/KI for DGY-06-116 was 174 M−1 s−1, consistent with covalent bond formation. However, when comparing inactivation rate constant (kinact), DGY-06-116 showed a rate of 5.7 × 10−7 s−1, several orders of magnitude slower than for other validated covalent compounds such as neratinib (2 × 10−3 s−1) and afatinib (1 × 10−3 s−1) (Gierse et al., 1999; Papp-Wallace et al., 2014; Schwartz et al., 2014), showing modest irreversible inhibition. Given that the p-loop must shift to form a covalent bond with DGY-06-116, we speculate that this slow kinact occurs because it depends on protein dynamics at the p-loop. This is in agreement with prior ideas about the importance of p-loop movement for p-loop targeted inhibitors (Kwarcinski et al., 2012).
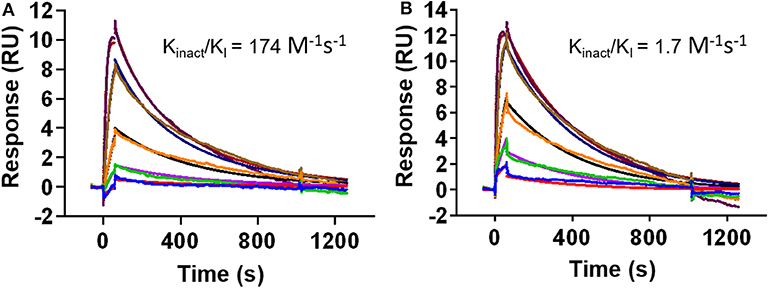
Figure 4. DGY-06-116 shows a slow inactivation rate. (A) Covalent DGY-06-116 and (B) non-covalent analog NJH-01-111. Data from each interaction were analyzed using both a 1:1 kinetic model and a two-state covalent interaction (p-loop movement) model for calculation of kinact/KI.
In these studies, we established that DGY-06-116 binds covalently to Src in a manner similar to SM1-71, where the p-loop must kink to establish the covalent bond. However, we also showed that a non-covalent analog, NJH-01-111, binds with a similar high affinity. We also showed that the covalent reaction is slow. Altogether, our interpretation of these findings is that a strong reversible interaction is required to allow sufficient time for the p-loop to sample a kinked conformation compatible with covalent bond formation.
We speculate that that optimization of the interaction between the p-loop and compound may increase the inactivation rate, leading to further improvements in compound selectivity in biological systems, since covalent bond formation appears to be the major driver of selectivity (Du et al., 2020). One way to do this would be to increase the length of the linker to the covalent warhead so the p-loop does not have to kink. Computationally, this appears to be a viable strategy since simulated docking shows that extended linkers retain existing interactions and may even add additional hydrogen bonding with the main-chain oxygen of Gln-275 (Figure S4).
Despite the slow inactivation rate of DGY-06-116, this compound is a selective Src inhibitor that will enable laboratory studies of Src-driven biology, with applications in cancer. One potential application may relate to the subject of acquired resistance, which is a major challenge with kinase inhibitors (Lovly and Shaw, 2014). Src has been implicated in mechanisms that underlie the development of hormone therapy resistance in breast cancer (McDonnell and Norris, 2002; Hiscox et al., 2006) and resistance to chemotherapy in triple-negative breast cancer (Wu et al., 2016). Src is also involved in drug resistance to Her2-directed therapy and for certain head and neck and lung cancers (Carretero et al., 2010; Sen et al., 2011). Src has also been implicated in non-small cell lung cancers harboring mutations in EGFR where Src is activated via Cripto-1 (Park et al., 2014). Indeed, this concept is being tested in a clinical trial (NCT02954523) where dasatinib and osimertinib are delivered together. Another possible application is in KRAS-mutated lung cancer, where loss of the Lkb1 tumor suppressor activates Src signaling. In mouse cancer models that mimic this cancer state, the combined inhibition of Src, PI3K, and Mek showed synergistic tumor regression (Carretero et al., 2010). Finally, a lack of predictive biomarkers has limited prior Src-directed trials (Puls et al., 2011). Our tool compound may allow us to identify cancer populations that are sensitive to Src inhibition through chemistry-first biomarker discovery approaches (McMillan et al., 2018).
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank archive (PDB) for human proto-oncogene tyrosine-protein kinase Src in complex with DGY-06-116 (PDB ID: 6E6E). SRC_HUMAN P12931.
This article contains previously unpublished data. The name of the repository and accession number(s) are not available.
NG, TZ, KW, and DG: Conceptualization. GD and NH: Chemistry. DG: Structure determination and biochemistry. TZ, DG, KW, and NG: Writing. The manuscript was edited through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by Welch I-1829, ACS RSG-18-039-01-DMC, and a Career Enhancement Grant through NIH P50CA07090720 to KW and by Linde Center for medicinal chemistry to NG.
KW is on the SAB for Vibliome Therapeutics. NG is a founder, SAB, and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives/has received funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield, and Sanofi. GD, NH, SR, DG, TZ, KW, and NG are inventors on a Src covalent inhibitor patent. Information regarding Src is accessible via UniProtKB P12931.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank Eric Roush for technical assistance with the Biacore S200 instrument and data analysis. We also thank Dr. Damiana Chiavolini for editing the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00081/full#supplementary-material
Figure S1. Src kinase domain architecture and location of targetable cysteine the p-loop.
Figure S2. SrcC280S efficiently phosphorylates the peptide substrate. Percent conversion from non-phosphorylated to phosphorylated substrate is shown over time for a range of Src concentrations. Each data point represents a triplicate measurement. (A) WT Src. (B) SrcC280S.
Figure S3. Differences between protomers in the asymmetric crystallographic unit. All 8 protomers were superimposed and each colored differently.
Figure S4. Covalent docking model predicts hydrogen bonding interactions for linker optimization.
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66(Pt 2), 213–221. doi: 10.1107/S0907444909052925
Algazi, A. P., Weber, J. S., Andrews, S. C., Urbas, P., Munster, P. N., DeConti, R. C., et al. (2012). Phase I clinical trial of the Src inhibitor dasatinib with dacarbazine in metastatic melanoma. Br. J. Cancer 106, 85–91. doi: 10.1038/bjc.2011.514
Ammer, A. G., Kelley, L. C., Hayes, K. E., Evans, J. V., Lopez-Skinner, L. A., Martin, K. H., et al. (2009). Saracatinib impairs head and neck squamous cell carcinoma invasion by disrupting invadopodia function. J. Cancer Sci. Ther. 1, 52–61. doi: 10.4172/1948-5956.1000009
Anderson, G. R., Winter, P. S., Lin, K. H., Nussbaum, D. P., Cakir, M., Stein, E. M., et al. (2017). A landscape of therapeutic cooperativity in KRAS mutant cancers reveals principles for controlling tumor evolution. Cell Rep. 20, 999–1015. doi: 10.1016/j.celrep.2017.07.006
Appel, C. K., Gallego-Pedersen, S., Andersen, L., Blancheflor Kristensen, S., Ding, M., Falk, S., et al. (2017). The Src family kinase inhibitor dasatinib delays pain-related behaviour and conserves bone in a rat model of cancer-induced bone pain. Sci. Rep. 7:4792. doi: 10.1038/s41598-017-05029-1
Araujo, J., and Logothetis, C. (2009). Targeting Src signaling in metastatic bone disease. Int. J. Cancer. 124, 1–6. doi: 10.1002/ijc.23998
Araujo, J., and Logothetis, C. (2010). Dasatinib: a potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 36, 492–500. doi: 10.1016/j.ctrv.2010.02.015
Araujo, J. C., Mathew, P., Armstrong, A. J., Braud, E. L., Posadas, E., Lonberg, M., et al. (2012). Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1-2 study. Cancer. 118, 63–71. doi: 10.1002/cncr.26204
Buglio, D., Palakurthi, S., Byth, K., Vega, F., Toader, D., Saeh, J., et al. (2012). Essential role of TAK1 in regulating mantle cell lymphoma survival. Blood 120, 347–355. doi: 10.1182/blood-2011-07-369397
Carretero, J., Shimamura, T., Rikova, K., Jackson, A. L., Wilkerson, M. D., Borgman, C. L., et al. (2010). Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 17, 547–559. doi: 10.1016/j.ccr.2010.04.026
Chen, J., Elfiky, A., Han, M., Chen, C., and Saif, M. W. (2014). The role of Src in colon cancer and its therapeutic implications. Clin. Colorectal Cancer. 13, 5–13. doi: 10.1016/j.clcc.2013.10.003
Copeland, R. A. (2013). Evaluation of Enzyme Inhibitors In Drug Discovery: A Guide For Medicinal Chemists And Pharmacologists. Hoboken, NJ: John Wiley & Sons.
Cortes, J. E., Gambacorti-Passerini, C., Deininger, M. W., Mauro, M. J., Chuah, C., Kim, D. W., et al. (2019). Patient-reported outcomes in the phase 3 BFORE trial of bosutinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia. J. Cancer Res. Clin. Oncol. 145, 1589–1599. doi: 10.1007/s00432-019-02894-3
Daud, A. I., Krishnamurthi, S. S., Saleh, M. N., Gitlitz, B. J., Borad, M. J., Gold, P. J., et al. (2012). Phase I study of bosutinib, a src/abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin. Cancer Res. 18, 1092–1100. doi: 10.1158/1078-0432.CCR-11-2378
de Felice, M., Lambert, D., Holen, I., Escott, K. J., and Andrew, D. (2016). Effects of Src-kinase inhibition in cancer-induced bone pain. Mol. Pain. 12, 1–14. doi: 10.1177/1744806916643725
Du, G., Rao, S., Gurbani, D., Henning, N. J., Jiang, J., Che, J., et al. (2020). Structure-based design of a potent and selective covalent inhibitor for SRC kinase that targets a P-loop cysteine. J Med Chem. 63, 1624–1641. doi: 10.1021/acs.jmedchem.9b01502
Duan, Y. Q., Ma, Y., Wang, X. J., Jin, Y. Y., Wang, R. L., Dong, W. L., et al. (2014). Design potential selective inhibitors for treating cancer by targeting the SRC homology 2 (SH2) domain-containing phosphatase 2 (Shp2) with core hopping approach. Protein Pept. Lett. 21, 556–563. doi: 10.2174/0929866521666131223143913
Elias, D., and Ditzel, H. J. (2015). The potential of Src inhibitors. Aging (Albany. NY). 7, 734–735. doi: 10.18632/aging.100821
Elkins, J. M., Fedele, V., Szklarz, M., Abdul Azeez, K. R., Salah, E., Mikolajczyk, J., et al. (2016). Comprehensive characterization of the published kinase inhibitor set. Nat. Biotechnol. 34, 95–103. doi: 10.1038/nbt.3374
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60(Pt 12 Pt 1), 2126–2132. doi: 10.1107/S0907444904019158
Gierse, J. K., Koboldt, C. M., Walker, M. C., Seibert, K., and Isakson, P. C. (1999). Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem. J. 339 (Pt 3), 607–614.
Hiscox, S., Morgan, L., Green, T., and Nicholson, R. I. (2006). Src as a therapeutic target in anti-hormone/anti-growth factor-resistant breast cancer. Endocr. Relat. Cancer 13(Suppl. 1), S53–S59. doi: 10.1677/erc.1.01297
Keskin, D., Sadri, S., and Eskazan, A. E. (2016). Dasatinib for the treatment of chronic myeloid leukemia: patient selection and special considerations. Drug Des. Devel. Ther. 10, 3355–3361. doi: 10.2147/DDDT.S85050
Kwarcinski, F. E., Fox, C. C., Steffey, M. E., and Soellner, M. B. (2012). Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem. Biol. 7, 1910–1917. doi: 10.1021/cb300337u
Lovly, C. M., and Shaw, A. T. (2014). Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 20, 2249–2256. doi: 10.1158/1078-0432.CCR-13-1610
Marcotte, R., Smith, H. W., Sanguin-Gendreau, V., McDonough, R. V., and Muller, W. J. (2012). Mammary epithelial-specific disruption of c-Src impairs cell cycle progression and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 2808–2813. doi: 10.1073/pnas.1018861108
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40(Pt 4), 658–674. doi: 10.1107/S0021889807021206
McDonnell, D. P., and Norris, J. D. (2002). Connections and regulation of the human estrogen receptor. Science. 296, 1642–1644. doi: 10.1126/science.1071884
McMillan, E. A., Ryu, M. J., Diep, C. H., Mendiratta, S., Clemenceau, J. R., Vaden, R. M., et al. (2018). Chemistry-first approach for nomination of personalized treatment in lung cancer. Cell 173, 864–878. doi: 10.1016/j.cell.2018.03.028
Minor, W., Cymborowski, M., Otwinowski, Z., and Chruszcz, M. (2006). HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62(Pt 8), 859–866. doi: 10.1107/S0907444906019949
Miyahisa, I., Sameshima, T., and Hixon, M. S. (2015). Rapid determination of the specificity constant of irreversible inhibitors (kinact/KI) by means of an endpoint competition assay. Angew. Chem. Int. Ed. Engl. 54, 14099–14102. doi: 10.1002/anie.201505800
Papp-Wallace, K. M., Mallo, S., Bethel, C. R., Taracila, M. A., Hujer, A. M., Fernandez, A., et al. (2014). A kinetic analysis of the inhibition of FOX-4 beta-lactamase, a plasmid-mediated AmpC cephalosporinase, by monocyclic beta-lactams and carbapenems. J. Antimicrob. Chemother. 69, 682–690. doi: 10.1093/jac/dkt434
Park, K. S., Raffeld, M., Moon, Y. W., Xi, L., Bianco, C., Pham, T., et al. (2014). CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J. Clin. Invest. 124, 3003–3015. doi: 10.1172/JCI73048
Puls, L. N., Eadens, M., and Messersmith, W. (2011). Current status of SRC inhibitors in solid tumor malignancies. Oncologist 16, 566–578. doi: 10.1634/theoncologist.2010-0408
Rao, S., Gurbani, D., Du, G. Y., Everley, R. A., Browne, C. M., Chaikuad, A., et al. (2019). Leveraging compound promiscuity to identify targetable cysteines within the kinome. Cell Chem. Biol. 26, 818–829. doi: 10.1016/j.chembiol.2019.02.021
Schott, A. F., Barlow, W. E., Van Poznak, C. H., Hayes, D. F., Moinpour, C. M., Lew, D. L., et al. (2016). Phase II studies of two different schedules of dasatinib in bone metastasis predominant metastatic breast cancer: SWOG S0622. Breast Cancer Res. Treat. 159, 87–95. doi: 10.1007/s10549-016-3911-z
Schwartz, P. A., Kuzmic, P., Solowiej, J., Bergqvist, S., Bolanos, B., Almaden, C., et al. (2014). Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. U.S.A. 111, 173–178. doi: 10.1073/pnas.1313733111
Secord, A. A., Teoh, D. K., Barry, W. T., Yu, M., Broadwater, G., Havrilesky, L. J., et al. (2012). A phase I trial of dasatinib, an SRC-family kinase inhibitor, in combination with paclitaxel and carboplatin in patients with advanced or recurrent ovarian cancer. Clin. Cancer Res. 18, 5489–5498. doi: 10.1158/1078-0432.CCR-12-0507
Sen, B., and Johnson, F. M. (2011). Regulation of SRC family kinases in human cancers. J. Signal Transduct. 2011:865819. doi: 10.1155/2011/865819
Sen, B., Peng, S., Saigal, B., Williams, M. D., and Johnson, F. M. (2011). Distinct interactions between c-Src and c-Met in mediating resistance to c-Src inhibition in head and neck cancer. Clin. Cancer Res. 17, 514–524. doi: 10.1158/1078-0432.CCR-10-1617
Shah, N. P., Kim, D. W., Kantarjian, H., Rousselot, P., Llacer, P. E., Enrico, A., et al. (2010). Potent, transient inhibition of BCR-ABL with dasatinib 100 mg daily achieves rapid and durable cytogenetic responses and high transformation-free survival rates in chronic phase chronic myeloid leukemia patients with resistance, suboptimal response or intolerance to imatinib. Haematologica. 95, 232–240. doi: 10.3324/haematol.2009.011452
Sharma, M. R., Wroblewski, K., Polite, B. N., Knost, J. A., Wallace, J. A., Modi, S., et al. (2012). Dasatinib in previously treated metastatic colorectal cancer: a phase II trial of the University of Chicago phase II consortium. Invest. New Drugs. 30, 1211–1215. doi: 10.1007/s10637-011-9681-x
Stehelin, D., Varmus, H. E., Bishop, J. M., and Vogt, P. K. (1976). DNA related to the transforming gene (s) of avian sarcoma viruses is present in normal avian DNA. Nature. 260, 170–173.
Tan, L., Gurbani, D., Weisberg, E. L., Hunter, J. C., Li, L., Jones, D. S., et al. (2017). Structure-guided development of covalent TAK1 inhibitors. Bioorg. Med. Chem. 25, 838–846. doi: 10.1016/j.bmc.2016.11.035
Trevino, J. G., Summy, J. M., Lesslie, D. P., Parikh, N. U., Hong, D. S., Lee, F. Y., et al. (2006). Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am. J. Pathol. 168, 962–972. doi: 10.2353/ajpath.2006.050570
Wu, Z.-H., Lin, C., Liu, M.-M., Zhang, J., Tao, Z.-H., and Hu, X.-C. (2016). Src inhibition can synergize with gemcitabine and reverse resistance in triple negative breast cancer cells via the AKT/c-jun pathway. PLoS ONE 11:e0169230. doi: 10.1371/journal.pone.0169230
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. (2011) 67, 235–242. doi: 10.1107/S0907444910045749
Zhang, X. H., Wang, Q., Gerald, W., Hudis, C. A., Norton, L., Smid, M., et al. (2009). Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 16, 67–78. doi: 10.1016/j.ccr.2009.05.017
The Src (UniProtKB P12931) kinase domain was purified as reported previously (Rao et al., 2019). The Cys280-Ser mutant Src version was generated via site-directed mutagenesis and purified similarly.
An automated microcapillary electrophoresis platform (PerkinElmer LabChip® EZ Reader) capable of separating and detecting fluorescently labeled peptides based on charge (Elkins et al., 2016) was used for measuring Src kinase activity. Briefly, purified Src enzyme, fluorescently labeled peptide-4 substrate (purchased from Perkin Elmer), ATP, and inhibitors (SM1-71, Bosutinib, DGY-06-116, and NJH-01-111) were combined into a single well, incubated for 1 h incubation following phosphorylation of the substrate. Substrate (non-phosphorylated peptide) to product (phosphorylated peptide as a result of Src kinase activity) conversion was measured over time. The kinase reaction buffer consisted of 100 mM Hepes, pH 7.3, 0.015% Brij-35, 0.004% Tween-20, and 10 mM MgCl2. Peptides were separated using running buffer made of 100 mM Hepes, pH 7.3, 0.015% Brij-35, 1 mM disodium EDTA, 0.1% coating reagent 3, 5% DMSO, and 1× coating reagent 8. Final reaction conditions included 1.25 nM of purified Src kinase, 100 μM ATP, and 1 μM peptide-4 substrate. The percent conversion was determined by EZReader 3.0 software and analyzed using GraphPad software version 7 (La Jolla, California, USA) for determination of IC50 values.
Purified Src kinase was concentrated to 10 mg/ml and incubated with three fold excess of inhibitor DGY-06-116 at room temperature for 1 h. Co-crystals of Src-DGY-06-116 complex were obtained by vapor diffusion from a hanging drop setup at 20°C using 0.1 M magnesium formate dihydrate and 15% PEG3350 as precipitant. Crystals appeared overnight and were harvested after 2 days in mother liquor with 30% glycerol before freezing in liquid nitrogen. Diffraction data were collected using beamline 19-ID of the Advanced Photon Source, Argonne National Laboratory and scaled using HKL3000 (Minor et al., 2006). Molecular replacement solution was obtained using Phaser (McCoy et al., 2007) and 4MXO as initial search model. Model building and refinement were carried out using Coot (Emsley and Cowtan, 2004) and Phenix (Adams et al., 2010).
A Biacore S200 was used to evaluate binding of covalent inhibitor DGY-06-116 and its non-covalent analog NJH-01-111 with Src kinase. Src was biotinylated using EZ-Link™ Sulfo-NHS-Biotinylation Kit (ThermoFisher). Biotinylated Src kinase was captured using the Biotin Capture Kit and Sensor Chip CAP (GE Healthcare). The Biotin Capture Kit works with a single-stranded DNA sequence on Sensor Chip CAP. A capture reagent consisting of a biotinylated complementary DNA strand and streptavidin is captured by the single-stranded DNA, allowing capture of other biotinylated molecules by streptavidin. At the end of the binding cycle, the DNA duplex is chemically disrupted, washing away all materials except the original single-stranded DNA sequence. The sequence is repeated for each binding cycle of the assay. Src was captured at surface densities between 800 and 900 RU for each cycle of the assay. Dilutions of DGY-06-116 and NJH-01-111 from 100 to 1.2 nM were made in running buffer (100 mM HEPES pH 7.3, 0.015% Brij-35, 0.004% Tween-20, 10 mM MgCl2, and 1% DMSO) and tested for binding using a multi-cycle assay. The SPR data were analyzed using Biacore™ Insight Evaluation Software (GE Healthcare Life Sciences, USA). Data from each interaction were analyzed using both a 1:1 kinetic model and a two-state covalent interaction model (involving p-loop movement) for calculation of kinact/KI using Biacore S200 Evaluation software (GE Healthcare Life Sciences, USA).
Keywords: src kinase, cancer, dasatinib, selectivity, irreversible inhibitor
Citation: Gurbani D, Du G, Henning NJ, Rao S, Bera AK, Zhang T, Gray NS and Westover KD (2020) Structure and Characterization of a Covalent Inhibitor of Src Kinase. Front. Mol. Biosci. 7:81. doi: 10.3389/fmolb.2020.00081
Received: 17 February 2020; Accepted: 08 April 2020;
Published: 19 May 2020.
Edited by:
Megha Agrawal, University of Illinois at Chicago, United StatesReviewed by:
Matthew Bryan Soellner, University of Michigan, United StatesCopyright © 2020 Gurbani, Du, Henning, Rao, Bera, Zhang, Gray and Westover. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathanael S. Gray, bmF0aGFuYWVsX2dyYXlAZGZjaS5oYXJ2YXJkLmVkdQ==; Tinghu Zhang, dGluZ2h1X3poYW5nQGRmY2kuaGFydmFyZC5lZHU=; Kenneth D. Westover, a2VubmV0aC53ZXN0b3ZlckB1dHNvdXRod2VzdGVybi5lZHU=
†These authors have contributed equally to this work
‡Lead Contact
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.