 Jinghua Ye
Jinghua Ye Jie Cheng2
Jie Cheng2 Yuanhang Ren
Yuanhang Ren Qiang Li
Qiang Li- 1College of Information Science & Technology, Chengdu University, Chengdu, China
- 2College of Food and Biological Engineering, Chengdu University, Chengdu, China
In this study, the mitogenome of artillery fungus, Sphaerobolus stellatus, was assembled and compared with other Basidiomycota mitogenomes. The Sphaerobolus stellatus mitogenome was composed of circular DNA molecules, with a total size of 152,722 bp. Accumulation of intergenic and intronic sequences contributed to the Sphaerobolus stellatus mitogenome becoming the fourth largest mitogenome among Basidiomycota. We detected large-scale gene rearrangements in Basidiomycota mitogenomes, and the Sphaerobolus stellatus mitogenome contains a unique gene order. The quantity and position classes of intron varied between 75 Basidiomycota species we tested, indicating frequent intron loss/gain events occurred in the evolution of Basidiomycota. A novel intron position classes (P1281) was detected in the Sphaerobolus stellatus mitogenome, without any homologous introns from other Basidiomycota species. A pair of fragments with a total length of 9.12 kb in both the nuclear and mitochondrial genomes of Sphaerobolus stellatus was detected, indicating possible gene transferring events. Phylogenetic analysis based on the combined mitochondrial gene set obtained well-supported tree topologies (Bayesian posterior probabilities ≥ 0.99; bootstrap values ≥98). This study served as the first report on the mitogenome from the order Geastrales, which will promote the understanding of the phylogeny, population genetics, and evolution of the artillery fungus, Sphaerobolus stellatus.
Introduction
The genus Sphaerobolus belongs to the family Sphaerobolaceae and the order Geastrales. It is widely distributed globally, and most commonly found on wood mulches (Geml et al., 2005b). The Sphaerobolus species is called artillery fungus because it can eject a 1-mm diameter “gleba” (spore mass) up to 6 m toward the brightest light in its environment (Ingold, 1968, 1969). Since the first documentation of Sphaerobolus nearly 300 years ago, many mycologists have focused on investigations of the growth and reproduction of the fungus (Geml et al., 2005a). According to morphological characteristics, the Sphaerobolus genus has been classified as a member of the class Gasteromycetes along with other fungi having passive spore discharge, including puffballs, earth balls, and earth stars. Molecular data assigned the genus Sphaerobolus into the gomphoid-phalloid clade with Phallus, Ramaria, and Gomphus as closest relatives (Geml et al., 2005a, b). Geml et al. (2005b) have classified the Sphaerobolus genus into three recognized species, including S. stellatus, S. iowensis, and S. ingoldii, based on molecular data and morphological characteristics. S. stellatus was the first reported and representative species of the genus Sphaerobolus. Several nuclear molecular markers and mitochondrial sequences have been used for the classification and identification of Sphaerobolus species, including the internal transcribed spacer regions of the nuclear ribosomal gene repeat (ITS), nuclear large ribosomal RNA subunit (LSU), translation elongation factor 1-a gene (EF 1-α), and mitochondrial ribosomal RNA small subunit (mtSSU) (Geml et al., 2005a, b). However, up till now, there is no complete mitochondrial genome (mitogenome) from the genus Sphaerobolus, or even from the order Geastrales has been reported, which limits understanding of the origin and evolution of this unique artillery fungus.
Mitochondria are important organelles of eukaryotes, which are the main sources of energy for the growth and development of eukaryotes (Ernster and Schatz, 1981; McBride et al., 2006). Mitochondria contain their own genomes, which are believed to have originated from Alphaproteobacteria by the ancestors of eukaryotes through endosymbiosis (Munoz-Gomez et al., 2017). Mitogenome has been widely used to analyze the evolution and phylogeny of eukaryotes due to the characteristics of uniparental inheritance, rapid evolution rate and several available molecular markers (Richards et al., 1998; Li et al., 2015, Li et al., 2020c). However, the current understanding of the mitogenome characteristics of Basidiomycota is limited, mainly due to the limited number of complete mitogenomes available. The available Basidiomycota mitogenomes (<120) were far less than the animal mitogenomes (>9,400) published, or even the number of Basidiomycota nuclear genomes (>5,700) reported1. The phylum Basidiomycota is the largest group of mushroom-forming fungi on earth. Analysis of Basidiomycota mitogenomes will help us to understand the origin and evolution of mushrooms (Li et al., 2020a). Previous studies have shown that the evolution rate of fungal mitogenome was intermediate between animals (the highest) and plants (the lowest) (Aguileta et al., 2014). In addition, the genome size, gene content, base composition, intron number, repeat sequences and gene arrangement varied greatly in the mitogenome of fungi (Mardanov et al., 2014; Li et al., 2018a; Sandor et al., 2018). However, most Basidiomycota mitogenomes contained a set of protein coding genes (PCGs), including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, and nad6 for energy metabolism and rps3 for transcriptional regulation, which we called core PCGs of Basidiomycota (Li et al., 2018b, 2019a, 2020b).
In the present study, the mitogenome of representative species from the genus Sphaerobolus, S. stellatus, was assembled, annotated and compared with other basidiomycete mitogenomes. The aims of this study are: (1) to reveal the characterization of the S. stellatus mitogenome; (2) to reveal the variations or similarities between S. stellatus and other Basidiomycota mitogenomes in genome size, base composition, gene content and gene arrangement by comparative mitogenomic analysis; (3) to reveal the intron dynamics of cox1 genes in Basidiomycota mitogenomes; (4) to understand the phylogenetic status of S. stellatus in the phylum Basidiomycota based on the combined mitochondrial gene set. This study served as the first report on the mitogenome from the order Geastrales, which will promote the understanding of the origin, evolution and genetics of Geastrales species.
Materials and Methods
Mitogenome Assembly and Annotations
The raw sequencing data of S. stellatus used for mitogenome assembly were downloaded from the Sequence Read Archive (SRA) (acc. SRR3928187) (Kohler et al., 2015). The raw sequencing data were firstly passed through a series of quality control steps to generate clean reads, which included filtering low-quality sequences and removing adapter reads by using AdapterRemoval v 2 (Schubert et al., 2016). The mitogenome of S. stellatus was de novo assembled using SPAdes 3.9.0 software with a kmer size of 17 (Bankevich et al., 2012). Gaps between contigs obtained were filled using MITObim V1.9 (Hahn et al., 2013), as well as separate PCR and Sanger sequencing. Then, we obtained a circular mitochondrial genome for S. stellatus. We annotated the complete mitogenome of S. stellatus according to our previously described methods (Li et al., 2018a, c). Briefly, the protein-coding genes (PCGs), rRNA genes, tRNA genes, and introns of the S. stellatus mitogenome were initially annotated using MITOS (Bernt et al., 2013) and MFannot (Valach et al., 2014), both based on the genetic code 4 (the Mold, Protozoan, and Coelenterate Mitochondrial Code). This was followed by modification and prediction of PCGs using the NCBI Open Reading Frame Finder (NCBI Resource Coordinators, 2017) under the genetic code 4, and further annotation by BLASTP searches against the NCBI non-redundant protein sequence database (Bleasby and Wootton, 1990). Intron-exon borders of PCGs were verified using the exonerate v2.2 software (Slater and Birney, 2005). The tRNA genes in the S. stellatus mitogenome were also predicted with tRNAscan-SE v1.3.1 (Lowe and Chan, 2016). Graphical map of the S. stellatus mitogenome was drawn with OGDraw v1.2 (Lohse et al., 2013).
Sequence and Repetitive Element Analyses of the S. stellatus Mitogenome
Base compositions of the S. stellatus mitogenome and other Basidiomycota mitogenomes were calculated using the DNASTAR Lasergene v7.12. We calculated strand asymmetries of the Basidiomycota mitogenomes according to the following formulas: AT skew = [A – T]/[A + T], and GC skew = [G – C]/[G + C] (Wang et al., 2017). BLASTN searches (Chen et al., 2015) of the S. stellatus mitogenome against itself were conducted to identify any intra-genomic duplications of large fragments or interspersed repeats throughout the S. stellatus mitogenome, using an E-value of <10–10 as a threshold. Tandem Repeats Finder (Benson, 1999) was used to detect tandem repeats (>10 bp) within the S. stellatus mitogenome. Repeated sequences in the S. stellatus mitogenome were also detected by REPuter to identify forward (direct), reverse, complemented, and palindromic (revere complemented) repeats (Kurtz et al., 2001). We also conducted BLASTN searches of the mitogenome against its published nuclear genome (CVRD00000000.1) to identify if there was natural gene segments transferred between the S. stellatus nuclear genome and its mitogenome.
Comparative Mitogenomic Analysis and Intron Analysis
To assess conservations and variations between the reported Basidiomycota mitogenomes, we conducted comparative mitogenomic analyses of mitogenome sizes, GC content, base composition, gene and intron numbers, and gene arrangement. Introns of cox1 genes of the 75 Basidiomycota mitogenomes tested were classified into different position classes (Pcls) according to previously described methods (Ferandon et al., 2010). The cox1 genes of 75 Basidiomycota species were first aligned with the cox1 gene of the medical fungus, Ganoderma calidophilum, by Clustal W (Thompson et al., 1994), which served as the reference (Li et al., 2019d). The Pcls were named according to its insertion site in the corresponding reference sequence. The same Pcls from different species were considered homologous and usually had high sequence similarities (Ferandon et al., 2010).
Phylogenetic Analysis
The phylogenetic status of the S. stellatus mitogenome within the phylum Basidiomycota was analyzed based on the combined mitochondrial gene set (15 core PCGs + 2 rRNA genes) (Li et al., 2018c). A total of 75 Basidiomycota species was included in the phylogenetic analysis, and Annulohypoxylon stygium from the phylum Ascomycota was used as the outgroup (Deng et al., 2018). We first aligned individual mitochondrial genes using the MAFFT v7.037 software (Katoh et al., 2017), and then concatenated the aligned mitochondrial genes into a combined mitochondrial gene set using the SequenceMatrix v1.7.8 (Vaidya et al., 2011). Potential phylogenetic conflicts between different mitochondrial genes were detected using a preliminary partition homogeneity test. The best-fit models of evolution and partitioning schemes for the mitochondrial gene set was determined using PartitionFinder 2.1.1 (Lanfear et al., 2017). Both Bayesian inference (BI) and maximum likelihood (ML) methods were used to construct the phylogenetic tree. RAxML v 8.0.0 (Stamatakis, 2014) was used to perform the ML analysis. We used MrBayes v3.2.6 (Ronquist et al., 2012) to conduct the BI analysis. Two independent runs with four chains (three heated and one cold) each were conducted simultaneously for 2 × 106 generations. Each run was sampled every 100 generations. We assumed that stationarity had been reached when the estimated sample size (ESS) was greater than 100, and the potential scale reduction factor (PSRF) approached 1.0. The first 25% samples were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (BPP) in a 50% majority-rule consensus tree.
Results
Characterization and PCGs of the S. stellatus Mitogenome
The complete mitogenome of S. stellatus was composed of circular DNA molecules with a total size of 152,722 bp (Figure 1). The GC content of the S. stellatus mitogenome was 27.05%. The mitogenome of S. stellatus had negative AT skew and GC skew (Supplementary Table S1). The S. stellatus mitogenome was found containing 43 PCGs, of which 22 belonged to non-intronic PCGs and the other 21 located in introns (Supplementary Table S2). The 22 non-intronic PCGs included a whole set of core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3) and 7 non-conserved PCGs. Non-conserved PCGs in the S. stellatus mitogenome mainly encoded proteins with unknown functions. A total of 34 introns were detected in the mitogenome of S. stellatus, 32 of which belonged to the group I (Supplementary Table S2). The 21 intronic ORFs in the S. stellatus mitogenome included 14 ORFs encoding LAGLIDADG endonuclease, 6 ORFs encoding GIY-YIG endonuclease, and 1 ORFs with unknown functions.
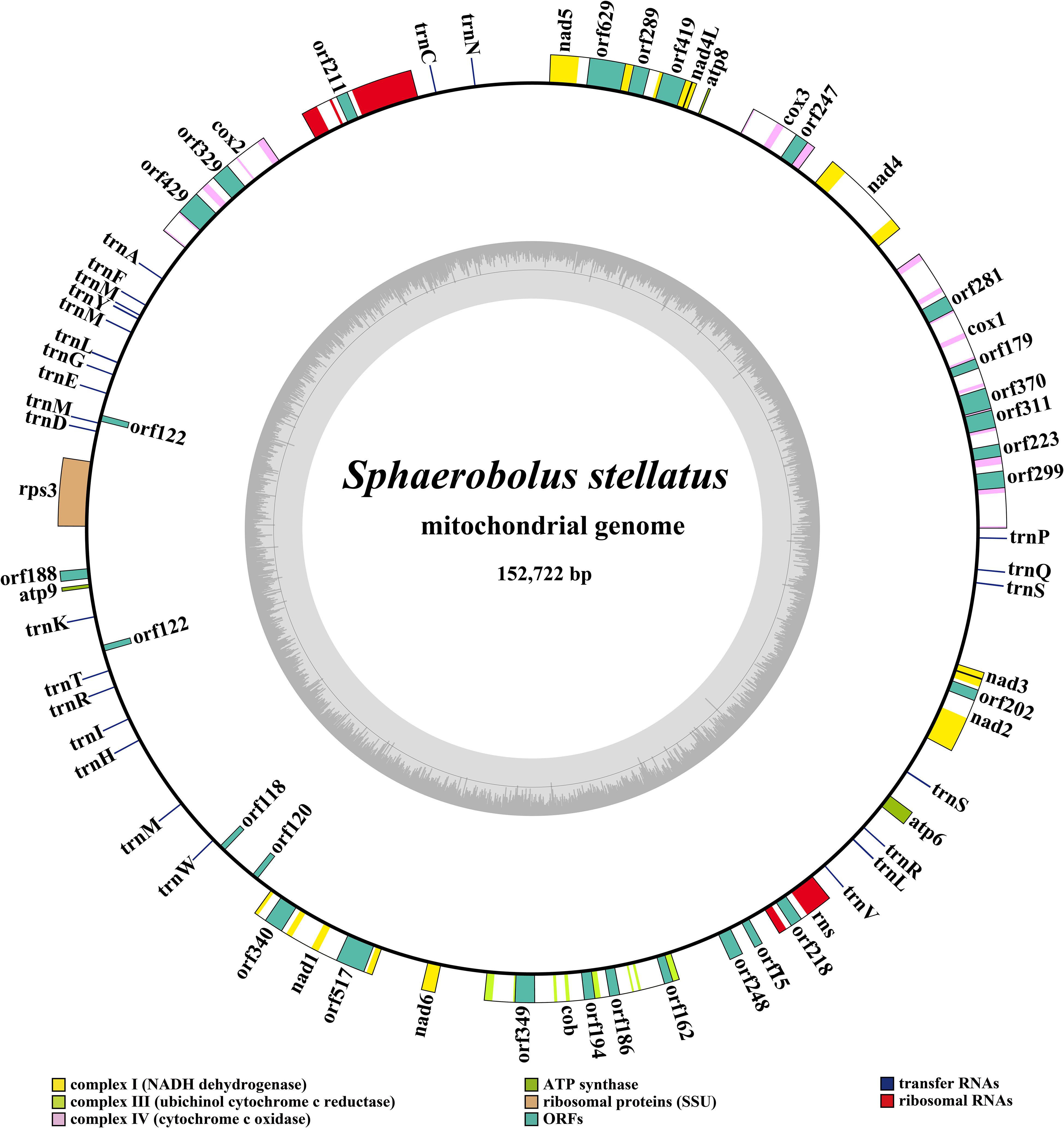
Figure 1. Circular map of the mitochondrial genome of Sphaerobolus stellatus. Genes are represented by different colored blocks. Colored blocks outside each ring indicate that the genes are on the direct strand, while colored blocks within the ring indicates that the genes are located on the reverse strand.
rRNA and tRNA Genes
Two rRNA genes were detected in the S. stellatus mitogenome, including the large subunit ribosomal RNA (rnl) and the small subunit ribosomal RNA (rns) (Supplementary Table S2). The mitogenome of S. stellatus contained 26 tRNA genes, which were folded into classical cloverleaf structures (Figure 2). The mitogenome of S. stellatus contained 2 tRNAs with different anticodons coding for leucine, arginine and serine, 4 tRNAs with the same anticodons coding for methionine. The length variations of extra arms contributed to the size variations of tRNA genes, with each ranging from 71 to 87 bp. Of all the 71 genes detected in the S. stellatus mitogenome, 67 genes were located on the direct strand and the other 4 genes were on the reverse strand.
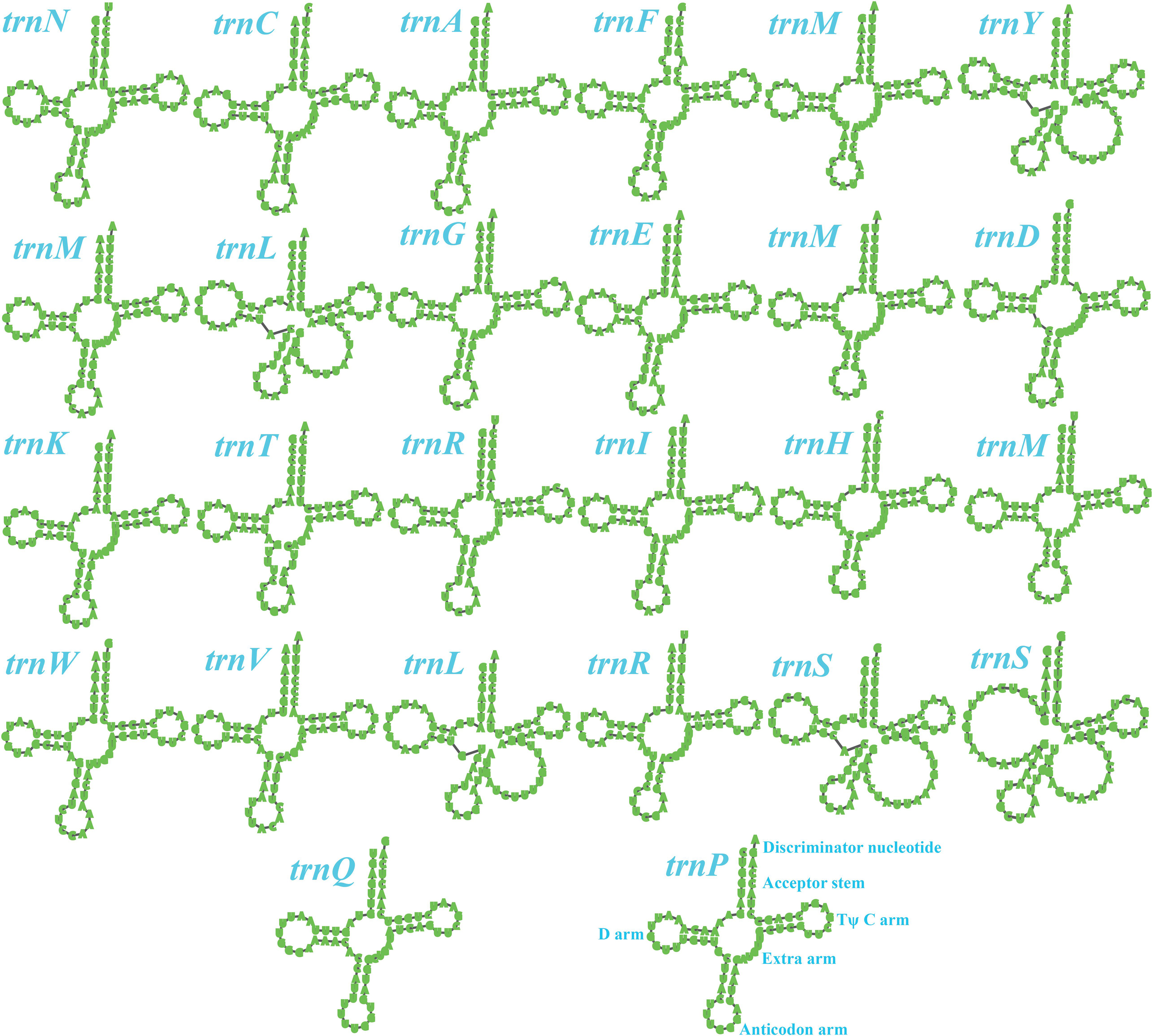
Figure 2. Putative secondary structures of the 26 tRNA genes identified in the mitochondrial genome of Sphaerobolus stellatus. All genes are shown in order of occurrence in the mitochondrial genome of Sphaerobolus stellatus, starting from trnN.
Mitogenome Compositions and Codon Usage
Intergenic region was the largest among all the regions in the S. stellatus mitogenome, which accounted for 47.69% of the entire mitogenome (Figure 3). The result indicated that the mitogenome of S. stellatus had a loose structure. A total of 51,399 bp of intronic sequences were detected in the mitogenome of S. stellatus, which comprised 33.66% of the S. stellatus mitogenome. Protein coding regions was the third largest part of the S. stellatus mitogenome, accounting for 13.57% of the entire mitogenome. RNA coding region accounted for 5.08% of the S. stellatus mitogenome.
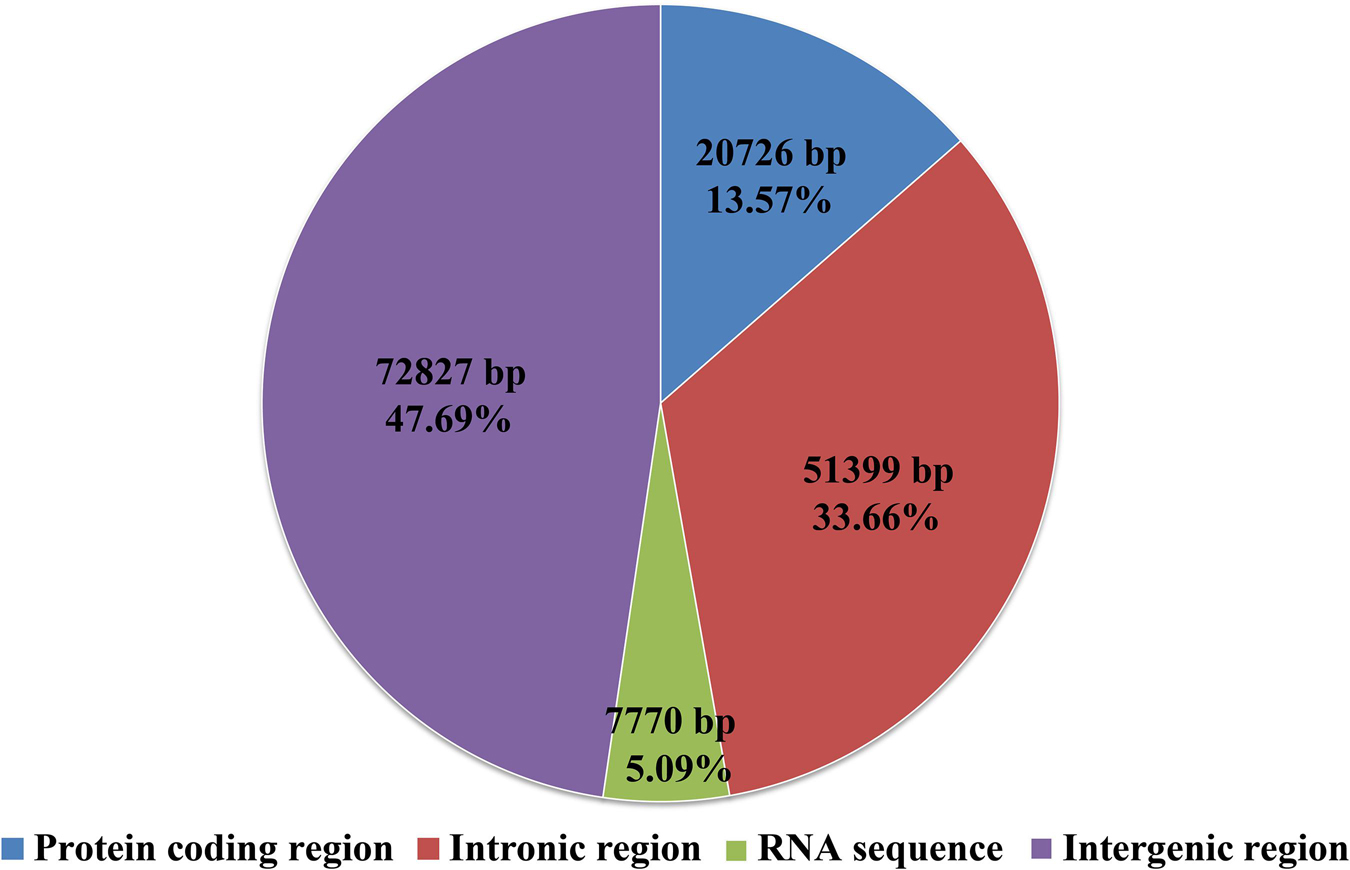
Figure 3. The protein-coding, intronic, intergenic, and RNA gene region proportions of the entire mitochondrial genome of Sphaerobolus stellatus.
Codon usage analysis indicated that the most frequently used codons in the S. stellatus mitogenome were TTA (for leucine; Leu), AAT (for asparagine; Asn), ATT (for isoleucine; Ile), TAT (for tyrosine; Tyr) and AAA (for lysine; Lys) (Figure 4). The high frequency of A and T used in codons contributed to the high AT content of the S. stellatus mitogenome (72.95%) (Supplementary Table S3).
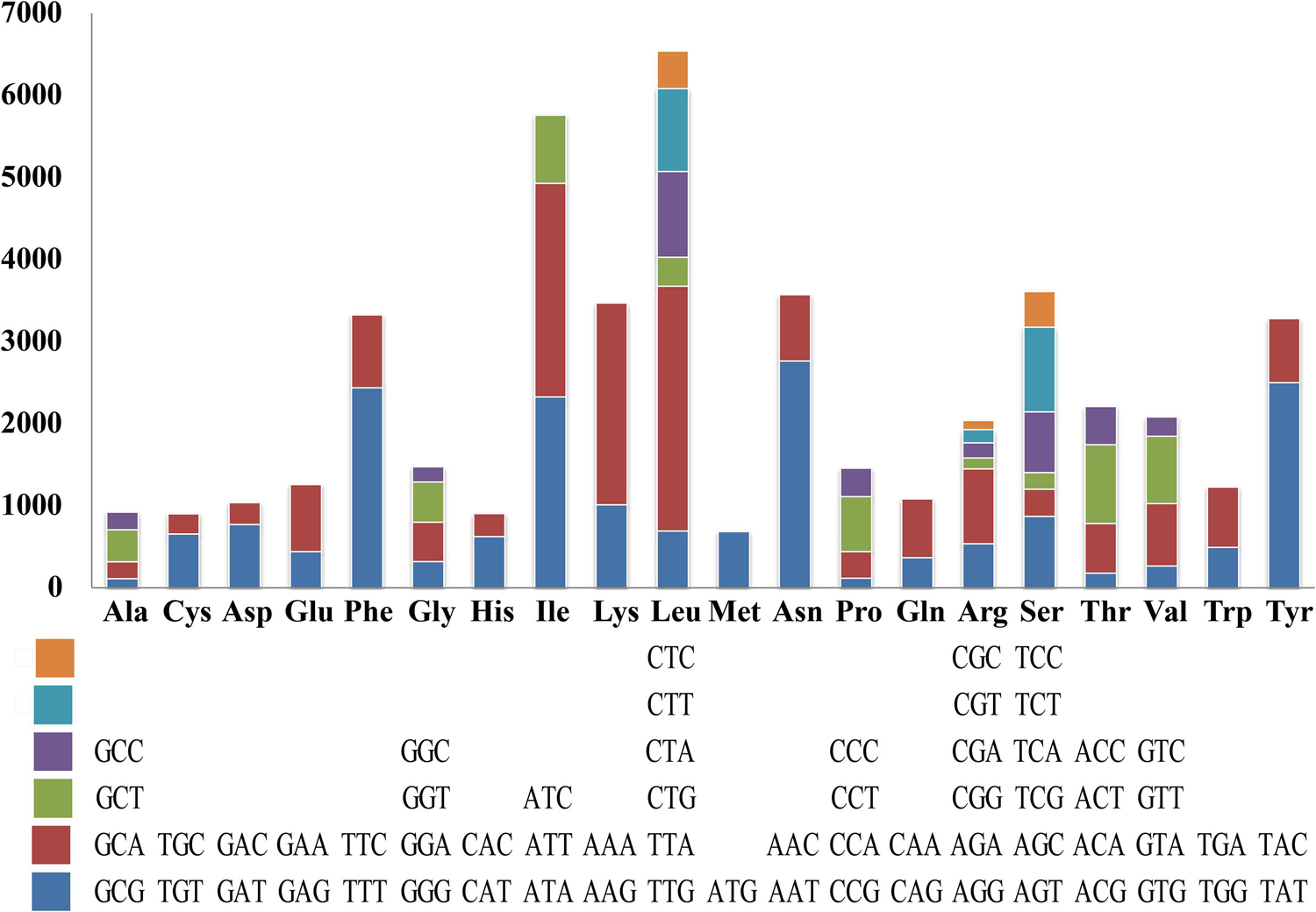
Figure 4. Codon usage in the mitochondrial genome of Sphaerobolus stellatus. Frequency of codon usage is plotted on the y-axis.
Repeat Elements in the S. stellatus Mitogenome
BLASTN searches of the S. stellatus mitogenome against itself identified 95 repetitive sequences in the S. stellatus mitogenome (Supplementary Table S4). The length of these repetitive sequences ranged from 28 bp to 101 bp, with pair-wise nucleotide similarities ranging from 87.76% to 100%. The longest repetitive sequences were detected in the coding region of ORF157. A total of 3,823 bp of repetitive sequences were detected in the mitogenome of S. stellatus, which accounted for 2.50% of the entire mitogenome.
A total of 329 tandem repeats were detected in the mitogenome of S. stellatus, which accounted for 2.95% of the entire mitogenome (Supplementary Table S5). The longest tandem sequence was found between the neighboring genes cox2 and trnA, with a size of 55 bp. Most tandem repeat sequences in the mitogenome of S. stellatus were copied 2 - 5 times, with the highest copy number of 26. We also identified 32 forward, 2 palindromic and 16 reverse repeats in the mitogenome of S. stellatus though REPuter (Kurtz et al., 2001) (Supplementary Table S6), which accounted for 2.24% of the whole mitogenome.
To identify if there were gene segments that transferred between the nuclear and mitochondrial genomes, we blasted the S. stellatus mitogenome against its nuclear genome. A total of 53 aligned fragments were detected in the mitogenome of S. stellatus, with each aligned fragment ranging from 34 bp to 1,468 bp (Supplementary Table S7). The sequence identities of these aligned fragments were between 89.13% and 100%. The largest aligned fragment was found located in the intergenic region between trnG and orf122, and encompassed the coding region of trnE. The presence of large fragments aligned between the nuclear and mitochondrial genomes of the S. stellatus mitogenome indicated that genetic transfer between nuclear and mitochondrial genome may have occurred in the evolution of S. stellatus.
Intron Dynamics of cox1 Genes in Basidiomycota
We calculated correlations between mitogenome sizes and intron numbers of the 75 Basidiomycota species. The results showed that the number of intron was closely related to the mitogenome size of Basidiomycota, with the pearson correlation coefficient of 0.81 (Figure 5). Therefore, the dynamics of intron could significantly promote the size variations of Basidiomycota mitogenomes. According to the insertion position of introns in the coding region of host genes, we could classify fungal introns into different position classes (Pcls). In the present study, a total of 1046 introns were detected in the 75 Basidiomycota mitogenomes, with each Basidiomycota species containing 0 – 46 mitochondrial introns. Large variations in intron number indicated that intron gain/loss events have occurred in the evolution of Basidiomycota species. These introns were harbored in 14 host genes: atp6, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, rns, and rnl genes.
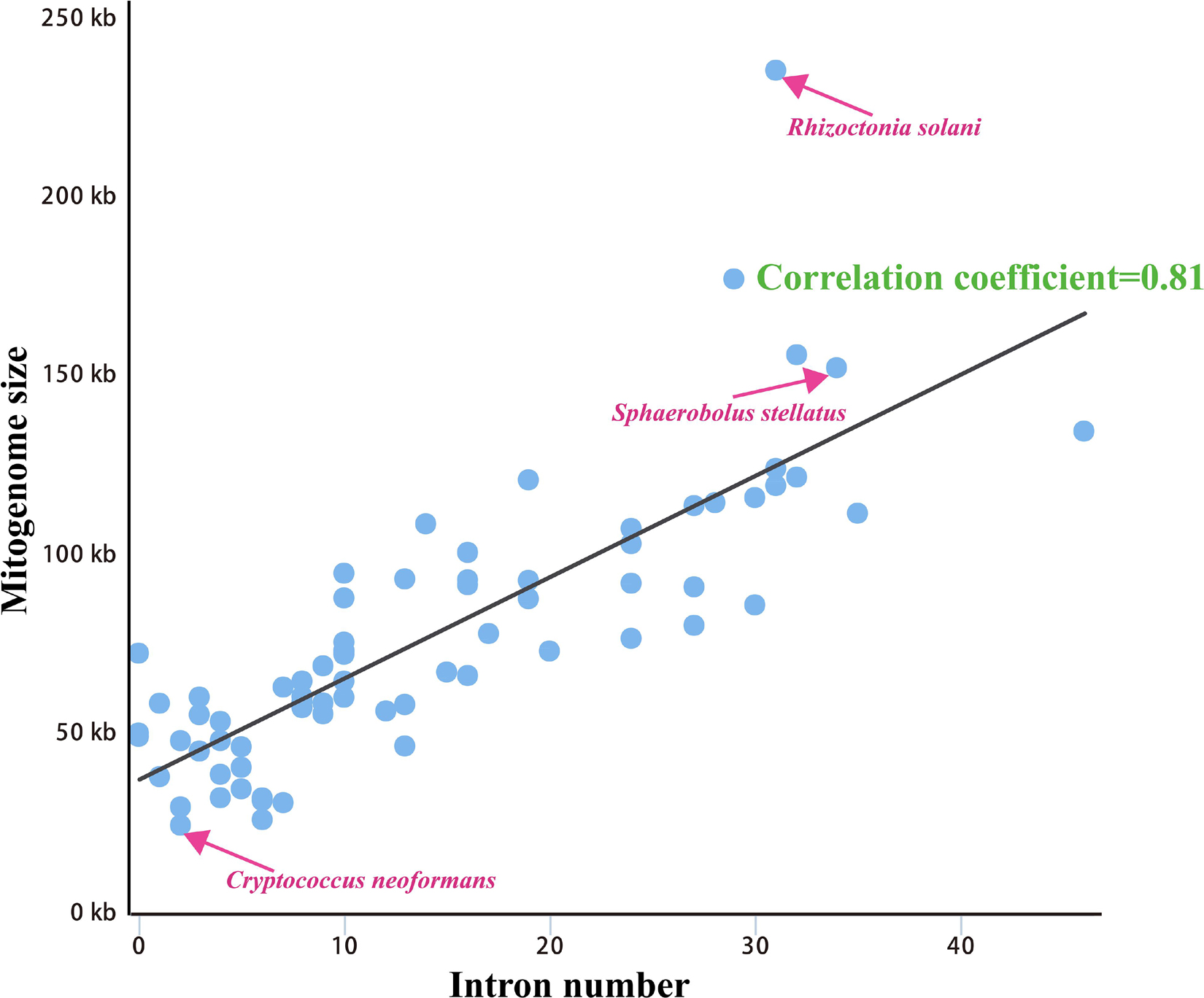
Figure 5. Pearson correlation analysis between mitogenome sizes and intron numbers of 75 Basidiomycota species.
The largest host gene of Basidiomycetes introns was the cox1 gene, which harbored 33.93% of the mitochondrial introns. We analyzed the intron dynamics of cox1 gene in the present study. A total of 43 Pcls were detected in cox1 genes of the 75 Basidiomycota species, of which 13 were considered as widely distributed Pcls (present in more than 1/5 Basidiomycota species) (Figure 6). P383 was the most common Pcl in Basidiomycota mitogenomes, which was distributed in 40 of the 75 Basidiomycota species (Figure 7). Followed by the P1107, it could be detected in 35 of the 75 Basidiomycota mitogenomes. Thirty Pcls were considered as rare Pcls in Basidiomycota, which were distributed in less than 1/5 Basidiomycota species. Several Pcls, including P166, P193, P218, P309, P318, P623, P701, P726, P1058, P1117, and P1281, were only distributed in one of the 75 Basidiomycota species. However, some of these rare Pcls were detected in mitogenomes from distant species, such as Chaetosphaeridium globosum (Turmel et al., 2002) and Rhizophydium sp. 136 (Forget et al., 2002), indicating possible horizontal gene transfer events occurred in evolution. The mitogenome of S. stellatus contained 1 rare Pcls (P1281) never detected in other species, indicating high diversities of introns in the S. stellatus mitogenome. The origin and evolution of introns in S. stellatus and other Basidiomycota species needs further studies.
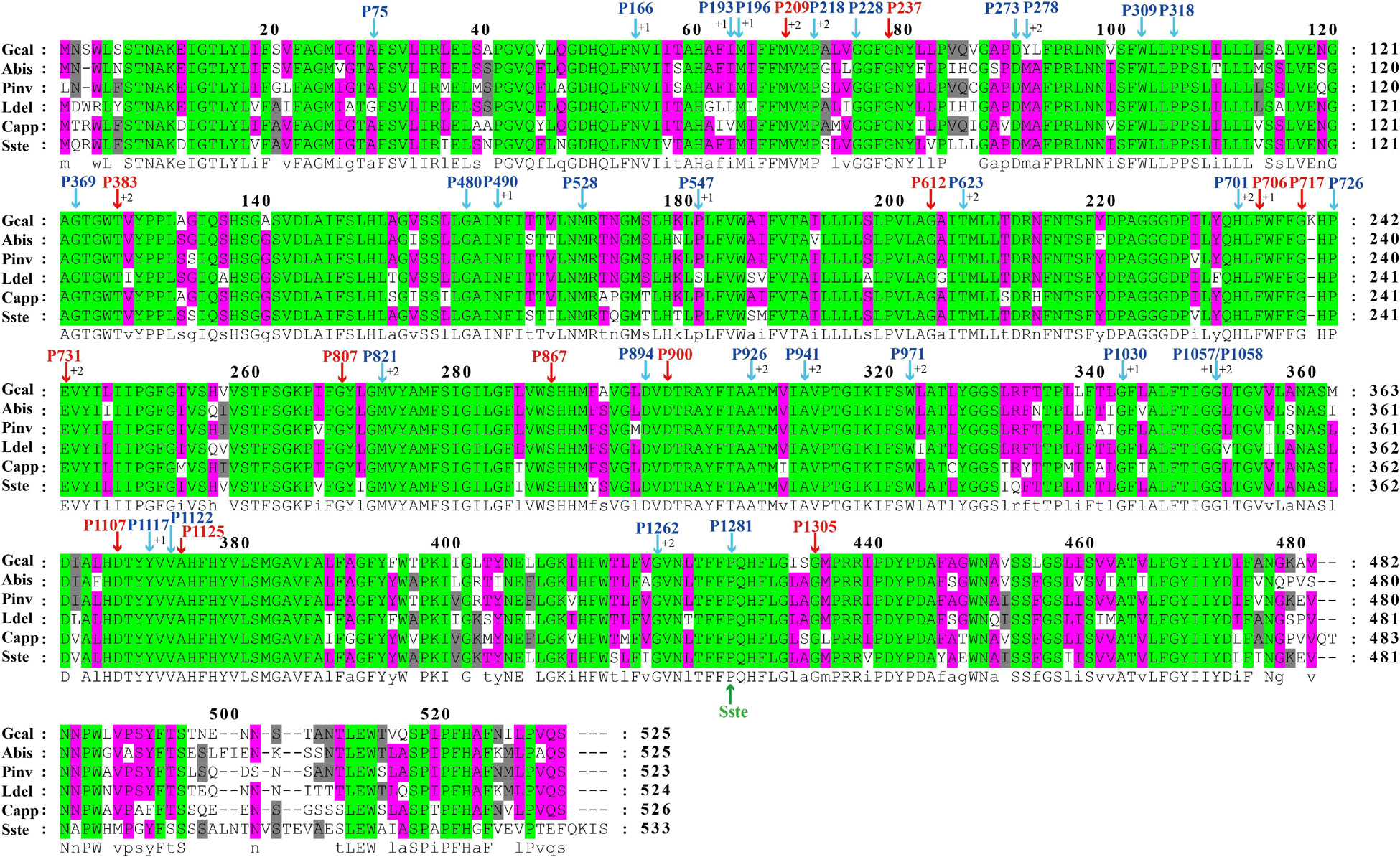
Figure 6. The insertion sites of different Pcls in cox1 coding regions of 6 basidiomycete species from different orders. The cox1 amino acids of the 6 basidiomycete species were first aligned using Clustal W. The Pcls in red indicate that they are widely distributed introns in the 75 basidiomycetes, while Pcls in blue indicate they are rare Pcls in the 75 basidiomycetes. The symbol ‘Sste’ in green font represents the novel intron identified in Sphaerobolus stellatus. The symbols ‘+1’ and ‘+2’ indicate that the insertion of the intron occurs inside the indicated codon: between the nt 1 and nt 2 of this codon for ‘+1’ and between the nt 2 and nt 3 for ‘+2’. Species ID are shown in Supplementary Table S1.
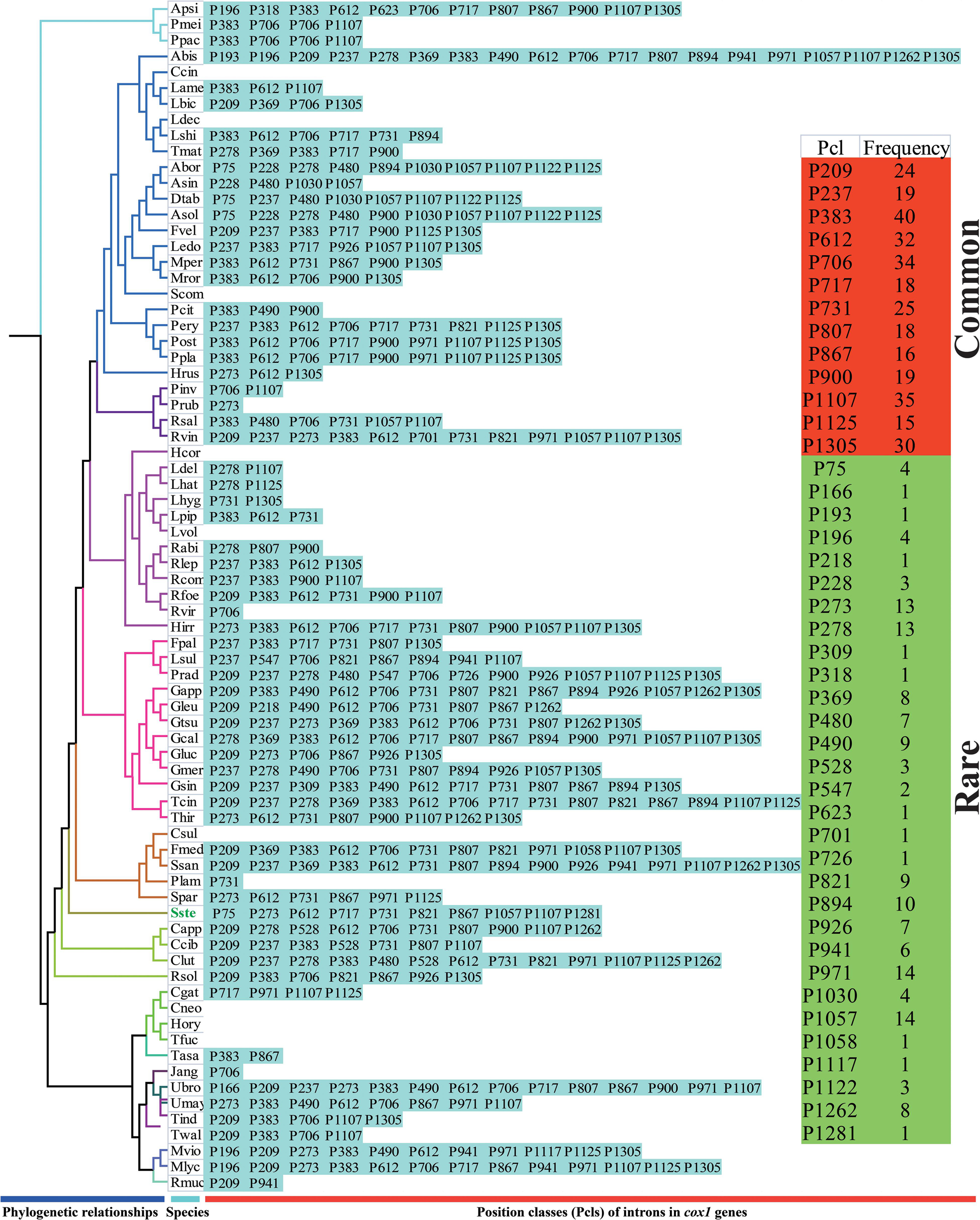
Figure 7. Pcl information of cox1 gene of the 75 Basidiomycota species. The Pcls were name according to its insertion site in the corresponding reference sequence (Ganoderma calidophilum: MH252535). Introns present in more than 1/5 Basidiomycota species was considered as common introns. The phylogenetic positions of the 75 Basidiomycota species were established using the Bayesian inference (BI) method and Maximum Likelihood (ML) method based on 15 concatenated mitochondrial core proteins and 2 rRNA genes. Species ID are shown in Supplementary Table S1.
Comparative Mitogenome and Gene Arrangement Analyses
The sizes of 75 Basidiomycota mitogenomes we tested varied greatly, ranging from 24,874 bp to 235,849 bp, with an average size of 77,184 bp (Supplementary Table S1). The 152,722 bp mitogenome of S. stellatus was the fourth largest among the 75 Basidiomycota mitogenomes detected, which was only smaller than Phlebia radiata (156, 348 bp) (Salavirta et al., 2014) from the order Polyporales, Rhizoctonia solani (235,849 bp) (Losada et al., 2014) from the order Cantharellales, and Ustilago bromivora (177,540 bp) (acc. LT558140 in the NCBI database) from the order Ustilaginales. The GC content of the S. stellatus mitogenome was about the average (27.34%) of that of the 75 Basidiomycota species. Forty-one of the 75 Basidiomycota mitogenomes had negative AT skews, and the remaining 34 had positive AT skews. Among the 75 Basidiomycota mitogenomes we detected, 57 species had positive GC skews. The mitogenome of Rhizoctonia solani contained the most PCGs, with each Basidiomycota species containing 14-127 PCGs. The S. stellatus mitogenome contained the third most introns among the 75 Basidiomycota mitogenomes detected, which was only less than Agaricus bisporus (Ferandon et al., 2013) and Sanghuangporus sanghuang (Han et al., 2018). All the 75 Basidiomycota species contained two rRNA genes. In addition, 20–35 tRNA genes were detected in the 75 Basidiomycota species.
The arrangements of 15 core PCGs and 2 rRNA genes varied greatly at family levels (Figure 8). Any species from different families had different gene orders, indicating large-scale gene rearrangements occurred in the evolution of Basidiomycota mitogenomes. Even within the same genera, we also observed large-scale gene rearrangements, including Laccaria, Rhizopogon, Lyophylum, Armillaria, Ustilago, and Microbotryum. Mitochondrial gene shifts and inversions were observed in the mitogenome of S. stellatus compared with other mitogenomes, which showed the S. stellatus mitogenome had a unique gene order among all Basidiomycota species detected.
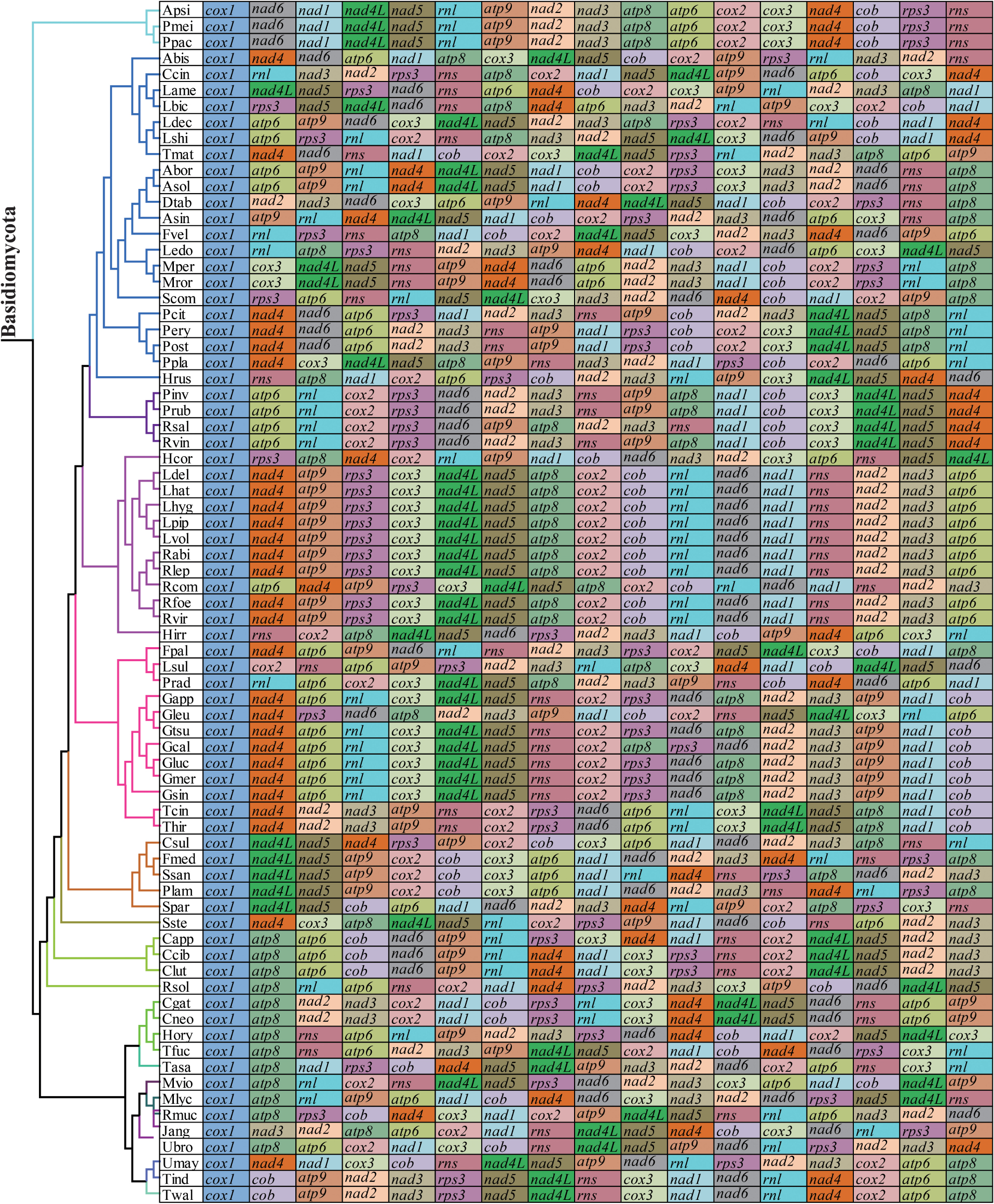
Figure 8. Mitochondrial gene arrangement analysis of 75 Basidiomycota mitogenomes. 15 core protein coding genes and 2 rRNA genes were included in the gene arrangement, starting from cox1 gene.
Phylogenetic Analysis
Identical and well-supported tree topologies were obtained using maximum likelihood (ML) and Bayesian inference (BI) methods based on the combined mitochondrial gene set (15 core PCGs + 2 rRNA genes) (Figure 9). All major clades within the trees had good support values (BPP ≥ 0.99; BS ≥ 98). Based on the phylogenetic analysis, the 75 Basidiomycota species could be divided into 15 major clades, corresponding to the orders Pucciniales, Agaricales, Boletales, Russulales, Polyporales, Hymenochaetales, Geastrales, Cantharellales, Tremellales, Trichosporonales, Microbotryales, Sporidiobolales, Microstromatales, Ustilaginales, and Tilletiales. The phylogenetic analysis indicated that S. stellatus had close relationships with Hymenochaetales and Cantharellales. The results showed that mitochondrial genes were effective molecular markers for phylogenetic analysis of Basidiomycota species.
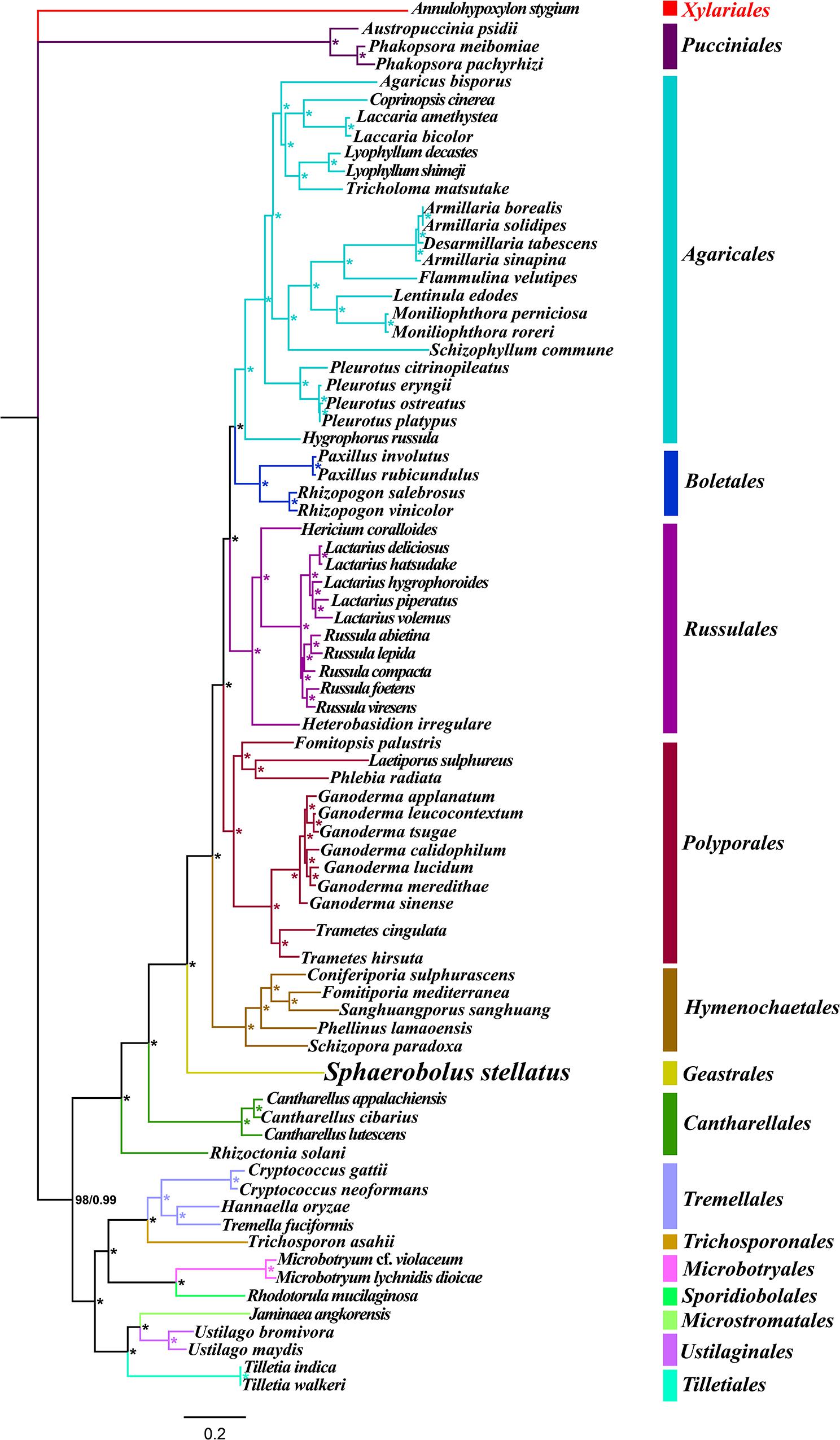
Figure 9. Molecular phylogeny of 75 Basidiomycota species based on Bayesian inference (BI) and Maximum Likelihood (ML) analyses of 15 protein coding genes and two rRNA genes. Support values are bayesian posterior probabilities (BPP, before slash) and bootstrap values (BS, after slash). The asterisk indicates that the BPP and BS values are 1 and 100, respectively. Species and NCBI accession numbers for genomes used in the phylogenetic analysis are provided in Supplementary Table S1.
Discussion
Size Variations and Intron Dynamics of Basidiomycota Mitogenomes
Compared with conservative mitogenome sizes of animals, fungal mitogenome sizes varied greatly. The mitogenome size of 75 Basidiomycota species we tested ranged from 24,874 bp to 235,849 bp. The mitogenome of S. stellatus was the fourth largest in Basidiomycota. Correlation analysis showed that the number of introns was closely related to the size of mitogenomes in Basidiomycota. The results indicated that the variation of introns was the main factor leading to the size variations of mitogenomes in Basidiomycota, which was consistent with previous studies (Mardanov et al., 2014; Liu et al., 2019). In some mitogenomes, such as Rhizoctonia solani, Phlebia radiata, and S. stellatus, we found accumulation of intergenic sequences and plasmid-derived genes (Losada et al., 2014; Salavirta et al., 2014), which also led to the size variations of mitogenomes. The cox1 gene was the largest host gene of basidiomycete introns (Wang et al., 2020), which harbored 33.93% of the total inrons in the 75 basidiomycetes. So the dynamics of introns in cox1 genes could markedly affect mitogenome size of basidiomycetes. In addition, the exon-intron borders of rRNA genes in Basidiomycota are difficult to identify accurately. So we analyzed the intron dynamics of cox1 gene in the present study. We found that the quantity and Pcl of intron varied greatly between different Basidiomycota species, even between species from the same genera, which indicated that intron loss/gain events occurred in the evolution of Basidiomycota. However, some rare Pcls in Basidiomycota were detected from distinct species in other phyla (Forget et al., 2002; Turmel et al., 2002), showing potential horizontal gene transferring events. The S. stellatus mitogenome contained a novel intron, which was never detected from other species. Further studies are needed to reveal the origin and evolution of introns in S. stellatus.
Gene Rearrangements in Basidiomycota Mitogenomes
In the present study, we found that Basidiomycota species from different families had different mitochondrial gene arrangements, indicating large-scale gene rearrangements occurred in the evolution of Basidiomycota (Li et al., 2019b). Compared with the mitogenome of Basidiomycota, the arrangement of mitochondrial genes in animals is more conservative (Aguileta et al., 2014). However, with the rapid development of next generation sequencing, mitochondrial genome rearrangements were also detected in some animal species and several models were proposed to reveal rearrangements of mitogenomes in animals (Sankoff et al., 1992; Lavrov et al., 2002; Xia et al., 2016). The mechanism of mitogenome rearrangement in fungi was less studied and the accumulation of repetitive sequences was believed to be closely related to the rearrangement of fungal mitogenomes (Aguileta et al., 2014). The average repeat content of the 75 Basidiomycota mitogenomes was >2.5%, which may lead to gene rearrangements in Basidiomycota mitogenomes. In addition, we found that the S. stellatus mitogenome had a unique gene arrangement, which was different from other species in Basidiomycota. More mitogenomes from the order Geastrales need to be sequenced and analyzed to assess conservations and variations of mitochondrial gene arrangement in the order Geastrales.
Gene Transfer Between Mitochondrial and Nuclear Genomes
Most mitochondrial genes have been transferred to nuclear genomes in evolution, which was considered to have many advantages (Adams and Palmer, 2003). So far, only a dozen to one hundred mitochondrial genes have been retained in eukaryotic mitogenomes (Allen, 2015). Genes naturally transferring between nuclear and mitochondrial genome have been observed in various organisms, which plays an important role in species evolution and environmental adaptation (Adams et al., 2002; Zhao et al., 2018). In the present study, large aligned fragments between the mitochondrial and nuclear genomes of S. stellatus were observed, which included gene coding regions and intergenic regions. The effects of natural gene transferring between mitochondrial and nuclear genomes on the evolution and development or growth of S. stellatus need to be further studied.
Phylogenetic Analysis of Basidiomycota
Limited morphological characters and the overlapping of some morphological features make it difficult to identify Basidiomycota species accurately (Li et al., 2019c). With the rapid development of next generation sequencing technology, mitochondrial genes have been widely used as molecular markers to analyze population genetics, taxonomy and biogeography of animals (Cameron, 2014; Li et al., 2015; Wang and Xu, 2020; Wang et al., 2020). However, phylogenetic studies of Basidiomycota species based on combined mitochondrial gene sets were few due to limited number of fungal mitogenomes available in public databases. In the present study, over two thirds of Basidiomycota mitogenomes available were included in the phylogenetic study. A well-supported phylogenetic tree was obtained based on the combined mitochondrial gene set. The result indicted that the mitogenome was suitable for study of phylogeny of Basidiomycota species. More mitogenomes of Basidiomycota need to be studied to reveal the evolution and phylogeny of Basidiomycetes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
QL, YR, and JY conceived and designed the experiments and contributed reagents, materials, and analysis tools. QL and JC performed the experiments. QL, WL, and JC analyzed the data. QL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01970/full#supplementary-material
Footnotes
References
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/s1055-7903(03)00194-5
Adams, K. L., Qiu, Y. L., Stoutemyer, M., and Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 9905–9912. doi: 10.1073/pnas.042694899
Aguileta, G., de Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Allen, J. F. (2015). Why chloroplasts and mitochondria retain their own genomes and genetic systems: colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. U.S.A. 112, 10231–10238. doi: 10.1073/pnas.1500012112
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bernt, M., Donath, A., Juhling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Bleasby, A. J., and Wootton, J. C. (1990). Construction of validated, non-redundant composite protein sequence databases. Protein Eng. 3, 153–159. doi: 10.1093/protein/3.3.153
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Chen, Y., Ye, W., Zhang, Y., and Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Deng, Y., Hsiang, T., Li, S., Lin, L., Wang, Q., Chen, Q., et al. (2018). Comparison of the mitochondrial genome sequences of six Annulohypoxylon stygium isolates suggests short fragment insertions as a potential factor leading to larger genomic size. Front. Microbiol. 9:2079. doi: 10.3389/fmicb.2020.02079
Ernster, L., and Schatz, G. (1981). Mitochondria: a historical review. J. Cell Biol. 91, 227s–255s. doi: 10.1083/jcb.91.3.227s
Ferandon, C., Moukha, S., Callac, P., Benedetto, J. P., Castroviejo, M., and Barroso, G. (2010). The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I Introns. PLoS One 5:e14048. doi: 10.1371/journal.pone.0014048
Ferandon, C., Xu, J., and Barroso, G. (2013). The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 55, 85–91. doi: 10.1016/j.fgb.2013.01.009
Forget, L., Ustinova, J., Wang, Z., Huss, V. A., and Lang, B. F. (2002). Hyaloraphidium curvatum: a linear mitochondrial genome, tRNA editing, and an evolutionary link to lower fungi. Mol. Biol. Evol. 19, 310–319. doi: 10.1093/oxfordjournals.molbev.a004084
Geml, J., Davis, D. D., and Geiser, D. M. (2005a). Phylogenetic analyses reveal deeply divergent species lineages in the genus Sphaerobolus (Phallales: Basidiomycota). Mol. Phylogenet. Evol. 35, 313–322. doi: 10.1016/j.ympev.2005.01.014
Geml, J., Davis, D. D., and Geiser, D. M. (2005b). Systematics of the genus Sphaerobolus based on molecular and morphological data, with the description of Sphaerobolus ingoldii sp. nov. Mycologia 97, 680–694. doi: 10.3852/mycologia.97.3.680
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–a baiting and iterative mapping approach. Nucleic Acids Res. 41, e129. doi: 10.1093/nar/gkt371
Han, J. G., Oh, J., Jo, J. W., Kim, C. S., Kwag, Y. N., Han, S. K., et al. (2018). The complete mitochondrial genome of Sanghuangporus sanghuang (Hymenochaetaceae, Basidiomycota). Mitochond. DNA Part B Resour. 3, 456–457. doi: 10.1080/23802359.2018.1462116
Ingold, C. T. (1968). Fruiting in Sphaerobolus: an effect of yellow light reversed by blue. Nature 219:1265. doi: 10.1038/2191265a0
Ingold, C. T. (1969). Effect of blue and yellow light during the later developmental stages of Sphaerobolus. Am. J. Bot. 56, 759–766. doi: 10.1002/j.1537-2197.1969.tb09723.x
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kohler, A., Kuo, A., Nagy, L. G., Morin, E., Barry, K. W., Buscot, F., et al. (2015). Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in Mycorrhizal mutualists. Nat. Genet. 47, 410–415. doi: 10.1038/ng.3223
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773.
Lavrov, D. V., Boore, J. L., and Brown, W. M. (2002). Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol. Biol. Evol. 19, 163–169. doi: 10.1093/oxfordjournals.molbev.a004068
Li, H., Shao, R., Song, N., Song, F., Jiang, P., Li, Z., et al. (2015). Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5:8527.
Li, Q., Chen, C., Xiong, C., Jin, X., Chen, Z., and Huang, W. (2018a). Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Appl. Microbiol. Biotechnol. 102, 6143–6153. doi: 10.1007/s00253-018-9082-6
Li, Q., Wang, Q., Chen, C., Jin, X., Chen, Z., Xiong, C., et al. (2018b). Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 119, 792–802. doi: 10.1016/j.ijbiomac.2018.07.197
Li, Q., Yang, M., Chen, C., Xiong, C., Jin, X., Pu, Z., et al. (2018c). Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus. Sci. Rep. 8:9104.
Li, Q., He, X., Ren, Y., Xiong, C., Jin, X., Peng, L., et al. (2020a). Comparative mitogenome analysis reveals mitochondrial genome differentiation in ectomycorrhizal and asymbiotic amanita species. Front. Microbiol. 11:1382. doi: 10.3389/fmicb.2020.01382
Li, Q., Ren, Y., Shi, X., Peng, L., Zhao, J., Song, Y., et al. (2019a). Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (Rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the subphylum Agaricomycotina. Int. J. Mol. Sci. 20:5167. doi: 10.3390/ijms20205167
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019b). Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 121, 249–260. doi: 10.1016/j.ijbiomac.2018.10.029
Li, Q., Wang, Q., Jin, X., Chen, Z., Xiong, C., Li, P., et al. (2019c). Characterization and comparison of the mitochondrial genomes from two Lyophyllum fungal species and insights into phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 121, 364–372. doi: 10.1016/j.ijbiomac.2018.10.037
Li, Q., Xiang, D., Wan, Y., Wu, Q., Wu, X., Ma, C., et al. (2019d). The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. Int. J. Biol. Macromol. 139, 397–408. doi: 10.1016/j.ijbiomac.2019.08.003
Li, Q., Ren, Y., Xiang, D., Shi, X., Zhao, J., Peng, L., et al. (2020b). Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 11:12.
Li, Q., Yang, L., Xiang, D., Wan, Y., Wu, Q., Huang, W., et al. (2020c). The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 145, 974–984. doi: 10.1016/j.ijbiomac.2019.09.188
Liu, W., Cai, Y., Zhang, Q., Chen, L., Shu, F., Ma, X., et al. (2019). The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 143, 373–381. doi: 10.1016/j.ijbiomac.2019.12.056
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581.
Losada, L., Pakala, S. B., Fedorova, N. D., Joardar, V., Shabalina, S. A., Hostetler, J., et al. (2014). Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 352, 165–173. doi: 10.1111/1574-6968.12387
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57.
Mardanov, A. V., Beletsky, A. V., Kadnikov, V. V., Ignatov, A. N., and Ravin, N. V. (2014). The 203 kbp mitochondrial genome of the phytopathogenic fungus Sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS One 9:e107536. doi: 10.1371/journal.pone.0107536
McBride, H. M., Neuspiel, M., and Wasiak, S. (2006). Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560.
Munoz-Gomez, S. A., Wideman, J. G., Roger, A. J., and Slamovits, C. H. (2017). The origin of mitochondrial cristae from Alphaproteobacteria. Mol. Biol. Evol. 34, 943–956.
NCBI Resource Coordinators (2017). Database resources of the national center for biotechnology information. Nucleic Acids Res. 44, D7–D19.
Richards, M. B., Macaulay, V. A., Bandelt, H. J., and Sykes, B. C. (1998). Phylogeography of mitochondrial DNA in western Europe. Ann. Hum. Genet. 62, 241–260. doi: 10.1046/j.1469-1809.1998.6230241.x
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Salavirta, H., Oksanen, I., Kuuskeri, J., Makela, M., Laine, P., Paulin, L., et al. (2014). Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS One 9:e97141. doi: 10.1371/journal.pone.097141
Sandor, S., Zhang, Y., and Xu, J. (2018). Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102, 9433–9448. doi: 10.1007/s00253-018-9350-5
Sankoff, D., Leduc, G., Antoine, N., Paquin, B., Lang, B. F., and Cedergren, R. (1992). Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proc. Natl. Acad. Sci. U.S.A. 89, 6575–6579. doi: 10.1073/pnas.89.14.6575
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Slater, G. S., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6:31. doi: 10.1186/s13104-016-1900-31
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Thompson, J. D., Higgins, D. G., Gibson, T. J., and Clustal, W. (1994). improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Turmel, M., Otis, C., and Lemieux, C. (2002). The chloroplast and mitochondrial genome sequences of the charophyte Chaetosphaeridium globosum: insights into the timing of the events that restructured organelle DNAs within the green algal lineage that led to land plants. Proc. Natl. Acad. Sci. U.S.A. 99, 11275–11280. doi: 10.1073/pnas.162203299
Vaidya, G., Lohman, D. L., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Valach, M., Burger, G., Gray, M. W., and Lang, B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Wang, J., Zhang, L., Zhang, Q. L., Zhou, M. Q., Wang, X. T., Yang, X. Z., et al. (2017). Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PeerJ 5:e3661. doi: 10.7717/peerj.3661
Wang, X., Song, A., Wang, F., Chen, M., Li, X., Li, Q., et al. (2020). The 206 kbp mitochondrial genome of Phanerochaete carnosa reveals dynamics of introns, accumulation of repeat sequences and plasmid-derived genes. Intern. J. Biol. Macromol. 162, 209–219. doi: 10.1016/j.ijbiomac.2020.06.142
Wang, Y., and Xu, J. (2020). Mitochondrial genome polymorphisms in the human pathogenic fungus Cryptococcus neoformans. Front. Microbiol. 11:706. doi: 10.3389/fmicb.2020.0706
Xia, Y., Zheng, Y., Murphy, R. W., and Zeng, X. (2016). Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genomics 17:965. doi: 10.1186/s13104-016-1900-965
Keywords: mitochondrial genome, protein coding gene, repeat sequence, intron, gene rearrangement, phylogenetic analysis
Citation: Ye J, Cheng J, Ren Y, Liao W and Li Q (2020) The First Mitochondrial Genome for Geastrales (Sphaerobolus stellatus) Reveals Intron Dynamics and Large-Scale Gene Rearrangements of Basidiomycota. Front. Microbiol. 11:1970. doi: 10.3389/fmicb.2020.01970
Received: 24 April 2020; Accepted: 24 July 2020;
Published: 11 August 2020.
Edited by:
Mohamed Hijri, Université de Montréal, CanadaReviewed by:
Abbot Okotie Oghenekaro, University of Manitoba, CanadaShanshan Liu, The First Affiliated Hospital of Bengbu Medical College, China
Copyright © 2020 Ye, Cheng, Ren, Liao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Li, bGVlcTExMEAxMjYuY29t
†Present address: Qiang Li, College of Food and Biological Engineering, Chengdu University, Chengdu, China