 Laura Orellana1,2*
Laura Orellana1,2*- 1Institutionen för Biokemi och Biofysik, Stockholms Universitet, Stockholm, Sweden
- 2Science for Life Laboratory, Solna, Sweden
Large-scale conformational changes are essential to link protein structures with their function at the cell and organism scale, but have been elusive both experimentally and computationally. Over the past few years developments in cryo-electron microscopy and crystallography techniques have started to reveal multiple snapshots of increasingly large and flexible systems, deemed impossible only short time ago. As structural information accumulates, theoretical methods become central to understand how different conformers interconvert to mediate biological function. Here we briefly survey current in silico methods to tackle large conformational changes, reviewing recent examples of cross-validation of experiments and computational predictions, which show how the integration of different scale simulations with biological information is already starting to break the barriers between the in silico, in vitro, and in vivo worlds, shedding new light onto complex biological problems inaccessible so far.
Conformational Changes: Linking Shape and Function
Protein structure and dynamics are essential to understand living organisms at the molecular level. Already 60 years ago Feynman envisioned that life is, roughly speaking, not only about atomic organization, but also about the “jiggling and wiggling of atoms” (Feynman et al., 1963). The central paradigm of structural biology stated that the 3D-fold of a protein is encoded in the sequence (Dill and Chan, 1997; Wright and Dyson, 2015); the explosion of structural data in the past decades has dramatically expanded this classical view, confirming Feynman's prediction. Far from being static structures, it is now clear that proteins rather behave as living entities (Henzler-Wildman and Kern, 2007), ever-changing on temporal and spatial scales spanning several orders of magnitude: from local loop fluctuations in enzyme active sites (Aglietti et al., 2013; Pal et al., 2016) to concerted beta-sheets motions (Fenwick et al., 2014) or large-scale allosteric motions in transmembrane receptors (Bugge et al., 2016). Importantly, growing evidence indicates that these large conformational changes are intrinsically encoded in the overall 3D-shape (Bahar et al., 2010), and that external stimuli –binding, post-translational modifications, electrochemical gradients, etc.—just drive these “natural” motions further to trigger output responses. Signal transduction, membrane transport or synaptic communication, almost every cell process relies on switches that cycle between distinct states to allow for bioregulation (Figure 1A). The way that proteins change to sense and respond to such stimuli is therefore central to connect the micro-, meso-, and macro-scales in biology. However, their elucidation from atomic “jiggling and wiggling” is far from trivial.
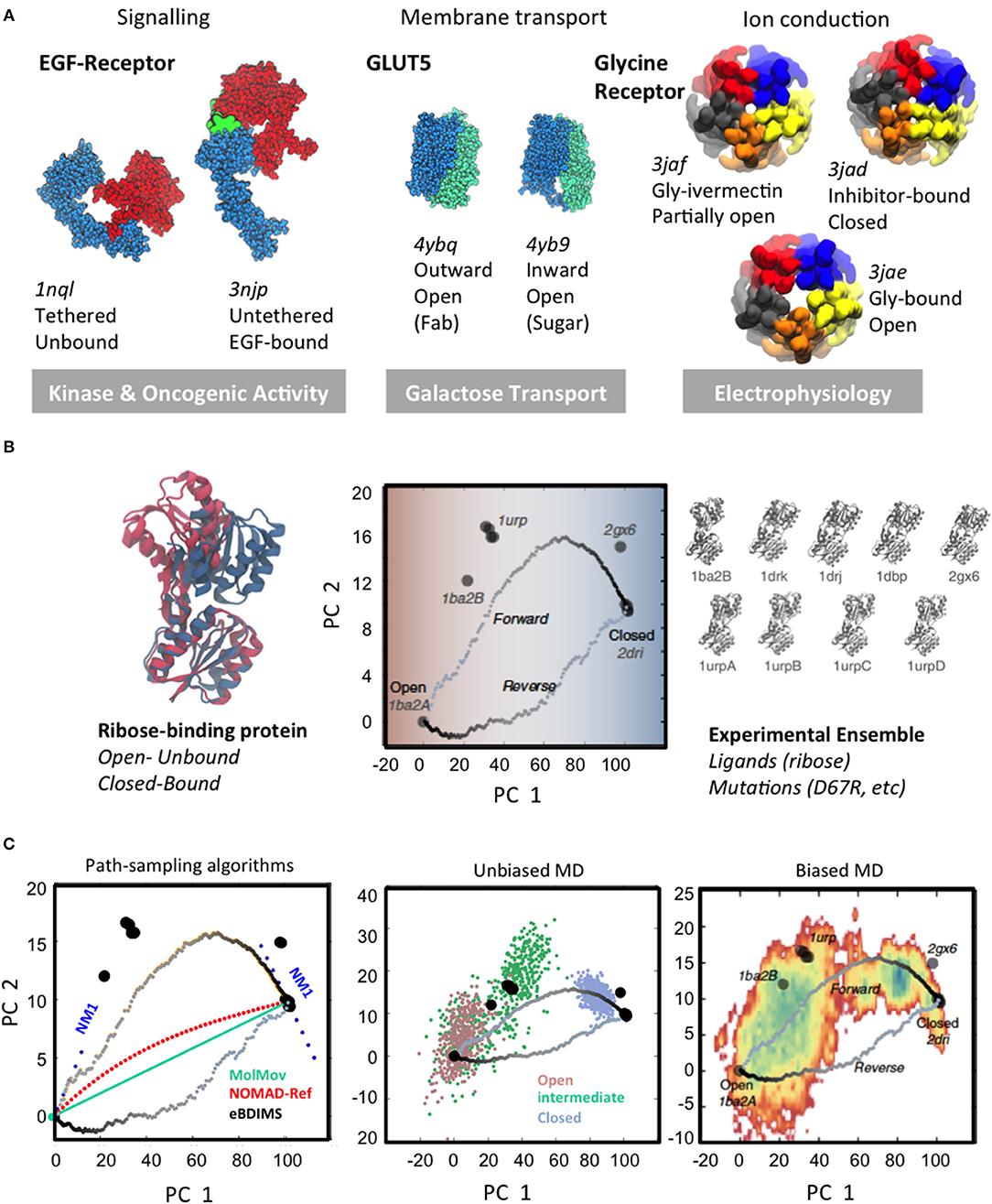
Figure 1. Large-scale conformational changes and different scale sampling methods. (A) Three examples of transitions of different scales linked to biological function: left, large-scale domain rearrangement in EGFR upon ligand binding; center, rearrangement of tandem repeats in sugar porters; right, cooperative pentamer motions in pentameric ligand-gated ion channels. The majority of conformations trapped by structural techniques correspond to the extreme, lowest-energy states of biological cycles. (B) Experimental conformational landscapes for the hinge-bending transition of the Ribose Binding Protein (RBP) as computed from Principal Component Analysis: the open to closed RBP conformational change upon ribose binding (Left); RBP conformational landscape and eBDIMS coarse-grained (CG) transitions (Center) as projected onto the PCs derived from the 9 solved structures (Right). Note how eBDIMS paths approach the sequence of experimental intermediates. (C) Comparison of sampling strategies: NMs and path-finding CG-methods (Left); atomistic MD unbiased (500 ns from each unbound state) (Center) and 1 μs-biasing to the closed state (Right). Note how the first NM derived from both RBP end-states (Left) points to the experimental intermediates; note also how eBDIMS paths (gray) roughly follow the MD/X-ray sampled area. Adapted by the author with CC BY license from Orellana et al. (2016).
During the past decade, structural determination techniques have made incredible progresses in resolving structures of increasing complexity and flexibility. Currently, high-throughput time-resolved X-ray crystallography (Levantino et al., 2015; Neutze et al., 2015; Ourmazd, 2019), cryo-Electron Microscopy (cryo-EM; Nogales and Scheres, 2015; Murata and Wolf, 2018; Shoemaker and Ando, 2018), and Nuclear Magnetic Resonance (NMR; Baker and Baldus, 2014; Jiang and Kalodimos, 2017; Opella and Marassi, 2017), together with complimentary techniques like Small Angle X-ray Scattering (SAXS; Vestergaard, 2016), Förster Resonance Energy Transfer (FRET; Okamoto and Sako, 2017), double electron-electron resonance (DEER; Jeschke, 2012), mass spectrometry (Kahsai et al., 2014) or fluorescence microscopy (Lewis and Lu, 2019) are allowing to resolve and gain dynamic information from extremely challenging systems. Despite such advances, the experimental study of protein transitions is still demanding. A complete understanding of equilibrium dynamics requires sampling both the structure space available and the underlying free-energy landscape (FEL; Frauenfelder et al., 1991; Zhuravlev and Papoian, 2010a; Nussinov and Wolynes, 2014; Röder et al., 2019) along its relevant dimensions (Figures 1B,C). Ideally, a completely rational and quantitative FEL characterization should stem from first principles, for example, using methods like Molecular Dynamics (MD; Karplus and McCammon, 2002; Orozco, 2014), in which Newton's equations are integrated over time for an atomistic model of the system based on physical potentials. In practice, atomistic-level sampling of the functional FEL of biomolecules poses by itself a huge conceptual and technical problem in silico. Collective rearrangements and allosteric events in proteins can involve scales around ms-μs and up to 102Å. Note that this is far beyond what classical MD can address in terms of time and size: roughly two orders of magnitude larger than average simulated interatomic distances (~1–10Å), and up to 9–12 orders of magnitude larger than the smallest simulated timestep (fs oscillations) (Sweet et al., 2013). Importantly, functional transitions often occur in this blurred frontier between theory and experimentation.
Scalable codes, graphic processing units (GPUs), parallelization and optimized simulation algorithms (Pierce et al., 2012; Sweet et al., 2013; Kutzner et al., 2015; Páll et al., 2015; Pouya et al., 2017) are however making increasingly feasible to simulate systems with millions of atoms for few μs, or even whole bacterial cytoplasms in the submicrosecond range (Yu et al., 2016). Still, for most proteins, these timescales cover a small part of the structural landscape, and longer simulations are only accessible with special-purpose supercomputers like Anton (Shaw et al., 2009; Dror et al., 2012). Apart from these technical aspects, there is a fundamental “sampling problem,” not efficiently addressed by long simulations: transition paths in a multidimensional landscape are intrinsically stochastic—there are multiple possible transition routes, subject to random fluctuations that unpredictably push over energy barriers. Multiple evidences indicate that the way in which the configuration space is sampled is thus more critical than simulation length. For example, while in μs-long MD, full transitions are still rarely observed, in certain conditions e.g., upon relaxation after removing ligands (Nury et al., 2010; Calimet et al., 2013; Degiacomi, 2019) or introducing mutations (Smolin and Robia, 2015; Orellana et al., 2019b) they can occur in significantly shorter times. Similarly, coarse-grained (CG) methods like Elastic Network Models (ENMs; Mahajan and Sanejouand, 2015), are also capable to predict with striking accuracy, just from the overall shape of a protein, not only the conformational changes observed experimentally but also entire sequences of on-pathway intermediates (Orellana et al., 2016). This suggests that large-scale motions like those defining protein functional FELs may be better understood as collective, supra-atomistic and higher-scale phenomena. Whatever the theoretical framework chosen to explore this issue, the validation of in-silico predicted mechanisms is becoming a central question, as quantitative analysis become essential to rationalize the growing dynamical information from techniques like cryo-EM (Frank, 2018; Bonomi and Vendruscolo, 2019).
Let's now imagine the reader wants to know how a series of conformations for a given protein are related, to get insight into some biological mechanism. It is appropriate then to ask: Can in silico methods really predict conformational transitions? Have such in-silico transitions been validated and how? This review is intended to provide the non-specialist with some answers to these questions, first raised by Weiss and Levitt (2009). On the first part (Table 1), we will briefly review theoretical methods to predict transition pathways, focusing on the two most common approaches to explore the FEL between two states: either increase atomistic MD sampling (Maximova et al., 2016) or coarse-grain the model of the system (Zheng and Wen, 2017). On the second part (Table 2), we will discuss recent examples from our group and others attempting cross-validation between theory and experiments in this context. This review does not aim to provide an in-depth description of specific methods which can be found elsewhere (Bernardi et al., 2015; Maximova et al., 2016; Mori et al., 2016; Zheng and Wen, 2017; Harpole and Delemotte, 2018). We rather intend to provide general readers, and specially experimentalists, with a broad overview of the most accessible approaches to explore a transition for a typical protein, along with possible validation strategies. Our goal is to help the reader grasp the current potential of in silico methods to explain biological phenomena from microscopic scales, and the exciting boundaries we are reaching.
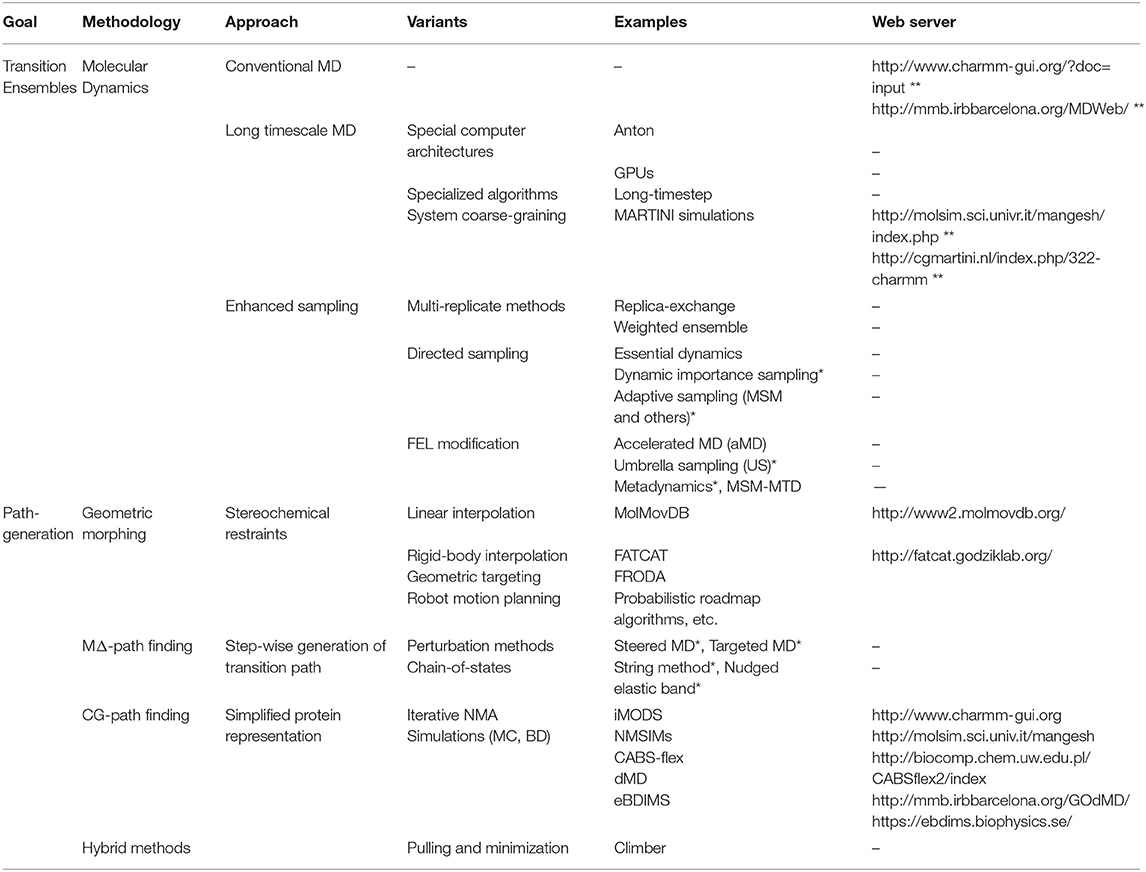
Table 1. Summary of common in silico methods to explore protein conformational changes (*CV-based, **only for setup/short run).
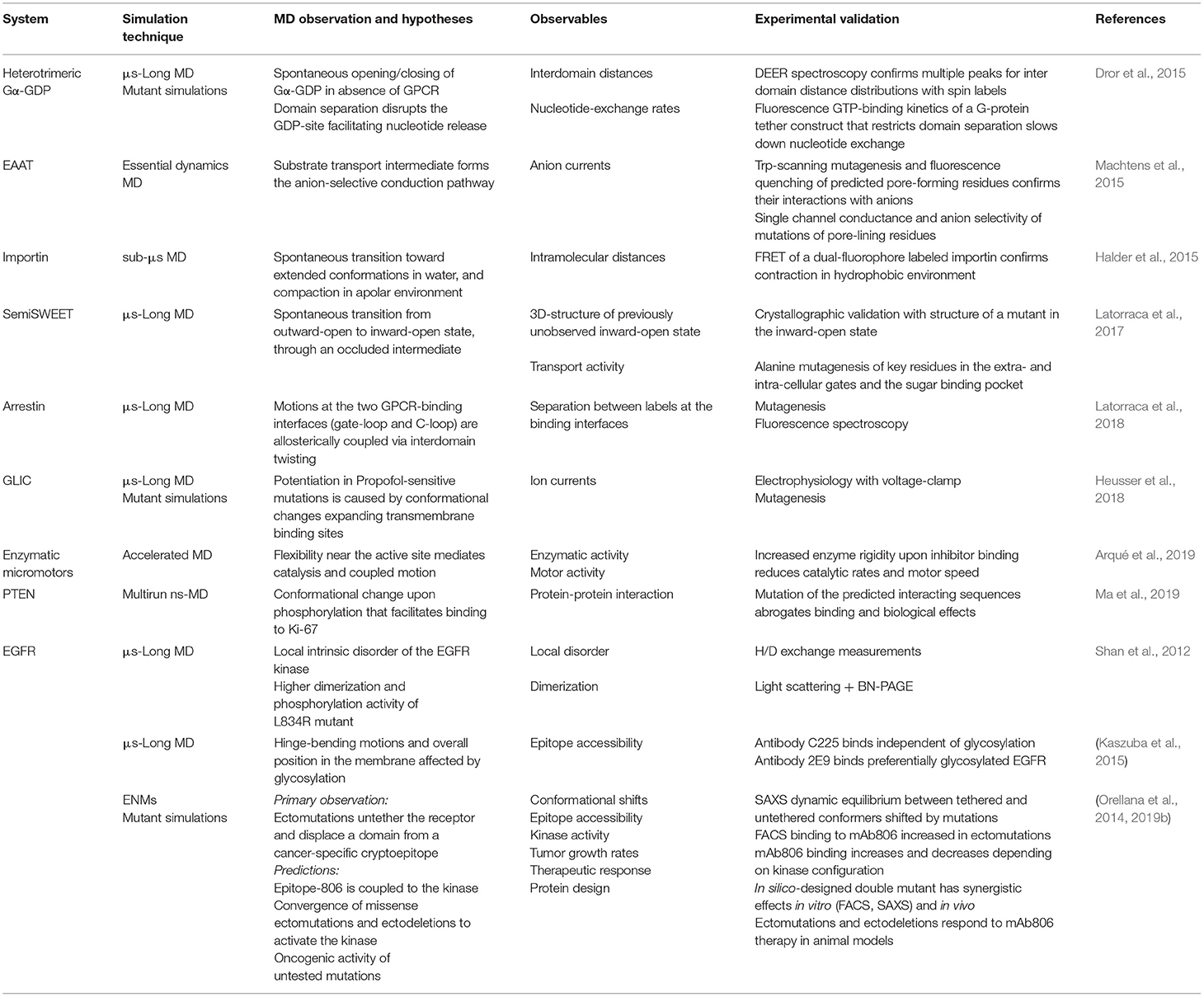
Table 2. Examples of cross-validation of in silico-predicted properties with experiments to specifically probe conformational changes.
From Static Snapshots to Multi-State Structural Ensembles
Since the first structure was determined by X-ray crystallography in the late 50s (Kendrew et al., 1958), the number of protein structures deposited every year in the Protein Data Bank (Berman et al., 2000) has been growing exponentially, from a few dozens in the 90s up to over 10,000 structures/year in the past 2 years. As of 2019, we know around 140 thousand native-like protein structures, with resolutions as low as 0.5Å. For a majority of them however, the conformers solved represent the equilibrium end-structures along their functional cycles, typically composed of at least two different meta-stable states (Figure 1A): active/inactive, bound/unbound, open/closed, etc. For such average proteins (Figure 1B), the native apo state frequently populates the deepest basin and spontaneously samples another of comparable or reduced depth, favored by stimuli like binding, post-translational modifications, etc. that shift the population (Nussinov and Wolynes, 2014). Structural determination techniques usually trap conformations near one of such low-energy basins, while the short-lived intermediates connecting them—which can be key to grasp mechanisms (see e.g., Machtens et al., 2015; Orellana et al., 2019b)—are often elusive both experimental and computationally.
To explore the conformational space, structures are typically solved in multiple conditions e.g., introducing mutations, modulating pH, ions, or complexing with molecules—from ligands to antibodies, affibodies, or small drugs. This contributes to enormous redundancy in the PDB, but at the same time, it is a powerful approach to catch intermediates along transitions. For a growing number of intensely studied proteins the multitude of conditions that has been used to determine their 3D-structures has gradually covered the entire conformational landscape. Especially cryo-EM is allowing to routinely obtain protein snapshots in multiple states with each data deposition [see e.g., the Glycine Receptor (GlyR; Du et al., 2015) in Figure 1A] and, although limited to a few protein families, this is revealing the first glimpses into structural ensembles that cover nearly-complete conformational landscapes (Frank, 2018; Bonomi and Vendruscolo, 2019; Hofmann et al., 2019).
Obtaining multiple snapshots of a protein is however just the first step to characterize its transitions. The second consists on understanding their relationships, which also implies identifying the relevant collective variables (CVs; Kitao and Go, 1999; Zhuravlev and Papoian, 2010b; Noé and Clementi, 2017) for each system. This task is comparable to taking multiple pictures of a moving animal in diverse situations, and then trying to reconstruct its biomechanics; one needs first to find a way to measure, classify and organize the images, so that an ordered sequence can be reconstructed. How are we going to efficiently describe the system? What are we going to measure to detect changes from one functional state to another? Fortunately, large-scale transitions can be often described by a remarkably low number of CVs (Henzler-Wildman and Kern, 2007). This is not surprising since, for most proteins, functional movements are collective: each level of protein motion translates into the next, creating wider and slower movements. For example, local atomic vibrations are transmitted via hydrogen bond networks that make up secondary structures, creating higher amplitude motions; as shown in Fenwick et al. (2011, 2014) the coupled movements of interacting atoms in beta-sheet motifs create collective bending and twisting motions, which propagate to higher collective movements linked to allosteric regulation. Another recurring motif in large-scale protein transitions are open-to-closed motions upon binding (Flores et al., 2006; Amemiya et al., 2011), which in their simplest version consist in rigid-body displacements around a cracking hinge (Figure 1A, left). The hinge region, often located near a binding pocket, is typically an interdomain linker; in more complex transitions wider intra/inter-molecular surfaces can reshape as hinges e.g., in the “rocker-switch” motions between tandem repeats of solute transporters (Drew and Boudker, 2016; Figure 1A, center). Linker or interface reshaping propagate across structures triggering large-scale rearrangements. Usually, such rigid-body transitions are tracked with ad-hoc defined angles, distances, etc. However, while for simple hinge-bending transitions, an angle defined by moving rigid bodies can render a fair description of the process, the situation changes when systems undergo complex concerted changes: to accurately describe e.g., gating for ion channels like GlyR (Figure 1A, right) typically demands multiple variables describing extra- and intra-cellular motion features, much harder to define. In such cases, if the protein in question has solved structures in different basins, Principal Component Analysis (PCA; Jolliffe, 2002; Abdi and Williams, 2010), can provide a “natural” representation of the conformational landscape (Sankar et al., 2015) in the form of experimentally-encoded CVs. Compared to other approaches for semi-automated conformer annotation (e.g., based on machine learning; Ung et al., 2018), PCA does not need a priori system-tailored structural descriptors, requiring minimal user intervention PCA. PC-projections approach was recently applied in spliceosome cryo-EM to perform conformer classification, understand its dynamics and obtain a fist assessment of the FEL straight from experimental data (Haselbach et al., 2017, 2018). Moreover, PCs from multi-state ensembles behave as intrinsic complex coordinates that “contain” the heuristic CVs typically defined for each system. As we will discuss later, when such ensemble-analysis are combined with path-sampling, they can illuminate relationships between multiple basins and accurately assign intermediate states, allowing reconstruction of the landscape and its transitions into its experimentally-defined CVs (Figure 2). For example, the Ca2+ pump SERCA, with over 70 structures and at least four different states along its complex pumping cycle, constitutes an exceptional example of a multi-basin ensemble where such analysis is critical to unambiguously assign and order experimental on-pathway intermediates (see Orellana et al., 2016). Importantly, PCA of such “structurally-rich” or multi-state ensembles provides a much needed and stringent test for any modeling technique to explore protein FELs. In the next two sections, we will review the two most popular and accessible approaches to perform such in silico exploration to “connect” experimental basins and “fill in” the conformational landscape: first, sampling with classical MD and its many derivatives, and second, path-finding with computationally simpler methods.
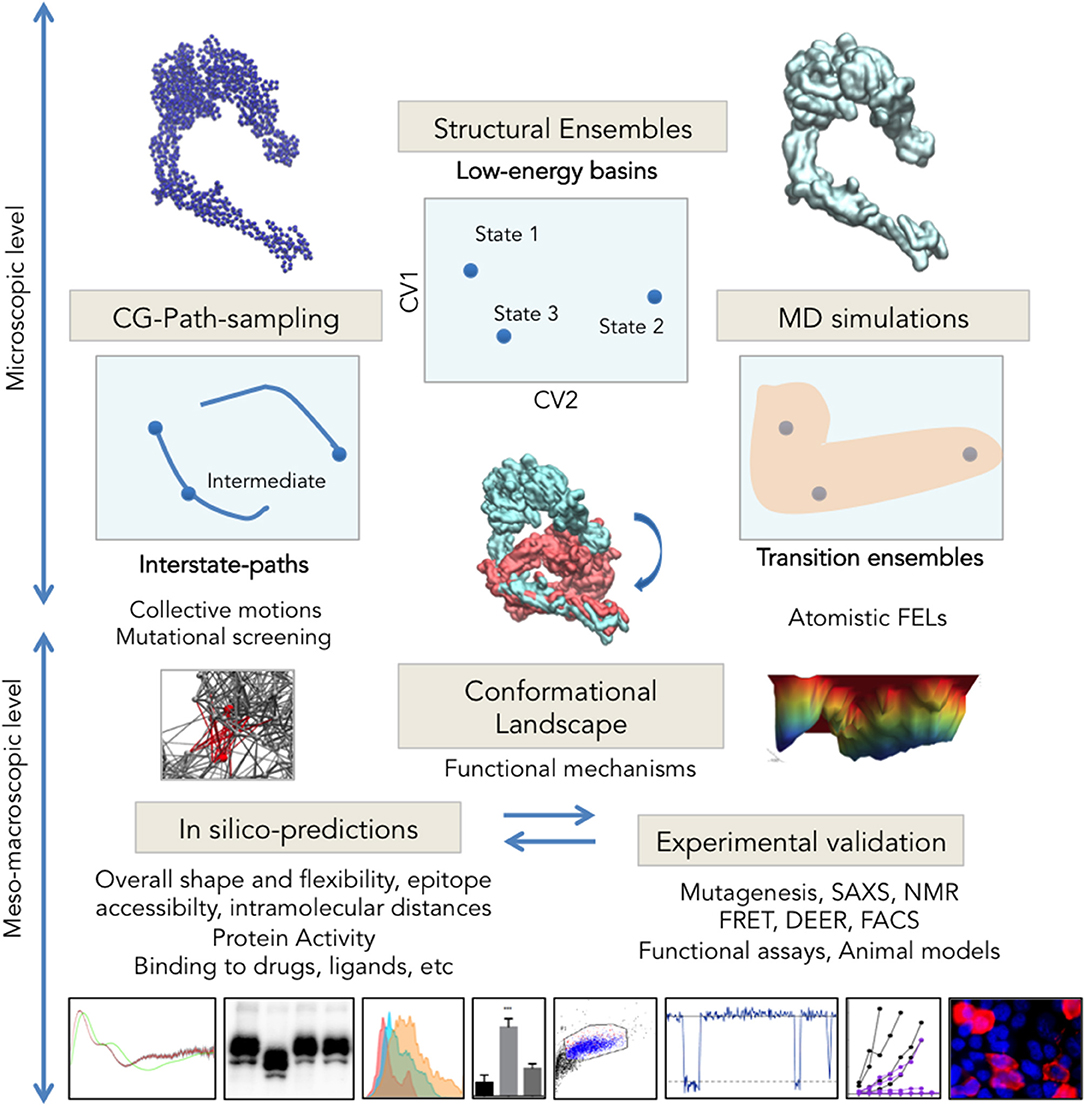
Figure 2. Integrative multi-scale structural biology. Experimental techniques are solving an increasing number of structures trapped in different energy basins, which through ensemble-analysis like PCA can yield intrinsic CVs for landscape exploration (Top Center). Transition pathways computed e.g., from CG-models (Right) connect experimental states allowing for intermediate assignment, provide insights into collective motions and can facilitate identification of key regions for mutational analysis. Long or enhanced MD simulations sample the configuration space with atomistic detail and allow reconstructing the complete FEL. Integration of microscopic data on conformational changes generates higher-scale predictions on protein shape, activity and interactions (Bottom) that can be tested through structural and molecular biology (microscopy techniques, SAXS, functional assays, etc. See examples in Table 2).
Exploring the Landscape: Sampling Long vs. Tricked
MD simulations, based on the rigorous formalism of molecular physics, constitute possibly the most accurate and accessible approach to model protein motions with atomic detail. Although still an idealized description of reality—proteins diffusing in a crowded and complex cellular soup—MD is based on a careful parameterization of covalent and non-covalent forces on the atomic scale (Beauchamp et al., 2012; Lindorff-Larsen et al., 2012; Monticelli and Tieleman, 2013). Since the first eye blink 9.2 ps simulation of the small BPTI (McCammon et al., 1977), MD has evolved dramatically over the past 40 years up to become almost a “computational microscope” (Dror et al., 2012): it is expected that for relatively small systems like GPCRs, MD will reach the second scale within 5 years (Martínez-Rosell et al., 2017). Nevertheless, for average protein machines, transitions are difficult to sample due to inherent stochasticity and high-energy barriers, involving challenging time and length scales. Although specialized computers like Anton allow simulations of ever-larger systems, longer than ever, and have indeed brought novel insights for key drug targets like GPCRs (Dror et al., 2015), Voltage-gated channels (Jensen et al., 2012) or Kinases (Shan et al., 2012, 2014), conformational changes are still hard to catch. As a rule of thumb, “everyday” simulations invariably require algorithmic “tricks” to explore transitions with reasonable efficiency. More than computational power or simulation length, efficient sampling remains a bottleneck.
The next brief enumeration of MD-strategies to overcome this problem and explore large transitions should provide the reader with a clear picture of its complexity and its many potential pitfalls. Without aiming to be exhaustive (for detailed reviews see e.g., Bernardi et al., 2015; Maximova et al., 2016; Mori et al., 2016; Harpole and Delemotte, 2018), the most common “tricks” (Pietrucci, 2017) to explore transitions are broadly: first, to speed up or optimize exploration of the FEL, without modifying it; second, to actually change the FEL to easily move and jump across its “hills and valleys” (Table 1). In both cases, the search can be biased or directly pushed along some a priori “direction,” i.e., a CV. Among the first group are many multi-replicate methods, well-suited for highly scalable software implementations thanks to their intrinsically parallel algorithms. Replica exchange MD (REMD) often called “parallel tempering” [first applied to MD in Sugita and Okamoto (1999), reviewed in Ostermeir and Zacharias (2013)], exchanges multiple trajectories run in parallel (typically at different temperatures) to escape local minima. Weighted ensemble methods (WEM) originally developed for simpler Brownian Dynamics (BD; Huber and Kim, 1996; see also Zuckerman and Chong, 2017), use quasi-independent trajectories in which individual runs spawn daughter trajectories upon reaching new “bins” of the configuration space. Mention apart deserves adaptive-MD, a general term which includes a wide array of multi-run schemes aimed to speed up rare events without explicit biasing (Bowman et al., 2010; Pronk et al., 2011; Doerr et al., 2016). The main idea behind adaptive-MD is that simulations are guided toward underexplored FEL regions via iterative on-the-fly analysis; similarly, WEM partition of the FEL into bins also needs previous CV-reduction. Therefore, to identify meaningful CVs to check how simulations proceed becomes central, with risks to generate overly smooth landscapes or distort transition mechanisms (see Hruska et al., 2018; Zimmerman et al., 2018). One analysis approach used to guide sampling in adaptive-MD, are Markov State Models (MSMs; Pande et al., 2010), a statistical method to describe dynamics as memory-less transitions between states. MSMs can infer long-timescale dynamics from sets of shorter simulations, providing yet another shortcut to the sampling conundrum (Chodera and Noé, 2014). In contrast to these costly multi-replicate schemes, biasing methods directly guide single simulations through relevant CVs. For example, Essential Dynamics (Amadei et al., 1993; Daidone and Amadei, 2012) extracts with PCA the “essential” CVs (Essential Modes), which are used to bias the sampling toward collective motions. In Dynamic importance sampling (DIMS; Zuckerman and Woolf, 2000; Perilla et al., 2011) a progress variable or CV is used to select the most productive movement toward the target in a MC-scheme, while in Temperature-Accelerated MD (TAMD; Maragliano and Vanden-Eijnden, 2006) temperature is increased specifically along selected CVs.
A completely different approach is taken in FEL-modifying approaches like Umbrella sampling (US; Torrie and Valleau, 1977), which introduces harmonic biasing potentials along CVs in overlapping “umbrella” windows. Accelerated MD methods (aMD; Hamelberg et al., 2004; Pierce et al., 2012) change the relative height of the basins by adding “boost” potentials when the system's energy falls, locally flattening the FEL. In metadynamics (MTD), free energy wells are filled with “computational sand” to prevent returning back to previously explored CV-regions (Laio and Parrinello, 2002; Laio and Gervasio, 2008). The accelerated weight histogram (AWH; Lindahl et al., 2014, 2018) adaptively bias simulations to fit a target distribution, filling up energy minima in a similar spirit as MTD (see Figure 1C), while in conformational flooding (Grubmüller, 1995), a destabilizing potential is added to the starting state, lowering the transition barrier. From all above methods, MTD has been maybe the most widely applied to study large transitions in a number of pioneering works, from the opening/closing of kinases (Berteotti et al., 2009) or actin monomers (Pfaendtner et al., 2009) to flexible binding and dissociation events (Limongelli et al., 2010, 2012; Formoso et al., 2015).
Moreover, all these different approaches can be combined in virtually infinite ways, giving rise to hybrid methods like Bias-Exchange MTD (Piana and Laio, 2007), MSM-driven MTD (Sultan and Pande, 2017), and many others. The main shared concern for the above listed methodologies is that trajectories may not accurately reproduce the biologically relevant motions (i.e., trapped experimentally), since they either modify the way sampling is done by decreasing its randomness, or directly change the underlying landscape, which can require re-scaling to remove biasing. Particularly, the bias-introducing methods require extra caution to not produce unrealistic high-energy intermediates (Ma and Karplus, 2002; Ovchinnikov and Karplus, 2012). A tightly connected issue stems from the choice of CVs, which is critical (Pan et al., 2014) but nevertheless, is frequently defined ad-hoc for each system. Typically, CVs are defined in terms of e.g., radius of gyration, distances, angles, rMSDs changing across sets of trial trajectories, which are expected to correlate or “describe” the transition. MSMs (Sultan and Pande, 2017), or machine-learning (Chen et al., 2018) can also be applied to solve this “dimensionality reduction” problem and identify relevant CVs. Another alternative is to define CVs from experimental data e.g., NMR chemical shifts (Granata et al., 2013) or SAXS intensities (Kimanius et al., 2015). In summary, CV definition is a non-trivial problem. For all these reasons, unbiased long simulations, which neither perturb the FEL nor require previous CV knowledge, are often preferred alternatives in many studies aiming for experimental validation, as we will review in the last section.
Path-Finding Methods: Throwing Ropes Over Mountains
Apart from the host of methods to enhance MD conformational sampling, there is another fundamental strategy to explore protein transitions: to simplify either the simulation algorithm or the system, in order to obtain just a feasible pathway between states. Finding transition paths has been compared to “throwing ropes over mountain passes in the dark” (Bolhuis et al., 2002; Dellago and Bolhuis, 2007), since indeed, such methods produce one-dimensional trajectories, like ropes in the conformation space (Figure 1C). Instead of sampling transition ensembles covering broad areas of the FEL, the goal of path-sampling methods is to generate sequences of structures connecting end-states. Such rope-like transitions, apart of providing first mechanistic insights, can serve as seeds for further MD (e.g., with US, MTD or “swarms-of-trajectories” Pan et al., 2008; Maragliano et al., 2014) to reconstruct the FEL.
Very broadly, path-generating methods (Weiss and Koehl, 2014; Table 1) can be also classified into two groups: (i) geometric morphing algorithms, which generate stereochemically correct morphs between structures, without any potential function, and (ii) those methods based on some potential energy, that actually attempt to approach minimum energy paths (MEPs) connecting basins. Among the latter, there are path-finding schemes based on MD inspired by the same ideas of enhanced sampling, along with a series of CG-methods, which take a entirely different approach, simplifying description of structures and their interactions.
The first online tool to compute transition pathways appeared within the MolMov Database (MolMovDB; Gerstein and Krebs, 1998; Krebs and Gerstein, 2000), and applied the simplest possible morphing: a linear interpolation in Cartesian coordinates, followed by energy minimization. As could be expected, MolMovDB paths project as perfectly straight lines in the experimental PC-landscape, and thus do not correspond at all to realistic transitions (Figure 1C, left). FATCAT also uses a interpolation of rigid-body motions (Ye and Godzik, 2004). More sophisticated are methods like FRODA (Wells et al., 2005) or geometric targeting (Farrell et al., 2010), which move atoms toward the target by enforcing geometric constraints to keep stereochemistry, while robot motion-planning algorithms (Cortés et al., 2005; Haspel et al., 2010; Al-Bluwi et al., 2012) exploit analogies between molecular bonds and robot links to perform fast molecular kinematics. Note that none of these geometric path-finding methods, which usually generate atomistic paths thanks to high computational efficiency, aims to provide a physical approximation to the FEL. This is not the case for MD-derived perturbation methods (Huang et al., 2009) like targeted (Schlitter et al., 1994), steered (Izrailev et al., 1997), or adiabatic MD (Marchi and Ballone, 1999; Paci and Karplus, 1999), where an MD simulation is directly pushed to the target by time-dependent potentials along a CV. In the so-called “chain-of-states” methods (Tao et al., 2012) like the nudged elastic band (Maragakis et al., 2002) or the string methods (Ren and Vanden-Eijnden, 2005; Ren et al., 2005; Ovchinnikov et al., 2011), serial images of the system are minimized to find MEPs; in the “path-method,” a guess path coordinate and two CVs that are functions of it are introduced to locally explore and optimize pathways (Branduardi et al., 2007; Bonomi et al., 2008). Although all these enhanced MD-derived path- sampling methods can be faster than conventional MD, finding proper CVs, biasing definitions or initial paths is again critical, and thus their implementation is not straightforward.
In contrast with the MD-inspired methods, CG-approaches, more than as alternative methods, should be rather considered a different way of looking at the sampling problem, literally, from a more collective scale. Coarse-graining simplifies the description of a system to capture its behavior with a minimum of parameters (Tozzini, 2010; Orozco et al., 2011; Saunders and Voth, 2013). By simplifying both potentials and structure description (Kmiecik et al., 2016), CG-methods accelerate computation increasing orders of magnitude the accessible scales; metaphorically speaking, they would be analogs to approaches like cryo-EM or SAXS, in which detail can be sacrificed to gain information from very large or flexible systems. Although hampered by loss-of-resolution regarding time and chemical properties, CG-methods can thus provide deep insights into complex systems behavior, as they distill multidimensional information to its very essential features. Although there are CG-force fields like the popular MARTINI implemented into real MD schemes (Marrink and Tieleman, 2013; Ingólfsson et al., 2014), in general CG-models are used in the context of much simpler algorithms, typically produce one-dimensional pathways, and are often available as webservers (Table 1).
To generate quick and efficient transitions, CG path-finding methods (Zheng and Wen, 2017) use a host of conceptually diverse protein representations: from a few heavy atoms (e.g., CABS model; Jamroz et al., 2013; Kmiecik et al., 2016) to residue beads (typical of ENMs) or rigid domains; and the same holds true for algorithmic approaches, that span from matrix diagonalization to MC or BD simulations. The only thing they have in common is skipping MD computational limitations, at the cost of losing information about time and energy. Among CG-methods, ENMs (Chennubhotla et al., 2005; Bahar et al., 2010) stand out due to conceptual simplicity and power to predict large changes through Normal Mode Analysis (NMA; Case, 1994). NMA is a molecular mechanics technique based on harmonic potentials, which was first used to predict infrared spectra and soon became also applied to analytically compute near-equilibrium protein atomic oscillations (Brooks, 1983; Levitt et al., 1985): solving a simple eigenvalue problem, a vector describing the directions of movement for every atom could be obtained. Inspired by “beads-and-springs” polymer models (Flory et al., 1976; Go and Scheraga, 1976), further coarse-graining of the protein description up to the C-alpha backbone lead to the minimalist ENM-NMA (Tirion, 1996; Bahar et al., 1997; Atilgan et al., 2001). Typically, ENMs reduce protein architecture to a network of Cα-carbons connected by springs, which model covalent and non-covalent interactions. In spite of this simplicity, it soon became evident that ENMs can not only predict residue fluctuations, but are also capable of guessing with striking precision the directions of large-scale transitions between e.g., X-ray open and closed pairs (Tama and Sanejouand, 2001). Later work has shown that ENMs reproduce as well the flexibility from experimental X-ray and NMR ensembles, or long MD simulations (Rueda et al., 2007; Orellana et al., 2010; Mahajan and Sanejouand, 2015; Sankar et al., 2018) and importantly, track the pathways for conformational change (Orellana et al., 2016; see NM projections, Figure 1C, left). Therefore, ENMs have been at the core of CG-strategies to find transition paths; however, being limited to an equilibrium basin, pathway generation requires iterative deformation along selected NMs, or implementation into some simulation scheme. Iterative ENMs range from simple interpolations like NOMAD-Ref and others (Kim et al., 2002; Lindahl et al., 2006; Seo and Kim, 2012) to more complex two-state approaches like iENM or ANMPathway (Yang et al., 2009; Tekpinar and Zheng, 2010; Das et al., 2014) or MinActPath (Franklin et al., 2007; Chandrasekaran et al., 2016), which assumes harmonic potential at the end-states and solves the action minimization problem to find the crossing points. A common issue for such CG-methods is that they typically produce stereochemical distortions, which can be reduced using internal coordinates like in iMODS (López-Blanco et al., 2014), structure corrections in NMSIMs (Ahmed et al., 2011; Krüger et al., 2012), or conjugate peak refinement like in the plastic network model (PNM; Maragakis and Karplus, 2005). In general, these approaches share the ENM power to capture allosteric transitions, but also display a shared weakness: a trend to reproduce similar one-dimensional paths rather than random pathway ensembles (Figure 1C, center). One solution to this problem is using NMs to bias simple e.g., Discrete Dynamics (dMD) simulations (Sfriso et al., 2012, 2013) in order to obtain a wider ensemble, although still, mode selection, as CV selection in enhanced MD schemes, poses a problem. Recently, we proposed an ENM-driven simulation approach, eBDIMS (Orellana et al., 2016, 2019a), also performing in parallel a thorough validation of path-finding algorithms against multi-state ensemble PCA. Based on a refined ENM force-field (Orellana et al., 2010), eBDIMS generates paths driven by interresidue distances, using a DIMS-Langevin scheme with a friction term mimicking solvent. This avoids unrealistic deformations, at the same time that ENM-modes are spontaneously sampled, generating random and non-linear trajectories.
Mention apart deserve hybrid methods like Climber (Weiss and Levitt, 2009), which iteratively pulls the interresidue distances adding harmonic restraints to an internal energy function, based on the atomistic ENCAD atomistic force-field (Levitt et al., 1995). In our comparative studies we found that eBDIMS and Climber, starting from entirely different approaches (CG- vs. atomistic, Langevin integration vs. iterative pulling/minimization), generate surprisingly convergent, non-linear, and asymmetric paths in PC-space. Remarkably, these paths closely overlap with solved experimental intermediates, which delimit the areas typically sampled by MD (see Figure 1C, center and right). Overall, our findings strongly suggested that these non-linear path-finding methods converge to actual MEPs, which are populated by trapped experimental intermediates. This raises a important question: how is it possible that such simple C-alpha based harmonic models like eBDIMS, can predict the directions of non-equilibrium conformational changes, while MD often requires powerful computing or enhanced sampling? On one hand, it has been suggested that dynamical systems theory assures the conservation of quasi-periodic motions upon small perturbations (Bastolla, 2014), and thus, ENMs are valid beyond the equilibrium, and in a wider set of conditions than was previously thought. On the other hand, the evident power of CG-methods to predict large-scale transitions and intermediates trapped by cryo-EM and crystallography, not only demonstrates such validity, but more importantly, it confirms that the collective shape-encoded dynamics of proteins, is maybe an essential determinant driving their underlying biologically functional transitions. Therefore, CG-methods are not just a quicker alternative to MD, but can provide an essential tool to dissect multi-scale problems like protein large transitions (Voth, 2009), specially in schemes where they are integrated with MD and experiments [see e.g., our recent experience (Orellana et al., 2019b) briefly discussed below].
Cross-Validation of Simulations and Experiments: Toward Integrative Biology
Although the “raison d'être” of most theoretical methods to model protein transitions is to gain insight into molecular mechanisms and connect them to biology, attempts to validate them are still rare, and thus, any in silico predictions usually remain in the computational realm as mere hypotheses and are looked at with suspicion by experimentalists (see Lowe, 2015 critique on Kohlhoff et al., 2014). Traditionally, MD provided dynamic information on microscopic scales often inaccessible by experimental probes (e.g., atomic details on hydrogen bonding, loop fluctuations, etc.), and thus were un-testable. As larger scale events like conformational changes are simulated, MD can generate semi-quantitative estimates of observables that can be more easily measured experimentally. Therefore, current MD can significantly contribute to the understanding and interpretation of experimental data; and alternatively, it can also be driven by experiments (Hollingsworth and Dror, 2018). However, in comparison with the large efforts concentrated on pushing the simulation length and sampling, little has been done to systematize and validate in parallel the information obtained, especially when approaching the scales in which transitions happen and propagate over.
Simulating the physical world always involves a degree of approximation and uncertainty (Berendsen, 2007); but the same is true for biological experiments. This constitutes maybe the core reason separating the in silico world from actual biology: the extraordinary difficulty posed by integration of atomic-level data on motion with higher-scale experiments, which typically average out dynamic properties over time and space. Recently, a thorough critical analysis of factors influencing the agreement of simulations and experimental data was presented by van Gunsteren et al. (2018). We will not discuss here related issues associated to force-field parameterization (Lindorff-Larsen et al., 2012), convergence of the simulations (Knapp et al., 2011; Sawle and Ghosh, 2016), prediction of microscopic observables (Childers and Daggett, 2018), or the multiple caveats of modeling more realistic e.g., crowded complex environments (Chavent et al., 2016), electrochemical gradients (Delemotte et al., 2008; Khalili-Araghi et al., 2013), etc. We aim rather to revisit some experimental approaches that have recently provided hands-on direct or indirect validation of in silico predicted large-scale transitions (Table 2).
While there has been extensive work on force-field parameterization e.g., benchmarking predictions about microscopic properties, studies benchmarking the performance of atomistic simulation methods to sample conformational transitions are limited and often reduced to small proteins (Pan et al., 2016). A related issue with MD- benchmarking is also the abovementioned difficulty to identify relevant CVs for complex systems, especially when only one of the conformational states is known. Note that, in contrast to MD, benchmarking against large-scale changes not only has routinely been done for CG-methods, but also constituted the main basis for their parameterization and in consequence, are extremely effective at predicting transitions along with their CVs. Independent of the strengths and weaknesses of each method, however, the main issues to validate transition pathways are essentially two: on one hand, the scarcity of experimental data about on-pathway intermediates; on the other, the uncertainty determining the relevant CVs to monitor changes and their associated observables. Although a transition pathway should be ideally supported by direct structural data (either crystallography, cryo-EM, NMR, or SAXS), this is often difficult and the only feasible option is to attempt indirect “soft” validation, either from distance parameters e.g., via single-molecule FRET, FACS, or from functional assays, which can test predictions about protein activity, as we briefly review next.
Direct Path-Validation: Protein Data Bank Ensembles and Landscapes
Classically, in silico pathways like those generated by path-sampling were evaluated on the sole basis of stereochemical quality, or by tracking progression along ad-hoc system-defined coordinates (Das et al., 2014; Seyler and Beckstein, 2014). As mentioned above, the selection of heuristic CVs for dimensionality reduction is problematic (Seyler et al., 2015), and in practice, structural quality or progression along user-defined CVs does not assure that a pathway samples the biologically relevant routes. Weiss and Levitt clearly stated this question a decade ago: “Can morphing methods predict intermediate structures?” (Weiss and Levitt, 2009), proposing for the first time to benchmark against proteins with at least three distinct states solved, and asses how close sampled pathways spontaneously approach known intermediates in terms of rMSD. Although this procedure definitely poses a more accurate test for in silico pathways, it cannot assess the feasibility of the movements or to what extent they correspond to biological motions. Based on these ideas, we proposed to go beyond two- or three-state benchmarking by introducing ensemble-level analyses that consider all structural information available in the PDB for a given protein, extracting at the same time their intrinsic CVs using PCA (Orellana et al., 2016). This kind of validation provides an extremely stringent test to evaluate sampling both by MD and path-finding algorithms and, thanks to the increasing amount of multi-state structural data available, we foresee that it could become widely applicable in the near future with cryo-EM. As a case apart of “hard” pathway validation, it is necessary to mention the study on a SWEET transporter by Latorraca et al. (2017), in which the spontaneous transition toward the inward-open state was first observed in silico with Anton simulations, and subsequently validated by determining an X-ray structure trapped in the same conformation. Although such an approach is not feasible to routinely validate pathways, it has provided maybe the strongest evidence to date in favor of the power of MD simulations to accurately sample the conformational space of proteins.
Soft Validation: From FRET and Antibody Binding to Functional Assays and Animal Models
MD simulations have been traditionally validated and compared with microscopic information on relatively local protein flexibility like NMR couplings, B-Factors, etc. During the last years however, simulations have started to generate predictions of a scale that is suitable for experimental validation through non-structural techniques, finally crossing the boundaries with molecular biology. A quick glimpse into recent examples of cross-validation of conformational changes between simulation and experiments (Table 2) clearly shows how finally, we are starting to break the barriers separating both, providing new insights into biomedically relevant systems, including key drug targets. Functional conformational changes usually involve either large rigid-body motions of structural elements or more local unfolding, loop fluctuation transitions. While the first can expose or bury molecular surfaces for dimerization, interaction with other proteins or ligands etc., the second may have more subtle effects on structure-function relationship e.g., at enzyme active sites. Observing such changes in silico, has given rise to quantitative or qualitative predictions that mainly fall into two categories: concerning interactions with other proteins or small molecules (dimerization, binding), and/or regarding activity—catalysis, phosphorylation, ion transport, etc.
Maybe one of the first examples attempting soft validation of in silico transitions aroused from short simulations of open/closed changes in Hsp70, confirmed by Trp-fluorescence changes upon ATP binding (Woo et al., 2009). A more complex validation strategy was taken by Laine et al. (2010), designing and testing a series of inhibitors against the different conformations of an allosteric site throughout an in silico transition path. In a groundbreaking study of the EGF-Receptor (EGFR kinase domain; Shan et al., 2012), Anton simulations revealed a third intermediate state characterized by local αC-helix disorder; further simulations of mutations indicated that they suppress this disorder to enhance dimerization and activation. In this case, proving intrinsic disorder and mutation effects required Hydrogen/Deuterium (H/D) exchange mass spectrometry (Wales and Engen, 2006), while enhanced dimerization was shown by Blue Native Gel electrophoresis (Wittig et al., 2006). Later work by the Shaw group, cross-validating NMR data and simulations (Arkhipov et al., 2013; Endres et al., 2013), provided new insights into EGFR transmembrane dimerization. Shorter μs-simulations by Kaszuba et al. (2015) also led to predictions about the impact of glycans on EGFR conformation, which were tested by monitoring the accessibility of glycosylation-sensitive surface-epitopes. Recently, we combined first a mutational screening partly based on ENMs, followed by MD simulations of “dynamically” hot EGFR ectodomain mutations (Orellana et al., 2014, 2019b) in a multiscale CG-MD scheme similar to that proposed by Saunders and Voth (2013). This approach highlighted how, as happens often experimentally, mutagenesis can help to trap intermediate states. In this case, the MD-trapped transition state, happened to be the target for a therapeutically relevant antibody, mAb806, which had been long hypothesized to bind a third ectodomain conformer distinct from the known crystal structures and enriched in tumor cells. This provided a rare opportunity to directly extrapolate an MD prediction to animal models by testing mAb806 therapeutic impact, with surprising success (Binder et al., 2018; Orellana, 2019); moreover, the integration of functional experiments, SAXS and MD revealed unsuspected functional and allosteric convergence of ectodomain deletions and missense mutations. A similar example, in which a protein is known to perform a certain biological activity but the corresponding conformation remains elusive, is illustrated by the work by Machtens et al. (2015), which extended previous MTD findings by Grazioso et al. (2012). In this case, excitatory aminoacid transporters (EAATs) were known to transport anions but the specific conduction path was not obvious in end-state X-ray structures. ED simulations of a prokaryotic glutamate transporter homolog, Gltph, revealed a potential channel in an intermediate state (independently trapped with crystallography), and the predicted pore-lining residues were confirmed with Trp-scanning mutagenesis, fluorescence quenching, and electrophysiology. Another indirect approach to validate MD-predicted changes consists on assessing intra or intermolecular distances with FRET, used e.g., to confirm the compaction of importin in apolar solvents (Halder et al., 2015) or DEER, an approach that allowed to prove the opening/closing dynamics in heterotrimeric G-proteins (Dror et al., 2015) and its modulation by nucleotide binding.
Although not the subject of this review, it is worth to mention the advances on simulations of spontaneous ligand binding events and protein-protein interactions, which constitute a special case regarding experimental validation and can occasionally provide indirect validation for conformational changes related to binding. For example, either long simulations or enhanced sampling techniques like aMD or MTD have captured spontaneous binding of small molecules to protein kinases or GPCRs (Dror et al., 2011, 2013; Shan et al., 2011, 2012; Kappel et al., 2015), dimerization in membranes (Lelimousin et al., 2016), or protein-protein interactions (Ma et al., 2019), approaching or reproducing crystallographic binding poses or NMR ensembles. In these cases, the PDB coordinates of known complexes, together with free energies of binding, drug efficacies, etc. (Shukla et al., 2015) can provide a hard-validation for MD.
Concluding Remarks
We have provided a brief overview of the multiple approaches that are used to explore the conformational landscapes of proteins and their transitions in silico, and reviewed different methods used for their validation. On one hand, it becomes clear that the accumulated structural information and flexibility-capturing techniques like cryo-EM are revealing first glimpses on functional landscapes. On the other hand, computational methods have reached maturity and are entering a stage in which they can start to contribute to real biology, modeling longer and larger scales. We have revisited the many approaches available to explore the FEL of proteins, optimizing hardware, software and algorithms pursuing the dream of the seconds-long sampling. From a completely different standpoint, simulations in crowded cell-like soups of multiple copies of the same protein, although still in the ns-scale, are already a reality that holds promise to reveal dynamical complexity in local microenvironments, providing yet another approach to the sampling problem (Yu et al., 2016; Feig et al., 2018). We have also briefly mentioned machine learning algorithms, paradigmatic of a series of novel fast-developing non-physically based strategies which are gaining ground to study transitions, either alone or in combination with MD or CG-methods: from co-evolution analysis (Morcos et al., 2013; Sutto et al., 2015; Sfriso et al., 2016) to cross-correlation, network and community approaches (Potestio et al., 2009; Morra et al., 2012; Rivalta et al., 2012; Papaleo, 2015; Negre et al., 2018), neural networks and deep learning (Ung et al., 2018; Degiacomi, 2019), or integrative sequence and structural analysis (Flock et al., 2015). These approaches, not primarily intended to generate conformational pathways or obtain a physical FEL, have shown their power to reveal new alternative conformations and dissect allosteric mechanisms, and thus are also greatly contributing to the exploration of protein flexibility space. We have reviewed some of the many flavors of CG- models and algorithms, and how they can provide low-resolution but stunningly accurate pathways. Finally, we have discussed recent examples where simulations have trapped intermediate states before confirmation by X-ray crystallography (Latorraca et al., 2017), or by in vivo tumor models (Orellana et al., 2019b). Altogether, the explosion of structural data, along with the ever expanding toolkit of in silico methods, computer capabilities and growing integration between simulations and experiments—driving or being driven by them—are beginning to fulfill the dream of connecting the micro-, meso-, and macro- scales in the study of life phenomena. It also becomes evident that this enterprise requires careful integration of a multitude of techniques and approaches, to connect the atomistic level with the emerging collective behaviors that rule conformational changes. The times ahead are exciting, as we are approaching a critical mass of information on protein structures, and experimental techniques allow exploring their dynamics with ever-increasing detail. The challenge will be to merge the ever-growing data into a coherent picture, which has certainly the potential to revolutionize biology, medicine and drug discovery.
Author Contributions
LO conceived and wrote the manuscript and prepared figures.
Funding
LO was supported by grants from the Knut and Alice Wallenberg (KAW) Foundation and the O.E. and Edla Johanssons Stiftelse.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdi, H., and Williams, L. J. (2010). Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2, 433–459. doi: 10.1002/wics.101
Aglietti, R. A., Floor, S. N., McClendon, C. L., Jacobson, M. P., and Gross, J. D. (2013). Active site conformational dynamics are coupled to catalysis in the mRNA decapping enzyme Dcp2. Structure. 21, 1571–1580. doi: 10.1016/j.str.2013.06.021
Ahmed, A., Rippmann, F., Barnickel, G., and Gohlke, H. (2011). A normal mode-based geometric simulation approach for exploring biologically relevant conformational transitions in proteins. J. Chem. Inf. Model. 51, 1604–1622. doi: 10.1021/ci100461k
Al-Bluwi, I., Siméon, T., and Cortés, J. (2012). Motion planning algorithms for molecular simulations: a survey. Comput. Sci. Rev. 6, 125–143. doi: 10.1016/j.cosrev.2012.07.002
Amadei, A., Linssen, A. B., and Berendsen, H. J. (1993). Essential dynamics of proteins. Proteins 17, 412–425. doi: 10.1002/prot.340170408
Amemiya, T., Koike, R., Fuchigami, S., Ikeguchi, M., and Kidera, A. (2011). Classification and annotation of the relationship between protein structural change and ligand binding. J. Mol. Biol. 408, 568–584. doi: 10.1016/j.jmb.2011.02.058
Arkhipov, A., Shan, Y., Das, R., Endres, N. F., Eastwood, M. P., Wemmer, D. E., et al. (2013). Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569. doi: 10.1016/j.cell.2012.12.030
Arqué, X., Romero-Rivera, A., Feixas, F., Patiño, T., Osuna, S., and Sánchez, S. (2019). Intrinsic enzymatic properties modulate the self-propulsion of micromotors. Nat. Commun. 10:2826. doi: 10.1038/s41467-019-10726-8
Atilgan, A. R., Durell, S. R., Jernigan, R. L., Demirel, M. C., Keskin, O., and Bahar, I. (2001). Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–15. doi: 10.1016/S0006-3495(01)76033-X
Bahar, I., Atilgan, A. R., and Erman, B. (1997). Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 2, 173–181. doi: 10.1016/S1359-0278(97)00024-2
Bahar, I., Lezon, T. R., Yang, L.-W., and Eyal, E. (2010). Global dynamics of proteins: bridging between structure and function. Annu. Rev. Biophys. 39, 23–42. doi: 10.1146/annurev.biophys.093008.131258
Baker, L. A., and Baldus, M. (2014). Characterization of membrane protein function by solid-state NMR spectroscopy. Curr. Opin. Struct. Biol. 27, 48–55. doi: 10.1016/j.sbi.2014.03.009
Bastolla, U. (2014). Computing protein dynamics from protein structure with elastic network models. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 488–503. doi: 10.1002/wcms.1186
Beauchamp, K. A., Lin, Y.-S., Das, R., and Pande, V. S. (2012). Are protein force fields getting better? A systematic Benchmark on 524 diverse NMR measurements. J. Chem. Theory Comput. 8, 1409–1414. doi: 10.1021/ct2007814
Berendsen, H. J. C. (2007). Simulating the Physical World: Hierarchical Modeling From Quantum Mechanics to Fluid Dynamics. doi: 10.1017/CBO9780511815348
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Bernardi, R. C., Melo, M. C. R., and Schulten, K. (2015). Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 1850, 872–877. doi: 10.1016/j.bbagen.2014.10.019
Berteotti, A., Cavalli, A., Branduardi, D., Gervasio, F. L., Recanatini, M., and Parrinello, M. (2009). Protein conformational transitions: the closure mechanism of a kinase explored by atomistic simulations. J. Am. Chem. Soc. 131, 244–50. doi: 10.1021/ja806846q
Binder, Z. A., Thorne, A. H., Bakas, S., Wileyto, E. P., Bilello, M., Akbari, H., et al. (2018). Epidermal growth factor receptor extracellular domain mutations in glioblastoma present opportunities for clinical imaging and therapeutic development. Cancer Cell 34, 163–177.e7. doi: 10.1016/j.ccell.2018.06.006
Bolhuis, P. G., Chandler, D., Dellago, C., and Geissler, P. L. (2002). Transition paths ampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 53, 291–318. doi: 10.1146/annurev.physchem.53.082301.113146
Bonomi, M., Branduardi, D., Gervasio, F. L., Buffi, V. G., and Lugano, C. (2008). The unfolded ensemble and folding mechanism of the C-terminal GB1 -hairpin. J. Am. Chem. Soc. 130, 13938–13944. doi: 10.1021/ja803652f
Bonomi, M., and Vendruscolo, M. (2019). Determination of protein structural ensembles using cryo-electron microscopy. Curr. Opin. Struct. Biol. 56, 37–45. doi: 10.1016/j.sbi.2018.10.006
Bowman, G. R., Ensign, D. L., and Pande, V. S. (2010). Enhanced modeling via network theory: adaptive sampling of markov state models. J. Chem. Theory Comput. 6, 787–794. doi: 10.1021/ct900620b
Branduardi, D., Gervasio, F. L., and Parrinello, M. (2007). From A to B in free energy space. J. Chem. Phys. 126:054103. doi: 10.1063/1.2432340
Brooks, B. (1983). Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. U.S.A. 80, 6571–6575. doi: 10.1073/pnas.80.21.6571
Bugge, K., Lindorff-Larsen, K., and Kragelund, B. B. (2016). Understanding single-pass transmembrane receptor signaling from a structural viewpoint—what are we missing? FEBS J. 283, 4424–4451. doi: 10.1111/febs.13793
Calimet, N., Simoes, M., Changeux, J.-P., Karplus, M., Taly, A., and Cecchini, M. (2013). A gating mechanism of pentameric ligand-gated ion channels. Proc. Natl. Acad. Sci. U.S.A. 110, E3987–E3996. doi: 10.1073/pnas.1313785110
Case, D. A. (1994). Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 4, 285–290. doi: 10.1016/S0959-440X(94)90321-2
Chandrasekaran, S. N., Das, J., Dokholyan, N. V., and Carter, C. W. (2016). A modified PATH algorithm rapidly generates transition states comparable to those found by other well established algorithms. Struct. Dyn. 3:012101. doi: 10.1063/1.4941599
Chavent, M., Duncan, A. L., and Sansom, M. S. P. (2016). Molecular dynamics simulations of membrane proteins and their interactions: from nanoscale to mesoscale. Curr. Opin. Struct. Biol. 40, 8–16. doi: 10.1016/j.sbi.2016.06.007
Chen, W., Tan, A. R., and Ferguson, A. L. (2018). Collective variable discovery and enhanced sampling using autoencoders: Innovations in network architecture and error function design. J. Chem. Phys. 149:072312. doi: 10.1063/1.5023804
Chennubhotla, C., Rader, A. J., Yang, L.-W., and Bahar, I. (2005). Elastic network models for understanding biomolecular machinery: from enzymes to supramolecular assemblies. Phys. Biol. 2, S173–S180. doi: 10.1088/1478-3975/2/4/S12
Childers, M. C., and Daggett, V. (2018). Validating molecular dynamics simulations against experimental observables in light of underlying conformational ensembles. J. Phys. Chem. B 122, 6673–6689. doi: 10.1021/acs.jpcb.8b02144
Chodera, J. D., and Noé, F. (2014). Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 25, 135–144. doi: 10.1016/j.sbi.2014.04.002
Cortés, J., Siméon, T., Ruiz de Angulo, V., Guieysse, D., Remaud-Siméon, M., and Tran, V. (2005). A path planning approach for computing large-amplitude motions of flexible molecules. Bioinformatics 21, i116–i125. doi: 10.1093/bioinformatics/bti1017
Daidone, I., and Amadei, A. (2012). Essential dynamics: foundation and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 762–770. doi: 10.1002/wcms.1099
Das, A., Gur, M., Cheng, M. H., Jo, S., Bahar, I., and Roux, B. (2014). Exploring the conformational transitions of biomolecular systems using a simple two-state anisotropic network model. PLoS Comput. Biol. 10:e1003521. doi: 10.1371/journal.pcbi.1003521
Degiacomi, M. T. (2019). Coupling molecular dynamics and deep learning to mine protein conformational space. Structure 27, 1034–1040.e3. doi: 10.1016/j.str.2019.03.018
Delemotte, L., Dehez, F., Treptow, W., and Tarek, M. (2008). Modeling membranes under a transmembrane potential. J. Phys. Chem. B 112, 5547–5550. doi: 10.1021/jp710846y
Dellago, C., and Bolhuis, P. (2007). Transition path sampling simulations of biological systems. Top. Curr. Chem. 268, 291–317. doi: 10.1007/128_085
Dill, K. A., and Chan, H. S. (1997). From Levinthal to pathways to funnels. Nat. Struct. Biol. 4, 10–19. doi: 10.1038/nsb0197-10
Doerr, S., Harvey, M. J., Noé, F., and De Fabritiis, G. (2016). HTMD: high-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput. 12, 1845–1852. doi: 10.1021/acs.jctc.6b00049
Drew, D., and Boudker, O. (2016). Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 85, 543–572. doi: 10.1146/annurev-biochem-060815-014520
Dror, R. O., Dirks, R. M., Grossman, J. P., Xu, H., and Shaw, D. E. (2012). Biomolecular simulation: a computational microscope for molecular biology. Annu. Rev. Biophys. 41, 429–52. doi: 10.1146/annurev-biophys-042910-155245
Dror, R. O., Green, H. F., Valant, C., Borhani, D. W., Valcourt, J. R., Pan, A. C., et al. (2013). Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299. doi: 10.1038/nature12595
Dror, R. O., Mildorf, T. J., Hilger, D., Manglik, A., Borhani, D. W., Arlow, D. H., et al. (2015). Structural basis for nucleotide exchange in heterotrimeric G proteins. Science. 348, 1361–1365. doi: 10.1126/science.aaa5264
Dror, R. O., Pan, A. C., Arlow, D. H., Borhani, D. W., Maragakis, P., Shan, Y., et al. (2011). Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 108, 13118–13123. doi: 10.1073/pnas.1104614108
Du, J., Lü, W., Wu, S., Cheng, Y., and Gouaux, E. (2015). Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 526, 224–229. doi: 10.1038/nature14853
Endres, N. F., Das, R., Smith, A. W., Arkhipov, A., Kovacs, E., Huang, Y., et al. (2013). Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556. doi: 10.1016/j.cell.2012.12.032
Farrell, D. W., Speranskiy, K., and Thorpe, M. F. (2010). Generating stereochemically acceptable protein pathways. Proteins 78, 2908–21. doi: 10.1002/prot.22810
Feig, M., Nawrocki, G., Yu, I., Wang, P. H., and Sugita, Y. (2018). Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 1036:012010. doi: 10.1088/1742-6596/1036/1/012010
Fenwick, R. B., Esteban-Martín, S., Richter, B., Lee, D., Walter, K. F. A, Milovanovic, D., et al. (2011). Weak long-range correlated motions in a surface patch of ubiquitin involved in molecular recognition. J. Am. Chem. Soc. 133, 10336–9. doi: 10.1021/ja200461n
Fenwick, R. B., Orellana, L., Esteban-Martín, S., Orozco, M., and Salvatella, X. (2014). Correlated motions are a fundamental property of β-sheets. Nat. Commun. 5:4070. doi: 10.1038/ncomms5070
Feynman, R. P., Leighton, R. B., and Sands, M. (1963). The Feynman Lectures on Physics. Reading, MA: Addison-Wesley.
Flock, T., Ravarani, C. N. J., Sun, D., Venkatakrishnan, A. J., Kayikci, M., Tate, C. G., et al. (2015). Universal allosteric mechanism for G a activation by GPCRs. Nature 524, 173–179. doi: 10.1038/nature14663
Flores, S., Echols, N., Milburn, D., Hespenheide, B., Keating, K., Lu, J., et al. (2006). The database of macromolecular motions: new features added at the decade mark. Nucleic Acids Res. 34, D296–D301. doi: 10.1093/nar/gkj046
Flory, P. J., Gordon, M., and McCrum, N. G. (1976). Statistical thermodynamics of random networks [and discussion]. Proc. R. Soc. A Math. Phys. Eng. Sci. 351, 351–380. doi: 10.1098/rspa.1976.0146
Formoso, E., Limongelli, V., and Parrinello, M. (2015). Energetics and structural characterization of the large-scale functional motion of Adenylate Kinase. Sci. Rep. 5:8425. doi: 10.1038/srep08425
Frank, J. (2018). New opportunities created by single-particle Cryo-EM: the mapping of conformational space. Biochemistry 57:888. doi: 10.1021/acs.biochem.8b00064
Franklin, J., Koehl, P., Doniach, S., and Delarue, M. (2007). MinActionPath: maximum likelihood trajectory for large-scale structural transitions in a coarse-grained locally harmonic energy landscape. Nucleic Acids Res. 35, W477–W482. doi: 10.1093/nar/gkm342
Frauenfelder, H., Sligar, S. G., and Wolynes, P. G. (1991). The energy landscapes and motions of proteins. Science 254, 1598–1603. doi: 10.1126/science.1749933
Gerstein, M., and Krebs, W. (1998). A database of macromolecular motions. Nucleic Acids Res. 26, 4280–90. doi: 10.1093/nar/26.18.4280
Go, N., and Scheraga, H. A. (1976). On the use of classical statistical mechanics in the treatment of polymer chain conformation. Macromolecules 9, 535–542. doi: 10.1021/ma60052a001
Granata, D., Camilloni, C., Vendruscolo, M., and Laio, A. (2013). Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc. Natl. Acad. Sci. U.S.A. 110, 6817–6822. doi: 10.1073/pnas.1218350110
Grazioso, G., Limongelli, V., Branduardi, D., Novellino, E., De Micheli, C., Cavalli, A., et al. (2012). Investigating the mechanism of substrate uptake and release in the glutamate transporter homologue Glt Ph through metadynamics simulations. J. Am. Chem. Soc. 134, 453–463. doi: 10.1021/ja208485w
Grubmüller, H. (1995). Predicting slow structural transitions in macromolecular systems: conformational flooding. Phys. Rev. E 52, 2893–2906. doi: 10.1103/PhysRevE.52.2893
Halder, K., Dölker, N., Van, Q., Gregor, I., Dickmanns, A., Baade, I., et al. (2015). MD simulations and FRET reveal an environment-sensitive conformational plasticity of importin-β. Biophys. J. 109, 277–286. doi: 10.1016/j.bpj.2015.06.014
Hamelberg, D., Mongan, J., and McCammon, J. A. (2004). Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 120, 11919–11929. doi: 10.1063/1.1755656
Harpole, T. J., and Delemotte, L. (2018). Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. Biochim. Biophys. Acta Biomembr. 1860, 909–926. doi: 10.1016/j.bbamem.2017.10.033
Haselbach, D., Komarov, I., Agafonov, D. E., Kastner, B., Haselbach, D., Komarov, I., et al. (2018). Structure and conformational dynamics of the human spliceosomal B act complex article structure and conformational dynamics of the human spliceosomal B act complex. Cell 172, 454–464. doi: 10.1016/j.cell.2018.01.010
Haselbach, D., Schrader, J., Lambrecht, F., Henneberg, F., Chari, A., and Stark, H. (2017). Long-range allosteric regulation of the human 26S proteasome by 20S proteasome-targeting cancer drugs. Nat. Commun. 8, 1–8. doi: 10.1038/ncomms15578
Haspel, N., Moll, M., Baker, M. L., Chiu, W., and Kavraki, L. E. (2010). Tracing conformational changes in proteins. BMC Struct. Biol. 10(Suppl. 1):S1. doi: 10.1186/1472-6807-10-S1-S1
Henzler-Wildman, K., and Kern, D. (2007). Dynamic personalities of proteins. Nature 450, 964–72. doi: 10.1038/nature06522
Heusser, S. A., Lycksell, M., Wang, X., McComas, S. E., Howard, R. J., and Lindahl, E. (2018). Allosteric potentiation of a ligand-gated ion channel is mediated by access to a deep membrane-facing cavity. Proc. Natl. Acad. Sci. U.S.A. 115, 10672–10677. doi: 10.1073/pnas.1809650115
Hofmann, S., Januliene, D., Mehdipour, A. R., Thomas, C., Stefan, E., Brüchert, S., et al. (2019). Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571, 580–583. doi: 10.1038/s41586-019-1391-0
Hollingsworth, S. A., and Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron 99, 1129–1143. doi: 10.1016/j.neuron.2018.08.011
Hruska, E., Abella, J. R., Nüske, F., Kavraki, L. E., and Clementi, C. (2018). Quantitative comparison of adaptive sampling methods for protein dynamics. J. Chem. Phys. 149:244119. doi: 10.1063/1.5053582
Huang, H., Ozkirimli, E., and Post, C. B. (2009). Comparison of three perturbation molecular dynamics methods for modeling conformational transitions. J. Chem. Theory Comput. 5, 1304–1314. doi: 10.1021/ct9000153
Huber, G. A., and Kim, S. (1996). Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys. J. 70:97–110. doi: 10.1016/S0006-3495(96)79552-8
Ingólfsson, H. I., Lopez, C. A., Uusitalo, J. J., de Jong, D. H., Gopal, S. M., Periole, X., et al. (2014). The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 225–248. doi: 10.1002/wcms.1169
Izrailev, S., Stepaniants, S., Balsera, M., Oono, Y., and Schulten, K. (1997). Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72, 1568–1581. doi: 10.1016/S0006-3495(97)78804-0
Jamroz, M., Kolinski, A., and Kmiecik, S. (2013). CABS-flex: server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 41:W427–W431. doi: 10.1093/nar/gkt332
Jensen, M., Jogini, V., Borhani, D. W., Leffler, A. E., Dror, R. O., and Shaw, D. E. (2012). Mechanism of voltage gating in potassium channels. Science 336, 229–233. doi: 10.1126/science.1216533
Jeschke, G. (2012). DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446. doi: 10.1146/annurev-physchem-032511-143716
Jiang, Y., and Kalodimos, C. G. (2017). NMR studies of large proteins. J. Mol. Biol. 429, 2667–2676. doi: 10.1016/j.jmb.2017.07.007
Kahsai, A. W., Rajagopal, S., Sun, J., and Xiao, K. (2014). Monitoring protein conformational changes and dynamics using stable-isotope labeling and mass spectrometry. Nat. Protoc. 9, 1301–1319. doi: 10.1038/nprot.2014.075
Kappel, K., Miao, Y., and McCammon, J. A. (2015). Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 48, 479–487. doi: 10.1017/S0033583515000153
Karplus, M., and McCammon, J. A. (2002). Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9, 646–652. doi: 10.1038/nsb0902-646
Kaszuba, K., Grzybek, M., Orłowski, A., Danne, R., Róg, T., Simons, K., et al. (2015). N-Glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc. Natl. Acad. Sci. U.S.A. 112, 4334–4339. doi: 10.1073/pnas.1503262112
Kendrew, J. C., Bodo, G., Dintzis, H. M., Parrish, R. G., Wyckoff, H., and Phillips, D. C. (1958). A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 181, 662–666. doi: 10.1038/181662a0
Khalili-Araghi, F., Ziervogel, B., Gumbart, J. C., and Roux, B. (2013). Molecular dynamics simulations of membrane proteins under asymmetric ionic concentrations. J. Gen. Physiol. 142, 465–475. doi: 10.1085/jgp.201311014
Kim, M. K., Jernigan, R. L., and Chirikjian, G. S. (2002). Efficient generation of feasible pathways for protein conformational transitions. Biophys. J. 83, 1620–30. doi: 10.1016/S0006-3495(02)73931-3
Kimanius, D., Pettersson, I., Schluckebier, G., Lindahl, E., and Andersson, M. (2015). SAXS-guided metadynamics. J. Chem. Theory Comput. 11, 3491–3498. doi: 10.1021/acs.jctc.5b00299
Kitao, A, and Go, N. (1999). Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 9, 164–9. doi: 10.1016/S0959-440X(99)80023-2
Kmiecik, S., Gront, D., Kolinski, M., Wieteska, L., Dawid, A. E., and Kolinski, A. (2016). Coarse-grained protein models and their applications. Chem. Rev. 116, 7898–7936. doi: 10.1021/acs.chemrev.6b00163
Knapp, B., Frantal, S., Cibena, M., Schreiner, W., and Bauer, P. (2011). Is an intuitive convergence definition of molecular dynamics simulations solely based on the root mean square deviation possible? J. Comput. Biol. 18, 997–1005. doi: 10.1089/cmb.2010.0237
Kohlhoff, K. J., Shukla, D., Lawrenz, M., Bowman, G. R., Konerding, D. E., Belov, D., et al. (2014). Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 6, 15–21. doi: 10.1038/nchem.1821
Krebs, W. G., and Gerstein, M. (2000). The morph server: a standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 28, 1665–1675. doi: 10.1093/nar/28.8.1665
Krüger, D. M., Ahmed, A., and Gohlke, H. (2012). NMSim web server: integrated approach for normal mode-based geometric simulations of biologically relevant conformational transitions in proteins. Nucleic Acids Res. 40, 310–316. doi: 10.1093/nar/gks478
Kutzner, C., Páll, S., Fechner, M., Esztermann, A., De Groot, B. L., and Grubmüller, H. (2015). Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem. 36, 1990–2008. doi: 10.1002/jcc.24030
Laine, E., Goncalves, C., Karst, J. C., Lesnard, A., Rault, S., Tang, W. J., et al. (2010). Use of allostery to identify inhibitors of calmodulin-induced activation of Bacillus anthracis edema factor. Proc. Natl. Acad. Sci. U.S.A. 107, 11277–11282. doi: 10.1073/pnas.0914611107
Laio, A., and Gervasio, F. L. (2008). Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 71:126601. doi: 10.1088/0034-4885/71/12/126601
Laio, A., and Parrinello, M. (2002). Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A. 99, 12562–12566. doi: 10.1073/pnas.202427399
Latorraca, N. R., Fastman, N. M., Venkatakrishnan, A. J., Frommer, W. B., Dror, R. O., and Feng, L. (2017). Mechanism of substrate translocation in an alternating access transporter. Cell 169, 96–107.e12. doi: 10.1016/j.cell.2017.03.010
Latorraca, N. R., Wang, J. K., Bauer, B., Townshend, R. J. L., Hollingsworth, S. A., Olivieri, J. E., et al. (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. doi: 10.1038/s41586-018-0077-3
Lelimousin, M., Limongelli, V., and Sansom, M. S. P. (2016). Conformational changes in the epidermal growth factor receptor: role of the transmembrane domain investigated by coarse-grained metadynamics free energy calculations. J. Am. Chem. Soc. 138, 10611–10622. doi: 10.1021/jacs.6b05602
Levantino, M., Yorke, B. A., Monteiro, D. C., Cammarata, M., and Pearson, A. R. (2015). Using synchrotrons and XFELs for time-resolved X-ray crystallography and solution scattering experiments on biomolecules. Curr. Opin. Struct. Biol. 35, 41–8. doi: 10.1016/j.sbi.2015.07.017
Levitt, M., Hirshberg, M., Sharon, R., and Daggett, V. (1995). Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput. Phys. Commun. 91, 215–231. doi: 10.1016/0010-4655(95)00049-L
Levitt, M., Sander, C., and Stern, P. S. (1985). Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 181, 423–447. doi: 10.1016/0022-2836(85)90230-X
Lewis, J. H., and Lu, Z. (2019). Integrating spatiotemporal features of a ligand-regulated, multi-state allosteric protein. Nat. Struct. Mol. Biol. 26, 816–822. doi: 10.1038/s41594-019-0276-0
Limongelli, V., Bonomi, M., Marinelli, L., Gervasio, F. L., Cavalli, A., Novellino, E., et al. (2010). Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 5411–5416. doi: 10.1073/pnas.0913377107
Limongelli, V., Marinelli, L., Cosconati, S., La Motta, C., Sartini, S., Mugnaini, L., et al. (2012). Sampling protein motion and solvent effect during ligand binding. Proc. Natl. Acad. Sci. U.S.A. 109, 1467–1472. doi: 10.1073/pnas.1112181108
Lindahl, E., Azuara, C., Koehl, P., and Delarue, M. (2006). NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 34, W52–W56. doi: 10.1093/nar/gkl082
Lindahl, V., Gourdon, P., Andersson, M., and Hess, B. (2018). Permeability and ammonia selectivity in aquaporin TIP2;1: linking structure to function. Sci. Rep. 8:2995. doi: 10.1038/s41598-018-21357-2
Lindahl, V., Lidmar, J., and Hess, B. (2014). Accelerated weight histogram method for exploring free energy landscapes. J. Chem. Phys. 141:044110. doi: 10.1063/1.4890371
Lindorff-Larsen, K., Maragakis, P., Piana, S., Eastwood, M. P., Dror, R. O., and Shaw, D. E. (2012). Systematic validation of protein force fields against experimental data. PLoS ONE 7:e32131. doi: 10.1371/journal.pone.0032131
López-Blanco, J. R., Aliaga, J. I., Quintana-Ortí, E. S., and Chacón, P. (2014). iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 42, W271–W276. doi: 10.1093/nar/gku339
Lowe, D. (2015). Odd structures, subjected to powerful computations. Sci. Transl. Med. In the pipeline. Available online at: https://blogs.sciencemag.org/pipeline/archives/2015/01/13/odd_structures_subjected_to_powerful_computations
Ma, J., Benitez, J. A., Li, J., Miki, S., Ponte de Albuquerque, C., Galatro, T., et al. (2019). Inhibition of nuclear PTEN tyrosine phosphorylation enhances glioma radiation sensitivity through attenuated DNA repair. Cancer Cell 35, 504–518.e7. doi: 10.1016/j.ccell.2019.04.011
Ma, J., and Karplus, M. (2002). Molecular switch in signal transduction: reaction paths of the conformational changes in ras p21. Proc. Natl. Acad. Sci. U.S.A. 94, 11905–11910. doi: 10.1073/pnas.94.22.11905
Machtens, J. P., Kortzak, D., Lansche, C., Leinenweber, A., Kilian, P., Begemann, B., et al. (2015). Mechanisms of anion conduction by coupled glutamate transporters. Cell 160, 542–553. doi: 10.1016/j.cell.2014.12.035
Mahajan, S., and Sanejouand, Y. H. (2015). On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys. 567, 59–65. doi: 10.1016/j.abb.2014.12.020
Maragakis, P., Andreev, S. A., Brumer, Y., Reichman, D. R., and Kaxiras, E. (2002). Adaptive nudged elastic band approach for transition state calculation. J. Chem. Phys. 117, 4651–4658. doi: 10.1063/1.1495401
Maragakis, P., and Karplus, M. (2005). Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J. Mol. Biol. 352, 807–822. doi: 10.1016/j.jmb.2005.07.031
Maragliano, L., Roux, B., and Vanden-Eijnden, E. (2014). Comparison between mean forces and swarms-of-trajectories string methods. J. Chem. Theory Comput. 10, 524–533. doi: 10.1021/ct400606c
Maragliano, L., and Vanden-Eijnden, E. (2006). A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 426, 168–175. doi: 10.1016/j.cplett.2006.05.062
Marchi, M., and Ballone, P. (1999). Adiabatic bias molecular dynamics: a method to navigate the conformational space of complex molecular systems. J. Chem. Phys. 110:3697. doi: 10.1063/1.478259
Marrink, S. J., and Tieleman, D. P. (2013). Perspective on the Martini model. Chem. Soc. Rev. 42, 6801–6822. doi: 10.1039/c3cs60093a
Martínez-Rosell, G., Giorgino, T., Harvey, M. J., and de Fabritiis, G. (2017). Drug discovery and molecular dynamics: methods, applications and perspective beyond the second timescale. Curr. Top. Med. Chem. 17, 2617–2625. doi: 10.2174/1568026617666170414142549
Maximova, T., Moffatt, R., Ma, B., Nussinov, R., and Shehu, A. (2016). Principles and overview of sampling methods for modeling macromolecular structure and dynamics. PLoS Comput. Biol. 12, 1–70. doi: 10.1371/journal.pcbi.1004619
McCammon, J. A., Gelin, B. R., and Karplus, M. (1977). Dynamics of folded proteins. Nature 267, 585–590. doi: 10.1038/267585a0
Monticelli, L., and Tieleman, D. P. (2013). Force fields for classical molecular dynamics. Methods Mol. Biol. 924, 197–213. doi: 10.1007/978-1-62703-017-5_8
Morcos, F., Jana, B., Hwa, T., and Onuchic, J. N. (2013). Coevolutionary signals across protein lineages help capture multiple protein conformations. Proc. Natl. Acad. Sci. U.S.A. 110, 20533–20538. doi: 10.1073/pnas.1315625110
Mori, T., Miyashita, N., Im, W., Feig, M., and Sugita, Y. (2016). Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Biophys. Acta Biomembr. 1858, 1635–1651. doi: 10.1016/j.bbamem.2015.12.032
Morra, G., Potestio, R., Micheletti, C., and Colombo, G. (2012). Corresponding functional dynamics across the Hsp90 chaperone family: Insights from a multiscale analysis of MD simulations. PLoS Comput. Biol. 8:e1002433. doi: 10.1371/journal.pcbi.1002433
Murata, K., and Wolf, M. (2018). Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta Gen. Subj. 1862, 324–334. doi: 10.1016/j.bbagen.2017.07.020
Negre, C. F. A., Morzan, U. N., Hendrickson, H. P., Pal, R., Lisi, G. P., Loria, J. P., et al. (2018). Eigenvector centrality for characterization of protein allosteric pathways. Proc. Natl. Acad. Sci. U.S.A. 115, E12201–E12208. doi: 10.1073/pnas.1810452115
Neutze, R., Brändén, G., and Schertler, G. F. X. (2015). Membrane protein structural biology using X-ray free electron lasers. Curr. Opin. Struct. Biol. 33, 115–125. doi: 10.1016/j.sbi.2015.08.006
Noé, F., and Clementi, C. (2017). Collective variables for the study of long-time kinetics from molecular trajectories: theory and methods. Curr. Opin. Struct. Biol. 43, 141–417. doi: 10.1016/j.sbi.2017.02.006
Nogales, E., and Scheres, S. H. W. (2015). Cryo-EM: a unique tool for the visualization of macromolecular complexity. Mol. Cell 58, 677–689. doi: 10.1016/j.molcel.2015.02.019
Nury, H., Poitevin, F., Van Renterghem, C., Changeux, J.-P., Corringer, P.-J., Delarue, M., et al. (2010). One-microsecond molecular dynamics simulation of channel gating in a nicotinic receptor homologue. Proc. Natl. Acad. Sci. U.S.A. 107, 6275–80. doi: 10.1073/pnas.1001832107
Nussinov, R., and Wolynes, P. G. (2014). A second molecular biology revolution? The energy landscapes of biomolecular function. Phys. Chem. Chem. Phys. 16, 6321–6322. doi: 10.1039/c4cp90027h
Okamoto, K., and Sako, Y. (2017). Recent advances in FRET for the study of protein interactions and dynamics. Curr. Opin. Struct. Biol. 46, 16–23. doi: 10.1016/j.sbi.2017.03.010
Opella, S. J., and Marassi, F. M. (2017). Applications of NMR to membrane proteins. Arch. Biochem. Biophys. 628, 92–101. doi: 10.1016/j.abb.2017.05.011
Orellana, L. (2019). Convergence of EGFR glioblastoma mutations: evolution and allostery rationalizing targeted therapy. Mol. Cell. Oncol. 6:e1630798. doi: 10.1080/23723556.2019.1630798
Orellana, L., Gustavsson, J., Bergh, C., Yoluk, O., and Lindahl, E. (2019a). eBDIMS server: protein transition pathways with ensemble analysis in 2D-motion spaces. Bioinformatics. 35, 3505–3507. doi: 10.1093/bioinformatics/btz104
Orellana, L., Hospital, A., and Orozco, M. (2014). Oncogenic mutations of the EGF-Receptor ectodomain reveal an unexpected mechanism for ligand-independent activation. bioRxiv 009068. doi: 10.1101/009068
Orellana, L., Rueda, M., Ferrer-Costa, C., Lopez-Blanco, J. R., Chacón, P., and Orozco, M. (2010). Approaching elastic network models to molecular dynamics flexibility. J. Chem. Theory Comput. 6, 2910–2923. doi: 10.1021/ct100208e
Orellana, L., Thorne, A. H., Lema, R., Gustavsson, J., Parisian, A. D., Hospital, A., et al. (2019b). Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. U.S.A. 116, 10009–10018. doi: 10.1073/pnas.1821442116
Orellana, L., Yoluk, O., Carrillo, O., Orozco, M., and Lindahl, E. (2016). Prediction and validation of protein intermediate states from structurally rich ensembles and coarse-grained simulations. Nat. Commun. 7:12575. doi: 10.1038/ncomms12575
Orozco, M. (2014). A theoretical view of protein dynamics. Chem. Soc. Rev. 43, 5051–5066. doi: 10.1039/C3CS60474H
Orozco, M., Orellana, L., Hospital, A., Naganathan, A. N., Emperador, A., Carrillo, O., et al. (2011). Coarse-grained Representation of Protein Flexibility. Foundations, Successes, and Shortcomings. Adv. Protein Chem. Struct. Biol. 85, 183–215. doi: 10.1016/B978-0-12-386485-7.00005-3
Ostermeir, K., and Zacharias, M. (2013). Advanced replica-exchange sampling to study the flexibility and plasticity of peptides and proteins. Biochim. Biophys. Acta 1834, 847–853. doi: 10.1016/j.bbapap.2012.12.016
Ourmazd, A. (2019). Cryo-EM, XFELs and the structure conundrum in structural biology. Nat. Methods. 16, 941–944. doi: 10.1038/s41592-019-0587-4
Ovchinnikov, V., and Karplus, M. (2012). Analysis and elimination of a bias in targeted molecular dynamics simulations of conformational transitions: application to calmodulin. J. Phys. Chem. B. 116, 8584–8603. doi: 10.1021/jp212634z
Ovchinnikov, V., Karplus, M., and Vanden-Eijnden, E. (2011). Free energy of conformational transition paths in biomolecules: the string method and its application to myosin VI. J. Chem. Phys. 134:085103. doi: 10.1063/1.3544209
Paci, E., and Karplus, M. (1999). Forced unfolding of fibronectin type 3 modules: An analysis by biased molecular dynamics simulations. J. Mol. Biol. 288, 441–459. doi: 10.1006/jmbi.1999.2670
Pal, N., Wu, M., and Lu, H. P. (2016). Probing conformational dynamics of an enzymatic active site by an in situ single fluorogenic probe under piconewton force manipulation. Proc. Natl. Acad. Sci. 113, 15006–15011. doi: 10.1073/pnas.1613404114
Páll, S., Abraham, M. J., Kutzner, C., Hess, B., and Lindahl, E. (2015). “Tackling exascale software challenges in molecular dynamics simulations with GROMACS,” in Solving Software Challenges for Exascale. EASC 2014. Lecture Notes in Computer Science, Vol. 8759, eds S. Markidis and E. Laure (Cham: Springer).
Pan, A. C., Sezer, D., and Roux, B. (2008). Finding transition pathways using the string method with swarms of trajectories. J. Phys. Chem. B 112, 3432–3440. doi: 10.1021/jp0777059
Pan, A. C., Weinreich, T. M., Piana, S., and Shaw, D. E. (2016). Demonstrating an order-of-magnitude sampling enhancement in molecular dynamics simulations of complex protein systems. J. Chem. Theory Comput. 12, 1360–1367. doi: 10.1021/acs.jctc.5b00913
Pan, A. C., Weinreich, T. M., Shan, Y., Scarpazza, D. P., and Shaw, D. E. (2014). Assessing the accuracy of two enhanced sampling methods using egfr kinase transition pathways: the influence of collective variable choice. J. Chem. Theory Comput. 10, 2860–2865.doi: 10.1021/ct500223p
Pande, V. S., Beauchamp, K., and Bowman, G. R. (2010). Everything you wanted to know about Markov State Models but were afraid to ask. Methods 52, 99–105. doi: 10.1016/j.ymeth.2010.06.002
Papaleo, E. (2015). Integrating atomistic molecular dynamics simulations, experiments, and network analysis to study protein dynamics: strength in unity. Front. Mol. Biosci. 2:28. doi: 10.3389/fmolb.2015.00028
Perilla, J. R., Beckstein, O., Denning, E. J., and Woolf, T. B. (2011). Computing ensembles of transitions from stable states: dynamic importance sampling. J. Comput. Chem. 32, 196–209. doi: 10.1002/jcc.21564
Pfaendtner, J., Branduardi, D., Parrinello, M., Pollard, T. D., and Voth, G. A. (2009). Nucleotide-dependent conformational states of actin. Proc. Natl. Acad. Sci. U.S.A. 106, 12723–12728. doi: 10.1073/pnas.0902092106
Piana, S., and Laio, A. (2007). A bias-exchange approach to protein folding. J. Phys. Chem. B 111, 4553–4559. doi: 10.1021/jp067873l
Pierce, L. C. T., Salomon-Ferrer, R., Augusto F. De Oliveira, C., McCammon, J. A., and Walker, R. C. (2012). Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 8, 2997–3002. doi: 10.1021/ct300284c
Pietrucci, F. (2017). Strategies for the exploration of free energy landscapes: unity in diversity and challenges ahead. Rev. Phys. 2, 32–45. doi: 10.1016/j.revip.2017.05.001
Potestio, R., Pontiggia, F., and Micheletti, C. (2009). Coarse-grained description of protein internal dynamics: an optimal strategy for decomposing proteins in rigid subunits. Biophys. J. 96, 4993–5002. doi: 10.1016/j.bpj.2009.03.051
Pouya, I., Pronk, S., Lundborg, M., and Lindahl, E. (2017). Copernicus: a hybrid dataflow and peer-to-peer scientific computing platform for efficient large-scale ensemble sampling. Future Gen. Comput. Sys. 71, 18–31. doi: 10.1016/j.future.2016.11.004
Pronk, S., Larsson, P., Pouya, I., Bowman, G. R., Haque, I. S., Beauchamp, K., et al. (2011). “Copernicus: a new paradigm for parallel adaptive molecular dynamics,” in Proceedings of 2011 SC - International Conference for High Performance Computing, Networking, Storage and Analysis. doi: 10.1145/2063384.2063465
Ren, W., and Vanden-Eijnden, E. (2005). Finite temperature string method for the study of rare events. J. Phys. Chem. B 109, 6688–6693. doi: 10.1021/jp0455430
Ren, W., Vanden-Eijnden, E., Maragakis, P., and E, W. (2005). Transition pathways in complex systems: Application of the finite-temperature string method to the alanine dipeptide. J. Chem. Phys. 123:134109. doi: 10.1063/1.2013256
Rivalta, I., Sultan, M. M., Lee, N. S., Manley, G. A., Loria, J. P., and Batista, V. S. (2012). Allosteric pathways in imidazole glycerol phosphate synthase. Proc. Natl. Acad. Sci. U.S.A. 109, E1428–E1436. doi: 10.1073/pnas.1120536109
Röder, K., Joseph, J. A., Husic, B. E., and Wales, D. J. (2019). Energy landscapes for proteins: from single funnels to multifunctional systems. Adv. Theory Simulat. 2:1800175. doi: 10.1002/adts.201800175
Rueda, M., Chacón, P., and Orozco, M. (2007). Thorough validation of protein normal mode analysis: a comparative study with essential dynamics. Structure 15, 565–75. doi: 10.1016/j.str.2007.03.013
Sankar, K., Liu, J., Wang, Y., and Jernigan, R. L. (2015). Distributions of experimental protein structures on coarse-grained free energy landscapes. J. Chem. Phys. 143:243153. doi: 10.1063/1.4937940
Sankar, K., Mishra, S. K., and Jernigan, R. L. (2018). Comparisons of protein dynamics from experimental structure ensembles, molecular dynamics ensembles, and coarse-grained elastic network models. J. Phys. Chem. B 122, 5409–5417. doi: 10.1021/acs.jpcb.7b11668
Saunders, M. G., and Voth, G. A. (2013). Coarse-graining methods for computational biology. Annu. Rev. Biophys. 42, 73–93. doi: 10.1146/annurev-biophys-083012-130348
Sawle, L., and Ghosh, K. (2016). Convergence of molecular dynamics simulation of protein native states : feasibility vs self-consistency dilemma. J. Chem. Theory Comput. 12, 861–869. doi: 10.1021/acs.jctc.5b00999
Schlitter, J., Engels, M., and Kruger, P. (1994). Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 12, 84–89. doi: 10.1016/0263-7855(94)80072-3
Seo, S., and Kim, M. K. (2012). KOSMOS: a universal morph server for nucleic acids, proteins and their complexes. Nucleic Acids Res. 40, W531–W536. doi: 10.1093/nar/gks525
Seyler, S. L., and Beckstein, O. (2014). Sampling large conformational transitions: adenylate kinase as a testing ground. Mol. Simul. 40, 1–23. doi: 10.1080/08927022.2014.919497
Seyler, S. L., Kumar, A., Thorpe, M. F., and Beckstein, O. (2015). Path similarity analysis: a method for quantifying macromolecular pathways. PLoS Comput. Biol. 11:e1004568. doi: 10.1371/journal.pcbi.1004568
Sfriso, P., Duran-Frigola, M., Mosca, R., Emperador, A., Aloy, P., and Orozco, M. (2016). Residues coevolution guides the systematic identification of alternative functional conformations in proteins. Structure. 24, 116–126. doi: 10.1016/j.str.2015.10.025
Sfriso, P., Emperador, A., Orellana, L., Hospital, A., and Orozco, M. (2012). Finding conformational transition pathways from discrete 2 molecular dynamics simulations. J. Chem. Theory Comput. 8, 4707–4718. doi: 10.1021/ct300494q
Sfriso, P., Hospital, A., Emperador, A., and Orozco, M. (2013). Exploration of conformational transition pathways from coarse-grained simulations. Bioinformatics 29, 1980–1986. doi: 10.1093/bioinformatics/btt324
Shan, Y., Eastwood, M. P., Zhang, X., Kim, E. T., Arkhipov, A., Dror, R. O., et al. (2012). Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 149, 860–870. doi: 10.1016/j.cell.2012.02.063
Shan, Y., Gnanasambandan, K., Ungureanu, D., Kim, E. T., Hammarén, H., Yamashita, K., et al. (2014). Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol. 21, 579–584. doi: 10.1038/nsmb.2849
Shan, Y., Kim, E. T., Eastwood, M. P., Dror, R. O., Seeliger, M. a, and Shaw, D. E. (2011). How does a drug molecule find its target binding site? J. Chem. Inf. Model. 24, 1981–1983. doi: 10.1021/ja202726y
Shaw, D. E., Bowers, K. J., Chow, E., Eastwood, M. P., Ierardi, D. J., Klepeis, J. L., et al. (2009). “Millisecond-scale molecular dynamics simulations on Anton,” in SC '09 Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis (Portland, OR). doi: 10.1145/1654059.1654126
Shoemaker, S. C., and Ando, N. (2018). X-rays in the cryo-electron microscopy era: structural biology's dynamic future. Biochemistry 57, 277–285. doi: 10.1021/acs.biochem.7b01031
Shukla, D., Lawrenz, M., and Pande, V. S. (2015). Elucidating ligand-modulated conformational landscape of GPCRs using cloud-computing approaches. Methods Enzymol. 557, 551–572. doi: 10.1016/bs.mie.2014.12.007
Smolin, N., and Robia, S. L. (2015). A structural mechanism for calcium transporter headpiece closure. J. Phys. Chem. B 119, 1407–1415. doi: 10.1021/jp511433v
Sugita, Y., and Okamoto, Y. (1999). Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. doi: 10.1016/S0009-2614(99)01123-9
Sultan, M. M., and Pande, V. S. (2017). TICA-metadynamics: accelerating metadynamics by using kinetically selected collective variables. J. Chem. Theory Comput. 13, 2440–2447. doi: 10.1021/acs.jctc.7b00182
Sutto, L., Marsili, S., Valencia, A., and Gervasio, F. L. (2015). From residue coevolution to protein conformational ensembles and functional dynamics. Proc. Natl. Acad. Sci. U. S. A. 112, 13567–13572. doi: 10.1073/pnas.1508584112
Sweet, J. C., Nowling, R. J., Cickovski, T., Sweet, C. R., Pande, V. S., and Izaguirre, J. A. (2013). Long timestep molecular dynamics on the graphical processing unit. J. Chem. Theory Comput. 9, 3267–3281. doi: 10.1021/ct400331r
Tama, F., and Sanejouand, Y. H. (2001). Conformational change of proteins arising from normal mode calculations. Protein Eng. 14, 1–6. doi: 10.1093/protein/14.1.1
Tao, P., Hodošček, M., Larkin, J. D., Shao, Y., and Brooks, B. R. (2012). Comparison of three chain-of-states methods: Nudged elastic band and replica path with restraints or constraints. J. Chem. Theory Comput. 8, 5035–5051. doi: 10.1021/ct3006248
Tekpinar, M., and Zheng, W. (2010). Predicting order of conformational changes during protein conformational transitions using an interpolated elastic network model. Proteins Struct. Funct. Bioinforma. 78, 2469–2481. doi: 10.1002/prot.22755
Tirion, M. (1996). Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77, 1905–1908. doi: 10.1103/PhysRevLett.77.1905
Torrie, G. M., and Valleau, J. P. (1977). Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23, 187–199. doi: 10.1016/0021-9991(77)90121-8
Tozzini, V. (2010). Multiscale modeling of proteins. Acc. Chem. Res. 43, 220–230. doi: 10.1021/ar9001476
Ung, P. M. U., Rahman, R., and Schlessinger, A. (2018). Redefining the protein kinase conformational space with machine learning. Cell Chem. Biol. 25, 916–924.e2 doi: 10.1016/j.chembiol.2018.05.002
van Gunsteren, W. F., Daura, X., Hansen, N., Mark, A. E., Oostenbrink, C., Riniker, S., et al. (2018). Validation of molecular simulation: an overview of issues. Angew. Chemie Int. Ed. 57, 884–902. doi: 10.1002/anie.201702945
Vestergaard, B. (2016). Analysis of biostructural changes, dynamics, and interactions - Small-angle X-ray scattering to the rescue. Arch. Biochem. Biophys. 602, 69–79. doi: 10.1016/j.abb.2016.02.029
G. A.. Voth (ed.). (2009). Coarse-Graining of Condensed Phase and Biomolecular Systems. Boca Raton, FL: CRC Press Taylor and Francis. doi: 10.1201/9781420059564
Wales, T. E., and Engen, J. R. (2006). Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170. doi: 10.1002/mas.20064
Weiss, D. R., and Koehl, P. (2014). Morphing methods to visualize coarse-grained protein dynamics. Methods Mol. Biol. 1084, 271–282. doi: 10.1007/978-1-62703-658-0_15
Weiss, D. R., and Levitt, M. (2009). Can morphing methods predict intermediate structures? J. Mol. Biol. 385, 665–674. doi: 10.1016/j.jmb.2008.10.064
Wells, S., Menor, S., Hespenheide, B., and Thorpe, M. F. (2005). Constrained geometric simulation of diffusive motion in proteins. Phys. Biol. 2, S127–S136.doi: 10.1088/1478-3975/2/4/S07
Wittig, I., Braun, H.-P., and Schagger, H. (2006). Blue native PAGE. Nat. Protoc. 1, 418–428. doi: 10.1038/nprot.2006.62
Woo, H. J., Jiang, J., Lafer, E. M., and Sousa, R. (2009). ATP-induced conformational changes in Hsp70: molecular dynamics and experimental validation of an in silico predicted conformation. Biochemistry 48, 11470–11477. doi: 10.1021/bi901256y
Wright, P. E., and Dyson, H. J. (2015). Intrinsically disordered proteins in cellular signaling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29. doi: 10.1038/nrm3920
Yang, Z., Májek, P., and Bahar, I. (2009). Allosteric transitions of supramolecular systems explored by network models: application to chaperonin GroEL. PLoS Comput. Biol. 5:e1000360. doi: 10.1371/journal.pcbi.1000360
Ye, Y., and Godzik, A. (2004). FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 32, W582–W585. doi: 10.1093/nar/gkh430
Yu, I., Mori, T., Ando, T., Harada, R., Jung, J., Sugita, Y., et al. (2016). Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 5:e19274. doi: 10.7554/eLife.19274
Zheng, W., and Wen, H. (2017). A survey of coarse-grained methods for modeling protein conformational transitions. Curr. Opin. Struct. Biol. 42, 24–30. doi: 10.1016/j.sbi.2016.10.008
Zhuravlev, P. I., and Papoian, G. A. (2010a). Functional versus folding landscapes: the same yet different. Curr. Opin. Struct. Biol. 20, 16–22. doi: 10.1016/j.sbi.2009.12.010
Zhuravlev, P. I., and Papoian, G. A. (2010b). Protein functional landscapes, dynamics, allostery: a tortuous path towards a universal theoretical framework. Q. Rev. Biophys. 43, 295–332. doi: 10.1017/S0033583510000119
Zimmerman, M. I., Porter, J. R., Sun, X., Silva, R. R., and Bowman, G. R. (2018). Choice of adaptive sampling strategy impacts state discovery, transition probabilities, and the apparent mechanism of conformational changes. J. Chem. Theory Comput. 14, 5459–5475. doi: 10.1021/acs.jctc.8b00500
Zuckerman, D. M., and Chong, L. T. (2017). Weighted ensemble simulation: review of methodology, applications, and software. Annu. Rev. Biophys. 46, 43–57. doi: 10.1146/annurev-biophys-070816-033834
Keywords: conformational change, proteins, molecular dynamics simulation, coarse-grained (CG) methods, structural ensemble
Citation: Orellana L (2019) Large-Scale Conformational Changes and Protein Function: Breaking the in silico Barrier. Front. Mol. Biosci. 6:117. doi: 10.3389/fmolb.2019.00117
Received: 20 July 2019; Accepted: 14 October 2019;
Published: 05 November 2019.
Edited by:
Alexandre M. J. J. Bonvin, Utrecht University, NetherlandsReviewed by:
Vittorio Limongelli, University of Lugano, SwitzerlandElodie Laine, Université Pierre et Marie Curie, France
Copyright © 2019 Orellana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Orellana, bGF1cmEub3JlbGxhbmEmI3gwMDA0MDtzY2lsaWZlbGFiLnNl; ZG9ibGUuaGVsaXgmI3gwMDA0MDtnbWFpbC5jb20=