 Mohamed M. H. Abdelbary1,2
Mohamed M. H. Abdelbary1,2 Edward J. Feil3
Edward J. Feil3 Laurence Senn1Christiane Petignat1Guy Prod’hom4
Laurence Senn1Christiane Petignat1Guy Prod’hom4 Jacques Schrenzel5Patrice François5
Jacques Schrenzel5Patrice François5 Guido Werner6
Guido Werner6 Franziska Layer6
Franziska Layer6 Birgit Strommenger6
Birgit Strommenger6 Annalisa Pantosti7
Annalisa Pantosti7 Monica Monaco7
Monica Monaco7 Olivier Denis8,9Ariane Deplano8Hajo Grundmann10
Olivier Denis8,9Ariane Deplano8Hajo Grundmann10 Dominique S. Blanc1*
Dominique S. Blanc1*- 1Service of Hospital Preventive Medicine, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 2Division of Oral Microbiology and Immunology, Department of Operative Dentistry, Periodontology and Preventive Dentistry, RWTH Aachen University Hospital, Aachen, Germany
- 3The Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, United Kingdom
- 4Institute of Microbiology, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 5Bacteriology Laboratory, Division of Infectious Diseases, Geneva University Hospitals, Geneva, Switzerland
- 6National Reference Centre for Staphylococci and Enterococci, Division of Nosocomial Pathogens and Antibiotic Resistances, Department of Infectious Diseases, Robert Koch Institute, Wernigerode, Germany
- 7Department of Infectious, Parasitic and Immune-Mediated Diseases, Istituto Superiore di Sanità, Rome, Italy
- 8National Reference Centre-Staphylococcus aureus, Department of Microbiology, Hôpital Erasme, Université libre de Bruxelles, Brussels, Belgium
- 9Laboratory of Microbiology, CHU UCL Namur, Université catholique de Louvain, Yvoir, Belgium
- 10Department of Infectious Diseases Epidemiology, The University of Groningen, Groningen, Netherlands
Background: Methicillin-resistant Staphylococcus aureus (MRSA) is a common healthcare-associated pathogen that remains a major public health concern. Sequence type 228 (ST228) was first described in Germany and spread to become a successful MRSA clone in several European countries. In 2000, ST228 emerged in Lausanne and has subsequently caused several large outbreaks. Here, we describe the evolutionary history of this clone and identify the genetic changes underlying its expansion in Switzerland.
Materials and Methods: We aimed to understand the phylogeographic and demographic dynamics of MRSA ST228/ST111 by sequencing 530 representative isolates of this clone that were collected from 14 European countries between 1997 and 2012.
Results: The phylogenetic analysis revealed distinct lineages of ST228 isolates associated with specific geographic origins. In contrast, isolates of ST111, which is a single locus variant of ST228 sharing the same spa type t041, formed a monophyletic cluster associated with multiple countries. The evidence points to a German origin of the sampled population, with the basal German lineage being characterized by spa type t001. The highly successful Swiss ST228 lineage diverged from this progenitor clone through the loss of the aminoglycoside-streptothricin resistance gene cluster and the gain of mupirocin resistance. This lineage was introduced first in Geneva and was subsequently introduced into Lausanne.
Conclusion: Our results reveal the radiation of distinct lineages of MRSA ST228 from a German progenitor, as the clone spread into different European countries. In Switzerland, ST228 was introduced first in Geneva and was subsequently introduced into Lausanne.
Introduction
Staphylococcus aureus colonizes the anterior nares of about 30% of healthy humans. However, methicillin-resistant Staphylococcus aureus (MRSA) remains a major global cause of healthcare associated infections (Stefani et al., 2012; Aanensen et al., 2016). In addition, infections due to MRSA are associated with higher mortality rates than infections caused by methicillin-susceptible strains and result in increased lengths of hospital stays as well as associated health care costs (Barrasa-Villar et al., 2017; Hassoun et al., 2017). Successful reduction of MRSA infection rates depends on preventing MRSA transmission and detecting and containing outbreaks (Byrne and Wilcox, 2011). Understanding the settings and circumstances under which MRSA spread in the hospital setting and in the community is central to design new strategies to reduce transmission.
Molecular typing methods play an important role in understanding the epidemiology of MRSA and tracking different outbreaks (Urwin and Maiden, 2003). For example, the multilocus sequence typing (MLST) method have assigned most of MRSA isolates into a limited number of clonal lineages (Robinson and Enright, 2003). In addition, it was previously shown that certain MRSA clones have disseminated worldwide, while others were found in geographically limited regions (Monecke et al., 2011). However, the epidemiology of MRSA is dynamic, and clonal replacement of predominant clones within a given hospital has been widely reported (Kuhn et al., 2007; Albrecht et al., 2011; Campanile et al., 2015; Zarfel et al., 2016). In contrast to MLST, whole-genome sequencing (WGS) provides high discriminatory power that reveals the diversity among isolates of the same clone (Sequence Type). Such data are proving very powerful for epidemiological surveillance on continental scales down to a single hospital, and are informing more efficient infection control (Aanensen et al., 2016; Raven et al., 2019).
Methicillin-resistant Staphylococcus aureus-ST228 was first described in Germany in 1992, and was initially called the southern German or Italian clone (Witte et al., 1997; Witte, 1999; Monecke et al., 2011). This clone first emerged in the Lausanne university hospital in 2000 and caused a long-term outbreak that lasted between 2008 and 2012. Senn et al. recently used WGS to study this outbreak and argued that carriers of this clone may have gone unrecognized in the hospital due to an atypically high rate of enteric carriage, combined with high transmissibility (Vogel et al., 2012; Senn et al., 2016).
In this study, we significantly expand on this work, and place the Lausanne outbreak within a phylogeographic context, by generating WGS data for 302 additional isolates representing a wide temporal and geographic spread. We combined these data with those of Senn et al. (2016) to give a total dataset of 530 isolates. These data point to a German origin of this clone and reveal detailed geographic and temporal information on subsequent inter- and intra-national spread and significant genomic events occurring during this spread.
Results
Phylogenetic Reconstruction of the ST228
We analyzed genome sequences of 530 MRSA isolates that were collected from health-care institutions of 14 European countries between 1997 and 2012 (Supplementary Table S1). In silico MLST analysis revealed that 487 isolates were assigned to ST228, while 43 belonged to ST111 (a single locus variant of ST228 sharing the same t041 spa type) (Supplementary Table S1). Mapping the 530 MRSA genomes to the N315 reference genome (accession number NC_002745.2) and subsequent Gubbins analysis identified 16 diverse genes exhibiting between 4 and 17 SNPs (Supplementary Table S2). These included surface antigens such as spa, adhesins such as clfA, clfB, ssrA, and MBL fold metallo-hydrolase genes. It is likely that this diversity reflects a history of duplication/deletion or recombination at these loci.
The phylogenetic analysis identified 3,899 SNPs within the core-genomes of the 530 isolates, after exclusion of mobile genetic elements (MGEs), recombinant and repetitive regions. These core-genome SNPs were used to reconstruct the phylogeny of all ST228 and ST111 isolates using a maximum-likelihood (ML) approach (Figure 1). Based on the phylogenetic tree, isolates were clustered into nine distinct clades that broadly correlated with geographical origin. The ST111 isolates formed a distinct cluster that was not obviously associated with a single geographic origin. The basal clade consisted exclusively of German ST228 isolates with spa type t001 (n = 18). Other clades of ST228 isolates also showed geographical association based on the country of origin. For example, the European clade included the Italian ST228 isolates (n = 41), which form two distinct sub-clades; 2 Polish isolates and 7 Austrian isolates are closely related to these Italian isolates. All Belgian ST228 isolates (n = 12) made up a distinct cluster among the European clade (Figure 1). Interestingly, the Swiss ST228 isolates (n = 402) corresponded to a monophyletic cluster that was divided into six clades (Geneva 1999, Geneva 2000, Lausanne 2001, Geneva 2001–2006, Geneva 2007–2012, and Lausanne 2008–2012) with a basal clade composed of Geneva isolates (Geneva 1999). Only three Swiss isolates were in other clusters. This suggests that the rate of transmission of MRSA ST228 into, and out of, Switzerland has been relatively low. Two Swiss isolates correspond to ST111 (La233 and Vd466) and clustered in clade ST111, and one isolate La005 clustered in the European clade close to an Italian isolate IT705. Of note, the epidemiological data revealed that isolate La005 was retrieved from a patient who was transferred from Italy. Clade Lausanne 2008–2012 (Figure 1) consists of isolates that were previously investigated and represent the Lausanne outbreak involving over 1600 patients from 2008 to 2012 (Senn et al., 2016). The phylogenetic structure of this clade showed 7 branches with low level of variation that was concordant with the previous observations, which suggested a clonal dissemination of this lineage within the university hospital of Lausanne (Senn et al., 2016). In contrast to ST228, the ST111 clade had a weak signal of geographical clustering, suggesting frequent transmission of ST111 strains between European countries.
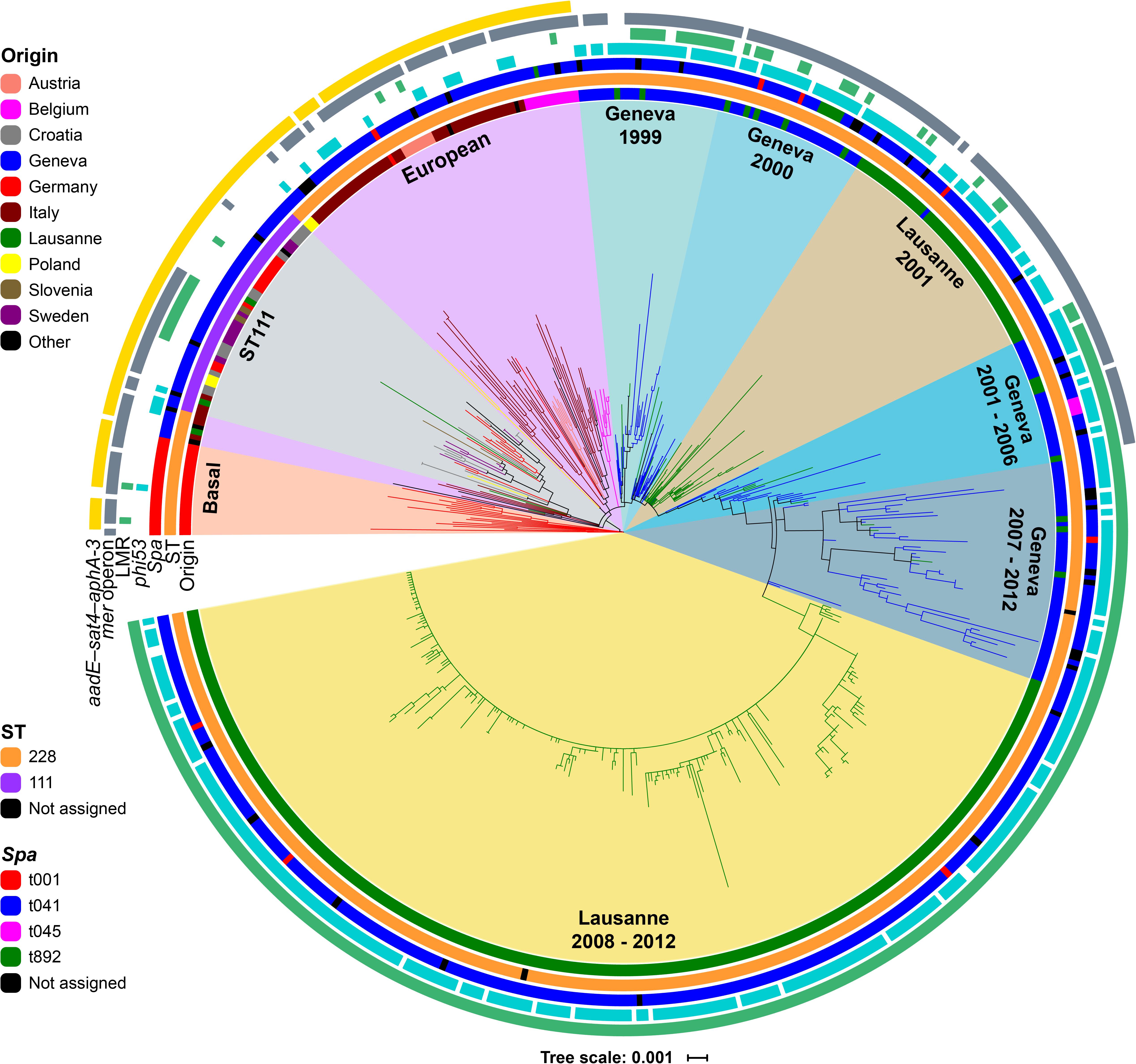
Figure 1. Rooted maximum likelihood (ML) phylogenetic tree of the 530 ST228 (n = 485) and ST111 (n = 43) MRSA isolates based on 3,899 core-genomes SNPs. The tree was rooted by using the distantly related S. aureus N315 as an outgroup. Branch colors represent country/cities of isolation. Rings from inner to outer; first ring (Origin) represent the geographical origin, second ring (ST) is assigned to the different STs, third ring (spa), fourth ring (phi53) detected, fifth ring (LMR) mutation responsible for the low level mupirocin resistance phenotype, sixth ring (mer operon) shows the isolates harboring the mercury operon gene cluster, and seventh ring (aadE-sat4-aphA-3) represents the acquisition of the gene cluster.
Genomic Diversity and Antibiotic Resistance
Mobile genetic elements are a substantial constituent of S. aureus genomes, and they may mediate the emergence of new MRSA lineages (Jamrozy et al., 2017). Pan-genome analysis detected a total of 3,271 genes among the 530 MRSA isolates, 2,531 (77%) of these were core genes (present in >95% of the isolates) while the remaining 740 genes (23%) are variably present or absent and hence correspond to the accessory genome (Supplementary Figure S1). Furthermore, 609 of the accessory genes were present in <15% of the isolates.
The number of putative prophages identified with each genome ranged from three to nine, only three of these were intact prophages and these were the most abundant. The presence of φ53 prophage (accession number NC_007049) was detected in 71% (n = 377) of isolates, which was mainly associated with the six Swiss ST228 clades (n = 356) (Figure 1), while φJB prophage (accession number KT344895) was linked to the non-Swiss isolates. A previous study showed that φ53 and φJB were genetically distinct and φJB prophage was able to transfer antibiotic resistance plasmids at higher frequencies compared with φ53 prophage during transduction experiments (Varga et al., 2016). φPT1028-like prophage (accession number NC_007045.1) was present in all the genomes so it was probably present in the common ancestor of the ST228/111 clone.
All the antimicrobial resistance and virulence genes detected using the approaches described in Methods are listed in Supplementary Table S3. Remarkably, all the Swiss isolates lacked the ant(6)-Ia, aph(3)-III and sat4 genes (the aminoglycoside-streptothricin resistance gene cluster) that encodes resistance to kanamycin/amikacin and streptothricin, while they were present in all remaining European ST228 and ST111 isolates. This gene cluster was part of a transposon structure, Tn5405, found previously on the chromosomes of staphylococci and other enterococci (Derbise et al., 1996; Werner et al., 2001). However, the bifunctional aminoglycoside modifying enzyme gene [aac(6′)-Ie-aph(2″)-Ia] was detected in all isolates (Supplementary Table S3). This gene confers high-level resistance to a wide range of aminoglycosides including gentamicin but, excluding streptomycin (Livermore, 2000).
Forty percent of isolates harbor the plasmid pTW20_1 that was previously described in MRSA ST239 (Holden et al., 2010). The pTW20_1 sequence was assembled on two contigs, one contig included the qacA gene (confers resistance to detergents and disinfectants: quaternary ammonia compounds – a marker for hospital adaptation) (Costa et al., 2013) and cadmium resistance operon (cadA and cadD), while the second contig carried genes encoding the mercury resistance operon (merA, merB, merR, merT). A variant of pTW20_1 that lacked the mer operon contig was detected in 55% (n = 296) of the isolates, the majority of which originated from Switzerland (230 from Lausanne and 39 from Geneva) and correspond to the clades Geneva 2007–2012 and Lausanne 2008–2012. However, this pTW20_1 variant (that lacked the mer operon) was detected in 27 isolates that were located in the Basal, the ST111 and the European clades and were collected from Germany (n = 11), Italy (n = 7), Croatia (n = 6), Sweden (n = 2), and Poland (n = 1) (Figure 1). These 27 isolates were collected between 1997 and 2012 and the oldest isolate (DE802) was collected in Germany during 1997 (Supplementary Table S1). We detected the β-lactamase operon (bla) in all 530 MRSA genomes that composed of blaZ, blaR1 and blaI genes, which was inserted between qacA gene and cadmium resistance operon. Furthermore, mapping the genomes against the plasmid pTW20_1 sequence (29,585 bp; accession number FN433597) confirmed that those genomes which contained this variant (lacking the mer operon) had only 74% of aligned bases to the plasmid sequence, while the other genomes that carried this operon had an average of 96% aligned bases. Hence, these findings suggest that ST228 acquired two plasmid variants on different occasions and these lineages were circulating in the Swiss hospitals. A similar observation was previously described for MRSA ST239, where pTW20_1 was detected among three phylogenetic clades (Tong et al., 2015). Two of these clades carried pTW20_1 with mer operon, while the third clade carried the pTW20_1 variant that lacked mer operon.
The point mutation (V588F) in the chromosomal isoleucyl-tRNA synthase gene (ileS) that encodes low-level mupirocin resistance (LMR) was detected in 65% (n = 344) of isolates. Among these isolates 95% (n = 326) were of Swiss origin that were collected between 2001 and 2012 and distributed over the six Swiss phylogenetic clades (Figure 1). The entire clades Geneva 2001–2006, Geneva 2007–2012, and Lausanne 2008–2012 were composed of isolates with LMR, with the exception of isolate Ge348 that was collected in Geneva during 2004 (Figure 1). Furthermore, the point mutation V588F was detected in 18 non-Swiss isolates that were collected between 1999 and 2011 (Supplementary Table S3). The iles-2 gene encoding high-level mupirocin resistance was detected in 26 isolates that were all from Switzerland.
Demography and Phylogeographic Spread of ST228
The root-to-tip regression analysis of all 530 genomes, and the sub-sample of 245 genomes (2,870 SNPs) both revealed a strong temporal signal (R2 = 0.68 and 0.58, respectively; p < 0.0001) (Supplementary Figure S2). Furthermore, we tested several demographic models on the sub-sample of 245 genomes and compared their marginal likelihood estimation (MLE). This revealed that the Hasegawa-Kishino-Yano (HKY) substitution model with relaxed uncorrelated molecular clock and a Bayesian skyline coalescent was the most supported combination (Supplementary Table S4). We estimated a mean mutation rate of 1.87 × 10–6 (95% highest posterior density interval (HPD), 1.63 × 10–6 to 2.13 × 10–6) substitutions per site per year, which is highly consistent with previous estimates for CC5 strains from the Western Hemisphere and other S. aureus clones (Glaser et al., 2016; Uhlemann et al., 2017; Challagundla et al., 2018). Based on this rate, the time of most common recent ancestor (TMRCA) of ST228 was estimated as 1988 (95% HPD, 1983 to 1992), and ST111 is estimated to have emerged in 1999 (95% HPD, 1998 to 2000). The TMRCA of the Swiss ST228 dates to 1998 (95% HPD, 1997 to 1999), and it was introduced first in Geneva and one month later it was circulated in Lausanne (Figure 2).
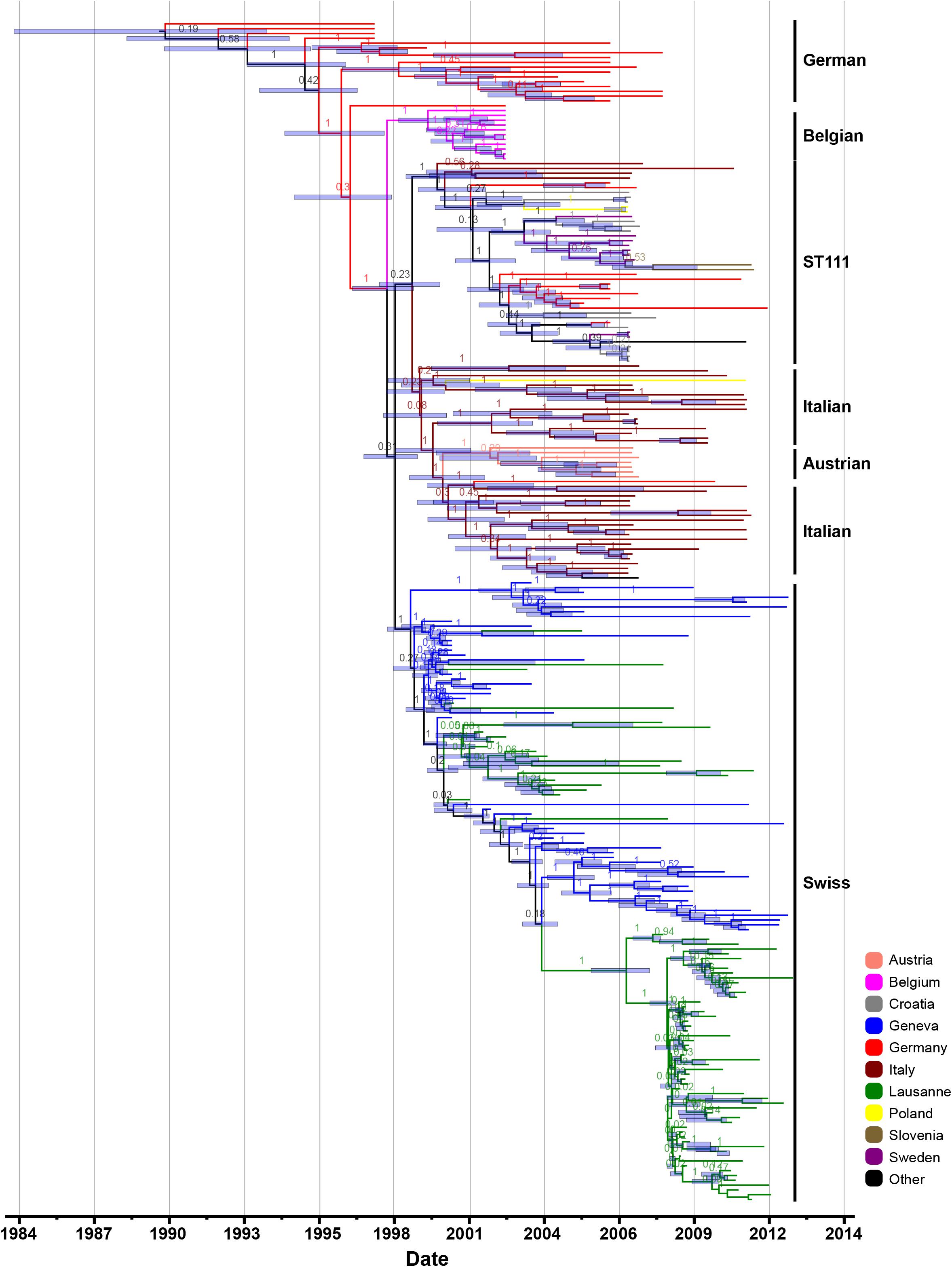
Figure 2. Maximum clade credibility (MCC) tree. Maximum clade credibility Bayesian phylogenetic tree reconstruction of the selected subset of 245 MRSA ST228 and ST111 MRSA genomes that were collected between 1993 and 2012. Tips of the tree are embarrassed by isolation dates; the time scale is shown at the base of the tree and countries/cities of origin are highlighted in different colors.
We then dated the acquisition and loss of various MGEs associated with antibiotic resistance. Our analysis suggests that the aminoglycoside-streptothricin resistance gene cluster was lost in 1989 (95% HPD, 1984 to 1993), while the acquisition of pTW20_1 plasmid variant (lacking the mer operon) and LMR were gained in 1990 (95% HPD, 1986 to 1993) and 1994 (95% HPD, 1991 to 1995), respectively.
The Bayesian skyline plot points to a steady effective population size of ST228 up until the first introduction into Switzerland in 1998 (Figure 3). Subsequently, a one order of magnitude increase in the ST228 population size was observed, which corresponds to the dissemination and expansion in Switzerland during the first outbreak in Geneva in 1999. The population size of ST228 continued to increase until a peak in around 2004, representing the different epidemic episodes in Geneva and Lausanne (Figure 3). During 2006, a sharp decline in ST228 population size was detected followed by a bottleneck in 2007. The introduction of ST228 isolates that lacked the mer operon into Switzerland gave rise to the last epidemic wave that lasted until 2012, when the ST228 population size became steady once again. These findings are concordant with our previous study (Senn et al., 2016), along with epidemiological data, which point to a decrease in MRSA ST228 incidence in Lausanne from 2005 till 2008, and the absence of large outbreaks in Switzerland after 2012. The inclusion of additional isolates from Geneva and Lausanne in the current study enabled a precise estimation of the demographic expansion of ST228 in Switzerland and revealed important details regarding the underlying genetic changes that are likely to have contributed to this spread.
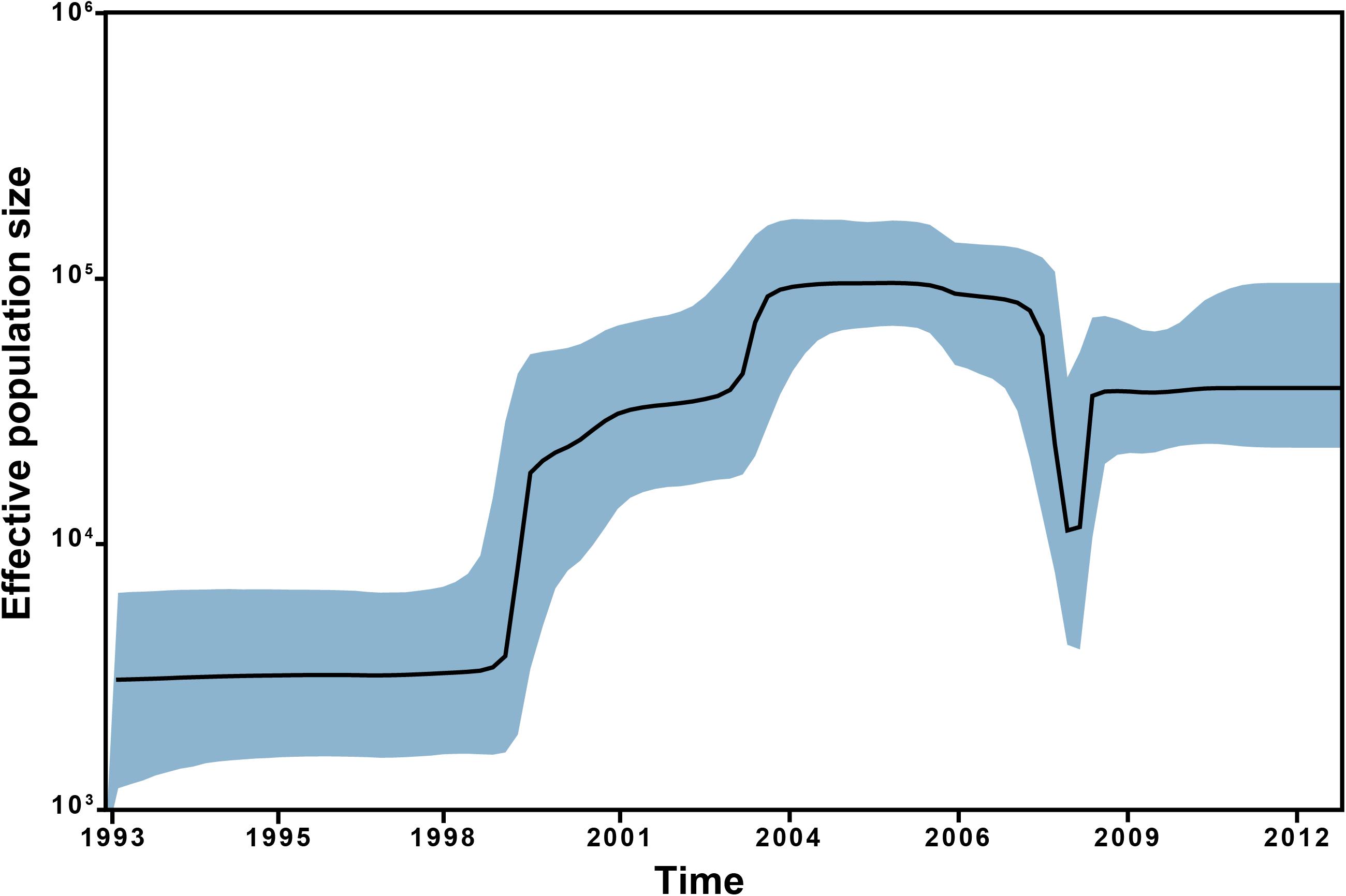
Figure 3. Bayesian Skyline plot of the analyzed selected subset of 245 MRSA ST228 and ST111 MRSA genomes. Bayesian Skyline plot with a relaxed molecular clock representing the effective population size of the ST228 over time with the shaded areas representing 95% confidence intervals.
The Bayesian phylogeographical analysis confirmed that Germany was the source of ST228, and that descendants of this progenitor populations were introduced into multiple countries. The spatial phylogenetic reconstruction (Figure 4) supports transmission from Germany to Bulgaria [Posterior probability (PP) = 1]. A direct introduction from Germany into Italy was observed but weakly supported (PP = 0.36) by the Bayesian stochastic search variable selection (BSSVS) analysis. Furthermore, we detected significant evidence of demographic expansion of ST228 from Geneva into Lausanne (PP = 0.97). In addition, transmission events from Austria, Belgium, Italy, and Germany into Lausanne were also statistically supported (PP = 0.98, 0.76, 0.99, and 0.67, respectively). These data implicate that ST228 has been introduced into Lausanne on at least five separate occasions.
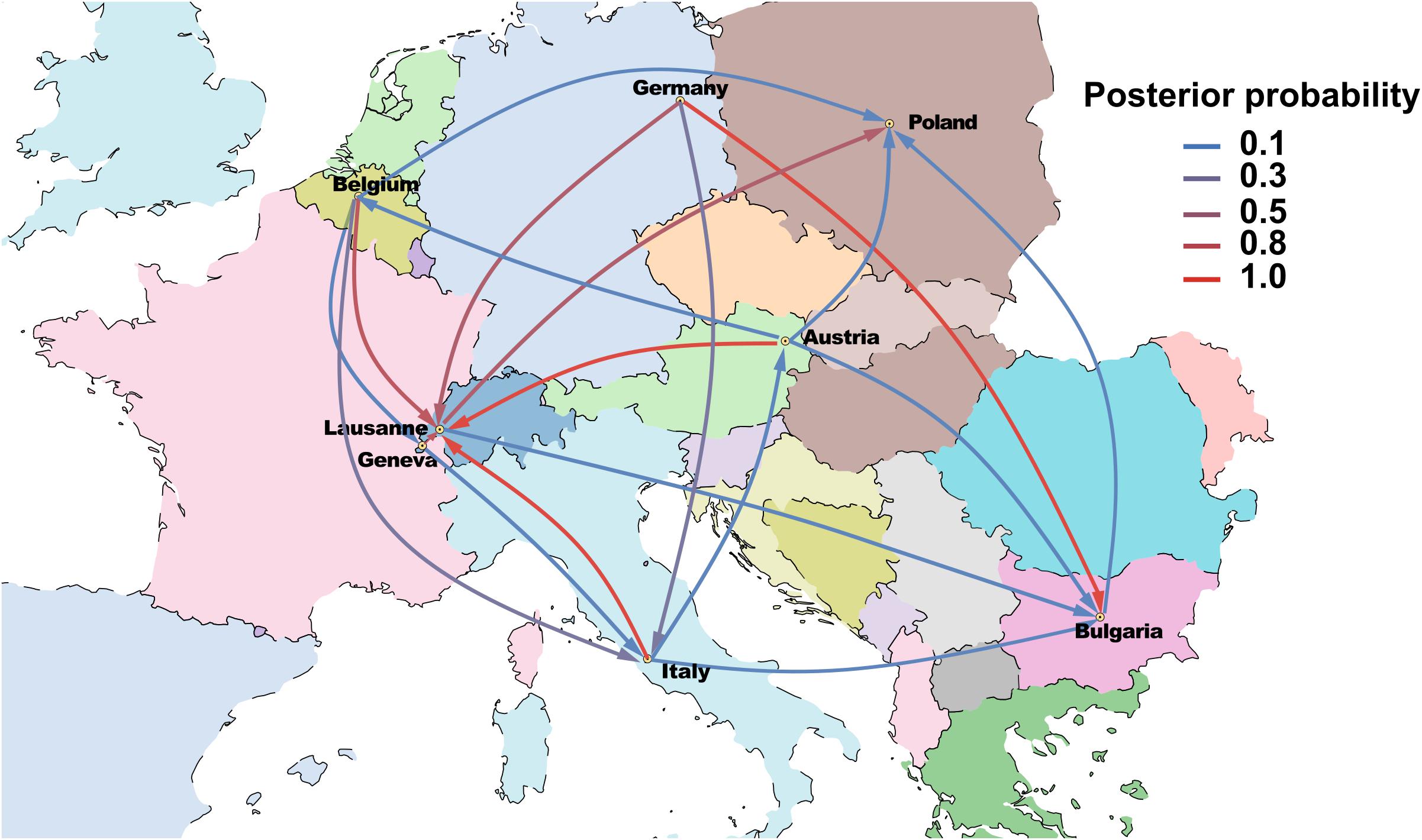
Figure 4. Phylogeographic reconstruction of ST228 representing the various transmission events among European countries. Lines are colored according to posterior probability support and arrows represent the general direction of the expansions.
Discussion
In this study, we described the core-genome variation and the accessory gene profiles of 530 isolates of ST228/111 representing 14 European countries and 15 years of clonal spread. The WGS data provided detailed evidence concerning the epidemiological and evolutionary dynamics of this clone and dated key transmission events from the original progenitor population in Germany.
Our analyses show that recombination played a limited role in the evolutionary dynamics of ST228, and that the majority of the core genome variation detected arose instead through de novo mutation. These findings agree with previous study that estimated a low ratio of recombination to mutation events (0.0046) among strains of CC5, which included ST228 and ST111 (Challagundla et al., 2018). Exceptions included genes encoding surface proteins such as spa, clfA and clfB, which is consistent with the view that such genes are subject to diversifying selection pressure. High rates of homologous recombination have previously been observed in S. aureus adhesion genes (clfA and clfB) (Kuhn et al., 2006; Senn et al., 2016) and in surface antigens such as spa (Santos-Júnior et al., 2016).
Our pan-genome analysis revealed that several ST228 lineages have recently acquired or lost MGEs. Notably, all the Swiss isolates lacked the aminoglycoside-streptothricin resistance gene cluster [ant(6)-Ia, aph(3)-III and sat4]. The loss of this cluster may be explained by the fact that aminoglycosides are primarily used to treat infections caused by Gram-negatives and are rarely used for treating MRSA. The selective pressure to maintain the ant(6)-Ia, aph(3)-III and sat4 gene cluster will be further reduced by the fact that an alternative gene cluster, aac(6′)-Ie-aph(2″)-Ia, which confers high-level resistance aminoglycosides (including gentamicin) was present in all of the isolates. We note that our Bayesian analysis revealed that the loss of the ant(6)-Ia, aph(3)-III and sat4 gene cluster occurred in 1989 (95% HPD, 1984 to 1993).
Our data also reveal that the related clusters Geneva 2007–2012 and Lausanne 2008–2012 have lost the pTW20_1 plasmid, which harbors the mer operon (conferring resistance to mercury) and which is present in the majority of strains from various other origins (Figure 1). The strains in these clusters instead harbor a distinct variant of this plasmid which does not harbor the mer operon. However, this plasmid variant (lacking the mer operon) was detected in 27 non-Swiss isolates that were collected between 1997 and 2012 and located in the Basal, ST111 and European clades. Furthermore, our analysis revealed that the loss of this operon occurred in 1990 (95% HPD, 1986 to 1993), suggesting that the acquisition of this plasmid variant occurred prior to the introduction into Switzerland.
A third change in resistance profile evidenced from our data concerns the low-level mupirocin resistance (LMR), which is conferred by the V588F mutation in the ileS gene. The phylogenetic tree (Figure 1) shows that it arose on several occasions during the evolution of the ST228 clone; the oldest LMR isolate (DE805) being of German origin and collected during 1999. However, the great majority of isolates in the three monophyletic clades Geneva 2001–2006, Geneva 2007–2012 and Lausanne 2008–2012 showed this mutation. As argued by Senn et al. (2016) the presence of LMR probably resulted from the topical use of mupirocin for the decolonization of MRSA carriers in both hospitals and thus can help to explain this sustained outbreaks. It is interesting to note that both clades Geneva 2007–2012 and Lausanne 2008–2012 lacked the mer operon and showed the LMR mutation, suggesting that both mutations were at the origin of a successful lineage responsible for the two largest outbreaks in Geneva and Lausanne.
The mutation rate estimated here of 1.87 × 10–6 (95% HPD, 1.63 × 10–6 to 2.13 × 10–6) substitutions per site per year is highly consistent with previous estimates of the Swiss ST228 outbreak and with other MRSA STs (Harris et al., 2013; Hsu et al., 2015; Senn et al., 2016). In addition, a previous study that investigated the evolution of 598 CC5 genomes in the Western Hemisphere estimated three mutations rates for the entire CC5 [1.55 × 10–6 (95% HPD, 1.39 × 10–6 to 1.71 × 10–6), 1.52 × 10–6 (95% HPD, 1.36 × 10–6 to 1.68 × 10–6) and 1.55 × 10–6 (95% HPD, 1.39 × 10–6 to 1.70 × 10–6)], which were comparable to our estimation (Challagundla et al., 2018). The Bayesian skyline plot indicates that the expansion of the ST228 population was mainly due to its dissemination in Switzerland. In addition, the skyline analysis revealed a sharp decline of ST228 population size from 2006 to 2007, reflecting the spatiotemporal change of this clone within central Europe. This finding agrees with previous studies that reported a decreasing incidence of ST228. For example, a decreased in the incidence of ST228 in German hospitals was reported since 2001 (Werner et al., 2004; Albrecht et al., 2011; Layer et al., 2012, 2018). In Italy, the replacement of ST228 by ST22-IV clone was documented during 2000–2007 (Baldan et al., 2012).
Our Bayesian phylogeographic analysis traced the progenitor population of ST228 to Germany in 1988 (95% HPD, 1983 to 1992), and that the other lineages subsequently diverged from this population. This result is consistent with the original nomenclature of ST228 as southern German epidemic strain (Witte et al., 1997). Challagundla et al. shown that European strains of ST228 and ST111 represented early-branching of CC5 clade with SCCmec I and the TMRCA was estimated as the early 1970s [1973 (95% HPD, 1986 to 1993)]. However, the aforementioned study included only five ST228 and four ST111 strains and the estimated TMRCA represented strains of ST5 (n = 135), which might explain the divergence between our TMRCA estimate and that of by Challagundla et al. (2018). Our phylogeographic analysis provides strong support for some international transmission events, such as from Germany to Bulgaria (Figure 4), but only weak support for other routes such as Germany to Italy. This is likely due to the paucity of isolates from Italy recovered after 2005.
However, we find strong evidence that ST228 disseminated between Geneva and Lausanne from 1998 onwards. These data agree with a previous study showing that the ST228 was first introduced into Switzerland via Geneva in 1998 (De Angelis et al., 2011; Kelley et al., 2015). Here we also show strong evidence that ST228 has been introduced into Lausanne on four additional occasions, from Austria, Belgium, Italy, and Germany. These findings suggest that some ST228 sub-lineages that evolved from the ancestor lineage were possibly disseminating in Switzerland through patient transfer or traveling and migration.
A limitation of our study is the lack of ST228 isolates from the remaining three European countries (Spain, Slovenia, and Denmark) where ST228 had been previously detected (Robinson and Enright, 2003; Alm et al., 2014). However, it is unlikely that this has seriously confounded our analyses, as only a single outbreak was reported for each country. In addition, most isolates in our collection are from Geneva and Lausanne, which may have introduced sampling bias during the phylogeographic reconstruction. However, this bias was considered prior to the Bayesian analysis, and a subset of 245 genomes was used for estimating the evolutionary history of ST228. Hence, we are confident that our data and analysis is largely representative for the ST228/111 clone as a whole.
In contrast to ST228 isolates, isolates of ST111 from different countries clustered together suggesting single introduction followed by diversification in these countries, which is probably due to human travel within Europe. ST111 is highly prevalent only in Croatia and was occasionally found in other European countries such as Denmark, Italy, and Sweden (Budimir et al., 2010; Aschbacher et al., 2012). However, the detection of ST111 in these countries has been associated with single introduction through human travel (Bartels et al., 2007; Aanensen et al., 2016).
In summary, this study revealed that MGEs have played an important role in the evolution of ST228 MRSA. Our results suggest that Germany was the main reservoir for ST228 lineages, and the dissemination of descendant lineages occurred into different European countries shortly after they emerged in Germany. Phylogeographical reconstruction suggests multiple introductions of ST228 into Switzerland that occurred on several occasions rather than a diverse local population. To our knowledge, this is the first study that represents the phylogeographic history of the ST228 MRSA clone.
Materials and Methods
Bacterial Isolates
We included a total of 530 MRSA isolates that were collected from health-care institutions of 14 European countries (Supplementary Table S1). The Swiss isolates were collected from the university hospital of Lausanne (n = 266), from long-term care institutions near Lausanne (n = 25) and from the university hospitals of Geneva (n = 114). Among this collection, 228 isolates from Lausanne were included in a previous study (Senn et al., 2016). All isolates were healthcare-associated MRSA. Other European isolates originated from two studies of the European Staphylococcal Reference Laboratory Working Group (Grundmann et al., 2010, 2014).
Whole Genome Sequencing and Phylogenetic Analysis
Whole-genome sequencing (WGS) was performed as previously described (Senn et al., 2016). Briefly, bacterial genomic DNA was extracted and sequenced using the Illumina HiSeq 2000 platform (San Diego, CA, United States) generating 100 base paired-end reads. In silico multi-locus sequence typing (MLST) analysis was performed via SRST2 pipeline using the sequence reads (Inouye et al., 2014). For the phylogenetic analysis, sequence reads were mapped against the distantly related S. aureus N315 reference genome (accession number NC_002745.2) using Stampy version 1.0 (Kuroda et al., 2001; Lunter and Goodson, 2011). The S. aureus N315 was chosen as reference due to being the closest sequenced genome to ST228 (both belong to CC5), to determine the ancestral node of ST228, and to ensure the reproducibility and comparability to the previously published study by Senn et al. (2016). Single nucleotide polymorphisms (SNPs) were called across the core-genome using SAMtools mpileup as previously described by Li et al. (2009) and Senn et al. (2016). The core-genome alignment was defined as nucleotide sites shared by all consensus sequences. Therefore, unmapped reads and sequences that were not present in all 530 genomes (including insertions/deletions and ambiguous sites) were not counted as part of the core-genome and were not included in the phylogenetic analysis. Similarly, SNPs falling within repetitive genomic regions and MGEs of N315 were excluded from the core-genome alignment (Supplementary Table S5). Briefly, the MGEs of N315 listed in Supplementary Table S5 were previously published by Kuroda et al. (2001) and their coordinates were identified using the annotation file, while the positions of repetitive regions were identified using the repeat-match algorithm, which is implemented in MUMmer package version 3.23 (Kurtz et al., 2004). Gubbins version 2.3.1 (Croucher et al., 2015) with default settings was used to identify regions of high SNPs densities and suspected recombination events within the investigated genomes. The final alignment of non-recombinant core-genome SNPs was generated and used to construct a maximum likelihood (ML) phylogenetic tree using PhyML that is implemented in Seaview software version 4.7 (Gouy et al., 2010). The ML tree was rooted by using the distantly related S. aureus N315 as an outgroup. Visualization and annotation of the tree was performed using iTOL1 (Letunic and Bork, 2011).
Demographic and Phylogeographic Analyses
To investigate the temporal signal in the data set, analyses of the correlation between root-to-tip genetic distance and year of sampling were performed on the maximum-likelihood tree using TempEst version 1.5.3 (Rambaut et al., 2016). In order to reduce the confounding effects of geographical biases, and to make the analyses more computationally efficient, we subsampled 245 representative genomes of ST111 and ST228 based on their isolation date and country of origin (Supplementary Table S1). Using this subsample, we performed a Bayesian analysis of evolutionary rates, divergence times estimation and phylogeographic inference using BEAST version 1.10.4 (Suchard et al., 2018) in combination with BEAGLE library version 3.1.2 (Ayres et al., 2012). A concatenated core SNPs alignment of all 245 MRSA genomes was used to infer the TMRCA and to reconstruct the phylogeographic spread. BEAST analysis was performed using the general-time reversible (GTR) and the HKY substitution models with gamma distributed among-site rate variation with four rate categories. A strict and uncorrelated relaxed molecular clock model with constant size, exponentially growing and Bayesian skyline coalescent was applied. For an accurate estimation of population size and branch lengths, unambiguous constant (invariable) sites were also included in the xml files. Three BEAST runs were performed, and for each run 100 million steps were generated, and the chain sampled every 10,000 states. A marginal likelihood estimation (MLE) using path sampling and stepping stone sampling was calculated for each run and the average values were used to compare the different combinations of clock and tree models and choosing the best supported model parameters. LogCombiner version 1.10.4 was used to combine the trees and log files from the multiple runs with 10% burn-in. Tracer version 1.7.1 (Rambaut et al., 2018) was used to visualize the results, examine the effective samples size (ESS) and to reconstruct the Bayesian Skyline plot. A maximum clade credibility (MCC) tree from the combined trees was obtained using TreeAnnotator version 1.10.4. Discrete trait model was applied to study the phylogeographic spread and the dissemination of antibiotic resistance and virulence determinants across the phylogeny. The phylogeographic tree was constructed using the ST228 sequences and their geographic coordinates in latitude and longitude as discrete traits under an asymmetric diffusion model implemented in BEAST. SPREAD3 version 0.9.7 (Bielejec et al., 2016) was used to visualize the spatial phylogenetic reconstruction of ST228, and to calculating Bayes factors (BFs) and posterior probabilities (PPs) from BSSVS analysis.
In silico Detection of Genetic Markers
Sequence reads were assembled de novo into contigs, using Velvet version 1.0.14 and VelvetOptimiser version 2.2.5 (Zerbino and Birney, 2008). The generated contigs were ordered using MAUVE version 2.4.0 (Darling et al., 2010) and then, annotated using Prokka pipeline version 1.13.0 (Seemann, 2014). These de novo assembled, and annotated genomes were used to analyze the Pan-genome via Roary version 3.11.2 (Page et al., 2015), while Phandango version 1.3.0 (Hadfield et al., 2018) was used to visualize the results. We determined the presence or absence of acquired resistance and putative virulence genes via ABRicate version 0.8.132 pipeline with default settings. The public databases ARG-ANNOT (Gupta et al., 2014) and CARD (Jia et al., 2017) were used as references for detecting the antimicrobial resistance determinants, while the VFDB (Chen et al., 2005) and PlasmidFinder (Carattoli et al., 2014) databases were used for identifying the virulence factor genes and plasmid sequences, respectively. Putative prophage regions among the assembled genomes were predicted using PHASTER (Arndt et al., 2016).
Data Availability Statement
All sequencing raw data have been deposited in the European Nucleotide Archive (ENA) under study number ERP117831.
Author Contributions
MA carried out the entire analysis and wrote the manuscript. DB and EF were the initiators of the study and edited the manuscript. The remaining authors contributed to the isolates collection with demographic and epidemiological data and commented on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the Swiss National Science Foundation (no. 31003A_150029, DB). We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant no. 090532/Z/09/Z) for the generation of the sequencing data.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work could not have been done without the background of European isolates, for that we are grateful to HG for the organization of the European Staphylococcal Reference Laboratory Working Group. A special thanks goes to members who provide us with ST228/ST111 isolates (G. Renzi, A. Budimir, S. Kalenic, Helmut Mittermayer, K. Krziwanek, S. Stumvoll, S. Haegman, B. Olsen, GW, FL, BS, AP, MM, OD, and AD).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.02063/full#supplementary-material
FIGURE S1 | Pan-genome alignment of the investigated 530 ST228/ST111 MRSA genomes using Roary.
FIGURE S2 | Root-to-tip regression compared to sampling date as estimated by TempEst. (A) Temporal signal in the entire dataset of 530 ST228/ST111 MRSA genomes. (B) Temporal signal in the entire sub-sampled dataset of 245 ST228/ST111 MRSA genomes.
TABLE S1 | Metadata of the 530 MRSA isolates included in this study.
TABLE S2 | Gubbins results representing detected genes with a high number of SNPs.
TABLE S3 | Resistance and virulence factors detected in the 530 MRSA genomes.
TABLE S4 | Marginal likelihood estimation for the 245 MRSA genomes sub-sampled dataset under various coalescent priors using path sampling and stepping stone.
TABLE S5 | Mobile genetic elements and repetitive regions identified in the reference genome N315.
Footnotes
References
Aanensen, D. M., Feil, E. J., Holden, M. T. G., Dordel, J., Yeats, C. A., Fedosejev, A., et al. (2016). Whole-genome sequencing for routine pathogen surveillance in public health: a population snapshot of invasive Staphylococcus aureus in Europe. mBio 7:e00444-16. doi: 10.1128/mBio.00444-16
Albrecht, N., Jatzwauk, L., Slickers, P., Ehricht, R., and Monecke, S. (2011). Clonal replacement of epidemic methicillin-resistant Staphylococcus aureus strains in a German University Hospital over a period of eleven years. PLoS One 6:e28189. doi: 10.1371/journal.pone.0028189
Alm, R. A., McLaughlin, R. E., Kos, V. N., Sader, H. S., Iaconis, J. P., and Lahiri, S. D. (2014). Analysis of Staphylococcus aureus clinical isolates with reduced susceptibility to ceftaroline: an epidemiological and structural perspective. J. Antimicrob. Chemother. 69, 2065–2075. doi: 10.1093/jac/dku114
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucl. Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aschbacher, R., Pichon, B., Wootton, M., Davies, L., Binazzi, R., Pike, R., et al. (2012). Molecular epidemiology of methicillin-resistant Staphylococcus aureus from bacteraemia in northern Italy. Infez. Med. 20, 256–264.
Ayres, D. L., Darling, A., Zwickl, D. J., Beerli, P., Holder, M. T., Lewis, P. O., et al. (2012). BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 61, 170–173. doi: 10.1093/sysbio/syr100
Baldan, R., Testa, F., Lorè, N. I., Bragonzi, A., Cichero, P., Ossi, C., et al. (2012). Factors contributing to epidemic MRSA clones replacement in a hospital setting. PLoS One 7:e43153. doi: 10.1371/journal.pone.0043153
Barrasa-Villar, J. I., Aibar-Remón, C., Prieto-Andrés, P., Mareca-Doñate, R., and Moliner-Lahoz, J. (2017). Impact on morbidity, mortality, and length of stay of hospital-acquired infections by resistant microorganisms. Clin. Infect. Dis. 65, 644–652. doi: 10.1093/cid/cix411
Bartels, M. D., Boye, K., Larsen, A. R., Skov, R., and Westh, H. (2007). Rapid increase of genetically diverse methicillin-resistant Staphylococcus aureus, Copenhagen, Denmark. Emerg. Infect. Dis. 13, 1533–1540. doi: 10.3201/eid1310.070503
Bielejec, F., Baele, G., Vrancken, B., Suchard, M. A., Rambaut, A., and Lemey, P. (2016). SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169. doi: 10.1093/molbev/msw082
Budimir, A., Deurenberg, R. H., Bošnjak, Z., Stobberingh, E. E., Cetkovic, H., and Kalenic, S. (2010). A variant of the Southern German clone of methicillin-resistant Staphylococcus aureus is predominant in Croatia. Clin. Microbiol. Infect. 16, 1077–1083. doi: 10.1111/j.1469-0691.2009.03042.x
Byrne, F. M., and Wilcox, M. H. (2011). MRSA prevention strategies and current guidelines. Injury 42, S3–S6. doi: 10.1016/S0020-1383(11)70125-5
Campanile, F., Bongiorno, D., Perez, M., Mongelli, G., Sessa, L., Benvenuto, S., et al. (2015). Epidemiology of Staphylococcus aureus in Italy: first nationwide survey, 2012. J. Glob. Antimicrob. Resist. 3, 247–254. doi: 10.1016/j.jgar.2015.06.006
Carattoli, A., Zankari, E., García-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Challagundla, L., Reyes, J., Rafiqullah, I., Sordelli, D. O., Echaniz-Aviles, G., Velazquez-Meza, M. E., et al. (2018). Phylogenomic classification and the evolution of clonal complex 5 methicillin-resistant Staphylococcus aureus in the Western Hemisphere. Front. Microbiol. 9:1901. doi: 10.3389/fmicb.2018.01901
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Costa, S. S., Viveiros, M., Amaral, L., and Couto, I. (2013). Multidrug efflux pumps in Staphylococcus aureus: an update. Open Microbiol. J. 7, 59–71. doi: 10.2174/1874285801307010059
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
De Angelis, G., Francois, P., Lee, A., Schrenzel, J., Renzi, G., Girard, M., et al. (2011). Molecular and epidemiological evaluation of strain replacement in patients previously harboring gentamicin-resistant MRSA: table 1. J. Clin. Microbiol. 49, 3880–3884. doi: 10.1128/JCM.01061-11
Derbise, A., Dyke, K. G. H., and El Solh, N. (1996). Characterization of a Staphylococcus aureus Transposon, Tn5405, located within Tn5404and carrying the aminoglycoside resistance genes, aphA-3andaadE. Plasmid 35, 174–188. doi: 10.1006/plas.1996.0020
Glaser, P., Martins-Simões, P., Villain, A., Barbier, M., Tristan, A., Bouchier, C., et al. (2016). Demography and intercontinental spread of the USA300 community-acquired methicillin-resistant Staphylococcus aureus lineage. mBio 7:e02183-15. doi: 10.1128/mBio.02183-15
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Grundmann, H., Aanensen, D. M., van den Wijngaard, C. C., Spratt, B. G., Harmsen, D., Friedrich, A. W., et al. (2010). Geographic distribution of Staphylococcus aureus causing invasive infections in europe: a molecular-epidemiological analysis. PLoS Med. 7:e1000215. doi: 10.1371/journal.pmed.1000215
Grundmann, H., Schouls, L. M., Aanensen, D. M., Pluister, G. N., Tami, A., Chlebowicz, M., et al. (2014). The dynamic changes of dominant clones of Staphylococcus aureus causing bloodstream infections in the European region: results of a second structured survey. Eurosurveillance 19:20987. doi: 10.2807/1560-7917.ES2014.19.49.20987
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Harris, S. R., Cartwright, E. J., Török, M. E., Holden, M. T., Brown, N. M., Ogilvy-Stuart, A. L., et al. (2013). Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 13, 130–136. doi: 10.1016/S1473-3099(12)70268-2
Hassoun, A., Linden, P. K., and Friedman, B. (2017). Incidence, prevalence, and management of MRSA bacteremia across patient populations—a review of recent developments in MRSA management and treatment. Critic. Care 21:211. doi: 10.1186/s13054-017-1801-3
Holden, M. T. G., Lindsay, J. A., Corton, C., Quail, M. A., Cockfield, J. D., Pathak, S., et al. (2010). Genome sequence of a recently emerged, highly transmissible, multi-antibiotic- and antiseptic-resistant variant of methicillin-resistant Staphylococcus aureus, sequence type 239 (TW). J. Bacteriol. 192, 888–892. doi: 10.1128/JB.01255-09
Hsu, L.-Y., Harris, S. R., Chlebowicz, M. A., Lindsay, J. A., Koh, T.-H., Krishnan, P., et al. (2015). Evolutionary dynamics of methicillin-resistant Staphylococcus aureus within a healthcare system. Genome Biol. 16:81. doi: 10.1186/s13059-015-0643-z
Inouye, M., Dashnow, H., Raven, L.-A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Jamrozy, D., Coll, F., Mather, A. E., Harris, S. R., Harrison, E. M., MacGowan, A., et al. (2017). Evolution of mobile genetic element composition in an epidemic methicillin-resistant Staphylococcus aureus: temporal changes correlated with frequent loss and gain events. BMC Genomics 18:684. doi: 10.1186/s12864-017-4065-z
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kelley, W. L., Jousselin, A., Barras, C., Lelong, E., and Renzoni, A. (2015). Missense mutations in PBP2A affecting ceftaroline susceptibility detected in epidemic hospital-acquired methicillin-resistant Staphylococcus aureus clonotypes ST228 and ST247 in Western Switzerland Archived since 1998. Antimicrob. Agents Chemother. 59, 1922–1930. doi: 10.1128/AAC.04068-14
Kuhn, G., Francioli, P., and Blanc, D. S. (2006). Evidence for clonal evolution among highly polymorphic genes in methicillin-resistant Staphylococcus aureus. J. Bacteriol. 188, 169–178. doi: 10.1128/JB.188.1.169-178.2006
Kuhn, G., Francioli, P., and Blanc, D. S. (2007). Double-locus sequence typing using clfB and spa, a fast and simple method for epidemiological typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 45, 54–62. doi: 10.1128/JCM.01457-06
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/S0140-6736(00)04403-2
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Layer, F., Cuny, C., Strommenger, B., Werner, G., and Witte, W. (2012). Aktuelle daten und trends zu methicillin-resistenten Staphylococcus aureus (MRSA). Bundesgesundheitsblatt-Gesundheitsforschung-Gesundheitsschutz 55, 1377–1386. doi: 10.1007/s00103-012-1560-x
Layer, F., Strommenger, B., Cuny, C., Klingeberg, A., Eckmanns, T., and Werner, G. (2018). Eigenschaften, Häufigkeit und Verbreitung von Methicillin-resistenten S. aureus in Deutschland – Update 2017 aus der Sicht des NRZ für Staphylokokken und Enterokokken. J. Environ. Occup. Health Sci. 23, 393–400.
Letunic, I., and Bork, P. (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. doi: 10.1093/nar/gkr201
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Livermore, D. M. (2000). Antibiotic resistance in staphylococci. Int. J. Antimicrob. Agents 16, 3–10. doi: 10.1016/S0924-8579(00)00299-5
Lunter, G., and Goodson, M. (2011). Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939. doi: 10.1101/gr.111120.110
Monecke, S., Coombs, G., Shore, A. C., Coleman, D. C., Akpaka, P., Borg, M., et al. (2011). A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6:e17936. doi: 10.1371/journal.pone.0017936
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rambaut, A., Lam, T. T., Max Carvalho, L., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. doi: 10.1093/ve/vew007
Raven, K. E., Blane, B., Leek, D., Churcher, C., Kokko-Gonzales, P., Pugazhendhi, D., et al. (2019). Methodology for whole-genome sequencing of methicillin-resistant Staphylococcus aureus isolates in a routine hospital microbiology laboratory. J. Clin. Microbiol. 57:e00180-19. doi: 10.1128/JCM.00180-19
Robinson, D. A., and Enright, M. C. (2003). Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 47, 3926–3934. doi: 10.1128/AAC.47.12.3926-3934.2003
Santos-Júnior, C. D., Veríssimo, A., and Costa, J. (2016). The recombination dynamics of Staphylococcus aureus inferred from spA gene. BMC Microbiol. 16:143. doi: 10.1186/s12866-016-0757-9
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Senn, L., Clerc, O., Zanetti, G., Basset, P., Prod’hom, G., Gordon, N. C., et al. (2016). The stealthy superbug: the role of asymptomatic enteric carriage in maintaining a long-term hospital outbreak of ST228 methicillin-resistant Staphylococcus aureus. mBio 7:e02039-15. doi: 10.1128/mBio.02039-15
Stefani, S., Chung, D. R., Lindsay, J. A., Friedrich, A. W., Kearns, A. M., Westh, H., et al. (2012). Meticillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int. Antimicrob. Agents 39, 273–282. doi: 10.1016/j.ijantimicag.2011.09.030
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4:vey016. doi: 10.1093/ve/vey016
Tong, S. Y. C., Holden, M. T. G., Nickerson, E. K., Cooper, B. S., Köser, C. U., Cori, A., et al. (2015). Genome sequencing defines phylogeny and spread of methicillin-resistant Staphylococcus aureus in a high transmission setting. Genome Res. 25, 111–118. doi: 10.1101/gr.174730.114
Uhlemann, A.-C., McAdam, P. R., Sullivan, S. B., Knox, J. R., Khiabanian, H., Rabadan, R., et al. (2017). Evolutionary dynamics of pandemic methicillin-sensitive Staphylococcus aureus ST398 and its international spread via routes of human migration. mBio 8:e01375-16. doi: 10.1128/mBio.01375-16
Urwin, R., and Maiden, M. C. J. (2003). Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11, 479–487. doi: 10.1016/j.tim.2003.08.006
Varga, M., Pantu̇ček, R., Ru̇žičková, V., and Doškař, J. (2016). Molecular characterization of a new efficiently transducing bacteriophage identified in meticillin-resistant Staphylococcus aureus. J. Gen. Virol. 97, 258–268. doi: 10.1099/jgv.0.000329
Vogel, V., Falquet, L., Calderon-Copete, S. P., Basset, P., and Blanc, D. S. (2012). Short term evolution of a highly transmissible methicillin-resistant Staphylococcus aureus clone (ST228) in a Tertiary Care Hospital. PLoS One 7:e38969. doi: 10.1371/journal.pone.0038969
Werner, G., Hildebrandt, B., and Witte, W. (2001). Aminoglycoside-streptothricin resistance gene cluster aadE-sat4-aphA-3 disseminated among multiresistant isolates of Enterococcus faecium. Antimicrob. Agents Chemother. 45, 3267–3269. doi: 10.1128/AAC.45.11.3267-3269.2001
Werner, G., Witte, W., Strommenger, B., and Cuny, C. (2004). Obvious lack of association between dynamics of epidemic methicillin-resistant Staphylococcus aureus in Central Europe and agr specificity groups. Eur. J. Clin. Microbiol. Infect. Dis. 23, 15–19. doi: 10.1007/s10096-003-1046-8
Witte, W. (1999). Antibiotic resistance in Gram-positive bacteria: epidemiological aspects. J. Antimicrob. Chemother. 44, 1–9. doi: 10.1093/jac/44.suppl_1.1
Witte, W., Kresken, M., Braulke, C., and Cuny, C. (1997). Increasing incidence and widespread dissemination of methicillin-resistant Staphylococcus aureus (MRSA) in hospitals in central Europe, with special reference to German hospitals. Clin. Microbiol. Infect. 3, 414–422. doi: 10.1111/j.1469-0691.1997.tb00277.x
Zarfel, G., Luxner, J., Folli, B., Leitner, E., Feierl, G., Kittinger, C., et al. (2016). Increase of genetic diversity and clonal replacement of epidemic methicillin-resistant Staphylococcus aureus strains in South-East Austria. FEMS Microbiol. Lett. 363:fnw137. doi: 10.1093/femsle/fnw137
Keywords: MRSA, ST228, WGS, phylogeography, phylogeny, genomic epidemiology, epidemiology
Citation: Abdelbary MMH, Feil EJ, Senn L, Petignat C, Prod’hom G, Schrenzel J, François P, Werner G, Layer F, Strommenger B, Pantosti A, Monaco M, Denis O, Deplano A, Grundmann H and Blanc DS (2020) Phylogeographical Analysis Reveals the Historic Origin, Emergence, and Evolutionary Dynamics of Methicillin-Resistant Staphylococcus aureus ST228. Front. Microbiol. 11:2063. doi: 10.3389/fmicb.2020.02063
Received: 01 May 2020; Accepted: 05 August 2020;
Published: 26 August 2020.
Edited by:
Zhi Ruan, Zhejiang University, ChinaReviewed by:
Mehul Jani, University of North Texas, United StatesSantiago Castillo Ramírez, National Autonomous University of Mexico, Mexico
Copyright © 2020 Abdelbary, Feil, Senn, Petignat, Prod’hom, Schrenzel, François, Werner, Layer, Strommenger, Pantosti, Monaco, Denis, Deplano, Grundmann and Blanc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique S. Blanc, RG9taW5pcXVlLkJsYW5jQGNodXYuY2g=