
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 30 September 2020
Sec. Terrestrial Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.560942
 Chenshuo Lin1,2Hang Xu2Wei Qin3Shaoyi Xu4Xiufeng Tang5Lu Kuang5Xinxin Wang5Bin Jiang6
Chenshuo Lin1,2Hang Xu2Wei Qin3Shaoyi Xu4Xiufeng Tang5Lu Kuang5Xinxin Wang5Bin Jiang6 Junhui Chen1
Junhui Chen1 Jun Shan5Jonathan Adams7
Jun Shan5Jonathan Adams7 Hua Qin1*
Hua Qin1* Baozhan Wang5,8*
Baozhan Wang5,8*After the discovery of complete ammonia-oxidizing (comammox) Nitrospira, detection and assessments of the contribution of comammox Nitrospira communities to nitrogen cycling are in great demand. PCR-based approach, a common method for the detection of comammox, depends strongly on accurate amplification of the amoA genes from the original DNA samples using appropriate primers. In this study, we reported an evaluation of the performance of two commonly used primer sets, Ntsp-amoA 162F/359R and comaA/B-244f/659r, for amplifying the comammox amoA genes from three representative wetland soils in China [Sangsang (SS), Sanjiang (SJ), and Xianghai (XH)]. Our results demonstrated the two primer sets could both successfully amplify the clades with high relative abundances (RA), and further revealed a broadly similar diversity and community composition of dominant comammox operational taxonomic units (OTUs) (RA ≥ 1%) in each of the three wetland soils. However, the clades with low RA, such as the clade A (1.26%) in SJ and the clade B (11.54%) in XH that were recovered by metagenomics analysis, failed to be amplified using comaA/B-244f/659r, but were successfully amplified and sequenced using Ntsp-amoA 162F/359R. It indicated that, compared to comaA/B-244f/659r, Ntsp-amoA 162F/359R was more sensitive to the clades with low RA. However, it is worth noting that Ntsp-amoA 162F/359R would overestimate the RA of some rare clades. For example, the RAs of clade B in XH were overestimated by 32-fold. Furthermore, high levels of non-target amplification were detected via gel electrophoresis using both primer sets, especially for comammox Clade B amoA genes, implying that we should treat qPCR results based on these primers with caution. Taken together, our study comprehensively compared the performance of the two primer sets on the sensitivity and specificity of amplifying comammox amoA genes in three wetland soils, pointing out the necessity of further development of new primers for the efficient and accurate detection of comammox in various environments.
Nitrification, the aerobic oxidation of ammonia via nitrite to nitrate, is an essential part of the global nitrogen (N) cycling process (Galloway et al., 2008). It was originally considered to be a two-step process catalyzed by two distinct functional groups of microorganisms, the ammonia-oxidizing microbes (AOM) including ammonia-oxidizing bacteria (AOB) and ammonia-oxidizing archaea (AOA) (Könneke et al., 2005), and nitrite-oxidizing bacteria (NOB). This perception was overturned in 2015 with the surprising discovery of complete ammonia oxidizers (comammox) (Daims et al., 2015; van Kessel et al., 2015), which encode all the necessary enzymes involved in ammonia oxidation and nitrite oxidation in their genomes and perform complete nitrification activity in a single organism (Santoro, 2016).
Comammox of the genus Nitrospira are widely distributed in both anthropogenic and natural habitats, including recirculating aquaculture systems (Bartelme et al., 2017), drinking water distribution systems (Pinto et al., 2016; Wang Y. et al., 2017), groundwater-fed rapid sand filters (Palomo et al., 2016; Bartelme et al., 2017; Fowler et al., 2018), pipe surface (Daims et al., 2015), wastewater treatment plants (Chao et al., 2016; Camejo et al., 2017; Fan et al., 2017; Annavajhala et al., 2018; Wu et al., 2019), estuarine waters, leaf surface and agricultural soils (Orellana et al., 2018), terrestrial subsurface (Poghosyan et al., 2019), forest soils (Pjevac et al., 2017), and freshwater environments (wetlands, river waters, lake waters, aquifers and lake sediments) (Yu et al., 2018; Jiang et al., 2019). These studies have highlighted considerable phylogenetic diversity among comammox Nitrospira. So far, the existing comammox Nitrospira sequences can be assigned to two clades, designated as clades A and B (Daims et al., 2015). Clade A is further divided into clades A.1 and A.2 (Xia et al., 2018). However, current knowledge about the ecological distribution of phylotypes and their potential contribution to nitrification is still limited.
Molecular surveys of the environmental distribution of comammox Nitrospira are primarily based on PCR amplification of the amoA gene that codes for the alpha subunit of ammonia monooxygenase (AMO) (Purkhold et al., 2000; Pester et al., 2011), the key enzyme of aerobic ammonia oxidation. However, the AMO of comammox Nitrospira represents a novel type of AMO that is distinct from its counterparts among the known AOB and AOA (Pjevac et al., 2017). Thus, the established primer sets that target the amoA genes of AOB or AOA cannot be used for the detection of comammox amoA genes in environments. Consequently, several primer sets specific for comammox amoA genes have been developed, including separate primer collections for clade A and clade B of comammox (Pjevac et al., 2017), a primer pair of high coverage simultaneously targeting all comammox within Nitrospira (Fowler et al., 2018) and a two-step PCR protocol primer giving broad coverage of copper containing membrane monooxygenase genes (Wang J.G. et al., 2017). Other newly developed primers only detect a certain fraction of the comammox amoA genes within the clade A (Bartelme et al., 2017; Wang et al., 2018; Xia et al., 2018; Beach and Noguera, 2019; Zheng et al., 2019). Among them, the primer sets of Ntsp-amoA 162F/359R (Fowler et al., 2018) and comaA/B-244f/659r (Pjevac et al., 2017) are the two most widely used primers for studying the environmental distribution of comammox amoA genes (Wang Y. et al., 2017; Fowler et al., 2018; Shi et al., 2018; Roots et al., 2019; Spasov et al., 2019; Wang et al., 2019). An appropriate primer set is crucial for achieving an unbiased representation and efficient assessment of comammox community composition in various environments. However, it is not yet clear if there are major differences between these two primer sets in terms of PCR amplification efficiency and specificity.
In this study, we investigated two widely used primer sets, comaA/B-244f/659r (Pjevac et al., 2017) and Ntsp-amoA 162F/359R (Fowler et al., 2018) targeting the comammox amoA genes. We used high-throughput amplicon sequencing and metagenomics sequencing on the Illumina platforms to characterize the composition and structure of comammox community in three typical wetland soils from the Tibetan Plateau and Northeast Plain in China. The amplification efficiency and specificity of the two primer sets was evaluated based on the comparative analysis of the phylogenetic distribution of comammox amoA amplicons and amoA fragments from directly sequenced metagenomes. The aim of this study was to assess the degree of caution that should be exercised when interpreting the comammox communities results obtained from the two PCR primers (Ntsp-amoA 162F/359R and comaA/B-244f/659r).
Soil samples were collected in August 2016 from Sanjiang (SJ) wetland (47°35′N, 133°38′E) and Xianghai (XH) wetland (44°35′N, 122°33′E) in the Northeast Plain and from Sangsang (SS) wetland (29°22′N, 86°31′E) in the Qinghai-Tibet Plateau. Bulk soil at a depth of 0–10 cm were collected in triplicate from each site. Soil samples were immediately transported on ice to the laboratory and stored at −20°C. The soils were freeze-dried and passed through a 2-mm sieve and then homogenized for DNA extraction. Total DNA was extracted using a FastDNA® spin kit (MP Biomedicals, Cleveland, OH, United States) according to the manufacturer’s instructions. Quantity and concentration of soil DNA were measured by a NanoDrop ND-1000 UV–vis spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States).
Amplification of comammox amoA genes was performed using the primer mixtures comaA/B-244f/659r and the primer set of Ntsp-amoA 162F/359R (Figure 1 and Table 1) on a T100 thermal cycler (Bio-Rad, United States). A 10-bp-long unique barcode was added to the forward primers to distinguish the samples. PCRs of total comammox amoA genes (Fowler et al., 2018) were performed in triplicate at a volume of 50 μl: 25 μl of 2 × Green Taq Mix (Vazyme, Nanjing, China), 2 μl of each primer (10 μM) and 10 ng of template DNA. Thermal cycling was carried out with an initial denaturation step at 95°C for 3 min followed by 30 cycles of initial denaturation at 95°C for 30 s, annealing at 48°C for 20 s, extension at 72°C for 30 s and a final elongation step at 72°C for 5 min.
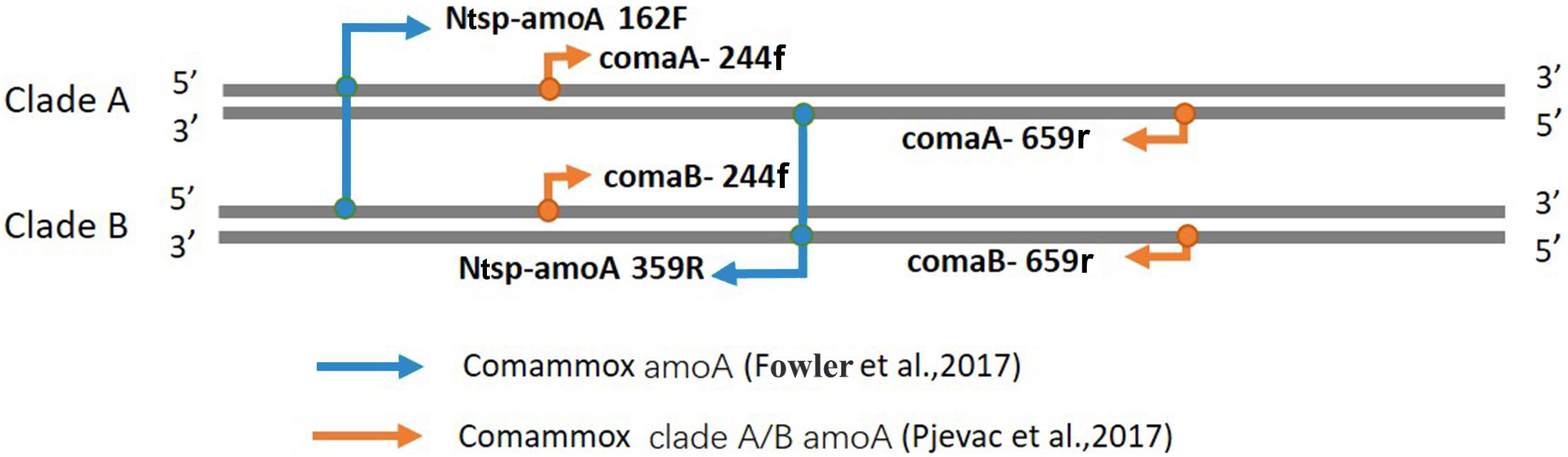
Figure 1. Visual representation of primer-binding sites with comammox clade A amoA genes and comammox clade B amoA genes. The primer set comaA/B -244f/359r, labeled with orange arrows, are located between positions 244–261 and 643–659 in comammox amoA genes. The Amplification with primers Ntsp-amoA 162F/359R (blue arrows) occurs at binding regions between positions between 162–182 and 339–359.
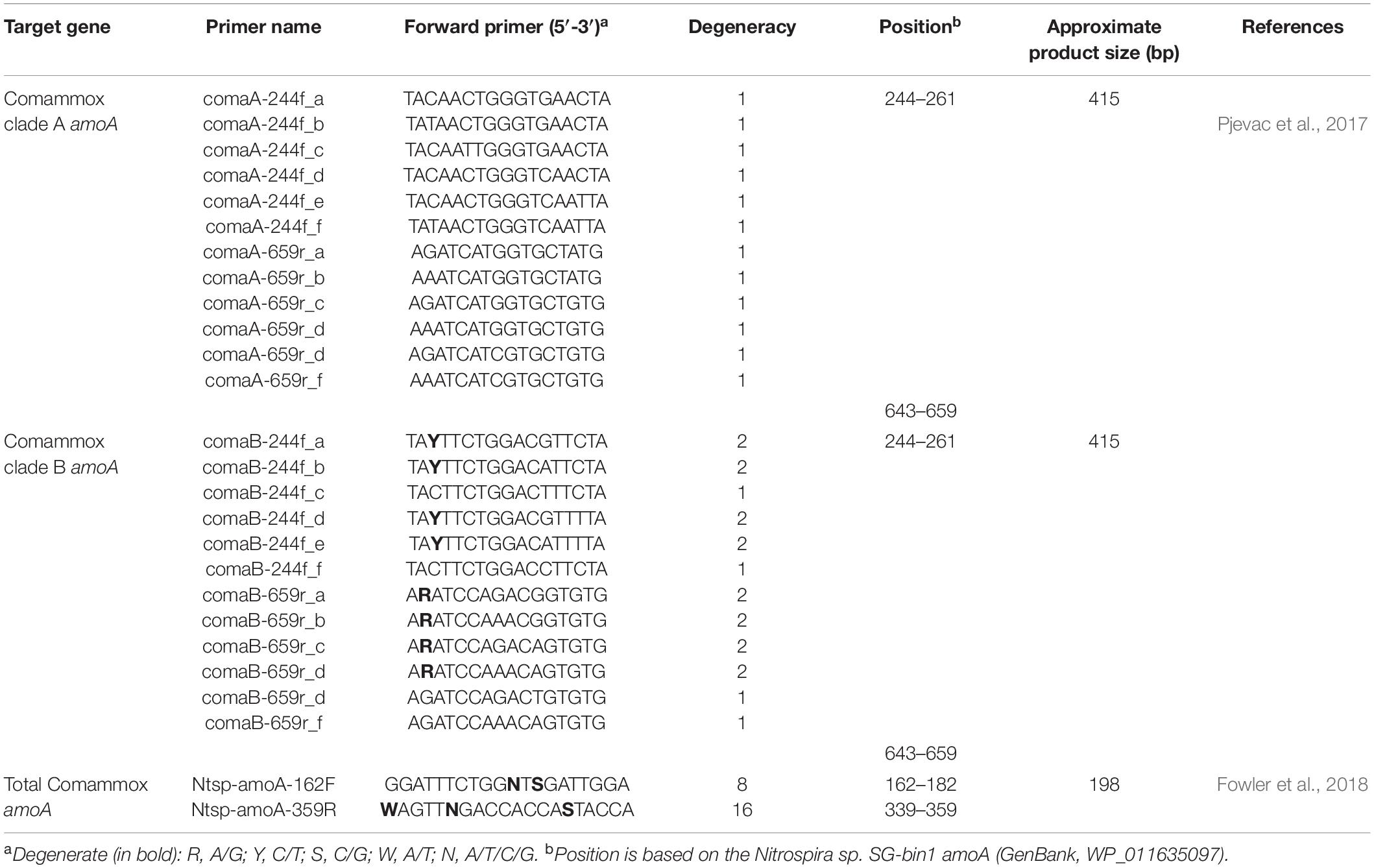
Table 1. Properties of the comammox amoA-targeted primer sets used for PCR in this study.
Amplifications of clade A and clade B comammox amoA genes (Pjevac et al., 2017) were performed with individual equimolar primer mixtures according to Pjevac et al. (2017) (Figure 1 and Table 1). The PCR reactions were performed as described above with the following thermocycling protocol: 95°C for 3 min, 30 cycles of 95°C for 15 s, 52°C for 20 s, and 72°C for 30 s, and a final elongation step 72°C for 5 min. After running on the 2% agarose gels, all PCR products of the potentially correct length were purified with an AxyPrep gel extraction kit (AxyGen, Hangzhou, China). NanoDrop ND-1000 spectrophotometer was used to quantify the purified amplicons.
The purified amplicons using primer set Ntsp-amoA 162F/359R were sequenced on an Illumina HiSeq platform (2 × 150 bp, Illumina, San Diego, CA, United States). The amplicons of the clade A and clade B comammox amoA genes using comaA/B-244f/659r were subjected to paired-end sequencing (2 × 300 bp) on an Illumina MiSeq platform (Illumina, San Diego, CA, United States) by Shanghai Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China).
Illumina data were checked for quality using FastQC1 and trimmed in Quantitative Insight into Microbial Ecology (QIIME)2 (Zhou et al., 2016), where sequences were filtered based on their Phred quality scores (Q = 20). Sequences amplified by comaA/B-244f/659r and Ntsp-amoA 162F/359R were removed if they were shorter than 300 and 150 bp, respectively. Chimeras were identified and eliminated based on both de novo and comammox amoA reference database3 (Edgar, 2010; Zhou et al., 2016). The remaining sequences were clustered into operational taxonomic units (OTUs) with UCLUST based on a 95% identity threshold. The representative sequence of each OTU was determined by blasting against NCBI-nr database, and the sequences with blast hits of comammox amoA genes or pmoA genes (potential comammox amoA gene but wrongly annotated as pmoA gene before 2015, since the two primer sets could not generate real AOA or AOB amoA genes or pmoA genes) were kept and exclusively determined as comammox amoA genes in the following phylogenetic analysis. To take advantage of the full potential of the data, Illumina data was also analyzed in QIIME 2 which provides quality control, merging of paired ends, and error correction with DADA2 (Callahan et al., 2016) to remove or correct sequencing reads with errors or chimeras and outputs the abundance of error-corrected amplicon sequence variants (ASVs).
The representative sequence of each OTU that clustered with the comammox amoA genes along with an outgroup of bacterial amoA were aligned using MEGA 6.0 (Tamura et al., 2013). Aligned sequences were employed to construct the maximum likelihood by IQ-TREE4 with 1,000 bootstrap iterations and neighbor-joining tree by MEGA 6.0 (Tamura et al., 2013).
The genomic DNA was extracted from each wetland soil as described above and sequenced on the Illumina HiSeq 4000 platform with a paired-end strategy (2 × 150 bp with an insert size of 350 bp) at Shanghai Majorbio Bio-Pharm Technology Co., Ltd (Shanghai, China). All raw reads were quality trimmed and filtered using Sickle5 with default parameters. A total of ∼40.0 Gbp of clean reads was obtained for each sample. Due to the very low coverage of each comammox genome in the metagenomic dataset, it was impossible to uncover with accuracy the community structures of comammox based on amoA genes assembled from the metagenome data [only 2–5 comammox amoA genes ≥338 bp were obtained by metagenomic assembly using metaSPAdes v3.13.0 (Nurk et al., 2017) from each sample]. Therefore, we used a hybrid annotation strategy for the short-read screening as described in Liu et al. (2020) to determine the RAs of clade A and clade B in each soil sample. Briefly, clean metagenomic reads were analyzed by BLASTx against the comammox amoA gene database from Liu et al. (2020) with E value ≤ 1 × 10–5. A read was annotated as a corresponding comammox amoA-like sequence only if its BLAST hit had ≥80% amino acid read identity for ≥25 amino acids.
The Shannon index, OTU richness (with RA ≥ 0.0%, ≥0.01%, ≥0.1%, ≥0.5%, ≥1% of the total comammox amoA sequences) and phylogenetic diversity (PD) were calculated using the QIIME package based on OTU thresholds of 95%. To compare diversity indices of wetland samples with different primer sets, one-way analysis of variance (ANOVA) was applied using Statistical Package of Social Sciences (SPSS, version 20.0). P values less than 0.05 were considered as statistically significant for all statistical test. To explore the potential similarity of sequences obtained by two primer sets, Compute Pairwise Distance with MEGA 6.0 (Tamura et al., 2013) was applied to calculated similarity of the overlapped areas from OTU representative sequences captured by different primer sets.
Amplicon data are deposited in NCBI Sequence Read Archive (SRA) under accession number PRJNA626576. Metagenome data are available from the NCBI SRA under SRR12444714, SRR12444882, and SRR12445281. The OTUs representative sequences are deposited in NCBI GenBank under accession numbers MT409044-MT409096.
Two widely used primer sets, Ntsp-amoA 162F/359R and comaA/B-244f/659r, were selected in this study. Ntsp-amoA 162F/359R contains high degeneracy (5 out of 24 base pairs) and is the first primer pair designed to simultaneously target both clade A and B comammox Nitrospira, generating a 198 bp-length fragment between the position of 162–182 and of 339–359 in comammox amoA gene (Figure 1 and Table 1). It has been widely used in comammox community analysis from various environments, such as freshwater biofilms, brackish lake sediments, rice rhizospheres, rice paddy soils, forest soils, wastewater treatment plants, and drinking water treatment plants (Wang Y. et al., 2017; Fowler et al., 2018).
The comaA/B-244f/659r primer mixtures consist of separately synthesized versions of the six respective sets of forward and reverse primers, with each primer containing only one or no base ambiguity (Table 1). They were designed to differentiate between (comaA-244f/659r for) clades A and (comaB-244f/659r for) clade B comammox amoA sequences within the Nitrospira genus (Table 1), amplifying a comammox amoA gene amplicons with a size of 415 bp at binding regions between positions 244–261 and 643–659 (Figure 1). This primer set has been used for the detection of comammox amoA genes in nitrification reactors (Roots et al., 2019), agricultural soils (Li et al., 2019; Liu et al., 2019; Wang et al., 2019), wetland (Shah and Wang, 2019), forest soil (Shi et al., 2018) and wastewater treatment plant (Spasov et al., 2019).
In order to compare the efficiency of two primer sets in amplifying comammox amoA genes and their ability to reveal the diversity of comammox community, PCR and agarose gel electrophoresis were conducted with the two primer sets for three typical wetland soils from the Tibetan Plateau and the Northeast Plain of China.
In the case of the primer set Ntsp-amoA 162F/359R, target PCR products with 198 bp length and non-specific amplification with around 500 bp length were both detected in all samples (Figure 2A). Increasing the annealing temperature could not eliminate this non-specific amplification (data not shown). This finding was similar to previous studies, showing that multiple products with correct and unexpected product sizes were both found in the agarose gel of qPCR products obtained with Ntsp-amoA 162F/359R (Beach and Noguera, 2019).
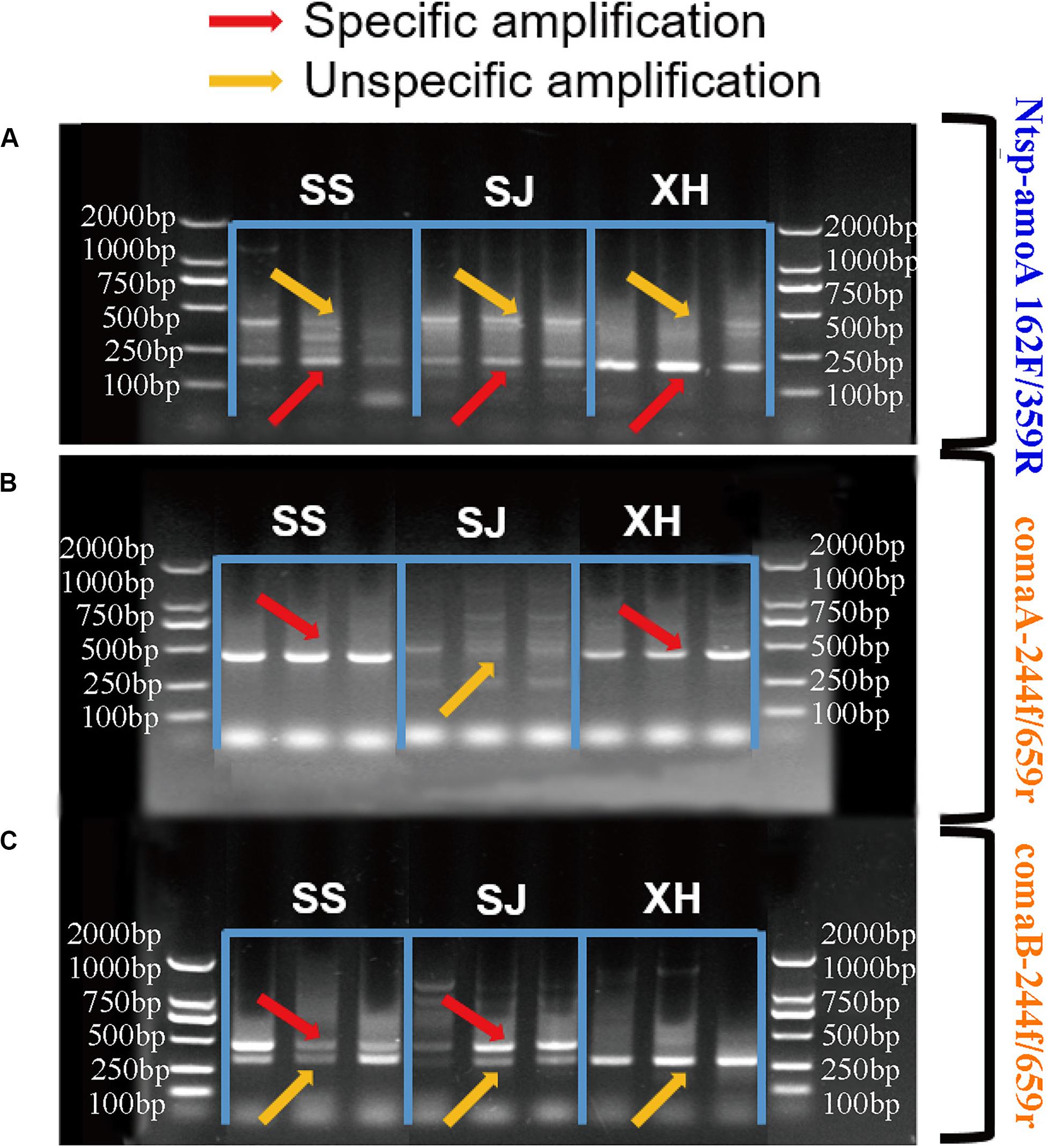
Figure 2. Agarose gel electrophoresis of PCR amplification products (A) with primer sets Ntsp-amoA 162F/359R, (B) comaA-244f/659r and (C) comaB-244f/659r in SS (Sangsang), SJ (Sanjiang) and XH (Xianghai) wetland soil samples. The red and yellow arrows represented specific amplification and non-specific amplification, respectively.
Regarding to the primer set comaA/B-244f/659r, comammox clade A amoA genes were detected in SS and XH wetland soils with high specificity (Figure 2B). The dim fragment with around 415 bp-long were observed in the SJ samples (Figure 2B), but no comammox amoA genes were identified by high-throughput sequencing from this dim fragment (Supplementary Table S3). In the clade B assay, the expected fragments were successfully amplified from SS and SJ samples with primer comaB-244f/659r (Figure 2C), but non-specific products of various sizes were also detected in the same samples (Supplementary Table S3), which was consistent with previous studies (Beach and Noguera, 2019). Primer comaB-244f/659r failed to amplify comammox clade B amoA genes in XH samples, while it produced a clear non-specific band approximately 350 bp in length, which was challenging to distinguish because it was very close to the expected 415 bp target size (Figure 2C). Numerous factors such as choice of annealing temperature, cycle times, DNA concentration and template preference might affect PCR efficiency (D’Amore et al., 2016; Gohl et al., 2016; Nilsson et al., 2018). However, after considerable efforts in adjusting the annealing temperature (varied in small increments from 48 to 60°C), concentration of template DNA (40, 20, and 1 ng DNA/reaction) and cycle times (from 30 to 35) of the PCR procedure, we still failed to get a clear target PCR band for the clade A in SJ and clade B in XH, and the non-specific products in our PCR results still could not be eliminated.
The comammox amoA gene PCR products amplified with the primer pairs of Ntsp-amoA 162F/359R and comaA/B-244f/659r were sequenced on the Illumina MiSeq and HiSeq platforms, respectively. After quality control, we obtained a total of 2,669,629 comammox amoA gene sequences (a mixture of clades A and B) with Ntsp-amoA 162F/359R, and 776,017 gene sequences of clade A and 802,915 gene sequences of clade B with comaA-244f/659r and comaB-244f/659r, respectively. Rarefaction curves showed that the sequencing depth was sufficient for diversity analysis of comammox amoA genes (Supplementary Figure S1). Each data set was then trimmed to 16,109 reads per sample for the following community composition analysis. In total, 1,115 OTUs at a cutoff of 95% sequence similarity were obtained for all comammox amoA genes with the primer pair of Ntsp-amoA 162F/359R for the three wetland soils, while 70 and 105 OTUs were detected for the clade A and clade B comammox with the primer pairs of comaA-244f/659r and comaB-244f/659r, respectively.
With primer set of Ntsp-amoA 162F/359R, the diversity indices (OTU numbers, Shannon index, phylogenetic diversity) indicated that the comammox community with the highest diversity was found in the SS site, followed by the SJ and XH sites (Figures 3A–C and Supplementary Table S1). Similar results were found with the primer set comaA/B-244f/659r (SSB > SSA > SJB > XHA) (Figures 3A–C and Supplementary Table S1), but as mentioned above, clade A and clade B failed to be amplified using comaA/B-244f/659r in SJ and XH, respectively (Figures 2B,C). However, compared to comaA/B-244f/659r, Ntsp-amoA 162F/359R tended to identify more ASVs (Supplementary Table S5) or detect a greater richness of OTUs for the common clades that both primer sets could successfully amplify (i.e., clades A and B in SS, clade B in SJ and clade A in XH) from the three wetland soils. To further test this pattern, we selected the OTUs with RA greater than 0.01, 0.1, 0.5, and 1% from the total reads of each sample, and then compared the coverage of dominant and rare OTUs of comammox amoA by using these two primer sets. Results showed that for the clades that can be captured by two primer sets, Ntsp-amoA 162F/359R had significantly higher (P < 0.05) number of OTUs (RA ≤ 0.5%) than comaA/B-244f/659r (Figure 3D). However, for the dominant OTUs with RA ≥ 1.0%, the two primer sets identified similar OTU numbers (P > 0.05) for the clade A from SS and XH and for the clade B from SJ (Figure 3D). This indicated that the two primer sets normally detected a similar number of dominant OTUs with RA ≥ 1.0%, and Ntsp-amoA 162F/359R tended to cover more rare OTUs with 0.1% ≤ RA < 0.5% than comaA/B-244f/659r.
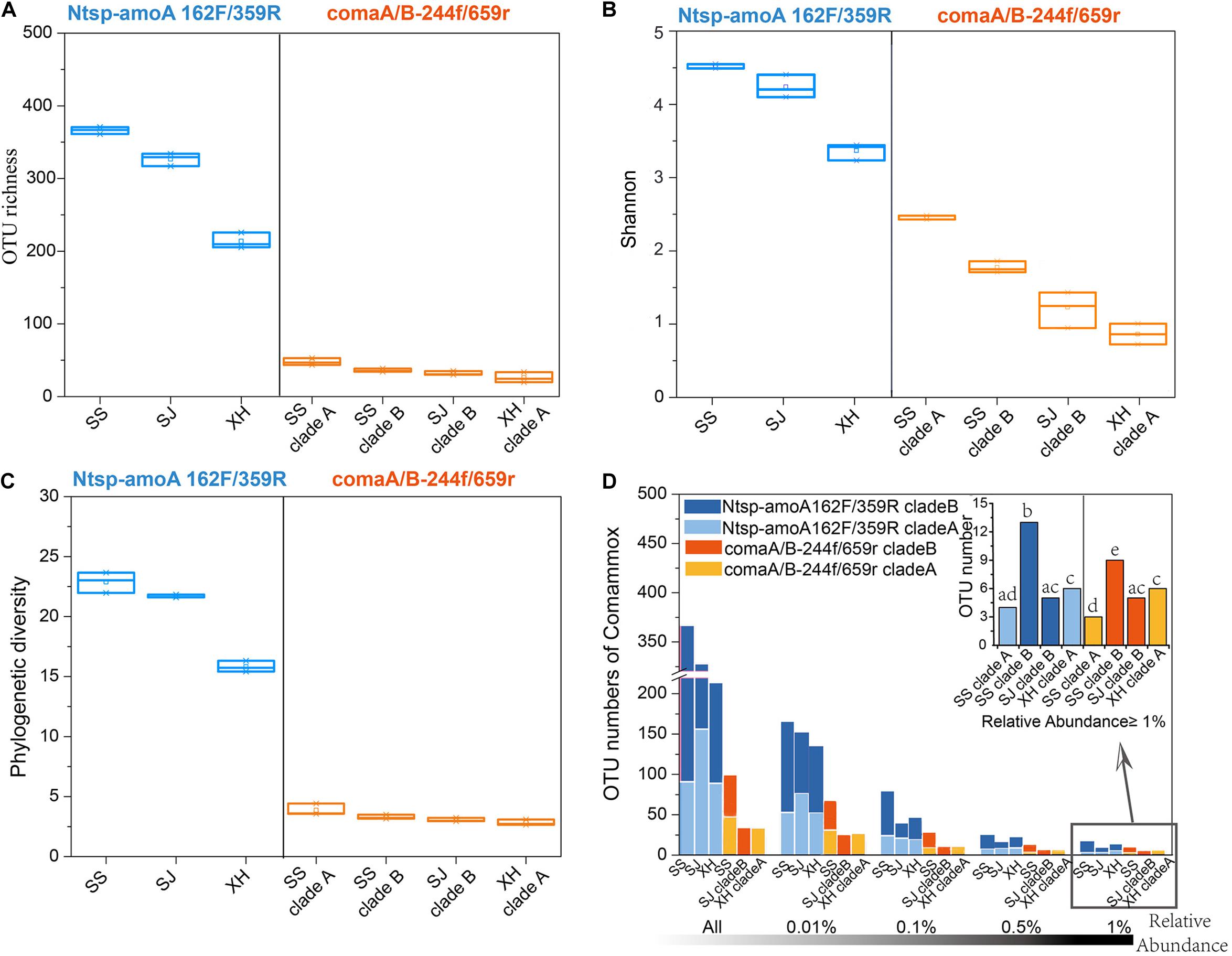
Figure 3. (A) OTU richness, (B) Shannon index, and (C) phylogenetic diversity of comammox communities in three wetland soils using primer Ntsp-amoA 162F/359R (blue) and comaA/B-244f/659r (orange). (D) OTU numbers of comammox amoA gene sequences amplified by primer Ntsp-amoA 162F/359R (blue) and comaA/B-244f/659r (orange) with RA greater than 0.01, 0.1, 0.5, and 1% in the total reads of each sample. A bar graph on the top right indicated the difference between OTU numbers of the dominant OTUs with RA ≥ 1.0% amplified by two primer sets. Bars do not share the same letter considered to be different (P < 0.05).
Phylogenetic trees of comammox amoA OTUs (RA > 0.5% of each sample) from three wetland samples obtained with the two primer sets were constructed separately using the maximum-likelihood method (Figure 4). Clearly, separation of the known clades A and B, with each clade being further divided into two monophyletic groups, sub-clades A.1, A.2 and sub-clades B.1, B.2, respectively, was observed in the phylogenetic trees (Figure 4). The division of clade B into two monophyletic groups was supported by high bootstrap values (ranging from 70 to 85%) and was also confirmed with neighbor-joining phylogenetic trees.
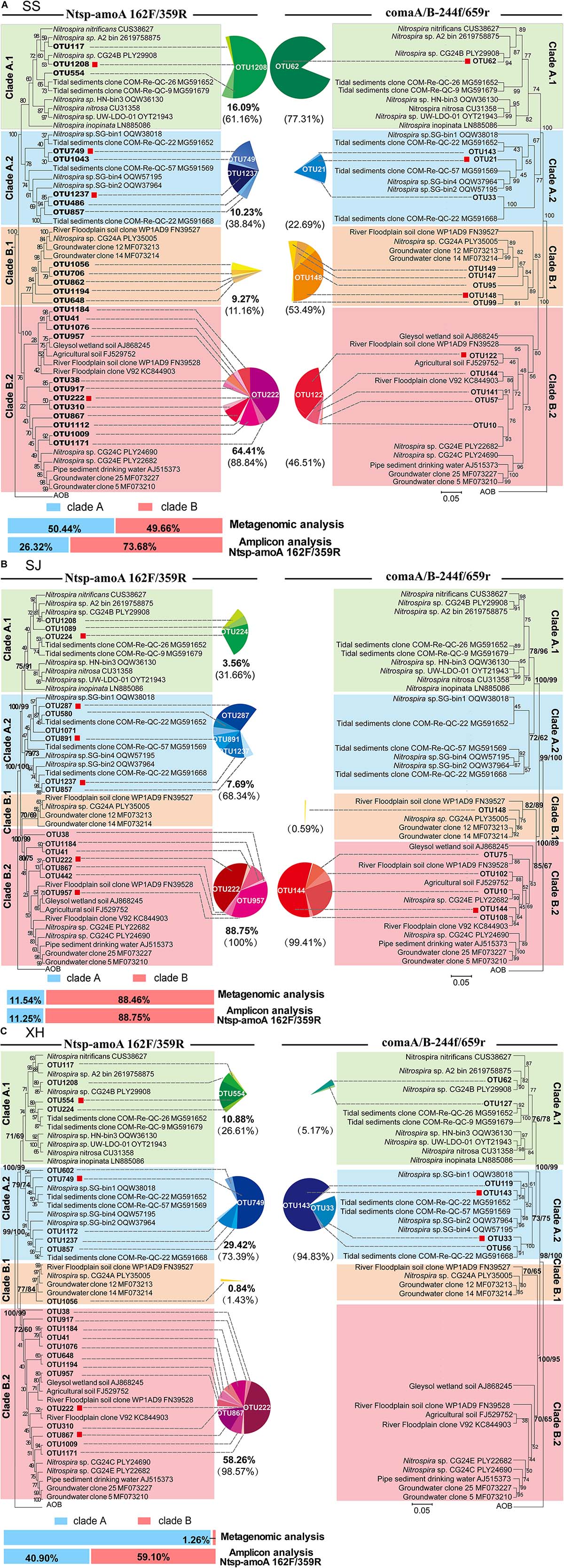
Figure 4. Maximum-likelihood phylogenetic trees based on representative sequences of the OTUs (RA > 0.5% of each sample) and 77 reference sequences using Ntsp-amoA 162F/359R (left) and comaA/B-244f/659r (right) from panel (A) SS, (B) SJ, and (C) XH samples. A Sequence from betaproteobacterial amoA was used as a outgroup. Bootstrap values in the maximum-likelihood and phylogenetic neighbor-joining trees calculated based on 1,000 trials were separated by a slash. Sequences obtained in this study were marked in bold. Red squares indicate the OTUs that its RA was larger than 0.5% of each clade. Sequences obtained from different clades are indicated by color symbols for clade A.1 (green), clade A.2 (blue), clade B.1 (orange) and clade B.2 (pink). Pie charts illustrate the proportion of OTUs in each clade with the bold data under the pie chart presenting the account of each group in the total sequences that RA was larger than 0.5%. The data in parentheses indicate the proportion of each group in clade A or B. Metagenomics and amplicon (Ntsp-amoA 162F/359R) results of clade A (blue) and B (red) proportion in each sample are in the lower-left corner.
Both clades A and B of comammox were detected in the SS samples with the two primer sets (Figure 4A). Results of Ntsp-amoA 162F/359R showed that clade B predominated at SS with 73.68% of the total comammox amoA gene sequences, within which 87.4% belonged to clade B.2. The proportion of clade A.1 was slightly higher than that of clade A.2, and there was one dominant OTU (OTU1208) in clade A.1 accounting for 52.1% of total clade A comammox amoA gene sequences in SS. Similarly, with the primer set of comaA/B-244f/659r, it showed higher proportion of clade A.1 than clade A.2 within clade A and a highly predominant OTU (OTU62) in clade A.1 (Figure 4A). The high sequence similarity (96.5%) between OTU1208 detected by Ntsp-amoA 162F/359R and OTU62 detected by comaA/B-244f/659r suggested they could most likely clustered into the same OTU (Supplementary Table S2). But unlike the results of former primer pair, the latter revealed a slightly higher proportion of clade B.1 than that of clade B.2 in the SS wetland.
As mentioned above the primer set of comaA/B-244f/659r failed to amplify clade A in SJ and clade B in XH. In contrast, both clades could be successfully amplified using the primer set of Ntsp-amoA 162F/359R (Figures 2B,C, 4B,C). However, the two primer sets demonstrated relatively similar structure patterns for the common clades (clade B in SJ and clade A in XH) that successfully amplified with both of the two primer sets (Figure 4B,C). For instance, both two primer sets revealed that, in SJ, > 99% comammox amoA gene sequences of the clade B belonged to clade B.2 that included the two most abundant OTUs (Figure 4B). Similarly, in XH, clade A.2 was revealed as the dominant lineage for the clade A comammox amoA genes using both of the two primer sets (Figure 4C). The most abundant OTU detected with Ntsp-amoA 162F/359R (OUT 749) and comaA/B-244f/659r (OUT 143) shared high sequence similarity (97.2%) in the overlapped areas (Supplementary Table S2), indicating the general consistency of two primer sets in identifying the most abundant OTU (Figure 4C).
Due to the low relative abundance of comammox in the soil microbial community in each sample, only 2–5 comammox amoA gene fragments with the length ≥338 bp were retrieved from the metagenomic data, which could not represent the diversity of comammox in each soil sample as shown above (Figure 4). Therefore, a hybrid annotation strategy was used for the short-read screening (Liu et al., 2020) to determine the relative abundances of clade A and clade B in each soil sample, and found that the relative abundance of clade A and clade B is 50.44% (74 reads) and 49.66% (73 reads) in SS (Figure 4A and Supplementary Table S4), 11.54% (3 reads) and 88.46% (23 reads) in SJ (Figure 4B and Supplementary Table S4), and 98.74% (314 reads) and 1.26% (4 reads) in XH (Figure 4C and Supplementary Table S4), respectively.
As mentioned above, comaA/B-244f/659r still failed to capture comammox amoA clade A in SJ samples and clade B in XH samples even after our great efforts in changing annealing temperature, concentration of template DNA and cycle times. Thus, we explored whether the mismatch of primer-template was the reason for the inability of comaA/B-244f/659r to amplify clade A in SJ and clade B in XH (Figures 2B,C) that, however, could be successfully amplified by the primer pair of Ntsp-amoA 162F/359R. Because there is an overlap between the PCR products obtained by the two primer sets (Figure 1), i.e., the 198 bp-long fragment generated by Ntsp-amoA 162F/359R contains the region of 244–261 bp of comammox amoA gene, which is exactly the targeted position designed for the forward primer of comaA/B-244f/659r (Figure 1). Thus, it allows us to analyze the sequence diversity of the region of 244–261 bp within 198 bp-long gene fragments amplified by Ntsp-amoA 162F/359R, comparing with the in silico coverage of forward primer of comaA/B-244f/659r.
It is odd that up to 72.03% of the 198 bp length fragments of clade A amoA gene produced by Ntsp-amoA 162F/359R in the SJ wetland harbor a region of 244–261 bp, sharing 100% sequence identity with the forward primer of comaA-244f/659r (Figure 5). Similarly, 86.41% of the 198 bp length gene fragments of clade B amoA gene amplified by Ntsp-amoA 162F/359R in XH contain a region of 244–261 bp, sharing 100% sequence identity with the forward primer of comaB-244f/659r (Figure 5). Allowing one mismatches, the forward primers comaA/B-244f could cover 100% of sequences produced by Ntsp-amoA 162F/359R. However, as mentioned above, comaA/B-244f/659r could not successfully amplify the clade A and clade B comammox amoA genes in the SJ and XH wetland, respectively (Figures 2B,C, 4C). The above results clearly suggest that partial amplification of comaA/B-244f/659r was not sorely attributed to the low in silico coverage of its forward primer mixture. Additionally, due to the limited length of PCR products of Ntsp-amoA 162F/359R, there is no information on sequence match between reverse primer of comaA/B-244f/659r and the position of 643–659 bp within the clade A/B amoA gene lineage detected by Ntsp-amoA 162F/359R.
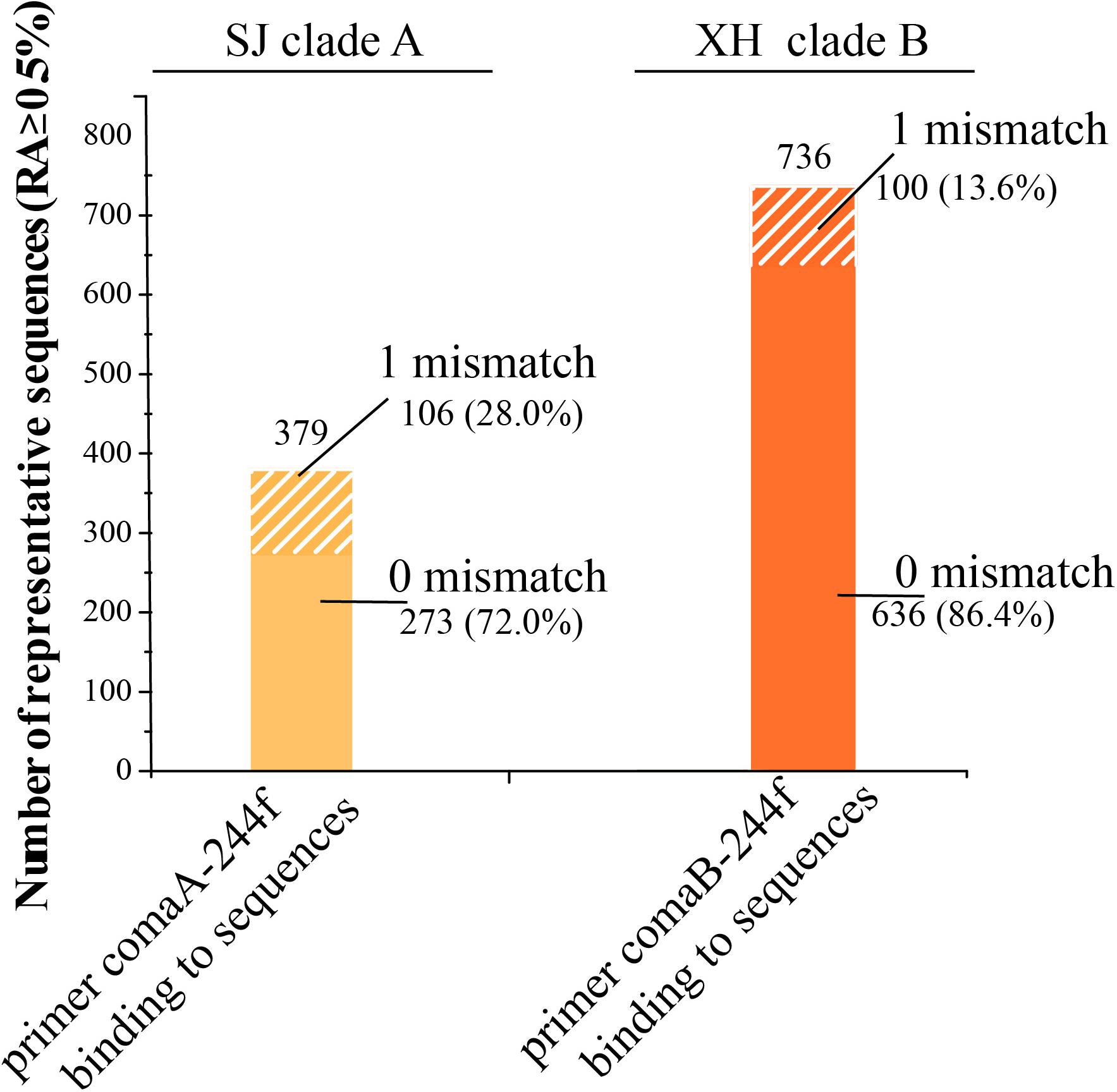
Figure 5. Primer-target mismatch analysis for former primer comaA/B-244f and the missed comammox amoA sequences (clade A in SJ and clade B in XH samples) amplified by Ntsp-amoA 162F/359R (RA > 0.5%). 0 and 1 mismatch indicate former primer comaA/B-244f binding to corresponding region in comammox amoA gene sequences amplified by Ntsp-amoA 162F/359R without base mismatch and with 1 base mismatch, respectively.
A critical step in understanding the ecological characteristics and functional importance of comammox communities is choosing suitable primers that have good coverage, high specificity, and sufficient amplification efficiency without bias. In order to provide reliable evidence for primer selection, we compared the performance of two of the most commonly used primer sets Ntsp-amoA 162F/359R and comaA/B -244f/659r for the detection of comammox communities in three typical wetland soils. Despite limited samples, we still found that two primer sets revealed a broadly similar diversity and community distribution of dominant OTUs, but there are also differences in terms of identifying the clades with relative low abundance and rare OTUs, as well as generating non-specific amplification. We then explored the possible reasons underlying the observed differences.
For the common clades that could successfully be amplified by both sets of primers (i.e., clade A and B in SS, clade B in SJ, and clade A in XH) from the three wetland soils, it was not surprising that the number of dominant OTUs obtained by Ntsp-amoA 162F/359R and comaA/B-244f/659r was similar (Figure 3D). However, larger OTU numbers, hence greater diversity indices were obtained when detecting with Ntsp-amoA 162F/359R compared to comaA/B-244f/659r (Figures 4A–C). Since all the amoA gene reads were strictly trimmed and quality-controlled before OTU clustering, and all of rare OTUs (0.1% ≤ RA < 0.5%) captured by Ntsp-amoA 162F/359R or by comaA/B-244f/659r contained more than 48 sequences, it appears that these rare OTUs were real biological sequences and not produced by sequencing errors. Furthermore, this conclusion was further supported by the analysis based on ASVs (Nearing et al., 2018; Forster et al., 2019), which also demonstrated much more ASVs were obtained by Ntsp-amoA 162F/359R than by comaA/B-244f/659r (Supplementary Table S5).
A reasonable supposition is that the different sequence variance of regions amplified by the two primer sets may have affected the OTU clustering and subsequent diversity analysis. Even if the sequences were the same, OTU numbers obtained in different amplified regions might be different due to their different average genetic distance. Therefore, we searched all the comammox amoA genes with length ≥503 bp from the NCBI-nr database covering both regions amplified by two primer sets to analyze the diversity of 198 bp length, 415 bp length amplification regions and 503 bp length regions. A total collection of 115 sequences of clade A and 112 sequences of clade B was then clustered into OTUs with 95% sequence similarity for the OTUs comparison analysis. We found that the number of OTUs (both clades A and B) among the two amplicon regions and the whole gene length showed little differences, indicating that it was not the differently amplified regions of the two primer sets resulted in higher diversity of the amoA gene sequences captured by Ntsp-amoA 162F/359R (Figure 6). Although only 237 sequences were insufficient to represent the global diversity of comammox amoA genes, we regarded this method as a random sampling. We also recognize that more comammox amoA gene sequences are required to further confirm our findings.
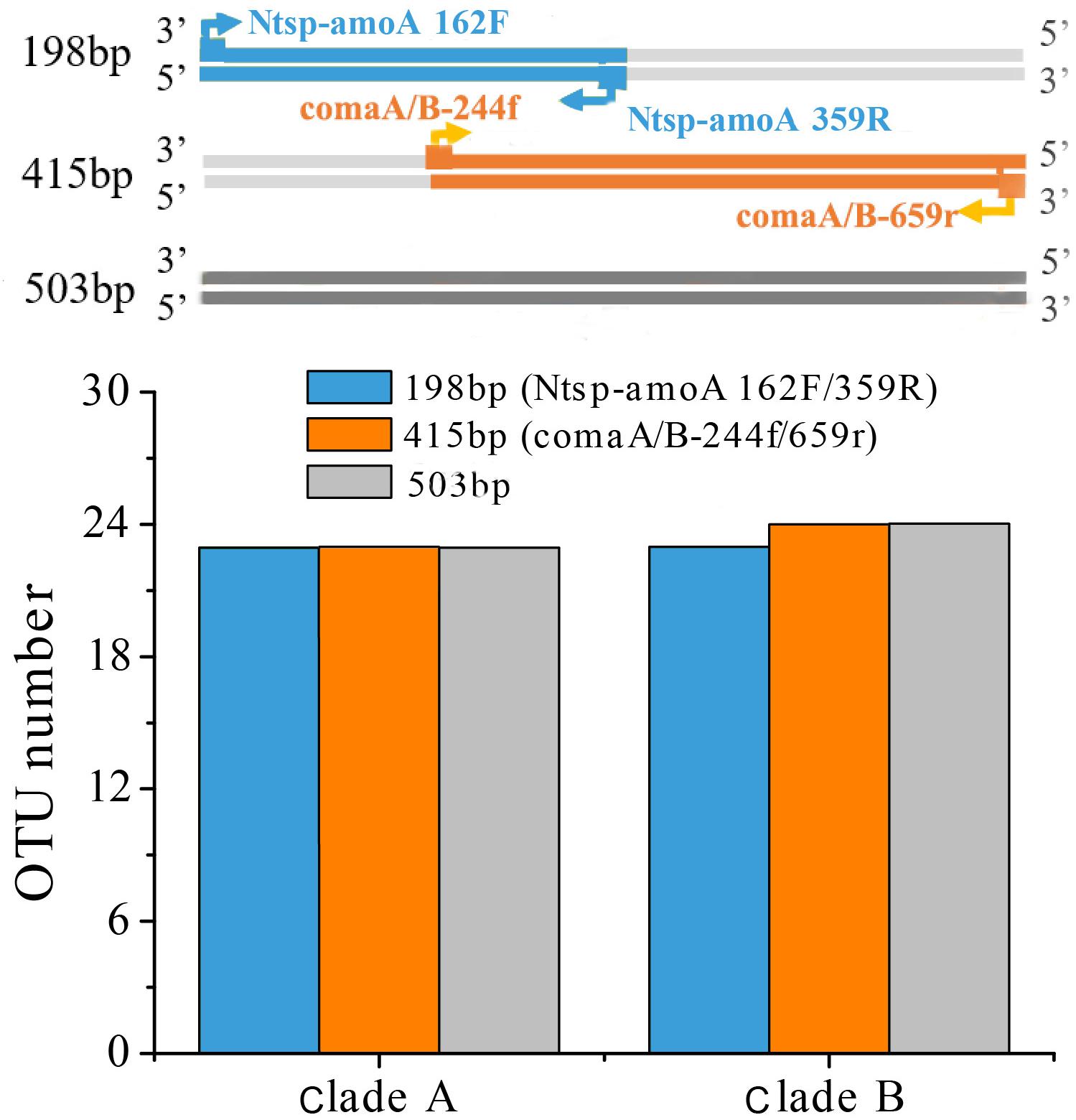
Figure 6. OTU cluster analysis with 95% similarity cutoffs of comammox amoA gene sequences for clade A (115 sequences) and B (112 sequences) with length ≥503 bp from the NCBI-nr database covering both regions amplified by two primer sets at 198 bp-length (Ntsp-amoA 162F/359R), 415 bp-length (comaA/B-244f/659r) amplification regions and 503 bp-length regions.
For the failure of comaA/B-244f/659r to amplify some comammox amoA clades in SJ and XH, metagenomic results suggested that this may be due to the low-abundance of target genes in wetland soil samples. For example, clade A in SJ and clade B in XH that were not detected with comaA/B-244f/659r, accounted for only 11.54 and 1.26% of the total comammox amoA genes in each sample, respectively, according to metagenomic results (Figures 4B,C), indicating that the unsuccessful amplification might be attributed to the low abundance of target clades. By contrast, Ntsp-amoA 162F/359R could detect these low-abundance clades, suggesting its potential advantages of identifying rare OTUs. Wang et al. (2019) reported a similar phenomenon that primer sets comaA/B -244f/659r failed to amplify comammox amoA for most DNA samples from fertilized soil where the abundance of comammox amoA genes ranged from 1.3 × 105 to 5.5 × 105 copies/g soil yielded with the primer set Ntsp-amoA 162F/359R (Wang et al., 2019). In addition, primer-target mismatch is likely another factor to prevent detection of clade A in SJ and clade B in XH. Silico analysis indicated that, compared to forward primer comaA/B-244f, 27.97% of clade A comammox amoA genes in SJ and 13.59% of clade B in XH captured by Ntsp-amoA 162F/359R contained one mismatch (Figure 5). A single mismatch in a primer sequence can affect the efficiency of PCR amplification and even lead to an underestimation of the gene copy number by 1,000 times (Bru et al., 2008). But given that majority of missed sequences shared 100% exact match with forward primer comaA/B-244f, primer-target mismatch might not the main cause. Moreover, the impact of reverse primers could not be ruled out as we were not able to analyze the silico binding region of reverse primers. But due to the limitation in sample type and numbers, this conclusion warrants further investigation.
It is worth noting that although the RA of each clade obtained with Ntsp-amoA 162F/359R was consistent with that retrieved from metagenome data in SJ samples (Figure 4B), Ntsp-amoA 162F/359R vastly overestimated the abundance of comammox clade B in XH samples. Comparison of metagenomic and amplicon data showed a dramatic increase in the RA of comammox clade B from 1.26% (metagenomic results) to 59.70% of comammox amoA genes in XH samples (Figure 4C). The overestimation of comammox clade B also appeared in SS samples, where clade B accounted for 73.68% with primer set Ntsp-amoA 162F/359R, whereas actually 49.66% of comammox amoA genes was detected as clade B using PCR-free metagenomic analysis (Figure 4A). Therefore, the results of relative abundance for clades A and B comammox amoA genes obtained by Ntsp-amoA 162F/359R should be regarded with caution unless verified by metagenomic analysis of the same samples.
Despite that comammox amoA genes can be successfully amplified in some samples using comaA/B -244f/659r and Ntsp-amoA 162F/359R, these two primers yielded non-specific amplification in all our tests, which could not be eliminated by adjusting annealing temperature, concentration of template DNA and cycle numbers of PCR procedure. This observation was consistent with previous studies (Pjevac et al., 2017; Beach and Noguera, 2019). The ubiquitous non-specific amplification suggested that we should be careful to avoid overestimation of comammox amoA genes when performing qPCR with the two primer sets. One possible reason for non-specific amplification was the high similarity between target fragments and some other naturally occurring gene fragments in the microbial community, and that both of them can match to the primer binding sites (Supplementary Table S3). Another possible reason would be the presence of ambiguous bases in the primer sets that could impair PCR specificity (Linhart and Shamir, 2002). For example, only one small non-target PCR band was observed (Figure 2B) in three wetland soil samples with primer sets comaA -244f/659r for clade A (without any ambiguous bases). While, several non-target PCR bands were observed with primer sets comaB -244f/659r (with one ambiguous base) for clade B and Ntsp-amoA 162F/359R (with five ambiguous bases) (Figures 2A,C).
Taken together, our study clearly demonstrated that the two widely used primer sets of comammox amoA genes performed equally in terms of revealing the composition of the clades with relative high abundance and showing similar distribution of the dominant OTUs in the wetland soil samples. The primer pair comaA/B-244f/659r has prominent advantage in the aspect of studying the separate clade of comammox amoA genes and in generating fewer non-target gene sequences. In comparison, Ntsp-amoA 162F/359R has better performance for capturing comammox clades with low RA and for revealing more information about rare OTUs in community diversity. However, it is also more prone to forming non-specific PCR-band and non-target gene sequences via Illumina sequencing, and often greatly overestimating the clades with low RA. Due to the generally very low abundance of comammox in soil microbial community, the metagenome sequencing as deep as 40 Gbp could only assemble a few long comammox amoA gene fragments from each sample, screening of the short reads proved to be a reliable alternative strategy for revealing the true community structures of comammox in each sample. All considered, we urge caution in interpreting results based on these primers and suggest the development of more efficient and specific primer sets, as well as the necessary verification work. Future studies may also consider the potential benefits of using PCR-free deep metagenomic sequencing to investigate highly diverse comammox communities in natural environments.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MT409044–MT409096; https://www.ncbi.nlm.nih.gov/, SRR12444714, SRR12444882, and SRR12445281.
CL wrote the manuscript. WQ, HX, and BJ revised the manuscript. SX analyzed experimental results. XT carried out the experiments. LK and XW analyzed sequencing data. JC assisted with Illumina sequencing. JS and JA revised the manuscript. HQ designed the experiments. BW substantial contributed to the conception and design. All authors contributed to the article and approved the submitted version.
This work was supported by the State Key Laboratory of Subtropical Silviculture (No. KF2017-02) and the National Science Foundation of China (Nos. 41977056, 41671252, 31971631, and 41701269).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Michael Wagner from University of Vienna for the valuable discussion. We thank Yu Shi and Haiyan Chu for assistance in wetland soil sampling. We are grateful to two reviewers and editor for constructive and very helpful comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.560942/full#supplementary-material
Annavajhala, M. K., Kapoor, V., Santo-Domingo, J., and Chandran, K. (2018). Comammox functionality identified in diverse engineered biological wastewater treatment systems. Environ. Sci. Technol. Lett. 5, 110–116. doi: 10.1021/acs.estlett.7b00577
Bartelme, R. P., McLellan, S. L., and Newton, R. J. (2017). Freshwater recirculating aquaculture system operations drive biofilter bacterial community shifts around a stable nitrifying consortium of ammonia-oxidizing archaea and comammox nitrospira. Front. Microbiol. 8:101. doi: 10.3389/fmicb.2017.00101
Beach, N. K., and Noguera, D. R. (2019). Design and assessment of species-level qpcr primers targeting comammox. Front. Microbiol. 10:36. doi: 10.3389/fmicb.2019.00036
Bru, D., Martin-Laurent, F., and Philippot, L. (2008). Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl. Environ. Microbiol. 74, 1660–1663. doi: 10.1128/Aem.02403-07
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). Dada2: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Camejo, P. Y., Santo Domingo, J., McMahon, K. D., and Noguera, D. R. (2017). Genome-enabled insights into the ecophysiology of the comammox bacterium “Candidatus Nitrospira nitrosa”. mSystems 2:e00059-17. doi: 10.1128/mSystems.00059-17
Chao, Y., Mao, Y., Yu, K., and Zhang, T. (2016). Novel nitrifiers and comammox in a full-scale hybrid biofilm and activated sludge reactor revealed by metagenomic approach. Appl. Microbiol. Biotechnol. 100, 8225–8237. doi: 10.1007/s00253-016-7655-9
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by Nitrospira bacteria. Nature 528, 504–509. doi: 10.1038/nature16461
D’Amore, R., Ijaz, U. Z., Schirmer, M., Kenny, J. G., Gregory, R., Darby, A. C., et al. (2016). A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics 17:55. doi: 10.1186/s12864-015-2194-9
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Fan, X. Y., Gao, J. F., Pan, K. L., Li, D. C., and Dai, H. H. (2017). Temporal dynamics of bacterial communities and predicted nitrogen metabolism genes in a full-scale wastewater treatment plant. RSC Adv. 7, 56317–56327. doi: 10.1039/C7RA10704H
Forster, D., Lentendu, G., Filker, S., Dubois, E., Wilding, T. A., and Stoeck, T. (2019). Improving edna-based protist diversity assessments using networks of amplicon sequence variants. Environ. Microbiol. 21, 4109–4124. doi: 10.1111/1462-2920.14764
Fowler, S. J., Palomo, A., Dechesne, A., Mines, P. D., and Smets, B. F. (2018). Comammox Nitrospira are abundant ammonia oxidizers in diverse groundwater-fed rapid sand filter communities. Environ. Microbiol. 20, 1002–1015. doi: 10.1111/1462-2920.14033
Galloway, J. N., Townsend, A. R., Erisman, J. W., Bekunda, M., Cai, Z., Freney, J. R., et al. (2008). Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320, 889–892. doi: 10.1126/science.1136674
Gohl, D. M., Vangay, P., Garbe, J., MacLean, A., Hauge, A., Becker, A., et al. (2016). Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 34, 942–949. doi: 10.1038/nbt.3601
Hu, H. W., and He, J. Z. (2017). Comammox—a newly discovered nitrification process in the terrestrial nitrogen cycle. J. Soils Sedim. 17, 2709–2717. doi: 10.1007/s11368-017-1851-9
Jiang, Q., Xia, F., Zhu, T., Wang, D., and Quan, Z. (2019). Distribution of comammox and canonical ammonia-oxidizing bacteria in tidal flat sediments of the Yangtze River estuary at different depths over four seasons. J. Appl. Microbiol. 127, 533–543. doi: 10.1111/jam.14337
Könneke, M., Bernhard, A. E., de la Torre, J. R., Walker, C. B., Waterbury, J. B., and Stahl, D. A. (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437, 543–546. doi: 10.1038/nature03911
Li, C. Y., Hu, H. W., Chen, Q. L., Chen, D. L., and He, J. Z. (2019). Comammox Nitrospira play an active role in nitrification of agricultural soils amended with nitrogen fertilizers. Soil Biol. Biochem. 138:107609. doi: 10.1016/j.soilbio.2019.107609
Linhart, C., and Shamir, R. (2002). The degenerate primer design problem. Bioinformatics 18, S172–S181. doi: 10.1093/bioinformatics/18.suppl_1.S172
Liu, S., Wang, H., Chen, L., Wang, J., and Ni, J. (2020). Comammox nitrospira within the yangtze river continuum: community, biogeography, and ecological drivers. ISME J. 14, 2488–2504. doi: 10.1038/s41396-020-0701-8
Liu, T., Wang, Z., Wang, S., Zhao, Y., Wright, A. L., and Jiang, X. (2019). Responses of ammonia-oxidizers and comammox to different long-term fertilization regimes in a subtropical paddy soil. Eur. J. Soil Biol. 93:103087. doi: 10.1016/j.ejsobi.2019.103087
Nearing, J. T., Douglas, G. M., Comeau, A. M., and Langille, M. G. I. (2018). Denoising the denoisers: an independent evaluation of microbiome sequence error- correction approaches. Peerj 6:e5364. doi: 10.7717/peerj.5364
Nilsson, R. H., Anslan, S., Bahram, M., Wurzbacher, C., Baldrian, P., and Tedersoo, L. (2018). Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 17, 95–109. doi: 10.1038/s41579-018-0116-y
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). Metaspades: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Orellana, L. H., Chee-Sanford, J. C., Sanford, R. A., Loffler, F. E., and Konstantinidis, K. T. (2018). Year-round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl. Environ. Microbiol. 84:e01646-17. doi: 10.1128/AEM.01646-17
Palomo, A., Jane Fowler, S., Gulay, A., Rasmussen, S., Sicheritz-Ponten, T., and Smets, B. F. (2016). Metagenomic analysis of rapid gravity sand filter microbial communities suggests novel physiology of Nitrospira spp. ISME J. 10, 2569–2581. doi: 10.1038/ismej.2016.63
Pester, M., Rattei, T., Flechl, S., Gröngröft, A., and Wagner, M. (2011). amoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions. Environ. Microbiol. 42, 525–539. doi: 10.1111/j.1462-2920.2011.02666.x
Pinto, A. J., Marcus, D. N., Ijaz, U. Z., Bautista-de Lose Santos, Q. M., Dick, G. J., and Raskin, L. (2016). Metagenomic evidence for the presence of comammox nitrospira-like bacteria in a drinking water system. mSphere 1:e00054-15. doi: 10.1128/mSphere.00054-15
Pjevac, P., Schauberger, C., Poghosyan, L., Herbold, C. W., van Kessel, M., Daebeler, A., et al. (2017). AmoA-targeted polymerase chain reaction primers for the specific detection and quantification of comammox nitrospira in the environment. Front. Microbiol. 8:1508. doi: 10.3389/fmicb.2017.01508
Poghosyan, L., Koch, H., Lavy, A., Frank, J., van Kessel, M., Jetten, M., et al. (2019). Metagenomic recovery of two distinct comammox Nitrospira from the terrestrial subsurface[J]. Environ. Microbiol. 21, 3627–3637. doi: 10.1111/1462-2920.14691
Purkhold, U., Pommereningröser, A., Juretschko, S., Schmid, M. C., Koops, H. P., and Wagner, M. (2000). Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl. Environ. Microbiol. 66, 5368–5382. doi: 10.1128/AEM.66.12.5368-5382.2000
Roots, P., Wang, Y., Rosenthal, A. F., Griffin, J. S., Sabba, F., Petrovich, M., et al. (2019). Comammox Nitrospira are the dominant ammonia oxidizers in a mainstream low dissolved oxygen nitrification reactor. Water Res. 157, 396–405. doi: 10.1016/j.watres.2019.03.060
Shah, P., and Wang, Z. W. (2019). Using digital polymerase chain reaction to characterize microbial communities in wetland mesocosm soils under different vegetation and seasonal nutrient loadings. Sci. Total Environ. 689, 269–277. doi: 10.1016/j.scitotenv.2019.06.305
Shi, X., Hu, H.-W., Wang, J., He, J.-Z., Zheng, C., Wan, X., et al. (2018). Niche separation of comammox Nitrospira and canonical ammonia oxidizers in an acidic subtropical forest soil under long-term nitrogen deposition. Soil Biol. Biochem. 126, 114–122. doi: 10.1016/j.soilbio.2018.09.004
Spasov, E., Tsuji, J. M., Hug, L. A., Doxey, A. C., Sauder, L. A., Parker, W. J., et al. (2019). Comammox bacteria are dominant ammonia oxidizers in tertiary rotating biological contactors of a municipal wastewater treatment plant. bioRxiv [Preprint]. doi: 10.1101/529826
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
van Kessel, M. A., Speth, D. R., Albertsen, M., Nielsen, P. H., Op den Camp, H. J., Kartal, B., et al. (2015). Complete nitrification by a single microorganism. Nature 528, 555–559. doi: 10.1038/nature16459
Wang, J., Wang, J., Rhodes, G., He, J. Z., and Ge, Y. (2019). Adaptive responses of comammox Nitrospira and canonical ammonia oxidizers to long-term fertilizations: implications for the relative contributions of different ammonia oxidizers to soil nitrogen cycling. Sci. Total Environ. 668, 224–233. doi: 10.1016/j.scitotenv.2019.02.427
Wang, J. G., Xia, F., Zeleke, J., Zou, B., Rhee, S. K., and Quan, Z. X. (2017). An improved protocol with a highly degenerate primer targeting copper-containing membrane-bound monooxygenase genes for community analysis of methane- and ammonia-oxidizing bacteria. FEMS Microbiol. Ecol. 93:fiw244. doi: 10.1093/femsec/fiw244
Wang, Y., Ma, L., Mao, Y., Jiang, X., Xia, Y., Yu, K., et al. (2017). Comammox in drinking water systems. Water Res. 116, 332–341. doi: 10.1016/j.watres.2017.03.042
Wang, M., Huang, G., Zhao, Z., Dang, C., Liu, W., and Zheng, M. (2018). Newly designed primer pair revealed dominant and diverse comammox amoA gene in full-scale wastewater treatment plants. Bioresour. Technol. 270, 580–587. doi: 10.1016/j.biortech.2018.09.089
Wu, L., Shen, M., Li, J., Huang, S., Li, Z., Yan, Z., et al. (2019). Cooperation between partial-nitrification, complete ammonia oxidation (comammox), and anaerobic ammonia oxidation (anammox) in sludge digestion liquid for nitrogen removal. Environ. Pollut. 254:112965. doi: 10.1016/j.envpol.2019.112965
Xia, F., Wang, J. G., Zhu, T., Zou, B., Rhee, S. K., and Quan, Z. X. (2018). Ubiquity and diversity of complete ammonia oxidizers (Comammox). Appl. Environ. Microbiol. 84:e01390-18. doi: 10.1128/AEM.01390-18
Yu, C., Hou, L., Zheng, Y., Liu, M., Yin, G., and Gao, J. (2018). Evidence for complete nitrification in enrichment culture of tidal sediments and diversity analysis of clade a comammox Nitrospira in natural environments. Appl. Microbiol. Biotechnol. 102, 9363–9377. doi: 10.1007/s00253-018-9274-0
Zheng, M., Wang, M., Zhao, Z., Zhou, N., He, S., Liu, S., et al. (2019). Transcriptional activity and diversity of comammox bacteria as a previously overlooked ammonia oxidizing prokaryote in full-scale wastewater treatment plants. Sci. Total Environ. 656, 717–722. doi: 10.1016/j.scitotenv.2018.11.435
Keywords: comammox, amoA, Ntsp-amoA 162F/359R, comaA/B-244f/659r, PCR
Citation: Lin C, Xu H, Qin W, Xu S, Tang X, Kuang L, Wang X, Jiang B, Chen J, Shan J, Adams J, Qin H and Wang B (2020) Evaluation of Two Primer Sets for Amplification of Comammox Nitrospira amoA Genes in Wetland Soils. Front. Microbiol. 11:560942. doi: 10.3389/fmicb.2020.560942
Received: 11 May 2020; Accepted: 10 September 2020;
Published: 30 September 2020.
Edited by:
Aymé Spor, INRA UMR 1347 Agroécologie, FranceReviewed by:
Arnaud Dechesne, Technical University of Denmark, DenmarkCopyright © 2020 Lin, Xu, Qin, Xu, Tang, Kuang, Wang, Jiang, Chen, Shan, Adams, Qin and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Qin, cWluaHVhQHphZnUuZWR1LmNu; Baozhan Wang, Ynp3YW5nQG5qYXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.