 Steve Agajanian1
Steve Agajanian1 Gennady M. Verkhivker
Gennady M. Verkhivker- 1Graduate Program in Computational and Data Sciences, Schmid College of Science and Technology, Chapman University, Orange, CA, United States
- 2Department of Biomedical and Pharmaceutical Sciences, Chapman University School of Pharmacy, Irvine, CA, United States
Development of machine learning solutions for prediction of functional and clinical significance of cancer driver genes and mutations are paramount in modern biomedical research and have gained a significant momentum in a recent decade. In this work, we integrate different machine learning approaches, including tree based methods, random forest and gradient boosted tree (GBT) classifiers along with deep convolutional neural networks (CNN) for prediction of cancer driver mutations in the genomic datasets. The feasibility of CNN in using raw nucleotide sequences for classification of cancer driver mutations was initially explored by employing label encoding, one hot encoding, and embedding to preprocess the DNA information. These classifiers were benchmarked against their tree-based alternatives in order to evaluate the performance on a relative scale. We then integrated DNA-based scores generated by CNN with various categories of conservational, evolutionary and functional features into a generalized random forest classifier. The results of this study have demonstrated that CNN can learn high level features from genomic information that are complementary to the ensemble-based predictors often employed for classification of cancer mutations. By combining deep learning-generated score with only two main ensemble-based functional features, we can achieve a superior performance of various machine learning classifiers. Our findings have also suggested that synergy of nucleotide-based deep learning scores and integrated metrics derived from protein sequence conservation scores can allow for robust classification of cancer driver mutations with a limited number of highly informative features. Machine learning predictions are leveraged in molecular simulations, protein stability, and network-based analysis of cancer mutations in the protein kinase genes to obtain insights about molecular signatures of driver mutations and enhance the interpretability of cancer-specific classification models.
Introduction
Deep sequencing studies have enabled a detailed characterization of cancer genomes and unveiled important gene-specific signatures of somatic mutations (Davies et al., 2002; Bardelli et al., 2003; Futreal et al., 2004; Samuels et al., 2004; Stephens et al., 2004, 2005; Wang et al., 2004; Sjoblom et al., 2006; Greenman et al., 2007; Wood et al., 2007; Vogelstein et al., 2013; Watson et al., 2013). The steadily growing amount of data generated in cancer genomic studies and next-generation sequencing (NGS) have been the impetus behind formation of international cancer genomic projects and development of large bioinformatics data resources such as Cancer Genome Atlas (TCGA), Genomics Data Commons Portal (https://portal.gdc.cancer.gov/) (Weinstein et al., 2013; Jensen et al., 2017), COSMIC database (http://cancer.sanger.ac.uk) (Forbes et al., 2015), and the International Cancer Genome Consortium (ICGC) (Hudson et al., 2010; Zhang et al., 2011; Klonowska et al., 2016; Hinkson et al., 2017). The Cancer Gene Census of the Catalog of Somatic Mutations in Cancer (COSMIC) database has grown from 291 well-characterized cancer genes (Futreal et al., 2004) to more than 500 entries (Forbes et al., 2015) where some cancer genes can be commonly mutated across cancer types, while other genes are predominantly cancer-specific. The cBio Cancer Genomics Portal (https://www.cbioportal.org/) is an open-access resource for exploration of large cancer genomics data sets (Cerami et al., 2012; Gao et al., 2013). These datasets have allowed for comprehensive genome-wide analyses of genetic alterations in multiple tumor types (Poulos and Wong, 2018). A relatively small fraction of somatic variants known as driver mutations have considerable functional effects and can be acquired over time as a result of a range of mutational processes, rather than inherited (Haber and Settleman, 2007; Lawrence et al., 2013; Vogelstein et al., 2013). A comprehensive analysis of cancer driver genes and mutations has provided classification of 751,876 unique missense mutations, producing a dataset of 3,442 functionally validated driver mutations (Bailey et al., 2018). Another significant dataset of 1,049 experimentally tested and functionally validated driver mutations (Ng et al., 2018) has expanded our knowledge of cancer-causing variants in oncogenes and tumor suppressor genes. TCGA organized the Multi-Center Mutation Calling in Multiple Cancers (MC3) network project which generated a comprehensive and consistent collection of somatic mutation calls for the 10,437 tumor samples dataset (Ellrott et al., 2018). Computational approaches that assess the impact of somatic mutations are often characterized by different basic assumptions, types of input information, models, and prediction targets such as driver gene or driver mutation (Gonzalez-Perez et al., 2013; Cheng et al., 2016).
A number of somatic variant callers based on various statistical and machine learning approaches are now available for somatic mutation detection, including MuTect2 (Cibulskis et al., 2013), MuSE (Fan et al., 2016), VarDict (Lai et al., 2016), VarScan2 (Koboldt et al., 2012), Strelka2 (Kim et al., 2018), SomaticSniper (Larson et al., 2012), and SNooPer (Spinella et al., 2016). A deep convolutional neural network (CNN) approach termed DeepVariant can identify genetic variation in NGS data by discerning statistical relationships around putative variant sites (Poplin et al., 2018). To facilitate systematic and standardized somatic variant refinement from cancer sequencing data, random forest (RF) models and deep learning (DL) approach were utilized, showing that these machine learning techniques could achieve high and similar classification performance across all variant refinement classes (Ainscough et al., 2018). A machine learning approach called Cerebro increased the accuracy of calling validated somatic mutations in tumor samples and outperformed several other somatic mutation detection methods (Wood et al., 2018).
Many computational methods have been proposed for prediction of cancer driver genes. Some of these approaches use cohort-based analysis to detect driver genes, including ActiveDriver (Reimand and Bader, 2013), MutSigCV (Lawrence et al., 2013), MuSiC (Dees et al., 2012), OncodriveCLUST (Tamborero et al., 2013), OncodriveFM (Gonzalez-Perez and Lopez-Bigas, 2012), and OncodriveFML (Mularoni et al., 2016). The success of hybrid methods for scoring coding variants has indicated that integration of different tools may enhance predictive accuracy for both coding and non-coding variants (Li et al., 2015). A deep learning-based method (deepDriver) predicts driver genes by CNN trained with mutation-based feature matrix constructed using similarity networks (Luo et al., 2019). Since many methods are often found to predict distinct or partially overlapping subsets of cancer driver genes, a consensus-based strategy was recently proposed, showing considerable promise and outperforming the individual approaches (Bertrand et al., 2018). A unified machine learning-based evaluation framework for analysis of driver gene predictions compared the performance of these methods, showing that the driver genes predicted by individual tools can vary widely (Tokheim C. et al., 2016; Tokheim C. J. et al., 2016).
Computational methods designed to identify driver mutations have become increasingly important to facilitate an automated assessment of functional and clinical impacts (Gnad et al., 2013; Ding et al., 2014; Martelotto et al., 2014; Raphael et al., 2014; Cheng et al., 2016). Functional computational prediction methods include Sorted Intolerant From Tolerant (SIFT) (Sim et al., 2012), PolyPhen-2 (Adzhubei et al., 2010), Mutation Assessor (Reva et al., 2011), MutationTaster (Schwarz et al., 2010), CONsensus DELeteriousness score of missense mutations (Condel) (Gonzalez-Perez and Lopez-Bigas, 2011), Protein Variation Effect Analyzer (PROVEAN) (Choi et al., 2012), and Functional Analysis Through Hidden Markov Models (FATHMM) (Shihab et al., 2013). Cancer-specific High-throughput Annotation of Somatic Mutations (CHASM) (Carter et al., 2009; Douville et al., 2013; Masica et al., 2017), Cancer Driver Annotation (CanDrA) (Mao et al., 2013), and FATHMM (Shihab et al., 2013). Many new approaches have recently addressed a problem of locating driver mutations within the non-coding genome regions (Piraino and Furney, 2016). The identification of cancer mutation hotspots in protein structures has been a fruitful approach for identifying driver mutations (Dixit et al., 2009; Dixit and Verkhivker, 2011; Gao et al., 2013; Gauthier et al., 2016; Niu et al., 2016; Tokheim C. et al., 2016; Tokheim C. J. et al., 2016). To consolidate functional annotation for SNVs discovered in exome sequencing studies, a database of human non-synonymous SNVs (dbNSFP) was developed (Liu et al., 2011, 2013, 2016; Dong et al., 2015; Wu et al., 2016). This resource allows for computation of a total of 48 functional prediction scores for each SNV, including 32 functional prediction scores by 13 approaches and 15 conservation features (Wu et al., 2016). In our recent investigation, two cancer-specific machine learning classifiers were proposed that utilized 48 functional scores from dbWGFP server in classification of cancer driver mutations (Agajanian et al., 2018).
In this work, we explore and integrate RF and DL/CNN machine learning approaches for prediction and classification of cancer driver mutations. We first explore the ability of CNN models to identify and classify cancer driver mutations directly from raw nucleotide sequence information without relying on specific functional scores. The performance of these classifiers was compared to RF and gradient boosted tree (GBT) methods to provide a comparative analysis of various classification models. These raw sequence-derived scores are advantageous because they can be obtained for any mutation with a known chromosome and position, whereas the functional scoring features can be limited to subsets of genomic mutations. By developing a successful classification scheme that could leverage information from raw DNA sequences, the universe of classifiable mutations can be greatly expanded leading to more general and robust machine learning tools. The results of this study reveal that CNN models can learn high importance features from genomic information that are complementary to the ensemble-based predictor scores traditionally employed in machine learning classification of cancer mutations. We show that integration of the DL-derived predictor score with only several ensemble-based features can recapitulate the results obtained with a large number of functional features and improve performance in capturing driver mutations across a spectrum of machine learning classifiers. Machine learning predictions are leveraged in biophysical simulations and network analysis of protein kinase oncogenes to obtain more detailed functional information about molecular signatures of activating driver mutations, aiding in the interpretability of cancer mutation classifiers.
Materials and Methods
Mutational Datasets and Feature Selection
In our earlier study (Agajanian et al., 2018) we used RF classifier to predict cancer driver mutations using a combination of two golden datasets (Mao et al., 2013; Martelotto et al., 2014). Here, we expanded this dataset by adding the predicted cancer driver mutations and passengers from the analysis of missense mutations in Cbioportal database (Agajanian et al., 2018). By leveraging the earlier analysis, we created a dataset consisting of functionally validated 6,389 cancer driver mutations and 12,941 passenger mutations. The driver/passenger classifications for 2,570 of these mutations were present in the two aforementioned golden datasets, and our RF classifier made predictions on the remaining 16,760 missense mutations from the Cbioportal database. Given the performance level of our model (Agajanian et al., 2018), we conjectured that a combination of the two golden datasets and the missense mutations in the Cbioportal database would yield an informative dataset for the current study. The initially selected features for RF predictions were obtained from dbWGFP web server (Wu et al., 2016) of functional predictions for human whole-genome single nucleotide variants (Supplementary Table S1). A total of 32 sequence-based, evolutionary and functional features identified in our previous study (Agajanian et al., 2018) were initially used for machine learning experiments with the new dataset of cancer mutations. In cancer driver mutation predictions, traditional input data contain distinct features that cannot be directly applied to CNN models due to their lack of spatial meaning. Using the chromosome and the position on that chromosome that corresponded to the mutated nucleotide, we could retrieve the surrounding nucleotides of the mutation of interest to perform classification with only this raw string of nucleotides. To represent the original nucleotide and its mutated version, we placed two nucleotide sequences on top of each other, one containing the original string, and the other contained the mutated version. This would only result in a one nucleotide difference between the two, allowing to effectively utilizing the sliding window format of the CNN models. The schematic workflow diagram of the CNN approach employed in this study is presented in Figure 1.
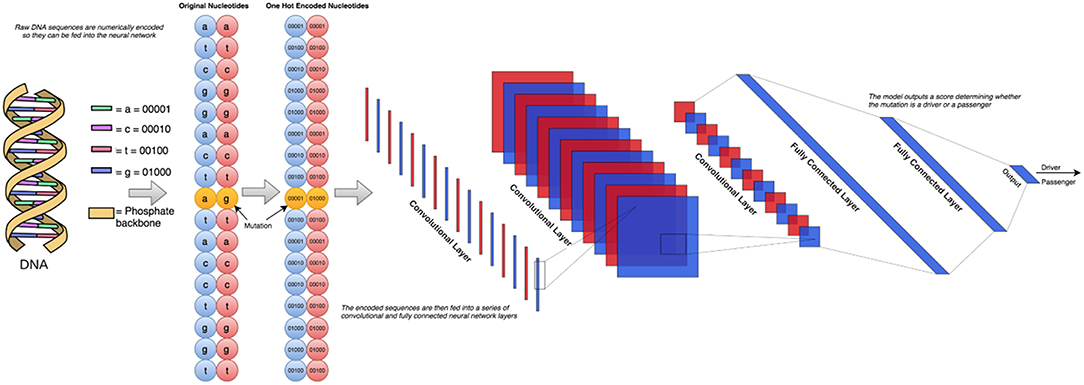
Figure 1. The schematic workflow diagram of the CNN approach employed in this study. To determine the optimal architecture, we performed a grid search over a total of 72 different neural network architectures. These 72 architectures consisted of between 1 and 3 convolutional layers and 1–3 fully connected layers following. The number of nodes in each of these layers was also varied between 2 and 256 in powers of 2. The simplest architecture covered in this search contains 1 convolutional layer with 2 filters feeding into 1 fully connected layer with 2 nodes, and the most complex would have 3 convolutional layers feeding into 3 fully connected layers, all containing 256 nodes.
To create this dataset, we parsed information from University of California, Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/) (Tyner et al., 2017) which takes a chromosome (CHR) and a position (POS) on that chromosome as arguments and returns back all nucleotides within the sequence. Using the dataset consisting of 6,389 driver mutations and 12,941 passengers, we created 5 different datasets of various window sizes around each given CHR/POS pair. The explored window sizes (10, 50, 100, 500, and 5,000) produced nucleotide strings of length 21, 101, 201, 1,001, and 10,001, respectively. To represent the type of mutation (A->C, A->G, etc.) we stacked two of the same nucleotide sequences on top of each other, having one contain the original nucleotide at the position passed in initially, and the other containing the mutated version (Figure 2A). This operation resulted in a total input matrix size of (2, 21), (2, 101), (2, 201), (2, 1001), and (2, 10001), respectively. Three different preprocessing techniques were then applied to the dataset to allow it to be passed into the CNN model in the numerical form: label encoding (Figure 2B), one-hot encoding (Figure 2C; Goh et al., 2017), and embedding (Figure 2D). Label encoding involves assigning each nucleotide its own unique ID (A->0, C->1, etc.) This imposes an ordering on the nucleotide sequences that may have implications for the neural network learning (Figure 2B). This technique was implemented using the Scikit-learn LabelEncoder package for the Python programming language. We also tried one-hot encoding the dataset by assigning each nucleotide its own bit encoded string (A -> [0,0,0,0,1], C-> [0,0,0,1,0]) (Figure 2C). This tends to be a favorable preprocessing function for weight-based classifiers because no artificial ordering is imposed on the samples. This technique tends to be the default representation choice for categorical variables due to how it is interpreted. Because each nucleotide gets its own index in a 5 bit string, a 1 in any particular index means that nucleotide is present in that location. For example, since A->[0,0,0,0,1], this can essentially be read as “There are 0 ‘n,' 0 ‘g,' 0 ‘t,' 0 ‘c,' and 1 ‘a' nucleotides present at this location.” Since the one-hot encoding preprocessing technique lengthens the string, the resulting dimensionalities were (2, 105), (2, 505), (2, 1005), (2, 5005), and (2, 50005), respectively. The final preprocessing technique employed for the DNA sequences involved learned embeddings created with the word2vec algorithm (Mikolov et al., 2013). This technique analyzes the sequential context of the nucleotides assigning them a numeric representation in vector space. Using this representation, the nucleotide segments with similar meaning in the word2vec model would yield similar vectors in an N-dimensional representation. This technique was implemented using the Word2Vec model from the genism library for the Python programming language. Since the vocabulary in this application is fairly small, consisting of only 5 bit components, we chose to convert the nucleotide to 2 dimensional vectors which is sufficient to effectively encode this set. This resulted in the input sizes (2, 42), (2, 202), (2, 402), (2, 2002), and (2, 20002), respectively (Figures 1, 2). The implementation and execution of these three preprocessing techniques provides adequate and efficient nucleotide representations for the CNN classifier.
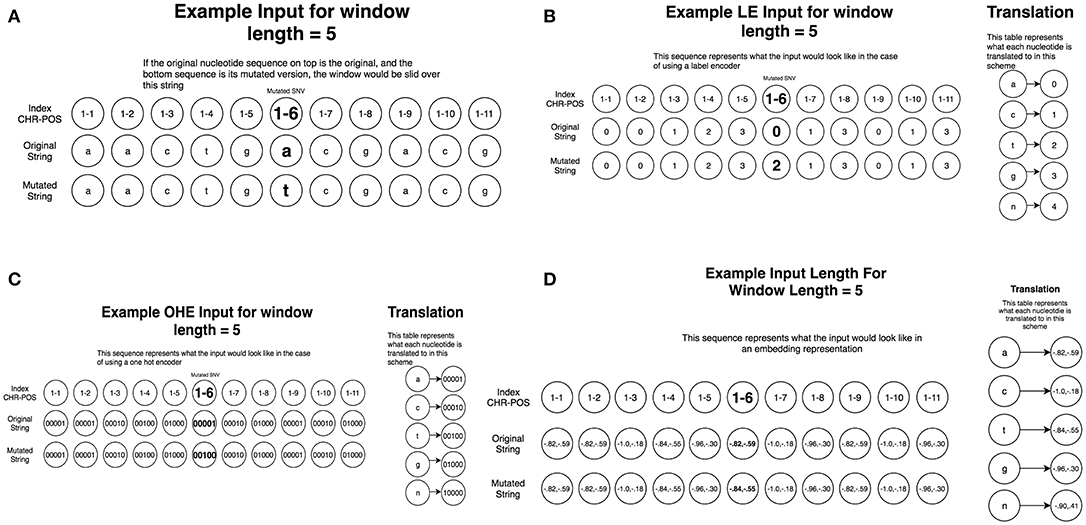
Figure 2. Preprocessing of the nucleotide information for CNN machine learning of cancer driver mutations. Two different preprocessing techniques were then applied to the dataset to allow it to be passed into the CNN model in the numerical form: label encoding and one-hot encoding. (A) A schematic diagram of window sliding protocol. To represent the original nucleotide and its mutated version, two nucleotide sequences are placed on top of each other, one containing the original string, and the other contained the mutated version. This representation allows to utilize the sliding window format of the CNN models. (B) A schematic diagram of label encoding preprocessing protocol. Label encoding assigns each nucleotide its own unique ID (A->0, C->1 etc.) This imposes an ordering on the nucleotide sequences. (C) A schematic diagram of one hot encoding preprocessing protocol. One-hot encoding assigns each nucleotide its own bit encoded string (A -> [0,0,0,0,1], C-> [0,0,0,1,0]). This tends to be a favorable preprocessing function for weight-based classifiers because no artificial ordering is imposed on the samples. (D) A schematic diagram of embedding preprocessing scheme created with the word2vec algorithm.
Machine Learning Models
We used and compared performance of tree based classifiers and DL/CNN machine learning models. For the tree based methods, we used previously established protocol for obtaining hyper-parameters (Agajanian et al., 2018). The model training and tuning was done using Scikit-learn free software machine learning library for the Python programming language (Pedregosa et al., 2011; Biau, 2012). The Keras framework was used for training, validation and testing of CNN models (Erickson et al., 2017). We initially held out 20% of the data in a stratified manner as a testing set so that it had the same distribution of passengers/drivers as the total dataset. We then used the remaining 80% of the dataset as the training set to learn and tune its hyper-parameters. To choose between the hyper-parameters attempted, we test our model out on unseen data so that we have an unbiased estimate of its performance. To do this, we performed 3-fold cross validation, splitting the training set up into three equal sized portions. The model trains on two of them, and makes predictions on the third. This is repeated three times so that each of the three portions has been predicted on. A workflow diagram of the CNN approach (Figure 1) was carefully engineered to determine the optimal architecture. For this, we performed a grid search over a total of 72 different neural network architectures. These 72 architectures consisted of between 1 and 3 convolutional layers and 1–3 fully connected layers following. The number of nodes in each of these layers was also varied between 2 and 256 in powers of 2. The simplest architecture covered in this search contains 1 convolutional layer with 2 filters feeding into 1 fully connected layer with 2 nodes, and the most complex would have 3 convolutional layers feeding into 3 fully connected layers, all containing 256 nodes. The ReLU activation function was used, which returns max (0, X). All 72 different architectures (Table 1) were tested using this cross-validation algorithm and the architecture that had the highest F1 score across all 3-folds was chosen. Our neural networks were trained for 100 epochs, which means that they will pass through the entire dataset 100 times to complete their training. In between each epoch, the model recorded its predictions on the validation fold, and the epoch with the best performance on the validation set was recorded. Dropout was applied in between layers, so that inputs into a layer are randomly set to 0 with a certain probability. This prevents the neural network from overfitting, forcing it to learn without random features present. The best architecture was used for predictions on the test set.
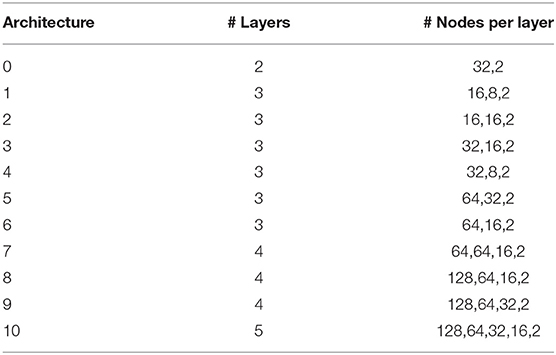
Table 1. The parameters of displayed CNN architectures in classification of cancer driver mutations.
To assess the performance of each model, Accuracy, Recall, Precision, and F1 score were calculated to measure the performance of classification models. These parameters are defined as follows:
True Positive (TP) and True Negative (TN) are defined as the number of mutations that are classified correctly as driver and passenger mutations, respectively. False Positive (FP) and False Negative (FN) are defined as the number of mutations that are misclassified into the other mutational classes. Precision is defined as the amount of positive samples the model predicts correctly (true positives) divided by the true positives plus the false positives. Recall is defined as true positives divided by true positives plus false negatives. The model performance was evaluated using receiver operating characteristic area under the curve. The receiver operating curve (ROC) is a graph where sensitivity is plotted as a function of 1-specificity. The area under the ROC is denoted AUC. The sensitivity or true positive rate (TPR) is defined as the percentage of non-neutral mutations that are correctly identified as driver mutations:
The specificity or true negative rate (TNR) is defined as the percentage of mutations that are correctly identified as passengers:
In combination, these scores allow us to differentiate models by providing evaluation options to properly asses a model's performance. We relied on the F1 score, precision and recall as the primary discriminatory measures that can assess the quality of classification more reliably than accuracy. Under this data distribution, a model that only predicted passenger would yield an accuracy of 66.95%, but an F1 score of 0. In the case that two model's exhibited the same F1 score, we used the AUC measure to break the tie. The AUC measure is derived from the fact that the output of these classification models is a likelihood value between 0 and 1. A powerful classifier learns a likelihood function that consistently maps instances of the negative class to likelihoods lower than the positive class. A model that is reliable able to do this would receive an AUC of 1, whereas a model that only predicted the negative class would also receive an AUC of 0.
Bimolecular Simulations of Cancer Mutation Effects: Rigidity Decomposition and Protein Stability Analysis
We used FIRST (Floppy Inclusion and Rigid Substructure Topography) approach (Jacobs et al., 2001; Rader et al., 2002; Chubynsky and Thorpe, 2007) and the Python-based Constraint Network Analysis (CNA) interface (Hespenheide et al., 2002; Kruger et al., 2013; Pfleger et al., 2013a,b) to analyze partition of rigid and flexible regions in a set of protein kinases with the predicted cancer driver mutations. The employed parameters are consistent with our previous studies of protein kinases (Stetz et al., 2017). Protein stability computations that evaluated the effect of cancer driver mutations on the functional forms of the ErbB kinases were performed using CUPSAT (Cologne University Protein Stability Analysis Tool) (Parthiban et al., 2006, 2007). This approach was successfully adopted for the energetic analysis of cancer mutation hotspots (Dixit et al., 2009; Dixit and Verkhivker, 2011). We also employed the Foldx method (Guerois et al., 2002; Schymkowitz et al., 2005; Tokuriki et al., 2007; Van Durme et al., 2011) that allows for robust assessment of mutational effects on protein stability. These calculations were done with the user interface for the FoldX force field calculations (Schymkowitz et al., 2005) implemented as a plugin for the YASARA molecular graphics suite (Van Durme et al., 2011).
Protein Structure Network Analysis
For network-based analysis, a graph-based representation of protein structures is employed in which residues are treated as network nodes and inter-residue edges represent residue interactions (Sethi et al., 2009; Vijayabaskar and Vishveshwara, 2010; Stetz and Verkhivker, 2017). NAPS approach (Chakrabarty and Parekh, 2016) was used for construction of the residue interaction networks and subsequent residue-based network centrality analysis. For our analysis, an interaction strength-based graph representation of protein structures was used in which a residue is considered as node in the network and an edge is constructed if the interaction strength between two residues is more than the threshold of 4%. The pair of residues with the interaction Iij greater than a user-defined cut-off (Imin) are connected by edges and produce a protein structure network graph for a given interaction cutoff Imin. The interaction strength Iij is considered as edge weight. The edges in the residue interaction networks were weighted based on the defined interaction strength and dynamic residue correlations couplings (Sethi et al., 2009; Stetz and Verkhivker, 2017). Using the constructed protein structure networks, the residue-based betweenness parameters were also computed with the NAPS server (Chakrabarty and Parekh, 2016). The betweenness of residue i is defined to be the sum of the fraction of shortest paths between all pairs of residues that pass through residue i:
gjk denotes the number of shortest geodesics paths connecting j and k, and gjk(i) is the number of shortest paths between residues j and k passing through the node ni. Residues with high occurrence in the shortest paths connecting all residue pairs have a higher betweenness values. For each node n, the betweenness value is normalized by the number of node pairs excluding n given as (N−1)(N−2)/2, where N is the total number of nodes in the connected component that node n belongs to.
Results
Deep Learning Classification of Cancer Driver Mutations From Nucleotide Information
We began with an attempt to recapitulate our predictions by using various DL/CNN architectures informed by raw nucleotide sequence data evaluated the ability to make predictions based solely on raw genomic information. The inclusion of the three different preprocessing techniques allowed us to select the most informative representation of the nucleotides. The one hot encoded sequences yielded the model with the best performance, and for clarity of presentation we report only the dimensions and performance of the one hot encoded model. This preprocessing model resulted in input matrices of size (2, 105), (2, 505), (2, 1005), (2, 5005), and (2, 50005) corresponding to the different window sizes (10, 50, 100, 500, 1,000) surrounding the original nucleotide. It is worth noting that the embedding algorithm also learned meaningful representations of the nucleotides. The missing place indicator, “n,” was predictably separated from the original nucleotides, which were arranged in 2 neat clusters (Figure 2D). Cluster 1 consisted of the adenine and tyrosine nucleotides, and cluster 2 consisted of the guanine and cytosine nucleotides. These two clusters are easily identified due to the fact that their constituent components are very close to each other while simultaneously being far away from the other cluster.
We employed 72 different DL architectures (Table 1) and the results for the window size of 10 are presented since they revealed more variance (Figure 3). The figures below display the 10 best performing models out of the 72 attempted. The training accuracy continued to increase for the duration of training (Figure 3A), while on the validation testing set of cancer mutations, the best DL/CNN architecture achieved an average validation accuracy of 86.68% with an F1 score of 0.61 (Figure 3B). Interestingly, we found that the DL model seemed to learn early on, overfitting with each successive epoch (Figure 3B). In fact, the model achieved its highest validation accuracy on the first epoch, and proceeds to decline as learning proceeds in subsequent epochs. Furthermore, the AUC score of the model as well as the F1 score consistently stayed the same throughout all of the process. This is further contextualized by the tree based method's performance on the same dataset. The GBT classifier exhibited an F1 score of 0.57 with an average validation accuracy of 66.59%, and the RF classifier exhibited an F1 score of 0.58 and an average validation accuracy of 69.86%. We analyzed predictions by the DL/CNN model by assigning the predicted values for the entire dataset as a separate new feature termed DL score. Although we probed a variety of different architectures and several nucleotide-encoding protocols, a direct brute-force application of DL/CNN models to predict driver mutations only as a function of surrounding nucleotides appeared to be challenging. As a result, we suggested that a diverse set of more informative features may be required to recapitulate the level of robust performance achieved in our earlier work with sequence-based conservation and functional features (Agajanian et al., 2018).
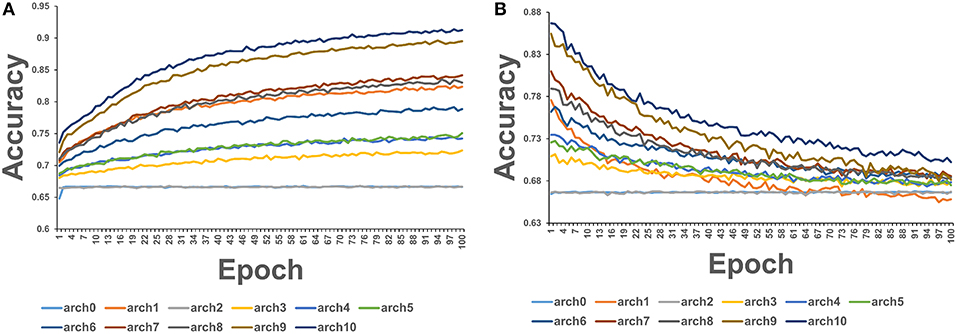
Figure 3. The average accuracy of CNN model using exclusively nucleotide information. (A) Average accuracy across all 3-folds on an epoch by epoch basis on the training set with the sliding window size = 10. (B) Average accuracy across all 3-folds on an epoch by epoch basis on the validation set with the sliding window size = 10.
We first used the RF classifier on the cancer mutation dataset with functional and conservation features obtained from dbWGFP server and adopted in our previous study (Agajanian et al., 2018). A database of human non-synonymous SNVs (dbNSFP) was developed as a one-stop resource for analysis of disease-causing mutations (Liu et al., 2011, 2013, 2016; Dong et al., 2015; Wu et al., 2016) storing 8.58 billion possible human whole-genome SNVs, with capabilities to compute a total of 48 functional prediction scores for each SNV, including 32 functional prediction scores by 13 approaches, 15 conservation features from 4 different tools including ensemble-based predictors RadialSVM, LR, and MSRV scores. The initially selected features were obtained from dbWGFP web server of functional predictions for human whole-genome single nucleotide variants that provided 32 functional prediction scores and 15 evolutionary features (Agajanian et al., 2018). Functional prediction scores refer to scores that predict the likelihood of a given SNV to cause a deleterious functional change in the protein, and evolutionary scores refer to scores providing different conservation measures of a given nucleotide site across multiple species (Supplementary Table S1). Some of the score features (SIFT, PolyPhen, LRT, Mutation Assessor, MutationTaster, FATHMM, RadialSVM, LR, MSRV, and SinBaD) can be applied only to SNVs in the protein coding regions, while other scores (Gerp++, SiPhy, PhyloP, Grantham, CADD, and GWAVA) can evaluate SNVs spreading over the whole genome (Supplementary Table S1). The ensemble-based scores RadialSVM and LR are integrated features that used machine learning approaches to combine information from 10 individual component scores (SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, Gerp++, MutationTaster, Mutation Assessor, FATHMM, LRT, SiPhy, PhyloP) (Agajanian et al., 2018).
In this baseline experiment we evaluated feature performance of 32 input features on the expanded dataset (Figure 4A). Similar to our previous investigation (Agajanian et al., 2018), we found that the ensemble-based scores LR and RadialSVM considerably overshadowed the contributions of other features (Figure 4). By adding DL score to the original 32 features, we applied the RF model for predicting cancer driver mutations with this expanded set of features. The first question was to analyze feature importance of the RF model with the DL score included and determine whether the nucleotide-based scoring feature can contribute to the prediction performance in a meaningful and appreciable way (Figure 4). In the second round of RF classification experiments, we added DL score to the original list of 32 features (Figure 4B). Strikingly, the DL score ranked third following the ensemble-based LR and RadialSVM scores (Figure 4B). Moreover, it was evident that these three feature scores completely dominated feature importance distribution, with the DL score contributing almost as much as the ensemble-based RadialSVM feature (Figure 4B). Quite remarkably, the DL-based score derived by CNN exclusively from primary nucleotide information can deliver significant information content and enrich predictions.
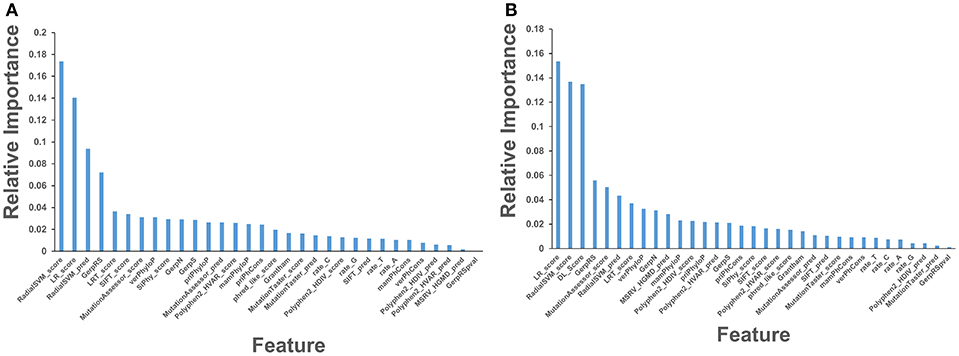
Figure 4. Feature importance of the RF machine learning model on the cancer mutation dataset. The dataset consists of functionally validated 6,389 cancer driver mutations and 12,941 passenger mutations. The initially selected features for RF predictions were obtained from dbWGFP web server (Wu et al., 2016) of functional predictions for human whole-genome single nucleotide variants (Supplementary Table S1). The test set contained 20% of the samples from the original dataset, ensuring that the distribution of drivers and passengers was equivalent to that of the original dataset. The training set was subjected to recursive feature elimination process, resulting in a final dataset of 32 features. (A) Feature importance of 32 functional and sequence conservation features with DL score feature produced by CNN model excluded. (B) Feature importance of 33 features with the DL score included in the RF classification. The feature importance values are shown in blue filled bars and annotated. Feature importance is measured using the information value and weight of evidence criteria.
Using Spearman's rank correlation coefficient, we computed the pairwise correlations between different prediction scores (Figure 5). In this analysis, we found that the two dominant feature scores RadialSVM and LR are only moderately correlated with DL score, with the correlation coefficient of 0.486 and 0.423, respectively. Interestingly, RadialSVM and LR scores are more significantly correlated, suggesting that these ensemble-based features could be complementary with the nucleotide-based DL score. Accordingly, we argued that a combination of these dominant and yet complementary scores may allow for feature reduction and more robust performance of the RF classification models.
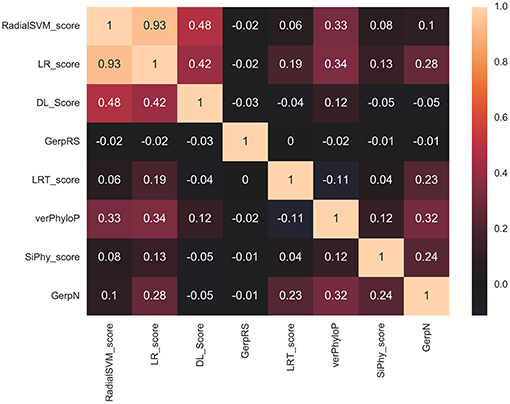
Figure 5. The pairwise Spearman's rank correlation heat map between different prediction scores. The heat map of pairwise Spearman's rank correlation coefficients is shown for top 8 ranking features in the RF classification of cancer mutations with a total of 33 features with DL score included. The high ranking features include ensemble-based RadialSVM, LR scores along with DL score produced by CNN model solely from the raw nucleotide information.
Integration of CNN Predictions With Ensemble-Based Features in Classification Models of Cancer Driver Mutations
Based on these findings, we evaluated feature selection again aiming to recreate the same accuracy with only 8 features: RadialSVM score, LR score, DL score, GerpRS, LRT score, verPhyloP, SiPhy score, GerpN (Figure 6A). The RF model with only 8 features produced a similar ranking in which the ensemble-based scores and DL score contributed the most (Figure 6A). Other contributing features included evolutionary conservation scores derived from multiple sequence alignments and reflecting functional specificity, such as GerpRS (Davydov et al., 2010), SiPhy (Garber et al., 2009), and PhyloP (Garber et al., 2009) also showed appreciable information score values (Figure 6A). We then tested the performance of the RF model and feature importance by performing machine learning of cancer driver mutations using only 3 top features (Figure 6B).
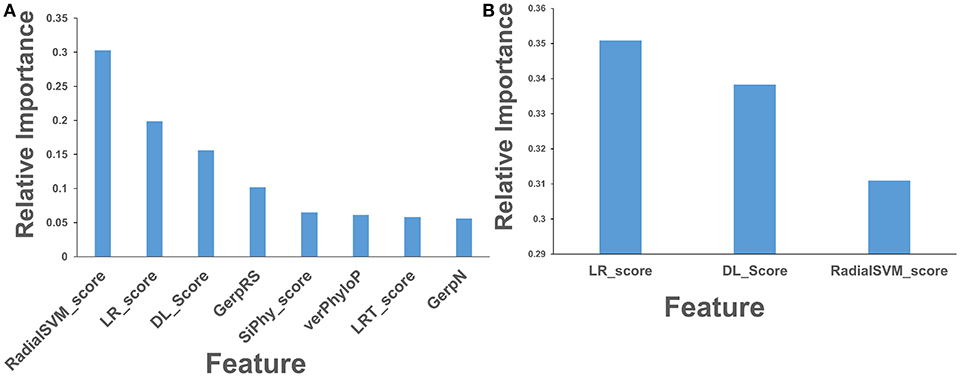
Figure 6. Feature importance of the RF model on the cancer mutation dataset with the reduced number of features. (A) Feature importance ranking based on RF classification with only 8 most informative features. (B) Feature importance ranking based on RF classification with only 3 top features that included ensemble-based RadialSVM, LR scores, and DL score produced by CNN model. The feature importance values are shown in blue filled bars and annotated. Feature importance is measured using the information value and weight of evidence criteria.
The predictive performance of the RF models with different set of features was examined using area under the curve (AUC) plots (Figure 7). First, we examined difference in the AUC curves for RF-based classification with 32 functional features and with additional DL score (Figure 7A). The results showed a very similar high-level prediction performance with AUC = 0.95–0.96. It is worth noting that due to high AUC value for RF classification with 32 informative functional features, the addition of DL could not significantly enhance it. However, we showed that this nucleotide-derived predictor score provides an additional information content and is complementary to the ensemble-based RadialSVM score and LR score. In this context, it was instructive to observe that addition of DL score may marginally improve separation between TPR and FPR at higher values of these parameters (Figure 7A).
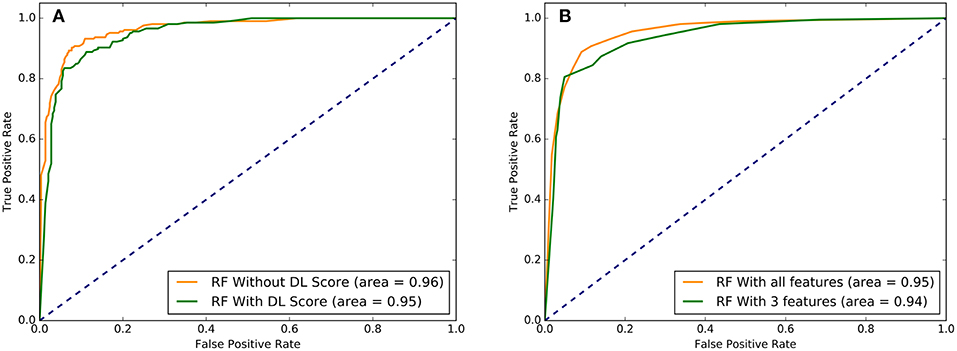
Figure 7. The ROC plots of sensitivity (TPR) as a function of 1-specificity, where specificity (TNR). (A) The ROC curves for overall performance of the RF model with 32 functional features excluding DL score (in green) and 33 features that included DL score (in red). (B) The ROC curves for the RF model with all 33 features (in green) and with the top 3 performing features that included LR score, Radial_SVM score, and DL score (in red). Higher AUC score indicates better performance. These plots illustrated a comparative performance of machine learning models for top prediction scores.
Strikingly, RF learning model that relied on only 3 top features (RadialSVM score, LR score, and DL score) yielded AUC = 0.94, thereby showing that these features may be sufficient to achieve robust classification of cancer driver mutations on a fairly large dataset of somatic mutations employed in this study. Combined with the findings that DL score only weakly correlated with the ensemble-based scores, we concluded that unexpectedly few highly informative parameters can achieve high level of performance (Figure 7). We then tested several machine learning models including RF, GBTs and support vector machine (SVM) on the dataset with the top 8 features to benchmark performance against the original RF model with 32 features (Agajanian et al., 2018). The performance of classification models was carefully assessed (Table 2). All methods achieved a high classification accuracy of ~90%. The sensitivity values were higher for the SVM and RF models, but all methods yielded similar high performance classification on the dataset with only limited number of major features that included DL score (Table 2).
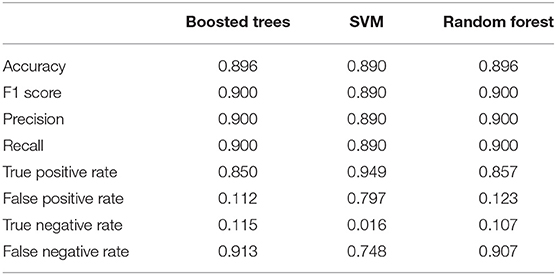
Table 2. The relative performance metrics and statistics of various machine learning models in classification of cancer driver mutations with the top 8 features.
To summarize, our results supported the notion that machine learning-derived ensemble functional predictors may play a central role in classification of cancer driver mutations. The central finding of these machine learning experiments was that combination of ensemble-based features and DL score derived by CNN model from nucleotide information are complementary and when combined can yield classification accuracy comparable and often exceeding the one obtained with a full set of features. The important lesson from this analysis is that integrated high-level features derived by machine learning approaches from primary nucleotide and protein sequence information may be sufficient to predict an important functional phenotype. Although structure-derived features and other functional scores contribute to feature importance ranking and tightly linked with the mutational phenotype, the success of machine learning tools in deciphering predictive features from primary sequence information is encouraging and should be further explored in other applications.
Leveraging Machine Learning Predictions in Structure-Functional Analysis of Molecular Signatures of Driver Mutations in Oncogenic Protein Kinases
Machine learning driver/passenger classifications typically consider activating, inactivating and inhibitory (or resistant) mutations as drivers, often leaving aside a more detailed characterization and assignment of driver positions. Direct predictions of these specific classes may not be adequately suited for machine learning tools due to smaller datasets. To expand our predictions and aim at extracting a more granular functional information about driver mutations, we conducted rigidity decomposition simulations and analyzed conformational flexibility of the predicted driver positions in protein kinase genes. The objective of this analysis was to facilitate functional validation and interpretation of machine learning results through coarse-grained biophysical simulations as an effective post-processing tool of machine learning classification. In fact, the proposed simulation analysis of mobility at the driver positions allows to expand classification of driver mutations further and characterize activating drivers. Previous studies have suggested that conformational mobility of many oncogenic kinases may be linked with preferential localization of activating cancer mutations in flexible functional regions (Paladino et al., 2015; Kiel et al., 2016; Stetz et al., 2017).
We examined flexibility of specific functional regions targeted by driver mutations in oncogenic protein kinases and probed functional propensity of these drivers to promote transitions to constitutively active states. The primary focus of this analysis is on the family of the ErbB protein tyrosine kinases (Lemmon and Schlessinger, 2010; Roskoski, 2014). A number of human cancers are associated with mutations causing the increased expression of the ErbB kinases. A large number of activating and drug resistance EGFR mutations have been extensively studied at the molecular and functional levels (Paez et al., 2004; Kobayashi et al., 2005; Zhou et al., 2009; Eck and Yun, 2010). Oncogenic kinase mutants are known to act by destabilizing the inactive dormant kinase form while promoting conformational transitions and stabilization of a constitutively active kinase state—a salient functional characteristic linked with the initiation or progression of cancer (Carey et al., 2006; Wang et al., 2011). We used the crystal structures of the EGFR, ErbB2, ErbB3, and ErbB4 kinases that constitute this family to perform rigidity decomposition and then align the positions of the predicted cancer driver mutations with the structural mobility maps (Figure 8). We examined how the predicted driver mutations for ErbB protein kinases are distributed on the rigidity/flexibility map of the catalytic core and whether the dynamic preferences of mutational sites can be linked with their primary function as activating drivers. To explore these questions, we examined the predicted cancer driver mutations for the ErbB kinase family. Structural mapping of these cancer mutations onto the crystallographic ErbB conformations showed that activating driver mutations are preferentially localized in the flexible regions and target positions where they can readily promote conformational changes to the active form without severely compromising thermodynamic stability (Figure 8).
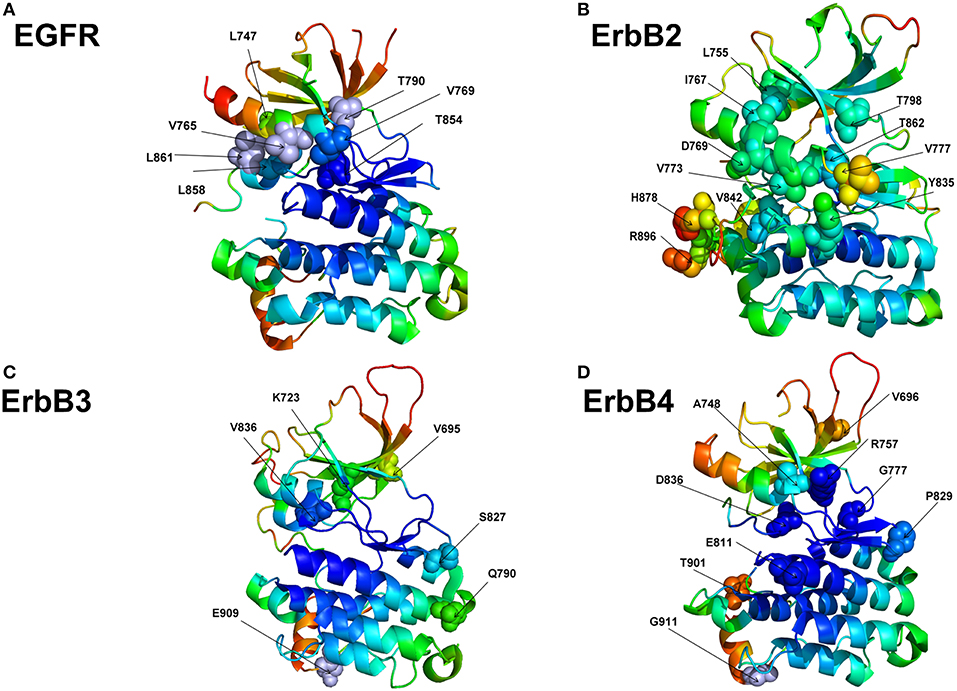
Figure 8. Structural maps of rigidity decomposition and mobility signatures of cancer mutation drivers in the ErbB protein kinases. Structural mapping of rigidity and flexibility regions in the crystal structure of EGFR (pdb id 1XKK) (A), crystal structure of ErbB2 kinase (pdb id 3PP0) (B), crystal structure of ErbB3 kinase (pdb id 3KEX) (C), and crystal structure of ErbB4 kinase (pdb id 3BBT) (D). Crystallographic conformations are colored using a color range from red (highest flexibility) to blue (highest rigidity). The positions of predicted in machine learning cancer driver mutations are shown in spheres (colored according to their mobility level) and annotated.
To quantify these arguments further, we also characterized the free energy differences between wild-type and cancer-driver mutations for the ErbB proteins in both inactive and active kinase forms (Figure 9). Since both CUPSAT and FoldX approaches yielded similar results, we illustrated our findings by presenting FoldX-derived protein stability changes (Figure 9). The results of this simulation-driven functional classification of predicted driver mutations were compared with the biochemical and mutagenesis data. The analysis of driver mutations in EGFR confirmed that L858 and L861 positions target flexible regions as can be manifested by classical activating driver mutations L858R and L861Q (Littlefield and Jura, 2013; Red Brewer et al., 2013). The energetics of these activating drivers is consistent with a common mechanism of the constitutive activation of kinases by driver mutations (Figure 9A). This mechanism reflects a combined effect of activating mutations producing a more significant destabilization of the inactive state as compared to the active state, triggering shift of the thermodynamic equilibrium toward the active conformation. We found that some EGFR mutations such as T854A are mapped onto more stable regions of the kinase (Figure 8A) and showed similar destabilization in the inactive and active forms. Accordingly, this predicted cancer driver mutation is likely not activating but rather may be attributed to inhibitory or resistant mutations. Indeed, the recent experimental studies showed that T854A mutation is the acquired mutation causing resistance to known drugs (Bean et al., 2008). Another EGFR mutation V769M/L showed an intermediate level of mobility (Figure 8A) and greater stabilization of the active state. These results are in line with recent functional experiments showing that EGFR-V769M mutation is indeed activating that may explain the role of this driver mutation in the development of multiple lung cancers in a pool of lung cancer patients (Deng et al., 2018).
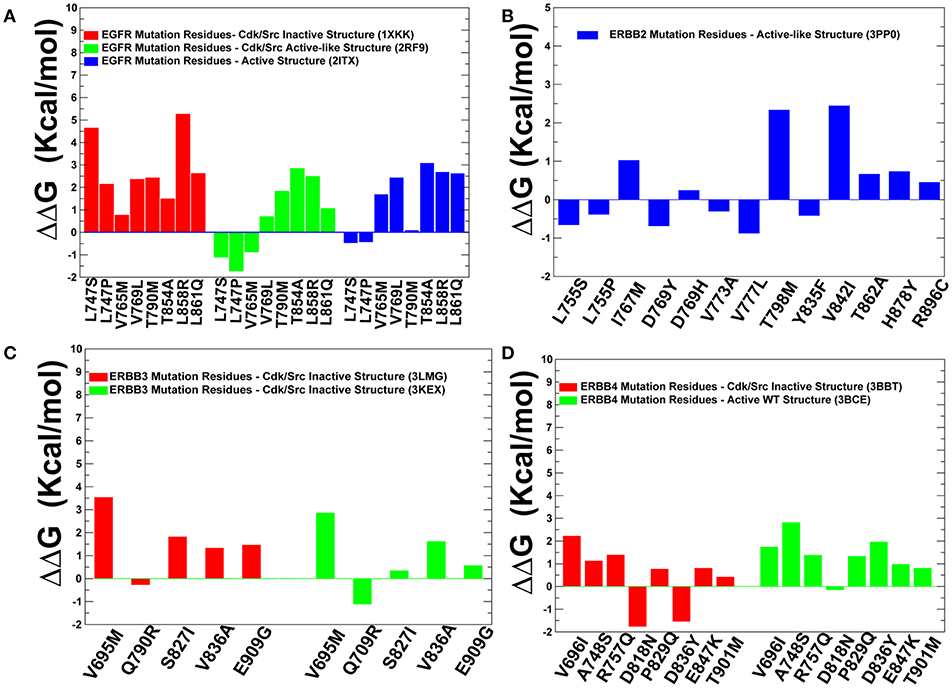
Figure 9. Protein stability analysis of the predicted cancer driver mutations. Protein stability differences calculated between the wild-type and mutants for predicted cancer driver mutations in the ErbB kinases using FOLDx approach. Protein stability changes induced by cancer driver mutations in the inactive and active states of EGFR kinase (A), ErbB2 kinase (B), ErbB3 kinase (C), and ErbB4 kinase (D). Positive values of protein stability changes correspond to destabilizing mutations.
The positions of almost all predicted driver mutations in ErbB2 kinase target highly flexible regions and can be assigned in our model to activating driver mutations (Figures 8B, 9B). Our previous biophysical simulations and network analysis of activation mechanisms in the ErbB proteins similarly indicated that almost all oncogenic ErbB2 variants are localized in the mobile αC-β4 loop and highly dynamic in their inactive states promoting transition to the active form and causing an uncontrollable activity (James and Verkhivker, 2014). These findings are consistent with the experimental studies (Fan et al., 2008; Aertgeerts et al., 2011). While the majority of somatic mutations in the EGFR and ErbB2 kinases increase the kinase activity, a number of the classified ErbB4 cancer mutants have been shown to inhibit or reduce the kinase activity (Tvorogov et al., 2009). In particular, some cancer-associated mutations of ErbB4 can promote loss of ErbB4 kinase activity as these alterations weaken the important functional interactions in the catalytic core and may interfere with the protein stability. According to experimental data, some cancer mutations have only minor or no effect on kinase activity (V696I, E785K, A748S, P757Q, P829Q, and T901M), while K726R abolishes kinase activity and D818N and D836Q are known as kinase-dead mutations (Tvorogov et al., 2009). We found that predicted cancer driver mutations are mapped onto more stable regions in ErbB4, owing to the greater rigidity of this catalytic domain (Figures 8D, 9D). Accordingly, the respective driver mutations cannot function as activating but rather may cause significant distortions of the kinase structure, causing abolishment of kinase activity which is the functional signature of most cancer drivers in ErbB4 kinase. The performed simulation-driven post-processing of machine learning predictions facilitated in silico functional characterization of cancer mutations and allowed to properly assign activating or inhibiting phenotypic effects to a pool of pathogenic kinase variants.
To provide more quantitative insights, we used the predicted cancer mutations in the ErbB kinases and conducted protein structure network analysis to identify whether positions of deleterious mutations would overlap with the global mediating nodes in the interaction networks. The betweenness of a residue node is defined as the number of shortest paths that can go through that node, thus estimating the contribution of the node to the global communication flow in the system. High betweenness nodes can influence the spread of information through the network by facilitating, hindering, or altering the communication between others. According to our hypothesis, cancer mutations may preferentially target the essential mediating residues with a high centrality that play an important role in activity and signaling of protein kinase genes.
The centrality analysis revealed important differences in the distribution of mediating centers in the ErbB kinase structures (Figure 10). We particularly observed that the betweenness of the active form of EGFR (Figure 10A) and ErbB4 (Figure 10D) was on average higher than for the inactive states. Importantly, the location of the properly classified EGFR mutations with the highest oncogenic potential (L858R, T790M, L838V, V742A, V851A, I853T) corresponds to some of the high centrality peaks of the profile (Figure 10A). In addition, these residues showed appreciable differences in the betweenness values between the inactive to the active states, as the residue centrality in these positions typically increased in the functional active form (Figures 10A,D). These findings suggested that a number of key activating mutations in the ErbB kinases target mediating sites of global allosteric communication in the protein structures. We believe that by adding this significant additional component to our study, we have been able to further quantify and explain the protein rigidity/flexibility analysis of predicted cancer mutations in the kinase genes. In our view, by complementing machine learning predictions with the structural and network-based analyses we can obtain useful insights into mechanisms underlying effects of cancer mutations and also identify limitations of classification models and ways to improve interpretability and trustability of machine learning model approaches.
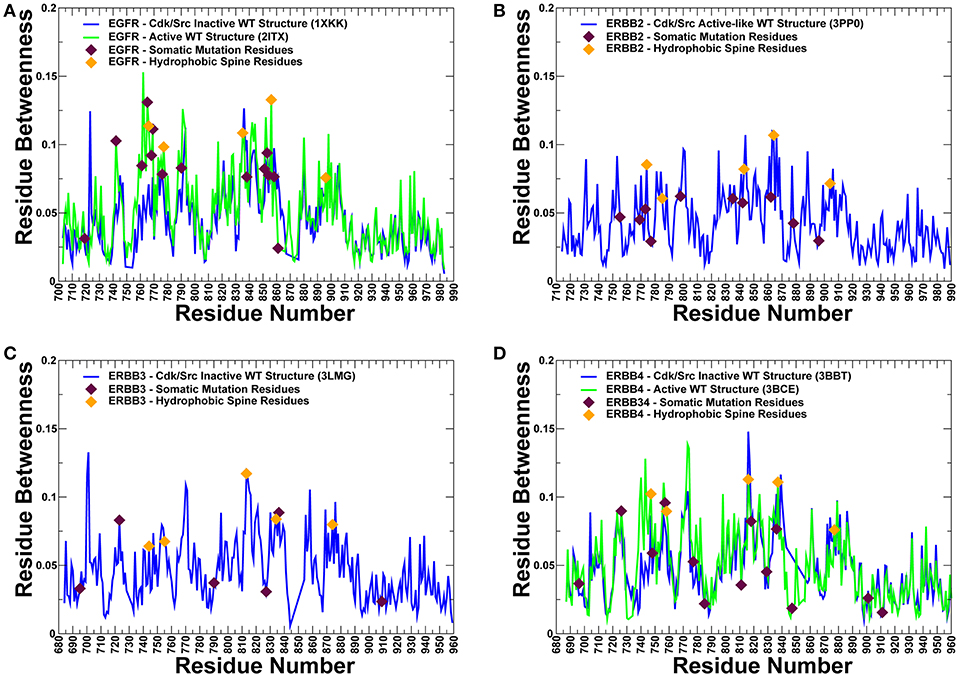
Figure 10. The residue-based betweenness profiles of the ErbB kinase structures. The residue betweenness (residue centrality) profiles for the inactive and active crystal structure states of EGFR (A), ErbB2 (B), ErbB3 (C), and ErbB4 kinases (D). For ErbB2 and ErbB3 only crystal structures of the inactive-type states were available for the analysis. The positions of somatic mutations predicted by machine learning experiments are shown in maroon-colored filled triangles, and residue positions corresponding to the hydrophobic spine residues are shown in orange-colored filled triangles. Protein kinase activation is controlled by two networks of mostly hydrophobic residues that form a regulatory spine (R-spine) and a catalytic spine (C-spine). The EGFR R-spine residues include L777 from the β4-strand, M766 from the C-terminal end of the αC-helix, F856 of the DFG motif in the activation segment, H835 of the HRD motif of the catalytic loop, and D896 of the αF-helix.). The R-spine residues in ErbB2 are M774, L785, F864, H843, and D904. The R-spine residues in ErbB3 are I744, L755, F843, H813, and D874. The R-spine residues in ErbB4 are M747, L758, H816, F837, and D877.
Discussion
As large-scale biological data are available from high-throughput assays, and methods for learning the thousands of network parameters have matured, we can now assess feasibility and practicality of using specialized neural network architectures as classification tools for recognizing cancer-causing variants and associated cancer types. Given rapid proliferation and increasing popularity of deep learning tools to address various biological problems, there are several fundamental questions arising in the context of classification of cancer driver mutations. Will deep learning make all other models obsolete? Can deep learning models achieve robust classification and recognition of cancer driver mutations based solely on nucleotide information? What is the role of many functional and structural predictors derived from biophysical perspective in this context? In this work, we have explored and integrated different machine learning approaches for prediction and classification of cancer driver mutations. We first explored the ability of CNN models to identify and classify cancer driver mutations directly from raw nucleotide sequence information without relying on specific functional scores.
The results of this study have demonstrated that while CNN models can learn high level features from genomic information that has sufficiently high importance, accurate classification of cancer mutation driver phenotype using exclusively nucleotide data continues to be challenging. This problem is admittedly more complex than the experimental design suggests, due to the complex nature of protein interactions in the human body. This experimental setup considered only the primary sequence form of the nucleotides, which could only ever partially explain the onset of cancer. The secondary, tertiary, and quaternary form of these same strings would certainly contain more information, due to the folding processes that occur in these steps. Additionally, this technique ignores all of the possible interactions that can be had with other structures in the body, which further dilutes the informational value present in the dataset. As such it's unreasonable to assume that our solely primary sequence based dataset would be able to explain all of the variance present in a complex problem like determining a single mutation's level of effect on the onset of cancer. The experimental inclusion of the different window sizes was also an attempt to allow increasing numbers of surrounding nucleotides to have an influence on our chosen mutation's effect. An obvious assumption here is that more nucleotides would in fact bring in more information. This, however, proved not to hold up as the only dataset that provided any significant variance in performance was the window size = 10 dataset. This suggests that more nucleotides only confuse the model and disallow it from learning informative patterns. This problem could possibly be combatted in future research by testing out larger architectures.
The benefits of integrating CNN-derived predictors obtained from nucleotide information with protein sequence features, evolutionary and functional scores were then carefully examined. By exploring various encoding techniques and an array of different CNN architectures, we have found that neural networks can quickly learn an important functional signal, but can rarely steadily improve the initial performance spike with the number of additional epochs. The juxtaposition of monotonically increasing training accuracy with monotonically decreasing validation accuracy is a telltale sign of overfitting. This suggests that there is only a small amount of useful information that can be learned very early on, and subsequent epochs only cause the model to learn noisy patterns that are only exhibited in the training set. It is difficult to determine exactly what was learned by the model due to the black box nature of neural networks, however due to the short path to optimality it is safe to say that any learned concepts cannot be overly complex. We have pursued a synergistic strategy in which the prediction score generated by CNN models was integrated with physics-based functional, structural and evolutionary conservation features. The important lesson of this analysis was the revelation that CNN-derived features may be complementary to the ensemble-based predictors often employed for classification of cancer mutations. These other scores are not calculated from raw sequence based techniques, which supports this DL score as a novel inclusion into a portfolio of scores due to its unique derivation.
By combining deep learning-generated score with only two main ensemble-based functional features, we were able to achieve a high performance level for cancer driver mutations. The robustness of this approach was verified by several traditional machine learning classifiers, including RF, SVM, and GBTs. We have found that integration of CNN-derived predictor score with only several ensemble-based features can recapitulate the results obtained with a large number of functional features and improve performance in capturing driver mutations across a spectrum of machine learning classifiers. Our findings have also demonstrated that synergy of nucleotide-based deep learning scores and integrated metrics derived from protein sequence conservation scores can allow for robust classification of cancer driver mutations with a reduced number of highly informative features. This is an interesting and highly informative result, as the law of parsimony holds for machine learning models so simpler models with comparable performance are typically preferred over their more complex counterparts. Part of this model complexity includes the number of features that a model relies on. As such a reduction in features is a universally positive outcome. In addition to the improved quality of the model, it also expands the universe of predictable nucleotides that are available to us since we depend only on the presence of two ensemble-based scores. The DL score can be derived for any mutation with known coordinates so this is not a limiting factor. In this respect our initial goal of expanding the nucleotides we can make predictions for was partially achieved. This increase in the generalization of these models facilitates the logical conclusion of driver classification efforts, accurately classifying all known nucleotides.
While machine learning approaches can often produce robust and accurate predictors, the ultimate goal of research is fundamental understanding of the underlying phenomena which requires a mechanistic model of the world. In this context, machine learning predictions are leveraged in biomolecular simulations to enable analysis of cancer mutation mechanisms and obtain a more specific information about an important subset of cancer mutations, activating drivers. The results of our investigation suggested that through integration of machine learning classification and biomolecular simulations of cancer mutations we can often validate the predictions and facilitate a more detailed functional analysis of activating driver mutations. These findings can provide insight and new angle to the problem of interpretability of “black box” machine learning results. By carefully inspecting predictions of machine learning models in the context of dynamic and energetic signatures of mutational sites for oncogenic protein kinases, this study offered instructive strategy for simulation-based post-processing of machine learning predictions and detailed functional specification of cancer driver mutations. The proposed synergistic integration of machine learning and biomolecular simulations into a single computational platform allows to rapidly process large datasets and make robust predictions on functionally significant cancer drivers. The results of this study may also inform and guide design of targeted and personalized therapeutic agents combating a spectrum of mutational changes occurring in cancer.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: https://www.cbioportal.org/.
Author Contributions
GV and SA conceived and designed the research. SA and OO performed the research. SA, OO, and GV analyzed the results and wrote the manuscript. GV wrote the final version of the manuscript and supervised the project.
Funding
This work was partly supported by institutional funding from Chapman University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the technical assistance of Schmid College Grand Challenge Initiative Postdoctoral Fellow Dr. Anne Sonnenschein.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2019.00044/full#supplementary-material
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Aertgeerts, K., Skene, R., Yano, J., Sang, B. C., Zou, H., Snell, G., et al. (2011). Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765. doi: 10.1074/jbc.M110.206193
Agajanian, S., Odeyemi, O., Bischoff, N., Ratra, S., and Verkhivker, G. M. (2018). Machine learning classification and structure-functional analysis of cancer mutations reveal unique dynamic and network signatures of driver sites in oncogenes and tumor suppressor genes. J. Chem. Inf. Model. 58, 2131–2150. doi: 10.1021/acs.jcim.8b00414
Ainscough, B. J., Barnell, E. K., Ronning, P., Campbell, K. M., Wagner, A. H., Fehniger, T. A., et al. (2018). A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat. Genet. 50, 1735–1743. doi: 10.1038/s41588-018-0257-y
Bailey, M. H., Tokheim, C., Porta-Pardo, E., Sengupta, S., Bertrand, D., Weerasinghe, A., et al. (2018). Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e318. doi: 10.1016/j.cell.2018.02.060
Bardelli, A., Parsons, D. W., Silliman, N., Ptak, J., Szabo, S., Saha, S., et al. (2003). Mutational analysis of the tyrosine kinome in colorectal cancers. Science 300:949. doi: 10.1126/science.1082596
Bean, J., Riely, G. J., Balak, M., Marks, J. L., Ladanyi, M., Miller, V. A., et al. (2008). Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin. Cancer Res. 14, 7519–7525. doi: 10.1158/1078-0432.CCR-08-0151
Bertrand, D., Drissler, S., Chia, B. K., Koh, J. Y., Li, C., Suphavilai, C., et al. (2018). Consensus driver improves upon individual algorithms for predicting driver alterations in different cancer types and individual patients. Cancer Res. 78, 290–301. doi: 10.1158/0008-5472.CAN-17-1345
Biau, G. (2012). Analysis of a random forest model. J. Mach. Learn. Res. 13, 1063–1095. Available online at: http://www.jmlr.org/papers/volume13/biau12a/biau12a.pdf
Carey, K. D., Garton, A. J., Romero, M. S., Kahler, J., Thomson, S., Ross, S., et al. (2006). Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 66, 8163–8171. doi: 10.1158/0008-5472.CAN-06-0453
Carter, H., Chen, S., Isik, L., Tyekucheva, S., Velculescu, V. E., Kinzler, K. W., et al. (2009). Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 69, 6660–6667. doi: 10.1158/0008-5472.CAN-09-1133
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. doi: 10.1158/2159-8290.CD-12-0095
Chakrabarty, B., and Parekh, N. (2016). NAPS: network analysis of protein structures. Nucleic Acids Res. 44, W375–W382. doi: 10.1093/nar/gkw383
Cheng, F., Zhao, J., and Zhao, Z. (2016). Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief. Bioinformatics 17, 642–656. doi: 10.1093/bib/bbv068
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7:e46688. doi: 10.1371/journal.pone.0046688
Chubynsky, M. V., and Thorpe, M. F. (2007). Algorithms for three-dimensional rigidity analysis and a first-order percolation transition. Phys. Rev. E. Stat. Nonlin. Soft. Matter. Phys. 76:041135. doi: 10.1103/PhysRevE.76.041135
Cibulskis, K., Lawrence, M. S., Carter, S. L., Sivachenko, A., Jaffe, D., Sougnez, C., et al. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. doi: 10.1038/nbt.2514
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. doi: 10.1038/nature00766
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
Dees, N. D., Zhang, Q., Kandoth, C., Wendl, M. C., Schierding, W., Koboldt, D. C., et al. (2012). MuSiC: identifying mutational significance in cancer genomes. Genome Res. 22, 1589–1598. doi: 10.1101/gr.134635.111
Deng, Q., Xie, B., Wu, L., Ji, X., Li, C., Feng, L., et al. (2018). Competitive evolution of NSCLC tumor clones and the drug resistance mechanism of first-generation EGFR-TKIs in Chinese NSCLC patients. Heliyon 4:e01031. doi: 10.1016/j.heliyon.2018.e01031
Ding, L., Wendl, M. C., McMichael, J. F., and Raphael, B. J. (2014). Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet. 15, 556–570. doi: 10.1038/nrg3767
Dixit, A., and Verkhivker, G. M. (2011). The energy landscape analysis of cancer mutations in protein kinases. PLoS ONE 6:13. doi: 10.1371/journal.pone.0026071
Dixit, A., Yi, L., Gowthaman, R., Torkamani, A., Schork, N. J., and Verkhivker, G. M. (2009). Sequence and structure signatures of cancer mutation hotspots in protein kinases. PLoS ONE 4:14. doi: 10.1371/journal.pone.0007485
Dong, C., Wei, P., Jian, X., Gibbs, R., Boerwinkle, E., Wang, K., et al. (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137. doi: 10.1093/hmg/ddu733
Douville, C., Carter, H., Kim, R., Niknafs, N., Diekhans, M., Stenson, P. D., et al. (2013). CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics 29, 647–648. doi: 10.1093/bioinformatics/btt017
Eck, M. J., and Yun, C. H. (2010). Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 1804, 559–566. doi: 10.1016/j.bbapap.2009.12.010
Ellrott, K., Bailey, M. H., Saksena, G., Covington, K. R., Kandoth, C., Stewart, C., et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e277. doi: 10.1016/j.cels.2018.03.002
Erickson, B. J., Korfiatis, P., Akkus, Z., Kline, T., and Philbrick, K. (2017). Toolkits and libraries for deep learning. J. Digit Imag. 30, 400–405. doi: 10.1007/s10278-017-9965-6
Fan, Y., Xi, L., Hughes, D. S., Zhang, J., Futreal, P. A., Wheeler, D. A., et al. (2016). MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 17:178. doi: 10.1186/s13059-016-1029-6
Fan, Y. X., Wong, L., Ding, J., Spiridonov, N. A., Johnson, R. C., and Johnson, G. R. (2008). Mutational activation of ErbB2 reveals a new protein kinase autoinhibition mechanism. J. Biol. Chem. 283, 1588–1596. doi: 10.1074/jbc.M708116200
Forbes, S. A., Beare, D., Gunasekaran, P., Leung, K., Bindal, N., Boutselakis, H., et al. (2015). COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–811. doi: 10.1093/nar/gku1075
Futreal, P. A., Coin, L., Marshall, M., Down, T., Hubbard, T., Wooster, R., et al. (2004). A census of human cancer genes. Nat. Rev. Cancer 4, 177–183. doi: 10.1038/nrc1299
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6:pl1. doi: 10.1126/scisignal.2004088
Garber, M., Guttman, M., Clamp, M., Zody, M. C., Friedman, N., and Xie, X. (2009). Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25, i54–62. doi: 10.1093/bioinformatics/btp190
Gauthier, N. P., Reznik, E., Gao, J., Sumer, S. O., Schultz, N., Sander, C., et al. (2016). MutationAligner: a resource of recurrent mutation hotspots in protein domains in cancer. Nucleic Acids Res.. 44, D986–991. doi: 10.1093/nar/gkv1132
Gnad, F., Baucom, A., Mukhyala, K., Manning, G., and Zhang, Z. (2013). Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics 14 (Suppl 3):S7. doi: 10.1186/1471-2164-14-S8-S7
Goh, G. B., Hodas, N. O., and Vishnu, A. (2017). Deep learning for computational chemistry. J. Comput. Chem. 38, 1291–1307. doi: 10.1002/jcc.24764
Gonzalez-Perez, A., and Lopez-Bigas, N. (2011). Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 88, 440–449. doi: 10.1016/j.ajhg.2011.03.004
Gonzalez-Perez, A., and Lopez-Bigas, N. (2012). Functional impact bias reveals cancer drivers. Nucleic Acids Res. 40:e169. doi: 10.1093/nar/gks743
Gonzalez-Perez, A., Mustonen, V., Reva, B., Ritchie, G. R., Creixell, P., Karchin, R., et al. (2013). Computational approaches to identify functional genetic variants in cancer genomes. Nat. Methods 10, 723–729. doi: 10.1038/nmeth.2562
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. doi: 10.1038/nature05610
Guerois, R., Nielsen, J. E., and Serrano, L. (2002). Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387. doi: 10.1016/S0022-2836(02)00442-4
Haber, D. A., and Settleman, J. (2007). Cancer: drivers and passengers Nature 446, 145–146. doi: 10.1038/446145a
Hespenheide, B. M., Rader, A. J., Thorpe, M. F., and Kuhn, L. A. (2002). Identifying protein folding cores from the evolution of flexible regions during unfolding. J. Mol. Graph. Model. 21, 195–207. doi: 10.1016/S1093-3263(02)00146-8
Hinkson, I. V., Davidsen, T. M., Klemm, J. D., Kerlavage, A. R., and Kibbe, W. A. (2017). A comprehensive infrastructure for big data in cancer research: accelerating cancer research and precision medicine. Front. Cell. Dev. Biol. 5:83. doi: 10.3389/fcell.2017.00083
Hudson, T. J., Anderson, W., Artez, A., Barker, A. D., Bell, C., Bernabe, R. R., et al. (2010). International network of cancer genome projects. Nature 464, 993–998. doi: 10.1038/nature08987
Jacobs, D. J., Rader, A. J., Kuhn, L. A., and Thorpe, M. F. (2001). Protein flexibility predictions using graph theory. Proteins 44, 150–165. doi: 10.1002/prot.1081
James, K. A., and Verkhivker, G. M. (2014). Structure-based network analysis of activation mechanisms in the ErbB family of receptor tyrosine kinases: the regulatory spine residues are global mediators of structural stability and allosteric interactions. PLoS ONE 9:e113488. doi: 10.1371/journal.pone.0113488
Jensen, M. A., Ferretti, V., Grossman, R. L., and Staudt, L. M. (2017). The NCI Genomic Data Commons as an engine for precision medicine. Blood 130, 453–459. doi: 10.1182/blood-2017-03-735654
Kiel, C., Benisty, H., Llorens-Rico, V., and Serrano, L. (2016). The yin-yang of kinase activation and unfolding explains the peculiarity of Val600 in the activation segment of BRAF. Elife 5:e12814. doi: 10.7554/eLife.12814
Kim, S., Scheffler, K., Halpern, A. L., Bekritsky, M. A., Noh, E., Kallberg, M., et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. doi: 10.1038/s41592-018-0051-x
Klonowska, K., Czubak, K., Wojciechowska, M., Handschuh, L., Zmienko, A., Figlerowicz, M., et al. (2016). Oncogenomic portals for the visualization and analysis of genome-wide cancer data. Oncotarget 7, 176–192. doi: 10.18632/oncotarget.6128
Kobayashi, S., Boggon, T. J., Dayaram, T., Janne, P. A., Kocher, O., Meyerson, M., et al. (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792. doi: 10.1056/NEJMoa044238
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Kruger, D. M., Rathi, P. C., Pfleger, C., and Gohlke, H. (2013). CNA web server: rigidity theory-based thermal unfolding simulations of proteins for linking structure, (thermo-)stability, and function. Nucleic Acids Res. 41, W340–W348. doi: 10.1093/nar/gkt292
Lai, Z., Markovets, A., Ahdesmaki, M., Chapman, B., Hofmann, O., McEwen, R., et al. (2016). VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44:e108. doi: 10.1093/nar/gkw227
Larson, D. E., Harris, C. C., Chen, K., Koboldt, D. C., Abbott, T. E., Dooling, D. J., et al. (2012). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317. doi: 10.1093/bioinformatics/btr665
Lawrence, M. S., Stojanov, P., Polak, P., Kryukov, G. V., Cibulskis, K., Sivachenko, A., et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. doi: 10.1038/nature12213
Lemmon, M. A., and Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. doi: 10.1016/j.cell.2010.06.011
Li, J., Drubay, D., Michiels, S., and Gautheret, D. (2015). Mining the coding and non-coding genome for cancer drivers. Cancer Lett. 369, 307–315. doi: 10.1016/j.canlet.2015.09.015
Littlefield, P., and Jura, N. (2013). EGFR lung cancer mutants get specialized. Proc. Natl. Acad. Sci. U.S.A. 110, 15169–15170. doi: 10.1073/pnas.1314719110
Liu, X., Jian, X., and Boerwinkle, E. (2011). dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899. doi: 10.1002/humu.21517
Liu, X., Jian, X., and Boerwinkle, E. (2013). dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 34, E2393–2402. doi: 10.1002/humu.22376
Liu, X., Wu, C., Li, C., and Boerwinkle, E. (2016). dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241. doi: 10.1002/humu.22932
Luo, P., Ding, Y., Lei, X., and Wu, F. X. (2019). deepDriver: predicting cancer driver genes based on somatic mutations using deep convolutional neural networks. Front. Genet. 10:13. doi: 10.3389/fgene.2019.00013
Mao, Y., Chen, H., Liang, H., Meric-Bernstam, F., Mills, G. B., and Chen, K. (2013). CanDrA: cancer-specific driver missense mutation annotation with optimized features. PLoS ONE 8:e77945. doi: 10.1371/journal.pone.0077945
Martelotto, L. G., Ng, C. K., De Filippo, M. R., Zhang, Y., Piscuoglio, S., Lim, R. S., et al. (2014). Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 15:484. doi: 10.1186/s13059-014-0484-1
Masica, D. L., Douville, C., Tokheim, C., Bhattacharya, R., Kim, R., Moad, K., et al. (2017). CRAVAT 4: cancer-related analysis of variants toolkit. Cancer Res. 77, e35–e38. doi: 10.1158/0008-5472.CAN-17-0338
Mikolov, T., Chen, K., Corrado, G., and Dean, J. (2013). Efficient Estimations of Word Representations in Vector Space. arXiv:1301.3781 [cs.CL]. Available online at: https://arxiv.org/abs/1301.3781
Mularoni, L., Sabarinathan, R., Deu-Pons, J., Gonzalez-Perez, A., and Lopez-Bigas, N. (2016). OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 17:128. doi: 10.1186/s13059-016-0994-0
Ng, P. K., Li, J., Jeong, K. J., Shao, S., Chen, H., Tsang, Y. H., et al. (2018). Systematic functional annotation of somatic mutations in cancer. Cancer Cell 33, 450–462.e410. doi: 10.1016/j.ccell.2018.01.021
Niu, B., Scott, A. D., Sengupta, S., Bailey, M. H., Batra, P., Ning, J., et al. (2016). Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet. 48, 827–837. doi: 10.1038/ng.3586
Paez, J. G., Janne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. doi: 10.1126/science.1099314
Paladino, A., Morra, G., and Colombo, G. (2015). Structural stability and flexibility direct the selection of activating mutations in epidermal growth factor receptor kinase. J. Chem. Inf. Model. 55, 1377–1387. doi: 10.1021/acs.jcim.5b00270
Parthiban, V., Gromiha, M. M., Abhinandan, M., and Schomburg, D. (2007). Computational modeling of protein mutant stability: analysis and optimization of statistical potentials and structural features reveal insights into prediction model development. BMC Struct. Biol. 7:54. doi: 10.1186/1472-6807-7-54
Parthiban, V., Gromiha, M. M., and Schomburg, D. (2006). CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 34, W239–242. doi: 10.1093/nar/gkl190
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830. Available online at: http://www.jmlr.org/papers/volume12/pedregosa11a/pedregosa11a.pdf
Pfleger, C., Radestock, S., Schmidt, E., and Gohlke, H. (2013a). Global and local indices for characterizing biomolecular flexibility and rigidity. J. Comput. Chem. 34, 220–233. doi: 10.1002/jcc.23122
Pfleger, C., Rathi, P. C., Klein, D. L., Radestock, S., and Gohlke, H. (2013b). Constraint Network Analysis (CNA): a python software package for efficiently linking biomacromolecular structure, flexibility, (thermo-)stability, and function. J. Chem. Inf. Model. 53, 1007–1015. doi: 10.1021/ci400044m
Piraino, S. W., and Furney, S. J. (2016). Beyond the exome: the role of non-coding somatic mutations in cancer. Ann. Oncol. 27, 240–248. doi: 10.1093/annonc/mdv561
Poplin, R., Chang, P. C., Alexander, D., Schwartz, S., Colthurst, T., Ku, A., et al. (2018). A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987. doi: 10.1038/nbt.4235
Poulos, R. C., and Wong, J. W. H. (2018). Finding cancer driver mutations in the era of big data research. Biophys. Rev. 11, 21–29. doi: 10.1007/s12551-018-0415-6
Rader, A. J., Hespenheide, B. M., Kuhn, L. A., and Thorpe, M. F. (2002). Protein unfolding: rigidity lost. Proc. Natl. Acad. Sci. U.S.A. 99, 3540–3545. doi: 10.1073/pnas.062492699
Raphael, B. J., Dobson, J. R., Oesper, L., and Vandin, F. (2014). Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med. 6:5. doi: 10.1186/gm524
Red Brewer, M., Yun, C. H., Lai, D., Lemmon, M. A., Eck, M. J., and Pao, W. (2013). Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. U.S.A. 110, E3595–3604. doi: 10.1073/pnas.1220050110
Reimand, J., and Bader, G. D. (2013). Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 9:637. doi: 10.1038/msb.2012.68
Reva, B., Antipin, Y., and Sander, C. (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39:e118. doi: 10.1093/nar/gkr407
Roskoski, R. Jr. (2014). The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 79, 34–74. doi: 10.1016/j.phrs.2013.11.002
Samuels, Y., Wang, Z., Bardelli, A., Silliman, N., Ptak, J., Szabo, S., et al. (2004). High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554. doi: 10.1126/science.1096502
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., and Serrano, L. (2005). The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–388. doi: 10.1093/nar/gki387
Sethi, A., Eargle, J., Black, A. A., and Luthey-Schulten, Z. (2009). Dynamical networks in tRNA: protein complexes. Proc. Natl. Acad. Sci. U.S.A. 106, 6620–6625. doi: 10.1073/pnas.0810961106
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi: 10.1002/humu.22225
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–457. doi: 10.1093/nar/gks539
Sjoblom, T., Jones, S., Wood, L. D., Parsons, D. W., Lin, J., Barber, T. D., et al. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274. doi: 10.1126/science.1133427
Spinella, J. F., Mehanna, P., Vidal, R., Saillour, V., Cassart, P., Richer, C., et al. (2016). SNooPer: a machine learning-based method for somatic variant identification from low-pass next-generation sequencing. BMC Genomics 17:912. doi: 10.1186/s12864-016-3281-2
Stephens, P., Edkins, S., Davies, H., Greenman, C., Cox, C., Hunter, C., et al. (2005). A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat. Genet. 37, 590–592. doi: 10.1038/ng1571
Stephens, P., Hunter, C., Bignell, G., Edkins, S., Davies, H., Teague, J., et al. (2004). Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526. doi: 10.1038/431525b
Stetz, G., Tse, A., and Verkhivker, G. M. (2017). Ensemble-based modeling and rigidity decomposition of allosteric interaction networks and communication pathways in cyclin-dependent kinases: differentiating kinase clients of the Hsp90-Cdc37 chaperone. PLoS ONE 12:e0186089. doi: 10.1371/journal.pone.0186089
Stetz, G., and Verkhivker, G. M. (2017). Computational analysis of residue interaction networks and coevolutionary relationships in the Hsp70 chaperones: a community-hopping model of allosteric regulation and communication. PLoS Comput. Biol. 13:e1005299. doi: 10.1371/journal.pcbi.1005299
Tamborero, D., Gonzalez-Perez, A., and Lopez-Bigas, N. (2013). OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 29, 2238–2244. doi: 10.1093/bioinformatics/btt395
Tokheim, C., Bhattacharya, R., Niknafs, N., Gygax, D. M., Kim, R., Ryan, M., et al. (2016). Exome-scale discovery of hotspot mutation regions in human cancer using 3D protein structure. Cancer Res. 76, 3719–3731. doi: 10.1158/0008-5472.CAN-15-3190
Tokheim, C. J., Papadopoulos, N., Kinzler, K. W., Vogelstein, B., and Karchin, R. (2016). Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. U.S.A. 113, 14330–14335. doi: 10.1073/pnas.1616440113
Tokuriki, N., Stricher, F., Schymkowitz, J., Serrano, L., and Tawfik, D. S. (2007). The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 369, 1318–1332. doi: 10.1016/j.jmb.2007.03.069
Tvorogov, D., Sundvall, M., Kurppa, K., Hollmen, M., Repo, S., Johnson, M. S., et al. (2009). Somatic mutations of ErbB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. J. Biol. Chem. 284, 5582–5591. doi: 10.1074/jbc.M805438200
Tyner, C., Barber, G. P., Casper, J., Clawson, H., Diekhans, M., Eisenhart, C., et al. (2017). The UCSC genome browser database: 2017 update. Nucleic Acids Res. 45, D626–D634. doi: 10.1093/nar/gkw1134
Van Durme, J., Delgado, J., Stricher, F., Serrano, L., Schymkowitz, J., and Rousseau, F. (2011). A graphical interface for the FoldX forcefield. Bioinformatics 27, 1711–1712. doi: 10.1093/bioinformatics/btr254
Vijayabaskar, M. S., and Vishveshwara, S. (2010). Interaction energy based protein structure networks. Biophys. J. 99, 3704–3715. doi: 10.1016/j.bpj.2010.08.079
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A. Jr., and Kinzler, K. W. (2013). Cancer genome landscapes. Science 339, 1546–1558. doi: 10.1126/science.1235122
Wang, Z., Longo, P. A., Tarrant, M. K., Kim, K., Head, S., Leahy, D. J., et al. (2011). Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 18, 1388–1393. doi: 10.1038/nsmb.2168
Wang, Z., Shen, D., Parsons, D. W., Bardelli, A., Sager, J., Szabo, S., et al. (2004). Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 304, 1164–1166. doi: 10.1126/science.1096096
Watson, I. R., Takahashi, K., Futreal, P. A., and Chin, L. (2013). Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 14, 703–718. doi: 10.1038/nrg3539
Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R., Ozenberger, B. A., Ellrott, K., et al. (2013). The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi: 10.1038/ng.2764
Wood, D. E., White, J. R., Georgiadis, A., Van Emburgh, B., Parpart-Li, S., Mitchell, J., et al. (2018). A machine learning approach for somatic mutation discovery. Sci. Transl. Med. 10:eaar7939. doi: 10.1126/scitranslmed.aar7939
Wood, L. D., Parsons, D. W., Jones, S., Lin, J., Sjoblom, T., Leary, R. J., et al. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113. doi: 10.1126/science.1145720
Wu, J., Wu, M., Li, L., Liu, Z., Zeng, W., and Jiang, R. (2016). dbWGFP: a database and web server of human whole-genome single nucleotide variants and their functional predictions. Database 2016:baw024. doi: 10.1093/database/baw024
Zhang, J., Baran, J., Cros, A., Guberman, J. M., Haider, S., Hsu, J., et al. (2011). International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data. Database 2011:bar026. doi: 10.1093/database/bar026
Keywords: cancer driver mutations, machine learning classifiers, ensemble-based machine learning features, random forest, deep learning, convolutional neural networks, drug discovery
Citation: Agajanian S, Oluyemi O and Verkhivker GM (2019) Integration of Random Forest Classifiers and Deep Convolutional Neural Networks for Classification and Biomolecular Modeling of Cancer Driver Mutations. Front. Mol. Biosci. 6:44. doi: 10.3389/fmolb.2019.00044
Received: 28 February 2019; Accepted: 23 May 2019;
Published: 11 June 2019.
Edited by:
Shozeb Haider, University College London, United KingdomReviewed by:
Arvind Ramanathan, Argonne National Laboratory (DOE), United StatesDebsindhu Bhowmik, Oak Ridge National Laboratory (DOE), United States
Copyright © 2019 Agajanian, Oluyemi and Verkhivker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gennady M. Verkhivker, dmVya2hpdmsmI3gwMDA0MDtjaGFwbWFuLmVkdQ==