 Talya Shacham
Talya Shacham Neeraj Sharma
Neeraj Sharma Gerardo Z. Lederkremer
Gerardo Z. Lederkremer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci. , 03 April 2019
Sec. Protein Folding, Misfolding and Degradation
Volume 6 - 2019 | https://doi.org/10.3389/fmolb.2019.00020
This article is part of the Research Topic Protein Misfolding in Ageing and Neurodegeneration: From Basic Biology to Drug Development View all 5 articles
Increasing evidence in recent years indicates that protein misfolding and aggregation, leading to ER stress, are central factors of pathogenicity in neurodegenerative diseases. This is particularly true in Huntington's disease (HD), where in contrast with other disorders, the cause is monogenic. Mutant huntingtin interferes with many cellular processes, but the fact that modulation of ER stress and of the unfolded response pathways reduces the toxicity, places these mechanisms at the core and gives hope for potential therapeutic approaches. There is currently no effective treatment for HD and it has a fatal outcome a few years after the start of symptoms of cognitive and motor impairment. Here we will discuss recent findings that shed light on the mechanisms of protein misfolding and aggregation that give origin to ER stress in neurodegenerative diseases, focusing on Huntington's disease, on the cellular response and on how to use this knowledge for possible therapeutic strategies.
The acquisition of a correct three-dimensional native structure is a crucial step during the biosynthesis of a functional cellular protein (Vendruscolo et al., 2003). Protein folding is an intrinsic property of a polypeptide chain, which is strongly influenced by the cellular environment and is under regulation of other partner proteins in oligomeric assemblies and of folding catalysts and molecular chaperones (Hartl and Hayer-Hartl, 2002; Ellis and Minton, 2006). During co-translational folding, exposed regions of partially folded proteins are at risk of interaction with other cellular molecules (Hartl and Hayer-Hartl, 2002; Balchin et al., 2016; Chiti and Dobson, 2017). Therefore, short non-native intermediate forms develop to protect the regions which are susceptible to aggregation (Mogk et al., 2018). Proteins undergo a stochastic search for more stable conformations with lowest free energy by a trial and error process (Dinner et al., 2000; Vendruscolo et al., 2003; Chiti and Dobson, 2017). In general, the marginally stable native state of a folded protein and the crowded cellular environment are responsible for the error proneness of protein folding, despite being always under inspection of the protein quality control system, the ubiquitin-proteasome complex and molecular chaperones (Vendruscolo et al., 2003; Labbadia and Morimoto, 2015; Miller et al., 2015; Balchin et al., 2016). Failure of any of these protective checkpoints can initiate a protein misfolding process, which can generate protein oligomers or larger aggregates. The failure can be caused, among others, by high temperature, low pH, oxidative stress, abnormal presence of metal ions, mutations, transcriptional, translational or posttranslational errors, and aging (Sekijima et al., 2005; Chu et al., 2007; Chiti and Dobson, 2009; Koga et al., 2011; Brehme et al., 2014; Mckinnon and Tabrizi, 2014; Labbadia and Morimoto, 2015; Martinez-Lopez et al., 2015; Balchin et al., 2016).
A protein can form amorphous aggregates or amyloid fibrils with well-ordered straight fibrillar structure, composed of protofilaments (Chiti and Dobson, 2017; Hartl, 2017). Conditions that support high net protein charge favor the formation of fibrillar aggregates.
Protein aggregation can follow several pathways that are not mutually exclusive. Native protein monomers may have a natural tendency to reversibly associate and to form small oligomers, which can occasionally associate into large aggregates. Alternatively, the native monomer may have a very low tendency to reversibly associate but it can undergo a conformational change resulting in an aggregation-prone misfolded form. This misfolded conformation may also arise from mutations. In a surface-induced aggregation mechanism, aggregation begins with the binding of a native monomer to the surface of the container in vitro or to intracellular membranes in cells. These interactions cause conformational changes in the monomer which increase its tendency to aggregate (Philo and Arakawa, 2009). For any of these mechanisms, it is widely accepted that progression into large aggregates involves a nucleation and seeding mechanism. Independently of the lower or higher tendencies to form small oligomers, once an aggregate of sufficient size (critical nucleus) forms, then it grows exponentially. Two kinetic phases are observed: a lag phase generating a seed and an elongation phase where visible particulates appear (Soto and Pritzkow, 2018). Seeding activity was recently found at early stages, before appearance of visible aggregates, in pre-symptomatic HD model mice (Ast et al., 2018).
Genetic, biochemical, mouse model studies, and neuropathological evidence have shown that in neurodegenerative diseases misfolded proteins form aggregates, in most cases amyloid fibrils, which may be deposited intracellularly or extracellularly. In diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), Amyotrophic Lateral Sclerosis (ALS), and HD, protein aggregates vary, but all forms have a similar intermolecular beta-sheet–rich structure, both in small oligomers and in large fibrillar aggregates. The implicated polypeptides are different but they can form similar structures of amyloid fibrils (Eisenberg and Jucker, 2012; Chiti and Dobson, 2017; Hartl, 2017). The aggregation process triggers cellular dysfunction, loss of synaptic connections and is responsible for brain damage or disease (Soto, 2003; Ross and Poirier, 2004; Goedert, 2015; Soto and Pritzkow, 2018). Initial studies hypothesized that the large insoluble aggregates lead to cell death, and therefore prevention of aggregate formation might be a strategy to help reduce the toxicity (Bucciantini et al., 2002). However, there is increasing evidence that in general, smaller soluble toxic misfolded oligomers are the main causative agent for neurodegeneration (Jarrett et al., 1993; Morris et al., 2009; Leitman et al., 2013; Chiti and Dobson, 2017; Hartl, 2017; Moily et al., 2017). The 3D structure of the oligomeric species appears to determine their toxic character (Hoffner and Djian, 2015; Smith, 2018). The insoluble aggregates could then be a result of a strategy of the cells to reduce the toxicity by shielding the toxic species in these large structures called dry steric zippers (Eisenberg and Jucker, 2012) (Figure 1).
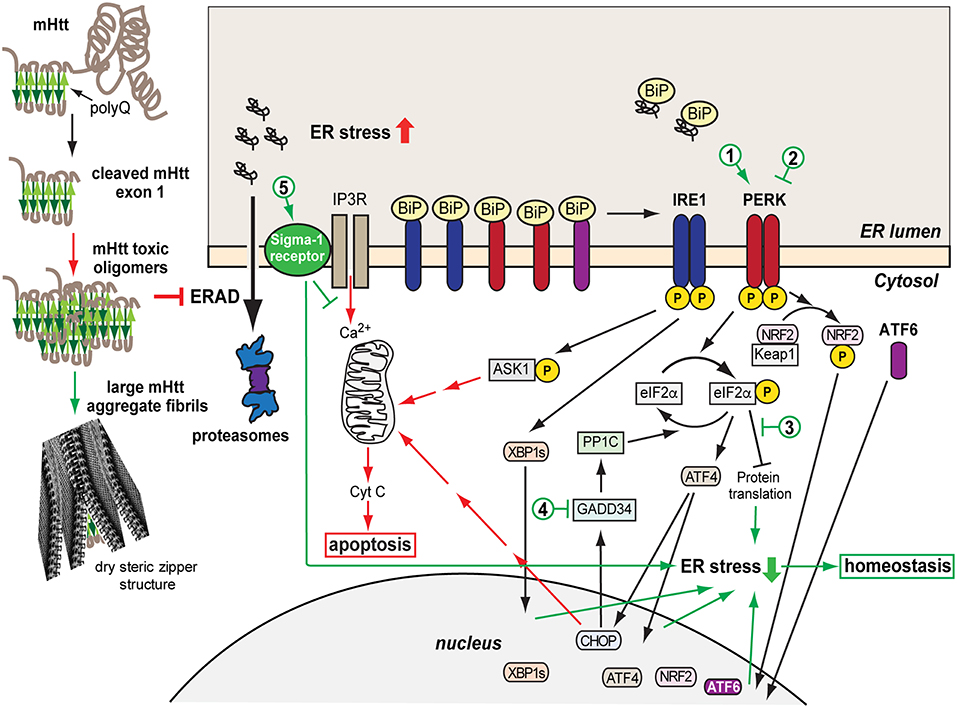
Figure 1. Model of mHtt aggregation, generation of ER stress and the consequent UPR protective and later pro-apoptotic responses. A misfolded mHtt monomer, with the aggregation prone polyQ domain indicated (beta sheets in green) is cleaved and associates with other monomers to form toxic oligomers, which among other effects cause sequestration and depletion of ERAD factors such as p97, inhibiting ERAD. mHtt oligomers can associate into larger aggregate fibrils with dry steric zipper structure that shield the toxicity of mHtt. However, the transient presence of toxic mHtt oligomers, inhibiting ERAD, causes accumulation of unfolded secretory proteins and ER stress, activating the UPR sensors IRE1, PERK, and ATF6, starting an initial protective or adaptive phase of the UPR. This includes upregulation and translocation to the nucleus of transcription factors, XBP1s, ATF4, NRF2, ATF6, which induce expression of chaperones, ERAD machinery, anti-oxidative response components. Concurrently, eIF2α phosphorylation by PERK causes transient arrest in translation, reducing the ER load. If the ER stress remains unresolved, the pro-apoptotic stage of the UPR is initiated, causing upregulation of ASK1-P and CHOP and Ca2+ exit from the ER, inducing the intrinsic apoptotic pathway with the mitochondrial release of cytochrome C. Sigma-1 receptor is upregulated and modulates Ca2+ release, with a protective effect. Cytotoxic pathways are indicated in red and cell protective ones in green. The numbers indicate possible points of therapeutic intervention, with activation (1) or inhibition (2) of PERK, inhibition of the downstream effects of eIF2α-P (3), inhibition of eIF2α-P dephosphorylation (4), and Sigma-1 receptor activation (5).
HD is a result of mutation in the gene encoding for the huntingtin protein (Htt), the expansion of CAG repeats that encode for a polyglutamine (polyQ) stretch, which is pathogenic when it contains more than about 35 glutamines (Zoghbi and Orr, 2000; Sakahira et al., 2002). A similar phenomenon occurs in other polyQ diseases, such as spinocerebellar ataxias (Shao and Diamond, 2007). In HD, the mutation results in mutant Htt (mHtt) misfolding and aggregation. The polyQ stretch is on exon 1, which is cleaved off, with the resulting N-terminal fragment enough to cause aggregation. A recent report shows also the presence of aberrantly spliced mHtt forms containing only exon 1 in samples from HD patients (Neueder et al., 2017). The 17-residue-long N-terminus preceding the polyQ stretch interacts with intracellular membranes and has a strong influence on mHtt aggregation (Pandey et al., 2017). Ubiquitination at the N-terminus appears to be an important determinant for mHtt degradation (Wang et al., 2017). In a recent study, a mutation in the N17 N-terminal region prevented the formation of large aggregates but not oligomers in a Drosophila HD model and led to an increase in toxicity. This suggested neurotoxicity of the oligomers and perhaps protection by the large aggregates (Branco-Santos et al., 2017). This follows a growing number of reports linking mHtt oligomers and not the final large aggregates to cytotoxicity (Schaffar et al., 2004; Takahashi et al., 2008; Lajoie and Snapp, 2010; Leitman et al., 2013). In fact, no connection was found between large mHtt aggregates and neuronal death in HD patients (Kuemmerle et al., 1999). In cellular HD models, the transition from oligomers to large aggregates correlates with a reduction in ER stress (Leitman et al., 2013) and in apoptosis (Ramdzan et al., 2017). However, the formation of mHtt inclusions still affects to some degree cellular function (Ramdzan et al., 2017). As mentioned above, the toxicity of soluble oligomers is not unique to HD. No correlation was found between large insoluble aggregates and memory loss in an AD mouse model (Lesné et al., 2008) and AD patients do not necessarily present brain aggregates (Petersen et al., 2013). Targeting oligomeric forms of Aβ instead of large aggregates provides better results in the reduction of AD toxicity (Sengupta et al., 2016).
Misfolded proteins trigger cellular responses, such as the heat shock response (Kakkar et al., 2014; Kampinga and Bergink, 2016) and the ER or mitochondrial unfolded protein responses (UPR). These responses attempt to increase the capacity to unfold and refold the misfolded proteins through upregulation of molecular chaperones and to improve the ability to degrade the misfolded proteins by upregulation of the proteasome machinery. These responses are cell autonomous but a non-cell autonomous regulation has been described, especially in studies in Caenorhabditis elegans (Prahlad and Morimoto, 2011; Volovik et al., 2014).
Failure of the UPR or of the heat-shock response are found to be associated with several pathologic conditions such as AD, PD, HD, prion diseases, Type II diabetes, and ALS (Douglas and Dillin, 2010; Lee et al., 2011; Hipp et al., 2014; Labbadia and Morimoto, 2015; Chiti and Dobson, 2017; Hartl, 2017; Shamsi et al., 2017). Progressive decline in the efficiency of these pathways with age has been linked to the late age of onset of these diseases (Taylor and Dillin, 2011). The UPR and its failure in disease are discussed in the following chapters.
ER stress develops in many neurodegenerative diseases such as AD, PD, ALS, and HD (Ogen-Shtern et al., 2016; Xiang et al., 2016; Remondelli and Renna, 2017) and there is increasing evidence that it is a main factor in the degeneration of the cells (Hoozemans et al., 2005; Vidal et al., 2011; Stutzbach et al., 2013; Heman-Ackah et al., 2017). Many of these diseases are a result of mutation in specific genes (e.g., Htt, SOD1) leading to the accumulation of a misfolded protein. As mentioned above, misfolded proteins have a propensity to interact and aggregate in the cell, giving rise to toxic species that generate ER stress and compromise cell function. One of the ways by which ER stress is generated in this process, in many cases by cytosolic proteins such as mHtt, is by interference of toxic oligomers with ER-associated degradation (ERAD) components (Figure 1). This has been observed in HD (Duennwald and Lindquist, 2008; Leitman et al., 2013) and AD models (Abisambra et al., 2013; Soejima et al., 2013; Fonseca et al., 2014). ER stress triggers activation of the UPR, with the aim of removing or refolding the damaged proteins (Hetz and Papa, 2017). The UPR is activated through three main transmembrane proteins acting as UPR sensors: IRE1, PERK, and ATF6 (Figure 1). Inositol-requiring protein 1α (IRE1) is a protein kinase associated in its inactive form with the chaperone Binding immunoglobulin Protein (BiP). Upon accumulation of unfolded proteins, IRE1 dissociates from BiP/GRP78 and becomes active by autophosphorylation. When active, IRE1 splices the mRNA encoding for the transcription factor XBP-1, which is then translated to the active form XBP-1s. XBP-1s translocates into the nucleus, inducing transcription of BiP and other chaperones and target genes related to ERAD regulation (Chen and Brandizzi, 2013). A second sensor associated with BiP is PKR-like ER-localized eIF2α kinase (PERK). Activation of PERK by autophosphorylation, after dissociation from BiP, results in phosphorylation of eukaryotic initiation factor 2α (eIF2α), leading to inhibition of protein translation. Paradoxically, translation of the transcription factor ATF4 increases upon phosphorylation of eIF2α (Ron, 2002; McQuiston and Diehl, 2017), leading to expression of many genes, among them Growth arrest and DNA damage-inducible gene 34 (GADD34, also called PPP1R15A), a regulatory subunit of protein phosphatase PP1, which dephosphorylates eIF2α, resulting in recovery from the PERK-mediated translational block (Novoa et al., 2001). Besides eIF2α, PERK phosphorylates nuclear erythroid 2 p45-related factor 2 (NRF2), causing dissociation of the cytosolic NRF2-Keap1 complex and leading to release of NRF2 to the nucleus where it promotes transcription of RNAs encoding components of the antioxidant response (Cullinan et al., 2003). The third UPR sensor is activating transcription factor 6 (ATF6) which is regulated by BiP as well. Upon BiP dissociation, ATF6 translocates from the ER to the Golgi, where it is cleaved by proteases that detach ATF6 from the Golgi membrane. The released soluble cytosolic domain of ATF6 is a transcription factor that enters the nucleus and activates expression of UPR target genes such as BiP and XBP1 (Yoshida et al., 2001; Shen et al., 2002; Hetz, 2012). ER stress activates the three UPR branches, leading to inhibition of protein translation, increase in chaperone production and enhanced degradation. However, a chronic activation of the UPR leads to a fatal outcome. In case of failure to restore protein homeostasis, the UPR initiates an apoptotic pathway leading to cell death. IRE1 mediates activation of tumor necrosis factor receptor associated factor 2 (TRAF2), activating apoptotic factors such as ASK1. A long-term phosphorylated status of eIF2α induces CHOP/GADD153, a transcription factor that activates expression of pro-apoptotic genes (Oyadomari and Mori, 2004; Sano and Reed, 2013). PERK activation and eIF2α phosphorylation without resolution of ER stress have been particularly implicated in neurodegeneration (Ohno, 2017; Taalab et al., 2018).
HD is a genetic disorder characterized by movement disorder, cognitive decline, and behavioral difficulties (Walker, 2007; Wright et al., 2017; Pandey and Rajamma, 2018). As found in post-mortem and MRI studies, HD patients suffer from neuronal cell death, initially and mostly in the striatum but also in the cortex and other areas of the brain (Reiner et al., 1988; Rosas et al., 2003). Although the involvement of protein aggregation in neurodegenerative disease is well established (Murphy, 2002; Taylor, 2002; Ross and Poirier, 2004), toxicity could arise through a number of pathways. In HD, protein aggregation in the cytoplasm interferes for example with nucleocytoplasmic transport, causing aberrant redistribution of nuclear shuttle factors to the cytosol (Woerner et al., 2016). Over-expression of molecular chaperones, such as Hsp70 and Hsp40, was shown to reduce the toxicity of mHtt aggregates in yeast and fly HD models, without preventing their formation, possibly by sheltering the exposed hydrophobic regions or by a conformational effect on the misfolded protein (Kazemi-Esfarjani and Benzer, 2000; Meriin et al., 2002). Another important consequence of mHtt aggregation is the activation of the UPR (Duennwald and Lindquist, 2008; Reijonen et al., 2008; Carnemolla et al., 2009; Leitman et al., 2013, 2014; Jiang et al., 2016). Interference with the ubiquitin-proteasome system (UPS) is important in HD models and apparently also in HD patients (Bennett et al., 2007; Ortega et al., 2007; Finkbeiner and Mitra, 2008; Hipp et al., 2012). However, some studies using cytosolic GFP-based reporters of proteasomal activity did not find a global UPS impairment (Bowman et al., 2005; Bett et al., 2009). In fact, an important interference was attributed to sequestration and depletion of p97/VCP and its cofactors Npl4 and Ufd1 by mHtt, which would not cause global UPS deficiency, but rather a crippling dysfunction of ERAD (Duennwald and Lindquist, 2008; Yang et al., 2010; Leitman et al., 2013). Homocysteine-induced endoplasmic reticulum protein (Herp), an important factor in ERAD, was recently reported to be directly involved in targeting of mHtt for degradation (Luo et al., 2018). The inhibition of ERAD leads to accumulation of unfolded proteins in the ER, ER stress, and UPR induction, which were observed in HD models in yeast and mammalian cells (Duennwald and Lindquist, 2008; Reijonen et al., 2008; Carnemolla et al., 2009; Leitman et al., 2013, 2014), in animal HD models (Carnemolla et al., 2009; Cho et al., 2009; Noh et al., 2009; Vidal et al., 2012), and in post-mortem samples from HD patients (Carnemolla et al., 2009). Other cellular factors, such as ubiquitin-specific protease-14 and ATF5, are important for reduction of ER stress, and were recently found to be sequestered and depleted during mHtt aggregation (Hyrskyluoto et al., 2014; Hernández et al., 2017). One of the consequences of ER stress is to affect mitochondrial function and exacerbate oxidative stress, a key element in mHtt cytotoxicity (reviewed in Zheng et al., 2018).
One important ER stress-activated protein, with a cell-protective function is the Sigma-1 receptor (S1R). The S1R is an evolutionarily conserved ligand–operated molecular chaperone, especially involved in brain function, including neuromodulation and neuroplasticity (Hayashi, 2015). The S1R is expressed in a wide range of tissues, with higher levels in the central nervous system (CNS), where it is linked to diverse pathologies (Nguyen et al., 2017). The S1R is a transmembrane protein (25 KD) consisting of 223 amino acids and largely localized to a specialized ER subdomain that contacts mitochondria and is called the mitochondrion–associated ER membrane (MAM) (Hanner et al., 1996; Hayashi and Su, 2007; Hayashi et al., 2009; Fujimoto and Hayashi, 2011; Kourrich et al., 2012). The overall topology of this receptor is still unclear and there are studies that suggest a single or two transmembrane domains (Hayashi and Su, 2007; Ortega-Roldan et al., 2015; Schmidt et al., 2016; Mavylutov et al., 2017). A recent crystal structure of the S1R suggests a single transmembrane domain and oligomerization into trimeric and up to hexameric assemblies (Schmidt et al., 2016). The S1R is activated by both ER stress and agonists, which might lead to its translocation to different subcellular compartments, such as the MAM, nuclear envelope, and plasma membrane (Hayashi and Su, 2003; Su et al., 2010; Kourrich et al., 2012). The association of BiP with the S1R keeps it in an inactive conformation (Hayashi and Su, 2007; Tsai et al., 2014). Upon ER stress, accumulation of misfolded proteins in the ER causes S1R dissociation from BiP and its activation. Calcium levels and S1R agonists also modulate its activity (Hayashi and Su, 2007; Wang et al., 2012), and S1R expression is upregulated in response to PERK pathway activation (Mitsuda et al., 2011). The “chaperone” activity of S1R is through modulation of inositol 1,4,5-triphosphate receptor (IP3R) activity. IP3R is a ligand-gated calcium channel that upon activation releases calcium from the ER, especially at the MAMs (Li et al., 2009; Gilady et al., 2010). Therefore, S1R modulation of IP3R activity regulates calcium transfer to mitochondria (Hayashi and Su, 2007) promoting ATP generation and attenuating the mitochondrial apoptotic pathway (Hayashi and Su, 2007; Bernard-Marissal et al., 2015; Hayashi, 2015). Studies have also demonstrated a suppressor effect of S1R activity on ROS and oxidative damage (Meunier and Hayashi, 2010; Pal et al., 2012; Wang et al., 2012).
A decreased level of S1R or its activity is found associated with neurodegenerative diseases (Nguyen et al., 2015). A S1R mutation was linked to a familial form of ALS (Al-Saif et al., 2011). The mutation impairs MAMs and affects calcium signaling (Bernard-Marissal et al., 2015). Several in vivo and in vitro studies have shown evidence of S1R as a target for treating neuropsychiatric as well as neurodegenerative disorders (Katnik et al., 2006; Meunier et al., 2006; Vagnerova et al., 2006; Mancuso et al., 2012; Behensky et al., 2013; Hyrskyluoto et al., 2013; Francardo et al., 2014; Nguyen et al., 2014). S1R was reported to have a protective effect in HeLa cells expressing mHtt, increasing proteasome activity and mHtt degradation (Miki et al., 2015). In the next section we will discuss recent studies on HD models, which reported a beneficial effect of S1R modulation.
There is currently no cure or effective therapy for HD. There are two main recent experimental approaches to try to develop a therapy (reviewed in Huang et al., 2016; Wright et al., 2017; Caron et al., 2018; Saavedra et al., 2018). One involves gene therapy, which includes knockdown of mHtt (Aguiar et al., 2017; Datson et al., 2017; Southwell et al., 2018) and expression of miRNAs (Miniarikova et al., 2017; Evers et al., 2018) (Table 1). Htt is necessary in embryonic development but it might be possible to eliminate it in the adult brain, (reviewed in Liu and Zeitlin, 2017). Allele-specific CRISPR/Cas9-mediated gene editing has been recently reported in adult HD140Q-knockin mice (Yang et al., 2017). CRISPR/Cas9 can also be used to specifically remove the CAG repeats (Dabrowska et al., 2018). A main set-back of gene therapy approaches is attaining efficient delivery, but there is huge progress in this direction in recent years. Therapy involving fetal cell transplantation has also been attempted (Bachoud-Lévi et al., 2006; Mazzocchi-Jones et al., 2009; Schackel et al., 2013; Precious et al., 2017). The other approach focuses on blocking the cellular toxicity of mHtt. This includes many strategies, for example inhibiting mHtt cleavage by caspase 6 (Aharony et al., 2015), targeting or modulating heat shock proteins (Kampinga and Bergink, 2016; Scior et al., 2018), neurotrophic factors such as CNTF (Emerich et al., 1997), HDACs (Suelves et al., 2017), proteasome activity (Jeon et al., 2016), and mitochondrial oxidative phosphorylation (Ruetenik et al., 2016). Compounds that promote daf-16/FOXO function and in turn stimulate insulin/IGF1 signaling and extend longevity (Hesp et al., 2015), such as resveratrol, metformin and some steroids, have shown beneficial effects in C. elegans (Farina et al., 2017) and mouse HD models (Arnoux et al., 2018). Metformin also appeared protective in a statistical analysis of HD patients participating in the Enroll-HD database (Hervas et al., 2017).
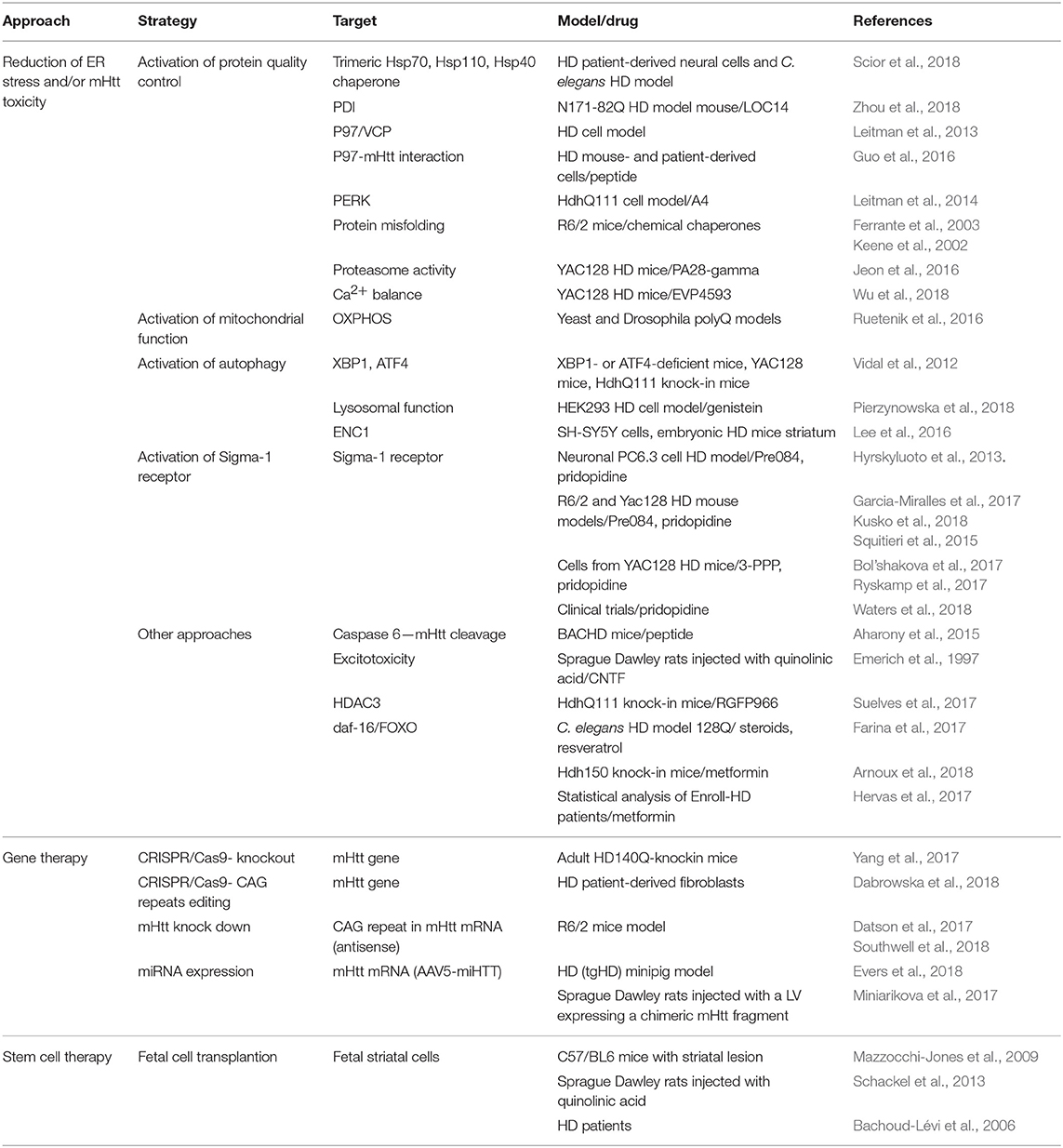
Table 1. Therapeutic approaches for HD.
Reduction of ER stress is protective in HD, and can be accomplished for example with the use of chemical chaperones (Keene et al., 2002; Ferrante et al., 2003). As mentioned above, p97/VCP depletion is an important factor in HD pathogenicity, through inhibition of ERAD and development of ER stress (Leitman et al., 2013). It was recently reported that VCP interacts with mHtt on mitochondria, enhancing mitophagy and cell death; a peptide was developed to inhibit the interaction (Guo et al., 2016). There is a cross-talk between ER stress and autophagy and for example, XBP1 deficiency leads to an enhancement of autophagy and overall improvement in HD model mice (Vidal et al., 2012). Induction of autophagy by genistein was protective in a cellular HD model (Pierzynowska et al., 2018). An ER stress upregulated protein, ectodermal-neural cortex 1 (ENC1), was recently shown to inhibit autophagy in an HD cellular model, and its knockdown increased autophagy and cell survival, suggesting it as a possible target for therapy (Lee et al., 2016). mHtt-induced ER stress also increases expression of protein disulfide isomerase (PDI) (Duennwald and Lindquist, 2008), a small molecule PDI modulator caused improved motor function and survival in the N171-82Q HD model mouse (Zhou et al., 2018). ER Ca2+ depletion causes protein misfolding in the ER and ER stress. Recent studies targeted Ca2+ balance, showing beneficial effects, with a small molecule activator of SERCA in a rat model of PD (Dahl, 2016) and with inhibition of TRPC1-Dependent Store-Operated Calcium Entry in an HD mouse model (Wu et al., 2018).
One strategy with encouraging results so far for several neurodegenerative diseases is to target the UPR pathways (Shenkman et al., 2015; Xiang et al., 2016; Pérez-Arancibia et al., 2017). PERK, one of the three UPR sensors, is an interesting and promising target (recently reviewed in Halliday et al., 2017a; McQuiston and Diehl, 2017; Ohno, 2017; Hughes and Mallucci, 2018; Taalab et al., 2018). A very low activity of PERK-mediated eIF2α phosphorylation in striatal neurons in culture and in the mouse brain striatum was found connected to the higher mHtt toxicity in this region (Leitman et al., 2014). As explained above, initial activation of PERK is beneficial, through phosphorylation of eIF2α, resulting in transient inhibition of protein synthesis and by activation of NRF2 and its anti-oxidant effects. However, chronic ER stress leads to upregulation of CHOP through the PERK pathway, which triggers a series of events ending in apoptosis. Therefore, inhibition of PERK can be beneficial, by reducing the activation of the apoptotic pathway, but this will preclude the initial protective role of PERK (Figure 1). Conversely, activating or prolonging eIF2α phosphorylation can improve the initial protective stage but might lead to apoptosis if it is not controlled. Both strategies have been attempted, with positive results, but also with some negative reports. The PERK inhibitor GSK2606414 was protective in prion (Moreno et al., 2013), frontotemporal dementia (Radford et al., 2015) AD (Yang et al., 2016) and PD (Mercado et al., 2018) mouse models but secondary side effects of pancreatic toxicity appeared as well, as high levels of insulin production in the pancreas require regulation by a functional PERK pathway. Compounds that inhibit the pathway downstream of eIF2α-P, and restore protein synthesis, were developed and showed protective effects in neurodegeneration caused by prion infection and in frontotemporal dementia, without the secondary toxicity (ISRIB, Trazodone) (Sidrauski et al., 2013; Halliday et al., 2015, 2017b). In several tauopathies, PERK variants with reduced activity are a genetic risk factor. Expression of these variant alleles in iPSC-derived neurons, or inhibition of PERK, showed high vulnerability to ER stress in these cells (Yuan et al., 2018), suggesting that an approach involving PERK pathway activation would be beneficial. In a strategy to increase eIF2α-P, GADD34 inhibitors (salubrinal, guanabenz, sephin 1) were tested, showing beneficial effects in mouse models of prion disease (Das et al., 2015), PD (Sun et al., 2018), and ALS (Tsaytler et al., 2011), but accelerated disease progression in another ALS study (Vieira et al., 2015). Another approach is to directly activate PERK. A PERK activator, CCT020312, was developed (Stockwell et al., 2012) and recently tested in mouse and cellular tauopathy models, showing beneficial effects and no toxicity (Bruch et al., 2017). PERK modulation has only been reported in HD cellular models (Leitman et al., 2014), but our own unpublished studies in mouse HD models, using a PERK activator that we have developed, showed significant protection. A combinatorial drug approach, targeting different UPR pathways, or concomitantly modulating other mechanisms has not been tried and may be advantageous.
As mentioned above, S1R, activated by ER stress, could be a promising target for ameliorating symptoms in HD (Nguyen et al., 2015; Bol'shakova et al., 2017). The treatment of an mHtt expressing neuronal cell line (PC6.3) with the S1R agonist PRE-084 had a neuroprotective effect by restoring the S1R deficit, upregulating antioxidant activity, and decreasing ROS levels via NF-kB signaling (Hyrskyluoto et al., 2013). This agonist and also 3-PPP showed a neuroprotective effect by increasing the density in neuronal cultures from HD model mice (Bol'shakova et al., 2017). Another S1R agonist, pridopidine (also known as a dopamine stabilizer) showed a neuroprotective effect in the R6/2 and Yac128 HD mouse models, improving motor performance and survival (Squitieri et al., 2015; Garcia-Miralles et al., 2017; Kusko et al., 2018), mainly through normalization of calcium homeostasis (Ryskamp et al., 2017). Our own unpublished results in HD cellular models suggest that the effect of pridopidine through S1R activation is by modulation of ER stress, affecting especially the PERK pathway. A series of clinical trials have found some encouraging results for pridopidine therapy in HD (Squitieri and De Yebenes, 2015; Waters et al., 2018).
Recent evidence increases our understanding of the consequences of protein misfolding and aggregation in HD on the generation of ER stress and cytotoxicity. Given the similarities in the origin of ER stress in multifactorial neurodegenerative diseases such as AD, the findings on HD have important wider implications. It is becoming more clear that the toxicity originates mainly from soluble oligomeric assemblies rather than the large protein inclusions that develop with time. Therefore, there is less interest in the development of inhibitors of aggregation, a strategy that has failed so far. Late onset has been linked to failure with age of protein homeostasis and the consequent development of uncontrolled ER stress. HD therapy is not yet imminent, but recent work points the way, with modulation of the UPR, especially of the PERK pathway. The question of whether to inhibit or activate this pathway should probably be rephrased, as the solution might be different for each disease and the aim should be to restore PERK pathway activity to an optimal middle point. Other therapeutic strategies could be the targeting of UPR-regulated downstream factors, such as the promising recent reports of modulation of the S1R in HD.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Work related to this article was supported by grants from the KAMIN program (grant 57748) from the Israel authority for Technology Innovation, the Recanati Medical Fund and the Israel Science Foundation Legacy Heritage Fund (2394/17).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the members of the Lederkremer lab that critically read the manuscript.
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O'leary, J. C. III., Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 33, 9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013
Aguiar, S., Van der Gaag, B., and Cortese, F. A. B. (2017). RNAi mechanisms in Huntington's disease therapy: siRNA versus shRNA. Transl. Neurodegener. 6:30. doi: 10.1186/s40035-017-0101-9
Aharony, I., Ehrnhoefer, D. E., Shruster, A., Qiu, X., Franciosi, S., Hayden, M. R., et al. (2015). A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum. Mol. Genet. 24, 2604–2614. doi: 10.1093/hmg/ddv023
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Arnoux, I., Willam, M., Griesche, N., Krummeich, J., Watari, H., Offermann, N., et al. (2018). Metformin reverses early cortical network dysfunction and behavior changes in Huntington's disease. Elife 7:e38744. doi: 10.7554/eLife.38744
Ast, A., Buntru, A., Schindler, F., Hasenkopf, R., Schulz, A., Brusendorf, L., et al. (2018). mHTT seeding activity: a marker of disease progression and neurotoxicity in models of Huntington's disease. Mol. Cell 71, 675–688.e676. doi: 10.1016/j.molcel.2018.07.032
Bachoud-Lévi, A. C., Gaura, V., Brugières, P., Lefaucheur, J. P., Boissé, M. F., Maison, P., et al. (2006). Effect of fetal neural transplants in patients with Huntington's disease 6 years after surgery: a long-term follow-up study. Lancet Neurol. 5, 303–309. doi: 10.1016/S1474-4422(06)70381-7
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Behensky, A. A., Yasny, I. E., Shuster, A. M., Seredenin, S. B., Petrov, A. V., and Cuevas, J. (2013). Stimulation of sigma receptors with afobazole blocks activation of microglia and reduces toxicity caused by amyloid-beta25-35. J. Pharmacol. Exp. Ther. 347, 458–467. doi: 10.1124/jpet.113.208348
Bennett, E. J., Shaler, T. A., Woodman, B., Ryu, K. Y., Zaitseva, T. S., Becker, C. H., et al. (2007). Global changes to the ubiquitin system in Huntington's disease. Nature 448, 704–708. doi: 10.1038/nature06022
Bernard-Marissal, N., Médard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890. doi: 10.1093/brain/awv008
Bett, J. S., Cook, C., Petrucelli, L., and Bates, G. P. (2009). The ubiquitin-proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's disease. PLoS ONE 4:e5128. doi: 10.1371/journal.pone.0005128
Bol'shakova, A. V., Kraskovskaya, N. A., Gainullina, A. N., Kukanova, E. O., Vlasova, O. L., and Bezprozvanny, I. B. (2017). Neuroprotective effect of sigma1-receptors on the cell model of Huntington's disease. Bull. Exp. Biol. Med. 164, 252–258. doi: 10.1007/s10517-017-3968-7
Bowman, A. B., Yoo, S. Y., Dantuma, N. P., and Zoghbi, H. Y. (2005). Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 14, 679–691. doi: 10.1093/hmg/ddi064
Branco-Santos, J., Herrera, F., Poças, G. M., Pires-Afonso, Y., Giorgini, F., Domingos, P. M., et al. (2017). Protein phosphatase 1 regulates huntingtin exon 1 aggregation and toxicity. Hum. Mol. Genet. 26, 3763–3775. doi: 10.1093/hmg/ddx260
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper, J. H., Zhu, Y., et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. doi: 10.1016/j.celrep.2014.09.042
Bruch, J., Xu, H., Rösler, T. W., De Andrade, A., Kuhn, P. H., Lichtenthaler, S. F., et al. (2017). PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol. Med. 9, 371–384. doi: 10.15252/emmm.201606664
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., et al. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. doi: 10.1038/416507a
Carnemolla, A., Fossale, E., Agostoni, E., Michelazzi, S., Calligaris, R., De Maso, L., et al. (2009). Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J. Biol. Chem. 284, 18167–18173. doi: 10.1074/jbc.M109.018325
Caron, N. S., Dorsey, E. R., and Hayden, M. R. (2018). Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat. Rev. Drug Discov. 17, 729–750. doi: 10.1038/nrd.2018.133
Chen, Y., and Brandizzi, F. (2013). IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555. doi: 10.1016/j.tcb.2013.06.005
Chiti, F., and Dobson, C. M. (2009). Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 5, 15–22. doi: 10.1038/nchembio.131
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi: 10.1146/annurev-biochem-061516-045115
Cho, K. J., Lee, B. I., Cheon, S. Y., Kim, H. W., Kim, H. J., and Kim, G. W. (2009). Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease. Neuroscience 163, 1128–1134. doi: 10.1016/j.neuroscience.2009.07.048
Chu, C. T., Plowey, E. D., Wang, Y., Patel, V., and Jordan-Sciutto, K. L. (2007). Location, location, location: altered transcription factor trafficking in neurodegeneration. J. Neuropathol. Exp. Neurol. 66, 873–883. doi: 10.1097/nen.0b013e318156a3d7
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., and Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003
Dabrowska, M., Juzwa, W., Krzyzosiak, W. J., and Olejniczak, M. (2018). Precise excision of the CAG tract from the Huntingtin gene by Cas9 nickases. Front. Neurosci. 12:75. doi: 10.3389/fnins.2018.00075
Dahl, R. (2016). A new target for Parkinson's disease: small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg. Med. Chem. 25, 53–57. doi: 10.1016/j.bmc.2016.10.008
Das, I., Krzyzosiak, A., Schneider, K., Wrabetz, L., D'antonio, M., Barry, N., et al. (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242. doi: 10.1126/science.aaa4484
Datson, N. A., González-Barriga, A., Kourkouta, E., Weij, R., Van de Giessen, J., Mulders, S., et al. (2017). The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS ONE 12:e0171127. doi: 10.1371/journal.pone.0171127
Dinner, A. R., Sali, A., Smith, L. J., Dobson, C. M., and Karplus, M. (2000). Understanding protein folding via free-energy surfaces from theory and experiment. Trends Biochem. Sci. 25, 331–339. doi: 10.1016/S0968-0004(00)01610-8
Douglas, P. M., and Dillin, A. (2010). Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 190, 719–729. doi: 10.1083/jcb.201005144
Duennwald, M. L., and Lindquist, S. (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319. doi: 10.1101/gad.1673408
Eisenberg, D., and Jucker, M. (2012). The amyloid state of proteins in human diseases. Cell 148, 1188–1203. doi: 10.1016/j.cell.2012.02.022
Ellis, R. J., and Minton, A. P. (2006). Protein aggregation in crowded environments. Biol. Chem. 387, 485–497. doi: 10.1515/BC.2006.064
Emerich, D. F., Cain, C. K., Greco, C., Saydoff, J. A., Hu, Z. Y., Liu, H., et al. (1997). Cellular delivery of human CNTF prevents motor and cognitive dysfunction in a rodent model of Huntington's disease. Cell Transplant. 6, 249–266. doi: 10.1177/096368979700600308
Evers, M. M., Miniarikova, J., Juhas, S., Vallès, A., Bohuslavova, B., Juhasova, J., et al. (2018). AAV5-miHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington's disease minipig model. Mol. Ther. 26, 2163–2177. doi: 10.1016/j.ymthe.2018.06.021
Farina, F., Lambert, E., Commeau, L., Lejeune, F. X., Roudier, N., Fonte, C., et al. (2017). The stress response factor daf-16/FOXO is required for multiple compound families to prolong the function of neurons with Huntington's disease. Sci. Rep. 7:4014. doi: 10.1038/s41598-017-04256-w
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., et al. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 23, 9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003
Finkbeiner, S., and Mitra, S. (2008). The ubiquitin-proteasome pathway in Huntington's disease. ScientificWorldJournal 8, 421–433. doi: 10.1100/tsw.2008.60
Fonseca, A. C., Oliveira, C. R., Pereira, C. F., and Cardoso, S. M. (2014). Loss of proteostasis induced by amyloid beta peptide in brain endothelial cells. Biochim. Biophys. Acta 1843, 1150–1161. doi: 10.1016/j.bbamcr.2014.02.016
Francardo, V., Bez, F., Wieloch, T., Nissbrandt, H., Ruscher, K., and Cenci, M. A. (2014). Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 137, 1998–2014. doi: 10.1093/brain/awu107
Fujimoto, M., and Hayashi, T. (2011). New insights into the role of mitochondria-associated endoplasmic reticulum membrane. Int. Rev. Cell Mol. Biol. 292, 73–117. doi: 10.1016/B978-0-12-386033-0.00002-5
Garcia-Miralles, M., Geva, M., Tan, J. Y., Yusof, N. A. B. M., Cha, Y., Kusko, R., et al. (2017). Early pridopidine treatment improves behavioral and transcriptional deficits in YAC128 Huntington disease mice. JCI Insight 2:95665. doi: 10.1172/jci.insight.95665
Gilady, S. Y., Bui, M., Lynes, E. M., Benson, M. D., Watts, R., Vance, J. E., et al. (2010). Ero1α requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 15, 619–629. doi: 10.1007/s12192-010-0174-1
Goedert, M. (2015). Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349:1255555. doi: 10.1126/science.1255555
Guo, X., Sun, X., Hu, D., Wang, Y. J., Fujioka, H., Vyas, R., et al. (2016). VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease. Nat. Commun. 7:12646. doi: 10.1038/ncomms12646
Halliday, M., Hughes, D., and Mallucci, G. (2017a). Fine tuning PERK signaling for neuroprotection. J. Neurochem. 142, 812–826. doi: 10.1111/jnc.14112
Halliday, M., Radford, H., Sekine, Y., Moreno, J., Verity, N., Le Quesne, J., et al. (2015). Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6:e1672. doi: 10.1038/cddis.2015.49
Halliday, M., Radford, H., Zents, K. A. M., Molloy, C., Moreno, J. A., Verity, N. C., et al. (2017b). Repurposed drugs targeting eIF2andalpha;-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783. doi: 10.1093/brain/awx074
Hanner, M., Moebius, F. F., Flandorfer, A., Knaus, H. G., Striessnig, J., Kempner, E., et al. (1996). Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. U.S.A. 93, 8072–8077. doi: 10.1073/pnas.93.15.8072
Hartl, F. U. (2017). Protein misfolding diseases. Annu. Rev. Biochem. 86, 21–26. doi: 10.1146/annurev-biochem-061516-044518
Hartl, F. U., and Hayer-Hartl, M. (2002). Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858. doi: 10.1126/science.1068408
Hayashi, T. (2015). Sigma-1 receptor: the novel intracellular target of neuropsychotherapeutic drugs. J. Pharmacol. Sci. 127, 2–5. doi: 10.1016/j.jphs.2014.07.001
Hayashi, T., Rizzuto, R., Hajnoczky, G., and Su, T. P. (2009). MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88. doi: 10.1016/j.tcb.2008.12.002
Hayashi, T., and Su, T. P. (2003). Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108-15 cells. J. Pharmacol. Exp. Ther. 306, 726–733. doi: 10.1124/jpet.103.051292
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Heman-Ackah, S. M., Manzano, R., Hoozemans, J. J. M., Scheper, W., Flynn, R., Haerty, W., et al. (2017). Alpha-synuclein induces the unfolded protein response in Parkinson's disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 26, 4441–4450. doi: 10.1093/hmg/ddx331
Hernández, I. H., Torres-Peraza, J., Santos-Galindo, M., Ramos-Morón, E., Fernández-Fernández, M. R., Peréz-Álvarez, M. J., et al. (2017). The neuroprotective transcription factor ATF5 is decreased and sequestered into polyglutamine inclusions in Huntington's disease. Acta Neuropathol. 134, 839–850. doi: 10.1007/s00401-017-1770-2
Hervás, D., Fornés-Ferrer, V., Gómez-Escribano, A. P., Sequedo, M. D., Peiró, C., Millán, J. M., et al. (2017). Metformin intake associates with better cognitive function in patients with Huntington's disease. PLoS ONE 12:e0179283. doi: 10.1371/journal.pone.0179283
Hesp, K., Smant, G., and Kammenga, J. E. (2015). Caenorhabditis elegans DAF-16/FOXO transcription factor and its mammalian homologs associate with age-related disease. Exp. Gerontol. 72, 1–7. doi: 10.1016/j.exger.2015.09.006
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., and Papa, F. R. (2017). The unfolded protein response and cell fate control. Mol. Cell. 69, 169–181. doi: 10.1016/j.molcel.2017.06.017
Hipp, M. S., Park, S. H., and Hartl, F. U. (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514. doi: 10.1016/j.tcb.2014.05.003
Hipp, M. S., Patel, C. N., Bersuker, K., Riley, B. E., Kaiser, S. E., Shaler, T. A., et al. (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington's disease. J. Cell Biol. 196, 573–587. doi: 10.1083/jcb.201110093
Hoffner, G., and Djian, P. (2015). Polyglutamine aggregation in Huntington disease: does structure determine toxicity? Mol. Neurobiol. 52, 1297–1314. doi: 10.1007/s12035-014-8932-1
Hoozemans, J. J. M., Veerhuis, R., Van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., et al. (2005). The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. 110, 165–172. doi: 10.1007/s00401-005-1038-0
Huang, W. J., Chen, W. W., and Zhang, X. (2016). Huntington's disease: molecular basis of pathology and status of current therapeutic approaches. Exp. Ther. Med. 12, 1951–1956. doi: 10.3892/etm.2016.3566
Hughes, D., and Mallucci, G. R. (2018). The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J. 286, 342–355. doi: 10.1111/febs.14422
Hyrskyluoto, A., Bruelle, C., Lundh, S. H., Do, H. T., Kivinen, J., Rappou, E., et al. (2014). Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: involvement of the proteasome and ER stress-activated kinase IRE1. Hum. Mol. Genet. 23, 5928–5939. doi: 10.1093/hmg/ddu317
Hyrskyluoto, A., Pulli, I., Tornqvist, K., Ho, T. H., Korhonen, L., and Lindholm, D. (2013). Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-kappaB pathway. Cell Death Dis. 4:e646. doi: 10.1038/cddis.2013.170
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jeon, J., Kim, W., Jang, J., Isacson, O., and Seo, H. (2016). Gene therapy by proteasome activator, PA28gamma, improves motor coordination and proteasome function in Huntington's disease YAC128 mice. Neuroscience 324, 20–28. doi: 10.1016/j.neuroscience.2016.02.054
Jiang, Y., Chadwick, S. R., and Lajoie, P. (2016). Endoplasmic reticulum stress: the cause and solution to Huntington's disease? Brain Res. 1648(Pt B), 650–657. doi: 10.1016/j.brainres.2016.03.034
Kakkar, V., Meister-Broekema, M., Minoia, M., Carra, S., and Kampinga, H. H. (2014). Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis. Model. Mech. 7, 421–434. doi: 10.1242/dmm.014563
Kampinga, H. H., and Bergink, S. (2016). Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 15, 748–759. doi: 10.1016/S1474-4422(16)00099-5
Katnik, C., Guerrero, W. R., Pennypacker, K. R., Herrera, Y., and Cuevas, J. (2006). Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J. Pharmacol. Exp. Ther. 319, 1355–1365. doi: 10.1124/jpet.106.107557
Kazemi-Esfarjani, P., and Benzer, S. (2000). Genetic suppression of polyglutamine toxicity in Drosophila. Science 287, 1837–1840. doi: 10.1126/science.287.5459.1837
Keene, C. D., Rodrigues, C. M., Eich, T., Chhabra, M. S., Steer, C. J., and Low, W. C. (2002). Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10671–10676. doi: 10.1073/pnas.162362299
Koga, H., Kaushik, S., and Cuervo, A. M. (2011). Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215. doi: 10.1016/j.arr.2010.02.001
Kourrich, S., Su, T. P., Fujimoto, M., and Bonci, A. (2012). The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 35, 762–771. doi: 10.1016/j.tins.2012.09.007
Kuemmerle, S., Gutekunst, C. A., Klein, A. M., Li, X. J., Li, S. H., Beal, M. F., et al. (1999). Huntington aggregates may not predict neuronal death in Huntington's disease. Ann. Neurol. 46, 842–849. doi: 10.1002/1531-8249(199912)46:6<842::AID-ANA6>3.0.CO;2-O
Kusko, R., Dreymann, J., Ross, J., Cha, Y., Escalante-Chong, R., Garcia-Miralles, M., et al. (2018). Large-scale transcriptomic analysis reveals that pridopidine reverses aberrant gene expression and activates neuroprotective pathways in the YAC128 HD mouse. Mol. Neurodegener. 13:25. doi: 10.1186/s13024-018-0259-3
Labbadia, J., and Morimoto, R. I. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. doi: 10.1146/annurev-biochem-060614-033955
Lajoie, P., and Snapp, E. L. (2010). Formation and toxicity of soluble polyglutamine oligomers in living cells. PLoS ONE 5:e15245. doi: 10.1371/journal.pone.0015245
Lee, H., Ahn, H.-H., Lee, W., Oh, Y., Choi, H., Shim, S. M., et al. (2016). ENC1 modulates the aggregation and neurotoxicity of mutant huntingtin through p62 under ER stress. Mol. Neurobiol. 53, 6620–6634. doi: 10.1007/s12035-015-9557-8
Lee, S. J., Lim, H. S., Masliah, E., and Lee, H. J. (2011). Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci. Res. 70, 339–348. doi: 10.1016/j.neures.2011.05.008
Leitman, J., Barak, B., Benyair, R., Shenkman, M., Ashery, U., Hartl, F. U., et al. (2014). ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS ONE 9:e90803. doi: 10.1371/journal.pone.0090803
Leitman, J., Ulrich Hartl, F., and Lederkremer, G. Z. (2013). Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 4:2753. doi: 10.1038/ncomms3753
Lesné, S., Kotilinek, L., and Ashe, K. H. (2008). Plaque-bearing mice with reduced levels of oligomeric amyloid-β assemblies have intact memory function. Neuroscience 151, 745–749. doi: 10.1016/j.neuroscience.2007.10.054
Li, G., Mongillo, M., Chin, K. T., Harding, H., Ron, D., Marks, A. R., et al. (2009). Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J. Cell Biol. 186, 783–792. doi: 10.1083/jcb.200904060
Liu, J. P., and Zeitlin, S. O. (2017). Is huntingtin dispensable in the adult brain? J. Huntingtons. Dis. 6, 1–17. doi: 10.3233/JHD-170235
Luo, H., Cao, L., Liang, X., Du, A., Peng, T., and Li, H. (2018). Herp promotes degradation of mutant huntingtin: involvement of the proteasome and molecular chaperones. Mol. Neurobiol. 55, 7652–7668. doi: 10.1007/s12035-018-0900-8
Mancuso, R., Olivan, S., Rando, A., Casas, C., Osta, R., and Navarro, X. (2012). Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9, 814–826. doi: 10.1007/s13311-012-0140-y
Martinez-Lopez, N., Athonvarangkul, D., and Singh, R. (2015). Autophagy and aging. Adv. Exp. Med. Biol. 847, 73–87. doi: 10.1007/978-1-4939-2404-2_3
Mavylutov, T., Chen, X., Guo, L., and Yang, J. (2017). APEX2- tagging of Sigma 1-receptor indicates subcellular protein topology with cytosolic N-terminus and ER luminal C-terminus. Protein Cell. 9, 733–737. doi: 10.1007/s13238-017-0468-5
Mazzocchi-Jones, D., Dobrossy, M., and Dunnett, S. B. (2009). Embryonic striatal grafts restore bi-directional synaptic plasticity in a rodent model of Huntington's disease. Eur. J. Neurosci. 30, 2134–2142. doi: 10.1111/j.1460-9568.2009.07006.x
Mckinnon, C., and Tabrizi, S. J. (2014). The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 21, 2302–2321. doi: 10.1089/ars.2013.5802
McQuiston, A., and Diehl, J. A. (2017). Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Res 6:1897. doi: 10.12688/f1000research.12138.1
Mercado, G., Castillo, V., Soto, P., López, N., Axten, J. M., Sardi, S. P., et al. (2018). Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol. Dis. 112, 136–148. doi: 10.1016/j.nbd.2018.01.004
Meriin, A. B., Zhang, X., He, X., Newnam, G. P., Chernoff, Y. O., and Sherman, M. Y. (2002). Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004. doi: 10.1083/jcb.200112104
Meunier, J., and Hayashi, T. (2010). Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J. Pharmacol. Exp. Ther. 332, 388–397. doi: 10.1124/jpet.109.160960
Meunier, J., Ieni, J., and Maurice, T. (2006). The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br. J. Pharmacol. 149, 998–1012. doi: 10.1038/sj.bjp.0706927
Miki, Y., Tanji, K., Mori, F., and Wakabayashi, K. (2015). Sigma-1 receptor is involved in degradation of intranuclear inclusions in a cellular model of Huntington's disease. Neurobiol. Dis. 74, 25–31. doi: 10.1016/j.nbd.2014.11.005
Miller, S. B., Ho, C. T., Winkler, J., Khokhrina, M., Neuner, A., Mohamed, M. Y., et al. (2015). Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 34, 778–797. doi: 10.15252/embj.201489524
Miniarikova, J., Zimmer, V., Martier, R., Brouwers, C. C., Pythoud, C., Richetin, K., et al. (2017). AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington's disease. Gene Ther. 24, 630–639. doi: 10.1038/gt.2017.71
Mitsuda, T., Omi, T., Tanimukai, H., Sakagami, Y., Tagami, S., Okochi, M., et al. (2011). Sigma-1Rs are upregulated via PERK/eIF2alpha/ATF4 pathway and execute protective function in ER stress. Biochem. Biophys. Res. Commun. 415, 519–525. doi: 10.1016/j.bbrc.2011.10.113
Mogk, A., Bukau, B., and Kampinga, H. H. (2018). Cellular handling of protein aggregates by disaggregation machines. Mol. Cell 69, 214–226. doi: 10.1016/j.molcel.2018.01.004
Moily, N. S., Ormsby, A. R., Stojilovic, A., Ramdzan, Y. M., Diesch, J., Hannan, R. D., et al. (2017). Transcriptional profiles for distinct aggregation states of mutant Huntingtin exon 1 protein unmask new Huntington's disease pathways. Mol. Cell. Neurosci. 83, 103–112. doi: 10.1016/j.mcn.2017.07.004
Moreno, J. A., Halliday, M., Molloy, C., Radford, H., Verity, N., Axten, J. M., et al. (2013). Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5:206ra138. doi: 10.1126/scitranslmed.3006767
Morris, A. M., Watzky, M. A., and Finke, R. G. (2009). Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim. Biophys. Acta 1794, 375–397. doi: 10.1016/j.bbapap.2008.10.016
Murphy, R. M. (2002). Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 4, 155–174. doi: 10.1146/annurev.bioeng.4.092801.094202
Neueder, A., Landles, C., Ghosh, R., Howland, D., Myers, R. H., Faull, R. L. M., et al. (2017). The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington's disease patients. Sci. Rep. 7:1307. doi: 10.1038/s41598-017-01510-z
Nguyen, L., Kaushal, N., Robson, M. J., and Matsumoto, R. R. (2014). Sigma receptors as potential therapeutic targets for neuroprotection. Eur. J. Pharmacol. 743, 42–47. doi: 10.1016/j.ejphar.2014.09.022
Nguyen, L., Lucke-Wold, B. P., Mookerjee, S., Kaushal, N., and Matsumoto, R. R. (2017). Sigma-1 receptors and neurodegenerative diseases: towards a hypothesis of Sigma-1 receptors as amplifiers of neurodegeneration and neuroprotection. Adv. Exp. Med. Biol. 964, 133–152. doi: 10.1007/978-3-319-50174-1_10
Nguyen, L., Lucke-Wold, B. P., Mookerjee, S. A., Cavendish, J. Z., Robson, M. J., Scandinaro, A. L., et al. (2015). Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 127, 17–29. doi: 10.1016/j.jphs.2014.12.005
Noh, J. Y., Lee, H., Song, S., Kim, N. S., Im, W., Kim, M., et al. (2009). SCAMP5 links endoplasmic reticulum stress to the accumulation of expanded polyglutamine protein aggregates via endocytosis inhibition. J. Biol. Chem. 284, 11318–11325. doi: 10.1074/jbc.M807620200
Novoa, I., Zeng, H., Harding, H. P., and Ron, D. (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 153, 1011–1022. doi: 10.1083/jcb.153.5.1011
Ogen-Shtern, N., Ben David, T., and Lederkremer, G. Z. (2016). Protein aggregation and ER stress. Brain Res. 1648, 658–666. doi: 10.1016/j.brainres.2016.03.044
Ohno, M. (2017). PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer's disease. Brain Res. Bull. 141, 72–78. doi: 10.1016/j.brainresbull.2017.08.007
Ortega, Z., Diaz-Hernandez, M., and Lucas, J. J. (2007). Is the ubiquitin-proteasome system impaired in Huntington's disease? Cell. Mol. Life Sci. 64, 2245–2257. doi: 10.1007/s00018-007-7222-8
Ortega-Roldan, J. L., Ossa, F., Amin, N. T., and Schnell, J. R. (2015). Solution NMR studies reveal the location of the second transmembrane domain of the human sigma-1 receptor. FEBS Lett. 589, 659–665. doi: 10.1016/j.febslet.2015.01.033
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389. doi: 10.1038/sj.cdd.4401373
Pal, A., Fontanilla, D., Gopalakrishnan, A., Chae, Y. K., Markley, J. L., and Ruoho, A. E. (2012). The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharmacol. 682, 12–20. doi: 10.1016/j.ejphar.2012.01.030
Pandey, M., and Rajamma, U. (2018). Huntington's disease: the coming of age. J. Genet. 97, 649–664. doi: 10.1007/s12041-018-0957-1
Pandey, N. K., Isas, J. M., Rawat, A., Lee, R. V., Langen, J., Pandey, P., et al. (2017). The 17-residue-long N terminus in huntingtin controls step-wise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem. 293, 2597–2605 doi: 10.1074/jbc.M117.813667
Pérez-Arancibia, R., Rivas, A., and Hetz, C. (2017). (off)Targeting UPR signaling: the race toward intervening ER proteostasis. Expert Opin. Ther. Targets. 22, 97–100. doi: 10.1080/14728222.2018.1420169
Petersen, R. C., Aisen, P., Boeve, B. F., Geda, Y. E., Ivnik, R. J., Knopman, D. S., et al. (2013). Mild cognitive impairment due to Alzheimer disease in the community. Ann. Neurol. 74, 199–208. doi: 10.1002/ana.23931
Philo, J. S., and Arakawa, T. (2009). Mechanisms of protein aggregation. Curr. Pharm. Biotechnol. 10, 348–351. doi: 10.2174/138920109788488932
Pierzynowska, K., Gaffke, L., Hac, A., Mantej, J., Niedzialek, N., Brokowska, J., et al. (2018). Correction of Huntington's disease phenotype by genistein-induced autophagy in the cellular model. Neuromolecular Med. 20, 112–123. doi: 10.1007/s12017-018-8482-1
Prahlad, V., and Morimoto, R. I. (2011). Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 14204–14209. doi: 10.1073/pnas.1106557108
Precious, S. V., Zietlow, R., Dunnett, S. B., Kelly, C. M., and Rosser, A. E. (2017). Is there a place for human fetal-derived stem cells for cell replacement therapy in Huntington's disease? Neurochem. Int. 106, 114–121. doi: 10.1016/j.neuint.2017.01.016
Radford, H., Moreno, J. A., Verity, N., Halliday, M., and Mallucci, G. R. (2015). PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642. doi: 10.1007/s00401-015-1487-z
Ramdzan, Y. M., Trubetskov, M. M., Ormsby, A. R., Newcombe, E. A., Sui, X., Tobin, M. J., et al. (2017). Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep. 19, 919–927. doi: 10.1016/j.celrep.2017.04.029
Reijonen, S., Putkonen, N., Norremolle, A., Lindholm, D., and Korhonen, L. (2008). Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 314, 950–960. doi: 10.1016/j.yexcr.2007.12.025
Reiner, A., Albin, R. L., Anderson, K. D., D'amato, C. J., Penney, J. B., and Young, A. B. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 85, 5733–5737. doi: 10.1073/pnas.85.15.5733
Remondelli, P., and Renna, M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10:187. doi: 10.3389/fnmol.2017.00187
Ron, D. (2002). Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388. doi: 10.1172/JCI0216784
Rosas, H. D., Koroshetz, W. J., Chen, Y. I., Skeuse, C., Vangel, M., Cudkowicz, M. E., et al. (2003). Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology 60, 1615–1620. doi: 10.1212/01.WNL.0000065888.88988.6E
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10 Suppl, S10–17. doi: 10.1038/nm1066
Ruetenik, A. L., Ocampo, A., Ruan, K., Zhu, Y., Li, C., Zhai, R. G., et al. (2016). Attenuation of polyglutamine-induced toxicity by enhancement of mitochondrial OXPHOS in yeast and fly models of aging. Microb. Cell 3, 338–351. doi: 10.15698/mic2016.08.518
Ryskamp, D., Wu, J., Geva, M., Kusko, R., Grossman, I., Hayden, M., et al. (2017). The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol. Dis. 97, 46–59. doi: 10.1016/j.nbd.2016.10.006
Saavedra, A., Garcia-Diaz Barriga, G., Perez-Navarro, E., and Alberch, J. (2018). Huntington's disease: novel therapeutic perspectives hanging in the balance. Expert Opin. Ther. Targets 22, 385–399. doi: 10.1080/14728222.2018.1465930
Sakahira, H., Breuer, P., Hayer-Hartl, M. K., and Hartl, F. U. (2002). Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci U.S.A. 99 (Suppl. 4), 16412–16418. doi: 10.1073/pnas.182426899
Sano, R., and Reed, J. C. (2013). ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833, 3460–3470. doi: 10.1016/j.bbamcr.2013.06.028
Schackel, S., Pauly, M. C., Piroth, T., Nikkhah, G., and Dobrossy, M. D. (2013). Donor age dependent graft development and recovery in a rat model of Huntington's disease: histological and behavioral analysis. Behav. Brain Res. 256, 56–63. doi: 10.1016/j.bbr.2013.07.053
Schaffar, G., Breuer, P., Boteva, R., Behrends, C., Tzvetkov, N., Strippel, N., et al. (2004). Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol. Cell 15, 95–105. doi: 10.1016/j.molcel.2004.06.029
Schmidt, H. R., Zheng, S., Gurpinar, E., Koehl, A., Manglik, A., and Kruse, A. C. (2016). Crystal structure of the human sigma1 receptor. Nature 532, 527–530. doi: 10.1038/nature17391
Scior, A., Buntru, A., Arnsburg, K., Ast, A., Iburg, M., Juenemann, K., et al. (2018). Complete suppression of Htt fibrilization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 37, 282–299. doi: 10.15252/embj.201797212
Sekijima, Y., Wiseman, R. L., Matteson, J., Hammarstrom, P., Miller, S. R., Sawkar, A. R., et al. (2005). The biological and chemical basis for tissue-selective amyloid disease. Cell 121, 73–85. doi: 10.1016/j.cell.2005.01.018
Sengupta, U., Nilson, A. N., and Kayed, R. (2016). The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6, 42–49. doi: 10.1016/j.ebiom.2016.03.035
Shamsi, T. N., Athar, T., Parveen, R., and Fatima, S. (2017). A review on protein misfolding, aggregation and strategies to prevent related ailments. Int. J. Biol. Macromol. 105, 993–1000. doi: 10.1016/j.ijbiomac.2017.07.116
Shao, J., and Diamond, M. I. (2007). Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 16 Spec No. 2, R115–123. doi: 10.1093/hmg/ddm213
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/S1534-5807(02)00203-4
Shenkman, M., Eiger, H., and Lederkremer Gerardo, Z. (2015). Genesis of ER Stress in Huntington's Disease. ER Stress Dis. 2, 94–106. doi: 10.1515/ersc-2015-0007
Sidrauski, C., Acosta-Alvear, D., Khoutorsky, A., Vedantham, P., Hearn, B. R., Li, H., et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2:e00498. doi: 10.7554/eLife.00498
Smith, D. M. (2018). Could a common mechanism of protein degradation impairment underlie many neurodegenerative diseases? J. Exp. Neurosci. 12:1179069518794675. doi: 10.1177/1179069518794675
Soejima, N., Ohyagi, Y., Nakamura, N., Himeno, E., Iinuma, K. M., Sakae, N., et al. (2013). Intracellular accumulation of toxic turn amyloid-beta is associated with endoplasmic reticulum stress in Alzheimer's disease. Curr. Alzheimer Res. 10, 11–20. doi: 10.2174/1567205011310010003
Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. doi: 10.1038/nrn1007
Soto, C., and Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340. doi: 10.1038/s41593-018-0235-9
Southwell, A. L., Kordasiewicz, H. B., Langbehn, D., Skotte, N. H., Parsons, M. P., Villanueva, E. B., et al. (2018). Huntingtin suppression restores cognitive function in a mouse model of Huntington's disease. Sci. Transl. Med. 10:eaar3959. doi: 10.1126/scitranslmed.aar3959
Squitieri, F., and De Yebenes, J. G. (2015). Profile of pridopidine and its potential in the treatment of Huntington disease: the evidence to date. Drug Des. Devel. Ther. 9, 5827–5833. doi: 10.2147/DDDT.S65738
Squitieri, F., Di Pardo, A., Favellato, M., Amico, E., Maglione, V., and Frati, L. (2015). Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 19, 2540–2548. doi: 10.1111/jcmm.12604
Stockwell, S. R., Platt, G., Barrie, S. E., Zoumpoulidou, G., Te Poele, R. H., Aherne, G. W., et al. (2012). Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS ONE 7:e28568. doi: 10.1371/journal.pone.0028568
Stutzbach, L. D., Xie, S. X., Naj, A. C., Albin, R., Gilman, S., Lee, V. M. Y., et al. (2013). The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol. Commun. 1:31. doi: 10.1186/2051-5960-1-31
Su, T. P., Hayashi, T., Maurice, T., Buch, S., and Ruoho, A. E. (2010). The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 31, 557–566. doi: 10.1016/j.tips.2010.08.007
Suelves, N., Kirkham-Mccarthy, L., Lahue, R. S., and Gines, S. (2017). A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal CAG repeat expansions in Huntington's disease mice. Sci. Rep. 7:6082. doi: 10.1038/s41598-017-05125-2
Sun, X., Aime, P., Dai, D., Ramalingam, N., Crary, J. F., Burke, R. E., et al. (2018). Guanabenz promotes neuronal survival via enhancement of ATF4 and parkin expression in models of Parkinson disease. Exp. Neurol. 303, 95–107. doi: 10.1016/j.expneurol.2018.01.015
Taalab, Y. M., Ibrahim, N., Maher, A., Hassan, M., Mohamed, W., Moustafa, A. A., et al. (2018). Mechanisms of disordered neurodegenerative function: concepts and facts about the different roles of the protein kinase RNA-like endoplasmic reticulum kinase (PERK). Rev. Neurosci. 29, 387–415. doi: 10.1515/revneuro-2017-0071
Takahashi, T., Kikuchi, S., Katada, S., Nagai, Y., Nishizawa, M., and Onodera, O. (2008). Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 17, 345–356. doi: 10.1093/hmg/ddm311
Taylor, J. P. (2002). Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. doi: 10.1126/science.1067122
Taylor, R. C., and Dillin, A. (2011). Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3:a004440. doi: 10.1101/cshperspect.a004440
Tsai, S. Y., Pokrass, M. J., Klauer, N. R., De Credico, N. E., and Su, T. P. (2014). Sigma-1 receptor chaperones in neurodegenerative and psychiatric disorders. Expert Opin. Ther. Targets 18, 1461–1476. doi: 10.1517/14728222.2014.972939
Tsaytler, P., Harding, H. P., Ron, D., and Bertolotti, A. (2011). Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94. doi: 10.1126/science.1201396
Vagnerova, K., Hurn, P. D., Bhardwaj, A., and Kirsch, J. R. (2006). Sigma 1 receptor agonists act as neuroprotective drugs through inhibition of inducible nitric oxide synthase. Anesth. Analg. 103, 430–434. doi: 10.1213/01.ane.0000226133.85114.91
Vendruscolo, M., Zurdo, J., Macphee, C. E., and Dobson, C. M. (2003). Protein folding and misfolding: a paradigm of self-assembly and regulation in complex biological systems. Philos. Trans. A Math. Phys. Eng. Sci. 361, 1205–1222. doi: 10.1098/rsta.2003.1194
Vidal, R., Caballero, B., Couve, A., and Hetz, C. (2011). Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington's disease. Curr. Mol. Med. 11, 1–12. doi: 10.2174/156652411794474419
Vidal, R. L., Figueroa, A., Court, F. A., Thielen, P., Molina, C., Wirth, C., et al. (2012). Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262. doi: 10.1093/hmg/dds040
Vieira, F. G., Ping, Q., Moreno, A. J., Kidd, J. D., Thompson, K., Jiang, B., et al. (2015). Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS ONE 10:e0135570. doi: 10.1371/journal.pone.0135570
Volovik, Y., Moll, L., Marques, F. C., Maman, M., Bejerano-Sagie, M., and Cohen, E. (2014). Differential regulation of the heat shock factor 1 and DAF-16 by neuronal nhl-1 in the nematode C. elegans. Cell Rep. 9, 2192–2205. doi: 10.1016/j.celrep.2014.11.028
Wang, B., Zeng, L., Merillat, S. A., Fischer, S., Ochaba, J., Thompson, L. M., et al. (2017). The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington's disease protein, huntingtin. Neurobiol. Dis. 109, 127–136. doi: 10.1016/j.nbd.2017.10.002
Wang, L., Eldred, J. A., Sidaway, P., Sanderson, J., Smith, A. J., Bowater, R. P., et al. (2012). Sigma 1 receptor stimulation protects against oxidative damage through suppression of the ER stress responses in the human lens. Mech. Ageing Dev. 133, 665–674. doi: 10.1016/j.mad.2012.09.005
Waters, S., Tedroff, J., Ponten, H., Klamer, D., Sonesson, C., and Waters, N. (2018). Pridopidine: overview of pharmacology and rationale for its use in Huntington's disease. J. Huntingtons. Dis. 7, 1–16. doi: 10.3233/JHD-170267
Woerner, A. C., Frottin, F., Hornburg, D., Feng, L. R., Meissner, F., Patra, M., et al. (2016). Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176. doi: 10.1126/science.aad2033
Wright, D. J., Renoir, T., Gray, L. J., and Hannan, A. J. (2017). Huntington's disease: pathogenic mechanisms and therapeutic targets. Adv. Neurobiol. 15, 93–128. doi: 10.1007/978-3-319-57193-5_4
Wu, J., Ryskamp, D., Birnbaumer, L., and Bezprozvanny, I. (2018). Inhibition of TRPC1-dependent store-operated calcium entry improves synaptic stability and motor performance in a mouse model of Huntington's disease. J. Huntingtons. Dis. 7, 35–50. doi: 10.3233/JHD-170266
Xiang, C., Wang, Y., Zhang, H., and Han, F. (2016). The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 22, 1–26. doi: 10.1007/s10495-016-1296-4
Yang, H., Liu, C., Zhong, Y., Luo, S., Monteiro, M. J., and Fang, S. (2010). Huntingtin interacts with the cue domain of gp78 and inhibits gp78 binding to ubiquitin and p97/VCP. PLoS ONE 5:e8905. doi: 10.1371/journal.pone.0008905
Yang, S., Chang, R., Yang, H., Zhao, T., Hong, Y., Kong, H. E., et al. (2017). CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease. J. Clin. Invest. 127, 2719–2724. doi: 10.1172/JCI92087
Yang, W., Zhou, X., Zimmermann, H. R., Cavener, D. R., Klann, E., and Ma, T. (2016). Repression of the eIF2alpha kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer's disease. Neurobiol. Aging 41, 19–24. doi: 10.1016/j.neurobiolaging.2016.02.005
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Yuan, S. H., Hiramatsu, N., Liu, Q., Sun, X. V., Lenh, D., Chan, P., et al. (2018). Tauopathy-associated PERK alleles are functional hypomorphs that increase neuronal vulnerability to ER stress. Hum. Mol. Genet. 27, 3951–3963. doi: 10.1093/hmg/ddy297
Zheng, J., Winderickx, J., Franssens, V., and Liu, B. (2018). A Mitochondria-associated oxidative stress perspective on Huntington's disease. Front. Mol. Neurosci. 11:329. doi: 10.3389/fnmol.2018.00329
Zhou, X., Li, G., Kaplan, A., Gaschler, M. M., Zhang, X., Hou, Z., et al. (2018). Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington's disease. Hum. Mol. Genet. 27, 1545–1555. doi: 10.1093/hmg/ddy061
Keywords: Huntington's disease, neurodegeneration, protein misfolding, protein aggregation, ER stress, unfolded protein response
Citation: Shacham T, Sharma N and Lederkremer GZ (2019) Protein Misfolding and ER Stress in Huntington's Disease. Front. Mol. Biosci. 6:20. doi: 10.3389/fmolb.2019.00020
Received: 15 January 2019; Accepted: 11 March 2019;
Published: 03 April 2019.
Edited by:
Anat Ben-Zvi, Ben-Gurion University of the Negev, IsraelReviewed by:
Ehud Cohen, Hebrew University of Jerusalem, IsraelCopyright © 2019 Shacham, Sharma and Lederkremer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerardo Z. Lederkremer, Z2VyYXJkb2xAdGF1ZXgudGF1LmFjLmls
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.