 Suzan Kors
Suzan Kors Karlijne Geijtenbeek
Karlijne Geijtenbeek Eric Reits
Eric Reits Sabine Schipper-Krom
Sabine Schipper-Krom- Department of Medical Biology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
Intracellular protein synthesis, folding, and degradation are tightly controlled processes to ensure proper protein homeostasis. The proteasome is responsible for the degradation of the majority of intracellular proteins, which are often targeted for degradation via polyubiquitination. However, the degradation rate of proteins is also affected by the capacity of proteasomes to recognize and degrade these substrate proteins. This capacity is regulated by a variety of proteasome modulations including (1) changes in complex composition, (2) post-translational modifications, and (3) altered transcription of proteasomal subunits and activators. Various diseases are linked to proteasome modulation and altered proteasome function. A better understanding of these modulations may offer new perspectives for therapeutic intervention. Here we present an overview of these three proteasome modulating mechanisms to give better insight into the diversity of proteasomes.
Introduction
Protein degradation by proteasomes plays a major role in the regulation of a wide range of basic cellular processes (Rock et al., 1994). Therefore, it is not surprising that aberrations in this pathway have been linked to several diseases. Some diseases are due to the increased lifetime of disease-related proteins, whereas others are caused by accelerated protein degradation (Ciechanover and Schwartz, 2004; Hanna et al., 2019). This altered degradation capacity by the proteasome can be caused by a change in the expression of proteasome subunits or by an aberrant proteasome composition (Ciechanover and Schwartz, 2004; Dahlmann et al., 2007). Processes to enhance proteasome activity and induce expression of proteasome(-related) components have been implicated in several cancers and muscle wasting condition (Chen and Madura, 2005; Dahlmann et al., 2007; Klaude et al., 2007; Cohen et al., 2015; Zhang et al., 2015; Chen et al., 2017). In contrast, neurodegenerative disorders and cardiac dysfunction have been related to accumulation of proteins and/or decreased proteasome activity (Keller et al., 2000; Tsukamoto et al., 2006; Dahlmann et al., 2007; Dantuma and Bott, 2014; Gilda and Gomes, 2017). This emphasizes the importance of properly functioning proteasomes and the relevance for therapeutic interference. The use of proteasome inhibitors in cancer treatment is a well-known example of using the proteasome as a therapeutic target (Orlowski and Kuhn, 2008; Schlafer et al., 2017), which raises the question whether intervention in the proteasome system would also be beneficial in other diseases (Njomen and Tepe, 2019).
In order to cope with particular stress conditions, cells have their own mechanisms to inhibit and activate the proteasome. These proteasome modulations include (1) changes in the composition of proteasome complexes, (2) post-translational modifications (PTMs), or (3) alterations at the transcriptional level (Figure 1). A better understanding of these diverse endogenous modulations of the proteasome may give more insight into new possibilities for therapeutic interventions. Here we review various mechanisms used by cells to modify proteasome abundance, composition, and consequently activity.
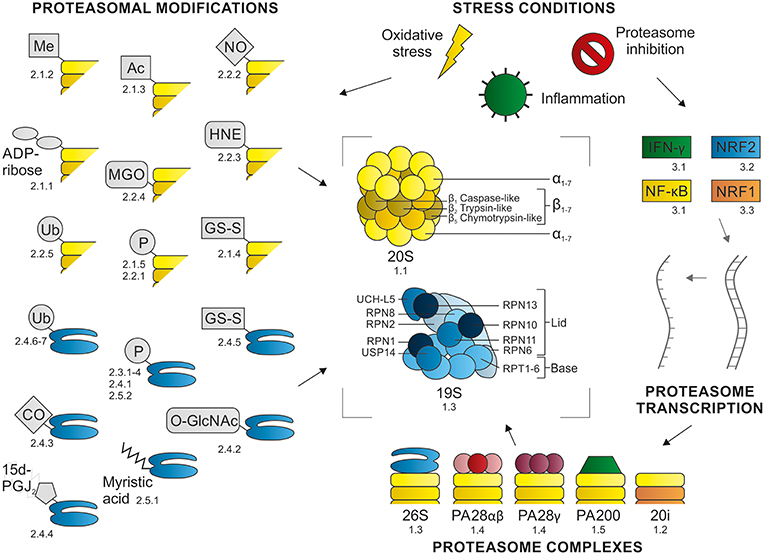
Figure 1. Proteasome modulations. Proteasome function can be modulated at different levels: (1) changes in proteasome complex composition, (2) post-translational modifications, and (3) alterations at transcriptional level. These modulation are induced in response to specific (stress) conditions. This figure summarizes the content of this review, with the numbers referring to the section where the modulation is described.
1. Modulating Proteasome Activity by Changing Proteasome Composition
Proteasomes are multicatalytic complexes containing a cylindrical 20S core, which is composed of four heteroheptameric rings (Harshbarger et al., 2015). The two inner β-rings contain the six proteolytic sites where substrates are cleaved; each ring has caspase-like (C-L), trypsin-like (T-L), and chymotrypsin-like (ChT-L) activity (Figure 1). The two outer rings consist of α-subunits, which act as gatekeepers, controlling the accessibility of substrates into the catalytically active β-chamber (Kisselev et al., 2002). Proteasomes are not static complexes and the activity of the proteasome can be modulated by the binding of various proteasome activators (PAs): 19S, PA28, and PA200 (Mao et al., 2008; Savulescu and Glickman, 2011; Liu and Jacobson, 2013; Cascio, 2014). These regulators can bind symmetrically and asymmetrically to the α-rings of the 20S core, forming single or double capped proteasomes. However, the free 20S proteasome unit remains a very abundant conformation in cells (Fabre et al., 2014).
The 19S regulatory particle is the main PA, forming the 19S-20S (26S) proteasome complex (Fabre et al., 2014). This cap is essential in the ubiquitin-proteasome system (UPS); this pathway is responsible for the degradation of misfolded as well as short-lived regulatory proteins such as cell cycle regulators and transcriptional activators (Glickman and Ciechanover, 2002). Though, when mimicking starvation in cell culture via mTOR inhibition, the majority of long-lived proteins is also degraded via the UPS (Zhao et al., 2015). Folded proteins destined for degradation by the UPS are tagged by a polyubiquitin chain (Liu C. W. et al., 2006). After substrate binding to the proteasome, the 19S regulatory particle deubiquitinates, translocates and unfolds the substrate protein in an ATP-dependent manner, so that it can be degraded by the 20S core (Navon and Goldberg, 2001; Liu C. W. et al., 2006; Liu and Jacobson, 2013; Collins and Goldberg, 2017). Alternatively, the 20S proteasome can bind to PA28αβ, PA28γ, and PA200 (Rechsteiner and Hill, 2005); these PAs open the 20S core but lack deubiquitinating enzymes (DUBs) and ATPase activity. In addition, alternative forms of the 20S proteasome exist. The proteolytic active β-subunits (β1, β2, and β5) of the 20S proteasome can all or partly be replaced by so-called immunosubunits (β1i, β2i, and β5i), resulting in three 20S proteasome subpopulations: standard (or constitutive), immuno and intermediate proteasomes (Dahlmann, 2016). Finally, cell-type specific proteasome subpopulations have also been identified: thymoproteasomes (β5t) and spermatoproteasomes (α4s) which vary in catalytic activity or preference for specific PAs, respectively (Murata et al., 2007; Florea et al., 2010; Qian et al., 2013; Kniepert and Groettrup, 2014).
These various proteasome compositions can change as a consequence of various stimuli and diseases (Mishto et al., 2006; Zheng et al., 2012; Ali et al., 2013), thereby affecting their substrate specificity and the protein homeostasis in cells. In this section we give an overview of various proteasome complexes and the consequences on proteasome activity (summarized in Table 1).
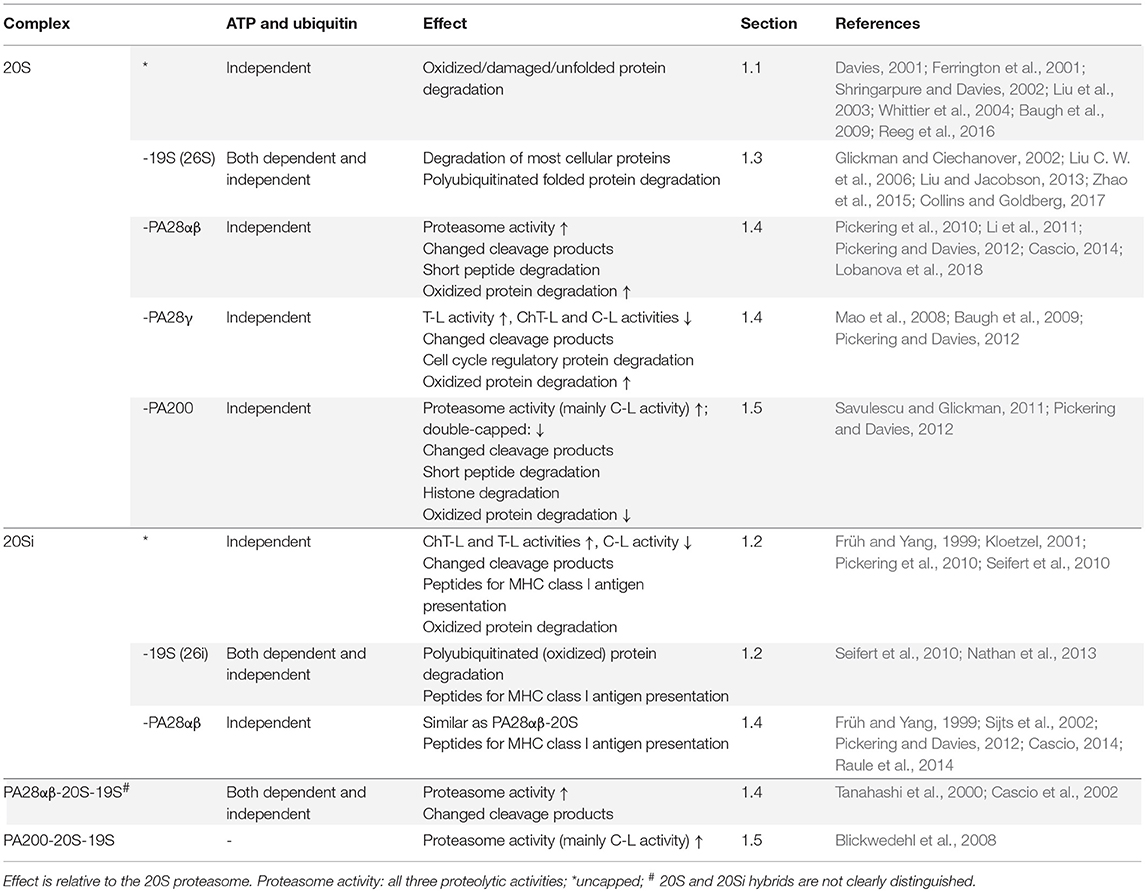
Table 1. The different proteasome complexes with their specific properties.
1.1. The 20S Proteasome
Substrate entrance into the proteolytic core of 20S proteasomes is physically blocked by the N-termini of the α-subunits (Groll et al., 2000). Binding of a PA relieves this barrier by opening the α-ring. The free 20S proteasome is therefore often described as a latent complex. Interestingly, damaged and especially oxidized proteins, which can be induced by exposure to environmental toxins, cellular stresses, diseases and aging, can be degraded by the 20S proteasome in vitro (Davies, 2001; Shringarpure and Davies, 2002; Whittier et al., 2004; Reeg et al., 2016). Protein oxidation results in conformational changes, and subsequently in the exposure of hydrophobic domains that were previously shielded (Ferrington et al., 2001; Lasch et al., 2001). These hydrophobic sites can bind to purified 20S proteasomes and stimulate proteasome activities by opening the barrel (Kisselev et al., 2002).
However, intracellular protein degradation by the 20S proteasome has not been clearly demonstrated (reviewed by Demasi and da Cunha, 2018). Studies suggest that the 20S proteasome can degrade oxidized proteins in vivo (Grune et al., 1996; Pickering et al., 2010), but direct evidence is still lacking. In response to oxidative stress the 19S regulatory particle dissociates from the 26S proteasome in yeast and mammalian cells, increasing the pool of free 20S proteasomes (Wang et al., 2010; Grune et al., 2011), which suggests a rapid mechanism to increase the capacity to degrade oxidized proteins. Though, studies show different results on whether oxidized proteins are generally ubiquitinated (Shang et al., 2001; Dudek et al., 2005; Medicherla and Goldberg, 2008) or non-ubiquitinated (Shringarpure et al., 2003; Kastle and Grune, 2011; Kastle et al., 2012), i.e., the involvement of the UPS.
Based on biochemical analysis of mammalian lysates, it was predicted that 20% of the cellular proteins is degraded by the 20S proteasome (Baugh et al., 2009). This seems a relatively high number if only damaged and oxidized proteins would be substrates for the 20S proteasome (Baugh et al., 2009). An explanation for this high number would be that the 20S proteasome also degrades native proteins. For example, p21 and α-synuclein have been linked to 20S proteasome degradation (Liu et al., 2003). Surprisingly, these proteins were even degraded in vitro when they lacked exposed termini. This endoproteolytic activity of the 20S proteasome was also confirmed in a study that reported cleavage in unfolded regions outside structured domains of various proteins (Baugh et al., 2009). This supports the suggestion that unfolded regions of proteins can promote gate opening and translocation into the proteolytic core. Therefore, in addition to oxidized proteins, the 20S proteasome may degrade a broad spectrum of native proteins, including tumor suppressors p21, p53, and p27 (Sheaff et al., 2000; Liu et al., 2003; Asher et al., 2005) and proteins associated with neurodegenerative diseases such as α-synuclein [Parkinson's Disease (PD)] and tau [Alzheimer's disease (AD)] (David et al., 2002; Liu et al., 2003). Though, one cannot be conclusive on this issue as most studies were performed using purified proteasomes that may degrade damaged and denatured proteins differently when compared to the UPS in living cells. Some of these proteins are indeed reported to be ubiquitinated and therefore subjected to 26S proteasome degradation. For instance, p53 is ubiquitinated and targeted for proteasomal degradation by E3 ligase MDM2 (Fang et al., 2000) and the E3 ligase CHIP was recently shown to be responsible for the ubiquitination of p21 (Biswas et al., 2017). This discrepancy may be explained by the different experimental setups, as purified proteasomes may degrade proteins independent of ubiquitination whereas intracellular degradation is largely dependent on selective protein ubiquitination followed by degradation by the 26S proteasome.
1.2. The Immunoproteasome
The 20S immunoproteasome (20Si) differs from the standard 20S proteasome by its proteolytic activity as the constitutive subunits β1, β2, and β5 are replaced by its immune counterparts β1i (LMP2), β2i (MECL1), and β5i (LMP7), respectively. Lymphoid tissue constitutively expresses the immunoproteasome at high levels (Sijts and Kloetzel, 2011). In non-lymphoid tissue, the immunoproteasome abundance is rather low and requires induction by cytokines, such as interferon γ (IFN-γ) (Früh and Yang, 1999; Kloetzel, 2001; Sijts and Kloetzel, 2011). The immunoproteasome has a higher ChT-L and T-L activity and lower C-L activity than the standard 20S proteasome, resulting in alternative cleavage of proteins (Gaczynska et al., 1993; Cascio et al., 2001). In general, peptides with hydrophobic or basic C-termini are generated, which are preferred by major histocompatibility complex (MHC) class I molecules that are important for the initiation of an immune response by infection (Gaczynska et al., 1993; Kloetzel, 2001).
For a long time the immunoproteasome was almost exclusively linked to peptide production for MHC class I antigen presentation. However, studies have elucidated roles for the immunoproteasome in macrophage activation and T-cell differentiation, and also in the differentiation of non-immune cells like skeletal muscle cells (Kimura et al., 2015). In addition, it has been proposed that the immunoproteasome is also involved in the preservation of general homeostasis. First, hydrogen peroxide (H2O2) treatment, which induces oxidative damage, enhanced the expression of immunoproteasomes in mouse cells (Pickering et al., 2010). Secondly, IFN-γ does not only induce immunoproteasome expression but also oxidative stress, resulting in oxidatively damaged proteins (Watanabe et al., 2003; Pickering et al., 2010; Seifert et al., 2010). Upon depletion of immunoproteasomes, formation of aggresome-like induced structures (ALIS) was accelerated in IFNγ treated cells compared to non-treated cells, indicating a role in the clearance of oxidatively damaged proteins (Seifert et al., 2010). The role of 26S immunoproteasomes in degrading oxidatively damaged proteins has however been challenged by others who did not observe improved degradation of ubiquitinated proteins by immunoproteasomes or the subsequent protective effects (Nathan et al., 2013; Lundh et al., 2017).
1.3. The 26S Proteasome
The 26S proteasome degrades the majority of cellular proteins and therefore plays an important role in a wide range of cellular processes, such as transcriptional regulation, the cell cycle, differentiation, DNA repair, the secretory pathway, and the biogenesis of organelles, designating the 26S proteasome as a key regulator in cellular quality control (Glickman and Ciechanover, 2002). Interestingly, it has been reported that the 26S proteasome is not very effective in degrading oxidized proteins in vitro, even in the presence of ATP and ubiquitin (Davies, 2001). Again, many studies were performed using purified proteasomes that may degrade damaged and denatured proteins differently when compared to the UPS in living cells.
A general overview of the most relevant 19S subunits and their function in the 26S proteasome will be discussed here. For an extensive review on the 26S proteasome's multistep degradation mechanisms we refer to Collins and Goldberg (2017) and Bard et al. (2018).
The 19S regulatory particle contains six regulatory triple-ATPase particles (RPT1–6) forming the base of the cap and 13 regulatory non-ATPase particles (RPN1–3, RPN5–13, and RPN15), which constitute the so-called lid (Figure 1) (Lander et al., 2012; Schweitzer et al., 2016). The RPN10- and RPN13-subunits are the ubiquitin-receptors, which bind ubiquitinated substrates with their ubiquitin interacting motif (UIM) or pleckstrin-like receptor for ubiquitin (PRU) domain, respectively (Elsasser et al., 2004; Husnjak et al., 2008). Recently, the RPN1 subunit was also identified as ubiquitin binding site (Shi et al., 2016). In addition to the intrinsic ubiquitin-receptors, ubiquitinated substrates can also bind to extrinsic UBL (ubiquitin-like)-UBA (ubiquitin-associated) ubiquitin-receptors, including DSK2, RAD23, and DDI1 (Elsasser and Finley, 2005). These UBL-UBA proteins interact via their UBL-domain with the proteasomal ubiquitin binding sites (Husnjak et al., 2008; Shi et al., 2016), functioning as ubiquitin shuttling proteins. However, not only the presence of ubiquitin regulates the selective and efficient degradation of proteins, the recognition of a loosely folded region also plays an important role (Peth et al., 2010).
After substrate recognition, a conformational switch of RPN11 stimulates its DUB activity, resulting in the removal and recycling of ubiquitins (Worden et al., 2017). In addition, two other DUBs, ubiquitin carboxyl-terminal hydrolase 14 (USP14) and ubiquitin carboxyl-terminal hydrolase isozyme L5 (UCH-L5) are associated with a minor pool of 26S proteasomes (de Poot et al., 2017; Kuo and Goldberg, 2017). These DUBs bind the proteasome via RPN1 and RPN10/RPN13, respectively, and trim the polyubiquitin chain into monoubiquitins and short ubiquitin chains, which consequently can either promote or prevent substrate degradation (Liu and Jacobson, 2013). Contrarily, the proteasome-associated ubiquitin-protein ligase E3C (UBE3C) extends the ubiquitin chain on substrates (Crosas et al., 2006). The exact role of ubiquitin remodeling by chain trimming and extending by these different enzymes is unclear, but it may regulate proteasome specificity (Crosas et al., 2006; Liu and Jacobson, 2013). In addition to the deubiquitinating role of USP14, it also plays a role in regulating proteasome activities. Although the 26S proteasome has a preference for polyubiquitinated proteins, the purified complex can also degrade non-ubiquitinated unfolded proteins, without ATP hydrolysis (Liu C. W. et al., 2006), but this activity is mainly inhibited by USP14 (Kim and Goldberg, 2017). When bound to ubiquitinated substrates, USP14 activates the proteasome via its UBL domain, otherwise it suppresses several activities to prevent unnecessary ATP consumption and non-specific hydrolysis (Peth et al., 2009, 2013; Kim and Goldberg, 2017, 2018). The ATP hydrolysis by the RPT-subunits in the base drives protein translocation through the α-ring gate, which forces protein unfolding (Navon and Goldberg, 2001; Kenniston et al., 2003; Snoberger et al., 2017). Gate opening is particular induced by the C-termini of RPT2 and RPT5 through binding between the 20S α-subunits (Smith et al., 2007). Though, RPT3 is also important for gate opening, since point mutations in the C-terminus inhibited gate opening of the 20S core. Other 19S-subunits are involved in stabilizing (RPN2) and structuring (RPN8) the 19S regulatory particle, or stabilizing the association between the 19S regulatory particle and the 20S core (RPN6) (Chen et al., 2016; Schweitzer et al., 2016). The role of the other subunits is not fully understood. They may support the function of other 19S-subunits (e.g., RPN3) (Chen et al., 2016; Schweitzer et al., 2016).
1.4. The PA28-20S Proteasome
PA28 is another regulatory particle that can associate with the 20S proteasome. The PA28 family exist of three members: PA28α, PA28β, and PA28γ. PA28α and PA28β assemble into a heteroheptamer, while PA28γ forms a homoheptamer (Mao et al., 2008; Cascio, 2014). The cap can also be formed by PA28α alone, but its affinity for and stimulation of the 20S core is lower (Huber and Groll, 2017). The localization of PA28α and PA28β, and the localization of PA28y are mutually exclusive (Wójcik et al., 1998). Whereas, PA28α and PA28β are primarily located in the cytoplasm, PA28γ is mainly present in the nucleus.
PA28αβ associates with the 20S proteasome and enhances all three proteolytic activities, stimulating its ability to degrade short peptides, rather than proteins or ubiquitinated substrates in vitro (Cascio, 2014). Indeed, overexpression of PA28α did not affect the turnover of some bona fide substrates like GATA4, AKT, and PTEN in rat cardiomyocytes or the degradation of polyubiquitinated protein/peptide substrates in retina lysates of transgenic mice (Li et al., 2011; Lobanova et al., 2018). However, PA28 overexpression does increase degradation of the UPS substrate GFPu or oxidized proteins in cells (Li et al., 2011). This is supported by in vitro data showing increased ability of purified proteasomes to degrade oxidized proteins in the presence of PA28αβ (Pickering et al., 2010). In addition, PA28αβ binds to the 20S proteasome immediately upon H2O2 treatment, followed by increased PA28αβ expression during oxidative stress adaptation (Pickering et al., 2010; Pickering and Davies, 2012). Therefore, PA28αβ seems to function in retaining cellular proteostasis. This is exemplified in a study on retinitis pigmentosa, where overexpression of PA28α in mice slowed retina degeneration caused by insufficient proteasome capacity to degrade misfolded mutant rhodopsin (Lobanova et al., 2018). Alternatively, the effect of PA28αβ may also be proteasome activity-independent, as chaperone-like functions have been suggested (Minami et al., 2000; Adelöf et al., 2018).
Since PA28αβ is induced by IFN-γ, the role of PA28αβ in MHC class I antigen presentation and the immune response has been studied most extensively (Tanahashi et al., 1997; Früh and Yang, 1999; Cascio, 2014). Expression of PA28αβ has been reported to selectively upregulate MHC class I antigen presentation, whereas downregulation impaired the presentation of specific antigens (Sijts et al., 2002). PA28αβ-20Si proteasomes generate higher amounts of very short products, and favors the release of several longer more hydrophilic peptides, probably by the enhanced proteolytic activities (Raule et al., 2014). While these products are not preferred by MHC class I molecules, some may be critical for an effective immune response. Mice lacking both PA28α and PA28β showed also that PA28αβ is required for the processing of certain antigens (Murata et al., 2001). However, PA28 knockout mice showed normal immune responses against an influenza virus infection, and normal disease progression during viral myocarditis, suggesting a small impact of PA28αβ in general antigen presentation (Murata et al., 2001; Respondek et al., 2017). Nevertheless, a phylogenetic analyses of proteasome subunits links the presence of PA28αβ with the IFN-γ-inducible MHC and immunoproteasome components, which would suggest an important role for PA28αβ in antigen processing (Fort et al., 2015). IFN-γ also induces the formation of PA28αβ-20S-19S hybrid proteasomes in human cells (Tanahashi et al., 2000). PA28αβ-20S-19S hybrid proteasomes generates an altered pattern of cleavage products, without altering the mean peptide length, in contrast to PA28αβ-20S proteasomes (Cascio et al., 2002; Raule et al., 2014).
PA28γ stimulates the T-L activity of the 20S proteasome, while it suppresses the other proteolytic activities (Realini et al., 1997; Mao et al., 2008). As a result, the PA28γ-20S proteasome has increased preference for cleavage after basic amino acids but enhances the degradation of short peptides only weakly. Recently, Jonik-Nowak et al. (2018) reported that in their study most PA28γ is bound to FAM192A/PIP30 in mammalian cells. This protein promotes PA28γ's interaction with the 20S core and impairs the entrance of some peptides in vitro, suggesting changed substrate selectivity. PA28γ-20S proteasomes can also degrade intact proteins which may occur via the cleavage of proteins in less structured domains (Baugh et al., 2009), although it remains poorly understood how these proteins can be unfolded and processed in an ATP-independent manner (Mao et al., 2008). In contrast to PA28αβ, PA28γ expression is not responsive to IFN-γ, which suggests a different role for this PA28 family member (Tanahashi et al., 1997). PA28γ is overexpressed in various type of cancers (Chai et al., 2014; Li J. et al., 2015) and several in vivo mice studies suggest that PA28γ is important in cell proliferation and apoptosis (Mao et al., 2008). In addition, the PA28γ-20S proteasome has been implicated in the degradation of important cell cycle regulatory proteins, including p21 and the steroid receptor SRC-3 (Li et al., 2006, 2007; Mao et al., 2008). PA28γ facilitates also the MDM2-dependent turnover of tumor suppressor p53 (Zhang and Zhang, 2008), and is involved in the regulation of chromosomal stability during mitosis (Zannini et al., 2008). Overall the PA28γ proteasome regulator is implied in cell cycle progression. PA28γ seems to have additional functions as it is recruited to sites of DNA double-strand breaks (Levy-Barda et al., 2011) and it has a role in the organization of nuclear bodies such as nuclear speckles and Cajal bodies (Cioce et al., 2006; Baldin et al., 2008). Association of PA28γ with a component of Cajal bodies is inhibited by PIP30, indicating that PIP30 can control multiple functions of the proteasome (Jonik-Nowak et al., 2018). However, the exact function of PA28γ remains unknown. There is also evidence that PA28γ enhances the ability of the 20S proteasome to degrade oxidized proteins, but in lesser extent than PA28αβ (Pickering and Davies, 2012).
1.5. The PA200-20S Proteasome
Like PA28γ, PA200 is a nuclear-localized proteasome regulator (Savulescu and Glickman, 2011). PA200 enhances the ability of purified 20S proteasome to degrade short peptides and unstructured proteins, especially by cleavage after acidic residues (C-L activity) (Dange et al., 2011; Savulescu and Glickman, 2011). PA200 strongly inhibited the ability of the 20S proteasome to degrade oxidized proteins in vitro (Pickering and Davies, 2012). The majority of PA200 regulatory particles is bound to 26S proteasomes in yeast and mammalian cells (Schmidt et al., 2005; Pickering and Davies, 2012) and in response to ionizing radiation, more PA200-hybrid proteasomes are formed, which accumulate on chromatin (Blickwedehl et al., 2008). PA200-containing proteasomes degrade acetylated core histones during DNA repair and replication stress, which is independent of ubiquitination (Qian et al., 2013; Mandemaker et al., 2018). Cells depleted from PA200 are more sensitive to DNA damage (Mandemaker et al., 2018). However, it seems that PA200 is not essential for DNA repair; in mice lacking PA200, processes that require DNA repair were not affected (Khor et al., 2006). Instead, PA200 knockout mice present with reduced fertility in male, which become complete infertile in combination with PA28γ knockout, caused by multiple defects in spermatogenesis e.g., PA200 has a role in acetylated histone degradation during spermatogenesis (Khor et al., 2006; Qian et al., 2013; Huang et al., 2016). Furthermore, PA200-capped proteasomes have been implicated in various other cell processes, such as mitochondrial fission, turnover of ribosome-related transcription factor Sfp1 and maintaining intracellular glutamine levels (Lopez et al., 2011; Blickwedehl et al., 2012; Tar et al., 2014).
2. Proteasome Modulation by Post-translational Modifications
Post-translational modifications (PTMs) of proteasome subunits include phosphorylation, methylation, acetylation, ubiquitination, and myristoylation. It has been shown that there is overlap between the modification sites, suggesting crosstalk in regulating proteasome function (Zong et al., 2014). To make the study of proteasome regulation by PTMs even more complex, the presence and sites of some PTMs differ between species, and there seems to be differences in PTMs between cell types and tissues. For most PTMs the specific target, effect or even relevance on proteasome functioning is unknown (Wang et al., 2007; Hirano et al., 2016). However, an increasing number of PTMs has been studied in the last decades, revealing their role in proteasome regulation. Proteasome modulation by phosphorylation has recently been reviewed by Guo et al. (2017) and VerPlank and Goldberg (2017), but an overview of all the different types of proteasomal PTMs was still missing. The following section on PTMs is subdivided in proteasome activating or inhibiting modifications of the 20S and 19S subunits (Tables 2, 3 respectively). For each PTM the class of modification is explained, followed by their consequences for proteasomal functioning and involvement in cellular processes. However, many studies show under a specific condition both altered proteasomal PTM(s) and altered proteasome activity, but a direct link between these alterations is often still lacking. In this section we will discuss PMT that are present in the 20S proteasome and PMT that are present in the 19S cap separately as to give a better overview in a broad range of modified subunits. However, post-translational modifications attributed to the 20S core could be part of larger complexes such as the 26S complex.
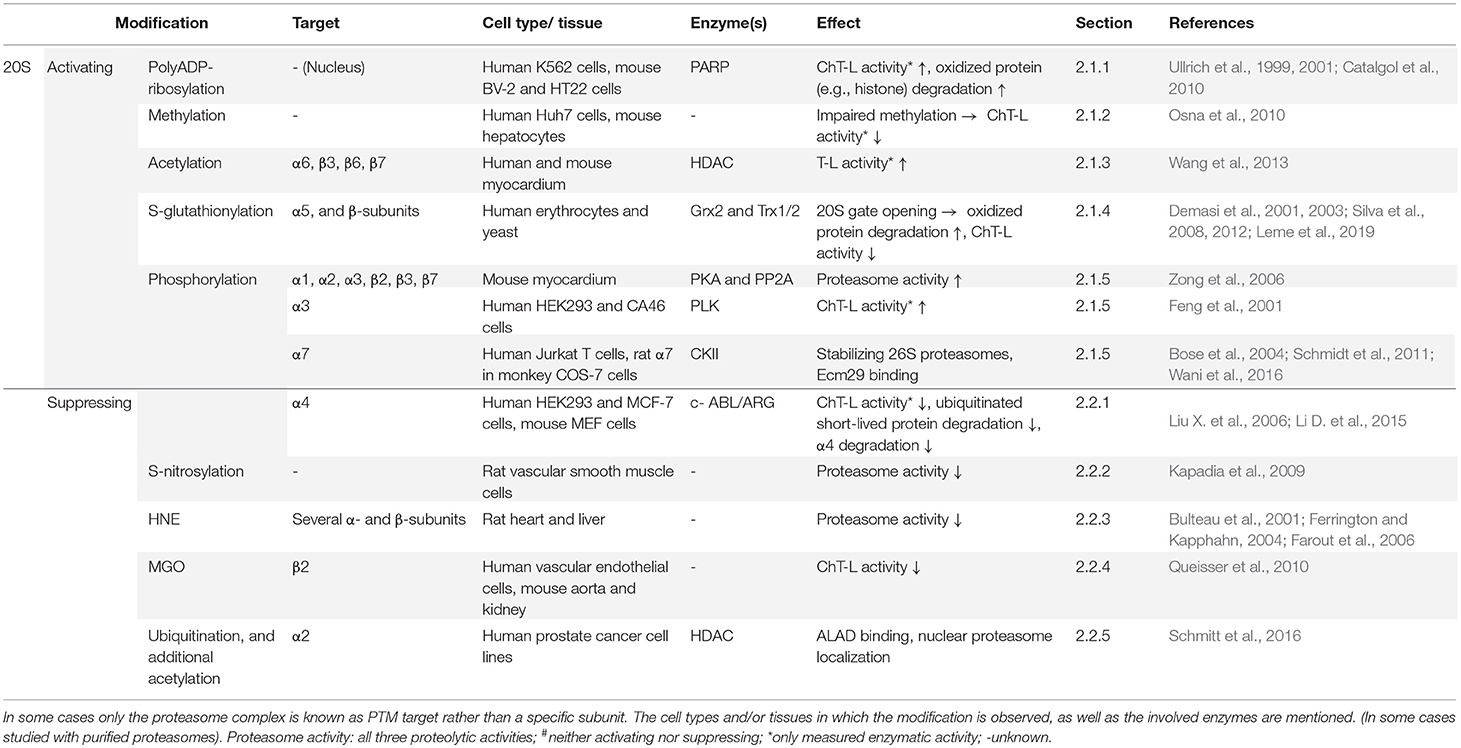
Table 2. An overview of the 20S proteasome PTMs with known target and effect.
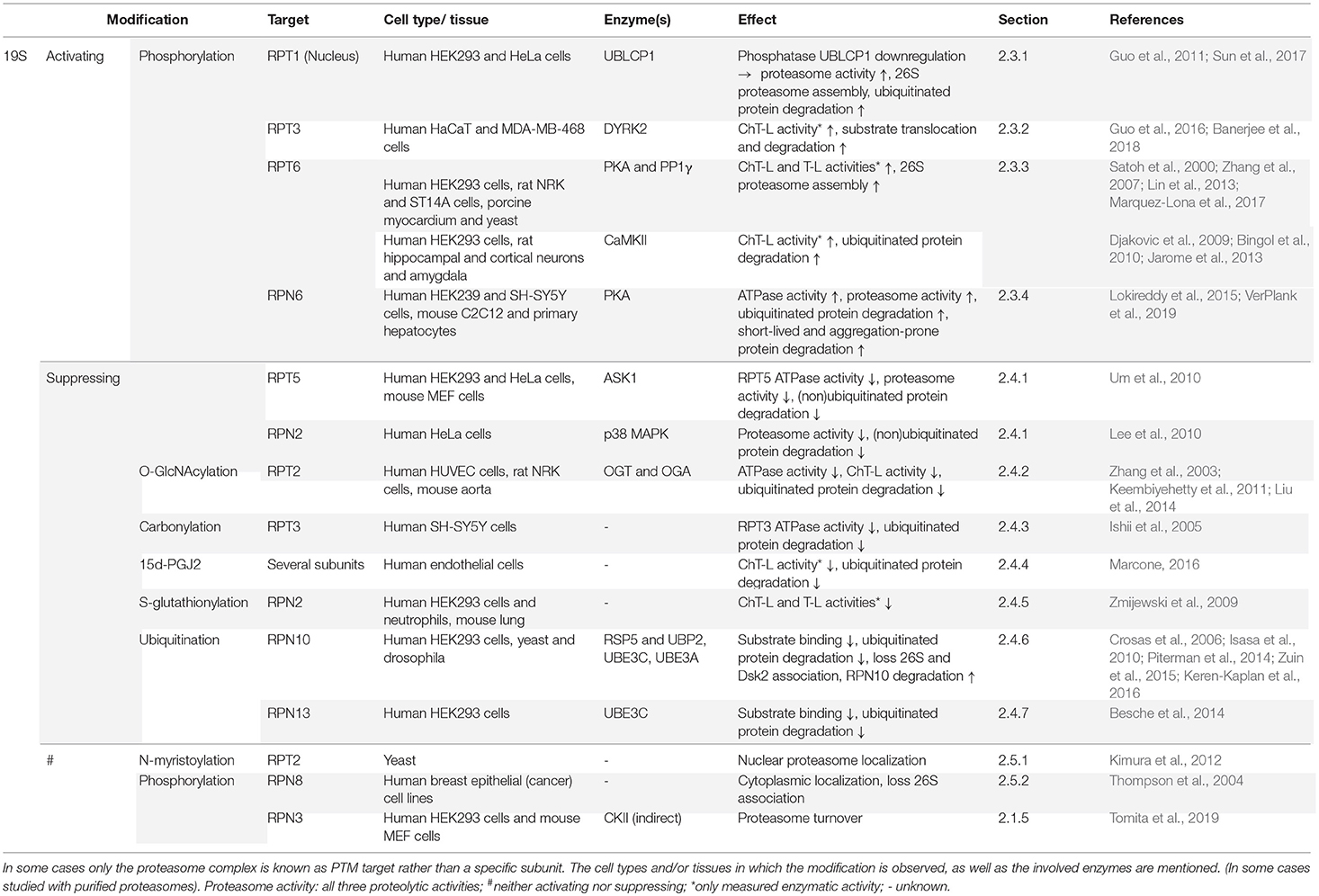
Table 3. An overview of the 19S related PTMs with known target and effect.
2.1. Activating Post-translational Modifications of the 20S Proteasome
2.1.1. PolyADP-Ribosylation
ADP-ribosylation is the addition of the ADP-ribose moiety of NAD+ to an acceptor protein (Ziegler, 2000). This covalent modification is implicated in several cellular processes, including DNA repair, apoptosis and gene regulation. Nuclear 20S proteasomes that are polyADP-ribosylated by PARP were also shown to be involved in DNA repair (Ullrich et al., 1999; Catalgol et al., 2010). H2O2-induced DNA damage activated PARP, which consequently bound DNA strand breaks and tightly interacted with proteasomes. ChT-L activity increased and the degradation of oxidatively damaged histones in the nucleus was elevated, which was dependent on the activation of the nuclear 20S proteasome by polyADP-ribosylation (Ullrich et al., 1999). In this way, proteasomes recognize and degrade the oxidized histones, which will otherwise cross-link with the DNA, making DNA repair impossible. Since antitumor chemotherapy generally causes oxidative stress in the nucleus, and subsequently DNA damage, polyADP-ribosylation of the nuclear 20S proteasome might be an adaptive response, and may be partly responsible for the development of long-term resistance to many of these drugs (Ozben, 2007). Therefore, PARP-inhibitors might improve antitumor chemotherapeutic treatment.
In addition, proteasome modification via ADP-ribosylation was also shown to be involved in neuroinflammation (Ullrich et al., 2001). Activated microglial cells release free radicals which can lead to neuronal cell death, which may have a role in neurodegenerative diseases (Liu and Hong, 2003). Microglial cells are more resistant toward free radicals. TNF-α induced activation of mouse microglial cells resulted in increased proteasomal degradation of an oxidatively damaged model substrate in lysates (Ullrich et al., 2001). This enhanced nuclear proteasome activity in activated microglial cells was dependent on active PARP, thereby protecting activated microglia from protein oxidation and cell death. Although the enhanced activity was attributed to the interaction between active PARP and the nuclear proteasome, polyADP-ribosylation of the nuclear 20S proteasome by PARP seems likely. In conclusion, the nuclear 20S proteasome can be polyADP-ribosylated by PARP, resulting in increased proteasome activity, which is probably reflected in the enhanced ability to degrade oxidized proteins, including histones.
2.1.2. Methylation
S-adenosylmethionine (SAM) is the principal methyl (-CH3) donor for methylation in many biological processes, and therefore, indicates the methylation potential of a cell (Chiang et al., 1996). It has been shown that ethanol exposure leads to a decrease in the methylation potential, and that this inhibits the ChT-L activity of the proteasome in mouse hepatocytes (Osna et al., 2010). Exposure of human hepatoma cells to a methylation inhibitor had a similar effect and incubation of purified 20S proteasomes at relatively low SAM levels reduced lysine methylation of the complex (Osna et al., 2010). This suggests that proteasome activity is directly regulated by the methylation potential via proteasomal subunits or via co-purified proteins with a SAM-dependent methyltransferase-like activity.
Since the methylation potential can be influenced by ethanol, the impaired proteasome activity due to a changed methylation state may be involved in the development and/or progression of diseases associated with alcohol consumption. Indeed a study by Bardag-Gorce et al. (2006) found an ethanol induced decrease in proteasome activity, leading to the formation of protein aggregates (Mallory bodies) in patients with alcoholic liver disease (ALD). Therefore, methyl group donors, such as SAM, might be potential as treatment to reverse the proteasome inhibition by correcting the methylation potential in the cells of ALD patients (Osna et al., 2010). In addition, alcohol abuse accelerates the progression of hepatitis C (HCV) infection, and increases the risk of death (Safdar and Schiff, 2004). Reduced proteasome methylation induced by alcohol consumption, is suggested to have a role in the accelerated pathogenesis since the decreased proteasome activity can dysregulate antigen presentation, and therefore the recognition of HCV infected cells by the immune system (Osna et al., 2012). This is further supported by the observation that immunoproteasomes seem to be more inhibited at low SAM levels than 20S proteasomes (Osna et al., 2010). Furthermore, the methylation potential was lower in ethanol-fed HCV+ mice than in ethanol-fed HCV− mice (Osna et al., 2012). This emphasizes the accumulating negative effect of ethanol on liver with inflammation. In short, an impaired cellular methylation potential suppresses proteasome activity, which is associated with pathogenesis.
2.1.3. Acetylation
Acetylation is the substitution of an acetyl group (–CH3CO) for an active hydrogen atom, and is an important modification of proteins in diverse cellular processes (Choudhary et al., 2009). It plays a central role in the control of gene expression, regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), which add and remove acetyl groups from lysine residues, respectively (Verdone et al., 2005).
These enzymes can also affect the proteasome; HDAC inhibitors enhanced the acetylation of the 20S proteasome, which correlated with an increase in the T-L activity of the proteasome in mouse and human myocardium (Wang et al., 2013). Examination of the acetylome of purified proteasomes of mouse myocardium treated with HDAC inhibitors in vivo revealed the inducible acetylation of α6 (Lys-30 and Lys-115), β3 (Lys-77), β6 (Lys-203), and β7 (Lys-201). The regions of these lysine residues are conserved in human (Wang et al., 2013).
Cardiac ischemia/reperfusion (I/R) injury is associated with suppressed proteasome activity, in which HNE modifications may have a role (section 2.2.3) (Bulteau et al., 2001). HDAC inhibition restored the proteasome activity in acutely I/R injured mice and end-stage ischemic failing human myocardium (Wang et al., 2013). Therefore, HDAC inhibitors might be potential drugs for regulating the proteasomal function in injured hearts. In summary, HDAC inhibitors enhance the proteolytic activity of the proteasome, likely by increased acetylation of 20S-subunits, although direct evidence for increased acetylationis lacking.
2.1.4. S-glutathionylation
S-glutathionylation is the reversible formation of disulfides (-S-S-) between the thiol group (-SH) of glutathione (GSSG or GSH) and cysteine residues, which can be activated by oxidants (Hill and Bhatnagar, 2012). Upon H2O2 treatment, yeast 20S proteasomes were S-glutathionylated both in vitro and in vivo (Demasi et al., 2003). This resulted also in decreased proteolytic proteasome function, especially the ChT-L activity. The ChT-L activity was also affected by addition of GSH (mM), but not by GSSG in vitro (Demasi et al., 2003). The activity of mammalian proteasomes was modulated by both glutathione redox forms; low concentrations (μM) of GSH or GSSG increased, and high concentrations (mM) of GSH or GSSG decreased the ChT-L activity (Demasi et al., 2001).
In contrast to reduced ChT-L activity, the degradation rate of oxidized and partially unstructured proteins was higher by the S-glutathionylated form of the purified yeast 20S proteasome than the reduced form (Silva et al., 2012). Examination of the S-glutathionylation state of the yeast proteasome revealed modified cysteine residues of the α5-subunit, of which Cys-76 is highly conserved from yeast to human. When this residue is S-glutathionylated, the 20S proteasome is in its maximal open gate conformation, increasing the accessibility for oxidized proteins (Silva et al., 2012; Demasi et al., 2014; Leme et al., 2019). Although the S-glutathionylated cysteine residues in the β-subunits could not be identified, S-glutathionylation of the proteasomal catalytic site promoted an allosteric modification, leading to changes in the length of the 20S proteasome, thereby probably inhibiting the ChT-L acticity (Silva et al., 2012). This last mechanism may also support the function of irreversible proteasome inhibitors, which increase the S-glutathionylation of purified human 20S proteasomes (Demasi et al., 2001). It is suggested that the binding of these inhibitors leads to a conformational change, opening the 20S proteasome, and subsequently allowing S-glutathionylation. This is in agreement with increased GSH incorporation in the proteasome upon heat-denaturation and treatment with detergents, which both trigger gate opening (Demasi et al., 2001). S-glutathionylation of the 20S proteasome is a reversible modification (Silva et al., 2008). The oxidoreductases glutaredoxin 2 and thioredoxins are able to enter the core particle, remove the S-glutathionylation and allow recovery of the proteolytic activity. In summary, S-glutathionylation of the 20S proteasome triggers gate opening, which likely increases the degradation of oxidized proteins, but reduces the ChT-L activity.
2.1.5. Phosphorylation
Protein phosphorylation is the addition of a phosphate group (P) to an amino acid residue, and is important in almost every cellular process (Cohen, 2002). Phosphorylation and its counterpart dephosphorylation are catalyzed by kinases and phosphatases, respectively, regulating protein function. The phosphorylation of several 20S-subunits can be regulated by cAMP-dependent protein kinase (PKA) and protein phosphatase 2A (PP2A). Active PKA enhanced the serine phosphorylation of the α1-, α2-, α3-, β2-, β3-, and β7-subunits, and the threonine phosphorylation of the α3-, β3-, and β7-subunits of purified mouse cardiac 20S proteasomes (Zong et al., 2006). These modifications elevated all three proteolytic activities of the 20S proteasome. The same study also showed that PP2A reduced serine phosphorylation on α1 and β7, and threonine phosphorylation on α1, which was linked to suppressed proteasome activity. It is not clear which role the different subunits have in the altered proteasome activity, but the study suggests that in general proteasome phosphorylation and dephosphorylation are associated with increased and decreased proteasome activity, respectively. In addition, Hirano et al. (2016) identified multiple phosphorylation sites on almost all subunits in yeast, though the question remained whether these sites are all functional.
In addition to PKA, α3-subunits can be phosphorylated by Polo-like kinase (PLK) (Feng et al., 2001). PLK interacted with 20S (and 26S) proteasomes, and subsequently, phosphorylated α3 in human cells, which resulted in higher ChT-L activity of the proteasome.
Phosphorylation seems a constitutive modification of the α7-subunit (Gersch et al., 2015). Casein kinase II (CKII) phosphorylates α7 at Ser-243 and Ser-250 (Bose et al., 2004). After IFN-γ treatment of monkey kidney-fibroblast cells, the α7 phosphorylation decreased, resulting in destabilization of the 26S proteasome (Bose et al., 2004). Decreased 26S proteasome levels were accompanied with increased levels of PA28-proteasomes. Thus, the α7 phosphorylation state may be involved in stabilizing the association of the 19S cap with the 20S core, and therefore, the regulation of proteasome complexes. The destabilizing effect of α7 dephosphorylation on 26S proteasomes may also have a role in binding to Ecm29, a proteasome quality control factor (Wani et al., 2016), and during apoptosis (Schmidt et al., 2011). Another target of CKII is RPN3, however this modification seems to be involved in the turnover of proteasomes (Tomita et al., 2019). Finally, the kinase Aurora B is now also identified as an enhancer of proteasome activity in cell cycle regulation. Although evidence for proteasome phosphorylation was not shown, a direct effect of Aurora B on the (26S) proteasome was demonstrated by interaction studies and in vitro activation (Fan et al., 2019).
2.2. Suppressing Post-translational Modifications of the 20S Proteasome
2.2.1. Phosphorylation
Phosphorylation of the α4-subunit by tyrosine kinases c-ABL and ARG has diverse effects (Liu X. et al., 2006; Li D. et al., 2015). First, phosphorylation at Tyr-153 (and maybe also at Tyr-106) led to the inhibition of the ChT-L activity of the 20S and 26S proteasome, and decreased degradation of ubiquitinated short-lived proteins by the 26S proteasome in human and mouse cells (Liu X. et al., 2006; Li D. et al., 2015). Activation of c-ABL by H2O2 or γ-irradiation increased its interaction with α4, and inhibited proteasome function (Liu X. et al., 2006). Expression of a phospho-dead α4 mutant at Tyr-153 in human cells resulted in downregulation of several cell cycle regulatory proteins, and G1/S cell cycle arrest, highlighting the role of proteasome tyrosine phosphorylation by c-ABL/ARG in cell cycle control.
Secondly, phosphorylation of α4 at Tyr-106 by c-ABL/ARG protected the 20S subunit from degradation due to suppressed polyubiquitination (Li D. et al., 2015). In addition, c-ABL/ARG upregulated α4, thereby increasing cellular proteasome abundance, under normal and oxidative stress conditions. This is consistent with the observation that cells expressing a BCR-ABL construct, a model for myeloid leukemia cells, had higher proteasome levels (Magill et al., 2004). However, this seems contradictory with the described decreased proteasome activity (Liu X. et al., 2006; Li D. et al., 2015). The authors explained the dual role of c-ABL/ARG on α4 via phosphorylation by the fact that the regulation is time-course dependent (Li D. et al., 2015). During oxidative stress, activated c-ABL/ARG initially inhibits the proteasome, preventing the degradation of short-lived regulatory cell cycle proteins, such as p53, and thereby inducing cell cycle arrest to prevent mitosis of oxidatively damaged cells. Although, this seems in disagreement with the observed G1/S cell cycle arrest in the absence of Tyr-153 phosphorylation (Liu X. et al., 2006). Meanwhile, the proteasome abundance is gradually increased via c-ABL/ARG to degrade oxidized proteins (Li D. et al., 2015). Thus, phosphorylation of the α4-subunit has various effects; it compromises proteasome activity and/or prevents ubiquitin-proteasome degradation of this subunit.
2.2.2. S-nitrosylation
S-nitrosylation is the transfer of a nitric oxide (NO) moiety on a free thiol group (-SH) of a protein to form nitrosothiol (-SNO) (Broillet, 1999). It was shown that recombinant 20S core particles can be S-nitrosylated at 10 cysteine residues (Kapadia et al., 2009). These modifications provide a mechanism where NO suppresses all three proteolytic activities of the 26S proteasome in rat vascular smooth muscle cells (VSMCs) (Kapadia et al., 2009). However, identification of the specific modified cysteine residues and mutational studies should provide evidence whether S-nitrosylation of the proteasome is indeed causing the reduction in proteasome activity upon NO exposure in vivo. NO induces the synthesis of cGMP and the resultant activation of GSK, which has been shown to enhance proteasome activity (Ranek et al., 2013), but inhibition of cGMP/cAMP synthesis or PKG/PKA did not affect the NO-mediated inhibition (Kapadia et al., 2009). In addition to the affected proteasome activity, the expression of the α5-, α6-, β1-, and β1i-subunits increased following NO exposure. This seems contradictory, because the C-L activity regulated by the β1-subunit was the most inhibited proteolytic activity in VSMCs exposed to NO. This increased expression might be an indirect response to the inhibition (a common autoregulatory process; see section 3.3), to limit the effect of NO by synthesis of additional proteasomes, and hence overcoming the suppression of the C-L activity. Since it was shown that NO inhibits the proteasome, and the proteasome regulates the cell cycle through protein degradation, it is likely that NO produced by endothelial cells suppresses the proliferation of VSMCs through S-nitrosylation of the 20S proteasome (Kapadia et al., 2009). Overall, NO can reversibly inhibit the 26S proteasome possibly by S-nitrosylation of the 20S core.
2.2.3. 4-hydroxy-2-nonenal Modification
4-hydroxy-2-nonenal (HNE), an α,β-unsaturated aldehyde (-CHO), is generated during lipid peroxidation by free radicals in response to oxidative stress (Esterbauer et al., 1991). HNE can react with cysteine, histidine and lysine residues to form a mixture of adduct types (which is a form of carbonylation; see section 2.4.3). Multiple 20S proteasome subunits have been identified that can be modified by HNE. Purified rat cardiac 20S proteasomes appeared to be modified after HNE treatment on the α1-, α2-, α4-, α5-, α6-, and β6-subunits (Farout et al., 2006). Another study found that three of these subunits (α1, α2 and α4) were HNE-modified in rat myocardium after I/R injury (Bulteau et al., 2001). In both studies, T-L activity of the purified cardiac 20S proteasomes was suppressed (Bulteau et al., 2001; Farout et al., 2006). Loss of ChT-L and C-L activities were observed after incubation at higher HNE concentrations and in cytosolic extracts of I/R injured myocardium (Bulteau et al., 2001; Ferrington and Kapphahn, 2004; Farout et al., 2006). Thus, it seems likely that in I/R injured myocardium the proteasome activity is reduced due to HNE modification.
The modification sites and the effects of HNE on the proteasome in heart differ from that in liver. Purified rat liver 20S proteasomes appeared to be modified after incubation with HNE on the α2-, α3, α4-, α5-, and β4-subunits, and at higher HNE concentrations also on the β3- and β1i-subunits (Farout et al., 2006). Another study observed a modification of α6, and suggested that α2 and α4 were already HNE-modified in vivo (Ferrington and Kapphahn, 2004). The ChT-L activity was reduced at low HNE concentrations, and inactivated rapidly, while for inhibition of the other proteolytic activities a higher concentration or prolonged exposure was required (Ferrington and Kapphahn, 2004; Farout et al., 2006). Thus, although the HNE-modification sites seem to be tissue specific and condition dependent, it generally results in downregulation of proteasome activity.
2.2.4. Methylglyoxal Modification
The reactive dicarbonyl methylglyoxal (MGO; CH3C(O)CHO) is a side-product of several metabolic pathways, with glycolysis as most important source (Allaman et al., 2015). MGO is one of the most potent glycating agents present in cells, and reacts with molecules, including lysine and arginine residues of proteins to form advanced glycation end products (AGEs), such as carboxyethyllysine and methylimidazolone, respectively (which is a form of carbonylation; see section 2.4.3). Normally, MGO is detoxified, but MGO levels are increased under intracellular hyperglycemia, a condition observed with diabetes mellitus (DM) (Queisser et al., 2010). Incubation of human vascular endothelial cells with high glucose or MGO reduced the proteasomal ChT-L activity, but the other proteolytic proteasome activities were not affected (Queisser et al., 2010). Downregulation of proteasome activity was also observed in kidneys of diabetic mice, and mice that exhibit high MGO levels, confirming that MGO alone can cause proteasome inhibition. In both mouse models MGO modification (methylimidazolone) of β2 was detected. However, the alteration in proteasome activity is tissue specific; in kidney of diabetic mice all three proteasome activities were reduced, while cardiac proteasome activity was not changed (Queisser et al., 2010). In addition, another important observation is that high glucose and MGO levels both reduced 19S protein content in cells. Overall, MGO can modify the proteasome, resulting in decreased activity.
2.2.5. Ubiquitination and Additional Acetylation of the α2-Subunit
Ubiquitination is the attachment of ubiquitin, a small 76-residue polypeptide, to lysine residues of protein substrates (Pickart, 2001). Although polyubiquitination is involved in the selective degradation of proteins by the proteasome, this modification can also affect proteins in other ways, such as altering activity, protein interactions, and cellular localization. The 20S proteasome can also be regulated by ubiquitination. δ-aminolevulinic acid dehydratase (ALAD) interacts with the 20S proteasome via ubiquitinated α2 in human cells and this interaction is enhanced after HDAC inhibition (Schmitt et al., 2016). Contradicting effects of ALAD on proteasome activity are reported. It was found that ALAD enhanced the ChT-L and T-L activities of 20S proteasomes purified from rat liver (Bardag-Gorce and French, 2011), whereas other studies observed that ALAD inhibited the degradation of a proteasome substrate and reduced the ChT-L in human cells (Guo et al., 1994; Schmitt et al., 2016). Since ALAD has been shown to be identical to proteasome inhibitor CF-2, it seems likely that the enzyme suppresses proteasome activity (Guo et al., 1994). ALAD might block the entrance of substrates into the 20S core.
2.3. Activating Post-translational Modifications of the 19S-Subunits
2.3.1. Phosphorylation of RPT1
The 26S proteasome is, like the 20S proteasome, regulated by phosphorylation. The phosphatase UBLCP1 has been shown to interact with the 19S regulatory particle via RPN1, preferentially of nuclear proteasomes (Guo et al., 2011; Sun et al., 2017). A dephosphorylation screen showed that RPT1 was the only 19S subunit that was dephosphorylated (Sun et al., 2017). As a consequence the ATPase activity of RPT1 was impaired. This resulted in the inhibition of the ChT-L activity and negatively regulated the assembly of the 26S proteasome in vitro and in vivo (Guo et al., 2011; Sun et al., 2017). Previously, it was reported that RPT1 with ATP binding mutations was unable to be incorporated into the 26S proteasome, showing that the ATPase activity is essential for 26S complex assambly (Liu C. W. et al., 2006; Kim et al., 2013). Furthermore, downregulation of UBLCP1 enhanced all three proteolytic activities and (polyubiquitinated) protein degradation in the nucleus (Guo et al., 2011). Therefore, UBLCP1 regulates the assembly of 26S proteasomes via dephosphorylation of RPT1.
2.3.2. Phosphorylation of RPT3
The RPT3-subunit is phosphorylated in a cell cycle-dependent manner: phosphorylation of Thr-25 by DYRK2 was low during G1 phase, became upregulated when cells transit form G1 to S phase, and remained thereafter constant in human cells (Guo et al., 2016). Thr-25 phosphorylation increased substrate-stimulated ATP-hydrolysis, without changing basal ATPase activity, indicating that the modification promotes substrate translocation and degradation (Guo et al., 2016). Overexpression of DYRK2 downregulated cell cycle inhibitors in human cells. Therefore, it is likely that increased RPT3 phosphorylation in cells entering the S phase results in the degradation of cell cycle inhibitors, promoting cell cycle progression. In addition, blocking RPT3 Thr-25 phosphorylation or knocking down DYRK2 resulted in slower proliferation of human cells, while overexpression of DYRK2, as seen in several cancer types (Santarius et al., 2010), showed opposite results (Guo et al., 2016). Interestingly, curcumin was recently identified to specifically inhibit DYRK2, diminishing RPT3 Thr-25 phosphorylation in human cells (Banerjee et al., 2018). The curcumin treatment inhibited all three proteolytic activities of the proteasome, impairing cell proliferation with induction of apoptosis, and resulted in reduced tumor growth in mice. In summary, RPT3 phosphorylation leads to increased substrate translocation into the proteasome and subsequent degradation, playing an important role in cell proliferation.
2.3.3. Phosphorylation of RPT6
Both PKA and Calcium-calmodulin-dependent protein kinase II (CaMKII) have been identified to phosphorylate the RPT6-subunit at Ser-120 (Zhang et al., 2007; Djakovic et al., 2009). It has been suggested that RPT6 can also be phosphorylated by PKG; Ranek et al. reported an acidic shift of RPT6 and β5 upon PKG activation in cardiac cells, although the phosphorylated residues were not identified (Ranek et al., 2013, 2014). In human cells, endogenous RPT6 was already phosphorylated at basal state, which increased after PKA activation (Zhang et al., 2007). Activated PKA stimulated the ChT-L and T-L activity of proteasomes in rat cells, and of purified 26S proteasomes (Zhang et al., 2007). How the modification affects proteasome function is unknown, but it may lead to a conformational change, enhancing the diffusion of small peptide substrates into the 20S core. The RPT6 phosphorylation may also initiate 26S assembly, by stimulating the association of the 19S particle with the 20S proteasome as shown in porcine cells (Satoh et al., 2000). Protein phosphatase 1γ (PP1γ) could reverse the RPT6 phosphorylation, and the effect of PKA on proteasome activity (Zhang et al., 2007). However, other phosphatases may also remove the modification.
In a mouse model for Huntington's Disease (HD), activation of PKA and a phospho-mimetic Ser-120 mutant both reduced mHTT aggregates, indicating increased proteasome activity (Lin et al., 2013). Furthermore, in a yeast HD model aggregates were larger in a phospho-dead Rpt6Ser-119 (Ser-120 in mammals) strain, which showed decreased proteasome activity (Marquez-Lona et al., 2017). This suggests that phosphorylation at Ser-119/120 has a role in counteracting proteotoxic stress including protein aggregation. Importantly, other studies could not identify phosphorylation of RPT6 by PKA in vitro and in vivo (Lokireddy et al., 2015; VerPlank et al., 2019). Decreased aggregation in the HD mouse model after PKA activation could therefore be a consequence of RPN6 phosphorylation (as discussed in section 2.3.4).
It was also reported that CaMKII phosphorylates RPT6 at Ser-120, which resulted in increased proteasome activity in rat neurons and human cells (Djakovic et al., 2009). Learning-induced enhancement of proteasome activity was associated with elevated phosphorylation of RPT6 Ser-120 by CaMKII, but not PKA, in the amygdala of rats (Jarome et al., 2013). The possible role of this modification in the formation of long-term memories has been shown in other studies; after neuronal activation, autophosphorylated CaMKII functioned as a scaffold to recruit proteasomes to dendritic spines, and increased their (ChT-L) activity by phosphorylating RPT6, leading to the degradation of polyubiquitinated proteins (Bingol et al., 2010). The enhanced RPT6 phosphorylation was sufficient to change synaptic strength and induce dendritic spine outgrowth (Djakovic et al., 2012; Hamilton et al., 2012). These findings suggest that CaMKII regulates proteasome activity in neurons. In conclusion, phosphorylation of RPT6 stimulates proteasome activity, and although the same residue is phosphorylated, the responsible kinase seems to be condition/cell type dependent.
2.3.4. Phosphorylation of RPN6
PKA is also responsible for the phosphorylation of RPN6 at Ser-14 (Lokireddy et al., 2015; VerPlank and Goldberg, 2017). Activated PKA promoted degradation of short-lived proteins, such as misfolded and regulatory proteins, and aggregation-prone proteins in soluble and insoluble state, associated with amyotrophic lateral sclerosis (ALS) and AD, in human cells (Lokireddy et al., 2015; VerPlank et al., 2019).
The increased degradation of ubiquitinated proteins is probably the result of the observed enhancement in ATPase activity. The stimulatory effect of PKA via RPN6 was confirmed with a phospho-mimetic RPN6 mutant (Lokireddy et al., 2015). In addition, all three proteolytic activities of purified 26S proteasomes were enhanced from cells treated with pharmacological agents that raise cAMP to activate PKA (Lokireddy et al., 2015). Raising cAMP levels also slightly increased the amount of double-capped 26S proteasomes, which suggests that RPN6 phosphorylation increases the association and stabilization of these complexes (Pathare et al., 2012; VerPlank et al., 2019).
All these findings show that activation of PKA may be useful in the treatment of neurodegenerative diseases to stimulate and increase the degradation of aggregation-prone proteins. Studies have already shown that raising cAMP levels reduced the aggregation of both mutant tau in a mouse model of tauopathy (associated with AD) (Myeku et al., 2016), and the aggregation of mutant huntingtin in an HD mouse model (as discussed in section 2.3.3) (Lin et al., 2013). Importantly, VerPlank et al. (2019) demonstrated that RPN6 phosphorylation and the consequent increased proteolysis is also initiated in response to various hormones and physiological conditions that raise cAMP, showing that cells can rapidly adapt to changing conditions when necessary.
2.4. Suppressing Post-translational Modifications of the 19S-Subunits
2.4.1. Phosphorylation
In contrast to the often observed increases in proteasome activity upon phosphorylation, phosphorylation of the RPT5-subunit reduces proteasome activity. Apoptosis-regulating kinase ASK1, a member of the MAP3K family, interacted with the 19S particle and phosphorylated RPT5 in human cells, thereby inhibiting the ATPase activity (Um et al., 2010). All three proteolytic activities of the proteasome were reduced in cells overexpressing ASK1. In addition, the degradation of poly- and nonubiquitinated substrates was slower. ASK1 negatively regulates the 26S proteasome under stress condition, since the enzyme was activated by H2O2 and an apoptosis-inducer, causing decreased 26S proteasomal activity in mouse cells (Um et al., 2010). Therefore, phosphorylation of RPT5 inhibits proteasome activity and seemingly plays a role in apoptosis.
The RPN2-subunit is phosphorylated by the kinase p38 MAPK (Lee et al., 2010). In human cells, p38 MAPK was activated by hyperosmotic stress, resulting in the phosphorylation of RPN2 at Thr-273, and stabilization of poly- and nonubiquitinated substrates (Lee et al., 2010). Purified 26S proteasomes from cells expressing activated p38 MAPK, had a reduction of all three proteolytic activities. Since a phospho-dead RPN2 mutant at Thr-273 antagonizes the inhibitory effect of p38 MAPK, it is likely that the modification on RPN2 plays an important role in the proteasome inhibition. Though, it was also reported that inhibition of the p38 MAPK pathway (by specific MAPK or MAP2K inhibitors) did induce increased proteasome activity but no difference in the phosphorylation state of the proteasome, including RPN2 Thr-273 (Leestemaker et al., 2017). The link between RPN2 and altered proteasome activities does not seem immediately clear, because RPN2 seems to be a scaffold for other proteasome subunits (Schweitzer et al., 2016). However, it is indicated that RPN2 interacts with 19S ATPases (Schweitzer et al., 2016), which regulate gate opening by their C-termini (Smith et al., 2007). The phosphorylation of RPN2 may cause a conformational change, affecting the accessibility of substrates to the 20S core via the ATPases. Therefore, phosphorylation of RPN2 negatively regulates proteasome activity.
2.4.2. O-GlcNAcylation
Protein O-GlcNAcylation is a form of glycosylation involving the addition of O-linked β-N-acetylglucosamine (O-GlcNAc) at serine and threonine residues (Love and Hanover, 2005). O-GlcNAc is derived from the hexosamine biosynthetic pathway, a nutrient-sensing pathway. The addition of O-GlcNAc is catalyzed by O-GlcNAc transferase (OGT). Exposure of purified mammalian 26S proteasomes to this transferase resulted in reduced ATPase and ChT-L activity of the proteasome, but not the T-L activities (Zhang et al., 2003). Human and rat cells treated with glucosamine (GlcN), which activates the hexosamine pathway, showed enhanced RPT2 O-GlcNAcylation and decreased proteasome ChT-L activity (Zhang et al., 2003; Wang et al., 2009; Liu et al., 2014). The GlcN-suppressed proteasome activity could be restored by O-GlcNAcase (OGA), which removes the O-GlcNAc (Zhang et al., 2003; Liu et al., 2014).
Downregulation of OGA resulted in accumulation of polyubiquitinated proteins and reduced the ChT-L activity of proteasomes in human cells (Keembiyehetty et al., 2011). Consistent with this, overexpression of OGA resulted in opposite findings (Liu et al., 2014). In both conditions, the T-L activity was not affected (Keembiyehetty et al., 2011; Liu et al., 2014). This effect on specific proteasome activities is in agreement with the mentioned effect of OGT (Zhang et al., 2003). The authors assign this selectivity to peptide hydrophobicity; since T-L and C-L substrates are more hydrophilic, they may not require the opening function of RPT2 to enter the 20S core.
Since O-GlcNAc is seen as a nutritional sensor, it may serve as a mechanism to control the availability of amino acids and regulatory proteins to metabolic changes, such as nutrient overload and starvation, by affecting proteasome activity via RPT2 modification (Zhang et al., 2003; Zachara and Hart, 2004). In addition, O-GlcNAcylation of the proteasome might play a role in lipid droplet metabolism, since a lipid droplet-associated OGA isoform and the proteasome regulated each other in a negative feedback loop (Keembiyehetty et al., 2011). Unfortunately, RPT2 O-GlcNAcylation was not analyzed in this study. In addition to the role of O-GlcNAcylation in metabolism, it has been shown that this modification on RPT2 can also be induced by NO in human vascular endothelial cells, which was confirmed in aortic tissue of mice (Liu et al., 2014). The subsequent reduced proteasome activity, similar as with OGT, may maintain, together with other NO-mediated PTMs such as the above described S-nitrosylation (section 2.2.2), the basal low proteasome activity in vascular endothelial cells (Kapadia et al., 2009; Liu et al., 2014). Together, several studies confirm that RPT2 can be modified by O-GlcNAcylation, which decreases proteasome activity.
2.4.3. Carbonylation
Carbonylation is an irreversible oxidative modification, which implies the introduction of a carbonyl group (-CO) into a protein (Dalle-Donne et al., 2006; Madian and Regnier, 2010). Protein carbonyl groups can be generated indirectly by forming adducts with lipid peroxidation products (e.g., aldehyde HNE; section 2.2.3) or reactive carbonyl derivatives produced by the reaction of reducing carbohydrates (e.g., ketoaldehyde MGO; section 2.2.4), or directly by oxidative cleavage of the protein backbone or amino acid side chain with free radicals. Direct carbonylation can be induced by 15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2; see section 2.4.4), and caused RPT3 carbonylation in human neuroblastoma cells (Ishii et al., 2005). This modification impaired RPT3 ATPase activity, and decreased degradation of ubiquitinated proteins in cell lysates, accompanied with enhanced accumulation of these proteins. In conclusion, carbonylation of proteasomes seems to have a negative effect on 26S proteasome activity.
2.4.4. 15-Deoxy-Δ12,14-prostaglandin J2 Modification
15d-PGJ2 is an active lipid compound derived from the prostaglandin PGD2 (Surh et al., 2011). The α,β-unsaturated carbonyl group located in the cyclopentenone ring can form a covalent bond with the thiol group of cysteine residues. Treatment of human aortic endothelial cells with 15d-PGJ2 resulted in 15d-PGJ2-modified 19S-subunits, e.g., RPN1, RPN2, RPN3, and RPN6, while 20S-subunits were unmodified (Marcone, 2016). Further examination showed that the ChT-L activity of the proteasome was inhibited and ubiquitin-conjugated proteins were accumulated. 15d-PGJ2 has anti-inflammatory actions, for instance by inhibiting nuclear factor-κB (NF-κB) activation (Surh et al., 2011). A possible mechanism for 15d-PGJ2 to suppress the NF-κB pathway is by modifying and inhibiting the proteasome, which regulates the processing of NF-κB inhibitors (Marcone, 2016). 15d-PGJ2 treatment reduced the adhesion and migration of monocytes toward TNF-α-exposed endothelial cells, a key process in vascular inflammation, and similar effects were observed upon proteasome inhibition. This suggest that 15d-PGJ2 regulates inflammatory processes by modifying proteasomal 19S-subunits.
2.4.5. S-glutathionylation
The RPN2-subunit can be S-glutathionylated, which can be induced by incubation of purified human 26S proteasomes with both GSH and H2O2, causing decreased ChT-L and T-L activities (Zmijewski et al., 2009). The S-glutathionylation of RPN2 was also observed after exposure of human cells to H2O2 as well as in lung extracts of mice with enhanced intracellular H2O2 levels. RPN2 may directly affect the 20S activity, since incubation of purified human 20S proteasomes with non-oxidized (cysteine residues of) RPN2 enhanced its activity, which was not observed with oxidized RPN2 (Zmijewski et al., 2009). Although it was demonstrated that RPN1 was also S-glutathionylated in the above described conditions, there was no significant difference in 20S proteasome activity between oxidized and non-oxidized RPN1. Besides the direct effect on 20S activity, it is also possible that S-glutathionylation of RPN2 changes its interaction with other 19S subunits, resulting in reduced substrate supply, entry, or processing within the 20S core (Zmijewski et al., 2009).
2.4.6. Mono- and Polyubiquitination of RPN10
The ubiquitin-receptor RPN10 can be mono- and polyubiquitinated, both having different functions. Ubiquitin-protein ligase RSP5 catalyzed the monoubiquitination of RPN10 in yeast, whereas deubiquitinating enzyme UBP2 removed this monoubiquitination (Isasa et al., 2010). Interestingly, it has been shown that RSP5 and UBP2 form a complex (Kee et al., 2006). These two enzymes controlled the monoubiquitination on three lysine residues at the N-terminus, and one lysine residue at the C-terminus of purified yeast RPN10 (Isasa et al., 2010). In human cells, also three monoubiquitination sites were identified in the N-terminus, although the lysine residues differed from yeast (Piterman et al., 2014). The C-terminal UIM-domain of RPN10, involved in the binding of polyubiquitinated substrates, was necessary for the monoubiquitination by RSP5 (Isasa et al., 2010). Monoubiquitination inhibited the capacity of RPN10 to interact with polyubiquitinated substrates, consequently inhibiting proteasome activity (Isasa et al., 2010; Piterman et al., 2014). This suggests an intramolecular interaction between the UIM-domain and RSP5/monoubiquitin.
It seems that monoubiquitination of RPN10 results in its dissociation from the 26S proteasome in yeast (Zuin et al., 2015; Keren-Kaplan et al., 2016). This is consistent with the observation that monoubiquitinated RPN10 was present mainly in a proteasome-free state in human cells (Piterman et al., 2014). The free RPN10 may bind polyubiquitinated proteins for proteasomal degradation, indicating a shuttling model regulated by cycles of ubiquitination (Keren-Kaplan et al., 2016). This may compensate the low diffusion rate of the large proteasome complex, increasing its catalytic function (Keren-Kaplan et al., 2016). This would be important under stress conditions, such as heat shock and oxidative stress, in which RPN10 was essential for the enhanced degradation of damaged and newly synthesized proteins in yeast (Medicherla and Goldberg, 2008). However, monoubiquitination of RPN10 was decreased under these stress conditions. In addition, free RPN10 binds to the extrinsic ubiquitin-receptor DSK2, which shuttles polyubiquitinated proteins to the 26S proteasome, and thereby regulates the amount of DSK2 that interacts with the proteasome (Matiuhin et al., 2008). Monoubiquitination of RPN10 by RSP5 decreased its association with the proteasome as well as DSK2, facilitating the formation of DSK2-26S proteasomes in yeast (Zuin et al., 2015). This suggests a mechanism that regulates the distribution of proteasome ubiquitin-receptors. However, it is unknown what the effect is of the DSK2-26S proteasome association.
It appears that UBE3C, a proteasome-associated ubiquitin ligase that presumably extends ubiquitin chains on substrates, extends the monoubiquitination of RPN10, since in the absence of UBE3C polyubiquitination disappeared, but mono- and diubiquitination were not affected in yeast (Crosas et al., 2006; Isasa et al., 2010). The polyubiquitination of RPN10 resulted in the degradation of the subunit (Crosas et al., 2006; Lee et al., 2014). Nevertheless, in human cells UBE3C does not seem to influence RPN10 ubqituination (Besche et al., 2014). In summary, monoubiquitination of RPN10 regulates substrate binding, and its association with the 26S proteasome and DSK2, while polyubiquitination likely results in RPN10 degradation.
2.4.7. Polyubiquitination of RPN13
Ubiquitin-receptor RPN13 is also polyubiquitinated by UBE3C, although this does not regulate the degradation of the subunit. Proteasome inhibitors enhanced the association of UBE3C with the proteasome, and stimulated the polyubiquitination of RPN13 in human cells, and on purified 26S proteasomes at Lys-21 and Lys-34, which are located within or near the ubiquitin-binding PRU-domain of the subunit (Besche et al., 2014). Therefore, it is not surprising that the RPN13 modification decreased the binding of polyubiquitinated substrates, and consequently their degradation. The proteasome's ability to degrade ChT-L peptides or non-ubiquitinated unfolded substrates was not affected. Increased RPN13 polyubiquitination in cells was also observed during heat-shock, arsenite-induced oxidative stress, 19S ATPase inhibition, or RPN11 deubiquitination inhibition (Besche et al., 2014). These conditions have in common that they cause accumulation of polyubiquitinated and/or damaged proteins. Triggering ubiquitination of RPN13 by UBE3C, may prevent the binding of additional substrates to stalled proteasomes caused by protein overload, protein aggregates or damaged proteins. This might prevent damage to the proteasome, but under proteotoxic stress it may contribute to further accumulation of proteins.
The dramatic decrease in the degradation of polyubiquitinated proteins by purified 26S proteasomes upon RPN13 polyubiquitination was not expected, since substrates could still be recognized by the unaffected ubiquitin-receptor RPN10. In yeast, RPN10 and RPN13 contributed equally to the binding of ubiquitin chains (Peth et al., 2010), and in mice, RPN10 knockout was more severe than RPN13 knockout, indicating that the contribution of RPN10 is even more important than RPN13 in mammals (Hamazaki et al., 2007, 2015). Therefore, it is suggested that modified RPN13 may negatively affect RPN10. Thus, polyubiquitination of RPN13 results in decreased substrate binding, and thereby it suppresses the degradation of polyubiquitinated proteins.
2.5. Localization Related PMTs of the 19S Subunits
2.5.1. N-myristoylation of RPT2
N-myristoylation is the irreversible linkage of myristic acid, a 14-carbon saturated fatty acid, to N-terminal glycine residues of proteins (Martin et al., 2011). This protein lipidation allows hydrophobic interactions with other proteins or membrane lipids, and plays a role in intracellular localization. N-myristoylation of the RPT2-subunit is involved in the intracellular localization of the 26S proteasome in yeast, without affecting proteasome assembly or activity (Kimura et al., 2012, 2016). In yeast cells expressing a non-myristoylated RPT2 mutant, there was a redistribution of the 26S proteasome from the nucleus to the cytoplasm (Kimura et al., 2012). The altered localization caused an accumulation of polyubiquitinated proteins in the nucleus, especially accumulation of nucleo-cytoplasmic proteins (Kimura et al., 2012, 2016). In yeast the nucleus is the major site of proteasome activity, and therefore, misfolded proteins are generally degraded in the nucleus (Prasad et al., 2010). These findings indicate that nuclear RPT2 myristoylated proteasomes play a role in the degradation of proteins in the nucleus, including misfolded proteins localized in the cytoplasm (Kimura et al., 2016). Whether nuclear degradation of cytoplasmic proteins can also be observed in mammalian cells, is not known, although it has been shown that human and mice RPT2 can be N-myristoylated (Gomes et al., 2006; Wang et al., 2007). Thus, it seems likely that the N-myristoylation of RPT2 inhibits transport of the nuclear proteasome into the cytoplasm, and therefore, is involved in the degradation of proteins in the nucleus.
2.5.2. Phosphorylation of RPN8
It has been found that phosphorylation of RPN8 is involved in the localization of the subunit in breast epithelial cell lines (Thompson et al., 2004). Unmodified RPN8 was observed throughout the nucleus and cytoplasm, whereas the modified form was localized mainly in the cytoplasm. However, in malignant breast epithelial cell lines unmodified RPN8 was only present in the cytoplasm, and the modified form was not found. This suggests that there is only in the normal cell lines a mechanism that regulates nuclear localization of RPN8. Thus, RPN8 phosphorylation, and thereby localization seems to be dysregulated in cancer. Importantly, phosphorylation of RPN8 was in both cell lines induced by proteasome inhibition (Thompson et al., 2004).
Unmodified RPN8 associated with the 26S proteasome, whereas the modified form did not (Thompson et al., 2004). It has been shown that modified RPN8 can associate with (19S-unincorparated) RPN7, and likely also with other 19S-subunits (Thompson et al., 2004). Since there is a difference between the phosphorylation state of RPN8 in normal and malignant cells, the possibility raises that RPN8 regulates transcription in association with other 19S-subunits, supported by several studies that indicate that proteasome 19S-subunits have a role in gene expression, and interact with transcription regulators (Yanagi et al., 2000; Ferdous et al., 2002; Kang et al., 2002; Maganti et al., 2014). In conclusion, RPN8 can be phosphorylated, which is associated with cellular localization, and its association with the proteasome.
3. Modulation of the Proteasome at Transcriptional Level
Besides modifications and modulations acting directly on the proteasomal complexes, proteasome activity can also be regulated at the transcriptional level. Alterations in the expression level of proteasome subunits can modify the overall capacity and selectivity to degrade proteins dramatically. In this section important signaling pathways that affect proteasome gene expression under different conditions will be discussed: IFN-γ, NF-κB, NF-E2-related factor-2 (NRF2), and NRF1 (summarized in Table 4).

Table 4. Signaling pathways that effect proteasome transcription.
3.1. Inflammatory Pathways
IFN-γ is an important activator of the immune response. It induces the expression of genes encoding proteasome subunits, or proteins associated with proteasome function. The expression of immunoproteasome subunits, PA28αβ, and MHC class I genes seems to be induced by IFN-γ via the JAK-STAT pathway (Zhou, 2009; Johnston-Carey et al., 2016; Moritz et al., 2017). Binding of IFN-γ to its receptor results in the phosphorylation of JAK1 and JAK2 (Zhou, 2009). The activated JAKs phosphorylate the receptor on specific tyrosine residues, thereby recruiting STAT1, which will lead to the dimerization and phosphorylation of STAT1. Thereafter, STAT1 will translocate into the nucleus, where it initiates the expression of IFN regulatory factor-1 (IRF-1), which induces the expression of immunoproteasome subunits, PA28αβ and MHC class I genes (Zhou, 2009; Johnston-Carey et al., 2016). Importantly, STAT1 knockout mice have lower basal expression of immunoproteasomes and PA28αβ, suggesting that basal levels are also regulated by the JAK-STAT pathway in vivo (Barton et al., 2002).
IFN-γ can trigger oxidative stress in cells (Watanabe et al., 2003; Seifert et al., 2010). This may be the result of increased NADPH oxidase 1 and 4 (NOX1 and NOX4) expression, via the same JAK-STAT pathway, as shown in human aortic smooth muscle cells, large intestinal epithelial cells and renal mesangial cells (Kuwano et al., 2006; Moriwaki et al., 2006; Manea et al., 2009). It seems that oxidative stress can further stimulate the JAK-STAT pathway since H2O2 also activates this pathway (Yu et al., 2006; Shimizu et al., 2008; Johnston-Carey et al., 2016). Therefore, it may enhance, in addition to IFN-γ, the expression of immunoproteasome subunits, PA28αβ and MHC class I genes (Zhou, 2009; Grimm et al., 2012; Johnston-Carey et al., 2016).
In addition, IFN-γ enhances translation of various genes via the AKT-mTOR pathway, which will increase the levels of nascent proteins, but also that of incorrectly folded proteins, known as Defective Ribosomal Products (DRiPs) (Kaur et al., 2008; Seifert et al., 2010). Damaged (newly synthesized) proteins caused by IFN-γ-induced free radicals will further enhance the pool of aggregation prone DRiPs (Seifert et al., 2010; van Deventer and Neefjes, 2010). 26S immunoproteasomes are essential for the degradation of these DRiPs which are partly polyubiquitinated, likely through the IFN-γ-induced upregulation of E3 ligases and E2 ligase UBE2L6 (Schubert et al., 2000; Seifert et al., 2010). There is evidence that the peptides generated after DRiP degradation, are ligands for MHC class I molecules (Reits et al., 2000; Schubert et al., 2000; Qian et al., 2006; Cardinaud et al., 2010; Dolan et al., 2011), although it is not clear how much DRiPs contribute to antigen presentation (Rock et al., 2014). In conclusion, IFN-γ upregulates gene expression of proteasome subunits via several pathways, thereby supporting MHC class I antigen presentation and preventing aggregate formation, which results from the increased pool of oxidized and nascent proteins (van Deventer and Neefjes, 2010).
The immunoproteasome subunits may also be upregulated via the NF-κB pathway (Johnston-Carey et al., 2016). Oxidative stress provokes the phosphorylation of protein kinase D (PKD) via the SRC-ABL pathway, which then causes the degradation of inhibitor κB (IκBα) (Storz and Toker, 2003). This results in the activation of NF-κB, which translocates into the nucleus where it induces the expression of immunoproteasome genes in cooperation with IRF-1 (Moschonas et al., 2008). This also implicates that oxidative stress can increase the expression of immunoproteasomes in the absence of an immune response, which seems consistent with the role of immunoproteasomes in the degradation of oxidized proteins (Pickering et al., 2010). In addition, it was reported that during bacterial infection, Toll-like receptor (TLR) ligands upregulate the expression of PA28γ via the NF-κB pathway in macrophages (Sun et al., 2016). In turn, PA28γ promotes NF-κB transcriptional activity by destabilizing suppressor KLF2. This positive feedforward mechanism may be important for effective defense against bacterial pathogens.
3.2. NRF2
The NRF2 pathway is important in the protection against oxidative stress (Taguchi et al., 2011; Tonelli et al., 2018). Under normal conditions, NRF2 is constantly polyubiquitinated via KEAP1-dependent ubiquitin conjugation, and subsequently degraded by the 26S proteasome (Kobayashi et al., 2004). Upon oxidative stress, the highly reactive cysteine residues of KEAP1 are oxidized, which causes dissociation of the KEAP1-NRF2 complex (Sekhar et al., 2010). The free NRF2 translocates into the nucleus where it heterodimerizes with small MAF proteins and binds to antioxidant/electrophile response elements (AREs or EPREs) (Taguchi et al., 2011). AREs are located in the promoter of various stress response genes, including subunits of the proteasome (Kwak et al., 2003a; Steffen et al., 2010; Pickering et al., 2012). It was shown that H2O2 treatment induces binding of NRF2 to the ARE of the β5-subunit gene, which increased the mRNA levels of this subunit (Pickering et al., 2012). It seems likely that NRF2 will also bind to the AREs of other proteasome subunits, since the enhanced expression of the 20S proteasome and PA28αβ during oxidative stress is NRF2-dependent (Pickering et al., 2012). In addition, NRF2 inducers could upregulate proteasome subunit levels and activity in mammalian fibroblasts and mouse liver tissue (Kwak et al., 2003a,b; Kapeta et al., 2010). Thus, proteasome subunit induction by the NRF2-pathway seems to increase the capacity of the cell to degrade damaged and oxidized proteins. NRF2 levels were increased in different mouse tissues in response to oxidative damage by air pollution, which may explain the accompanied elevation of 20S and immunoproteasome levels, contributing to oxidative stress adaptation Pomatto et al. (2018). However, this model was contradicted by the study of Pickering et al. (2012) which concluded that the expression of immunosubunits was not regulated by NRF2.
3.3. NRF1
Mild proteasome inhibition results in enhanced expression of proteasome genes, accompanied by increased proteasome protein synthesis and complex formation, which may compensate for the reduced proteasome activity (Meiners et al., 2003; Welk et al., 2016). Transcription factor NRF1 has been described as an important regulator of proteasome gene expression in response to proteasome inhibition in mammalian cells (Radhakrishnan et al., 2010). Under normal conditions, NRF1 is ER-bound and is continuously degraded via the ER-associated degradation pathway (ERAD), requiring the E3 ligase HRD1, p97 and the 26S proteasome (Steffen et al., 2010). However, when the proteasome is partially inhibited, NRF1 is proteolytically processed into a transcriptionally active form and translocates into the nucleus (Steffen et al., 2010; Sha and Alfred Goldberg, 2014). Interestingly, high concentrations of proteasome inhibitors could not induce the expression of proteasome subunits, implying completely blocked NRF1 processing by the proteasome (Sha and Alfred Goldberg, 2014). However, Sha and Goldberg (2016) showed that high concentrations of proteasome inhibitors caused NRF1 to accumulate into aggregates, thereby losing its potency to initiate transcription. Instead, it was shown that NRF1 processing does not depend on proteasomes (Sha and Goldberg, 2016; Vangala et al., 2016), but requires aspartyl proteases DDI-1/2 (Koizumi et al., 2016; Lehrbach and Ruvkun, 2016; Xiang et al., 2018). Since the enhanced transcription is dependent on ubiquitination, it was proposed that mild proteasome inhibition caused accumulation of ubiquitin moieties on NRF1. DDI2 would then bind the ubiquitin via its UBL domain and facilitate NRF1 processing (Koizumi et al., 2016). After NRF1 processing and nuclear translocation, NRF1 and the TIP60 chromatin-regulatory complex are co-recruited to the ARE-containing promoter regions of proteasome genes (Vangala and Radhakrishnan, 2019). Thereby upregulating the mRNA levels of all 26S proteasome subunits and PA200 and subsequently increasing their protein levels (Meiners et al., 2003; Radhakrishnan et al., 2010; Steffen et al., 2010; Tsuchiya et al., 2013; Sha and Alfred Goldberg, 2014; Welk et al., 2016). NRF1 also regulates the expression of its regulators p97, HRD1 and DDI creating positive and negative feedback circuits between NRF1 and these target genes (Sha and Alfred Goldberg, 2014; Xiang et al., 2018). Since NRF1 regulates the proteasome content in the cell, it is a potential therapeutic target for diseases associated with impaired or enhanced proteasome degradation by inducing or suppressing NRF1 activation, respectively (Bott et al., 2016; Weyburne et al., 2017).
NRF2 levels are also increased under proteasome inhibition, although a significant impact of NRF2 on gene-expression of proteasome genes has not been detected under this condition (Steffen et al., 2010). However, NRF2 activation upon proteasome inhibition supports the survival of cancer cells, suggesting that the NRF2 attenuates the anti-tumor efficacy of proteasome inhibitors (Lee et al., 2018; Sun et al., 2018). NRF1 and NRF2 thus differ in function, which is manifested by their different effects on proteasome gene expression. The upregulation of distinct and overlapping proteasome subunits by NRF1 and NRF2 via AREs may be explained by a difference in the binding capacity of the transcription factors to the AREs, which might also be condition-dependent (Steffen et al., 2010; Koch et al., 2011).
Conclusion
As illustrated by the various possible modifications, proteasomes are far from being static complexes. Continuous stimuli from internal and external factors ensure a constant adaptation of proteasomes toward their cellular environment. As shown in Table 5, various conditions can lead to multiple PMTs of the proteasome. The modifications may act together to activate or inhibit the proteasome, but can also have opposite effects, illustrating the dynamic regulation of the proteasome. Since proteasome complex alterations lead to altered substrate specificities and activities to cope with specific conditions, it makes the proteasome a promising target for therapies. By forcing the proteasome pool toward specific complexes or alter activity via particular modifications, degradation capacity and specificity can be altered in particular cellular processes, thereby improving or reducing the degradation of disease-related proteins. In addition, focussing on key components involved in substrate targeting toward proteasomes would also be beneficial in a search for therapies involving proteasome degradation.
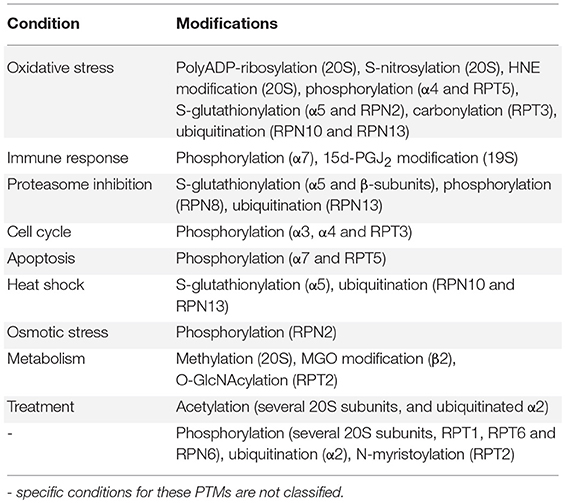
Table 5. Proteasome PTMs arranged according to their specific condition to which they are associated.
Author Contributions
SK wrote the review and made the figures and tables. KG updated literature and did last editing. ER was involved in revision. SS-K supervised and revised manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adelöf, J., Andersson, M., Porritt, M., Petersen, A., Zetterberg, M., Wiseman, J., et al. (2018). PA28αβ overexpression enhances learning and memory of female mice without inducing 20S proteasome activity. BMC Neurosci. 19:70. doi: 10.1186/s12868-018-0468-2
Ali, A., Wang, Z., Fu, J., Ji, L., Liu, J., Li, L., et al. (2013). Differential regulation of the REGγ-proteasome pathway by p53/TGF-β signalling and mutant p53 in cancer cells. Nat. Commun. 4, 8228–8242. doi: 10.1038/ncomms3667
Allaman, I., Bélanger, M., and Magistretti, P. J. (2015). Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 9, 23–23. doi: 10.3389/fnins.2015.00023
Asher, G., Tsvetkov, P., Kahana, C., and Shaul, Y. (2005). A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 19, 316–321. doi: 10.1101/gad.319905
Baldin, V., Militello, M., Thomas, Y., Doucet, C., Fic, W., Boireau, S., et al. (2008). A novel role for PA28gamma-proteasome in nuclear speckle organization and SR protein trafficking. Mol. Biol. Cell 19, 1706–1716. doi: 10.1091/mbc.e07-07-0637
Banerjee, S., Ji, C., Mayfield, J. E., Goel, A., Xiao, J., Dixon, J. E., et al. (2018). Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA. 115, 8155–8160. doi: 10.1073/pnas.1806797115
Bard, J. A. M., Goodall, E. A., Greene, E. R., Jonsson, E., Dong, K. C., and Martin, A. (2018). Structure and function of the 26S Proteasome. 87, 697–724. doi: 10.1146/annurev-biochem-062917-011931
Bardag-Gorce, F., French, B. A., Nan, L., Song, H., Nguyen, S. K., Yong, H., et al. (2006). CYP2E1 induced by ethanol causes oxidative stress, proteasome inhibition and cytokeratin aggresome (Mallory body-like) formation. Exp. Mol. Pathol. 81, 191–201. doi: 10.1016/j.yexmp.2006.07.007
Bardag-Gorce, F., and French, S. W. (2011). Delta-aminolevulinic dehydratase is a proteasome interacting protein. Exp. Mol. Pathol. 91, 485–489. doi: 10.1016/j.yexmp.2011.05.003
Barton, L. F., Cruz, M., Rangwala, R., Deepe, G. S. Jr., and Monaco, J. J. (2002). Regulation of immunoproteasome subunit expression in vivo following pathogenic fungal infection. J. Immunol. 169, 3046–3052. doi: 10.4049/jimmunol.169.6.3046
Baugh, J. M., Viktorova, E. G., and Pilipenko, E. V. (2009). Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol. 386, 814–827. doi: 10.1016/j.jmb.2008.12.081
Besche, H. C., Sha, Z., Kukushkin, N. V., Peth, A., Hock, E. M., Kim, W., et al. (2014). Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 33, 1159–1176. doi: 10.1002/embj.201386906
Bingol, B., Wang, C. F., Arnott, D., Cheng, D., Peng, J., and Sheng, M. (2010). Autophosphorylated CaMKIIα acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578. doi: 10.1016/j.cell.2010.01.024
Biswas, K., Sarkar, S., Du, K., Brautigan, D. L., Abbas, T., and Larner, J. M. (2017). The E3 ligase CHIP mediates p21 degradation to maintain radioresistance. Mol. Cancer Res. 15, 651–659. doi: 10.1158/1541-7786.MCR-16-0466
Blickwedehl, J., Agarwal, M., Seong, C., Pandita, R. K., Melendy, T., Sung, P., et al. (2008). Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc. Natl. Acad. 105, 16165–16170. doi: 10.1073/pnas.0803145105
Blickwedehl, J., Olejniczak, S., Cummings, R., Sarvaiya, N., Mantilla, A., Chanan-Khan, A., et al. (2012). The proteasome activator PA200 regulates tumor cell responsiveness to glutamine and resistance to ionizing radiation. Mol. Cancer Res. 10, 937–944. doi: 10.1158/1541-7786.MCR-11-0493-T
Bose, S., Stratford, F. L. L., Broadfoot, K. I., Mason, G. F., and Rivett, A. J. (2004). Phosphorylation of 20S proteasome alpha subunit C8 (α7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma-interferon. Biochem. J. 378, 177–184. doi: 10.1042/bj20031122
Bott, L. C., Badders, N. M., Chen, K. L., Harmison, G. G., Bautista, E., Shih, C. C., et al. (2016). A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 25, 1979–1989. doi: 10.1093/hmg/ddw073
Broillet, M. C. (1999). S-Nitrosylation of proteins. Cell. Mol. Life Sci. 55, 1036–1042. doi: 10.1007/s000180050354
Bulteau, A. L., Lundberg, K. C., Humphries, K. M., Sadek, H. A., Szweda, P. A., Friguet, B., et al. (2001). Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J. Biol. Chem. 276, 30057–30063. doi: 10.1074/jbc.M100142200
Cardinaud, S., Starck, S. R., Chandra, P., and Shastri, N. (2010). The synthesis of truncated polypeptides for immune surveillance and viral evasion. PLoS ONE 5:e8692. doi: 10.1371/journal.pone.0008692
Cascio, P. (2014). PA28αβ: the enigmatic magic ring of the proteasome? Biomolecules 4, 566–584. doi: 10.3390/biom4020566
Cascio, P., Call, M., Petre, B. M., Walz, T., Goldberg, A. L., Adams, G. M., et al. (2002). Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 21, 2636–2645. doi: 10.1093/emboj/21.11.2636
Cascio, P., Hilton, C., Kisselev, A. F., Rock, K. L., and Goldberg, A. L. (2001). 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J. 20, 2357–2366. doi: 10.1093/emboj/20.10.2357
Catalgol, B., Wendt, B., Grimm, S., Breusing, N., Özer, N. K., and Grune, T. (2010). Chromatin repair after oxidative stress: role of PARP-mediated proteasome activation. Free Rad. Biol. Med. 48, 673–680. doi: 10.1016/j.freeradbiomed.2009.12.010
Chai, F., Liang, Y., Bi, J., Chen, L., Zhang, F., Cui, Y., et al. (2014). High expression of REGgamma is associated with metastasis and poor prognosis of patients with breast cancer. Int. J. Clin. Exp. Pathol. 7, 7834–7843.
Chen, L., Brewer, M. D., Guo, L., Wang, R., Jiang, P., and Yang, X. (2017). Enhanced degradation of misfolded proteins promotes tumorigenesis. Cell Rep. 18, 3143–3154. doi: 10.1016/j.celrep.2017.03.010
Chen, L., and Madura, K. (2005). Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 65, 5599–5606. doi: 10.1158/0008-5472.CAN-05-0201
Chen, S., Wu, J., Lu, Y., Ma, Y. B., Lee, B. H., Yu, Z., et al. (2016). Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. 113, 12991–12996. doi: 10.1073/pnas.1614614113
Chiang, P. K., Gordon, R. K., Tal, J., Zeng, G. C., Docto, B. P., Pardhasaradhi, K., et al. (1996). S-Adenosylmethionine and methylation. FASEB J. 10, 471–480. doi: 10.1096/fasebj.10.4.8647346
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Ciechanover, A., and Schwartz, A. L. (2004). The ubiquitin system: Pathogenesis of human diseases and drug targeting. Biochim. et Biophys. Acta 1695, 3–17. doi: 10.1016/j.bbamcr.2004.09.018
Cioce, M., Boulon, S., Matera, A. G., and Lamond, A. I. (2006). UV-induced fragmentation of Cajal bodies. J. Cell Biol. 175, 401–413. doi: 10.1083/jcb.200604099
Cohen, P. (2002). The origins of protein phosphorylation. Nat. Cell Biol. 4 E127–E130. doi: 10.1038/ncb0502-e127
Cohen, S., Nathan, J. A., and Goldberg, A. L. (2015). Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug. Discov. 14, 58–74. doi: 10.1038/nrd4467
Collins, G. A., and Goldberg, A. L. (2017). The logic of the 26S proteasome. Cell 169, 792–806. doi: 10.1016/j.cell.2017.04.023
Crosas, B., Hanna, J., Kirkpatrick, D. S., Zhang, D. P., Tone, Y., Hathaway, N. A., et al. (2006). Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligasivitiese and deubiquitinating act. Cell 127, 1401–1413. doi: 10.1016/j.cell.2006.09.051
Dahlmann, B. (2016). Mammalian proteasome subtypes: their diversity in structure and function. Arch. Biochem. Biophys. 591, 132–140. doi: 10.1016/j.abb.2015.12.012
Dahlmann, B., Hershko, A., Ciechanover, A., Hough, R., Pratt, G., Rechsteiner, M., et al. (2007). Role of proteasomes in disease. BMC Biochem. 8, S3–S3. doi: 10.1186/1471-2091-8-S1-S3
Dalle-Donne, I., Aldini, G., Carini, M., Colombo, R., Rossi, R., and Milzani, A. (2006). Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 10, 389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x
Dange, T., Smith, D., Noy, T., Rommel, P. C., Jurzitza, L., Cordero, R. J., et al. (2011). Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem. 286, 42830–42839. doi: 10.1074/jbc.M111.300178
Dantuma, N. P., and Bott, L. C. (2014). The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci. 7:70. doi: 10.3389/fnmol.2014.00070
David, D. C., Layfield, R., Serpell, L., Narain, Y., Goedert, M., and Spillantini, M. G. (2002). Proteasomal degradation of tau protein. J. Neurochem. 83, 176–185. doi: 10.1046/j.1471-4159.2002.01137.x
Davies, K. J. A. (2001). Degradation of oxidized proteins by the 20S proteasome. Biochimie 83, 301–310. doi: 10.1016/S0300-9084(01)01250-0
de Poot, S. A. H., Tian, G., and Finley, D. (2017). Meddling with fate: the proteasomal deubiquitinating enzymes. J. Mol. Biol. 429, 3525–3545. doi: 10.1016/j.jmb.2017.09.015
Demasi, M., and da Cunha, F. M. (2018). The physiological role of the free 20S proteasome in protein degradation: a critical review. Biochim. et Biophys. Acta. Gen. Sub. 1862, 2948–2954. doi: 10.1016/j.bbagen.2018.09.009
Demasi, M., Hand, A., Ohara, E., Oliveira, C. L., Bicev, R. N., Bertoncini, C. A., et al. (2014). 20S proteasome activity is modified via S-glutathionylation based on intracellular redox status of the yeast Saccharomyces cerevisiae: implications for the degradation of oxidized proteins. Arch. Biochem. Biophys. 557, 65–71. doi: 10.1016/j.abb.2014.05.002
Demasi, M., Shringarpure, R., and Davies, K. J. A. (2001). Glutathiolation of the proteasome is enhanced by proteolytic inhibitors. Arch. Biochem. Biophys. 389, 254–263. doi: 10.1006/abbi.2001.2332
Demasi, M., Silva, G. M., and Netto, L. E. S. (2003). 20 S proteasome from Saccharomyces cerevisiae is responsive to redox modifications and is S-glutathionylated. J. Biol. Chem. 278, 679–685. doi: 10.1074/jbc.M209282200
Djakovic, S. N., Marquez-Lona, E. M., Jakawich, S. K., Wright, R., Chu, C., Sutton, M. A., et al. (2012). Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 32, 5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012
Djakovic, S. N., Schwarz, L. A., Barylko, B., DeMartino, G. N., and Patrick, G. N. (2009). Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 284, 26655–26665. doi: 10.1074/jbc.M109.021956
Dolan, B. P., Li, L., Veltri, C. A., Ireland, C. M., Bennink, J. R., and Yewdell, J. W. (2011). Distinct pathways generate peptides from defective ribosomal products for CD8+ T cell immunosurveillance. J Immunol. 186, 2065–72. doi: 10.4049/jimmunol.1003096
Dudek, E. J., Shang, F., Valverde, P., Liu, Q., Hobbs, M., and Taylor, A. (2005). Selectivity of the ubiquitin pathway for oxidatively modified proteins: relevance to protein precipitation diseases. FASEB J. 19, 1707–1709. doi: 10.1096/fj.05-4049fje
Elsasser, S., Chandler-Militello, D., Müller, B., Hanna, J., and Finley, D. (2004). Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822. doi: 10.1074/jbc.M404020200
Elsasser, S., and Finley, D. (2005). Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 7, 742–749. doi: 10.1038/ncb0805-742
Esterbauer, H., Schaur, R. J., and Zollner, H. (1991). Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Rad. Biol. Med. 11, 81–128. doi: 10.1016/0891-5849(91)90192-6
Fabre, B., Lambour, T., Garrigues, L., Ducoux-Petit, M., Amalric, F., Monsarrat, B., et al. (2014). Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 13, 3027–3037. doi: 10.1021/pr500193k
Fan, K., Wang, F., Li, Y., Chen, L., Gao, Z., Zhang, Y., et al. (2019). CRL4(DCAF2) is required for mature T-cell expansion via Aurora B-regulated proteasome activity. J. Autoimmun. 96, 74–85. doi: 10.1016/j.jaut.2018.08.006
Fang, S., Jensen, J. P., Ludwig, R. L., Vousden, K. H., and Weissman, A. M. (2000). Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 275, 8945–8951. doi: 10.1074/jbc.275.12.8945
Farout, L., Mary, J., Vinh, J., Szweda, L. I., and Friguet, B. (2006). Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch. Biochem. Biophys. 453, 135–142. doi: 10.1016/j.abb.2006.02.003
Feng, Y., Longo, D. L., and Ferris, D. K. (2001). Polo-like kinase interacts with proteasomes and regulates their activity. Cell Growth Diff. 12, 29–37.
Ferdous, A., Kodadek, T., and Johnston, S. A. (2002). A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 41, 12798–12805. doi: 10.1021/bi020425t
Ferrington, D. A., and Kapphahn, R. J. (2004). Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 578, 217–223. doi: 10.1016/j.febslet.2004.11.003
Ferrington, D. A., Sun, H., Murray, K. K., Costa, J., Williams, T. D., Bigelow, D. J., et al. (2001). Selective degradation of oxidized calmodulin by the 20 S proteasome. J. Biol. Chem. 276, 937–943. doi: 10.1074/jbc.M005356200
Florea, B. I., Verdoes, M., Li, N., van der Linden, W. A., Geurink, P. P. H., et al. (2010). Activity-based profiling reveals reactivity of the murine thymoproteasome-specific subunit beta5t. Chem. Biol. 17, 795–801. doi: 10.1016/j.chembiol.2010.05.027
Fort, P., Kajava, A. V., Delsuc, F., and Coux, O. (2015). Evolution of proteasome regulators in eukaryotes. Genome Biol. Evol. 7, 1363–1379. doi: 10.1093/gbe/evv068
Früh, K., and Yang, Y. (1999). Antigen presentation by MHC class I and its regulation by interferon γ. Curr. Opin. Immunol. 11, 76–81. doi: 10.1016/S0952-7915(99)80014-4
Gaczynska, M., Rock, K. L., and Goldberg, A. L. (1993). Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 365, 264–267. doi: 10.1038/365264a0
Gersch, M., Mathias Hackl, W., Dubiella, C., Dobrinevski, A., Groll, M., and Stephan Sieber, A. (2015). A mass spectrometry platform for a streamlined investigation of proteasome integrity, posttranslational modifications, and inhibitor binding. Chem. Biol. 22, 404–411. doi: 10.1016/j.chembiol.2015.01.004
Gilda, J. E., and Gomes, A. V. (2017). Proteasome dysfunction in cardiomyopathies. J. Physiol. 595, 4051–4071. doi: 10.1113/JP273607
Glickman, M. H., and Ciechanover, A. (2002). The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428. doi: 10.1152/physrev.00027.2001
Gomes, A. V., Zong, C., Edmondson, R. D., Li, X., Stefani, E., Zhang, J., et al. (2006). Mapping the murine cardiac 26S proteasome complexes. Circ. Res. 99, 362–371. doi: 10.1161/01.RES.0000237386.98506.f7
Grimm, S., Ott, C., Hörlacher, M., Weber, D., Höhn, A., and Grune, T. (2012). Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem. J. 448, 127–139. doi: 10.1042/BJ20120298
Groll, M., Glickman, M. H., Finley, D., Bajorek, M., Köhler, A., Moroder, L., et al. (2000). A gated channel into the proteasome core particle. Nat. Struct. Biol. 7, 1062–1067. doi: 10.1038/80992
Grune, T., Catalgol, B., Licht, A., Ermak, G., Pickering, A. M., Ngo, J. K., et al. (2011). HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 51, 1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015
Grune, T., Reinheckel, T., and Davies, K. J. (1996). Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. J. Biol. Chem. 271, 15504–15509. doi: 10.1074/jbc.271.26.15504
Guo, G. G., Gu, M., and Etlinger, J. D. (1994). 240-kDa proteasome inhibitor (CF-2) is identical to delta-aminolevulinic acid dehydratase. J. Biol. Chem. 269, 12399–12402.
Guo, X., Engel, J. L., Xiao, J., Tagliabracci, V. S., Wang, X., Huang, L., et al. (2011). UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc. Natl. Acad. Sci. U.S.A. 108, 18649–18654. doi: 10.1073/pnas.1113170108
Guo, X., Huang, X., and Chen, M. J. (2017). Reversible phosphorylation of the 26S proteasome. Protein Cell 8, 255–272. doi: 10.1007/s13238-017-0382-x
Guo, X., Wang, X., Wang, Z., Banerjee, S., Yang, J., Huang, L., et al. (2016). Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 18, 202–212. doi: 10.1038/ncb3289
Hamazaki, J., Hirayama, S., Murata, S., Glickman, M. H., Ciechanover, A., Glickman, M. H., et al. (2015). Redundant roles of Rpn10 and Rpn13 in recognition of ubiquitinated proteins and cellular homeostasis. PLoS Genet. 11:e1005401. doi: 10.1371/journal.pgen.1005401
Hamazaki, J., Sasaki, K., Kawahara, H., Hisanaga, S. I., Tanaka, K., and Murata, S. (2007). Rpn10-mediated degradation of ubiquitinated proteins is essential for mouse development. Mol. Cell. Biol. 27, 6629–6638. doi: 10.1128/MCB.00509-07
Hamilton, A. M., Oh, W. C., Vega-Ramirez, H., Stein, I. S., Hell, J. W., Patrick, G. N., et al. (2012). Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74, 1023–1030. doi: 10.1016/j.neuron.2012.04.031
Hanna, J., Guerra-Moreno, A., Ang, J., and Micoogullari, Y. (2019). Protein degradation and the pathologic basis of disease. Am. J. Pathol. 189, 94–103. doi: 10.1016/j.ajpath.2018.09.004
Harshbarger, W., Miller, C., Diedrich, C., and Sacchettini, J. (2015). Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 23, 418–424. doi: 10.1016/j.str.2014.11.017
Hill, B. G., and Bhatnagar, A. (2012). Protein S-glutathiolation: Redox-sensitive regulation of protein function. J. Mol. Cell. Cardiol. 52, 559–567. doi: 10.1016/j.yjmcc.2011.07.009
Hirano, H., Kimura, Y., and Kimura, A. (2016). Biological significance of co- and post-translational modifications of the yeast 26S proteasome. J. Proteomics 134, 37–46. doi: 10.1016/j.jprot.2015.11.016
Huang, L., Haratake, K., Miyahara, H., and Chiba, T. (2016). Proteasome activators, PA28gamma and PA200, play indispensable roles in male fertility. Sci. Rep. 6:23171. doi: 10.1038/srep23171
Huber, E. M., and Groll, M. (2017). The Mammalian Proteasome activator PA28 forms an asymmetric alpha4beta3 complex. Structure 25, 1473–1480.e3. doi: 10.1016/j.str.2017.07.013
Husnjak, K., Elsasser, S., Zhang, N., Chen, X., Randles, L., Shi, Y., et al. (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. doi: 10.1038/nature06926
Isasa, M., Katz, E. J., Kim, W., Yugo, V., González, S., Kirkpatrick, D. S., et al. (2010). Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome. Mol. Cell 38, 733–745. doi: 10.1016/j.molcel.2010.05.001
Ishii, T., Sakurai, T., Usami, H., and Uchida, K. (2005). Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry 44, 13893–13901. doi: 10.1021/bi051336u
Jarome, T. J., Kwapis, J. L., Ruenzel, W. L., and Helmstetter, F. J. (2013). CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 7, 115–115. doi: 10.3389/fnbeh.2013.00115
Johnston-Carey, H. K., Pomatto, L. C. D., and Davies, K. J. A. (2016). The immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 51, 268–281. doi: 10.3109/10409238.2016.1172554
Jonik-Nowak, B., Menneteau, T., Fesquet, D., Baldin, V., Bonne-Andrea, C., Méchali, F., et al. (2018). PIP30/FAM192A is a novel regulator of the nuclear proteasome activator PA28γ. Proc. Natl. Acad. Sci. U.S.A. 115:E6477. doi: 10.1073/pnas.1722299115
Kang, Z., Pirskanen, A., Jänne, O. A., and Palvimo, J. J. (2002). Involvement of proteasome in the dynamic assembly of the androgen receptor transcription complex. J. Biol. Chem. 277, 48366–48371. doi: 10.1074/jbc.M209074200
Kapadia, M. R., Eng, J. W., Jiang, Q., Stoyanovsky, D. A., and Kibbe, M. R. (2009). Nitric oxide regulates the 26S proteasome in vascular smooth muscle cells. Nitric Oxide 20, 279–288. doi: 10.1016/j.niox.2009.02.005
Kapeta, S., Chondrogianni, N., and Gonos, E. S. (2010). Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 285, 8171–8184. doi: 10.1074/jbc.M109.031575
Kastle, M., and Grune, T. (2011). Proteins bearing oxidation-induced carbonyl groups are not preferentially ubiquitinated. Biochimie 93, 1076–1079. doi: 10.1016/j.biochi.2011.03.004
Kastle, M., Reeg, S., Rogowska-Wrzesinska, A., and Grune, T. (2012). Chaperones, but not oxidized proteins, are ubiquitinated after oxidative stress. Free Radic. Biol. Med. 53, 1468–1477. doi: 10.1016/j.freeradbiomed.2012.05.039
Kaur, S., Sassano, A., Dolniak, B., Joshi, S., Majchrzak-Kita, B., Baker, D. P., et al. (2008). Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl. Acad. Sci. U.S.A. 105, 4808–4813. doi: 10.1073/pnas.0710907105
Kee, Y., Muñoz, W., Lyon, N., and Huibregtse, J. M. (2006). The deubiquitinating enzyme Ubp2 modulates Rsp5-dependent Lys63-linked polyubiquitin conjugates in Saccharomyces cerevisiae. J. Biol. Chem. 281, 36724–36731. doi: 10.1074/jbc.M608756200
Keembiyehetty, C. N., Krzeslak, A., Love, D. C., and Hanover, J. A. (2011). A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 124, 2851–2860. doi: 10.1242/jcs.083287
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer's disease. J. Neurochem. 75, 436–439. doi: 10.1046/j.1471-4159.2000.0750436.x
Kenniston, J. A., Baker, T. A., Fernandez, J. M., and Sauer, R. T. (2003). Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114, 511–520. doi: 10.1016/S0092-8674(03)00612-3
Keren-Kaplan, T., Zeev Peters, L., Levin-Kravets, O., Attali, I., Kleifeld, O., Shohat, N., et al. (2016). Structure of ubiquitylated-Rpn10 provides insight into its autoregulation mechanism. Nat. Commun. 7:12960. doi: 10.1038/ncomms12960
Khor, B., Bredemeyer, A. L., Huang, C. Y., Turnbull, I. R., Evans, R., Maggi, L. B., et al. (2006). Proteasome activator PA200 is required for normal spermatogenesis. Mol. Cell. Biol. 26, 2999–3007. doi: 10.1128/MCB.26.8.2999-3007.2006
Kim, H. T., and Goldberg, A. L. (2017). The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J. Biol. Chem. 292, 9830–9839. doi: 10.1074/jbc.M116.763128
Kim, H. T., and Goldberg, A. L. (2018). UBL domain of Usp14 and other proteins stimulates proteasome activities and protein degradation in cells. Proc. Natl. Acad. Sci. U.S.A. 115, E11642–e11650. doi: 10.1073/pnas.1808731115
Kim, Y. C., Li, X., Thompson, D., and DeMartino, G. N. (2013). ATP binding by proteasomal ATPases regulates cellular assembly and substrate-induced functions of the 26 S proteasome. J. Biol. Chem. 288, 3334–3345. doi: 10.1074/jbc.M112.424788
Kimura, A., Kato, Y., and Hirano, H. (2012). N-Myristoylation of the Rpt2 subunit regulates intracellular localization of the yeast 26S proteasome. Biochemistry 51, 8856–8866. doi: 10.1021/bi3007862
Kimura, A., Kurata, Y., Nakabayashi, J., Kagawa, H., and Hirano, H. (2016). N-Myristoylation of the Rpt2 subunit of the yeast 26S proteasome is implicated in the subcellular compartment-specific protein quality control system. J. Proteomics 130, 33–41. doi: 10.1016/j.jprot.2015.08.021
Kimura, H., Caturegli, P., Takahashi, M., and Suzuki, K. (2015). New insights into the function of the immunoproteasome in immune and nonimmune cells. J. Immunol. Res. 2015:541984. doi: 10.1155/2015/541984
Kisselev, A. F., Kaganovich, D., and Goldberg, A. L. (2002). Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the alpha-rings. J. Biol. Chem. 277, 22260–22270. doi: 10.1074/jbc.M112360200
Klaude, M., Fredriksson, K., Tjäder, I., Hammarqvist, F., Ahlman, B., Rooyackers, O., et al. (2007). Proteasome proteolytic activity in skeletal muscle is increased in patients with sepsis. Clin. Sci. 112, 499–506. doi: 10.1042/CS20060265
Kloetzel, P.-M. (2001). Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2, 179–188. doi: 10.1038/35056572
Kniepert, A., and Groettrup, M. (2014). The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 39, 17–24. doi: 10.1016/j.tibs.2013.10.004
Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., et al. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004
Koch, A., Steffen, J., and Krüger, E. (2011). TCF11 at the crossroads of oxidative stress and the ubiquitin proteasome system. Cell Cycle 10, 1200–1207. doi: 10.4161/cc.10.8.15327
Koizumi, S., Irie, T., Hirayama, S., Sakurai, Y., Yashiroda, H., Naguro, I., et al. (2016). The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 5:e18357. doi: 10.7554/eLife.18357
Kuo, C. L., and Goldberg, A. L. (2017). Ubiquitinated proteins promote the association of proteasomes with the deubiquitinating enzyme Usp14 and the ubiquitin ligase Ube3c. Proc. Natl. Acad. Sci. USA. 114 E3404–e3413. doi: 10.1073/pnas.1701734114
Kuwano, Y., Kawahara, T., Yamamoto, H., Teshima-Kondo, S., Tominaga, K., Masuda, K., et al. (2006). Interferon-γ activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 290, C433–C443. doi: 10.1152/ajpcell.00135.2005
Kwak, M. K., Wakabayashi, N., Greenlaw, J. L., Yamamoto, M., and Kensler, T. W. (2003a). Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 23, 8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003
Kwak, M. K., Wakabayashi, N., Itoh, K., Motohashi, H., Yamamoto, M., and Kensler, T. W. (2003b). Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 278, 8135–8145. doi: 10.1074/jbc.M211898200
Lander, G. C., Estrin, E., Matyskiela, M. E., Bashore, C., Nogales, E., and Martin, A. (2012). Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–196. doi: 10.1038/nature10774
Lasch, P., Petras, T., Ullrich, O., Backmann, J., Naumann, D., and Grune, T. (2001). Hydrogen peroxide-induced structural alterations of RNAse A. J. Biol. Chem. 276, 9492–9502. doi: 10.1074/jbc.M008528200
Lee, K. H., Lee, J., Woo, J., Lee, C. H., and Yoo, C. G. (2018). Proteasome inhibitor-induced IkappaB/NF-kappaB activation is mediated by Nrf2-dependent light chain 3B induction in lung cancer cells. Mol. Cells 41, 1008–1015. doi: 10.14348/molcells.2018.0277
Lee, S.-H., Park, Y., Yoon, S. K., and Yoon, J. B. (2010). Osmotic stress inhibits proteasome by p38 MAPK-dependent phosphorylation. J. Biol. Chem. 285, 41280–41289. doi: 10.1074/jbc.M110.182188
Lee, S. Y., Ramirez, J., Franco, M., Lectez, B., Gonzalez, M., Barrio, R., et al. (2014). Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life Sci. 71, 2747–2758. doi: 10.1007/s00018-013-1526-7
Leestemaker, Y., de Jong, A., Witting, K. F., Penning, R., Schuurman, K., Rodenko, B., et al. (2017). Proteasome activation by small molecules. Cell Chem. Biol. 24, 725–736.e7. doi: 10.1016/j.chembiol.2017.05.010
Lehrbach, N. J., and Ruvkun, G. (2016). Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 5:e17721. doi: 10.7554/eLife.17721
Leme, J. M. M., Ohara, E., Santiago, V. F., Barros, M. H., Netto, L. E. S., Pimenta, D. C., et al. (2019). Mutations of Cys and Ser residues in the α5-subunit of the 20S proteasome from Saccharomyces cerevisiae affects gating and chronological lifespan. Arch. Biochem. Biophys. 666, 63–72. doi: 10.1016/j.abb.2019.03.012
Levy-Barda, A., Lerenthal, Y., Davis, A. J., Chung, Y. M., Essers, J., Shao, Z., et al. (2011). Involvement of the nuclear proteasome activator PA28gamma in the cellular response to DNA double-strand breaks. Cell Cycle 10, 4300–4310. doi: 10.4161/cc.10.24.18642
Li, D., Dong, Q., Tao, Q., Gu, J., Cui, Y., Jiang, X., et al. (2015). c-Abl regulates proteasome abundance by controlling the ubiquitin-proteasomal degradation of PSMA7 subunit. Cell Rep. 10, 484–496. doi: 10.1016/j.celrep.2014.12.044
Li, J., Feng, X., Sun, C., Zeng, X., Xie, L., Xu, H., et al. (2015). Associations between proteasomal activator PA28gamma and outcome of oral squamous cell carcinoma: Evidence from cohort studies and functional analyses. EBioMedicine 2, 851–858. doi: 10.1016/j.ebiom.2015.07.004
Li, J., Powell, S. R., and Wang, X. (2011). Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J. 25, 883–93. doi: 10.1096/fj.10-160895
Li, X., Amazit, L., Long, W., Lonard, D. M., Monaco, J. J., and O'Malley, B. W. (2007). Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGγ-proteasome pathway. Mol. Cell 26, 831–842. doi: 10.1016/j.molcel.2007.05.028
Li, X., Lonard, D. M., Jung, S. Y., Malovannaya, A., Feng, Q., Qin, J., et al. (2006). The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGγ proteasome. Cell 124, 381–392. doi: 10.1016/j.cell.2005.11.037
Lin, J. T., Chang, W. C., Chen, H. M., Lai, H. L., Chen, C. Y., Tao, M. H., et al. (2013). Regulation of feedback between protein kinase A and the proteasome system worsens Huntington's disease. Mol. Cell. Biol. 33, 1073–1084. doi: 10.1128/MCB.01434-12
Liu, B., and Hong, J. S. (2003). Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for rherapeutic intervention. J. Pharmacol. Exp. Therapeut. 304, 1–7. doi: 10.1124/jpet.102.035048
Liu, C. W., Corboy, M. J., DeMartino, G. N., and Thomas, P. J. (2003). Endoproteolytic activity of the proteasome. Science 299, 408–411. doi: 10.1126/science.1079293
Liu, C. W., and Jacobson, A. D. (2013). Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 38, 103–110. doi: 10.1016/j.tibs.2012.11.009
Liu, C. W., Li, X., Thompson, D., Wooding, K., Chang, T. L., Tang, Z., et al. (2006). ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell 24, 39–50. doi: 10.1016/j.molcel.2006.08.025
Liu, H., Yu, S., Zhang, H., and Xu, J. (2014). Identification of nitric oxide as an endogenous inhibitor of 26S proteasomes in vascular endothelial cells. PLoS ONE 9:e98486. doi: 10.1371/journal.pone.0098486
Liu, X., Huang, W., Li, C., Li, P., Yuan, J., Li, X., et al. (2006). Interaction between c-Abl and Arg tyrosine kinases and proteasome subunit PSMA7 regulates proteasome degradation. Mol. Cell 22, 317–327. doi: 10.1016/j.molcel.2006.04.007
Lobanova, E. S., Finkelstein, S., Li, J., Travis, A. M., Hao, Y., Klingeborn, M., et al. (2018). Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration. Nat. Commun. 9:1738. doi: 10.1038/s41467-018-04117-8
Lokireddy, S., Kukushkin, N. V., and Goldberg, A. L. (2015). cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Nat. Acad. 112, E7176–E7185. doi: 10.1073/pnas.1522332112
Lopez, A. D., Tar, K., Krugel, U., Dange, T., Ros, I. G., and Schmidt, M. (2011). Proteasomal degradation of Sfp1 contributes to the repression of ribosome biogenesis during starvation and is mediated by the proteasome activator Blm10. Mol. Biol. Cell 22, 528–540. doi: 10.1091/mbc.e10-04-0352
Love, D. C., and Hanover, J. A. (2005). The hexosamine signaling pathway: deciphering the ‘O-GlcNAc code'. Sci. Signal. 2005:re13. doi: 10.1126/stke.3122005re13
Lundh, M., Bugliani, M., Dahlby, T., Chou, D. H., Wagner, B., Ghiasi, S. M., et al. (2017). The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J. Endocrinol. 233, 369–379. doi: 10.1530/JOE-17-0110
Madian, A. G., and Regnier, F. E. (2010). Proteomic identification of carbonylated proteins and their oxidation sites. J. Proteome Res. 9, 3766–3780. doi: 10.1021/pr1002609
Maganti, N., Moody, T. D., Truax, A. D., Thakkar, M., Spring, A. M., Germann, M. W., et al. (2014). Nonproteolytic roles of 19S ATPases in transcription of CIITApIV genes. PLoS ONE 9:e91200–e91200. doi: 10.1371/journal.pone.0091200
Magill, L., Lynas, J., Morris, T. C., Walker, B., and Irvine, A. E. (2004). Proteasome proteolytic activity in hematopoietic cells from patients with chronic myeloid leukemia and multiple myeloma. Haematologica 89, 1428–1433.
Mandemaker, I. K., Geijer, M. E., Kik, I., Bezstarosti, K., Rijkers, E., Raams, A., et al. (2018). DNA damage-induced replication stress results in PA200-proteasome-mediated degradation of acetylated histones. EMBO Rep. 19:e45566. doi: 10.15252/embr.201745566
Manea, A., Tanase, L. I., Raicu, M., and Simionescu, M. (2009). JAK/STAT signaling pathway pegulates Nox1 and Nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 30, 105–112. doi: 10.1161/ATVBAHA.109.193896
Mao, I., Liu, J., Li, X., and Luo, H. (2008). REGγ, a proteasome activator and beyond? Cell. Mol. Life Sci. 65, 3971–3980. doi: 10.1007/s00018-008-8291-z
Marcone, S. (2016). Evans, P., and Fitzgerald, D. J., 15-Deoxy-Delta(12,14)-prostaglandin J2 modifies components of the proteasome and inhibits inflammatory responses in human endothelial cells. Front. Immunol. 7:459. doi: 10.3389/fimmu.2016.00459
Marquez-Lona, E. M., Torres-Machorro, A. L., Gonzales, F. R., Pillus, L., and Patrick, G. N. (2017). Phosphorylation of the 19S regulatory particle ATPase subunit, Rpt6, modifies susceptibility to proteotoxic stress and protein aggregation. PLoS ONE 12:e0179893. doi: 10.1371/journal.pone.0179893
Martin, D. O., Beauchamp, E., and Berthiaume, L. G. (2011). Post-translational myristoylation: Fat matters in cellular life and death. Biochimie 93, 18–31. doi: 10.1016/j.biochi.2010.10.018
Matiuhin, Y., Kirkpatrick, D. S., Ziv, I., Kim, W., Dakshinamurthy, A., Kleifeld, O., et al. (2008). Extraproteasomal Rpn10 restricts access of the polyubiquitin-binding protein Dsk2 to proteasome. Mol. Cell 32, 415–425. doi: 10.1016/j.molcel.2008.10.011
Medicherla, B., and Goldberg, A. L. (2008). Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell Biol. 182, 663–673. doi: 10.1083/jcb.200803022
Meiners, S., Heyken, D., Weller, A., Ludwig, A., Stangl, K., Kloetzel, P. M., et al. (2003). Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J. Biol. Chem. 278, 21517–21525. doi: 10.1074/jbc.M301032200
Minami, Y., Kawasaki, H., Minami, M., Tanahashi, N., Tanaka, K., and Yahara, I. (2000). A critical role for the proteasome activator PA28 in the Hsp90-dependent protein refolding. J. Biol. Chem. 275, 9055–9061. doi: 10.1074/jbc.275.12.9055
Mishto, M., Bellavista, E., Santoro, A., Stolzing, A., Ligorio, C., Nacmias, B., et al. (2006). Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol. Aging 27, 54–66. doi: 10.1016/j.neurobiolaging.2004.12.004
Moritz, K. E., McCormack, N. M., Abera, M. B., Viollet, C., Yauger, Y. J., Sukumar, G., et al. (2017). The role of the immunoproteasome in interferon-gamma-mediated microglial activation. Sci. Rep. 7:9365. doi: 10.1038/s41598-017-09715-y
Moriwaki, K., Kiyomoto, H., Hitomi, H., Ihara, G., Kaifu, K., Matsubara, K., et al. (2006). Interferon-gamma enhances superoxide production in human mesangial cells via the JAK-STAT pathway. Kidney Int. 70, 788–793. doi: 10.1038/sj.ki.5001639
Moschonas, A., Kouraki, M., Knox, P. G., Thymiakou, E., Kardassis, D., and Eliopoulos, A. G. (2008). CD40 induces antigen transporter and immunoproteasome gene expression in carcinomas via the coordinated action of NF-kappaB and of NF-kappaB-mediated de novo synthesis of IRF-1. Mol. Cell. Biol. 28, 6208–6222. doi: 10.1128/MCB.00611-08
Murata, S., Sasaki, K., Kishimoto, T., Niwa, S., Hayashi, H., Takahama, Y., et al. (2007). Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 316, 1349–1353. doi: 10.1126/science.1141915
Murata, S., Udono, H., Tanahashi, N., Hamada, N., Watanabe, K., Adachi, K., et al. (2001). Immunoproteasome assembly and antigen presentation in mice lacking both PA28alpha and PA28beta. EMBO J. 20, 5898–5907. doi: 10.1093/emboj/20.21.5898
Myeku, N., Clelland, C. L., Emrani, S., Kukushkin, N. V., Yu, W. H., Goldberg, A. L., et al. (2016). Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22, 46–53. doi: 10.1038/nm.4011
Nathan, J. A., Spinnenhirn, V., Schmidtke, G., Basler, M., Groettrup, M., and Goldberg, A. L. (2013). Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell 152, 1184–1194. doi: 10.1016/j.cell.2013.01.037
Navon, A., and Goldberg, A. L. (2001). Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol. Cell 8, 1339–1349. doi: 10.1016/S1097-2765(01)00407-5
Njomen, E., and Tepe, J. J. (2019). Proteasome activation as a new therapeutic approach to target proteotoxic disorders. J. Med. Chem. doi: 10.1021/acs.jmedchem.9b00101. [Epub ahead of print].
Orlowski, R. Z., and Kuhn, D. J. (2008). Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin. Cancer Res. 14, 1649–1657. doi: 10.1158/1078-0432.CCR-07-2218
Osna, N. A., Bardag-Gorce, F., White, R. L., Weinman, S. A., Donohue, T. M., Kharbanda, K. K., et al. (2012). Ethanol and hepatitis C virus suppress peptide-MHC class I presentation in hepatocytes by altering proteasome function. Alcohol. Clin. Exp. Res. 36, 2028–2035. doi: 10.1111/j.1530-0277.2012.01813.x
Osna, N. A., White, R. L., Donohue, T. M., Beard, M. R., Tuma, D. J., Kharbanda, K. K., et al. (2010). Impaired methylation as a novel mechanism for proteasome suppression in liver cells. Biochem. Biophys. Res. Commun. 391, 1291–1296. doi: 10.1016/j.bbrc.2009.12.074
Ozben, T. (2007). Oxidative stress and apoptosis: impact on cancer therapy. J. Pharm. Sci. 96, 2181–2196. doi: 10.1002/jps.20874
Pathare, G. R., Nagy, I., Bohn, S., Unverdorben, P., Hubert, A., Körner, R., et al. (2012). The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl. Acad. Sci. USA. 109, 149–154. doi: 10.1073/pnas.1117648108
Peth, A., Besche, H. C., and Goldberg, A. L. (2009). Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 36, 794–804. doi: 10.1016/j.molcel.2009.11.015
Peth, A., Kukushkin, N., Bosse, M., and Goldberg, A. L. (2013). Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J. Biol. Chem. 288, 7781–7790. doi: 10.1074/jbc.M112.441907
Peth, A., Uchiki, T., and Goldberg, A. L. (2010). ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol. Cell 40, 671–681. doi: 10.1016/j.molcel.2010.11.002
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Pickering, A. M., and Davies, K. J. A. (2012). Differential roles of proteasome and immunoproteasome regulators Pa28αβ, Pa28γ and Pa200 in the degradation of oxidized proteins. Arch. Biochem. Biophys. 523, 181–190. doi: 10.1016/j.abb.2012.04.018
Pickering, A. M., Koop, A. L., Teoh, C. Y., Ermak, G., Grune, T., and Davies, K. J. A. (2010). The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 432, 585–594. doi: 10.1042/BJ20100878
Pickering, A. M., Linder, R. A., Zhang, H., Forman, H. J., and Davies, K. J. A. (2012). Nrf2-dependent induction of proteasome and Pa28αβ regulator are required for adaptation to oxidative stress. J. Biol. Chem. 287, 10021–10031. doi: 10.1074/jbc.M111.277145
Piterman, R., Braunstein, I., Isakov, E., Ziv, T., Navon, A., Cohen, S., et al. (2014). VWA domain of S5a restricts the ability to bind ubiquitin and Ubl to the 26S proteasome. Mol. Biol. Cell 25, 3988–3998. doi: 10.1091/mbc.e13-11-0697
Pomatto, L. C. D., Cline, M., Woodward, N., Pakbin, P., Sioutas, C., Morgan, T. E., et al. (2018). Aging attenuates redox adaptive homeostasis and proteostasis in female mice exposed to traffic-derived nanoparticles ('vehicular smog'). Free Rad. Biol. Med. 121, 86–97. doi: 10.1016/j.freeradbiomed.2018.04.574
Prasad, R., Kawaguchi, S., and Ng, D. T. W. (2010). A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 21, 2117–2227. doi: 10.1091/mbc.e10-02-0111
Qian, M. X., Pang, Y., Liu, C. H., Haratake, K., Du, B. Y., Ji, D. Y., et al. (2013). Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153, 1012–1024. doi: 10.1016/j.cell.2013.04.032
Qian, S. B., Reits, E., Neefjes, J., Deslich, J. M., Bennink, J. R., and Yewdell, J. W. (2006). Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J. Immunol. 177, 227–233. doi: 10.4049/jimmunol.177.1.227
Queisser, M. A., Yao, D., Geisler, S., Hammes, H. P., Lochnit, G., Schleicher, E. D., et al. (2010). Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 59, 670–678. doi: 10.2337/db08-1565
Radhakrishnan, S. K., Lee, C. S., Young, P., Beskow, A., Chan, J. Y., and Deshaies, R. J. (2010). Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 38, 17–28. doi: 10.1016/j.molcel.2010.02.029
Ranek, M. J., Kost, C. K. Jr., Hu, C., Martin, D. S., and Wang, X. (2014). Muscarinic 2 receptors modulate cardiac proteasome function in a protein kinase G-dependent manner. J. Mol. Cell. Cardiol. 69, 43–51. doi: 10.1016/j.yjmcc.2014.01.017
Ranek, M. J., Terpstra, E. J., Li, J., Kass, D. A., and Wang, X. (2013). Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128, 365–376. doi: 10.1161/CIRCULATIONAHA.113.001971
Raule, M., Cerruti, F., Benaroudj, N., Migotti, R., Kikuchi, J., Bachi, A., et al. (2014). PA28αβ reduces size and increases hydrophilicity of 20S immunoproteasome peptide products. Chem. Biol. 21, 470–480. doi: 10.1016/j.chembiol.2014.02.006
Realini, C., Jensen, C. C., Zhang, Z., Johnston, S. C., Knowlton, J. R., Hill, C. P., et al. (1997). Characterization of recombinant REGalpha, REGbeta, and REGgamma proteasome activators. J. Biol. Chem. 272, 25483–25492. doi: 10.1074/jbc.272.41.25483
Rechsteiner, M., and Hill, C. P. (2005). Mobilizing the proteolytic machine: Cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15, 27–33. doi: 10.1016/j.tcb.2004.11.003
Reeg, S., Jung, T., Castro, J. P., Davies, K. J. A., Henze, A., and Grune, T. (2016). The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Rad. Biol. Med. 99, 153–166. doi: 10.1016/j.freeradbiomed.2016.08.002
Reits, E. A. J., Vos, J. C., Grommé, M., and Neefjes, J. (2000). The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature 404, 774–778. doi: 10.1038/35008103
Respondek, D., Voss, M., Kuhlewindt, I., Klingel, K., Kruger, E., and Beling, A. (2017). PA28 modulates antigen processing and viral replication during coxsackievirus B3 infection. PLoS ONE 12:e0173259. doi: 10.1371/journal.pone.0173259
Rock, K. L., Farfan-Arribas, D. J., Colbert, J. D., and Goldberg, A. L. (2014). Re-examining class-I presentation and the DRiP hypothesis. Trends Immunol. 35, 144–152. doi: 10.1016/j.it.2014.01.002
Rock, K. L., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L., et al. (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. doi: 10.1016/S0092-8674(94)90462-6
Safdar, K., and Schiff, E. R. (2004). Alcohol and hepatitis C. Semin. Liver Dis. 24, 305–315. doi: 10.1055/s-2004-832942
Santarius, T., Shipley, J., Brewer, D., Stratton, M. R., and Cooper, C. S. (2010). A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 10, 59–64. doi: 10.1038/nrc2771
Satoh, K., Sasajima, H. K.-I, Nyoumura, Yokosawa, H., and Hitoshi, S. (2000). Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 40, 314–319. doi: 10.1021/bi001815n
Savulescu, A. F., and Glickman, M. H. (2011). Proteasome activator 200: the heat is on. Mol. Cell. Prot. 10:R110.006890. doi: 10.1074/mcp.R110.006890
Schlafer, D., Shah, K. S., Panjic, E. H., and Lonial, S. (2017). Safety of proteasome inhibitors for treatment of multiple myeloma. Expert Opin. Drug Saf. 16, 167–183. doi: 10.1080/14740338.2017.1259310
Schmidt, F., Dahlmann, B., Hustoft, H. K., Koehler, C. J., Strozynski, M., Klo,ß, A., et al. (2011). Quantitative proteome analysis of the 20S proteasome of apoptotic Jurkat T cells. Amino Acids 41, 351–361. doi: 10.1007/s00726-010-0575-6
Schmidt, M., Haas, W., Crosas, B., Santamaria, P. G., Gygi, S. P., Walz, T., et al. (2005). The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat. Struct. Mol. Biol. 12, 294–303. doi: 10.1038/nsmb914
Schmitt, S. M., Neslund-Dudas, C., Shen, M., Cui, C., Mitra, B., and Dou, Q. P. (2016). Involvement of ALAD-20S proteasome complexes in ubiquitination and acetylation of proteasomal α2 subunits. J. Cell. Biochem. 117, 144–151. doi: 10.1002/jcb.25259
Schubert, U., Antón, L. C., Gibbs, J., Norbury, C. C., and Bennink, J. R. (2000). Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. doi: 10.1038/35008096
Schweitzer, A., Aufderheide, A., Rudack, T., Beck, F., Pfeifer, G., Plitzko, J. M., et al. (2016). Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. U.S.A. 113, 7816–7821. doi: 10.1073/pnas.1608050113
Seifert, U., Bialy, L. P., Ebstein, F., Bech-Otschir, D., Voigt, A., Schröter, F., et al. (2010). Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142, 613–624. doi: 10.1016/j.cell.2010.07.036
Sekhar, K. R., Rachakonda, G., and Freeman, M. L. (2010). Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol. Appl. Pharmacol. 244, 21–26. doi: 10.1016/j.taap.2009.06.016
Sha, Z., and Alfred Goldberg, L. (2014). Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 24, 1573–1583. doi: 10.1016/j.cub.2014.06.004
Sha, Z., and Goldberg, A. L. (2016). Reply to Vangala et al.: Complete inhibition of the proteasome reduces new proteasome production by causing Nrf1 aggregation. Curr. Biol. 26:R836–r837. doi: 10.1016/j.cub.2016.08.030
Shang, F., Nowell, T. R. Jr., and Taylor, A. (2001). Removal of oxidatively damaged proteins from lens cells by the ubiquitin-proteasome pathway. Exp. Eye Res. 73, 229–238. doi: 10.1006/exer.2001.1029
Sheaff, R. J., Singer, J. D., Swanger, J., Smitherman, M., Roberts, J. M., and Clurman, B. E. (2000). Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol. Cell 5, 403–410. doi: 10.1016/S1097-2765(00)80435-9
Shi, Y., Chen, X., Elsasser, S., Stocks, B. B., Tian, G., Lee, B. H., et al. (2016). Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351:aad9421. doi: 10.1126/science.aad9421
Shimizu, S., Hiroi, T., Ishii, M., Hagiwara, T., Wajima, T., Miyazaki, A., et al. (2008). Hydrogen peroxide stimulates tetrahydrobiopterin synthesis through activation of the Jak2 tyrosine kinase pathway in vascular endothelial cells. Int. J. Biochem. Cell Biol. 40, 755–765. doi: 10.1016/j.biocel.2007.10.011
Shringarpure, R., and Davies, K. J. A. (2002). Protein turnover by the proteasome in aging and disease. Free Rad. Biol. Med. 32, 1084–1089. doi: 10.1016/S0891-5849(02)00824-9
Shringarpure, R., Grune, T., Mehlhase, J., and Davies, K. J. (2003). Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 278, 311–318. doi: 10.1074/jbc.M206279200
Sijts, A., Sun, Y., Janek, K., Kral, S., Paschen, A., Schadendorf, D., et al. (2002). The role of the proteasome activator PA28 in MHC class I antigen processing. Mol. Immunol. 39, 165–169. doi: 10.1016/S0161-5890(02)00099-8
Sijts, E. J., and Kloetzel, P. M. (2011). The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell. Mol. Life Sci. 68, 1491–1502. doi: 10.1007/s00018-011-0657-y
Silva, G. M., Netto, L. E., Discola, K. F., Piassa-Filho, G. M., Pimenta, D. C., Barcena, J. A., et al. (2008). Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J. 275, 2942–2955. doi: 10.1111/j.1742-4658.2008.06441.x
Silva, G. M., Netto, L. E. S., Simões, V., Santos, L. F. A., Gozzo, F. C., Demasi, M. A. A., et al. (2012). Redox control of 20S proteasome gating. Antioxid. Redox Signal. 16, 1183–1194. doi: 10.1089/ars.2011.4210
Smith, D. M., Chang, S.-C., Park, S., Finley, D., Cheng, Y., and Goldberg, A. L. (2007). Docking of the proteasomal ATPases' carboxyl termini in the 20S Proteasome's α ring opens the gate for substrate entry. Mol. Cell 27, 731–744. doi: 10.1016/j.molcel.2007.06.033
Snoberger, A., Anderson, R. T., and Smith, D. M. (2017). The Proteasomal ATPases use a slow but highly processive strategy to unfold proteins. Front. Mol. Biosci. 4:18. doi: 10.3389/fmolb.2017.00018
Steffen, J., Seeger, M., Koch, A., and Krüger, E. (2010). Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 40, 147–158. doi: 10.1016/j.molcel.2010.09.012
Storz, P., and Toker, A. (2003). Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 22, 109–120. doi: 10.1093/emboj/cdg009
Sun, J., Luan, Y., Xiang, D., Tan, X., Chen, H., Deng, Q., et al. (2016). The 11S proteasome subunit PSME3 is a positive feedforward regulator of NF-kappaB and important for host defense against bacterial pathogens. Cell Rep. 14, 737–749. doi: 10.1016/j.celrep.2015.12.069
Sun, S., Liu, S., Zhang, Z., Zeng, W., Sun, C., Tao, T., et al. (2017). Phosphatase UBLCP1 controls proteasome assembly. Open Biol. 7:170042. doi: 10.1098/rsob.170042
Sun, Y., Abdul Aziz, A., Bowles, K., and Rushworth, S. (2018). High NRF2 expression controls endoplasmic reticulum stress induced apoptosis in multiple myeloma. Cancer Lett. 412, 37–45. doi: 10.1016/j.canlet.2017.10.005
Surh, Y. J., Na, H. K., Park, J. M., Lee, H. N., Kim, W., Yoon, I. S., et al. (2011). 15-Deoxy-Delta(1)(2),(1)(4)-prostaglandin J(2), an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem. Pharmacol. 82, 1335–1351. doi: 10.1016/j.bcp.2011.07.100
Taguchi, K., Motohashi, H., and Yamamoto, M. (2011). Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140. doi: 10.1111/j.1365-2443.2010.01473.x
Tanahashi, N. K.-Y, Yokota, Ahn, J. Y., Chung, C. H., Fujiwara, T. E.-I., et al. (1997). Molecular properties of the proteasome activator PA28 family proteins and gamma-interferon regulation. Genes Cells 2, 195–211. doi: 10.1046/j.1365-2443.1997.d01-308.x
Tanahashi, N., Murakami, Y., Minami, Y., Shimbara, N., Hendil, K. B., and Tanaka, K. (2000). Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J. Biol. Chem. 275, 14336–14345. doi: 10.1074/jbc.275.19.14336
Tar, K., Dange, T., Yang, C., Yao, Y., Bulteau, A. L., Salcedo, E. F., et al. (2014). Proteasomes associated with the Blm10 activator protein antagonize mitochondrial fission through degradation of the fission protein Dnm1. J. Biol. Chem. 289, 12145–12156. doi: 10.1074/jbc.M114.554105
Thompson, H. G. R., Harris, J. W., and Brody, J. P. (2004). Post-translationally modified S12, absent in transformed breast epithelial cells, is not associated with the 26S proteasome and is induced by proteasome inhibitor. Int. J. Cancer 111, 338–347. doi: 10.1002/ijc.20261
Tomita, T., Hirayama, S., Sakurai, Y., Ohte, Y., Yoshihara, H., Saeki, Y., et al. (2019). Specific modification of aged proteasomes revealed by tag-exchangeable knock-in mice. Mol. Cell. Biol. 39:e00426–18. doi: 10.1128/MCB.00426-18
Tonelli, C., Chio, I. C., and Tuveson, D. A. (2018). Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 29, 1727–1745. doi: 10.1089/ars.2017.7342
Tsuchiya, Y., Taniguchi, H., Ito, Y., Morita, T., Karim, M. R., Ohtake, N., et al. (2013). The casein kinase 2-nrf1 axis controls the clearance of ubiquitinated proteins by regulating proteasome gene expression. Mol. Cell. Biol. 33, 3461–3472. doi: 10.1128/MCB.01271-12
Tsukamoto, O., Minamino, T. K.-I, Okada, Shintani, Y., Takashima, S., Kato, H., et al. (2006). Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem. Biophys. Res. Commun. 340, 1125–1133. doi: 10.1016/j.bbrc.2005.12.120
Ullrich, O., Diestel, A., Bechmann, I., Homberg, M., Grune, T., Hass, R., et al. (2001). Turnover of oxidatively damaged nuclear proteins in BV-2 microglial cells is linked to their activation state by poly-ADP-ribose polymerase. FASEB J. 15, 1460–1462. doi: 10.1096/fj.00-0540fje
Ullrich, O., Reinheckel, T., Sitte, N., Hass, R., Grune, T., and Davies, K. J. (1999). Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc. Natl. Acad. Sci. U.S.A. 96, 6223–6228. doi: 10.1073/pnas.96.11.6223
Um, J. W., Im, E., Park, J., Oh, Y., Min, B., Lee, H. J., et al. (2010). ASK1 negatively regulates the 26 S proteasome. J. Biol. Chem. 285, 36434–36446. doi: 10.1074/jbc.M110.133777
van Deventer, S., and Neefjes, J. (2010). The immunoproteasome cleans up after inflammation. Cell 142, 517–518. doi: 10.1016/j.cell.2010.08.002
Vangala, J. R., and Radhakrishnan, S. K. (2019). Nrf1-mediated transcriptional regulation of the proteasome requires a functional TIP60 complex. J. Biol. Chem. 294, 2036–2045. doi: 10.1074/jbc.RA118.006290
Vangala, J. R., Sotzny, F., Kruger, E., Deshaies, R. J., and Radhakrishnan, S. K. (2016). Nrf1 can be processed and activated in a proteasome-independent manner. Curr. Biol. 26, R834–r835. doi: 10.1016/j.cub.2016.08.008
Verdone, L., Caserta, M., and Mauro, E. D. (2005). Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 83, 344–353. doi: 10.1139/o05-041
VerPlank, J. J. S., and Goldberg, A. L. (2017). Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 474, 3355–3371. doi: 10.1042/BCJ20160809
VerPlank, J. S., Lokireddy, S., Zhao, J., and Goldberg, A. L. (2019). 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc. Natl. Acad. Sci. USA. 116, 4228–4237. doi: 10.1073/pnas.1809254116
Wang, D., Fang, C., Zong, N. C., Liem, D. A., Cadeiras, M., Scruggs, S. B., et al. (2013). Regulation of acetylation restores proteolytic function of diseased myocardium in mouse and human. Mol. Cell. Proteom. 12, 3793–3802. doi: 10.1074/mcp.M113.028332
Wang, K., Ho, S. R., Mao, W., Huang, P., Zhang, F., Schwiebert, E. M., et al. (2009). Increased O-GlcNAc causes disrupted lens fiber cell differentiation and cataracts. Biochem. Biophys. Res. Commun. 387, 70–76. doi: 10.1016/j.bbrc.2009.06.132
Wang, X., Chen, C. F., Baker, P. R. P.-L, Chen, Peter, K., and Huang, L. (2007). Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565. doi: 10.1021/bi061994u
Wang, X., Yen, J., Kaiser, P., and Huang, L. (2010). Regulation of the 26S proteasome complex during oxidative stress. Sci Signal 3:ra88. doi: 10.1126/scisignal.2001232
Wani, P. S., Suppahia, A., Capalla, X., Ondracek, A., and Roelofs, J. (2016). Phosphorylation of the C-terminal tail of proteasome subunit α7 is required for binding of the proteasome quality control factor Ecm29. Sci. Rep. 6:27873. doi: 10.1038/srep27873
Watanabe, Y., Suzuki, O., Haruyama, T., and Akaike, T. (2003). Interferon-γ induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J. Cell. Biochem. 89, 244–253. doi: 10.1002/jcb.10501
Welk, V., Coux, O., Kleene, V., Abeza, C., Trümbach, D., Eickelberg, O., et al. (2016). Inhibition of proteasome activity induces formation of alternative proteasome complexes. J. Biol. Chem. 291, 13147–13159. doi: 10.1074/jbc.M116.717652
Weyburne, E. S., Wilkins, O. M., Sha, Z., Williams, D. A., Pletnev, A. A., de Bruin, G., et al. (2017). Inhibition of the proteasome beta2 site sensitizes triple-negative breast cancer cells to beta5 inhibitors and suppresses Nrf1 activation. Cell Chem. Biol. 24, 218–230. doi: 10.1016/j.chembiol.2016.12.016
Whittier, J. E., Xiong, Y., Rechsteiner, M. C., and Squier, T. C. (2004). Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. J. Biol. Chem. 279, 46135–46142. doi: 10.1074/jbc.M406048200
Wójcik, C., Tanaka, K., Paweletz, N., Naab, U., and Wilk, S. (1998). Proteasome activator (PA28) subunits, α, β and γ (Ki antigen) in NT2 neuronal precursor cells and HeLa S3 cells. Eur. J. Cell Biol. 77, 151–160. doi: 10.1016/S0171-9335(98)80083-6
Worden, E. J., Dong, K. C., and Martin, A. (2017). An AAA motor-driven mechanical switch in Rpn11 controls deubiquitination at the 26S proteasome. Mol Cell 67, 799–811.e8. doi: 10.1016/j.molcel.2017.07.023
Xiang, Y., Wang, M., Hu, S., Qiu, L., Yang, F., Zhang, Z., et al. (2018). Mechanisms controlling the multistage post-translational processing of endogenous Nrf1alpha/TCF11 proteins to yield distinct isoforms within the coupled positive and negative feedback circuits. Toxicol. Appl. Pharmacol. 360, 212–235. doi: 10.1016/j.taap.2018.09.036
Yanagi, S., Shimbara, N., and Tamura, T.-A. (2000). Tissue and cell cistribution of a mammalian proteasomal ATPase, MSS1, and its complex formation with the basal transcription factors. Biochem. Biophys. Res. Commun. 279, 568–573. doi: 10.1006/bbrc.2000.3969
Yu, H. M., Zhi, J. L., Cui, Y., Tang, E. H., Sun, S. N., Feng, J. Q., et al. (2006). Role of the JAK-STAT pathway in protection of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress in PC12 cells. Apoptosis 11, 931–941. doi: 10.1007/s10495-006-6578-9
Zachara, N. E., and Hart, G. W. (2004). O-GlcNAc modification: A nutritional sensor that modulates proteasome function. Trends Cell Biol. 14, 218–221. doi: 10.1016/j.tcb.2004.03.005
Zannini, L., Lecis, D., Buscemi, G., Carlessi, L., Gasparini, P., Fontanella, E., et al. (2008). REGgamma proteasome activator is involved in the maintenance of chromosomal stability. Cell Cycle 7, 504–512. doi: 10.4161/cc.7.4.5355
Zhang, F., Hu, Y., Huang, P., Toleman, C. A., Paterson, A. J., and Kudlow, J. E. (2007). Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem. 282, 22460–22471. doi: 10.1074/jbc.M702439200
Zhang, F., Su, K., Yang, X., Bowe, D. B., Paterson, A. J., and Kudlow, J. E. (2003). O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115, 715–725. doi: 10.1016/S0092-8674(03)00974-7
Zhang, X., Schulz, R., Edmunds, S., Kruger, E., Markert, E., Gaedcke, J., et al. (2015). MicroRNA-101 suppresses tumor cell proliferation by acting as an endogenous proteasome inhibitor via targeting the proteasome assembly factor POMP. Mol. Cell 59, 243–257. doi: 10.1016/j.molcel.2015.05.036
Zhang, Z., and Zhang, R. (2008). Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 27, 852–864. doi: 10.1038/emboj.2008.25
Zhao, J., Zhai, B., Gygi, S. P., and Goldberg, A. L. (2015). mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. U.S.A. 112, 15790–15797. doi: 10.1073/pnas.1521919112
Zheng, J., Dasgupta, A., and Bizzozero, O. A. (2012). Changes in 20S subunit composition are largely responsible for altered proteasomal activities in experimental autoimmune encephalomyelitis. J. Neurochem. 121, 486–494. doi: 10.1111/j.1471-4159.2012.07699.x
Zhou, F. (2009). Molecular mechanisms of IFN-γ to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 28, 239–260. doi: 10.1080/08830180902978120
Ziegler, M. (2000). New functions of a long-known molecule. Emerging roles of NAD in cellular signaling. Eur. J. Biochem. 267, 1550–1564. doi: 10.1046/j.1432-1327.2000.01187.x
Zmijewski, J. W., Banerjee, S., and Abraham, E. (2009). S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J. Biol. Chem. 284, 22213–22221. doi: 10.1074/jbc.M109.028902
Zong, C., Gomes, A. V., Drews, O., Li, X., Young, G. W., Berhane, B., et al. (2006). Regulation of murine cardiac 20S proteasomes. Circ. Res. 99, 362–371. doi: 10.1161/01.RES.0000237389.40000.02
Zong, N., Ping, P., Lau, E., Choi, H. J. H., Ng, D. C. M., Meyer, D., et al. (2014). Lysine ubiquitination and acetylation of human cardiac 20S proteasomes. Prot. Clin. Appl. 8, 590–594. doi: 10.1002/prca.201400029
Keywords: proteasome, post translational modifications, 20S, 26S, proteasome complexes, proteasome activation/inhibition
Citation: Kors S, Geijtenbeek K, Reits E and Schipper-Krom S (2019) Regulation of Proteasome Activity by (Post-)transcriptional Mechanisms. Front. Mol. Biosci. 6:48. doi: 10.3389/fmolb.2019.00048
Received: 31 January 2019; Accepted: 11 June 2019;
Published: 16 July 2019.
Edited by:
Cordula Enenkel, University of Toronto, CanadaReviewed by:
Alfred L. Goldberg, Harvard Medical School, United StatesUmesh K. Jinwal, University of South Florida, United States
Copyright © 2019 Kors, Geijtenbeek, Reits and Schipper-Krom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabine Schipper-Krom, cy5rcm9tJiN4MDAwNDA7YW1zdGVyZGFtdW1jLm5s
†Present Address: Suzan Kors, Biosciences, University of Exeter, Exeter, United Kingdom