 Valentina Panzarin1*†
Valentina Panzarin1*† Argelia Cuenca2*†
Argelia Cuenca2*† Michele Gastaldelli1
Michele Gastaldelli1 Anna L. F. Alencar2
Anna L. F. Alencar2 Francesco Pascoli1
Francesco Pascoli1 Thierry Morin3
Thierry Morin3 Yannick Blanchard4Joëlle Cabon3Lénaïg Louboutin3
Yannick Blanchard4Joëlle Cabon3Lénaïg Louboutin3 David Ryder5
David Ryder5 Miriam Abbadi1
Miriam Abbadi1 Anna Toffan1
Anna Toffan1 Carlos P. Dopazo6
Carlos P. Dopazo6 Stéphane Biacchesi7Michel Brémont7
Stéphane Biacchesi7Michel Brémont7 Niels J. Olesen2
Niels J. Olesen2- 1Division of Comparative Biomedical Sciences, Istituto Zooprofilattico Sperimentale delle Venezie (IZSVe), Padua, Italy
- 2Unit for Fish and Shellfish Diseases, EURL for Fish and Crustacean Diseases, National Institute of Aquatic Resources, Technical University of Denmark (DTU), Kongens Lyngby, Denmark
- 3Unit of Viral Diseases in Fish, Laboratory of Ploufragan-Plouzané-Niort, French Agency for Food, Environmental and Occupational Health & Safety (ANSES), Plouzané, France
- 4Unit of Viral Genetics and Biosafety, Laboratory of Ploufragan-Plouzané-Niort, French Agency for Food, Environmental and Occupational Health & Safety (ANSES), Ploufragan, France
- 5International Centre of Excellence for Aquatic Animal Health, CEFAS Weymouth Laboratory, Weymouth, United Kingdom
- 6Departamento de Microbiología y Parasitología, Instituto de Acuicultura, Universidade de Santiago de Compostela, Santiago de Compostela, Spain
- 7Virologie et Immunologie Moléculaires, Université Paris-Saclay, Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement (INRAE), Université de Versailles Saint-Quentin-en-Yvelines, Jouy-en-Josas, France
The Viral Hemorrhagic Septicemia Virus (VHSV) is an OIE notifiable pathogen widespread in the Northern Hemisphere that encompasses four genotypes and nine subtypes. In Europe, subtype Ia impairs predominantly the rainbow trout industry causing severe rates of mortality, while other VHSV genotypes and subtypes affect a number of marine and freshwater species, both farmed and wild. VHSV has repeatedly proved to be able to jump to rainbow trout from the marine reservoir, causing mortality episodes. The molecular mechanisms regulating VHSV virulence and host tropism are not fully understood, mainly due to the scarce availability of complete genome sequences and information on the virulence phenotype. With the scope of identifying in silico molecular markers for VHSV virulence, we generated an extensive dataset of 55 viral genomes and related mortality data obtained from rainbow trout experimental challenges. Using statistical association analyses that combined genetic and mortality data, we found 38 single amino acid polymorphisms scattered throughout the complete coding regions of the viral genome that were putatively involved in virulence of VHSV in trout. Specific amino acid signatures were recognized as being associated with either low or high virulence phenotypes. The phylogenetic analysis of VHSV coding regions supported the evolution toward greater virulence in rainbow trout within subtype Ia, and identified several other subtypes which may be prone to be virulent for this species. This study sheds light on the molecular basis for VHSV virulence, and provides an extensive list of putative virulence markers for their subsequent validation.
Introduction
Viral Hemorrhagic Septicemia Virus (VHSV), member of genus Novirhabdovirus, species Piscine novirhabdovirus, possesses a bullet-shaped enveloped conformation and a negative-sense single-stranded RNA genome typical of the Rhabdoviridae family (Walker et al., 2018). Genes in the order 3′-N-P-M-G-NV-L-5′ encode five structural proteins with conserved functions among Rhabdoviruses (Schütze et al., 1999; Dietzgen et al., 2017). The nucleoprotein (N) is the major structural protein that tightly encapsidates the viral RNA. Together with its cofactor P (phosphoprotein), N binds the viral polymerase (L) to form with the viral RNA the ribonucleoprotein complex (RNP) responsible for transcription and replication of the viral genome. The matrix protein (M) coils and condenses the RNP complex and suppresses transcription of the host genome (Ke et al., 2017). The surface glycoprotein (G) is the main antigenic protein that binds to the cell surface permitting virus attachment and entry (Béarzotti et al., 1995; Lorenzen et al., 1999b). The genome of VHSV also encodes the non-virion protein (NV), which is exclusive to the Novirhabdovirus genus and has been shown to be required for in vivo pathogenicity (Biacchesi et al., 2010; Ammayappan et al., 2011) due to its role in inhibiting cell apoptosis and modulating the host immune response (Ammayappan and Vakharia, 2011; Kim and Kim, 2013; Biacchesi et al., 2017; Chinchilla and Gomez-Casado, 2017).
The nucleotide sequence encoding the G protein has been widely employed as the preferred molecular target to assess the genetic diversity and evolution of VHSV (Einer-Jensen et al., 2004; Kahns et al., 2012; He et al., 2014; Cieslak et al., 2016; Ghorani et al., 2016; Schönherz et al., 2018), although the N gene has also been adopted (Snow et al., 2004; Einer-Jensen et al., 2005). The phylogenetic studies undertaken so far have identified four genotypes over the Northern Hemisphere (I, II, III, and IV) encompassing nine subtypes (Ia, Ib, Ic, Id, Ie, IVa, IVb, IVc, IVd) and a number of clades (Einer-Jensen et al., 2004; Kahns et al., 2012; Pierce and Stepien, 2012; Vendramin et al., 2019).
In susceptible fish species, VHSV causes a severe systemic disease (i.e., the viral hemorrhagic septicemia, VHS), that often results in high mortality. Until the 1980s, the disease was thought to be restricted only to rainbow trout (Oncorhynchus mykiss) reared in freshwater. However, active and passive surveys have revealed that VHSV affects also other marine and freshwater fish, both farmed and wild, raising to more than 110 the number of species with evidence of susceptibility to infection (Skall et al., 2005; Anonymous, 2019; Dadar, 2020). VHSV plasticity in terms of host tropism, is a matter of concern because of the risk of host-jumps from wild fauna to fish species of relevance for the aquaculture industry, and in particular to rainbow trout. Indeed, it was shown that subtypes of VHSV adapted to rainbow trout reared in freshwater share a common ancestor with those subtypes from marine fish species, thus suggesting the occurrence of at least one successful spill over event that resulted in the establishment of subtypes Ia and Ic (Einer-Jensen et al., 2004; He et al., 2014; Schönherz et al., 2018).
In Europe, rainbow trout-adapted subtype Ia is responsible for the majority of disease outbreaks affecting this species, with severe sanitary and economic consequences (Einer-Jensen et al., 2004; Cieslak et al., 2016; Schönherz et al., 2018). In contrast, marine isolates of genotypes II and III, and of subtype Ib showed little or no pathogenicity in rainbow trout under experimental conditions (Skall et al., 2004; Ito et al., 2016). However, subsequent laboratory trials demonstrated that the degree of virulence of isolates belonging to subtype Ib and genotype III in rainbow trout depends, at least to some extent, on the route of infection (Campbell et al., 2009; Dale et al., 2009; Ito et al., 2018). Indeed, the survival rate of intra-peritoneal injected fish was lower, compared to that of bath challenged animals, highlighting that viral entry is critical for pathogenesis. Nevertheless, host adaptation, meaning the capacity of the pathogen to recognize, enter the cells and spread to target organs, is just one of the possible mechanisms that account for virulence. In addition to this, pathways involving genome replication and transcription, assembly and release of virions, and the interaction between viral and host macromolecules (e.g., those involved in the recruitment of cellular factors or in the inhibition of host antiviral defense) can also modulate virulence phenotype and host tropism. Thus, viral proteins and genetic motifs engaged in these processes might be virulence determinants (Baron et al., 1996; Cann, 2012). Consequently, in order to recognize molecular markers modulating VHSV pathogenicity and to understand their synergies, analytical approaches based on the study of the whole genome would be more informative than relying on individual genes.
Two studies carried out using the gain- or loss-of-function approach through reverse genetics, showed that the glycoprotein, the non-virion protein and the polymerase are not virulence determinants for VHSV by themselves, nor in combination (Einer-Jensen et al., 2014; Yusuff et al., 2019), although some authors disagree with this hypothesis (Kim et al., 2014; Baillon et al., 2017). In another study, the phenotype characterization of chimeric viruses resulting from the exchange of the N and P genes of viral clones with different pathogenicity revealed that the N gene plays an essential role in VHSV virulence to rainbow trout. More interestingly, the complete gain of function of chimeric viruses could have been achieved when N was combined with the P gene from the same ancestry (Vakharia et al., 2019). One common denominator of some of the above mentioned studies aiming to identify virulence markers in VHSV genome, is that they are based on the comparison of only two phylogenetically distant isolates (Vakharia et al., 2019; Yusuff et al., 2019). In addition, the virulence of chimeric VHSV was only evaluated in vivo by intra-peritoneal injection, which bypasses the natural infection barriers (Campbell et al., 2009; Dale et al., 2009; Ito et al., 2018). A recent study based on sequences comparison of 14 VHSV with different virulence phenotypes in trout has demonstrated that a unique amino acid change in the NV protein, repeatedly observed in several isolates from non-salmonid fish, had been the cause of a strong attenuation of mortality in rainbow trout (Baillon et al., 2017). Thus, it is likely that VHSV possesses different virulence mechanisms in rainbow trout, with other proteins possibly contributing to virulence and to the determination of the clinical outcome.
The combination of phenotype data and complete viral genomes through appropriate statistical models appears to be one of the most effective strategies to understand the molecular basis for VHSV virulence. However, a comprehensive study of this kind, which takes into account the genetic variability of VHSV is still lacking. To bridge this gap, within the Novimark project (Anhiwa ERA-Net) we generated the largest dataset of in vivo virulence records and associated whole genome sequences comprising 55 VHSV isolates from most of the known VHSV genotypes and subtypes. We used statistical association models that correlate phenotypic and genetic data to identify amino acid changes (i.e., single amino acid polymorphisms, SAPs) conserved within discrete virulence categories and, as such, potentially involved in VHSV virulence in trout. Secondly, we recognized specific amino acid signatures associated with different virulence phenotypes and mapped them across VHSV phylogenic reconstruction. This integrated approach allowed us to identify VHSV subtypes with potential for higher virulence in rainbow trout. Importantly, we also recognized 38 SAPs within the VHSV genome that are putative virulence determinants in this fish species. These polymorphic sites are candidates for subsequent validation to confirm their role in modulating virulence phenotype in vivo.
Materials and Methods
Selection of Viral Isolates
Fifty-five VHSV isolates (Table 1) were gathered together to identify putative virulence markers in the coding regions of the viral genome by coupling in vivo virulence data with related genetic sequences. Viruses were selected in order to encompass the largest possible genetic variability, according to previous molecular characterization studies based on analyses of partial genome sequences (Einer-Jensen et al., 2004; Abbadi et al., 2016; Baillon et al., 2017). The virulence phenotype (i.e., cumulative percent mortality in bath-challenged rainbow trout) of 35 strains was experimentally determined during the Novimark project, while 20 additional strains (DK-3345, DK-4635, DK-7054, DK-7300, DK-9895174, DK-5p276, DK-5p405, DK-5p26, DK-5e454, DK-5p393, DK-5p508, DK-5p795, DK-5p263, DK-5p11, DK-5p457, DK-5p785, SE-SVA-1033-9C, NO-2007-50-385, JF-JF00Ehil, M.rhabdo) were included in the analysis based on previous knowledge of their level of virulence (Ito et al., 2004, 2018; Skall et al., 2004; Dale et al., 2009). The collection covered a time period of more than 50 years (1962–2015) and comprised genotypes I (Ia–Ie), III and IVa. Most viruses (36/55) were isolated from rainbow trout (Oncorhynchus mykiss), while the remaining isolates originated from Anguilla anguilla (1/55), Clupea harengus (5/55), Esox lucius (1/55), Gadus morhua (1/55), Limanda limanda (2/55), Merlangius merlangus (1/55), Paralichthys olivaceus (1/55), Platichthys flesus (1/55), Pleuronectes platessa (2/55), Salmo trutta (2/55) and Sprattus sprattus (2/55).
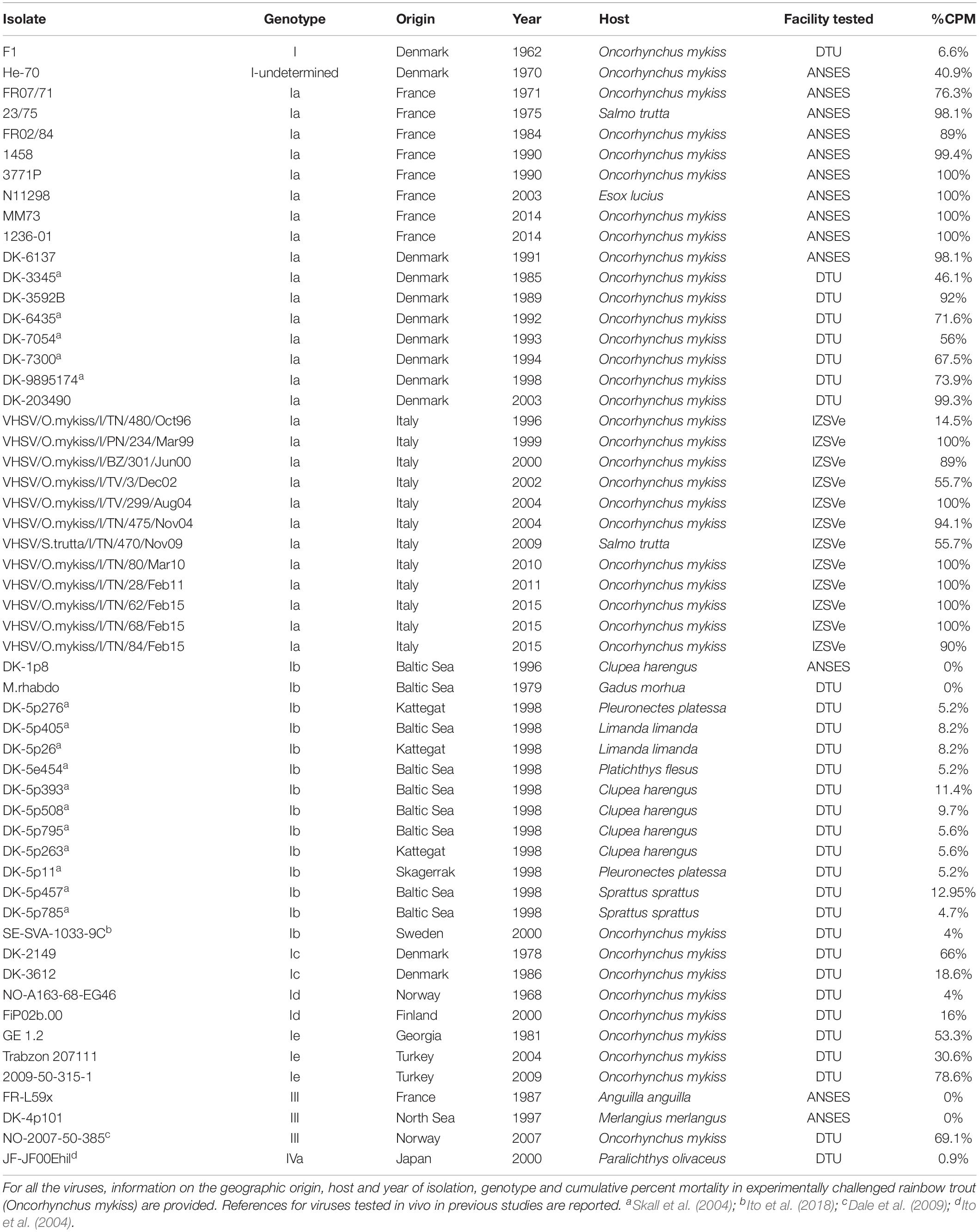
Table 1. List of VHSV isolates tested in vivo and subject to whole genome sequencing.
Viral Batches Production
Laboratory 1 – ANSES
Isolates He-70, FR07/71, 23/75, FR02/84, 1458, 3771P, N11298, MM73 and 1236-01 were propagated in bluegill fry (BF-2) cell monolayers (ATCC® CCL91TM) according to standard procedures (Lorenzen et al., 1999a; Anonymous, 2019). In detail, 1 ml of each viral strain was added to 75 cm2 polystyrene flasks (Falcon®) seeded with 24-h BF-2 cells and adsorbed for 1 h at 14 ± 1°C. After adsorption, 12 ml of pH 7.6 Tris-buffered L15 medium (Gibco) supplemented with 10% fetal calf serum (FCS, BioWest), 1% L-glutamine 200 mM (HycloneTM) and 1% penicillin–streptomycin solution 100X (PanTM Biotech) were added to each flask and viruses were incubated at 14 ± 1°C until completion of cytopathic effect (CPE). For each strain, cell culture supernatant was then collected, clarified at 4°C for 15 min at 2000 × g and checked by IFAT (Afnor, 2010; Anonymous, 2019). Viral stocks were titrated in 96-well plates using the 50% tissue culture infectious dose (TCID50) endpoint method (Kärber, 1931). Viral batches were finally aliquoted and stored at −80°C until use.
Laboratory 2 – DTU
Isolates F1, NO-A163-68-EG46, DK-2149, M.rhabdo, GE 1.2, DK-3612, DK-3592B, FiP02b.00, DK-203490, Trabzon 207111, and 2009-50-315-1 from the VHSV repository of the EURL for Fish and Crustacean Diseases were propagated in BF-2 cell monolayers in 25 cm2 flasks at 15°C for 7 days. Supernatant from the flasks was harvested, filtered using 0.45 μM Minisart® syringe filters (Sartorius) to remove cell debris and stored at −80°C until use.
Viruses were subject to plaque purification using methylcellulose in order to produce a homogeneous viral population. Briefly, all propagated isolates were ten-fold serially diluted and incubated with BF-2 cells monolayers in 96-well trays for 1 h at 15°C. The viral inoculum was removed and a mixture of methylcellulose and cell culture medium (Eagle’s MEM with L-glutamine, penicillin–streptomycin, Tris-HCl and 2% bovine fetal serum, Gibco) was added to each well. Cells were incubated at 15°C for 7 days and monitored for plaques formation. Due to the liquid nature of methylcellulose, plaques were collected only from wells containing single plaques to avoid mixed viral populations. Collected plaques were propagated in 75 cm2 flasks containing BF-2 monolayers, harvested as described above and frozen at −80°C in aliquots for subsequent titration, full genome sequencing and use in infection trials (see below). Determination of viral titer of the isolates was calculated through the method of Spearman-Karber (Kärber, 1931). Information regarding processing of the remaining isolates is available in the respective papers (Ito et al., 2004, 2018; Skall et al., 2004; Dale et al., 2009).
Laboratory 3 – IZSVe
All Italian VHSV isolates were ten-fold serially diluted (from 10–1 to 10–6) with MEM Eagle (Sigma-Aldrich) containing 2% FCS (Hyclone), 2 mM L-glutamine (Sigma-Aldrich) and 1X antibiotic/antimycotic (Sigma-Aldrich). Six-hundred microliters of each dilution were inoculated into Falcon® 12-well polystyrene culture plates (Corning) seeded with confluent 24-h-old EPC cells. Viral inoculum was adsorbed at 15°C for 3 h under gentle shaking and then removed. Subsequently, 2 ml of a 1:3 solution of 2% FCS-MEM Eagle (Sigma-Aldrich) and carboxymethyl cellulose (Sigma-Aldrich) were added to each well. Plates were then incubated at 15°C and checked daily for cytopathic effect. Upon observation of CPE, for each strain 10 μl of supernatant were collected from discrete plaques with defined edges with the aid of a pipette under the light microscope. Viruses originating from individual plaques were subject to two additional plaque-purification rounds as described above. Each viral clone was then propagated in 75 cm2 polystyrene flasks (Falcon®) seeded with 24-h-old EPC cells. Briefly, viruses were adsorbed for 1 h at 15°C under gently shaking. After adsorption, 15 ml of 10% FCS-MEM Eagle (Sigma-Aldrich), 2 mM L-glutamine (Sigma-Aldrich) and 1X antibiotic/antimycotic (Sigma-Aldrich) were added to each flask and viruses were incubated at 15°C until complete CPE. For each strain, cell culture supernatants were collected, clarified for 10 min at 2800 × g at 4°C, and checked by real-time reverse-transcription polymerase chain reaction PCR (rRT-PCR) (Jonstrup et al., 2013) and by G-gene sequencing (Abbadi et al., 2016). Viral titer was expressed as TCID50/ml according to the Reed and Müench formula (Reed and Müench, 1938). Viral batches were divided into aliquots and stocked at −80°C until use.
Experimental Challenges
Laboratory 1 – ANSES
The experimental protocol for fish challenges was conceived in compliance with the Directive 2010/63/EU and the transposition texts published in the Official Journal of the French Republic on the 6th January 2012 and on the 7th February 2013. The experimental design was evaluated by ANSES Ethics Committee (ANSES/ENVA/UPC n° 16) and finally approved by the Ministère de l’Éducation Nationale, de l’Enseignement Supérieur et de la Recherche, with the authorizations n° 08/04/14-10 and 14/06/16-8 (APAFiS: 2016053117453469).
In vivo tests were performed using pathogen-free rainbow trout of approximately 3 g originating from ANSES breeding facilities. Fish were maintained in filtered freshwater at a temperature of 10 ± 2°C and fed once a day with commercial feed. Infection trials were conducted in triplicate using 10 L tanks containing 50 fish each. Fish were exposed for 3 h to a static water 4 L bath containing 104 TCID50/ml of virus under enhanced aeration. One additional group (three tanks) was mock-infected with sterile L15 medium (Gibco) applying the same conditions. At the end of the challenge, water level was restored up to 10 L by adding clean freshwater and the flow was turned on (open water system, 10 L/h). Fish were monitored regularly for 32 days, and daily mortality was recorded. At the end of the observation period, survivor fish were euthanized by anesthetic overdose (Eugenol, Fili@vet). Bacteriological and virological analyses were conducted on dead fish as confirmatory diagnosis.
Laboratory 2 – DTU
Experimental infections were performed in accordance with the current animal welfare regulations (Directive 2010/63/EU) and approved by the Danish Animal Research Authority under the license 2013-15-2934-00976.
Rainbow trout (average size 1 g) reared at DTU facilities were transferred into aerated freshwater 8 L capacity tanks with constant flow-through system and 300% water renewal per day. Housing conditions were: 12 ± 1°C, a light to dark ratio of 12:12, a stocking density ≤ 70 kg/m3, feeding 1.5% of biomass. For each strain (i.e., F1, NO-A163-68-EG46, DK-2149, GE 1.2, DK-3612, DK-3592B, FiP02b.00, DK-203490, Trabzon 207111, 2009-50-315-1) the experimental infection was done in triplicate tanks containing 50 fish each. Challenge groups were bath-exposed for 5 h to 105 TCID50/ml of each VHSV isolate, except for the isolate DK-203490, whose infection dose was 104 TCID50/ml. Three additional 50-fish tanks were used to mock-infect animals with Eagle’s Essential Media (Gibco) with Tris-HCl and 10% fetal calf serum (Gibco), under the same conditions as above. The trial was terminated after 30 days. Fish were monitored daily for mortality, as well as for their welfare status. Animals presenting clinical signs (i.e., apathy, skin darkening, exophthalmos and swimming abnormal behavior) were euthanized by immersion in benzocaine chloride (800 mg/L) (Sigma-Aldrich), pooled by day and tank, and tested for VHSV by re-isolation in cell culture and ELISA identification (Anonymous, 2019).
In vivo virulence of strain M.rhabdo was assessed at the facilities of DTU in Aarhus (Denmark) in December 1990 and is presented here for the first time. Rainbow trout of 3 g were housed in duplicate 8 L tanks containing 40 fish each and exposed by immersion for 2 h to 105 TCID50/ml of virus. Two additional tanks were used to mock-challenge negative control fish with MEM (Minimum Essential Medium) containing 10% fetal bovine serum. Daily mortality was recorded for a 36-day period.
Additional in vivo virulence data related to 19 strains (i.e., DK-3345, DK-4635, DK-7054, DK-7300, DK-9895174, DK-5p276, DK-5p405, DK-5p26, DK-5e454, DK-5p393, DK-5p508, DK-5p795, DK-5p263, DK-5p11, DK-5p457, DK-5p785, NO-2007-50-385, SE-SVA-1033-9C, JF-JF00Ehil) were retrieved from previous papers (Skall et al., 2004; Ito et al., 2004, 2018; Dale et al., 2009) and used as metadata.
Laboratory 3 – IZSVe
The experimental protocol for fish challenges was designed in compliance with the Directive 2010/63/EU and the national Legislative Decree No. 26/2014. The experimental design was evaluated by the IZSVe Animal Welfare Body and Ethics Committee and finally approved by the Italian Ministry of Health with the authorization n° 735/2016-PR 22/7/2016.
The sample size for experimental challenges was determined by conditional Fisher’s exact test for two proportions with Walters’ normal approximation, assuming an α error of 0.05 (one tail) and power 1-β of 0.90 (SAS software).
Rainbow trout juveniles of approximately 0.6 g were purchased from an Italian commercial farm classified within Category I (EU Directive 88/2006). Fish were housed for 10 days in a 2500 L tank at 10 ± 2°C for acclimation, and fed with commercial feed. After this period, 12 groups of 70 specimens each were moved into individual tanks and challenged by immersion. Exposure with 104 TCID50 of each viral strain per ml of water was performed for 3 h in 20 L static water supplying additional aeration. One extra group was mock-infected with sterile MEM Eagle (Sigma-Aldrich) under the same conditions. At the end of the challenge, water level was resumed to 80 L by adding clean freshwater and the flow was turned on (open water system, 10 L/h). Fish were monitored regularly for a 4-week period, and daily mortality was recorded. At the end of the observation period, survivor fish were euthanized by anesthetic overdose (Tricaine, Pharmaq). Confirmatory diagnosis for VHSV detection was performed on dead specimens by rRT-PCR (Jonstrup et al., 2013).
Whole Genome Sequencing
All the viruses were subjected to high-throughput sequencing (HTS) to obtain their complete genome sequences. The isolates were sequenced by three different laboratories (ANSES, CEFAS, IZSVe) using two technologies, i.e., Illumina MiSeq (n = 28) and IonProtonTM (n = 27). Sample processing procedures, as well as sequencing and bioinformatic (BI) analyses are reported in detail in Zamperin et al. (2019). To assure data comparability among different laboratories performing HTS, a proficiency test focusing on the BI pipelines was carried out as described in the same paper. The test proved that the BI analyses of the three laboratories were 99.98% accurate and 99.94% repeatable.
Genome annotation was performed using a Blast-like algorithm1 (version 11.1.5) to identify regions with a similarity >80% to isolate 96-43 (Betts and Stone, 2000) available under the GenBank accession number AF143862.1. Coding regions were translated to protein sequences and visually inspected for shifts in the open reading frames (ORFs) and to detect premature/delayed stop codons.
For each strain, we produced a nucleotide (nt) sequence encompassing the complete open reading frame (ORFs) of every gene, without stop codons, later referred to as “concatamer.” The nt-concatamers, and their deduced amino acid (aa) sequences (aa-concatamers) were used for downstream analyses.
Phylogenetic Analysis
Full genome sequences were aligned using MAFFT v7.388 (Katoh, 2002; Katoh and Standley, 2013), with default settings. Two different datasets were created, the first including the whole genome sequences of VHSV isolates, except for the 3′ and 5′ UTRs (10,863 characters). In the second matrix, the nt-concatamers were included (10,338 characters). Phylogenetic analyses were carried out in both matrixes using RAxML v.8 (Stamatakis, 2014) with the GTR+G model, and all free parameters estimated by the program, with 1000 rapid bootstrap inferences and a search for the best-scoring maximum likelihood (ML) tree.
An additional phylogenetic reconstruction based on the nt-concatamer was carried out with IQ-Tree (Nguyen et al., 2015; Trifinopoulos et al., 2016) using the best-fitting nucleotide substitution model as estimated by ModelFinder (Kalyaanamoorthy et al., 2017), implementing the Bayesian Information Criterion (BIC), and with 1,000 replicates of the ultrafast bootstrap approximation (Hoang et al., 2018).
Statistical Analyses
All the analyses herein described were carried out using the R statistical programming environment (R Development Core Team, 2018).
Survivorship Analyses
Mortality data were analyzed by plotting Kaplan–Meier survival curves for all the isolates tested in vivo (n = 35) using the functions implemented in the package survival (Therneau, 2015).
Identification of VHSV Virulence Classes
Based on the multi-sequence alignment of the aa-concatamers, SAPs with a minimum frequency of 3% were selected. Among these SAPs, those significantly associated with variations in the cumulative percent of mortality (CPM) were identified applying a generalized linear model. The model assumed a quasi-binomial distribution of the mortality values to account for their over-dispersion. The strength of the association was then determined via a likelihood ratio test. SAPs for which we observed a p < 10–4 (significance limit = alpha/SAPs number = 0.05/499 ≈ 10–4) were considered statistically significant. Virulence classes were then defined based on CPM thresholds that maximized the genetic homogeneity among VHSV strains. In detail, for each polymorphism associated to CPM we conducted an iterative analysis in which at each cycle: (i) a CPM threshold (range: 1–99%; stepwise increase: 1%) identifying two viruses partitions was established; (ii) a contingency table reporting the number of strains segregating into each partition and displaying any possible aa signature was generated; (iii) Fisher’s exact test was performed on the contingency table to assess the dependency between the aa signatures and the two identified partitions; (iv) the observed p-value was acquired if smaller than the expected significance limit (alpha/number of tested thresholds = 0.05/99 ≈ 5 × 10–4). For each SAP, the CPM value associated with the smallest p-value was selected. The frequency of all candidate thresholds identified as above were plotted as histograms, and those showing the highest number of observations were used to designate three virulence classes, i.e., high “H”, moderate “M” and low “L”. Amino acid sequence homogeneity among VHSV strains belonging to the same virulence class were assessed by a quasi-binomial generalized linear model relating CPM to the variants observed at each SAP. Moderate viruses were not tested for their homogeneity because of dataset size constraints. The strength of the association was determined via a likelihood ratio test and the resulting p-value was retained only if smaller than 1.3 × 10–4 in the case of “H” (number of SAPs: 367) or 1.8 × 10–4 in the case of “L” (number of SAPs: 274).
Discovery of SAPs Implicated in Virulence and Host Tropism
Analyses were conducted in the context of conditional inference based on permutation tests with the package coin (Hothorn et al., 2008). In detail, the association between SAPs and the trait “virulence” (categorized as high/moderate/low) was evaluated by Cochran-Armitage or extended Cochran-Armitage test, for polymorphic sites presenting two or more than two different signatures in the aa-concatamers alignment, respectively. The association between SAPs and the variable “host tropism” (categorized as rainbow trout/other) was evaluated by Pearson’s chi square test. In all cases, p < 10–4 was considered significant (see above).
The correlation between “virulence” and “host tropism” was assessed with the Cramer’s index V, determined with the package vcd (Meyer et al., 2017). In order to infer the strength of the association of the variables “virulence” and “host tropism” to the SAPs herein identified, and to determine whether such association was significant for both traits or if a significant association between traits was present, a binomial or multinomial logistic regression model was constructed for each polymorphic site to relate the frequency of each amino acidic signature to both the aforementioned variables. In case of complete or quasi-complete separation, the regression model was based on penalized maximum likelihood implemented by the package brglm2 (Kosmidis, 2019). For both variables, the strength of the association was then determined via a likelihood ratio test.
Identification of Amino Acid Signatures Associated to Different Virulence Phenotypes
For the SAPs being relevant for virulence, we estimated the frequency of all the aa signatures within the different virulence classes previously identified. Based on these estimates, a pairwise proportion test was conducted to determine which of the aa signatures were significantly associated with the high, moderate and low virulence classes (package EnvStats) (Millard, 2013). The observed p-values were adjusted using the Bonferroni’s method and considered statistically significant when smaller than 0.05.
Results
Whole Genome Sequencing
In this study, we successfully sequenced the entire genome of 55 VHSV isolates. The average coverage ranged between 1,015 and 32,372X and was sufficient for variant calling and for producing high quality consensus sequences. Notably, the low coverage at the 3′ and 5′ ends of the viral genome (leader and trailer, respectively) impeded the assignment of unambiguous nucleotides (nt) in these regions and their full reconstruction, as previously reported (Zamperin et al., 2019). Illumina MiSeq and Ion ProtonTM raw data are available through NCBI’s Sequence Read Archive (SRA2). Consensus sequences can be retrieved from GenBank3 and the Fishpathogens database4 (Supplementary Table S1).
Annotation revealed the typical VHSV genome architecture, with six genes coding the N, P, M, G, NV and L proteins. Relatively numerous genome size differences among isolates were found at the intergenic regions because of the presence of deletions with variable extension. In contrast, only two peculiarities were found in the coding regions of the dataset analyzed in this study, both harbored by isolate VHSV/S.trutta/I/TN/470/Nov09. In the genome of this isolate, a premature stop codon was found in the M gene, rendering its deduced amino acid (aa) sequence eight residues shorter than expected. Additionally, if compared with the other isolates included within this study, the NV aa sequence of this isolate showed an extra eight residues near the C-terminus. Whether the size differences in both proteins had any influence on the phenotype of VHSV/S.trutta/I/TN/470/Nov09 was not further investigated.
Notably, as the presence of deletions within the intergenic regions impeded the comparison of all the isolates at these portions of the genome, their use in downstream association analyses was avoided. Thus, the search for VHSV virulence markers described below was carried out exclusively within the coding regions (i.e., aa-concatamers).
VHSV in vivo Phenotypes and Identification of Virulence Classes
Survival rate curves were plotted for the 35 VHSV isolates tested in vivo during the Novimark project (Figure 1). For the remaining strains, we referred to the specific publications (Ito et al., 2004, 2018; Skall et al., 2004; Dale et al., 2009). Altogether, these data pointed out a variety of different virulence phenotypes induced by this panel of isolates in experimentally challenged rainbow trout. Apart from the avirulent strains FR-L59x, DK-4p101 and DK-1p8, the first mortality records could be observed between 1 and 6 days post infection (dpi), depending on the isolate. Upon visual inspection of the Kaplan–Meier curves, it appeared that the viruses were clustered in different groups, based on survival rates over time. In general, the most lethal viruses determined a sharp decrease in fish survival during the first 10–15 days after challenge. On the contrary, less virulent viruses showed curves with a different trend, characterized by a slow and low decrease of the survival rate over time. Other viruses showed an intermediate behavior between these two groups, with no clear pattern.
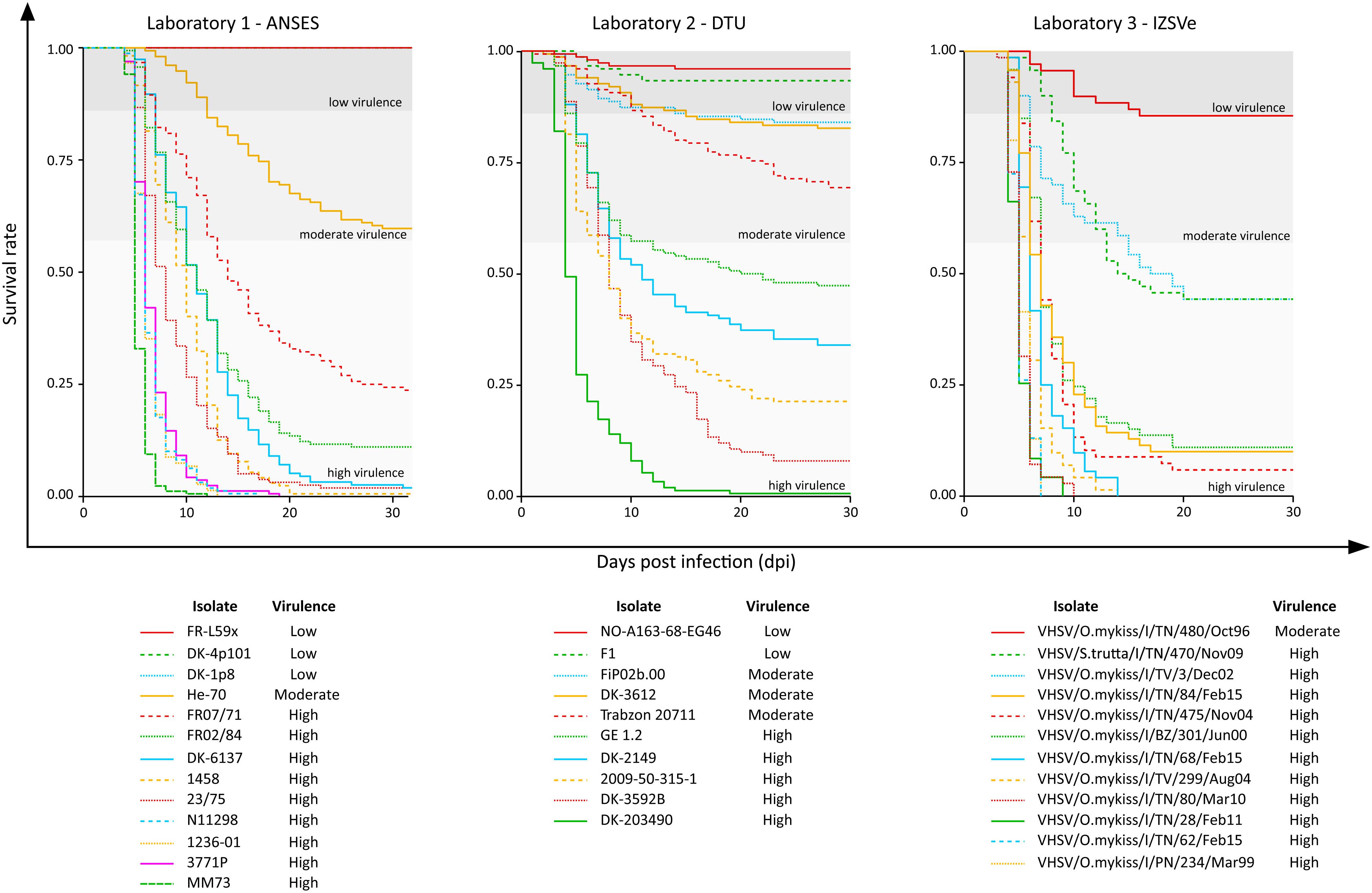
Figure 1. Kaplan–Meier curves of the 35 VHSV isolates tested for their virulence in rainbow trout in the current study. The y-axis reports the survival rate, and the x-axis reports the observation period expressed as days post infection (dpi). In each graph, the thresholds that distinguish the high “H” (CPM > 42%), moderate “M” (14% < CPM ≤ 42%) and low “L” (CPM ≤ 14%) virulent classes as assessed in this study are also reported.
Overall, the cumulative percentage of mortality recorded within the whole dataset (55 strains) was highly variable, ranging between 0% and 100% (Table 1). To assess the phenotypic differences observed in our virus collection using a more rigorous methodology, and to categorize each strain in terms of virulence, we used CPM and available protein sequence data. In detail, when considering the variable sites in the aa-concatamer multi-sequence alignment, we identified 41 SAPs that changed significantly among strains in response to variation in mortality (p < 10–4) (Supplementary Table S2). For each of these SAPs, we determined the CPM value that maximized polymorphism segregation. To do so, we used a simulation process that allowed viral partitioning into two distinct groups based on CPM cut offs. The mortality thresholds observed with the highest frequency among all 41 SAPs were adopted to outline virulence classes (Figure 2). Our data showed that all the viruses associated with a CPM of more than 42% (31 out of 55) were homogeneous at their aa sequences, when compared with isolates which caused a lower mortality (Supplementary Figure S1). This cluster of viruses was labeled as high virulent “H”. Similarly, isolates associated with a CPM of less or equal to 14% (19 out of 55) were characterized as having high aa homogeneity (Supplementary Figure S2) and were designated as low virulent strains “L”. Five isolates that did not fall into any of the “H” or “L” groups showed CPM > 14% and ≤42%, and were arbitrarily defined as moderate virulent strains “M”.
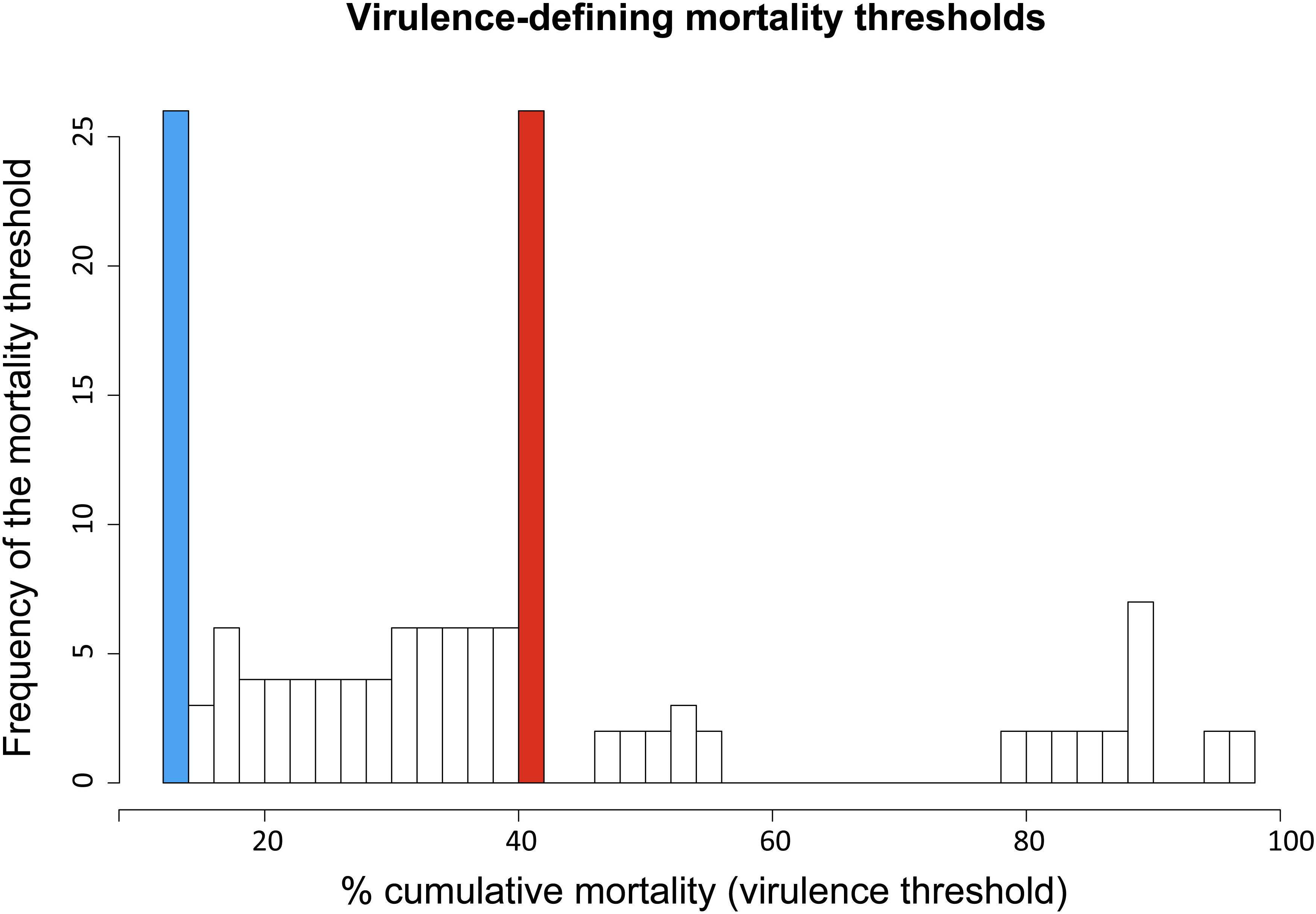
Figure 2. Frequency distribution of virulence-defining mortality thresholds. Each value derived from an iterative analysis conducted on SAPs associated to cumulative mortality variation. Ninety-nine different mortality thresholds, within the range 1–99%, were tested to identify the value maximizing VHSV strains segregation in terms of cumulative percent of mortality (CPM) and aa-signature. The CPM thresholds defining the virulence classes “L” (CPM ≤ 14%) and “H” (CPM > 42%) are highlighted in blue and red, respectively.
Identification of SAPs Associated to Virulence and Host Tropism
Based on the establishment of the “H”, “M”, and “L” virulence classes, we recognized 38 SAPs among the variable sites in the aa multi-sequence alignment that varied significantly among the three virulence groups (p < 10–4) (Table 2). These SAPs, putatively linked to VHSV virulence in trout and later referred to as “SAPs-virulence,” were scattered throughout the entire coding regions of VHSV (Figure 3). Eight SAPs-virulence were in the N protein, 4 in P, 2 in M, 8 in G, 7 in NV, and 9 in L. If taking into account the relative length of each protein, it was noticeable that the majority of SAPs-virulence were located in the nucleoprotein, the glycoprotein and the non-virion protein sequence.
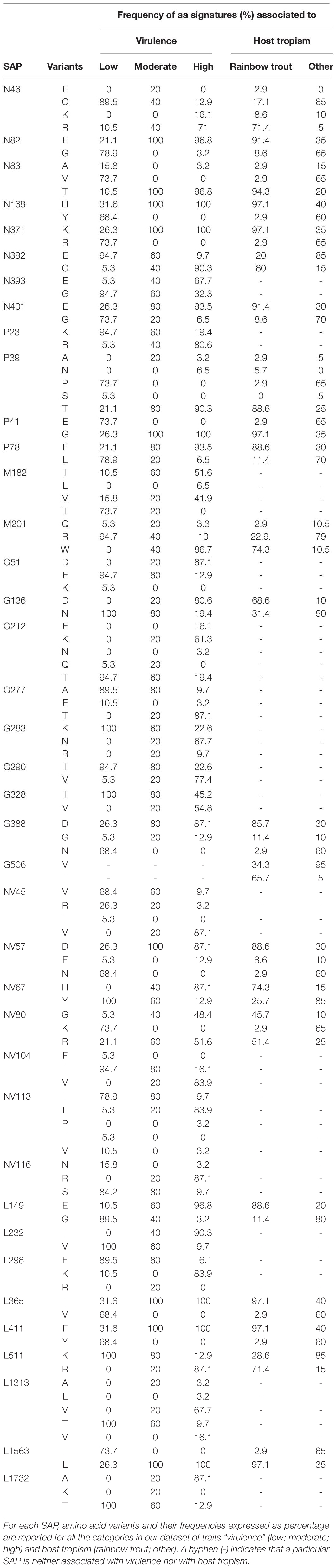
Table 2. Single amino acid polymorphisms (SAPs) associated to virulence and host tropism.
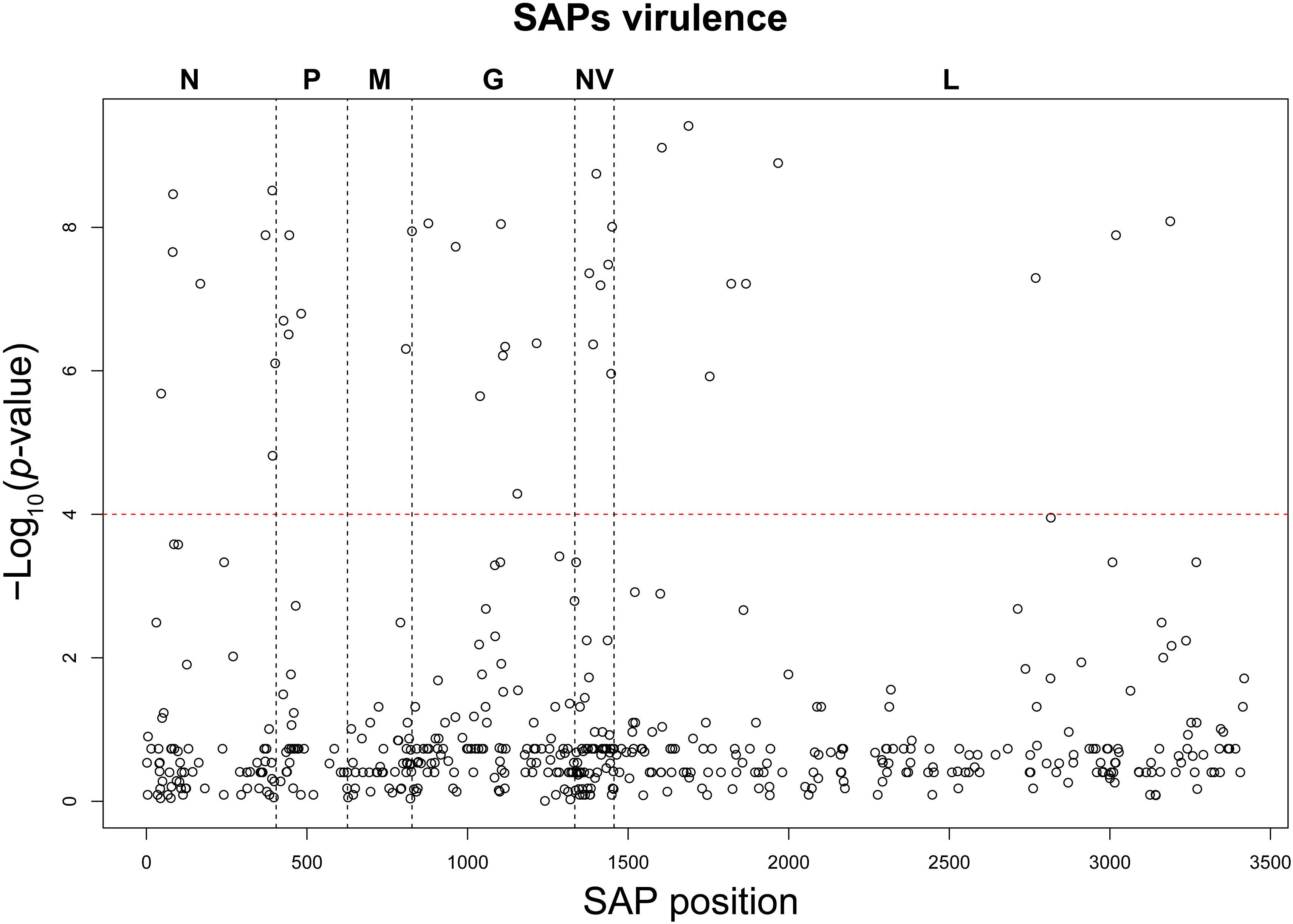
Figure 3. Manhattan plot of the association analysis conducted on VHSV SAPs and the trait “virulence.” The SAP position along the genome coding regions is displayed on the x-axis, while the y-axis reports the negative logarithm of the association p-value. The red dashed line identifies the association test significance limit.
To understand whether VHSV virulence might be related to host tropism, we applied the same analytical criteria as above to identify polymorphic sites (later referred as “SAPs-host”) that showed significant variations among isolates in respect to the host of origin, classing such isolates as either originating from rainbow trout or from other species (Table 2 and Supplementary Figure S3). Twenty-two SAPs-host putatively linked to the host of origin were subsequently identified. Notably, all of them were associated also with virulence, with the exception of G506, which was unique to this category. To rule out any possible bias in our analysis, we assessed the level of collinearity of the variables virulence and host tropism. The Cramer index V revealed that virulence and host tropism were, at least to some extent, co-dependent (0.73). However, a multi-trait association analysis using logistic binomial/multinomial models revealed the existence of SAPs (N46, N401, L149) significantly associated with both variables, individually, indicating that virulence and host tropism were sufficiently independent from each other to be considered simultaneously in the same model. Thus, we suggest that such polymorphisms are VHSV virulence markers acting as determinants of host tropism. In addition, the same analysis showed that, among SAPs that initially appeared associated with both traits (n = 21), all but N46, N401, and L149 could be considered significantly associated only to the trait “virulence” (Supplementary Table S3).
Identification of Amino Acid Signatures Associated to Virulence Phenotypes
For all the 38 SAPs-virulence, we estimated the association of the aa signatures observed at the polymorphic sites with the virulence classes “H”, “M”, and “L”. Significant associations (p < 0.05) were observed only in relation to the high and low virulent groups, while no aa signature was characteristic of moderate group (Supplementary Table S4).
Based on this association analysis, it was possible to identify, for all the VHSV herein studied, the presence of amino acid residues associated with the “H” or “L” phenotypes at the 38 SAPs implicated in virulence. The combination of amino acids obtained for all the strains is hereafter referred to as the “virulence haplotype.” Overall, we observed 23 unique virulence haplotypes out of 31 “H” strains, 5 “M” haplotypes among the five isolates of this group, and nine unique haplotypes within the 19 “L” viruses (Figure 4).
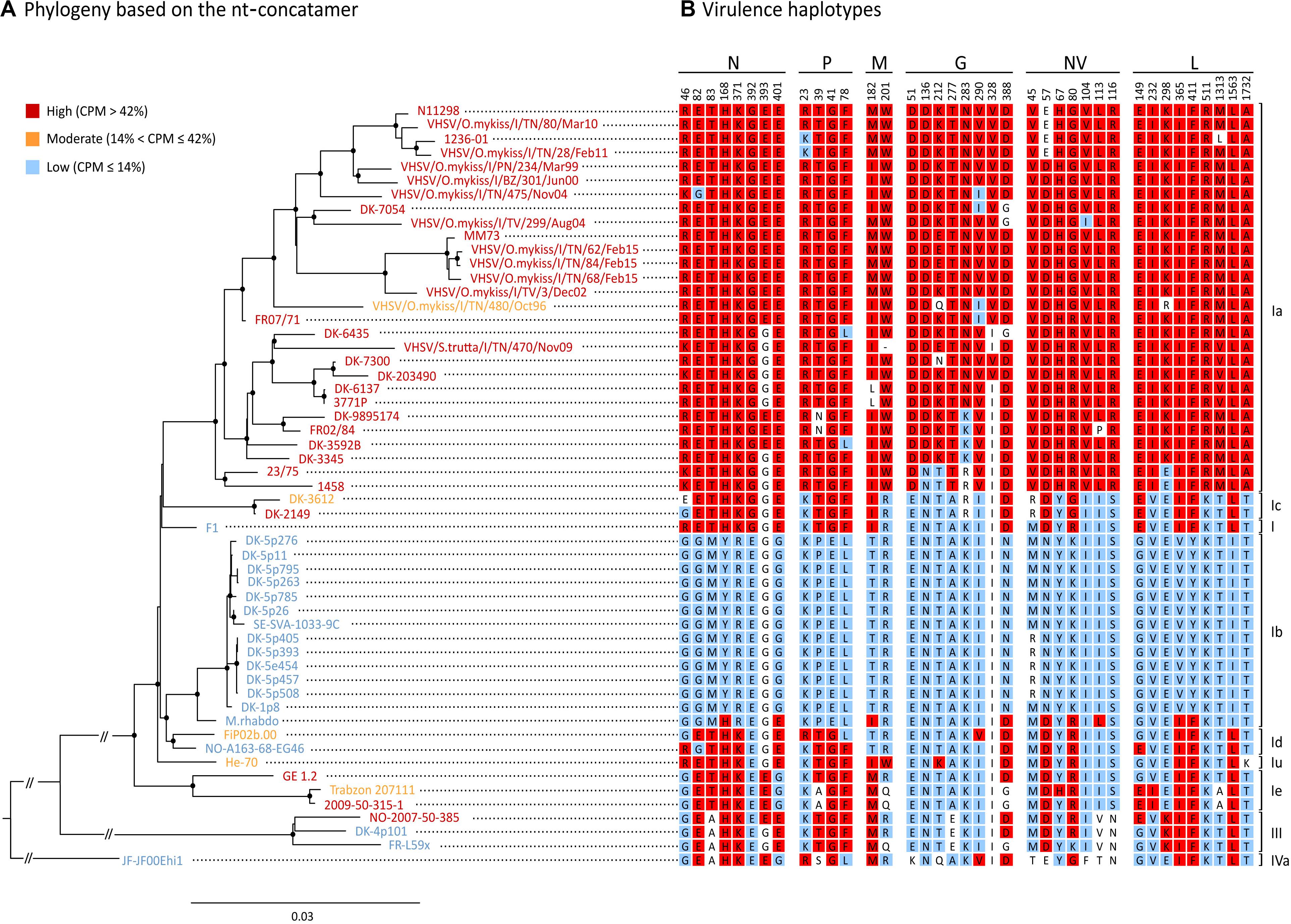
Figure 4. Phylogenetic reconstruction of the 55 isolates used in this study. (A) ML phylogenetic tree based on the nt-concatamer set. Isolates are color-coded depending which virulence class they belong to, where red is high (“H”, CPM > 42%), orange is moderate (“M”, 14% < CPM ≤ 42%) and blue is low (“L”, CPM ≤ 14%). Nodes with bootstrap support ≥ 70% are labeled with a dot. Branch lengths are scaled according to the number of nucleotide substitutions per site. Vertical bars on the right indicate genotypes/subtypes subdivision. (B) Amino acid residues at SAPs-virulence (i.e., virulence haplotypes) are shown for each isolate, indicating signatures associated either to the high (in red), or to the low virulence class (in blue) (p < 0.05). Amino acids with no significant association to any of the virulence classes are showed in white.
Phylogenetic Reconstruction
Overall, the level of similarity observed among the 55 isolates based on the nt-concatamer ranged between 86 and 100%. The percentage of similarity of individual clusters ranged between 95.7 and 100% for Ia, 99.2–100% for Ib, 97.5–99.8% for Ie, and 98.1–98.5% for genotype III, as well as being measured at around 99.7% and 99.2% for types Ic and Id, respectively, neither of which was represented by more than two isolates.
The tree topology based on the nt-concatamer was the same irrespective of using RAxML (Figure 4) or IQ-tree (not shown), and was consistent with the known phylogenetic reconstruction of VHSV based on the G gene. Genotypes I and III are monophyletic groups with 100% bootstrap support (BS) (Figure 4), as well as subtypes Ia, Ib, Ic, Id, and Ie (with 97 to 100% BS). With the exception of subtype Ie being the sister of other genotype I clusters (BS = 100%), phylogenetic relationships among subtypes of genotype I are difficult to identify, as they seem to be the result of a rapid radiation with very short branches and low BS. The only strongly supported relationship was the placement of Ib and Id as sister groups (BS > 90%). Ia and Ic were also recovered as sister groups but with low BS (<50%) forming a clade with the oldest VHSV isolate known (i.e., F1), although this relationship also showed low support (BS < 50%).
The phylogenetic tree shown in Figure 4 was annotated labeling taxa with a color code that identified different virulence classes established as above, where red was “H” (CPM > 42%), orange was “M” (14% < CPM ≤ 42%) and blue was “L” (CPM ≤ 14%). Notably, virulence in rainbow trout showed a strong phylogenetical component, except for “M” strains which were interspersed along the tree topology with no apparent pattern. The tree shows for each taxon the aa signatures associated with virulence (i.e., virulence haplotypes). Specific signatures were labeled with a color code to ease data visualization (red: aa significantly associated with the “H” phenotype; blue: aa significantly associated with the “L” phenotype; white: aa with no significant association). In general, SAPs-virulence appeared conserved among viruses belonging to the same virulence class, but we could observe several exceptions to this pattern. Subtype Ia consisted almost entirely of highly virulent isolates, apart from isolate VHSV/O.mykiss/I/TN/480/Oct96 (“M”; CPM = 14.5%) for which it was not possible to identify aa signatures that might explain its phenotype. Indeed, VHSV/O.mykiss/I/TN/480/Oct96 harbored Q and R residues at positions G212 and L298 which were unique to this strain. However, such signatures were not associated with any of the “H” or “L” groups, most likely due to limitations of our dataset; therefore, this observation should not be considered conclusive. Another exception is isolate DK-3612 (Ic), harboring an E at position N46, with no significant association with the “H” nor to the “L” categories. Similarly, Trabzon 207111 (Ie) showed a milder virulence phenotype in comparison to strains GE 1.2 and 2009-50-315-1 of the same subtype, but no relevant differences in SAPs-virulence were observed when compared to other “H” and “M” isolates within the same subtype. Viruses classified as Id, I, and IVa, encompassing only low virulent and one moderate strains, showed mixed “L” and “H” signatures, and none of the identified SAPs accounted for their diverse phenotypes. Similarly, strain M.rhabdo, a low virulent isolate within the Ib subtype, showed nine signatures significantly associated with the “H” phenotype, while causing no mortality. Finally, genotype III comprised two “L” and one “H” isolates. Differently from its close relatives, this latter isolate harbored highly virulent signatures at positions N393 and L149, which might determine its phenotype.
Discussion
Several studies have attempted to link VHSV virulence to specific molecular markers based on phenotype information and/or genetic data (Betts and Stone, 2000; Campbell et al., 2009; Dale et al., 2009; Einer-Jensen et al., 2014; Kim et al., 2014; Ito et al., 2016, 2018; Baillon et al., 2017; Vakharia et al., 2019; Yusuff et al., 2019). However, because most of these attempts have been based on the visual inspection of a restricted number of sequences, the mechanisms underlying virulence are still elusive and lack statistical support. The scarce availability of whole genome sequences data represents a major constraint to the discovery of VHSV virulence markers. This limit is currently being overcome by the advent of next generation sequencing (NGS). Equally crucial to the identification of virulence markers is the use of empirical research that implements in vitro/in vivo models to identify genome regions modulating viral phenotype. While the use of these two strategies alone might be insufficient because of their inherent limitations, their combination appears effective in pinpointing mutations and/or motifs involved in virulence (Geoghegan and Holmes, 2018). In our study, we adopted this approach and integrated the use of in vivo models with whole genome sequencing to identify putative virulence markers in VHSV coding regions. To the best of our knowledge, this is the first study based on such an extensive dataset of experimental records and sequence data that encompasses the highest possible known variability in VHSV phenotypes and genotypes. This, combined with a robust statistical methodology to associate CPM scores with genetic polymorphisms, allowed the identification of putative virulence markers within coding regions at an unprecedented resolution.
One of the major challenges in this study was to classify the VHSV isolates into virulence classes to assist the identification of aa polymorphisms (SAPs) as putative virulence markers in rainbow trout. While this classification was a requirement for the approach herein used, it is not a straight-forward process to decide how the different phenotype categories should be determined. Here, a consortium of laboratories joined in a common effort to generate and combine cumulative mortality and sequence data. While the comparability of the genomes produced at different facilities was assessed through an intra-consortium proficiency test (Zamperin et al., 2019), in vivo virulence data could not undergo the same validation process. Some of the trials were conducted over a time span of several years with different positive control viruses, thus it was not possible to conduct a normalization of CPM among facilities. On the other hand, the concerted action of different laboratories allowed collecting mortality data for 55 VHSV isolates. The size of this dataset and the statistical methodology adopted was intended to mitigate, to a large extent, the effect of potential bias in the study.
The “H” and “L” categories described herein encompassed viruses that, according to our data, were homogeneous in terms of phenotype (as assessed by CPM values) and protein sequences. Less well defined was the establishment of the moderate virulence class, which was arbitrarily formed by genetically heterogeneous isolates that were not associated with either low or high virulence classes. For this reason, results associated with this group might change when subject to greater sampling. Indeed, as part of the Novimark project, with another set of experiments focused on determining the steps in the viral replication cycle involved in virulence (López-Vázquez et al., unpublished), we observed that the boundary between CPM and virulence classes was not always as mathematically clean as indicated here. An example is strain VHSV/O.mykiss/I/TN/480/Oct96, classified as “M” based on the criteria of the present study (CPM 14.5%), but showing a replication phenotype in vitro typical of “L” strains. A relevant issue when defining the in vivo virulence phenotype is that mortality can be influenced by many factors such as water temperature, fish genetics and size, housing density, homogeneity of viral stock, viral propagation and number of passages in cell culture, as well as inherent variations from tank to tank (Chevassus and Dorson, 1990; Lorenzen et al., 1999c; Skall et al., 2004; Hawley and Garver, 2008; Polverino et al., 2016). Consequently, a virulence phenotype described by CPM is subject to fluctuations depending on the experimental setting specific to each trial. Therefore, the cut off thresholds used for different virulence classes (CPM 42% and 14%) should be taken as a descriptor of our dataset, as CPM values from other in vivo trials with the same isolates might differ from those reported in this study. Consequently, the translation of these thresholds to fish trials performed under different conditions must be cautious.
In our study, we identified an extensive list of polymorphic sites (SAPs) interspersed throughout VHSV coding regions that varied significantly with virulence as well as aa signatures specifically associated with the high and low virulent phenotypes. Clearly, their effect on the virulence to rainbow trout must be validated under experimental conditions. The putative virulence markers identified also include SAPs located in the N and P protein coding regions, which have recently been suggested to be major virulence determinants for VHSV in rainbow trout (Ito et al., 2016, 2018; Vakharia et al., 2019). On the other hand, our results are not fully in line with previous works that have ruled out the role of the glycoprotein, the non-virion protein and of the polymerase in determining VHSV virulence in rainbow trout. In fact, Einer-Jensen et al. (2014) showed that the replacement of the G and NV genes of the Infectious Hematopoietic Necrosis Virus (IHNV) with their VHSV homologs, deriving either from high (Ia) or low (Ib) virulent strains in rainbow trout, had no significant impact on the survival rate of rainbow trout fingerlings. This was in accordance with a more recent study where the G and NV genes alone, or in the G-NV and G-NV-L combinations, were interchanged between low (IVb) and high virulent (Ia) VHSV clones, showing that the virulence phenotype of chimeric viruses was not governed by the donor genotype of G, NV, and L genes (Yusuff et al., 2019). However, these studies do not completely rule out the presence of virulence determinants in the G, NV and L genes, but indicate that, if present, they may need to interact with additional determinants elsewhere in the genome. This is consistent with previous studies where VHSV clones whose NV gene had been knocked-out showed reduced mortality in yellow perch and trout, indicating that this gene is needed for in vivo pathogenicity (Biacchesi et al., 2010; Ammayappan et al., 2011). In fact, it has been shown that a change from Arginine (R) to Serine (S) at position 116 of the NV protein is enough for attenuating VHSV virulence in rainbow trout experimentally challenged by immersion (Baillon et al., 2017). Interestingly, this amino acid change was identified also in our analysis, where aa signatures R and S were significantly associated with the high and low virulent phenotype, respectively. Similarly, a recent study using a chimeric VHSV generated by reverse genetics suggested that positions N46, N82, N83, and P39, also identified in our study, were putative markers of virulence (Vakharia et al., 2019), highlighting once again the potential of the analytical model herein presented for predicting virulence determinants. Indeed, reverse genetics is the most appropriate tool to verify the SAPs identified in this work. However, studies carried out so far using this methodology are mostly based on chimeric constructs where entire genes are replaced with their homolog counterpart of another ancestry (Einer-Jensen et al., 2014; Vakharia et al., 2019; Yusuff et al., 2019). By contrast, in vivo testing of clones subject to site-directed mutagenesis is scarce. Kim et al. (2014) tested the effect of specific amino acid signatures on in vitro virulence at some of the positions identified within this study, namely NV57, NV80, L149, and L298, but no change in the ability of the recombinant virus to infect and replicate in rainbow trout gill epithelial cells was found. Moreover, this study found evidence that a single amino acid change in the polymerase gene (I1012F) was enough to change virulence in vitro using rainbow trout epithelial cells. It is not clear, however, how well these results can be correlated to in vivo studies, as a thorough evaluation of the usefulness of rainbow trout gill epithelial cells as a proxy for virulence in fish is lacking. So far, the correlation between VHSV virulence in vivo and in vitro has only been assessed by Brudeseth et al. (2008) who demonstrated that a high virulent isolate infected and caused cytotoxic effect in gill epithelial cells faster than a low virulent, marine derived isolate. Further studies using a wide range of VHSV isolates would be necessary to clarify and understand the differences among high and low virulence VHSV isolates in cell culture. The same remark could be made on studies investigating the effect of VHSV genes as virulence markers and challenging the trout by intra-peritoneal injection (Vakharia et al., 2019; Yusuff et al., 2019). It has been demonstrated that fins are the main portal of entry in trout of IHNV, a closely related virus, and that a NV knockout virus was blocked at this site, which explains in part its low pathogenicity (Harmache et al., 2006). Thus, there is a risk by injection of bypassing this natural infection barrier and excluding some important virulence markers essential in the entry and early spread of VHSV in its host.
The phylogenetic reconstruction of viral coding regions, coupled with in vivo phenotype information, clearly indicate that VHSV virulence is largely determined by viral genetic make-up. As all but one of the isolates included in genotype Ia are highly virulent, we may assume that many of the signatures associated to high virulence have originated on the branch leading to this clade. Although this seems true for many of the SAPs, particularly within the G-gene, for at least 12 of the identified polymorphisms the virulent variant seems to be the ancestral state in the phylogeny of VHSV, with the “H” variant present in both high and low virulent isolates (Figure 4). Indeed, only genotype Ib seems completely devoid of high virulence variants, except for the isolate M.rhabdo. Very few parallelisms in the way of clear-cut shared convergent mutations were found among highly virulent isolates through the whole phylogeny of VHSV. This may indicate that virulence in rainbow trout was not achieved in the same way in different genotypes and subtypes. Obviously, it is clear that our data proved insufficient to entirely describe the genotype-phenotype associations of the viruses in our dataset, as some of the phenotypes could not be explained by the aa signatures observed at the SAPs-virulence herein identified. In truth, some of the polymorphisms might not necessarily be determinants of virulence, but rather the result of a natural evolutionary process of VHSV. One obvious example is VHSV/O.mykiss/I/TN/480/Oct96, the lone Ia isolate with considerably reduced virulence in rainbow trout (Baillon et al., 2017). The only virulence markers in this isolate different from other Ia isolates were G212 (Q), and L298 (R). As both signatures were unique to this isolate, no statistical association with virulence was possible. Another strategy to investigate the reduced virulence of this isolate would be to compare it at sites other than SAPs-virulence, with relatives sharing the same ancestry but with contrasting phenotypic differences. When doing so, we found 15 additional aa differences with close relatives from subtype Ia. Indeed, there is evidence that one amino acid change from A to E in position 241 of the N protein may be involved in the reduction of virulence for this isolate. Similarly, genotype III isolate NO-2007-50-385 from rainbow trout (“H”) differs in only two of the SAPs-virulence from its lower virulence sister isolate DK-4p101 (N393 and L149). It is surprising to notice that only two SAPs may be responsible for such dramatic changes in virulence (0 to 69% CPM), indicating that we should be looking for genotype and subtype specific virulence determinants, as suggested previously (Campbell et al., 2009; Ito et al., 2016, 2018). Indeed, changes in N-T118A, N-D121N, and N-N123S were claimed to be responsible for shift of virulence to rainbow trout in genotype III (Ito et al., 2016), but they were not identified herein as virulence SAPs, possibly due to the few isolates of this genotype included in our dataset.
In our study, we also attempted to understand whether VHSV virulence might be related to adaptive mechanisms of the virus to the host, and in particular to rainbow trout. Initially, as many as 21 polymorphisms appeared to be related to both the variables “virulence” and “host tropism.” However, when the strength of the association was assessed including both variables in the multinomial regression models, only SAPs N46, N401, and L149 appeared significantly associated with both traits. This evidence leads us to speculate that these markers might be host tropism determinants and might act as virulence markers in a host-dependent manner. In this view, the study of these SAPs and their implication in virulence in terms of viral-host interactome appears particularly interesting. Differently, the remaining 35 SAPs resulted solely associated with virulence. Although such observation seems to be contradictory, it can be explained with the moderate co-dependence of the variables “virulence” and “host tropism,” with the former having the highest predictive strength.
One of the limitations of our work is that we were not able to provide any information on co-occurring mutations. This is relevant in light of the recent study by Vakharia et al. (2019) that hypothesizes a concerted action of the N and P proteins in VHSV virulence. In future studies, the frequencies observed for the aa signatures for SAPs-virulence could be one possible criterion to detect SAPs that are subject to genetic linkage. Indeed, SAPs showing the same frequency scores are more likely to be co-occurring, either when they are located within the same gene in order to produce a more stable and functional protein, in genes that encode for proteins that interact with each other (e.g., N and P), or in proteins that interact with specific RNA motifs (e.g., the RNP complex and the genome RNA). Another consideration that must be addressed is that virulence markers were investigated in the current study only in coding regions. Notably, it has been shown that a single substitution in the 3′-terminus of VHSV directly affects growth kinetics in vitro (Kim et al., 2015), but the role of the different intergenic regions, especially in in vivo studies, has gone completely unexplored in the literature so far.
To date, the molecular basis for VHSV virulence in rainbow trout is still largely undetermined because of the scarce availability of sequence and in vivo data. Within this study, we have generated the largest dataset of in vivo virulence records and complete VHSV genomes and correlated phenotype and genetic data applying statistical methods. This experimental strategy allowed us to identify 38 putative virulence markers for VHSV virulence in rainbow trout and to provide for the first time a wide and representative list of candidate virulence markers for their subsequent confirmation. With the availability of laboratories facilities for reverse genetics, the data presented here offer a challenging opportunity to explore VHSV virulence and verify the role of specific signatures in viral ecology, including replication, infectivity, pathogenicity, transmissibility and tropism. A selection of the most promising markers herein identified were investigated through the generation of recombinant viruses, and their implications in in vivo virulence using rainbow trout as a model are presented in a linked manuscript (Baillon et al., submitted).
Data Availability Statement
The sequence datasets presented in this study can be found in online repositories. The names of the repositories and accession numbers can be found in the Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Ministère de l’Éducation Nationale, de l’Enseignement Supérieur et de la Recherche, the Danish Animal Research Authority, and the Italian Ministry of Health.
Author Contributions
VP, AC, SB, MB, and NO conceived the study. VP, AC, MG, SB, and NO developed the experimental design. AA, YB, DR, and MA propagated and sequenced viral isolates. AA, FP, TM, JC, LL, and AT performed experimental trials. VP, AC, MA, MG, and AA curated the datasets. MG carried out statistical analyses. AC performed the phylogenetic reconstruction. VP, AC, MG, CD, SB, AT, and NO interpreted and analyzed data. VP, AC, MG, CD, and SB drafted the manuscript. MG and MA performed graphics editing. All authors have read and approved the final manuscript.
Funding
The present work was developed within the framework of the Novimark Project (G88F13000660001) funded by the Animal Health and Welfare (Anihwa) ERA-Net Consortium. DR was supported by the UK’s Department for Environment, Food and Rural Affairs (Defra) under contract C7277.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to: Irene Cano Cejas and David Stone from CEFAS Weymouth Laboratory for their help in sequencing; Estelle Vigouroux, Aurélie Leroux, and Pierrick Lucas from ANSES for their support in laboratory experiments and bioinformatic analyses; Giulia Bedendo, Alessandra Buratin, Francesca Ellero, and Gianpiero Zamperin from IZSVe for their contribution to viruses propagation, sequencing, and manuscript editing. The authors also wish to thank the official laboratories involved in first intention diagnostics of VHSV that kindly provided the strains.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01984/full#supplementary-material
Footnotes
- ^ https://www.geneious.com
- ^ www.ncbi.nlm.nih.gov/Traces/sra/
- ^ https://www.ncbi.nlm.nih.gov/genbank/
- ^ http://www.fishpathogens.eu/
References
Abbadi, M., Fusaro, A., Ceolin, C., Casarotto, C., Quartesan, R., Dalla Pozza, M., et al. (2016). Molecular evolution and phylogeography of co-circulating IHNV and VHSV in Italy. Front. Microbiol. 7:1306. doi: 10.3389/fmicb.2016.01306
Afnor (2010). Norme NF U 47 220: Isolement sur cultur cellulaire et identification par immunofluorescence du virus de la septicémie hémorragique virale des poisson. Available online at: https://www.boutique.afnor.org/norme/nf-u47-220/isolement-sur-culture-cellulaire-et-identification-par-immunofluorescence-du-virus-de-la-septicemie-hemorragique-virale-des-pois/article/777012/fa166656 (accessed November 4, 2019).
Ammayappan, A., Kurath, G., Thompson, T. M., and Vakharia, V. N. (2011). A reverse genetics system for the great lakes strain of viral hemorrhagic septicemia virus: the NV gene is required for pathogenicity. Mar. Biotechnol. 13, 672–683. doi: 10.1007/s10126-010-9329-4
Ammayappan, A., and Vakharia, V. N. (2011). Nonvirion protein of novirhabdovirus suppresses apoptosis at the early stage of virus infection. J. Virol. 85, 8393–8402. doi: 10.1128/jvi.00597-11
Anonymous (2019). OIE (World Organisation for Animal Health). On-line update of Manual of Diagnostic Tests for Aquatic Animals;. Chapter 2.3.10. Viral haemorrhagic septicaemia. Available online at: http://www.oie.int/en/standard-setting/aquatic-manual/access-online/ (accessed November 4, 2019).
Baillon, L., Mérour, E., Cabon, J., Louboutin, L., Quenault, H., Touzain, F., et al. (2017). A single amino acid change in the non-structural NV protein impacts the virulence phenotype of Viral hemorrhagic septicemia virus in trout. J. Gen. Virol. 98, 1181–1184. doi: 10.1099/jgv.0.000830
Baron, S., Fons, M., and Albrecht, T. (1996). “Viral pathogenesis,” in Medical Microbiology, 4th Edn, ed. S. Baron (Galveston, TX: Universityof Texas Medical Branch).
Béarzotti, M., Monnier, A. F., Vende, P., Grosclaude, J., de Kinkelin, P., and Benmansour, A. (1995). The glycoprotein of viral hemorrhagic septicemia virus (VHSV): antigenicity and role in virulence. Vet. Res. 26, 413–422.
Betts, A., and Stone, D. (2000). Nucleotide sequence analysis of the entire coding regions of virulent and avirulent strains of viral haemorrhagic septicaemia virus. Virus Genes 20, 259–262. doi: 10.1023/a:1008148813746
Biacchesi, S., Lamoureux, A., Merour, E., Bernard, J., and Brémont, M. (2010). Limited interference at the early stage of infection between two recombinant novirhabdoviruses: viral hemorrhagic septicemia virus and infectious hematopoietic necrosis virus. J. Virol. 84, 10038–10050. doi: 10.1128/JVI.00343-10
Biacchesi, S., Mérour, E., Chevret, D., Lamoureux, A., Bernard, J., and Brémont, M. (2017). NV proteins of fish novirhabdovirus recruit cellular PPM1Bb protein phosphatase and antagonize RIG-I-mediated IFN induction. Sci. Rep. 7:44025. doi: 10.1038/srep44025
Brudeseth, B. E., Skall, H. F., and Evensen, Ø (2008). Differences in virulence of marine and freshwater isolates of viral hemorrhagic septicemia virus in vivo correlate with in vitro ability to infect gill epithelial cells and macrophages of rainbow trout (Oncorhynchus mykiss). J. Virol. 82, 10359–10365. doi: 10.1128/JVI.01009-08
Campbell, S., Collet, B., Einer-Jensen, K., Secombes, C. J., and Snow, M. (2009). Identifying potential virulence determinants in viral haemorrhagic septicaemia virus (VHSV) for rainbow trout. Dis. Aquat. Organ. 86, 205–212. doi: 10.3354/dao02127
Cann, A. J. (2012). “Introduction, particles and genomes,” in Principles of Molecular Virology, 5th Edn (Walthman: Academic Press), 1–99.
Chevassus, B., and Dorson, M. (1990). Genetics of resistance to disease in fishes. Aquaculture 85, 83–107. doi: 10.1016/0044-8486(90)90009-C
Chinchilla, B., and Gomez-Casado, E. (2017). Identification of the functional regions of the viral haemorrhagic septicaemia virus (VHSV) NV protein: variants that improve function. Fish Shellfish Immunol. 70, 343–350. doi: 10.1016/j.fsi.2017.09.021
Cieslak, M., Mikkelsen, S. S., Skall, H. F., Baud, M., Diserens, N., Engelsma, M. Y., et al. (2016). Phylogeny of the viral hemorrhagic septicemia virus in European aquaculture. PLoS One 11:e0164475. doi: 10.1371/journal.pone.0164475
Dadar, M. (2020). “Viral hemorrhagic septicemia disease,” in Emerging and Reemerging Viral Pathogens, Volume 1: Fundamental and Basic Virology Aspects of Human, Animal and Plant Pathogens, 1st Edn, ed. M. M. Ennaji (New York, NY: Academic Press), 705–715.
Dale, O. B., Ørpetveit, I., Lyngstad, T. M., Kahns, S., Skall, H. F., Olesen, N. J., et al. (2009). Outbreak of viral haemorrhagic septicaemia (VHS) in seawater-farmed rainbow trout in Norway caused by VHS virus Genotype III. Dis. Aquat. Organ. 85, 93–103. doi: 10.3354/dao02065
Dietzgen, R. G., Kondo, H., Goodin, M. M., Kurath, G., and Vasilakis, N. (2017). The family Rhabdoviridae: mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 227, 158–170. doi: 10.1016/j.virusres.2016.10.010
Einer-Jensen, K., Ahrens, P., Forsberg, R., Lorenzen, N., and Einer-Jensen kej, K. (2004). Evolution of the fish rhabdovirus viral haemorrhagic septicaemia virus. J. Gen. Virol. 85, 1167–1179. doi: 10.1099/vir.0.79820-0
Einer-Jensen, K., Ahrens, P., and Lorenzen, N. (2005). Parallel phylogenetic analyses using the N, G or Nv gene from a fixed group of VHSV isolates reveal the same overall genetic typing. Dis. Aquat. Organ. 67, 39–45. doi: 10.3354/dao067039
Einer-Jensen, K., Harmache, A., Biacchesi, S., Brémont, M., Stegmann, A., and Lorenzen, N. (2014). High virulence differences among phylogenetically distinct isolates of the fish rhabdovirus viral hemorrhagic septicaemia virus are not explained by variability of the surface glycoprotein G or the nonvirion protein Nv. J. Gen. Virol. 95, 307–316. doi: 10.1099/vir.0.057448-0
Geoghegan, J. L., and Holmes, E. C. (2018). The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 19, 756–769. doi: 10.1038/s41576-018-0055-5
Ghorani, M., Adel, M., Dadar, M., Langeroudi, A. G., Kamyabi, R., Vakharia, V. N., et al. (2016). Phylogenetic analysis of the glycoprotein gene of viral hemorrhagic septicemia virus from Iranian trout farms points towards a common European origin. Vet. Microbiol. 186, 97–101. doi: 10.1016/j.vetmic.2016.02.019
Harmache, A., LeBerre, M., Droineau, S., Giovannini, M., and Brémont, M. (2006). Bioluminescence Imaging of Live Infected Salmonids Reveals that the Fin Bases Are the Major Portal of Entry for Novirhabdovirus. J. Virol. 80, 3655–3659. doi: 10.1128/jvi.80.7.3655-3659.2006
Hawley, L. M., and Garver, K. A. (2008). Stability of viral hemorrhagic septicemia virus (VHSV) in freshwater and seawater at various temperatures. Dis. Aquat. Organ. 82, 171–178. doi: 10.3354/dao01998
He, M., Yan, X. C., Liang, Y., Sun, X. W., and Teng, C. B. (2014). Evolution of the viral hemorrhagic septicemia virus: divergence, selection and origin. Mol. Phylogenet. Evol. 77, 34–40. doi: 10.1016/j.ympev.2014.04.002
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hothorn, T., Hornik, K., van de Wiel, M. A., and Zeileis, A. (2008). Implementing a class of permutation tests: The {coin} package. J. Stat. Softw. 28, 1–23. doi: 10.18637/jss.v028.i08
Ito, T., Kurita, J., Mori, K. I., and Olesen, N. J. (2016). Virulence of viral haemorrhagic septicaemia virus (VHSV) genotype III in rainbow trout. Vet. Res. 47, 1–13. doi: 10.1186/s13567-015-0303-z
Ito, T., Kurita, J., Mori, K. I., Skall, H. F., Lorenzen, N., Vendramin, N., et al. (2018). Virulence marker candidates in N-protein of viral haemorrhagic septicaemia virus (VHSV): virulence variability within VHSV Ib clones. Dis. Aquat. Organ. 128, 51–62. doi: 10.3354/dao03215
Ito, T., Mori, K. I., Arimoto, M., and Nakajima, K. (2004). Virulence of viral hemorrhagic septicemia virus (VHSV) isolates from Japanese flounder Paralichthys olivaceus in rainbow trout and several species of marine fish. Fish Pathol. 39, 103–104. doi: 10.3147/jsfp.39.103
Jonstrup, S. P., Kahns, S., Skall, H. F., Boutrup, T. S., and Olesen, N. J. (2013). Development and validation of a novel Taqman-based real-time RT-PCR assay suitable for demonstrating freedom from viral haemorrhagic septicaemia virus. J. Fish Dis. 36, 9–23. doi: 10.1111/j.1365-2761.2012.01416.x
Kahns, S., Skall, H. F., Kaas, R. S., Korsholm, H., Bang Jensen, B., Jonstrup, S. P., et al. (2012). European freshwater VHSV genotype Ia isolates divide into two distinct subpopulations. Dis. Aquat. Organ. 99, 23–35. doi: 10.3354/dao02444
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kärber, G. (1931). Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn. Schmiedebergs. Arch. Exp. Pathol. Pharmakol. 162, 480–483. doi: 10.1007/bf01863914
Katoh, K. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Ke, Q., Weaver, W., Pore, A., Gorgoglione, B., Wildschutte, J. H., Xiao, P., et al. (2017). Role of viral hemorrhagic septicemia virus matrix (M) protein in suppressing host transcription. J. Virol. 91, 1–24. doi: 10.1128/jvi.00279-17
Kim, M. S., and Kim, K. H. (2013). The role of viral hemorrhagic septicemia virus (VHSV) NV gene in TNF-α- and VHSV infection-mediated NF-κB activation. Fish Shellfish Immunol. 34, 1315–1319. doi: 10.1016/j.fsi.2013.02.026
Kim, S. H., Guo, T. C., Vakharia, V. N., and Evensen, Ø (2015). Specific nucleotides at the 3’-terminal promoter of viral hemorrhagic septicemia virus are important for virulence in vitro and in vivo. Virology 476, 226–232. doi: 10.1016/j.virol.2014.12.003
Kim, S. H., Thu, B. J., Skall, H. F., Vendramin, N., and Evensen, Ø (2014). A single amino acid mutation (I1012F) of the RNA polymerase of marine viral hemorrhagic septicemia virus changes in vitro virulence to rainbow trout gill epithelial cells. J. Virol. 88, 7189–7198. doi: 10.1128/jvi.00423-14
Kosmidis, I. (2019). {brglm2}: Bias Reduction in Generalized Linear Models. Available online at: https://cran.r-project.org/package=brglm2 (accessed November 4, 2019).
Lorenzen, E., Carstensen, B., and Olesen, N. J. (1999a). Inter-laboratory comparison of cell lines for susceptibility to three viruses: VHSV, IHNV and IPNV. Dis. Aquat. Organ. 37, 81–88. doi: 10.3354/dao037081
Lorenzen, N., Lorenzen, E., Einer-Jensen, K., Heppell, J., and Davis, H. L. (1999b). Genetic vaccination of rainbow trout against viral haemorrhagic septicaemia virus: Small amounts of plasmid DNA protect against a heterologous serotype. Virus Res. 63, 19–25. doi: 10.1016/S0168-1702(99)00054-4
Lorenzen, N., Olesen, N. J., and Koch, C. (1999c). Immunity to VHS virus in rainbow trout. Aquaculture 172, 41–61. doi: 10.1016/S0044-8486(98)00443-8
Meyer, D., Zeileis, A., and Hornik, K. (2017). vcd: Visualizing Categorical Data. R package. Available online at: https://CRAN.R-project.org/package=vcd (accessed November 4, 2019).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pierce, L. R., and Stepien, C. A. (2012). Evolution and biogeography of an emerging quasispecies: Diversity patterns of the fish Viral Hemorrhagic Septicemia virus (VHSv). Mol. Phylogenet. Evol. 63, 327–341. doi: 10.1016/j.ympev.2011.12.024
Polverino, G., Ruberto, T., Staaks, G., and Mehner, T. (2016). Tank size alters mean behaviours and individual rank orders in personality traits of fish depending on their life stage. Anim. Behav. 115, 127–135. doi: 10.1016/j.anbehav.2016.03.013
R Development Core Team (2018). R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
Reed, L. J., and Müench, H. (1938). A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497. doi: 10.1093/oxfordjournals.aje.a118408
Schönherz, A. A., Forsberg, R., Guldbrandtsen, B., Buitenhuis, A. J., and Einer-Jensen, K. (2018). Introduction of viral hemorrhagic septicemia virus into freshwater cultured rainbow trout is followed by bursts of adaptive evolution. J. Virol. 92, 1–20. doi: 10.1128/jvi.00436-18
Schütze, H., Mundt, E., and Mettenleiter, T. C. (1999). Complete genomic sequence of viral hemorrhagic septicemia virus, a fish rhabdovirus. Virus Genes 19, 59–65. doi: 10.1023/A:1008140707132
Skall, H. F., Olesen, N. J., and Mellergaard, S. (2005). Viral haemorrhagic septicaemia virus in marine fish and its implications for fish farming - a review. J. Fish Dis. 28, 509–529. doi: 10.1111/j.1365-2761.2005.00654.x
Skall, H. F., Slierendrecht, W. J., King, J. A., and Olesen, N. J. (2004). Experimental infection of rainbow trout Oncorhynchus mykiss with viral haemorrhagic septicaemia virus isolates from European marine and farmed fishes. Dis. Aquat. Organ. 58, 99–110. doi: 10.3354/dao058099
Snow, M., Bain, N., Black, J., Taupin, V., Cunningham, C. O., King, J. A., et al. (2004). Genetic population structure of marine viral haemorrhagic septicaemia virus (VHSV). Dis. Aquat. Organ. 61, 11–21. doi: 10.3354/dao061011
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Therneau, T. M. (2015). A Package for Survival Analysis in S. R package. Available online at: https://CRAN.R-project.org/package=survival (accessed November 4, 2019).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Vakharia, V. N., Li, J., McKenney, D. G., and Kurath, G. (2019). The Nucleoprotein and Phosphoprotein are Major Determinants of Virulence of Viral Hemorrhagic Septicemia Virus in Rainbow Trout. J. Virol. 93, 1–15. doi: 10.1128/jvi.00382-19
Vendramin, N., Guðmundsdóttir, S., Cuenca, A., Sigurðardóttir, H., Kristmundsson, A., Iburg, T. M., et al. (2019). Outbreak of viral haemorrhagic septicaemia (VHS) in lumpfish (Cyclopterus lumpus) in Iceland caused by VHS virus genotype IV. J. Fish Dis. 42, 47–62. doi: 10.1111/jfd.12910
Walker, P. J., Blasdell, K. R., Calisher, C. H., Dietzgen, R. G., Kondo, H., Kurath, G., et al. (2018). ICTV virus taxonomy profile: Rhabdoviridae. J. Gen. Virol. 99, 447–448. doi: 10.1099/jgv.0.001020
Yusuff, S., Kurath, G., Kim, M. S., Tesfaye, T. M., Li, J., McKenney, D. G., et al. (2019). The glycoprotein, non-virion protein, and polymerase of viral hemorrhagic septicemia virus are not determinants of host-specific virulence in rainbow trout. Virol. J. 16, 1–16. doi: 10.1186/s12985-019-1139-3
Keywords: VHSV, molecular markers, single amino acid polymorphism (SAP), virulence, rainbow trout
Citation: Panzarin V, Cuenca A, Gastaldelli M, Alencar ALF, Pascoli F, Morin T, Blanchard Y, Cabon J, Louboutin L, Ryder D, Abbadi M, Toffan A, Dopazo CP, Biacchesi S, Brémont M and Olesen NJ (2020) VHSV Single Amino Acid Polymorphisms (SAPs) Associated With Virulence in Rainbow Trout. Front. Microbiol. 11:1984. doi: 10.3389/fmicb.2020.01984
Received: 18 June 2020; Accepted: 27 July 2020;
Published: 27 August 2020.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Maryam Dadar, Razi Vaccine and Serum Research Institute, IranEduardo Gomez-Casado, National Institute for Agricultural and Food Research and Technology (INIA), Spain
Vikram N. Vakharia, University of Maryland, Baltimore, United States
Copyright © 2020 Panzarin, Cuenca, Gastaldelli, Alencar, Pascoli, Morin, Blanchard, Cabon, Louboutin, Ryder, Abbadi, Toffan, Dopazo, Biacchesi, Brémont and Olesen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Panzarin, dnBhbnphcmluQGl6c3ZlbmV6aWUuaXQ=; Argelia Cuenca, YXJjdW5AYXF1YS5kdHUuZGs=
†These authors have contributed equally to this work