 Thomas Wiegand1*†
Thomas Wiegand1*† Denis Lacabanne1†
Denis Lacabanne1† Anahit Torosyan1
Anahit Torosyan1 Julien Boudet2Riccardo Cadalbert1Frédéric H.-T. Allain2
Julien Boudet2Riccardo Cadalbert1Frédéric H.-T. Allain2 Beat H. Meier1
Beat H. Meier1 Anja Böckmann3
Anja Böckmann3- 1Physical Chemistry, ETH Zürich, Zurich, Switzerland
- 2Institute of Molecular Biology and Biophysics, ETH Zürich, Zurich, Switzerland
- 3Molecular Microbiology and Structural Biochemistry, UMR 5086 CNRS/Université de Lyon, Labex Ecofect, Lyon, France
Today, the sedimentation of proteins into a magic-angle spinning (MAS) rotor gives access to fast and reliable sample preparation for solid-state Nuclear Magnetic Resonance (NMR), and this has allowed for the investigation of a variety of non-crystalline protein samples. High protein concentrations on the order of 400 mg/mL can be achieved, meaning that around 50–60% of the NMR rotor content is protein; the rest is a buffer solution, which includes counter ions to compensate for the charge of the protein. We have demonstrated herein the long-term stability of four sedimented proteins and complexes thereof with nucleotides, comprising a bacterial DnaB helicase, an ABC transporter, an archaeal primase, and an RNA polymerase subunit. Solid-state NMR spectra recorded directly after sample filling and up to 5 years later indicated no spectral differences and no loss in signal intensity, allowing us to conclude that protein sediments in the rotor can be stable over many years. We have illustrated, using an example of an ABC transporter, that not only the structure is maintained, but that the protein is still functional after long-term storage in the sedimented state.
Introduction
Using solid-state Nuclear Magnetic Resonance (NMR), a wide variety of biological materials can be studied, such as micro- or nanocrystals, fibrillar aggregates, and proteins embedded in membranes. Sedimentation of dissolved proteins from the solution state directly into the magic-angle spinning (MAS) rotor has lately become the most important sample preparation approach for solid-state NMR (Bertini et al., 2011, 2012, 2013; Gardiennet et al., 2012; Ravera, 2014; van der Wel, 2018; Lacabanne et al., 2019a). This can be achieved in an ultracentrifuge (Böckmann et al., 2009; Bertini et al., 2012; Gardiennet et al., 2012) using specially designed rotor-filling tools (Böckmann et al., 2009; Mandal et al., 2017). With this approach, typical protein concentrations on the order of 400 mg/mL were achieved using the NMR rotor (Wiegand et al., 2016a) in which the protein was still hydrated and typically had around 50 weight percentages of water (Bertini et al., 2013). No long-range order in such sediments was observed by X-ray diffraction (Gardiennet et al., 2012). Sample preparation by sedimentation allowed for the investigation of proteins that are difficult (or even impossible) to crystallize. Theoretically, the molecular mass of the protein determines the success of sedimentation, and proteins, such as the RNA polymerase subunits Rpo4/7∗ (the ∗ indicates that the Rpo7 unit is uniformly 13C/15N labeled) with a molecular weight of 34 kDa (Torosyan et al., 2019), the pRN1 primase with 40 kDa (Boudet et al., 2019), the neonatal Fc receptor with 40 kDa (Stöppler et al., 2018) as well as the human superoxide dismutase with 32 kDa (Fragai et al., 2013), the bacterial helicase DnaB with 708 kDa (Gardiennet et al., 2012), the iron-storage protein ferritin with 480 kDa (Bertini et al., 2012), a variety of supramolecular assemblies (Lecoq et al., 2018; Gauto et al., 2019), and PEGylated proteins (Ravera et al., 2016), were shown to form sediments suitable for solid-state NMR. This is for some of these systems probably a consequence of oligomerization that has been induced by the high protein concentrations (Bertini et al., 2013; Fragai et al., 2013). Protein–protein complexes were studied by co-sedimentation (Gardiennet et al., 2016; Xiang et al., 2018) and protein–ligand complexes by sedimentation with the respective ligand (e.g. nucleic acids) directly into the NMR rotor (Wiegand et al., 2016a; Kaur et al., 2018; Stöppler et al., 2018; Lacabanne et al., 2019b).
Multiple 3D and/or 4D correlation experiments are usually collected to assign protein resonances. A basic set of 3D experiments requires, for larger proteins, around 1 month of solid-state NMR measurement time [e.g. 46 days for the 110 amino acids AL-09 VL immunoglobulin light chain fibrils (Piehl et al., 2017), 41 days for the 153 amino acids N-terminal domain of HpDnaB (Wiegand et al., 2016b), 42 days for the 350 amino acids TET2 (Gauto et al., 2019), and 29 days for the 488 amino acids DnaB (Wiegand et al., 2019)]. It is beneficial if the 3D set is recorded on the same sample. Consequently, the sample has to be stable over months for recording multidimensional solid-state NMR experiments.
The stability of a protein sample can in principle be affected by several factors, such as chemical processes (e.g. oxidation, deamination, and hydrolysis) (Celi and Gabai, 2015; Davies, 2016), temperature (Querol et al., 1996; Vogt et al., 1997; Bischof and He, 2006), pressure (Silva and Weber, 1993; Gross and Jaenicke, 1994; Hillson et al., 1999; de Oliveira and Silva, 2015), and freeze–thaw cycles (Manning et al., 1989, 2010). One of the major sources of protein instabilities induced by chemical processes involves oxidation of the protein, mainly at the side chains, such as thiol oxidation (cysteine and methionine), aromatic hydroxylation (tryptophane and tyrosine), deamination (lysine, arginine or backbone), and the formation of carbonyl groups (Celi and Gabai, 2015; Davies, 2016). The oxidation process is catalyzed by free radicals, radiation (high energy or UV-visible light), and also traces of metal ions (e.g. Fe3+ and Cu2+) (Zhang et al., 2013; Davies, 2016). Solvent-exposed cysteines can form intermolecular disulfide bonds, which can be a source of aggregation (Manning et al., 2010). Macromolecular crowding has also been discussed in the context of protein stability in the sense that a high concentration of a crowding reagent decreases the available space for the protein of interest (excluded volume effect), which favors the native protein state in certain cases (Minton, 1981; Wang et al., 2012). A further parameter that has been identified to influence the NMR spectral quality of proteins is the loss of water during a MAS NMR experiment (e.g. due to an incompletely sealed rotor cap), which results in broadened NMR resonances (Fragai et al., 2013).
Here, we describe our experimental observations regarding the stability of four sedimented samples from different protein systems: (i) the bacterial helicase DnaB from Helicobacter pylori in a complex with ADP:AlF4– and DNA (Wiegand et al., 2019), (ii) the archaeal pRN1 primase from Sulfolobus islandicus in a complex with DNA and ATP (Boudet et al., 2019), (iii) the membrane protein BmrA, an ABC transporter from Bacillus subtilis (Lacabanne et al., 2019b), and (iv) the two RNA polymerase subunits Rpo4/7∗ from Methanocaldococcus jannaschii. We use 2D DARR spectra and 31P cross polarization (CP) MAS experiments to assess the long-term stability of these sedimented protein samples. We show that the sedimented samples investigated are stable over several months to years in an NMR rotor.
Results and Discussion
Solid-state NMR protein samples were sedimented into the NMR rotors in an ultracentrifuge (typically overnight at 200,000 × g) using a home-made rotor-filling tool (Böckmann et al., 2009). Such tools withstand the high centrifugal forces during filling, which tend to press the rotor in the filling tool material (Lacabanne et al., 2019a). Figure 1A shows protein samples of the pRN1 primase, the ABC transporter BmrA, the DnaB helicase, and the RNA polymerase subunit Rpo4/7∗ which were drilled out of solid-state NMR rotors. These samples were stored for several years in the rotors after filling. The samples possess a gel-type appearance and are quite densely packed, as expected.
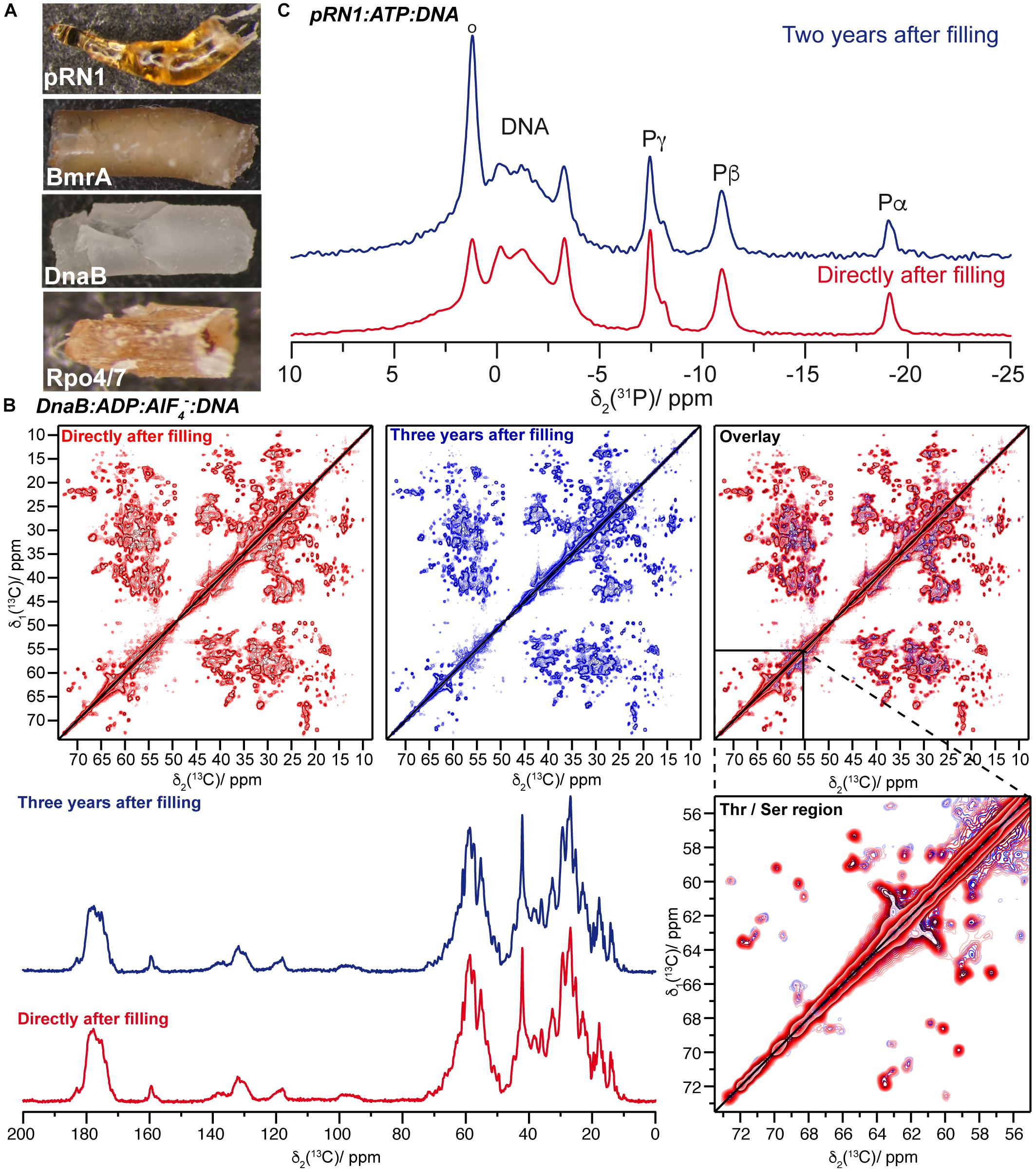
Figure 1. (A) Protein sediments drilled out of solid-state NMR rotors of Rpo4/7∗, DnaB, BmrA, and the pRN1 primase (bottom to top). These sediments belong to different protein samples and not systematically to the ones for which spectra are reported herein. (B) The top panel shows the comparison of 20 ms 13C-13C DARR correlation spectra of DnaB:ADP:AlF4–:DNA recorded directly after rotor filling (red, taken from reference Wiegand et al., 2019) and 3 years later (blue). The bottom panel presents a 13C,1H 1D CP MAS spectra recorded on DnaB:ADP:AlF4–:DNA. (C) 31P,1H CPMAS spectra of the quaternary pRN1:ATP:ATP:DNA complex collected directly after sample filling [red, adapted from reference (Boudet et al., 2019)] and 2 years later (blue).
We have recorded 13C-13C 20 ms Dipolar Assisted Rotational Resonance (DARR) (Takegoshi et al., 1999, 2001) fingerprint spectra on the proteins and complexes thereof. In all cases, one spectrum was recorded directly after filling the NMR rotor and the second one on the same protein sample several years later. In the meantime, the samples were stored at 4°C or −20°C (for pRN1) and were also frequently used for further NMR experiments.
The example of the DnaB:ADP:AlF4–:DNA complex, a hexamer in a complex with six ADP:AlF4– molecules and one DNA molecule, is shown in Figure 1B. The sample reveals nearly similar NMR spectra (linewidths, positions, and intensities – see Supplementary Figure S1 for the difference of the two DARR spectra and Supplementary Figure S2 for a peak-by-peak comparison of spectral features) indicating the long-term structural and conformational stability of the sample. Together with the nearly identical intensities of 1D 13C,1H CP-spectra (Figure 1B), we can conclude that almost no sample degradation took place.
BmrA-E504A is a non-catalytic mutant in which the ATP hydrolysis is abolished. Nevertheless, this variant can still operate conformational changes that are induced by ATP binding (Orelle et al., 2008). After 4 years of storage in the NMR rotor, the sample of the membrane protein BmrA-E504A in absence of ligand showed the same DARR spectrum with the same signal-to-noise ratio compared to the one measured directly after rotor filling [Figure 2. Note that LVIKH were selectively unlabeled to reduce spectral overlap in the spectrum (Lacabanne et al., 2018, 2019b)]. An interesting question is whether an aged sedimented sample can still undergo conformational changes, such as those previously detected by solid-state NMR reflecting the transition between the inward- and outward-facing state of the transporter (Lacabanne et al., 2019b). Therefore, a 4-month-old rotor containing the mutant BmrA-E504A apo (in its inward-facing state, see Figure 3A for the spectrum) was opened, and the sediment was unpacked, resuspended in the presence of ATP-Mg, and sedimented a second time into the solid-state NMR rotor. The new spectrum (Figure 3B, for an overlay with the spectrum obtained directly after rotor filling see Figure 3C) indeed indicated the conformational change to the outward-facing form as described previously by Lacabanne et al. (2019b).
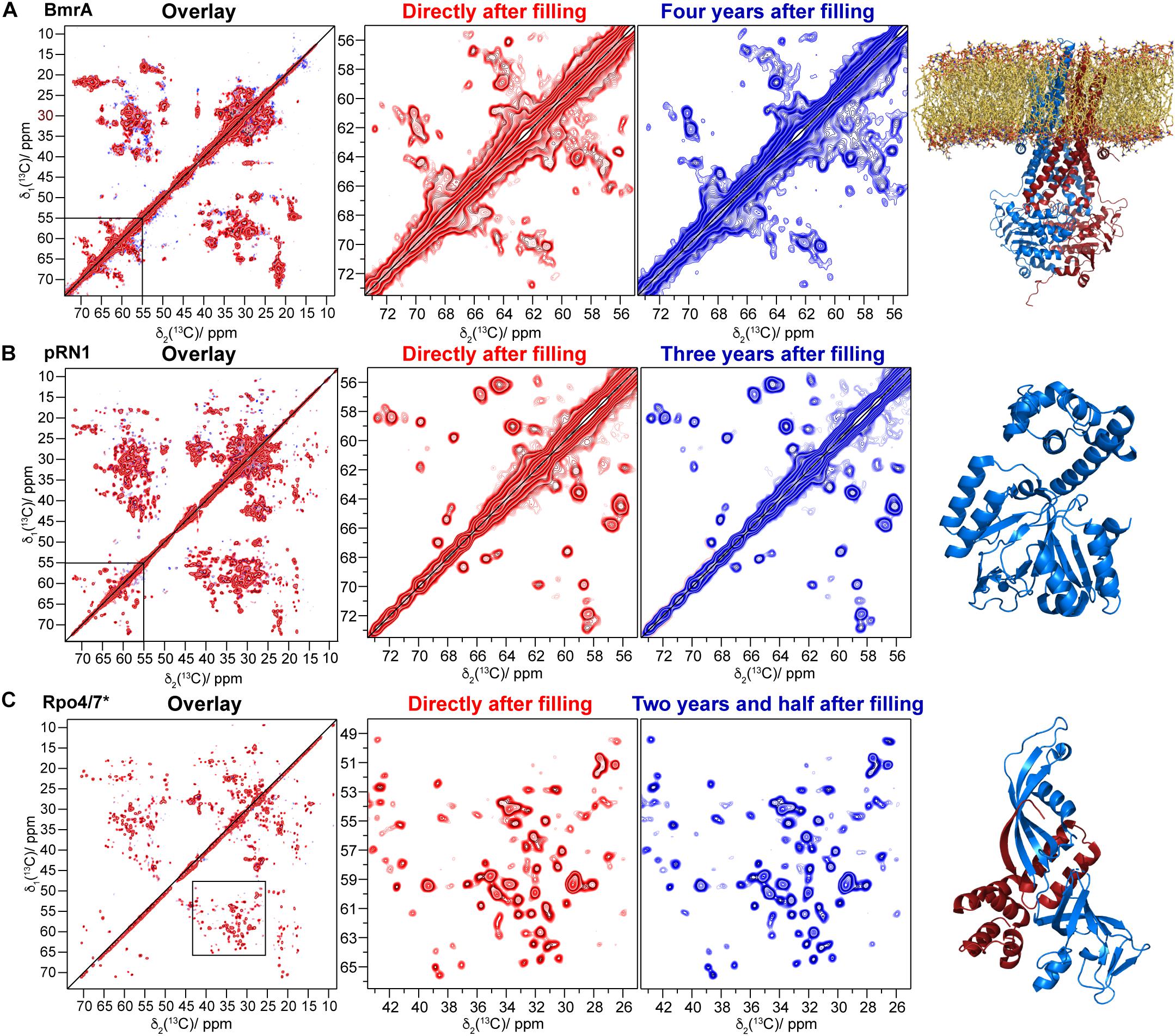
Figure 2. Comparison of 20 ms 13C-13C DARR correlation spectra recorded directly after filling (red) and up to several years later (blue): (A) BmrA E504 [a homology model based on SAV1866 is shown in the last column, PDB ID: 2HYD (Dawson and Locher, 2006), the red spectrum is taken from reference (Lacabanne et al., 2019b)], (B) the pRN1 primase (PDB ID: 6GVT), and (C) 13C/15N labeled Rpo7 (PDB ID: 1GO3, blue ribbons) in complex with unlabeled Rpo4 [red ribbons, where the red spectrum is taken from reference (Torosyan et al., 2019)]. The corresponding protein structures are shown on the right.
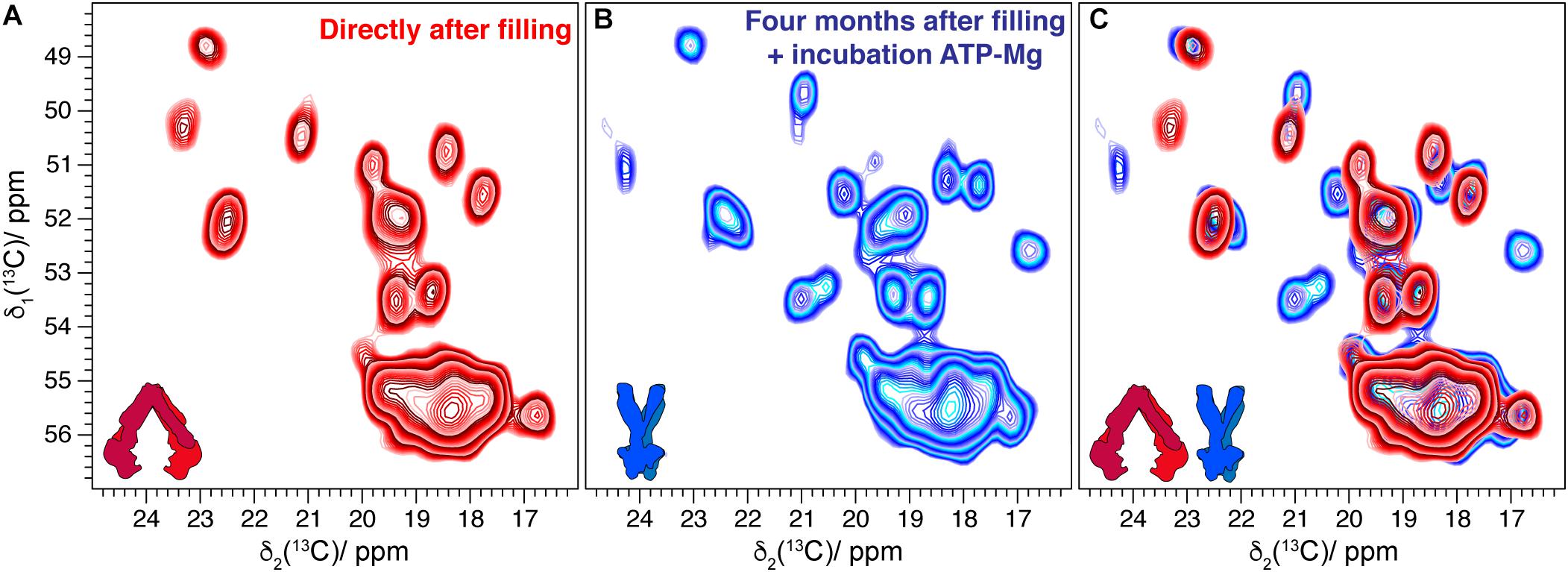
Figure 3. (A) Comparison of alanine region spectral fingerprints of 20 ms 13C-13C DARR correlation spectra of BmrA-E504A in its lipids environment recorded directly after rotor filling [taken from reference (Lacabanne et al., 2019b)]. (B) DARR spectrum collected on the same sample resuspended in buffer after being stored for 4 months, incubated with ATP-Mg, and sedimented again in the rotor. (C) Overlay indicating CSPs and new peaks corresponding to the conformational changes of the transporter [according to reference (Lacabanne et al., 2019b)].
For the pRN1 archaeal primase in complex with ATP and DNA (Boudet et al., 2019), we used 31P,1H cross-polarization spectra to probe whether the nucleotides remain bound to the enzyme after 2 years (Figure 1C). Such cross-polarization-based spectra only show immobilized ATP and DNA molecules, whereas such species dissolved in the supernatant are filtered out (Wiegand et al., 2016a); however, this could be detected by solution-state-like INEPT experiments (Wiegand et al., 2016a). The spectra reveal that ATP remains bound to the protein in its non-hydrolyzed form. In its active conformation, two ATP molecules should bind to one functional pRN1 primase (Boudet et al., 2015). This assumption is supported by the splitting of the ATP Pγ-resonance (Boudet et al., 2019). This allows us to conclude that the pRN1:ATP:DNA complex is stable over a time span of more than 2 years, although free ATP is not available in the rotor anymore due to ATP autohydrolysis. The hydrolysis of ATP also most likely explains the increasing resonance overtime at around 1.5 ppm, which is due to its typical 31P chemical shift toward a phosphate group assigned to some phosphate species that coordinate to the protein in an unspecified manner (resonance highlighted by a circle in Figure 1C, top panel). The pRN1 sample was stored at −20°C after the rotor filling. After 3 years, the 20 ms 13C-13C DARR spectrum is almost identical compared to the spectrum recorded directly after the rotor filling (Figure 2B). More specifically, no peak doubling indicating the formation of a second ATP- or DNA-free population is detected. The same conclusions can be drawn from the spectra of the Rpo4/7∗ RNA polymerase subunit for which the DARR spectra directly recorded after the rotor filling and 2.5 years later are highly similar (see Figure 2C).
The experimental observation of stable protein sediments over many years might be related to several precautions. To avoid cysteine oxidation of the protein sample, a sufficient amount of the redox reagent dithiothreitol (DTT) is typically added to the protein sample before sedimentation (except if the protein contains disulfide-bonds). Moreover, to avoid bacterial or fungal contamination, each rotor also contains a sufficient amount of sodium azide [0.01% (m/v)] (Snyder and Lichstein, 1940; Bowler et al., 2006). Depending on the time between protein–ligand complex formation and rotor filling, residual DTT might still be present in the rotor. Regarding the importance of oxidative processes, we estimated the amount of oxygen solubilized in the buffer within the NMR rotor, assuming that the rotor was sealed appropriately. This resulted in around 6 nmol of O2 in a 3.2 mm rotor that is ∼2 orders of magnitude less than the amount of protein. This estimation is based on a solubility of O2 of ∼8.5 mg/L at a O2 partial pressure of 0.21 atm (Rettich et al., 2000; Miyamoto et al., 2014), and the assumption that approximately half of the rotor is filled with the buffer (23 μL). A compelling reason for the absence of oxidation seems thus to be the low oxygen concentration compared to the high concentration of protein. As a higher temperature leads to faster denaturation of proteins, NMR measurements are typically performed at 5°C (sample temperature) (Böckmann et al., 2009), and the NMR rotor is stored at this temperature. An additional parameter altering protein stability is pressure. Indeed, many studies showed that high pressure induces dissociation and denaturation of proteins (Hillson et al., 1999; de Oliveira and Silva, 2015). However, the maximal centrifugal pressure in an NMR rotor acts on the ZrO2 rotor wall during MAS (∼1,500 bar at 17.0 kHz MAS in a 3.2 mm rotor, F. Engelke, lecture notes of EBSA solid-state NMR school 2014). The pressure value acting on the protein is even lower and is estimated between 60 and 91 bars (Kawamura et al., 2007; Asano et al., 2012). Consequently no pressure-induced protein degradation is expected (where this would, for example, occur at the 2,500 bar for ubiquitin (Charlier et al., 2018), 3,000 bar for PHS SNase (Roche et al., 2012), or 1,000 bar for spinach photosystem II (Tan et al., 2005)).
Our results on four sedimented protein samples show that proteins, protein–protein complexes, and protein–ligand complexes remain stable for years once sedimented into an NMR rotor. In the case of BmrA, the protein even retains its ability to transform into the outward-facing conformation upon addition of ATP. Our results are definitely of practical importance for solid-state NMR spectroscopists and participate in understanding better the properties of sedimented protein samples.
Materials and Methods
Solid-State NMR Experiments
13C-detected solid-state NMR spectra were acquired at 20.0 T static magnetic-field strength using a 3.2 mm Bruker Biospin E-free probe (Gor’kov et al., 2007). The MAS frequency was set to 17.0 kHz. The 2D spectra were processed with the software TOPSPIN (version 3.5, Bruker Biospin) with a shifted (3.0) squared cosine apodization function and automated baseline correction in the indirect and direct dimensions. The sample temperature was set to 278 K (Böckmann et al., 2009). All spectra were analyzed with the software CcpNmr (Fogh et al., 2002; Vranken et al., 2005; Stevens et al., 2011) and referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). Typical CP-conditions for all experiments described herein were ω(1H) = 60 kHz and ω(13C) = 45–49 kHz. 90 kHz SPINAL-64 1H decoupling (Fung et al., 2000) was applied during evolution and detection. Typical acquisition times in the direct dimension were 15.4 ms and, in the indirect dimension, 12.8 ms. 31P-detected experiments were acquired at 11.7 T in a Bruker 3.2 mm probe using a spinning frequency of 17.0 kHz. The spectra were referenced to 85% H3PO4. The CP conditions were ω(1H) = 60 kHz and ω(31P) = 45 kHz. 90 kHz SPINAL-64 1H decoupling was applied during detection.
Protein Sample Preparation
The preparations of the HpDnaB:ADP:AlF4–:DNA (Wiegand et al., 2019), the BmrA mutant E504A (Lacabanne et al., 2019b), pRN1:ATP:DNA (Boudet et al., 2019), and Rpo4/7∗ (only Rpo7 is uniformly labeled) (Torosyan et al., 2019) were prepared as described previously. In short, the proteins were recombinantly expressed in presence of 13C-glucose (2 g/L) and 15N-ammonium chloride (2 g/L) as sole sources of carbon-13 and nitrogen-15, respectively (for Rpo4/7∗ only Rpo7 was expressed in minimum medium supplemented with 13C-glucose and 15N-ammonium chloride).
HpDnaB
The purification of HpDnaB was achieved by a heparin-agarose affinity chromatography followed by an anion exchange chromatography with 2.5 mM sodium phosphate pH 7.5 and 130 mM NaCl. The complex formation with ADP:AlF4– and single-stranded DNA (20 thymidine nucleotides) is described in reference (Wiegand et al., 2019).
BmrA
Purification of BmrA-E504A was performed with a Ni-NTA purification system using a gravity column. The eluted protein was desalted using dedicated columns (PD10 – GE Healthcare Life Sciences) with 50 mM Tris–HCl pH 8.0, 100 mM NaCl, 10% glycerol, and 0.2% DDM (m/v). Bacillus subtilis lipids destabilized with Triton X-100 were added to the protein. The quantity of lipids follows a lipid-to-protein ratio of 0.5 (m/m). The DDM and Triton X-100 were removed using dialysis with Bio-beads (BioRad) for 9 days. The proteolipisomes were collected by centrifugation. LVIKH were selectively unlabeled, as described in references (Lacabanne et al., 2018, 2019b).
Rpo4/7∗
For the complex Rpo4/7, both proteins were expressed and purified separately, as described previously (Torosyan et al., 2019). In brief, Rpo4 was purified as a GST-fusion protein using glutathione agarose affinity chromatography in a P300 buffer (20 mM Tris/acetate pH 7.9, 300 mM potassium acetate, 10 mM magnesium acetate, 0.1 mM zinc sulfate, 5 mM DTT, and 10% glycerol) and eluted by P300 supplemented with 10 mM glutathione. The GST-tag was cleaved off by overnight incubation with thrombin at 37°C and subsequently removed by a 20-min heat shock of the cleaved elution fractions at 65°C followed by centrifugation.
The purification of uniformly 13C and 15N-labeled Rpo7∗ was carried out from inclusion bodies that were, after multiple rounds of washing, resuspended in 20 mM Tris/acetate, pH 7.9, 100 mM potassium acetate, 0.1 mM zinc sulfate, 10 mM magnesium acetate, 10% glycerol, and 6 M urea. The urea-soluble fraction containing Rpo7 was combined with Rpo4, and the complex was formed via urea-based unfolding-refolding dialysis using a slight molar excess of Rpo7∗ (1:1.2). Heat shock and centrifugation (as above) were used to remove incorrectly folded Rpo4/7∗ complexes as well as remaining sole Rpo7∗, yielding pure, heat-stable Rpo4/7∗ in the supernatant.
pRN1 Primase
The pRN1 primase was purified with an Ni-NTA purification system. The fractions containing the enzyme were dialyzed overnight with thrombin to remove the His-tag. The dialyzed protein was concentrated and loaded on a gel-filtration column with 25 mM Na2HPO4/NaH2PO4, NaCl 50 mM, and a pH of 7.
All proteins were concentrated up to 30 mg/mL by centrifugation and sedimented directly into the solid-state NMR rotor in an ultracentrifuge (overnight at 200,000 × g) using a homemade rotor filling tool (Böckmann et al., 2009).
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
DL, AT, JB, and RC prepared the samples. TW and DL performed the NMR experiments, analyzed the data, and wrote the manuscript with input from all authors. TW, DL, FA, BM, and AB designed and supervised the research. All authors approved the submitted version of the manuscript.
Funding
This work was supported by the ETH Career SEED-69 16-1, the ETH Research Grant ETH-43 17-2, the Swiss National Science Foundation (Grant 200020_159707), and by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (grant agreement no. 741863, FASTER). AB acknowledges support by the LABEX ECOFECT (ANR-11-LABX-0048) within the Université de Lyon program Investissements d’Avenir (ANR-11-IDEX-0007) and by the French Agence Nationale de Recherche (ANR-14-CE09-0024B, ANR-19-CE11-0023-01). FA acknowledges support from the Swiss National Science Foundation (grant no. 310030_163345).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00017/full#supplementary-material
References
Asano, A., Hori, S., Kitamura, M., Nakazawa, C. T., and Kurotsu, T. (2012). Influence of magic angle spinning on T1H of SBR studied by solid state 1H NMR. Polym. J. 44, 706–712. doi: 10.1038/pj.2012.10
Bertini, I., Engelke, F., Luchinat, C., Parigi, G., Ravera, E., Rosa, C., et al. (2012). NMR properties of sedimented solutes. Phys. Chem. Chem. Phys. 14, 439–447. doi: 10.1039/c1cp22978h
Bertini, I., Luchinat, C., Parigi, G., and Ravera, E. (2013). SedNMR: on the edge between solution and solid-state NMR. Acc. Chem. Res. 46, 2059–2069. doi: 10.1021/ar300342f
Bertini, I., Luchinat, C., Parigi, G., Ravera, E., Reif, B., and Turano, P. (2011). Solid-state NMR of proteins sedimented by ultracentrifugation. Proc. Natl. Acad. Sci. U.S.A. 108, 10396–10399. doi: 10.1073/pnas.1103854108
Böckmann, A., Gardiennet, C., Verel, R., Hunkeler, A., Loquet, A., Pintacuda, G., et al. (2009). Characterization of different water pools in solid-state NMR protein samples. J. Biomol. NMR 45, 319–327. doi: 10.1007/s10858-009-9374-3
Boudet, J., Devillier, J.-C., Allain, F. H. T., and Lipps, G. (2015). Structures to complement the archaeo-eukaryotic primases catalytic cycle description: what’s next? Comput. Struct. Biotechnol. J. 13, 339–351. doi: 10.1016/j.csbj.2015.04.006
Boudet, J., Devillier, J.-C., Wiegand, T., Salmon, L., Meier, B. H., Lipps, G., et al. (2019). A small helical bundle prepares primer synthesis by binding two nucleotides that enhance sequence-specific recognition of the DNA template. Cell 176, 154–166. doi: 10.1016/j.cell.2018.11.031
Bowler, M. W., Montgomery, M. G., Leslie, A. G. W., and Walker, J. E. (2006). How azide inhibits ATP hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. U.S.A. 103, 8646–8649. doi: 10.1073/pnas.0602915103
Celi, P., and Gabai, G. (2015). Oxidant/antioxidant balance in animal nutrition and health: the role of protein oxidation. Front. Vet. Sci. 2:48. doi: 10.3389/fvets.2015.00048
Charlier, C., Alderson, T. R., Courtney, J. M., Ying, J., Anfinrud, P., and Bax, A. (2018). Study of protein folding under native conditions by rapidly switching the hydrostatic pressure inside an NMR sample cell. Proc. Natl. Acad. Sci. U.S.A. 115, E4169–E4178. doi: 10.1073/pnas.1803642115
Davies, M. J. (2016). Protein oxidation and peroxidation. Biochem. J. 473, 805–825. doi: 10.1042/BJ20151227
Dawson, R. J. P., and Locher, K. P. (2006). Structure of a bacterial multidrug ABC transporter. Nature 443:180. doi: 10.1038/nature05155
de Oliveira, G. A. P., and Silva, J. L. (2015). A hypothesis to reconcile the physical and chemical unfolding of proteins. Proc. Natl. Acad. Sci. U.S.A. 112, E2775–E2784. doi: 10.1073/pnas.1500352112
Fogh, R., Ionides, J., Ulrich, E., Boucher, W., Vranken, W., Linge, J. P., et al. (2002). The CCPN project: an interim report on a data model for the NMR community. Nat. Struct. Mol. Biol. 9, 416–418. doi: 10.1038/nsb0602-416
Fragai, M., Luchinat, C., Parigi, G., and Ravera, E. (2013). Practical considerations over spectral quality in solid state NMR spectroscopy of soluble proteins. J. Biomol. NMR 57, 155–166. doi: 10.1007/s10858-013-9776-0
Fung, B. M., Khitrin, A. K., and Ermolaev, K. (2000). An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101. doi: 10.1006/jmre.1999.1896
Gardiennet, C., Schütz, A. K., Hunkeler, A., Kunert, B., Terradot, L., Böckmann, A., et al. (2012). A sedimented sample of a 59 kDa dodecameric helicase yields high-resolution solid-state NMR spectra. Angew. Chem. Int. Ed. 51, 7855–7858. doi: 10.1002/anie.201200779
Gardiennet, C., Wiegand, T., Bazin, A., Cadalbert, R., Kunert, B., Lacabanne, D., et al. (2016). Solid-state NMR chemical-shift perturbations indicate domain reorientation of the DnaG primase in the primosome of Helicobacter pylori. J. Biomol. NMR 64, 189–195. doi: 10.1007/s10858-016-0018-0
Gauto, D. F., Estrozi, L. F., Schwieters, C. D., Effantin, G., Macek, P., Sounier, R., et al. (2019). Integrated NMR and cryo-EM atomic-resolution structure determination of a half-megadalton enzyme complex. Nat. Commun. 10:2697. doi: 10.1038/s41467-019-10490-9
Gor’kov, P. L., Witter, R., Chekmenev, E. Y., Nozirov, F., Fu, R., and Brey, W. W. (2007). Low-E probe for 19F–1H NMR of dilute biological solids. J. Magn. Reson. 189, 182–189. doi: 10.1016/j.jmr.2007.09.008
Gross, M., and Jaenicke, R. (1994). Proteins under pressure. Eur. J. Biochem. 221, 617–630. doi: 10.1111/j.1432-1033.1994.tb18774.x
Hillson, N., Onuchic, J. N., and García, A. E. (1999). Pressure-induced protein-folding/unfolding kinetics. Proc. Natl. Acad. Sci. U.S.A. 96, 14848–14853. doi: 10.1073/pnas.96.26.14848
Kaur, H., Abreu, B., Akhmetzyanov, D., Lakatos-Karoly, A., Soares, C. M., Prisner, T., et al. (2018). Unexplored nucleotide binding modes for the ABC exporter MsbA. J. Am. Chem. Soc. 140, 14112–14125. doi: 10.1021/jacs.8b06739
Kawamura, I., Degawa, Y., Yamaguchi, S., Nishimura, K., Tuzi, S., Saitô, H., et al. (2007). Pressure-induced isomerization of retinal on bacteriorhodopsin as disclosed by fast magic angle spinning NMR†. Photochem. Photobiol. 83, 346–350. doi: 10.1562/2006-06-20-rc-941
Lacabanne, D., Fogeron, M.-L., Wiegand, T., Cadalbert, R., Meier, B. H., and Böckmann, A. (2019a). Protein sample preparation for solid-state NMR investigations. Prog. Nucl. Magn. Reson. Spectrosc. 110, 20–33. doi: 10.1016/j.pnmrs.2019.01.001
Lacabanne, D., Meier, B. H., and Bockmann, A. (2018). Selective labeling and unlabeling strategies in protein solid-state NMR spectroscopy. J. Biomol. NMR 71, 141–150. doi: 10.1007/s10858-017-0156-z
Lacabanne, D., Orelle, C., Lecoq, L., Kunert, B., Chuilon, C., Wiegand, T., et al. (2019b). Flexible-to-rigid transition is central for substrate transport in the ABC transporter BmrA from Bacillus subtilis. Commun. Biol. 2:149. doi: 10.1038/s42003-019-0390-x
Lecoq, L., Wang, S., Wiegand, T., Bressanelli, S., Nassal, M., Meier, B. H., et al. (2018). Localizing conformational hinges by NMR: where do hepatitis B virus core proteins adapt for capsid assembly? ChemPhysChem 19, 1336–1340. doi: 10.1002/cphc.201800211
Mandal, A., Boatz, J. C., Wheeler, T. B., and van der Wel, P. C. A. (2017). On the use of ultracentrifugal devices for routine sample preparation in biomolecular magic-angle-spinning NMR. J. Biomol. NMR 67, 165–178. doi: 10.1007/s10858-017-0089-6
Manning, M. C., Chou, D. K., Murphy, B. M., Payne, R. W., and Katayama, D. S. (2010). Stability of protein pharmaceuticals: an update. Pharm. Res. 27, 544–575. doi: 10.1007/s11095-009-0045-6
Manning, M. C., Patel, K., and Borchardt, R. T. (1989). Stability of protein pharmaceuticals. Pharm. Res. 6, 903–918.
Minton, A. P. (1981). Excluded volume as a determinant of macromolecular structure and reactivity. Biopolymers 20, 2093–2120. doi: 10.1002/bip.1981.360201006
Miyamoto, H., Yampolski, Y., and Young, C. L. (2014). Iupac-nist solubility data series. 103. Oxygen and ozone in water, aqueous solutions, and organic liquids (supplement to solubility data series volume 7. J. Phys. Chem. Ref. Data 43:033102. doi: 10.1063/1.4883876
Orelle, C., Gubellini, F., Durand, A., Marco, S., Lévy, D., Gros, P., et al. (2008). Conformational change induced by ATP binding in the multidrug atp-binding cassette transporter BmrA. Biochemistry 47, 2404–2412. doi: 10.1021/bi702303s
Piehl, D. W., Blancas-Mejía, L. M., Ramirez-Alvarado, M., and Rienstra, C. M. (2017). Solid-state NMR chemical shift assignments for AL-09 VL immunoglobulin light chain fibrils. Biomol. NMR Assign. 11, 45–50. doi: 10.1007/s12104-016-9718-3
Querol, E., Perez-Pons, J. A., and Mozo-Villarias, A. (1996). Analysis of protein conformational characteristics related to thermostability. Protein Eng. Des. Sel. 9, 265–271. doi: 10.1093/protein/9.3.265
Ravera, E. (2014). The bigger they are, the harder they fall: a topical review on sedimented solutes for solid-state NMR. Concepts Magn. Reson. A 43, 209–227. doi: 10.1002/cmr.a.21318
Ravera, E., Ciambellotti, S., Cerofolini, L., Martelli, T., Kozyreva, T., Bernacchioni, C., et al. (2016). Solid-state NMR of PEGylated proteins. Angew. Chem. Int. Ed. 55, 2446–2449. doi: 10.1002/anie.201510148
Rettich, T. R., Battino, R., and Wilhelm, E. (2000). Solubility of gases in liquids. 22. High-precision determination of Henry’s law constants of oxygen in liquid water fromT=274 K toT=328 K. J. Chem. Thermodyn. 32, 1145–1156. doi: 10.1006/jcht.1999.0581
Roche, J., Caro, J. A., Norberto, D. R., Barthe, P., Roumestand, C., Schlessman, J. L., et al. (2012). Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 6945–6950. doi: 10.1073/pnas.1200915109
Silva, J. L., and Weber, G. (1993). Pressure stability of proteins. Annu. Rev. Phys. Chem. 44, 89–113. doi: 10.1146/annurev.pc.44.100193.000513
Snyder, M. L., and Lichstein, H. C. (1940). Sodium azide as an inhibiting substance for gram-negative bacteria. J. Infect. Dis. 67, 113–115. doi: 10.1093/infdis/67.2.113
Stevens, T., Fogh, R., Boucher, W., Higman, V., Eisenmenger, F., Bardiaux, B., et al. (2011). A software framework for analysing solid-state MAS NMR data. J. Biomol. NMR 51, 437–447. doi: 10.1007/s10858-011-9569-2
Stöppler, D., Macpherson, A., Smith-Penzel, S., Basse, N., Lecomte, F., Deboves, H., et al. (2018). Insight into small molecule binding to the neonatal Fc receptor by X-ray crystallography and 100 kHz magic-angle-spinning NMR. PLoS Biol. 16:e2006192. doi: 10.1371/journal.pbio.2006192
Takegoshi, K., Nakamura, S., and Terao, T. (1999). 13C–13C polarization transfer by resonant interference recoupling under magic-angle spinning in solid-state NMR. Chem. Phys. Lett. 307, 295–302. doi: 10.1016/s0009-2614(99)00533-3
Takegoshi, K., Nakamura, S., and Terao, T. (2001). 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637. doi: 10.1016/s0009-2614(01)00791-6
Tan, C.-Y., Xu, C.-H., Wong, J., Shen, J.-R., Sakuma, S., Yamamoto, Y., et al. (2005). Pressure equilibrium and jump study on unfolding of 23-kDa protein from spinach photosystem II. Biophys. J. 88, 1264–1275. doi: 10.1529/biophysj.104.050435
Torosyan, A., Wiegand, T., Schledorn, M., Klose, D., Guntert, P., Bockmann, A., et al. (2019). Including protons in solid-state NMR resonance assignment and secondary structure analysis: the example of RNA polymerase II subunits Rpo4/7. Front. Mol. Biosci. 6:100. doi: 10.3389/fmolb.2019.00100
van der Wel, P. C. A. (2018). New applications of solid-state NMR in structural biology. Emerg. Top. Life Sci. 2, 57–67. doi: 10.1042/ETLS20170088
Vogt, G., Woell, S., and Argos, P. (1997). Protein thermal stability, hydrogen bonds, and ion pairs11Edited by F. E. Cohen. J. Mol. Biol. 269, 631–643. doi: 10.1006/jmbi.1997.1042
Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., et al. (2005). The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins Struct. Funct. Bioinform. 59, 687–696. doi: 10.1002/prot.20449
Wang, Y., Sarkar, M., Smith, A. E., Krois, A. S., and Pielak, G. J. (2012). Macromolecular crowding and protein stability. J. Am. Chem. Soc. 134, 16614–16618. doi: 10.1021/ja305300m
Wiegand, T., Cadalbert, R., Gardiennet, C., Timmins, J., Terradot, L., Böckmann, A., et al. (2016a). Monitoring ssDNA binding to the DnaB Helicase from Helicobacter pylori by solid-state NMR Spectroscopy. Angew. Chem. Int. Ed. 55, 14164–14168. doi: 10.1002/anie.201607295
Wiegand, T., Cadalbert, R., Lacabanne, D., Timmins, J., Terradot, L., Bockmann, A., et al. (2019). The conformational changes coupling ATP hydrolysis and translocation in a bacterial DnaB helicase. Nat. Commun. 10:31. doi: 10.1038/s41467-018-07968-3
Wiegand, T., Gardiennet, C., Ravotti, F., Bazin, A., Kunert, B., Lacabanne, D., et al. (2016b). Solid-state NMR sequential assignments of the N-terminal domain of HpDnaB helicase. Biomol. NMR Assign. 10, 13–23. doi: 10.1007/s12104-015-9629-8
Xiang, S., le Paige, U. B., Horn, V., Houben, K., Baldus, M., and van Ingen, H. (2018). Site-specific studies of nucleosome interactions by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 57, 4571–4575. doi: 10.1002/anie.201713158
Keywords: solid-state NMR, sedimentation, stability, proteins, nucleotides
Citation: Wiegand T, Lacabanne D, Torosyan A, Boudet J, Cadalbert R, Allain FH-T, Meier BH and Böckmann A (2020) Sedimentation Yields Long-Term Stable Protein Samples as Shown by Solid-State NMR. Front. Mol. Biosci. 7:17. doi: 10.3389/fmolb.2020.00017
Received: 18 December 2019; Accepted: 30 January 2020;
Published: 21 February 2020.
Edited by:
Anastasia S. Politou, University of Ioannina, GreeceReviewed by:
Enrico Ravera, University of Florence, ItalyMark Pfuhl, King’s College London, United Kingdom
Copyright © 2020 Wiegand, Lacabanne, Torosyan, Boudet, Cadalbert, Allain, Meier and Böckmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Wiegand, dGhvbWFzLndpZWdhbmRAcGh5cy5jaGVtLmV0aHouY2g=
†These authors have contributed equally to this work