 Dahe Zhao1†
Dahe Zhao1† Shengjie Zhang1,2†
Shengjie Zhang1,2† Qiong Xue1,2
Qiong Xue1,2 Junyu Chen1,2Jian Zhou1Feiyue Cheng1,2
Junyu Chen1,2Jian Zhou1Feiyue Cheng1,2 Ming Li1Yaxin Zhu1Haiying Yu1
Ming Li1Yaxin Zhu1Haiying Yu1 Songnian Hu1,2
Songnian Hu1,2 Yanning Zheng1
Yanning Zheng1 Shuangjiang Liu1,2
Shuangjiang Liu1,2 Hua Xiang1,2*
Hua Xiang1,2*- 1State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
Soda-saline lakes are a special type of alkaline lake in which the chloride concentration is greater than the carbonate/bicarbonate concentration. Due to the high pH and a usually higher osmotic pressure than that of a normal soda lake, the microbes may need more energy to thrive in such a double-extreme environment. In this study, we systematically investigated the microbiome of the brine and sediment samples of nine artificially separated ponds (salinities from 5.5% to saturation) within two soda-saline lakes in Inner Mongolia of China, assisted by deep metagenomic sequencing. The main inorganic ions shaped the microbial community in both the brines and sediments, and the chloride concentration exhibited the most significant effect. A total of 385 metagenome-assembled genomes (MAGs) were generated, in which 38 MAGs were revealed as the abundant species in at least one of the eighteen different samples. Interestingly, these abundant species also represented the most branches of the microbiome of the soda-saline lakes at the phylum level. These abundant taxa were close relatives of microorganisms from classic soda lakes and neutral saline environments, but forming a combination of both habitats. Notably, approximately half of the abundant MAGs had the potential to drive dissimilatory sulfur cycling. These MAGs included four autotrophic Ectothiorhodospiraceae MAGs, one Cyanobacteria MAG and nine heterotrophic MAGs with the potential to oxidize sulfur, as well as four abundant MAGs containing genes for elemental sulfur respiration. The possible reason is that reductive sulfur compounds could provide additional energy for the related species, and reductions of oxidative sulfur compounds are more prone to occur under alkaline conditions which support the sulfur cycling. In addition, a unique 1,4-alpha-glucan phosphorylation pathway, but not a normal hydrolysis one, was found in the abundant Candidatus Nanohaloarchaeota MAG NHA-1, which would produce more energy in polysaccharide degradation. In summary, this work has revealed the abundant taxa and favorable pathways in the soda-saline lakes, indicating that efficient energy regeneration pathway may increase the capacity for environmental adaptation in such saline-alkaline environments. These findings may help to elucidate the relationship between microbial metabolism and adaptation to extreme environments.
Introduction
A soda lake is a type of saline lake with extremely high pH and salinity mainly due to high concentrations (exceeding an equivalent percentage of 25) of carbonate/bicarbonate (Grant and Sorokin, 2011; Boros and Kolpakova, 2018). Recently, soda lakes were further divided into “soda” and “soda-saline” types based on the level of bicarbonate and carbonate. It is defined as “soda” type when the sum of bicarbonate and carbonate concentrations are the first in the rank of dominant ions, and is “soda-saline” type when the concentration of other ions is higher than that of bicarbonate/carbonate (Boros and Kolpakova, 2018). In these saline and alkaline environments, microorganisms exhibit surprisingly high biodiversity (Grant, 2006; Mesbah et al., 2007; Asao et al., 2011; Lanzen et al., 2013), relatively high primary productivity rates (Melack and Kilham, 1974; Melack, 1981; Kompantseva et al., 2009; Antony et al., 2013; Zorz et al., 2019), vigorous oxidation and reduction reactions of sulfur (Sorokin et al., 2010, 2011; Stam et al., 2010; Tourova et al., 2013; Vavourakis et al., 2019), and elevated metabolic activity of cellulose, methane, nitrogen and arsenic (Iversen et al., 1987; Carini and Joye, 2008; Oremland et al., 2017; Phitsuwan et al., 2019). High concentrations of inorganic ions, such as (bi)carbonate and phosphate provide adequate essential elements, while hydrogen sulfide exhibits low toxicity under alkaline conditions (Sorokin et al., 2015). This would support the microbes inhabiting such alkaline and saline environments, and playing important roles in the elemental cycling (Sorokin et al., 2014).
In the brines of alkaline soda lakes, Bacteroidetes, Alphaproteobacteria, Gammaproteobacteria, and Euryarchaeota were identified as taxa with the highest levels of abundance at different salinities (from 170 to 400 g/L) by both amplicon sequencing of the 16S rRNA gene and direct metagenomic sequencing (Vavourakis et al., 2016). The genomes of haloalkaliphilic members of the Candidate Phyla Radiation (CPR) and several hundred other novel prokaryote lineages were obtained from the metagenomic assembly of sequences from the sediment of soda lakes, and the Wood-Ljungdahl (WL) pathway for carbon fixation was detected in more taxa than already known groups from the same samples (Vavourakis et al., 2018). The autotrophic microbial community based on the detection of molecular markers, ribulose-1,5-bisphosphate carboxylase (RuBisCO) and ATP citrate lyase (Acl) in the Calvin-Benson-Bassham (CBB) and reductive tricarboxylic acid cycles, respectively, indicated that haloalkaliphilic cyanobacteria and sulfur-oxidizing bacteria of the genus Halorhodospira were predominant in soda lakes (Kovaleva et al., 2011; Tourova et al., 2011). Interestingly, even alkaline soda lakes separated by a large distance between Asia and North America share a similar core microbiome (Zorz et al., 2019).
As is well known, haloalkaliphiles from three domains of life thrive in the extreme environments with high salinity and alkalinity (Banciu and Muntyan, 2015). Monovalent cation/proton antiporters are widely present in archaea and bacteria and function in intracellular pH homeostasis (Krulwich et al., 2011). The biosynthesis or uptake of compatible solutes (such as glycine betaine and ectoine) by halophilic and haloalkaliphilic bacteria are commonly used as the primary mechanism to resist extracellular osmotic pressure (Roberts, 2005). Haloarchaea and anaerobic Natranaerobiaceae primarily maintain osmotic balance using K+ and Cl– import systems (Gunde-Cimerman et al., 2018). Because of relatively low solubility and incomplete ionization of carbonate/bicarbonate (comparing with chloride), haloalkaliphilic microbes (prefer NaCl) seemed to resist more osmotic pressure than natronophilic ones (prefer NaHCO3/Na2CO3) under alkaline conditions (Sorokin et al., 2015). The microbes inhabiting soda lakes, especially the soda-saline type, are considered to be the ideal materials to research the environmental adaptation to high salinity and alkalinity. Importantly, microbes must consume a large amount of energy to maintain neutral cytoplasm and osmotic balance in response to extreme conditions (Banciu and Muntyan, 2015). The favorable metabolic pathways of abundant taxa to produce sufficient energy need to be followed through at the metagenomics level.
Hundreds of small soda lakes and pans are located in the Inner Mongolia Autonomous Region of China (Zheng et al., 2002). Analysis of the physicochemical factors in these lakes indicated that many of them are soda-saline lakes of the chloride-carbonate-sulfate type, providing an applicable system for studying the coupling of carbon and sulfur cycling and microbial environmental adaptation. In this study, we collected brine and sediment samples from nine ponds associated with two soda-saline lakes and performed deep metagenomic sequencing. Combined with environmental characterizations, we dissected the microbial community structures and relationships based on metagenomic reads and assembled genomes, subsequently focusing on the abundant species representing most branches of a phylogenomic tree. The efficient energy regeneration pathways in the abundant MAGs may increase the capacity for environmental adaptation in such saline-alkaline environments. The superiority of energy production and thermodynamics in the abundant species was assessed to further understand the metabolic mechanism of adaptation to such extremely alkaline and saline conditions.
Materials and Methods
Physicochemical Characterization
Salinity was measured using a handheld refractometer (Beijing Wanchengbeizeng Precision Instrument Co., Ltd., Beijing, China). Conductivity, pH and the concentrations of CO32– and HCO3– were measured using a SensoDirect 150 and an MD600 Photometer (Lovibond® Water Testing, Dortmund, Germany). The concentrations of other inorganic ions (Cl–, SO42–, PO43–, Mg2+, Ca2+, and NH4+) and total organic nitrogen were measured using an AquakemTM 250 discrete photometric Autoanalyzer (Thermo Fisher Scientific, MA, United States).
DNA Extraction and Metagenomic Sequencing
Brine samples were prefiltered through four layers of gauze to eliminate eukaryotic animals and plants. The microorganisms in each sample were collected by 0.8- and 0.22-μm filters. Then, filters were used to extract DNA with a PowerWater® DNA Isolation kit (MoBio, CA, United States). DNA extracted from the same sample was mixed as a single sample. Total DNA of sediment samples was extracted using a PowerSoil® DNA Isolation kit (MoBio, CA, United States). The DNA concentration and purity were measured using a Qubit® dsDNA Assay kit with a Qubit® 2.0 Fluorometer (Life Technologies, CA, United States) and a NanoPhotometer® spectrophotometer (IMPLEN, CA, United States), respectively. The OD260/OD280 values of the samples were 1.8∼2.0, and DNA concentrations were >1 μg. Library construction and shotgun sequencing were performed using an Illumina HiSeq-2000 platform (Illumina, United States) to generate 150-bp paired-end reads.
Metagenomic Data Analysis Based on a Non-redundant Gene Catalog
Quality control of the raw reads was conducted using Readfq (V81) to remove low-quality reads and ensure that (a) each read had no more than 40 bases with a quality score smaller than 38, (b) each read had less than 10 ambiguous nucleotides, and (c) no reads shared more than a 15-bp overlap with the adapter. Clean reads were assembled into contigs using MEGAHIT (v1.1.2) (Li et al., 2015) with the following parameters: –presets meta-large (-min-count 2, –k-min 27, –k-max 127, and –k-step 10). Unassembled read pairs were retrieved by mapping paired-reads to contigs using BBMap (v37.57)2 with the following parameters: kfilter = 22, subfilter = 15, and maxindel = 80. Coassembly was conducted using MEGAHIT with the same parameters described above to acquire low-abundance reads. Contigs from single-assembled samples and one coassembly were merged together, and contigs with <500 bp were removed (Qin et al., 2014).
Open reading frames (ORFs) were predicted using MetaGeneMark (GeneMark.hmm v3.38) (Zhu et al., 2010) with default parameters. ORFs with lengths <100 bp were removed to reduce the number of pseudogenes (Qin et al., 2014). Redundancy removal was executed using CD-HIT (v4.7) (Li and Godzik, 2006) with the following parameters: -c 0.95, -G 0, -aS 0.9, -g 1, and -d 0. Clean reads from the 18 samples were mapped to the nr-ORF catalog using BWA mem (v0.7.17) (Li and Durbin, 2010) with the default parameters. ORFs with <2 reads aligned from the 18 samples were removed to prevent incorrect assembly. The abundance of each non-redundant gene in one sample was calculated based on the proportion of the mapped number of reads (counted by BamM3) divided by the gene length (Qin et al., 2012). Taxonomy assignments were performed by mapping the amino acid sequences against the NCBI non-redundant database (Version: 201709234) using Diamond (v0.9.10.111) (Buchfink et al., 2015) with the following parameters: –taxonmap, –taxonnodes, -e 1e-5, and –top 10. Non-redundant genes were assigned to the corresponding taxa calculated by the Lowest Common Ancestor (LCA) algorithm (Huson et al., 2007) in Diamond. The abundance of each metagenomic operational taxonomic unit (mOTU) (Sunagawa et al., 2013) was the sum of the abundances of all non-redundant genes assigned to that mOTU (Qin et al., 2012) and was supplied in Data Sheet 1 and 2. Functional annotation was performed using the KEGG databases by uploading to the Automatic Annotation Server (v2.1) (Moriya et al., 2007). The GENES dataset was set for Prokaryotes, while the Assignment method was set as BBH.
Binning and Post Analysis of MAGs
Single sample assembly and coassembly of three samples with same type and similar salinity (water and sediment samples of HC5, DK15, and HC17; water and sediment samples of HC22, DK20, and HC26; water and sediment samples of DK32, DK27, and HC27; total 6 groups) were conducted to obtain low abundance contigs. Contig datasets were binned using MetaBAT (v2.12.1) (Kang et al., 2015) with default parameters (contigs of less than 2500 bp were discarded) based on the tetranucleotide frequency and coverage values obtained by mapping the clean data onto the contig datasets using BWA mem (v0.7.17) (Li and Durbin, 2010). Genome completeness and contamination were estimated using CheckM (v1.0.12) (Parks et al., 2015) to generate genomes satisfying the minimum information about a MAG (Bowers et al., 2017). Draft genomes were dereplicated according to ANI using dRep (v2.2.1) (Olm et al., 2017). Gene and protein-coding sequences were predicted using Prodigal (v2.6.3) (Hyatt et al., 2010). The abundance of each MAG in each sample was calculated, equaling the sum of the coverage of all contigs multiplied by their respective length and divided by the genome size. Taxonomic assignments for each bin were performed using CheckM, the Diamond aligned to UniProt TrEMBL database (Bateman et al., 2017), and PhyloPhlan (v0.99) (Segata et al., 2013) and were manually curated afterward. A phylogenomic tree was constructed using reference genomes based on the most conserved 400 proteins across bacteria and archaea using PhyloPhlan and visualized using iTOL (v4) (Letunic and Bork, 2019). Functional annotation was conducted using GhostKOALA (‘genus_prokaryotes + family_eukaryotes + viruses’; v2.0) (Kanehisa et al., 2016) to reconstruct the metabolic pathways and was supplied in Data Sheet 4. The direction of dissimilatory sulfur metabolism by DsrAB was determined by the present or absent of dsrD and dsrEFH (Supplementary Table 8) (Anantharaman et al., 2018), and these genes were predicted based on the HMM profile from TIGRFAM (Haft et al., 2003) and Pfam (El-Gebali et al., 2019) using Hmmscan v3.1b2 (Eddy, 2011). For the phylogenetic analysis, proteins assigned to Ca. Nanohaloarchaeota and Ca. Woesearchaeota were retrieved from the corresponding 19 MAGs and aligned using MEGA X (Kumar et al., 2018) with the reference protein alpha-glucan phosphorylase from Escherichia coli (PWL89129.1).
Statistical Analysis
The datasets generated above were statistically analyzed using the free software R Project5. The sampling map was visualized using the leaflet package. The Shannon diversity indexes of samples were calculated using the vegan package6 and visualized by the ggpubr package7. Heatmap cluster analysis and principal component analysis (PCA) were visualized using the pheatmap8 and ggbiplot packages9, respectively. Redundancy analysis (RDA) was performed using the vegan package with all taxon abundances in genus level and physiochemical data. The co-occurrence network was based on the abundances of 385 MAGs across 18 samples (Supplementary Table 5). Pearson correlation coefficients were calculated using the psych package10, where a Pearson correlation coefficient > 0.9 and p-value < 0.01 were used. The network was visualized using Cytoscape (Shannon et al., 2003).
Data Availability
The raw sequence reads of 18 metagenomes were deposited in NCBI (see foot note 4) with the projectID PRJNA549802 and in gcMeta (Shi et al., 2019) with the projectID NMDC10010899. The binning results can be accessed at figshare11.
Results and Discussion
Multiple Environmental Factors Shape Microbial Community Composition
Habor Lake (DK) and Hutong Qagan Lake (HC) are located in the southwest of Inner Mongolia Autonomous Region of China (Supplementary Image 1A) and comprise numerous artificial ponds with and a typical depth of 1–5 m. The ponds from same lake share the similar ratio of inorganic salts but different total salinities. Brine and sediment samples of four DK ponds (DK15, DK20, DK27, and DK32) and five HC ponds (HC5, HC27, HC17, HC22, and HC26) were collected (Supplementary Images 1B,C). The salinities of these ponds ranged from 5.5% to saturation with pH values greater than 9.8. CO32– and HCO3– concentrations ranged from 78.33 to 820 mM and from 80.33 to 385.25 mM, respectively, while the chloride concentrations were 1.5–2.4 times as much as the sum of both. Therefore, both DK and HC were classified as soda-saline lakes of the chloride-carbonate-sulfate type (Boros and Kolpakova, 2018) and will simply be called soda-saline lakes in the text. The values of additional physicochemical parameters (magnesium ion, calcium ion, chloride, sulfate, phosphate, ammonia and total organic nitrogen concentrations) are shown in Supplementary Table 1.
Brine and sediment samples from the nine ponds were used to perform deep metagenomic sequencing (Supplementary Table 2). The average number of raw bases for the 18 samples was 15.98 Gb, and a total of 281.05 Gb of clean data was obtained after quality filtering. The bioinformatic analyses below were performed using these metagenomes.
The microbial community composition was determined based on the taxonomy assignment of total non-redundant gene catalog acquired from metagenomics assembly, open reading frame (ORF) prediction and redundancy removal, and the influences of environmental factors were analyzed by redundancy analysis (RDA) based on the microbial composition of 18 samples at the genus level. The brine and sediment samples clustered together in the RDA (Figure 1A), suggesting that the microbial community structures in the two sample types were significantly different. This conclusion was also supported by the principal component analysis (PCA) results (Supplementary Image 3A), whereas the microbial community structures of similar types of samples from different lakes (DK and HC) were generally similar (Supplementary Image 3B). Regarding microbial diversity, the Shannon-Weaver index value obtained for the sediment was considerably increased compared with that observed for the brine (Supplementary Image 4A). The sediment ecosystems, especially the anoxic environments, may provide more opportunities for niche diversification (Vavourakis et al., 2018). Again, the overall biodiversity in the DK and HC was similar (Supplementary Image 4B). For physicochemical factors, the Cl– concentration was the most influential environmental factor determining microbial composition (Figure 1A), to which pH, salinity, CO32–, HCO3–, SO42–, and conductivity were positively correlated. However, the Mg2+ concentration was negatively correlated (Figure 1A and Supplementary Image 2). The pH value, carbonate/bicarbonate concentrations and salinity appear to be of importance in shaping the prokaryotic communities in salt lakes (Pagaling et al., 2009; Simachew et al., 2016). As shown in Supplementary Image 2 and Figure 1, the Cl– concentration, which is positively correlated with pH, salinity, CO32–, HCO3–, SO42– and conductivity, was a key factor affecting microbial communities among these saline-alkaline lakes. Chloride and carbonate/bicarbonate, the primary anions and major contributors to osmotic pressure of the lakes (Supplementary Table 1), determine the microbial community composition (Banciu and Muntyan, 2015). Chloride-dominated brines exhibit approximately two times the osmotic pressure of the carbonate-dominated brines with the same Na+ molarity (Sorokin et al., 2015; Vavourakis et al., 2018), which explains why the chloride-dominated lakes had a stronger influence on microbial community than the typical (bi)carbonate-rich soda lakes (Zorz et al., 2019).
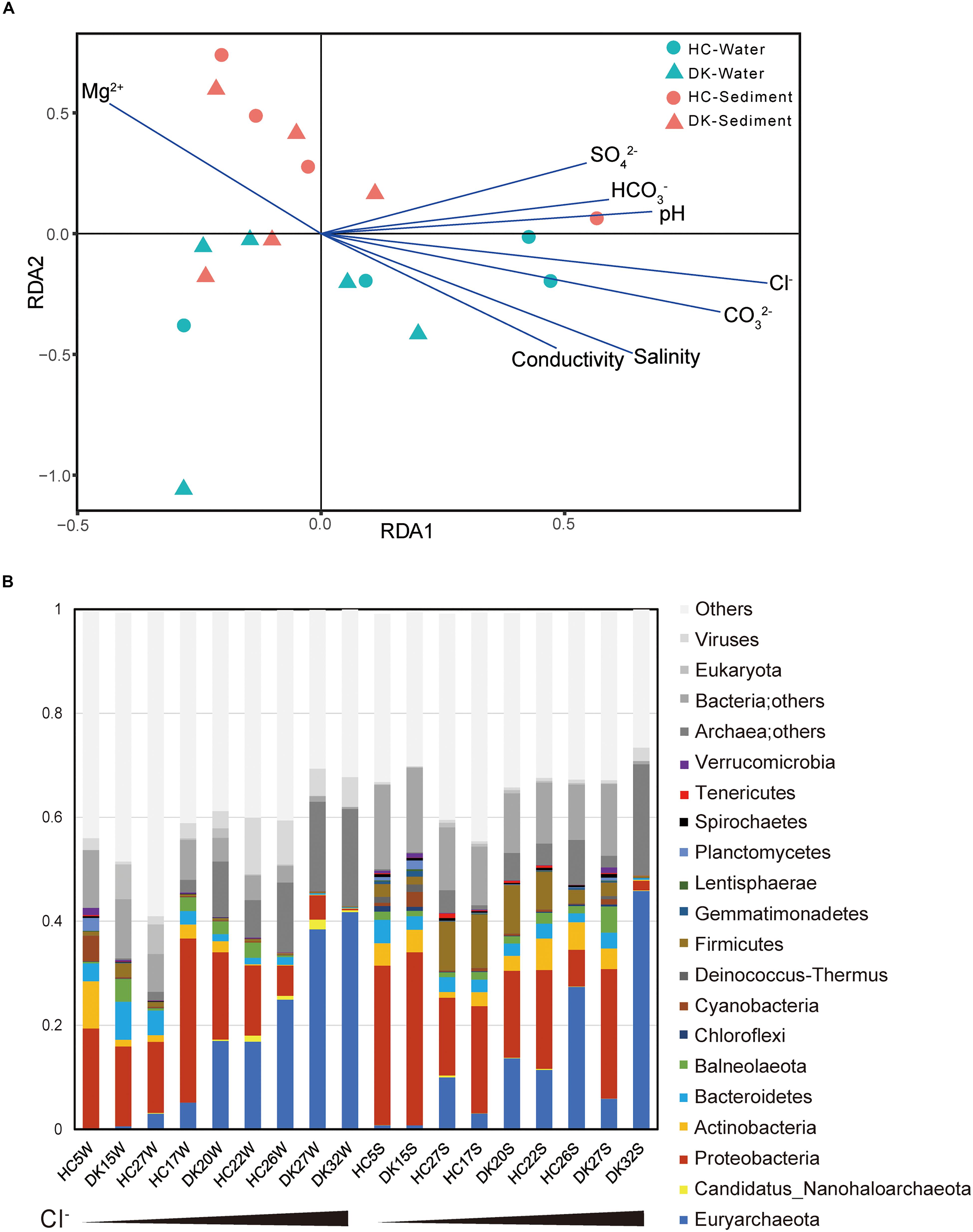
Figure 1. Relationship between environmental factors and microbial communities. (A) The influence of environmental factors on microbial community structure was analyzed by redundancy analysis. (B) The microbial composition of eighteen samples and the observed relative abundances (Supplementary Table 3) of taxa at the phylum level. The total relative abundances of taxa (greater than 0.1%) are shown. The first nine samples were water samples, whereas the last nine were sediment samples and were sorted by Cl– concentration.
We further assessed the microbial taxonomic profiles at the phylum level and their relative abundances in each of the samples based on the classification of non-redundant genes by alignment against the NCBI nr database (Figure 1B). Sixteen phyla were present at abundance levels (>0.1%) (Supplementary Table 3). Proteobacteria (bacteria) and/or Euryarchaeota (archaea) constituted the majority of phyla across the 18 samples. In brine samples, the abundance of Proteobacteria ranged from 13.6 to 31.5% in samples HC5W, DK15W, HC17W, HC27W, DK20W, and HC22W with relatively low Cl–, whereas this abundance was not greater than 5.5% in water samples containing extremely high Cl– levels (HC26W, DK27W, and DK32W). In contrast, the abundance of Euryarchaeota increased from 0.1 to 41.7% along with the increase of Cl– concentrations. Proteobacteria and Euryarchaeota were also the most abundant phyla in sediment samples. In addition, Firmicutes, Actinobacteria, Bacteroidetes, Cyanobacteria and Ca. Nanohaloarchaeota were all detected in abundance in soda-saline lakes (Figure 1B). A large variety of microorganisms were observed (Figures 1, 2, Supplementary Table 4), and a number of haloalkaliphilic microorganisms have been reported to be isolated (Grant, 2006; Antony et al., 2013; Sorokin et al., 2015).
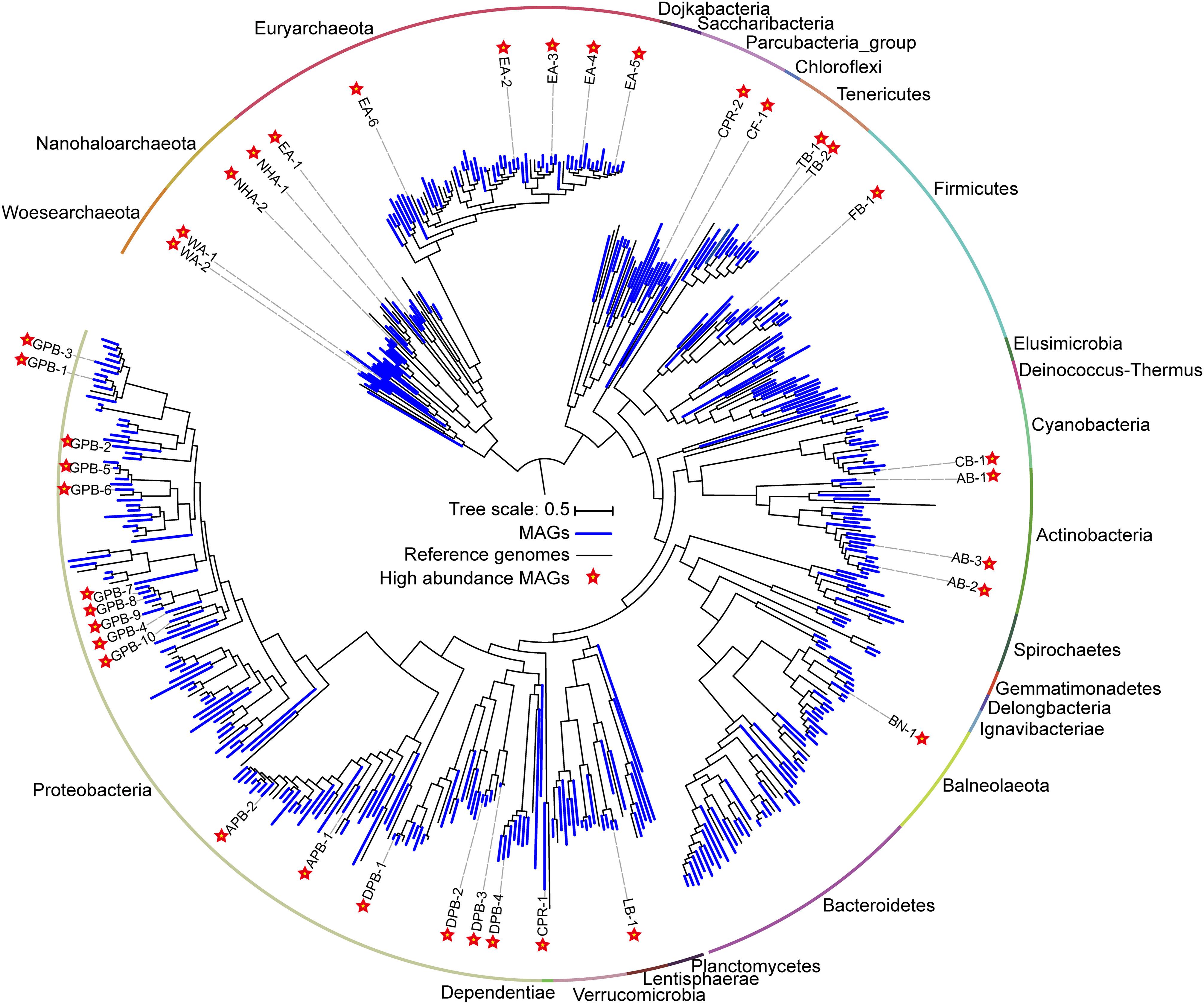
Figure 2. Phylogenomic tree of 385 metagenome-assembled genomes (MAGs) and the evolutionary distribution of abundant MAGs. The MAGs and reference genomes are colored blue and black, respectively. Abundant MAGs are marked with red five-pointed stars. The outer circle is colored by phylum. The complete tree is available with full bootstrap support values in Data Sheet 3.
MAGs Revealed Abundant Species
To determine the microbial composition and putative ecological function, contigs are binned into metagenome-assembled genomes (MAGs). We obtained 385 MAGs (completeness > 50% and contamination < 10%), in which 104 near-complete MAGs (completeness > 90% and contamination < 5%) were from 27 archaeal and bacterial phyla (Figure 2, Supplementary Table 4). The naming convention of MAGs is the abbreviation of class (in Proteobacteria), phylum (most phyla), or superphylum (CPR) with serial number (Supplementary Table 4). Of the 79 archaeal MAGs, 55 belonged to Euryarchaeota, while the remaining belonged to Ca. Nanohaloarchaeota (12 MAGs) and Ca. Woesearchaeota (12 MAGs). Of the 55 Euryarchaeota MAGs, 48 MAGs were from the class Halobacteria, and 7 were affiliated with the classes Methanomicrobia, Methanonatronarchaeia, Thermoplasmata and unclassified Euryarchaeota (Supplementary Table 4). In bacteria, most MAGs belonged to the phyla Proteobacteria (119 MAGs), Firmicutes (33 MAGs), Bacteroidetes (29 MAGs) and Actinobacteria (20 MAGs). Additionally, many other diverse MAGs were obtained, including Balneolaeota, Tenericutes, Verrucomicrobia, Cyanobacteria, Spirochetes, and CPR (Figure 2, Supplementary Table 4).
The relative abundances of the 385 MAGs in the 18 samples are shown in Figure 3. The microbiomes of the 18 brine and sediment samples comprised a small number of abundant species (relative abundance > 50% of MAGs with the highest coverage in the same niche) and a large number of rare MAGs (Figure 3). We observed more abundant bacterial MAGs in brine and sediment samples with relatively low chloride and salinity, especially Gammaproteobacteria (Thioalkalivibrio, Spiribacter, Thiohalomonas, and Halorhodospira), Deltaproteobacteria (Desulfuromusa and Desulfonatronospira) Alphaproteobacteria (Methylobacterium and Roseibaca), Actinobacteria (Ilumatobacter), and Tenericutes. Cyanobacteria MAG CB-1 was abundant in the brine with the lowest concentration of chloride (Figure 4). Two CPR MAGs were found in abundance, including CPR-1 in DK15W and CPR-2 in HC27S (Figure 4). Some bacterial MAGs were abundant in relatively high chloride samples, such as Balneolaeota MAG BN-1 in HC26S, Deltaproteobacteria MAG DPB-4 and Gammaproteobacteria MAG GPB-6 in HC26W (Figure 4). Thioalkalivibrio, Halorhodospira, Desulfonatronospira and Roseibaca were observed in classic soda lakes (Vavourakis et al., 2018). Thiohalomonas, Desulfuromusa, and Spiribacter were usually obtained from neutral saline environments (Vandieken et al., 2006; Lopez-Perez et al., 2013; Mori et al., 2015). Interestingly, GPB-6 (Spiribacter sp.) was an abundant microbe in brine and/or sediment samples from five ponds with different salinities (Figure 4), suggesting that GPB-6 exhibited excellent adaptation to saline and alkaline environments. In agreement with this observation, a previous study demonstrated that Spiribacter salinus M19-40 is one of the most predominant bacteria in neutral saline lakes (Leon et al., 2014), and the streamlined genome of this bacterium is considered to provide significant advantage in environmental adaptation (Lopez-Perez et al., 2013). This genus was not reported to be abundant in classic soda lakes (Vavourakis et al., 2016, 2018). We also observed archaeal MAGs were abundant in samples with relatively highest salinities, including Euryarchaeota (Salinarchaeum, Natronomonas, Halorubrum in Halobacteriaceae), Ca. Nanohaloarchaeota and Ca. Woesearchaeota (Figure 4). In the brine of soda lakes, Halorubrum- and Natrinema-related sequences were reported to be the most abundant taxa of Euryarchaeota (Vavourakis et al., 2016), while Thermoplasmata group KTK 4A and Halobacteria (Halohasta and Halorubrum) were detected in the sediment (Vavourakis et al., 2018). Most members of Salinarchaeum and Natronomonas were isolated from neutral solar salterns (Kamekura et al., 1997; Minegishi et al., 2017). These abundant taxa were the close relatives of microorganisms from both classic soda lakes and neutral saline environments, exhibiting a combination of aforementioned habitats.
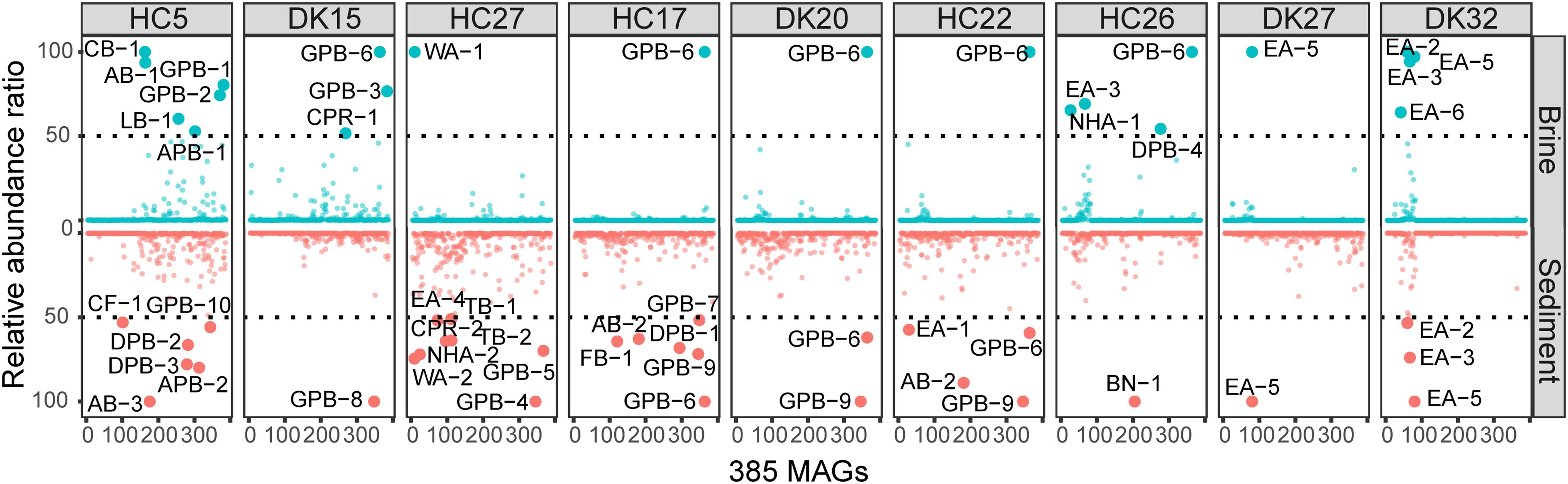
Figure 3. Relative abundances of 385 MAGs in 18 samples. The abundance was estimated by the average depth of all contigs in that MAG (Shown in Supplementary Table 5). The relative abundances of MAGs with the highest sequence read coverage in each sample were scaled to 100, and MAGs with relative abundances of greater than 50 were considered to be abundant. Blue and red indicate the abundances of MAGs in brine and sediment samples, respectively.
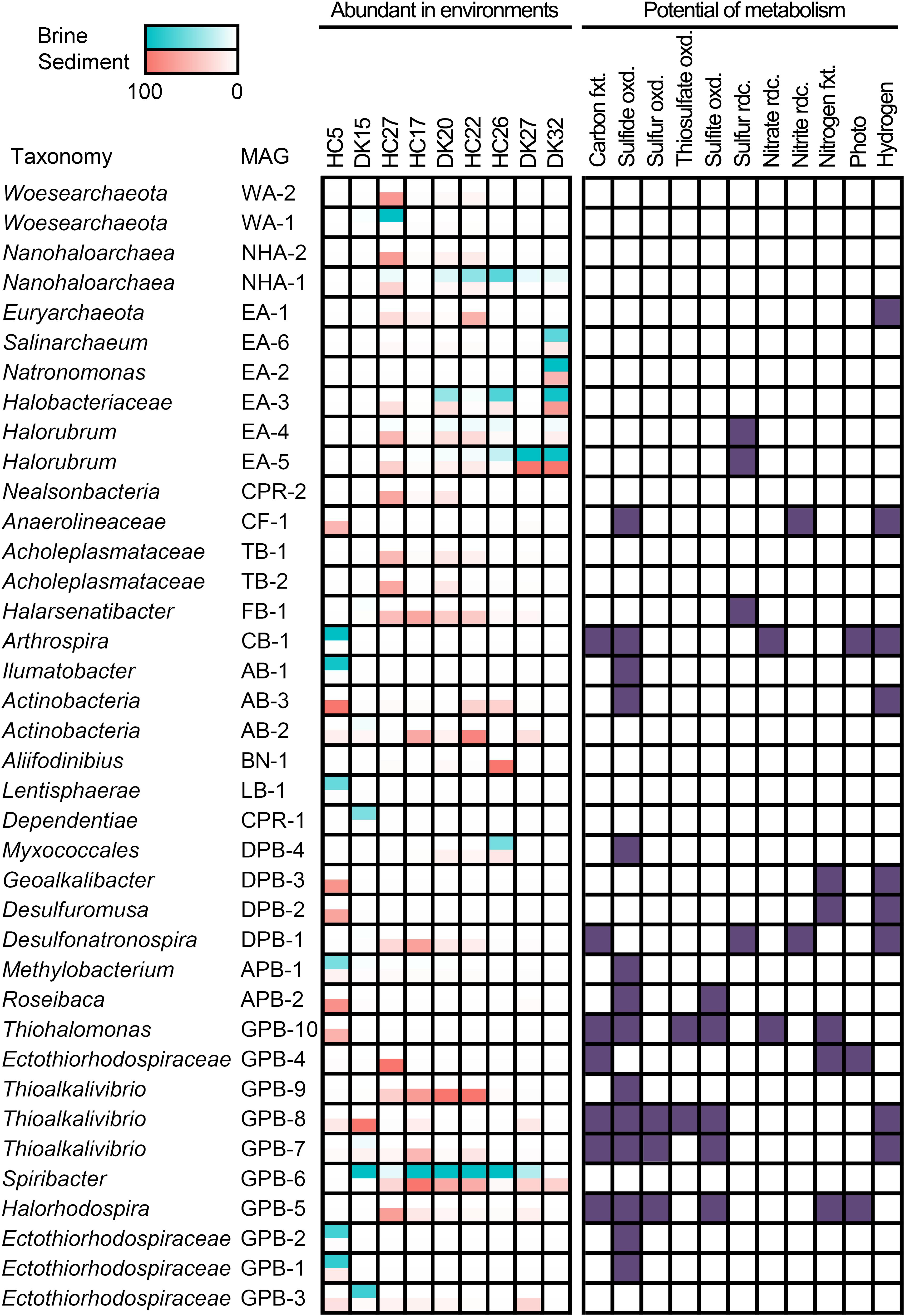
Figure 4. Distribution and metabolic potential of abundant MAGs. Abundant MAGs in brine and sediment samples are colored red and green, respectively; the relative abundance was shown in the legend. MAGs with metabolic potential are colored in purple. fxt., fixation; oxd., oxidation; rdc., reduction.
A total of 38 abundant MAGs were marked (with five-pointed star) in the phylogenomic tree based on the alignment of universal proteins across Bacteria and Archaea (Figure 2). Although these abundant MAGs accounted only approximately 10% of the total MAGs, they were from 13 phyla representing half of biodiversity at phylum level (Supplementary Table 7) and accounting for 24.81–69.23% of total coverage of 385 MAGs in 18 samples (Supplementary Table 5). Therefore, these taxa are ideal to understand the microbial functions and adaptation mechanisms in these specific alkaline chloride-carbonate-sulfate niches. To this end, we summarized the metabolic potential of these abundant MAGs in Supplementary Table 6.
Wide Distribution of Sulfur Oxidation in Autotrophic and Heterotrophic Bacteria
A total of 18 abundant MAGs have the potential to drive the dissimilatory cycling of sulfur element, and sulfide oxidation was present in 14 MAGs. In the autotrophic Ectothiorhodospiraceae family, two MAGs (GPB-4 and GPB-5) encoded bacterial photosynthetic reaction center and marker genes (prk encoding phosphoribulokinase, rbcLS encoding ribulose-bisphosphate carboxylase large and small chain) of the CBB cycle and were both abundant in the sediment of HC27 (Figure 4). Considering the potential for light energy utilization and anoxygenic photosynthetic lifestyle in these species, they may be dominant carbon fixing microbes at the surfaces of sediment. GPB-5 was observed to encode sulfide:quinone oxidoreductase (Sqr), flavocytochrome c sulfide dehydrogenase (FccB), reversed dissimilatory sulfite reductase (rDsrAB), and sulfite dehydrogenase (SoeABC) (Supplementary Table 6), exhibiting the metabolic potential to oxidize multiple sulfur compounds (Figure 4). GPB-5 was predicted to belong to the genus Halorhodospira and may be an anoxygenic purple sulfur bacterium. GPB-4 was identified as an unclassified member of the family Ectothiorhodospiraceae and did not show the ability to oxidize sulfur compounds or hydrogen (Supplementary Table 6). However, considering that the completeness of the MAG GPB-4 was relatively low (67.44%), partial genes encoding sulfur-oxidizing proteins (SoxBYZ), SoeC, coenzyme F420 hydrogenase subunit beta and hydrogenase expression/formation proteins in GPB-4 may indicate a putative ability of this organism to oxidize sulfur compound or hydrogen (Supplementary Table 6). Notably, both GPB-4 and GPB-5 possessed the nitrogenase gene nifHDK (Supplementary Table 6), indicating that they may be involved in nitrogen fixation in pond HC27. Two Thioalkalivibrio MAGs (GPB-7 and GPB-8) and one Thiohalomonas MAG (GPB-10) were abundant in sediment samples from ponds HC17, DK15 and HC5, respectively (Figure 4). These three MAGs had marker genes for the CBB cycle and the oxidization of multiple sulfur compounds (Supplementary Table 6), which is consistent with the metabolic characteristics of Thioalkalivibrio and Thiohalomonas. Apart from carbon fixation and sulfur cycling, GPB-7 and GPB-8 contained hydrogenase genes and may utilize hydrogen as energy source and reducing power, while GPB-10 harbored nifHDK genes (Supplementary Table 6) and exhibited the potential to fix nitrogen (Figure 4). In brief, our results suggest that carbon fixing and sulfur oxidation may be coupled in these anoxygenic photosynthetic and chemolithotrophic Ectothiorhodospiraceae species.
In addition, eight cyanobacterial MAGs were obtained, while only one MAG (CB-1) belonging to the cyanobacterial genus Arthrospira was detected as an abundant species in the brine of HC5 (Supplementary Table 4), which had the lowest salinity and chloride concentration (Figure 4). Cyanobacteria are typically the primary contributors to carbon fixation. CB-1 encoded Prk, RbcLS and the complete photosystems II and I (Supplementary Table 6), indicating that it has the potential to photosynthetically fix carbon via the CBB cycle (Figure 4). This MAG also encoded hydrogenase, nitrate reductase, and Sqr, exhibiting versatile metabolic capabilities. Four other cyanobacterial MAGs had the potential to perform photosynthesis, of which three encoded Sqr and two encoded nitrogenase (Supplementary Table 6). It was interesting to note that under alkaline conditions, most photosynthetic Cyanobacteria MAGs had the potential to oxidize sulfide (Supplementary Table 6) and may regulate photosynthesis and carbon fixing (Klatt et al., 2015). In addition, some cyanobacteria have been reported to be anoxygenic photosynthetic bacteria and capable of oxidizing sulfide by Sqr (Cohen et al., 1975; Sybesma et al., 1986; Grim and Dick, 2016; Hamilton et al., 2018). The electrons obtained from sulfide oxidation are further transferred to the reducing equivalent NADPH via quinone and electron transport chain components and are finally used for CO2 fixation (52).
Interestingly, we observed that many abundant bacteria from heterotrophic taxa also harbored Sqr, including AB-1 and AB-3 from Actinobacteria, CF-1 from Chloroflexi, APB-1 and APB-2 from Alphaproteobacteria, DPB-4 from Deltaproteobacteria, and GPB-1, GPB-2, and GPB-9 from Gammaproteobacteria (Figures 2, 4), indicating their potential in oxidation-mediated detoxification of sulfide with various salinities in the saline-alkaline environments. In addition, four Halobacteria MAGs EA-2, EA-4, EA-5, and EA-6 contain the thiosulfate dehydrogenase [quinone] large subunit (DoxD, KEGG Orthology: K16936). The UniProt list reports a wrong annotation (Vavourakis et al., 2019), but the lack of the small subunit (DoxA) suggested that the ability for thiosulfate oxidation might be absent. Sulfide and thiosulfate could provide accessory energy under nutrient-limited conditions to heterotrophs and increase the growth rate and flux of assimilatory carbon via anaplerotic reaction of oxaloacetate. This lifestyle is considered to be facultative lithoheterotrophy (Sorokin, 2003). Alkaline lakes, including soda-saline lakes (Figure 4) and typical soda lakes (Sorokin and Kuenen, 2005; Tourova et al., 2013), provide excess soluble phosphate and a low toxic form of sulfide (HS–), creating an advantage environment for the growth of diverse sulfur-oxidizing microbes (Sorokin et al., 2015; Vavourakis et al., 2018). The sqr gene was found in an abundance of heterotrophic taxa, such as Actinobacteria, Chloroflexi, Alphaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria, suggesting that these microbes play a role in the detoxification of sulfide. Interestingly, a large number of strictly organotrophic microbes inhabiting soda lakes or marine environments are capable of oxidizing thiosulfate to tetrathionate or sulfate (Sorokin, 2003; Sorokin et al., 2005). An increased growth rate of Limnobacter thiooxidans, a thiosulfate-oxidizing heterotrophic bacterium isolated from freshwater lake sediment, was observed by adding thiosulfate, suggesting that L. thiooxidans obtains an energy advantage via the oxidation of thiosulfate (Spring et al., 2001). Heterotrophic sulfur-oxidizing microbes (HSOB) provided thiosulfate restored the ATP synthesis in the starved cells (Sorokin, 2003) and increased the level of dark anaplerotic carbon dioxide assimilation (Tuttle and Jannasch, 1977; Perez and Matin, 1982). Since ATP synthesis is a rate-limiting step for the anaplerotic CO2 assimilation, the energy advantage obtained from sulfide oxidation by Sqr could be a reasonable explanation for the high coverage of Sqr-containing heterotrophs.
Potential Reduction of Sulfur Compounds in Abundant MAGs
Given that numerous autotrophic and heterotrophic abundant MAGs exhibited the potential to oxidize reductive sulfur compounds (RSCs), it would be interesting to investigate whether there were considerable sulfur-reducing microbes to complete the sulfur cycling in the chloride-carbonate-sulfate lakes. Notably, we found the presence of anaerobic respiration processes in abundant MAGs, including sulfur respiration (Figure 4). Halorubrum spp. EA-4, EA-5, Halarsenatibacter sp. FB-1, Desulfonatronospira sp. DPB-1 harbored the psrA/phsA gene (Supplementary Table 6), allowing for potential polysulfide reduction/thiosulfate reduction. The Desulfonatronospira MAG DPB-1 was an abundant species in the sediment of HC17 (Figure 4) and encoded thiosulfate reductase/polysulfide reductase chain A (psrA/phsA) and F420-non-reducing hydrogenase subunits (mvhADG) (Supplementary Table 6), indicating the lifestyle of chemolithotrophic sulfate-reducing bacterium. It is noteworthy that DPB-1 also had the marker genes of anaerobic carbon-monoxide dehydrogenase catalytic subunit (cooS), acetyl-CoA synthase (acsB), and acetyl-CoA decarbonylase/synthase subunits (cdhDE) in the WL pathway (Supplementary Table 6), indicating that it may fix inorganic carbon via the WL pathway.
In addition to elemental sulfur reduction, we also observed the potential of sulfate/sulfite and tetrathionate reduction in many MAGs. Seven MAGs from Desulfobacterales and Desulfovibrionales harbored the dsrAB gene for sulfate/sulfite respiration, and some of them had the potential for carbon fixing (Supplementary Table 6). The type strains from same taxonomies were reported facultative autotrophs using hydrogen and/or formate as substrates (Lien et al., 1998; Pikuta et al., 2003; Sorokin et al., 2008a, b; Sorokin and Chernyh, 2017). Halomonas sp. GPB-59, Marinobacter sp. GPB-11, and Aquisalimonas sp. GPB-38 harbored ttrABC genes for tetrathionate reductase, while another five MAGs from Actinobacter and one Desulfuromusa MAG had ttrAB (Supplementary Table 6) with the potential of tetrathionate respiration. Although the abundance was not high, all the above taxa could drive sulfur reduction and make sulfur cycling complete.
Interestingly, many elemental sulfur-reducing microbes, such as Halorubrum spp. EA-4, EA-5, Halarsenatibacter sp. FB-1, and Desulfonatronospira sp. DPB-1 (Figure 4), were abundant under extremely hypersaline conditions, given that the much lower concentrations of dissolved oxygen (Sherwood et al., 1991) and the notably reduced oxygen diffusion coefficients (Jamnongwong et al., 2010) boosted the anaerobic respiration of these microbes. In addition, diverse taxa were involved in sulfate and tetrathionate respiration (Supplementary Table 6). Using acetate as a substrate, the polysulfide/elemental sulfur reduction under alkaline conditions (pH 10) is much more exergonic (ΔG = -91.9 kJ/mol) than neutral pH conditions (ΔG0′ = -6.6 kJ/mol) (Reaction 1 in Table 1), suggesting that this reaction is energetically more favorable for dissimilatory elemental sulfur reduction at a higher pH value (Sorokin et al., 2010). However, for those microbes utilizing heterotrophic and chemolithotrophic sulfate respirations, approximately no differences in free energy changes are noted between neutral and alkaline conditions. Given that polysulfide/elemental sulfur-reducing microbes are able to obtain more free energy for ATP synthesis under alkaline conditions (Table 1), the difference in ATP yields between the two pH conditions could explain why polysulfide/elemental sulfur-reducing microbes could gain a growth advantage in alkaline environments.

Table 1. Summary of Gibbs free energy change in sulfur reactions#.
Symbionts Exist Abundant in Hypersaline Environments
Abundant MAGs, including 2 Ca. Nanohaloarchaeota MAGs and 2 Ca. Woesearchaeota MAGs in the DPANN superphylum as well as 1 Ca. Dependentiae MAG and 1 Ca. Nealsonbacteria MAG in the CPR superphylum, were also observed in these saline-alkaline lakes (Figures 2, 4). The metabolic potential of these microbes is typically limited due to the smaller genome size (Supplementary Table 4). All six MAGs did not show the potential for dissimilatory sulfur or nitrogen metabolism (Figure 4). Interestingly, most DPANN and CPR taxa are auxotrophic with respect to the biosynthesis of amino acids, purine and pyrimidine bases of nucleotides, and isoprenoids or fatty acids of the cell membrane (Castelle et al., 2018). The two Acholeplasmataceae MAGs TB-1 and TB-2 were observed to be abundant in the sediment of HC27 (Figure 4). Acholeplasmataceae is a class of cell-wall-free microbes (fried egg-like colony) that live together with plants or insects (Freundt et al., 1984).
Members of the DPANN and CPR superphyla have symbiotic lifestyles with other microbes (Castelle et al., 2018; Hamm et al., 2019). We constructed a co-occurrence network based on the coverage of 385 MAGs in the 18 samples to predict the putative associated symbiont (Figure 5). There were fifteen separate modules named M1 to M15 in the network (Figure 5A). M1, M2, and M4 contained most MAGs and existed almost in sediment samples, while M3, M5, M7, and M12 were observed in brine samples. MAGs in M6 and M8 were mainly found in hypersaline brine or sediment samples (Figure 5B). Among fifteen modules, only one abundant Ca. Nanohaloarchaeota MAG (NHA-1) was strongly co-present with the Halobacteriaceae MAG EA-19 in M12, while NHA-5 and NHA-3 correlated with Natronomonas sp. EA-16 and the abundant Halorubrum sp. EA-5 in M8, as well as with each other (Figure 5A). The above results suggested that the putative symbiont of Ca. Nanohaloarchaeota is likely a member of the taxon Halobacteria.
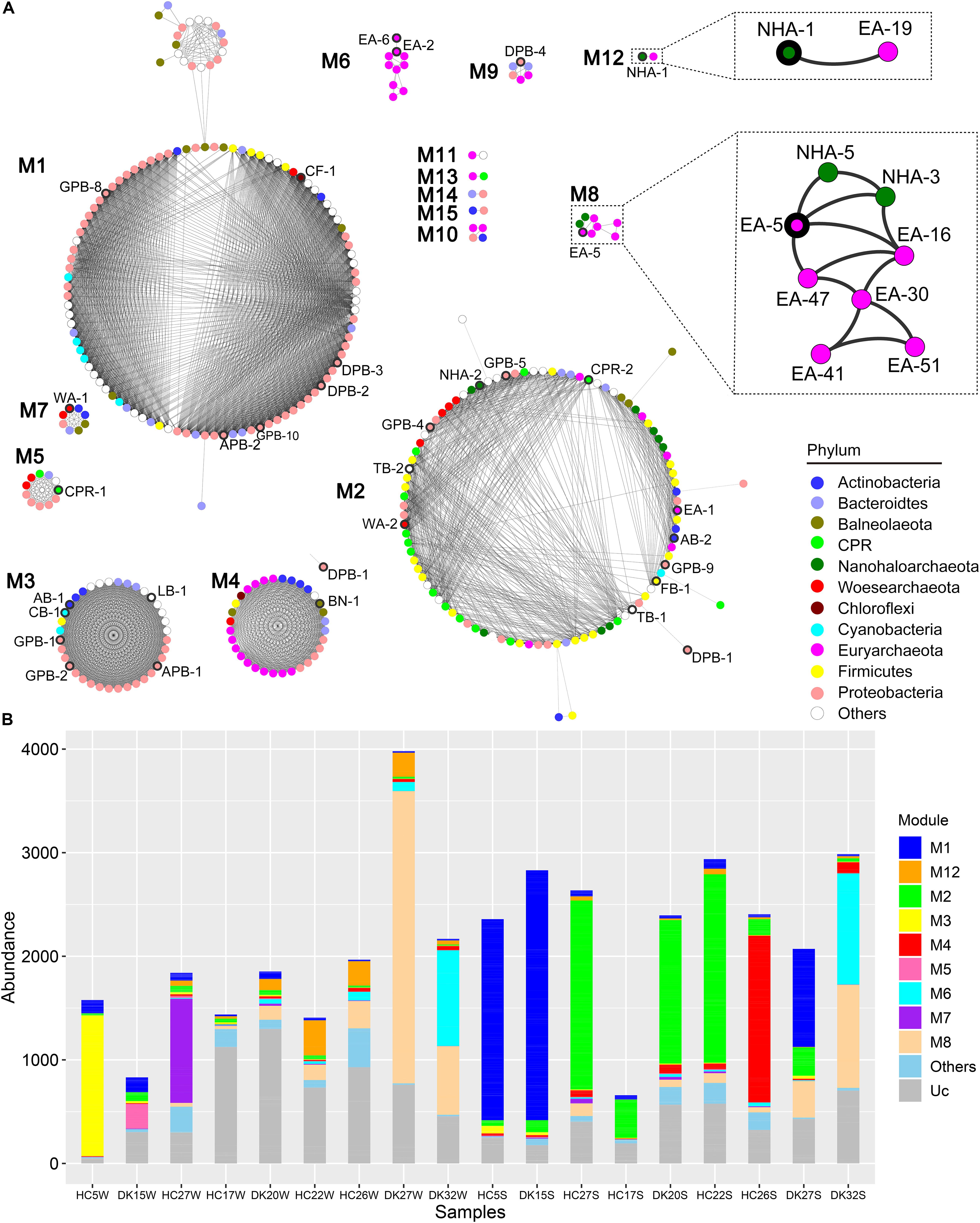
Figure 5. Co-occurrence network based on all 385 MAGs. (A) The coverage of 385 MAGs in 18 samples was used to construct a robust co-occurrence network (Pearson correlation coefficient > 0.9 and p-value < 0.01). The nodes in the network are colored by phylum, and the nodes representing abundant MAGs are labeled and bold in the outer ring. Two modules involved are zoomed in to show the robust co-occurrence of Candidatus Nanohaloarchaeota and Halobacteria. (B) The abundance of MAGs from the modules was summarized in different sample group. Other, sum of M9, M10, M11, M13, M14, and M15; Uc, unclassified in any module.
Ca. Nanohaloarchaeota and other DPANN members are difficult to culture in the laboratory (Castelle and Banfield, 2018; Liu et al., 2018; Hamm et al., 2019) given that their small sizes of genomes limit their anabolic abilities (Narasingarao et al., 2012; Castelle et al., 2018). Generally, these microbes still need to uptake nutrients from their symbionts for the biosynthesis of nucleic acids, proteins and lipids. Ca. Nanohaloarchaeum antarcticus was isolated together with Halorubrum lacusprofundi but away from other species (Hamm et al., 2019). The growth of Ca. Nanohaloarchaeota is probably dependent on a specific Halobacteria host (Hamm et al., 2019) (Figure 5), increasing the difficulty of culturing Ca. Nanohaloarchaeota. Although few Ca. Nanohaloarchaeota species have been cocultured with their hosts, they are abundant in multiple hypersaline environments, including neutral salterns (Ghai et al., 2011; Narasingarao et al., 2012), soda lakes (Vavourakis et al., 2016), and soda-saline lakes (Figures 2, 4). The “salt in” strategy Ca. Nanohaloarchaeota employs to resist osmotic force (Vavourakis et al., 2016) provides an effective solution of saving energy in hypersaline environments (Banciu and Muntyan, 2015; Gunde-Cimerman et al., 2018). This energy-saving mechanism may contribute to the observed high abundance of Ca. Nanohaloarchaeota in hypersaline environments.
Metabolic Advantage of Ca. Nanohaloarchaeota in Hypersaline Environments
The presence of symbiont must create mutually beneficial relationships; otherwise, the symbiont would be replaced by a separate existence. We constructed the energy generation pathway in DPANN superphylum to deeply understand the symbiotic lifestyle. DPANN and CPR cannot regenerate ATP via oxidative and photosynthetic phosphorylation due to the general absence of electron transport complexes and photosynthetic reaction center complexes (Supplementary Table 6). Some taxa (especially NHA-1 and NHA-3) are believed to gain energy by substrate level phosphorylation via carbohydrate fermentation for the complete glycolysis pathway from hexose to pyruvate (Figures 6A,B). Ca. Woesearchaeota in saline-alkaline lakes may ferment carbohydrates via a modified pathway or generate energy through another pathway considering the absence of 6-phosphofructose kinase (pfkAB or pfkC) (Figure 6A and Supplementary Table 6). Pyruvate:ferredoxin oxidoreductase, alcohol dehydrogenase and lactate dehydrogenase were not detected in Ca. Nanohaloarchaeota MAGs (Supplementary Table 6), indicating that pyruvate may be produced as final product for the symbiotic microbes (potentially Halobacteria). Consistently, no Halobacteria MAGs contained the polysaccharide phosphorylation pathway (Supplementary Table 6). Pyruvate is a key nutrient in hypersaline environments (Oren, 2015), indicating that the Ca. Nanohaloarchaeota and Ca. Woesearchaeota taxa may function as primary degraders of polysaccharides (Figure 6A) at least in the symbiotic system.
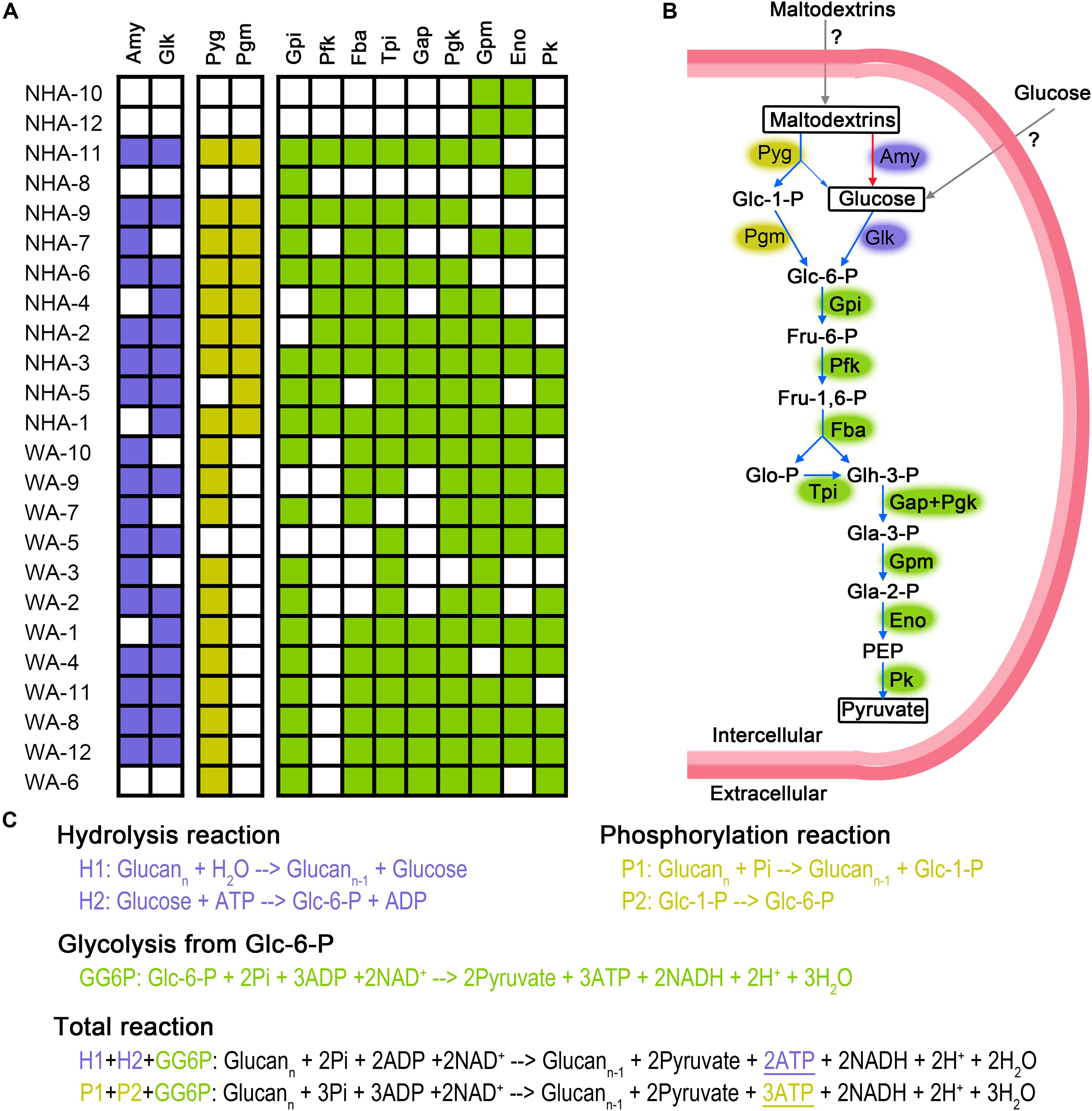
Figure 6. Putative fermentation pathways in Candidatus Nanohaloarchaeota and Candidatus Woesearchaeota MAGs. The presence of enzymes is exhibited in detailed (A). We draw the metabolic pathway of carbohydrate fermentation in Ca. Nanohaloarchaeota (B). Blue and red arrows represented the presence or absence, respectively, of that pathway in abundant MAG NHA-1. Gray arrow indicates that the specific transporters were not identified. Biochemical reaction and ATP production of glycolysis via phosphorylation and hydrolysis were compared (C). The metabolic potential of the phosphorylation, hydrolysis and glycolysis pathways are colored purple, yellow and green, respectively. Amy, alpha-Amylase; Glk, Glucokinase; Pyg, Glycogen phosphorylase (or named 1,4-alpha-Glucan phosphorylase); Pgm, Phosphoglucomutase; Gpi, Glucose-6-phosphate isomerase; Pfk, 6-Phosphofructokinase; Fba, Fructose-bisphosphate aldolase; Tpi, Triosephosphate isomerase; Gap, Glyceraldehyde 3-phosphate dehydrogenase; Pgk, Phosphoglycerate kinase; Gpm, Phosphoglycerate mutase; Eno, Enolase; Pk, Pyruvate kinase; Glc-1-P, Glucose 1-phosphate; Glc-6-P, Glucose 6-phosphate; Fru-6-P, Fructose 6-phosphate; Fru-1,6-P, Fructose 1,6-biphosphate; Glo-P, Glycerone phosphate; Glh-3-P, Glyceraldehyde 3-phosphate; Gla-3-P, Glycerate 3-phosphate; Gla-2-P, Glycerate 2-phosphate; PEP, Phosphoenolpyruvate; ATP, Adenosine 5’-triphosphate; ADP, Adenosine 5’-diphosphate; NAD+, Nicotinamide adenine dinucleotide; NADH, Reduced nicotinamide adenine dinucleotide; Pi, phosphate.
Most DPANN MAGs contained alpha-amylase and glucose kinase; thus, the 1,4-alpha-glucans (like starch) may be generally used as their carbon and energy sources (Figure 6A). Interestingly, eight of twelve Ca. Nanohaloarchaeota MAGs had genes encoding glycogen phosphorylase (or named 1,4-alpha-glucan phosphorylase) and phosphomannomutase/phosphoglucomutase enzymes (Pgm), which catalyze the phosphorylation of polysaccharides and transfer glucose-1-phosphate to glucose-6-phosphate (Figure 6B). In contrast, more Ca. Woesearchaeota MAGs encoded glycogen phosphorylase, but none of them harbored the pgm gene (Figure 6A). More interestingly, the abundant MAG NHA-1 contained 1,4-alpha-glucans phosphorylase and a complete glycolysis pathway, but not alpha-amylase (Figure 6A). Given the increased ATP produced compared with hydrolysis (Figure 6C), we infer that 1,4-alpha-glucans phosphorylation may play a significant role in maintaining the symbiotic lifestyle between Ca. Nanohaloarchaeota sp. NHA-1 and its Halobacteria host (discuss below).
To estimate the importance of 1,4-alpha-glucans phosphorylation, we compared the similarity of functional genes among DPANN. In the phylogenomic tree, four separate clades in Ca. Nanohaloarchaeota were observed (Figure 7A). The glycogen phosphorylase coding gene pyg was generally detected in clades II, III and IV but was not present in NHA-10 and NHA-12 from clade I (Figure 6A). Among the separate clades, Pyg from different MAGs with a high similarity were located together (Figure 7B), especially in NHA-1 and NHA-3. Interestingly, the amino acid sequences of the glycogen phosphorylase from clades II and III were located at closed branches, while that of clade IV was homologous with the enzyme from Ca. Woesearchaeota (Figure 7B). The evolutionary tree of Pyg indicated that phosphorylation was conserved in most taxa of Ca. Nanohaloarchaeota.
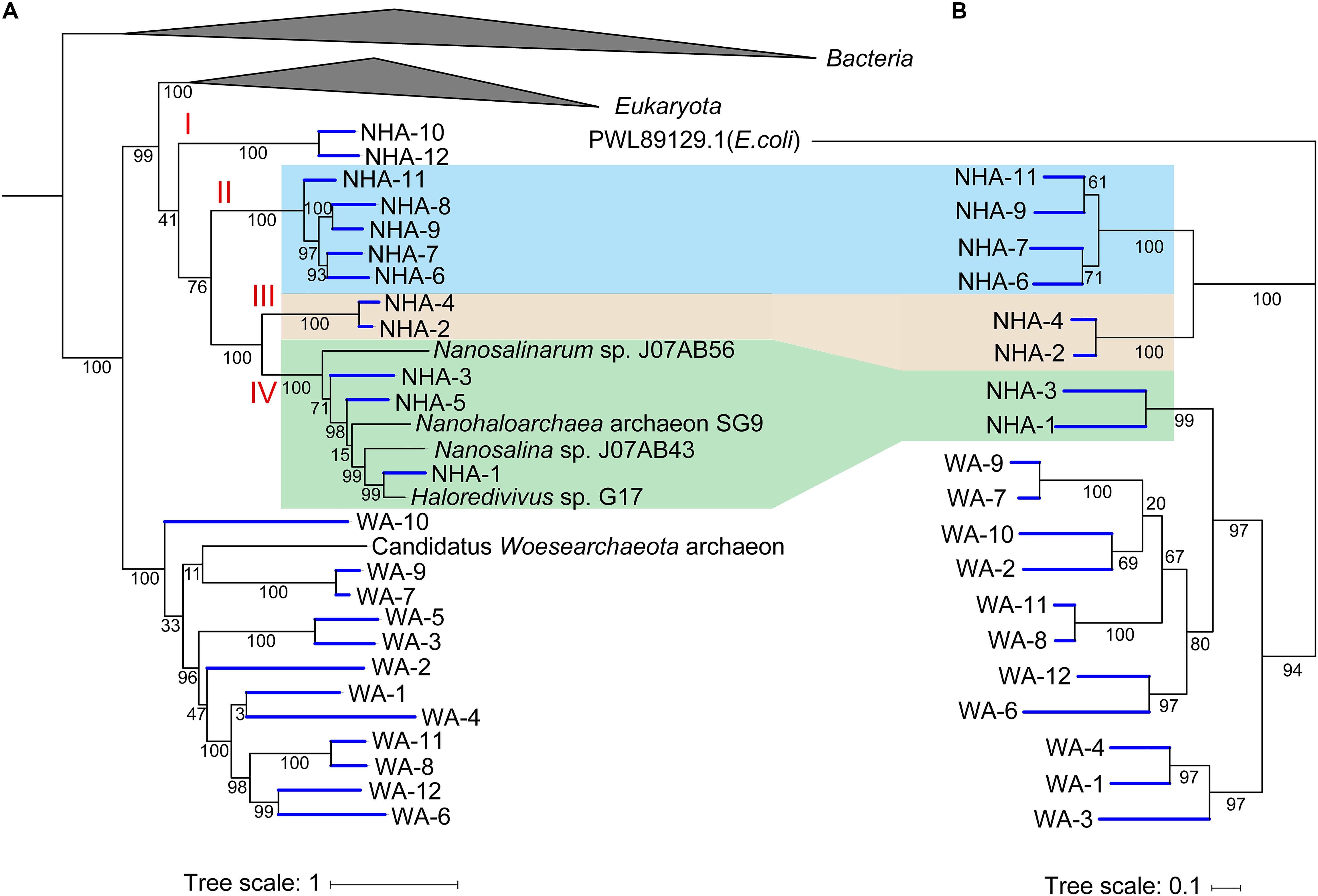
Figure 7. Phylogenetic analysis of amino acid sequences of glycogen phosphorylases obtained from Candidatus Nanohaloarchaeota and Candidatus Woesearchaeota MAGs. (A) The tree shown to the left is the phylogenomic tree of 385 MAGs with the reference genome, and the Bacteria and Euryarchaeota branches are collapsed. (B) Nineteen predicted glycogen phosphorylases in Ca. Nanohaloarchaeota and Ca. Woesearchaeota with the alpha-glucan phosphorylase from Escherichia coli (PWL89129.1) were retrieved, aligned and then used to infer the correct tree using the Maximum Likelihood method based on the JTT matrix-based model with MEGA. The tree with the highest log likelihood (-16393.43) is shown with bootstrap support.
Ca. Nanohaloarchaeota utilizes polysaccharide and generates ATP by substrate level phosphorylation during glycolysis (Vavourakis et al., 2016; Castelle et al., 2018). Alpha-amylase could be responsible for the hydrolysis of polysaccharide (Vavourakis et al., 2016; Castelle et al., 2018; Liu et al., 2018). More recently, Ca. Nanahalobium has been proven to be capable of hydrolyzing alpha-glucans (La Cono et al., 2019). Amylase genes were widely distributed in Ca. Nanohaloarchaeota and Ca. Woesearchaeota (Figure 6A), but they are absent in NHA-1, the most abundant Ca. Nanohaloarchaeota strain in hypersaline environments. The 1,4-alpha-glucan phosphorylation pathway, which is involved in maltose/maltodextrin/glycogen metabolism in both archaea and bacteria (Boos and Shuman, 1998; Xavier et al., 1999; Seibold et al., 2009), could be employed by the NHA-1 strain as an alternative strategy for polysaccharide degradation (Figures 6A,B). However, both 4-alpha-glucanotransferase catalyzing the conversion of maltose to maltodextrin and glycogen synthase responsible for the synthesis of glycogen were not available in NHA-1 (Supplementary Table 6). Therefore, we hypothesize that extracellular maltodextrin is a putative substrate. One less ATP molecule is used when one glucoside molecule is degraded via phosphorylation pathway rather than the hydrolysis pathway, so one more ATP molecule will be made by using maltodextrin substrate (Figure 6C). This relatively more efficient ATP generation system present in Ca. Nanohaloarchaeota MAG may provide the symbionts with a growth advantage in the competition with the free-living Halobacteria, which typically utilize starch via the alpha-amylase-based hydrolysis pathway (Perez-Pomares et al., 2003).
Conclusion
Chloride-carbonate-sulfate lakes (also known as soda-saline lake) is a double-extreme environment with high pH and high salinity, and diverse metabolic processes function well in such an environment. This study has revealed the microbial composition and their metabolic potential in the brines and sediments of the chloride-carbonate-sulfate lakes. The microbiomes from different habitats were composed of several abundant and numerous rare taxa. These abundant taxa represented most branches of the phylogenomic tree. The oxidation and reduction of sulfur and polysaccharide phosphorylation existed in certain abundant taxa, which may increase their adaptation to extreme alkaline and saline environments with unique advantages in terms of energy production and thermodynamics. Briefly, RSCs could be utilized as a putative accessory energy source for heterotrophs under nutrient limited conditions. Elemental sulfur respiration more easily occurs under high pH may due to thermodynamic advantages, which favors this type of sulfur reduction microbes in high abundance. More energy was produced by the phosphorylation pathway of 1,4-alpha-glucans compared with hydrolysis. The above results provide novel insights into the relationship between diverse lifestyles and adaptive characterizations of the prokaryotes thriving in such double-extreme environments.
Data Availability Statement
The datasets generated for this study can be found in the NCBI (projectID PRJNA549802), gcMeta (projectID NMDC10010899), and figshare (https://figshare.com/s/9c3cb76f0c9646a30e94).
Author Contributions
HX designed and supervised the study. DZ, SZ, QX, JC, JZ, FC, and HX collected water and sediment samples. QX and JC measured physicochemical characteristics. YZ extracted DNA from environmental samples. DZ and SZ performed bioinformatic and statistical analyses under the partial supervision of HY and SH. DZ and SZ prepared the figures and wrote the manuscript under the guidance of HX. ML, YZ, SL, and SH participated in discussions and revisions. All authors read and approved the final manuscript.
Funding
This study was funded by the National Natural Science Foundation of China (grant number 91751201), the National Key R&D Program of China (grant number 2020YFA0906800), and the Science and Technology Basic Resources Investigation Project (grant number 2017FY100300).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Wenyu Shi for data submission to NCBI and gcMeta. This manuscript was released as a preprint at Research Square (Zhao et al., 2019).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01740/full#supplementary-material
Footnotes
- ^ https://github.com/cjfields/readfq
- ^ https://sourceforge.net/projects/bbmap/
- ^ http://ecogenomics.github.io/BamM
- ^ https://www.ncbi.nlm.nih.gov/
- ^ https://www.r-project.org/
- ^ http://vegan.r-forge.r-project.org/
- ^ https://CRAN.R-project.org/package=ggpubr
- ^ https://CRAN.R-project.org/package=pheatmap
- ^ https://github.com/vqv/ggbiplot
- ^ https://CRAN.R-project.org/package=psych
- ^ https://figshare.com/s/9c3cb76f0c9646a30e94
References
Anantharaman, K., Hausmann, B., Jungbluth, S. P., Kantor, R. S., Lavy, A., Warren, L. A., et al. (2018). Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 12, 1715–1728. doi: 10.1038/s41396-018-0078-70
Antony, C. P., Kumaresan, D., Hunger, S., Drake, H. L., Murrell, J. C., and Shouche, Y. S. (2013). Microbiology of Lonar Lake and other soda lakes. ISME J. 7, 468–476. doi: 10.1038/ismej.2012.137
Asao, M., Pinkart, H. C., and Madigan, M. T. (2011). Diversity of extremophilic purple phototrophic bacteria in Soap Lake, a central Washington (USA) soda lake. Environ. Microbiol. 13, 2146–2157. doi: 10.1111/j.1462-2920.2011.02449.x
Banciu, H. L., and Muntyan, M. S. (2015). Adaptive strategies in the double-extremophilic prokaryotes inhabiting soda lakes. Curr. Opin. Microbiol. 25, 73–79. doi: 10.1016/j.mib.2015.05.003
Bateman, A., Martin, M. J., O’Donovan, C., Magrane, M., Alpi, E., Antunes, R., et al. (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169. doi: 10.1093/nar/gkw1099
Boos, W., and Shuman, H. (1998). Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev. 62, 204–229. doi: 10.1128/mmbr.62.1.204-229.1998
Boros, E., and Kolpakova, M. (2018). A review of the defining chemical properties of soda lakes and pans: an assessment on a large geographic scale of Eurasian inland saline surface waters. PLoS One 13:e0202205. doi: 10.1371/journal.pone.0202205
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Carini, S. A., and Joye, S. B. (2008). Nitrification in Mono Lake, California: activity and community composition during contrasting hydrological regimes. Limnol. Oceanogr. 53, 2546–2557. doi: 10.4319/lo.2008.53.6.2546
Castelle, C. J., and Banfield, J. F. (2018). Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 172, 1181–1197. doi: 10.1016/j.cell.2018.02.016
Castelle, C. J., Brown, C. T., Anantharaman, K., Probst, A. J., Huang, R. H., and Banfield, J. F. (2018). Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 16, 629–645. doi: 10.1038/s41579-018-0076-72
Cohen, Y., Jorgensen, B. B., Padan, E., and Shilo, M. (1975). Sulfide-dependent anoxygenic photosynthesis in cyanobacterium oscillatoria-limnetica. Nature 257, 489–492. doi: 10.1038/257489a0
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Freundt, E. A., Whitcomb, R. F., Barile, M. F., Razin, S., and Tully, J. G. (1984). Proposal for elevation of the family Acholeplasmataceae to Ordinal Rank - Acholeplasmatales. Int. J. Syst. Bacteriol. 34, 346–349. doi: 10.1099/00207713-34-3-346
Ghai, R., Pasic, L., Fernandez, A. B., Martin-Cuadrado, A. B., Mizuno, C. M., McMahon, K. D., et al. (2011). New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 1:135. doi: 10.1038/Srep00135
Grant, W. D. (2006). “Alkaline environments and biodiversity,” in Extremophiles Handbook, eds C. Gerday and N. Glansdorff (Berlin: Springer Science & Business Media).
Grant, W. D., and Sorokin, D. Y. (2011). ““Distribution and diversity of soda lake alkaliphiles,”,” in Extremophiles Handbook, ed. K. Horikoshi (Tokyo: Springer), 27–54. doi: 10.1007/978-4-431-53898-1_3
Grim, S. L., and Dick, G. J. (2016). Photosynthetic versatility in the genome of Geitlerinema sp. PCC 9228 (formerly Oscillatoria limnetica ‘Solar Lake’), a model anoxygenic photosynthetic cyanobacterium. Front. Microbiol. 7:1546. doi: 10.3389/Fmicb.2016.01546
Gunde-Cimerman, N., Plemenitas, A., and Oren, A. (2018). Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 42, 353–375. doi: 10.1093/femsre/fuy009
Haft, D. H., Selengut, J. D., and White, O. (2003). The TIGRFAMs database of protein families. Nucleic Acids Res. 31, 371–373. doi: 10.1093/nar/gkg128
Hamilton, T. L., Klatt, J. M., de Beer, D., and Macalady, J. L. (2018). Cyanobacterial photosynthesis under sulfidic conditions: insights from the isolate Leptolyngbya sp. strain hensonii. ISME J. 12, 568–584. doi: 10.1038/ismej.2017.193
Hamm, J. N., Erdmann, S., Eloe-Fadrosh, E. A., Angeloni, A., Zhong, L., Brownlee, C., et al. (2019). Unexpected host dependency of antarctic Nanohaloarchaeota. Proc. Natl. Acad. Sci. U.S.A. 116, 14661–14670. doi: 10.1073/pnas.1905179116
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Iversen, N., Oremland, R. S., and Klug, M. J. (1987). Big soda lake (Nevada) .3. pelagic methanogenesis and anaerobic methane oxidation. Limnol. Oceanogr. 32, 804–814. doi: 10.4319/lo.1987.32.4.0804
Jamnongwong, M., Loubiere, K., Dietrich, N., and Hebrard, G. (2010). Experimental study of oxygen diffusion coefficients in clean water containing salt, glucose or surfactant: consequences on the liquid-side mass transfer coefficients. Chem. Eng. J. 165, 758–768. doi: 10.1016/j.cej.2010.09.040
Kamekura, M., Dyall-Smith, M. L., Upasani, V., Ventosa, A., and Kates, M. (1997). Diversity of alkaliphilic halobacteria: proposals for transfer of Natronobacterium vacuolatum, Natronobacterium magadii, and Natronobacterium pharaonis to Halorubrum, Natrialba, and Natronomonas gen. nov., respectively, as Halorubrum vacuolatum comb. nov., Natrialba magadii comb. nov., and Natronomonas pharaonis comb. nov., respectively. Int. J. Syst. Bacteriol. 47, 853–857. doi: 10.1099/00207713-47-3-853
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165. doi: 10.7717/peerj.1165
Klatt, J. M., Haas, S., Yilmaz, P., de Beer, D., and Polerecky, L. (2015). Hydrogen sulfide can inhibit and enhance oxygenic photosynthesis in a cyanobacterium from sulfidic springs. Environ. Microbiol. 17, 3301–3313. doi: 10.1111/1462-2920.12791
Kompantseva, E. I., Komova, A. V., Rusanov, I. I., Pimenov, N. V., and Sorokin, D. Y. (2009). Primary production of organic matter and phototrophic communities in the soda lakes of the Kulunda Steppe (Altai Krai). Microbiology 78, 643–649. doi: 10.1134/S002626170905018x
Kovaleva, O. L., Tourova, T. P., Muyzer, G., Kolganova, T. V., and Sorokin, D. Y. (2011). Diversity of RuBisCO and ATP citrate lyase genes in soda lake sediments. FEMS Microbiol. Ecol. 75, 37–47. doi: 10.1111/j.1574-6941.2010.00996.x
Krulwich, T. A., Sachs, G., and Padan, E. (2011). Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol. 9, 330–343. doi: 10.1038/nrmicro2549
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
La Cono, V., Messina, E., Rohde, M., Arcadi, E., Ciordia, S., Crisafi, F., et al. (2019). Differential polysaccharide utilization is the basis for a nanohaloarchaeon : haloarchaeon symbiosis. bioRxiv [Preprint]. doi: 10.1101/794461
Lanzen, A., Simachew, A., Gessesse, A., Chmolowska, D., Jonassen, I., and Ovreas, L. (2013). Surprising prokaryotic and eukaryotic diversity, community structure and biogeography of Ethiopian soda lakes. PLoS One 8:e72577. doi: 10.1371/journal.pone.0072577
Leon, M. J., Fernandez, A. B., Ghai, R., Sanchez-Porro, C., Rodriguez-Valera, F., and Ventosa, A. (2014). From metagenomics to pure culture: isolation and characterization of the moderately halophilic bacterium Spiribacter salinus gen. nov., sp nov. Appl. Environ. Microbiol. 80, 3850–3857. doi: 10.1128/Aem.00430-414
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, D. H., Liu, C. M., Luo, R. B., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Lien, T., Madsen, M., Steen, I. H., and Gjerdevik, K. (1998). Desulfobulbus rhabdoformis sp. nov., a sulfate reducer from a water-oil separation system. Int. J. Syst. Bacteriol. 48, 469–474. doi: 10.1099/00207713-48-2-469
Liu, X. B., Li, M., Castelle, C. J., Probst, A. I., Zhou, Z. C., Pan, J., et al. (2018). Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 6:102. doi: 10.1186/S40168-018-0488-482
Lopez-Perez, M., Ghai, R., Leon, M. J., Rodriguez-Olmos, A., Copa-Patino, J. L., Soliveri, J., et al. (2013). Genomes of “Spiribacter”, a streamlined, successful halophilic bacterium. BMC Genomics 14:787. doi: 10.1186/1471-2164-14-787
Melack, J. M. (1981). Photosynthetic activity of phytoplankton in tropical African soda lakes. Hydrobiologia 81, 71–85. doi: 10.1007/Bf00048707
Melack, J. M., and Kilham, P. (1974). Photosynthetic rates of phytoplankton in East-African Alkaline, Saline Lakes. Limnol. Oceanogr. 19, 743–755. doi: 10.4319/lo.1974.19.5.0743
Mesbah, N. M., Abou-El-Ela, S. H., and Wiegel, J. (2007). Novel and unexpected prokaryotic diversity in water and sediments of the alkaline, hypersaline lakes of the wadi an natrun, egypt. Microb. Ecol. 54, 598–617. doi: 10.1007/s00248-006-9193-y
Minegishi, H., Enomoto, S., Echigo, A., Shimane, Y., Kondo, Y., Inoma, A., et al. (2017). Salinarchaeum chitinilyticum sp. nov., a chitin-degrading haloarchaeon isolated from commercial salt. Int. J. Syst. Evol. Microbiol. 67, 2274–2278. doi: 10.1099/ijsem.0.001941
Mori, K., Suzuki, K. I., Yamaguchi, K., Urabe, T., and Hanada, S. (2015). Thiogranum longum gen. nov., sp. nov., an obligately chemolithoautotrophic, sulfur-oxidizing bacterium of the family Ectothiorhodospiraceae isolated from a deep-sea hydrothermal field, and an emended description of the genus Thiohalomonas. Int. J. Syst. Evol. Microbiol. 65(Pt 1), 235–241. doi: 10.1099/ijs.0.070599-70590
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Narasingarao, P., Podell, S., Ugalde, J. A., Brochier-Armanet, C., Emerson, J. B., Brocks, J. J., et al. (2012). De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 6, 81–93. doi: 10.1038/ismej.2011.78
Olm, M. R., Brown, C. T., Brooks, B., and Banfield, J. F. (2017). dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868. doi: 10.1038/ismej.2017.126
Oremland, R. S., Saltikov, C. W., Stolz, J. F., and Hollibaugh, J. T. (2017). Autotrophic microbial arsenotrophy in arsenic-rich soda lakes. FEMS Microbiol. Lett. 364:fnx146. doi: 10.1093/femsle/fnx146
Oren, A. (2011). Thermodynamic limits to microbial life at high salt concentrations. Environ. Microbiol. 13, 1908–1923. doi: 10.1111/j.1462-2920.2010.02365.x
Oren, A. (2015). Pyruvate: a key nutrient in hypersaline environments? Microorganisms 3, 407–416. doi: 10.3390/microorganisms3030407
Pagaling, E., Wang, H. Z., Venables, M., Wallace, A., Grant, W. D., Cowan, D. A., et al. (2009). Microbial biogeography of six salt lakes in Inner Mongolia, China, and a salt lake in Argentina. Appl. Environ. Microbiol. 75, 5750–5760. doi: 10.1128/Aem.00040-49
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Perez, R. C., and Matin, A. (1982). Carbon dioxide assimilation by Thiobacillus novellus under nutrient-limited mixotrophic conditions. J. Bacteriol. 150, 46–51. doi: 10.1128/jb.150.1.46-51.1982
Perez-Pomares, F., Bautista, V., Ferrer, J., Pire, C., Marhuenda-Egea, F. C., and Bonete, M. J. (2003). alpha-amylase activity from the halophilic archaeon Haloferax mediterranei. Extremophiles 7, 299–306. doi: 10.1007/s00792-003-0327-326
Phitsuwan, P., Morais, S., Dassa, B., Henrissat, B., and Bayer, E. A. (2019). The cellulosome paradigm in an extreme alkaline environment. Microorganisms 7:347. doi: 10.3390/microorganisms7090347
Pikuta, E. V., Hoover, R. B., Bej, A. K., Marsic, D., Whitman, W. B., Cleland, D., et al. (2003). Desulfonatronum thiodismutans sp nov., a novel alkaliphilic, sulfate-reducing bacterium capable of lithoautotrophic growth. Int. J. Syst. Evol. Microbiol. 53, 1327–1332. doi: 10.1099/ijs.0.02598-2590
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Qin, N., Yang, F. L., Li, A., Prifti, E., Chen, Y. F., Shao, L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64. doi: 10.1038/nature13568
Roberts, M. F. (2005). Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Syst. 1:5. doi: 10.1186/1746-1448-1-5
Segata, N., Bornigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/Ncomms3304
Seibold, G. M., Wurst, M., and Eikmanns, B. J. (2009). Roles of maltodextrin and glycogen phosphorylases in maltose utilization and glycogen metabolism in Corynebacterium glutamicum. Microbiol. Sgm 155, 347–358. doi: 10.1099/mic.0.023614-23610
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sherwood, J. E., Stagnitti, F., Kokkinn, M. J., and Williams, W. D. (1991). Dissolved-oxygen concentrations in hypersaline waters. Limnol. Oceanogr. 36, 235–250. doi: 10.4319/lo.1991.36.2.0235
Shi, W. Y., Qi, H. Y., Sun, Q. L., Fan, G. M., Liu, S. J., Wang, J., et al. (2019). gcMeta: a global catalogue of metagenomics platform to support the archiving, standardization and analysis of microbiome data. Nucleic Acids Res. 47, D637–D648. doi: 10.1093/nar/gky1008
Simachew, A., Lanzen, A., Gessesse, A., and Ovreas, L. (2016). Prokaryotic community diversity along an increasing salt gradient in a soda ash concentration pond. Microb. Ecol. 71, 326–338. doi: 10.1007/s00248-015-0675-677
Sorokin, D. Y. (2003). Oxidation of inorganic sulfur compounds by obligately organotrophic bacteria. Microbiology 72, 641–653. doi: 10.1023/B:Mici.0000008363.24128.E5
Sorokin, D. Y., Banciu, H. L., and Muyzer, G. (2015). Functional microbiology of soda lakes. Curr. Opin. Microbiol. 25, 88–96. doi: 10.1016/j.mib.2015.05.004
Sorokin, D. Y., Berben, T., Melton, E. D., Overmars, L., Vavourakis, C. D., and Muyzer, G. (2014). Microbial diversity and biogeochemical cycling in soda lakes. Extremophiles 18, 791–809. doi: 10.1007/s00792-014-0670-679
Sorokin, D. Y., and Chernyh, N. A. (2017). Desulfonatronospira sulfatiphila sp nov., and Desulfitispora elongata sp nov., two novel haloalkaliphilic sulfidogenic bacteria from soda lakes. Int. J. Syst. Evol. Microbiol. 67, 396–401. doi: 10.1099/ijsem.0.001640
Sorokin, D. Y., and Kuenen, J. G. (2005). Haloalkaliphilic sulfur-oxidizing bacteria in soda lakes. FEMS Microbiol. Rev. 29, 685–702. doi: 10.1016/j.femsre.2004.10.005
Sorokin, D. Y., Kuenen, J. G., and Muyzer, G. (2011). The microbial sulfur cycle at extremely haloalkaline conditions of soda lakes. Front. Microbiol. 2:44. doi: 10.3389/Fmicb.2011.00044
Sorokin, D. Y., Rusanov, I. I., Pimenov, N. V., Tourova, T. P., Abbas, B., and Muyzer, G. (2010). Sulfidogenesis under extremely haloalkaline conditions in soda lakes of Kulunda Steppe (Altai, Russia). FEMS Microbiol. Ecol. 73, 278–290. doi: 10.1111/j.1574-6941.2010.00901.x
Sorokin, D. Y., Tourova, T. P., Henstra, A. M., Stams, A. J. M., Galinski, E. A., and Muyzer, G. (2008a). Sulfidogenesis under extremely haloalkaline conditions by Desulfonatronospira thiodismutans gen. nov., sp nov., and Desulfonatronospira delicata sp nov - a novel lineage of Deltaproteobacteria from hypersaline soda lakes. Microbiol. Sgm 154, 1444–1453. doi: 10.1099/mic.0.2007/015628-15620
Sorokin, D. Y., Tourova, T. P., Mussmann, M., and Muyzer, G. (2008b). Dethiobacter alkaliphilus gen. nov sp nov., and Desulfurivibrio alkaliphilus gen. nov sp nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles 12, 431–439. doi: 10.1007/s00792-008-0148-148
Sorokin, D. Y., Tourova, T. P., and Muyzer, G. (2005). Oxidation of thiosulfate to tetrathionate by an haloarchaeon isolated from hypersaline habitat. Extremophiles 9, 501–504. doi: 10.1007/s00792-005-0465-460
Spring, S., Kampfer, P., and Schleifer, K. H. (2001). Limnobacter thiooxidans gen. nov., sp nov., a novel thiosulfate-oxidizing bacterium isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 51, 1463–1470. doi: 10.1099/00207713-51-4-1463
Stam, M. C., Mason, P. R. D., Pallud, C., and Van Cappellen, P. (2010). Sulfate reducing activity and sulfur isotope fractionation by natural microbial communities in sediments of a hypersaline soda lake (Mono Lake. California). Chem. Geol. 278, 23–30. doi: 10.1016/j.chemgeo.2010.08.006
Sunagawa, S., Mende, D. R., Zeller, G., Izquierdo-Carrasco, F., Berger, S. A., Kultima, J. R., et al. (2013). Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 10, 1196–1199. doi: 10.1038/Nmeth.2693
Sybesma, C., Schowanek, D., Slooten, L., and Walravens, N. (1986). Anoxygenic photosynthetic hydrogen-production and electron-transport in the cyanobacterium oscillatoria-limnetica. Photosynth. Res. 9, 149–158. doi: 10.1007/Bf00029740
Thauer, R. K., Jungermann, K., and Decker, K. (1977). Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41, 100–180. doi: 10.1128/mmbr.41.1.100-180.1977
Tourova, T. P., Kovaleva, O. L., Bumazhkin, B. K., Patutina, E. O., Kuznetsov, B. B., Bryantseva, I. A., et al. (2011). Application of Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase genes as molecular markers for assessment of the diversity of autotrophic microbial communities inhabiting the upper sediment Horizons of the Saline and Soda Lakes of the Kulunda Steppe. Microbiology 80, 812–825. doi: 10.1134/S0026261711060221
Tourova, T. P., Slobodova, N. V., Bumazhkin, B. K., Kolganova, T. V., Muyzer, G., and Sorokin, D. Y. (2013). Analysis of community composition of sulfur-oxidizing bacteria in hypersaline and soda lakes using soxB as a functional molecular marker. FEMS Microbiol. Ecol. 84, 280–289. doi: 10.1111/1574-6941.12056
Tuttle, J. H., and Jannasch, H. W. (1977). Thiosulfate stimulation of microbial dark assimilation of carbon dioxide in shallow marine waters. Microb. Ecol. 4, 9–25. doi: 10.1007/BF02010426
Vandieken, V., Mussmann, M., Niemann, H., and Jorgensen, B. B. (2006). Desulfuromonas svalbardensis sp. nov. and Desulfuromusa ferrireducens sp. nov., psychrophilic, Fe(III)-reducing bacteria isolated from Arctic sediments, Svalbard. Int. J. Syst. Evol. Microbiol. 56(Pt 5), 1133–1139. doi: 10.1099/ijs.0.63639-63630
Vavourakis, C. D., Andrei, A. S., Mehrshad, M., Ghai, R., Sorokin, D. Y., and Muyzer, G. (2018). A metagenomics roadmap to the uncultured genome diversity in hypersaline soda lake sediments. Microbiome 6:168. doi: 10.1186/S40168-018-0548-547
Vavourakis, C. D., Ghai, R., Rodriguez-Valera, F., Sorokin, D. Y., Tringe, S. G., Hugenholtz, P., et al. (2016). Metagenomic insights into the uncultured diversity and physiology of microbes in four hypersaline soda lake brines. Front. Microbiol. 7:211. doi: 10.3389/Fmicb.2016.00211
Vavourakis, C. D., Mehrshad, M., Balkema, C., van Hall, R., Andrei, A. S., Ghai, R., et al. (2019). Metagenomes and metatranscriptomes shed new light on the microbial-mediated sulfur cycle in a Siberian soda lake. BMC Biol. 17:69. doi: 10.1186/s12915-019-0688-687
Xavier, K. B., Peist, R., Kossmann, M., Boos, W., and Santos, H. (1999). Maltose metabolism in the hyperthermophilic archaeon Thermococcus litoralis: Purification and characterization of key enzymes. J. Bacteriol. 181, 3358–3367. doi: 10.1128/jb.181.11.3358-3367.1999
Zhao, D. H., Zhang, S. J., Xue, Q., Chen, J. Y., Zhou. J., Cheng, F. Y., et al. (2019). Abundant taxa and favorable pathways in the microbiome of soda-saline lakes in Inner Mongolia. Res. Square [Preprint]. doi: 10.21203/rs.2.19124/v1
Zheng, X. Z., Minggang, Xu, C., and Li, B. (2002). Records of Salt Lakes in China. Beijing: Science Press (in Chinese).
Zhu, W. H., Lomsadze, A., and Borodovsky, M. (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38:e132. doi: 10.1093/nar/gkq275
Keywords: soda-saline lakes, deep metagenomic sequencing, microbiome, abundant taxa, sulfur cycling, glucan metabolism
Citation: Zhao D, Zhang S, Xue Q, Chen J, Zhou J, Cheng F, Li M, Zhu Y, Yu H, Hu S, Zheng Y, Liu S and Xiang H (2020) Abundant Taxa and Favorable Pathways in the Microbiome of Soda-Saline Lakes in Inner Mongolia. Front. Microbiol. 11:1740. doi: 10.3389/fmicb.2020.01740
Received: 02 February 2020; Accepted: 03 July 2020;
Published: 24 July 2020.
Edited by:
Brian D. Lanoil, University of Alberta, CanadaReviewed by:
Charlotte Dafni Vavourakis, Independent Researcher, Innsbruck, AustriaHongchen Jiang, China University of Geosciences, China
Copyright © 2020 Zhao, Zhang, Xue, Chen, Zhou, Cheng, Li, Zhu, Yu, Hu, Zheng, Liu and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Xiang, eGlhbmdoQGltLmFjLmNu
†These authors have contributed equally to this work