 Maria Mooshammer1†
Maria Mooshammer1† Ricardo J. E. Alves2†
Ricardo J. E. Alves2† Barbara Bayer2†
Barbara Bayer2† Michael Melcher2
Michael Melcher2 Michaela Stieglmeier2Lara Jochum3
Michaela Stieglmeier2Lara Jochum3 Simon K.-M. R. Rittmann2Margarete Watzka1
Simon K.-M. R. Rittmann2Margarete Watzka1 Christa Schleper2
Christa Schleper2 Gerhard J. Herndl2,4
Gerhard J. Herndl2,4 Wolfgang Wanek1*
Wolfgang Wanek1*- 1Centre for Microbiology and Environmental Systems Science, University of Vienna, Vienna, Austria
- 2Department of Functional and Evolutionary Ecology, University of Vienna, Vienna, Austria
- 3LMU – Max von Pettenkofer Institute for Hygiene and Medical Microbiology, Ludwig Maximilian University of Munich, Munich, Germany
- 4Department of Marine Microbiology and Biogeochemistry, Royal Netherlands Institute for Sea Research (NIOZ), Utrecht University, Utrecht, Netherlands
The naturally occurring nitrogen (N) isotopes, 15N and 14N, exhibit different reaction rates during many microbial N transformation processes, which results in N isotope fractionation. Such isotope effects are critical parameters for interpreting natural stable isotope abundances as proxies for biological process rates in the environment across scales. The kinetic isotope effect of ammonia oxidation (AO) to nitrite (NO2–), performed by ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB), is generally ascribed to the enzyme ammonia monooxygenase (AMO), which catalyzes the first step in this process. However, the kinetic isotope effect of AMO, or εAMO, has been typically determined based on isotope kinetics during product formation (cumulative product, NO2–) alone, which may have overestimated εAMO due to possible accumulation of chemical intermediates and alternative sinks of ammonia/ammonium (NH3/NH4+). Here, we analyzed 15N isotope fractionation during archaeal ammonia oxidation based on both isotopic changes in residual substrate (RS, NH4+) and cumulative product (CP, NO2–) pools in pure cultures of the soil strain Nitrososphaera viennensis EN76 and in highly enriched cultures of the marine strain Nitrosopumilus adriaticus NF5, under non-limiting substrate conditions. We obtained εAMO values of 31.9–33.1‰ for both strains based on RS (δ15NH4+) and showed that estimates based on CP (δ15NO2–) give larger isotope fractionation factors by 6–8‰. Complementary analyses showed that, at the end of the growth period, microbial biomass was 15N-enriched (10.1‰), whereas nitrous oxide (N2O) was highly 15N depleted (−38.1‰) relative to the initial substrate. Although we did not determine the isotope effect of NH4+ assimilation (biomass formation) and N2O production by AOA, our results nevertheless show that the discrepancy between εAMO estimates based on RS and CP might have derived from the incorporation of 15N-enriched residual NH4+ after AMO reaction into microbial biomass and that N2O production did not affect isotope fractionation estimates significantly.
Introduction
Knowledge of natural 15N abundances and of nitrogen (N) isotope fractionation effects associated with key microbial N transformation processes has contributed greatly to our understanding of the marine N cycle (Casciotti and Buchwald, 2012; Buchwald and Casciotti, 2013) and of terrestrial gaseous N emissions (Houlton and Bai, 2009), namely atmospheric N2O sources and sinks (Yoshida and Toyoda, 2000), and biological N fixation (Vitousek et al., 2013). The oxidation of NH4+ to NO2–—the first and rate-limiting step in nitrification—is a central process in the marine and terrestrial N cycles, as well as the major driver of a large N isotope effect that leads to formation of 15N-depleted products such as NO, N2O, NO2–, and NO3–, while residual NH4+ becomes 15N-enriched during that process (Mariotti et al., 1981; Sigman and Casciotti, 2001). A detailed understanding of N isotope effects of the range of N transformation processes is thus critical for adequate biological interpretation of natural 15N isotope patterns in the environment (Casciotti, 2016).
Besides the recently discovered comammox bacteria (Daims et al., 2015; van Kessel et al., 2015), ammonia oxidation is catalyzed by both ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB), with different relative contributions across ecosystems and environmental conditions (Prosser and Nicol, 2012; Prosser et al., 2019). On a cellular level, ammonia oxidation is a multi-step process that comprises different enzymatic reactions and chemical equilibrium processes, which can all contribute to the N isotope fractionation effects inferred from extracellular N pools (Casciotti et al., 2003; Santoro and Casciotti, 2011). The isotopic fractionation effect (ε) of ammonia oxidizers has been typically inferred based on changes in δ15N of the cumulative product (CP) NO2– (εCP), and attributed to the initial enzymatic step catalyzed by the ammonia monooxygenase (AMO) enzyme, defined as εAMO. However, εCP estimates reflect the combined fractionation effects of the isotope equilibrium between NH4+ and NH3 [NH3, the proposed substrate for ammonia oxidation, is depleted in 15N relative to NH4+ (Hermes et al., 1985)], the AMO-catalyzed reaction, and accumulation of several intermediates derived from subsequent enzymatic processes (Casciotti et al., 2003). For instance, εCP estimates may be affected by the accumulation of essential intermediates, such as hydroxylamine (NH2OH) and by the production of gaseous N by-products (nitric oxide, NO; and nitrous oxide, N2O), which may represent further 15N fractionation steps. Consequently, this could result in a difference of kinetic isotope effect estimates derived from residual substrate (RS) and CP (Casciotti et al., 2003). Not only could these “leakage” processes alter CP-based estimates of εAMO, but their different contributions to ammonia utilization and to εCP may also underlie the large differences observed in εAMObetween ammonia-oxidizing organisms (Mariotti et al., 1981; Yoshida, 1988; Casciotti et al., 2003; Santoro and Casciotti, 2011).
Estimates of isotope effects based on the change in δ15NH4+ (εRS) can circumvent many of the expected biases associated with εCP, as they are not affected by the multiple subsequent equilibria, enzymatic transformations, and intermediate N pools, as discussed but not quantified previously (Casciotti et al., 2003; Santoro and Casciotti, 2011). However, to our knowledge, only one study has determined the isotope fractionation factors based on concurrent measurements of changes in isotopic composition of RS and CP of ammonia oxidation, namely in cultures of the AOB Nitrosomonas europaea (Mariotti et al., 1981). This study found no difference between εRS and εCP, suggesting that ammonia oxidation can be effectively regarded as a “one-step process,” where the AMO-catalyzed reaction constitutes the rate-limiting and sole isotope fractionation step. On the other hand, AOB and AOA seem to harbor fundamentally distinct ammonia oxidation pathways and exhibit different yields of gaseous N compounds per mole of NH4+ consumed (Walker et al., 2010; Kozlowski et al., 2016). Importantly, the enzyme hydroxylamine dehydrogenase (HAO), which performs the second step in ammonia oxidation of AOB, has not been identified in AOA, and thus it remains unclear how NH2OH is converted to NO2– in AOA (Walker et al., 2010; Kerou et al., 2016). Moreover, a recent study has provided evidence that the bacterial HAO oxidizes NH2OH to NO rather than to NO2–, as generally assumed, with the latter resulting from non-enzymatic oxidation of NO by oxygen (Caranto and Lancaster, 2017). Previous studies have shown that NO is also an essential intermediate in ammonia oxidation by AOA, as their growth and activity is highly sensitive to exposure to an NO scavenger (Shen et al., 2013; Kozlowski et al., 2016).
Here, we tested whether the kinetic isotope effect of archaeal ammonia oxidation based on CP (δ15NO2–) alone might be biased, by comparing the isotope fractionation factors inferred from both RS and CP pools. For this, we determined the kinetic isotope effects during growth of two phylogenetically and ecologically distinct AOA: the axenic strain Nitrososphaera viennensis EN76 (Stieglmeier et al., 2014a), isolated from soil, and the highly enriched marine strain Nitrosopumilus adriaticus NF5 (Bayer et al., 2016). This is also the first study of 15N isotope fractionation of ammonia oxidation by an AOA strain in pure culture. All previous studies of kinetic isotope effects of AOA have been performed with enrichment cultures with varying degrees of enrichment (Santoro and Casciotti, 2011; Nishizawa et al., 2016) and bacterial contaminants that may have contributed to the variation in isotope effects through consumption of and inputs to the same N pools.
Materials and Methods
Pure cultures of N. viennensis EN76 were cultivated in freshwater medium and incubated at 37°C, as described by Tourna et al. (2011). In a first experiment, quadruplicate cultures were supplemented with 1 mM NH4+ and 0.1 mM pyruvate; in a second experiment, quadruplicate cultures were supplemented with 2 mM NH4+ and 0.5 mM pyruvate to generate higher cell biomass and sufficient N2O concentrations for isotopic analysis, in order to determine their potential effect on εAMO. Quadruplicate enrichment cultures of N. adriaticus NF5 were cultivated in Synthetic Crenarchaeota Medium (SCM) at 30°C as described by Bayer et al. (2016). The medium was supplemented with 1 mM NH4+ and 5% (v/v) autoclaved seawater, which was sterile-filtered (0.22 μm GTTP, Millipore). Kanamycin at a final concentration of 100 μg ml–1 was used to inhibit bacterial contaminants. At the time of the experiment (January 2013), the enrichment level of strain NF5 was ∼95%, as it contained a heterotrophic non-nitrifying/non-denitrifying contaminant of the alphaproteobacterial species Oceanicaulis alexandrii (Bayer et al., 2019).
Ammonia-oxidizing archaea growth was monitored by measuring nitrite production using the Griess method (Hood-Nowotny et al., 2010), coupled to NH4+ consumption determined using the Berthelot method for N. viennensis cultures (Hood-Nowotny et al., 2010) and the o-Phthalaldehyde (OPA) method for N. adriaticus cultures (Goyal et al., 1988). δ15NH4+ was quantified by microdiffusion (Sørensen and Jensen, 1991) with subsequent analysis on a continuous-flow isotope ratio mass spectrometer consisting of an elemental analyzer (EA1110, CE Instruments) coupled via a ConFlo III interface (Finnigan MAT, Thermo Fisher Scientific) to the isotope ratio mass spectrometer (IRMS; DeltaPLUS, Finnigan MAT, Thermo Fisher Scientific). δ15NO2– was determined based on the reduction of NO2– to N2O by azide under acidified conditions (Lachouani et al., 2010). Concentrations and isotopic ratios of N2O were determined using a purge-and-trap GC/IRMS system (PreCon - GasBench II headspace analyzer, Delta Advantage V IRMS; Thermo Fischer Scientific, Vienna, Austria). For NH4+ and NO2– isotope measurements, we included blanks, concentration standards, and isotope standards varying in natural 15N abundance together with the samples through the full microdiffusion and azide procedures to allow corrections for blank contribution, incomplete reaction, and procedural isotope fractionation (Lachouani et al., 2010). Nitrogen content and δ15N signature of AOA biomass were determined by EA-IRMS as described above. δ15N signatures [‰ vs. AIR] were calculated relative to the ratio R (15N:14N) of the atmospheric N2 standard (AIR), as δ15N = (Rsample/Rstandard− 1) × 1000.
Isotope fractionation factors (ε) were calculated based on the Rayleigh closed system isotope fractionation, based on changes in the isotopic compositions of RS (i.e., NH4+) and CP (i.e., NO2–) (Mariotti et al., 1981):
where δS0 is δ15N of initial NH4+, δRS is δ15NH4+, δCP is δ15NO2– and f is the fraction of the initial [NH4+] remaining in the culture. Plots of versus ln(f) and of δCP−δS0 versus yield linear relations, with the slope representing the kinetic isotope effect based on the isotopic change in substrate (εRS) and product (εAP), respectively. Uncertainties of ε are expressed as standard error of the slope. Differences in isotope fractionation effects between cultures were assessed by testing significant differences between their regression plots, using R (R Development Core Team, 2012).
Results and Discussion
Based on the oxidation of NH3/NH4+ to NO2–—a typical proxy for ammonia oxidizer growth, as it strongly correlates with growth rates (Stieglmeier et al., 2014a; Bayer et al., 2016)—all cultures showed growth curves typical for batch cultures of AOA, reaching stationary phase after 7 days for N. adriaticus, and after 3–4 days for N. viennensis cultures (Figures 1A–C). Nitrogen isotope fractionation was reflected in both the substrate (i.e., NH4+) and the product (i.e., NO2–) of ammonia oxidation, and followed typical Rayleigh isotope fractionation kinetics for closed systems (Figures 1D–F): NH4+ became increasingly 15N-enriched with the fraction of NH4+ oxidized, while NO2– was strongly 15N-depleted after correction for NO2– deriving from the inoculum. With an increasing fraction of NH4+ oxidized, δ15NO2– converged toward the isotopic signature of the initial NH4+. Both N. adriaticus and N. viennensis (including cultures grown on 1 and 2 mM NH4+) exhibited 15N isotope fractionation factors based on substrate (εRS) between 31.9 and 33.1‰, and based on product (εCP) between 37.7 and 49.1‰ (Figures 2A–F). We found no significant difference between the isotope fractionation factors of the different AOA cultures studied here based on δ15N evolution of the substrate (εRS; comparison of slopes, df = 2, F = 0.519, p = 0.598) or the product (εCP; comparison of slopes, df = 2, F = 2.380, p = 0.102). The N isotope fractionation factors based on δ15NO2– (εCP) were larger than those based on δ15NH4+ (εRS) by 8.0, 5.8, and 5.9‰ for N. adriaticus, and for N. viennensis grown on 1 mM or 2 mM NH4+, respectively.
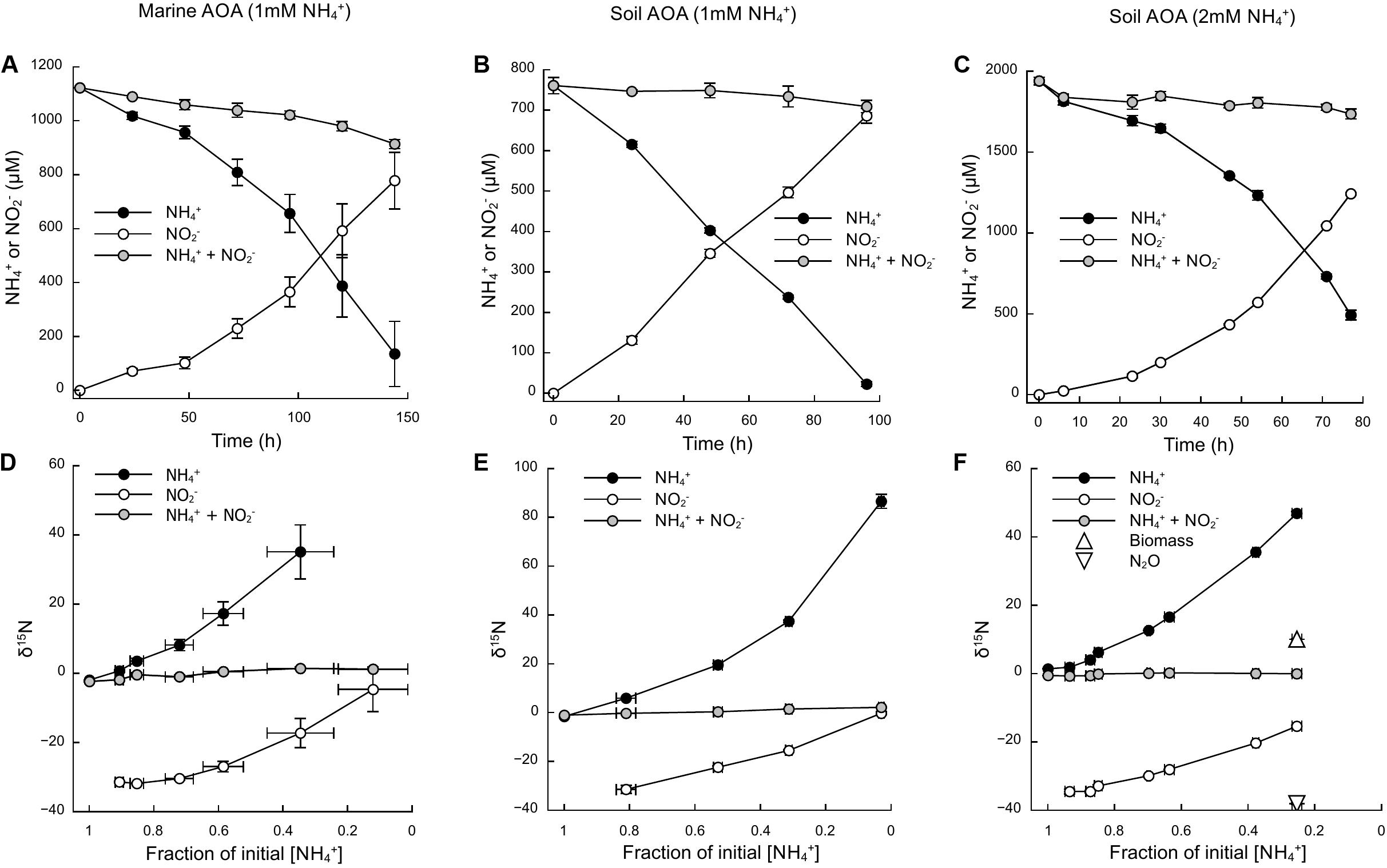
Figure 1. Ammonia oxidation and isotopic signature of NH4+ and NO2– of a marine (N. adriaticus NF5 enrichment culture) and a soil (N. viennensis EN76) AOA. (A–C) Time course of NH4+ oxidation to NO2–. (D–F) Isotopic composition of NH4+ and NO2– as a function of initial NH4+ oxidized. Some error bars are smaller than the symbol.
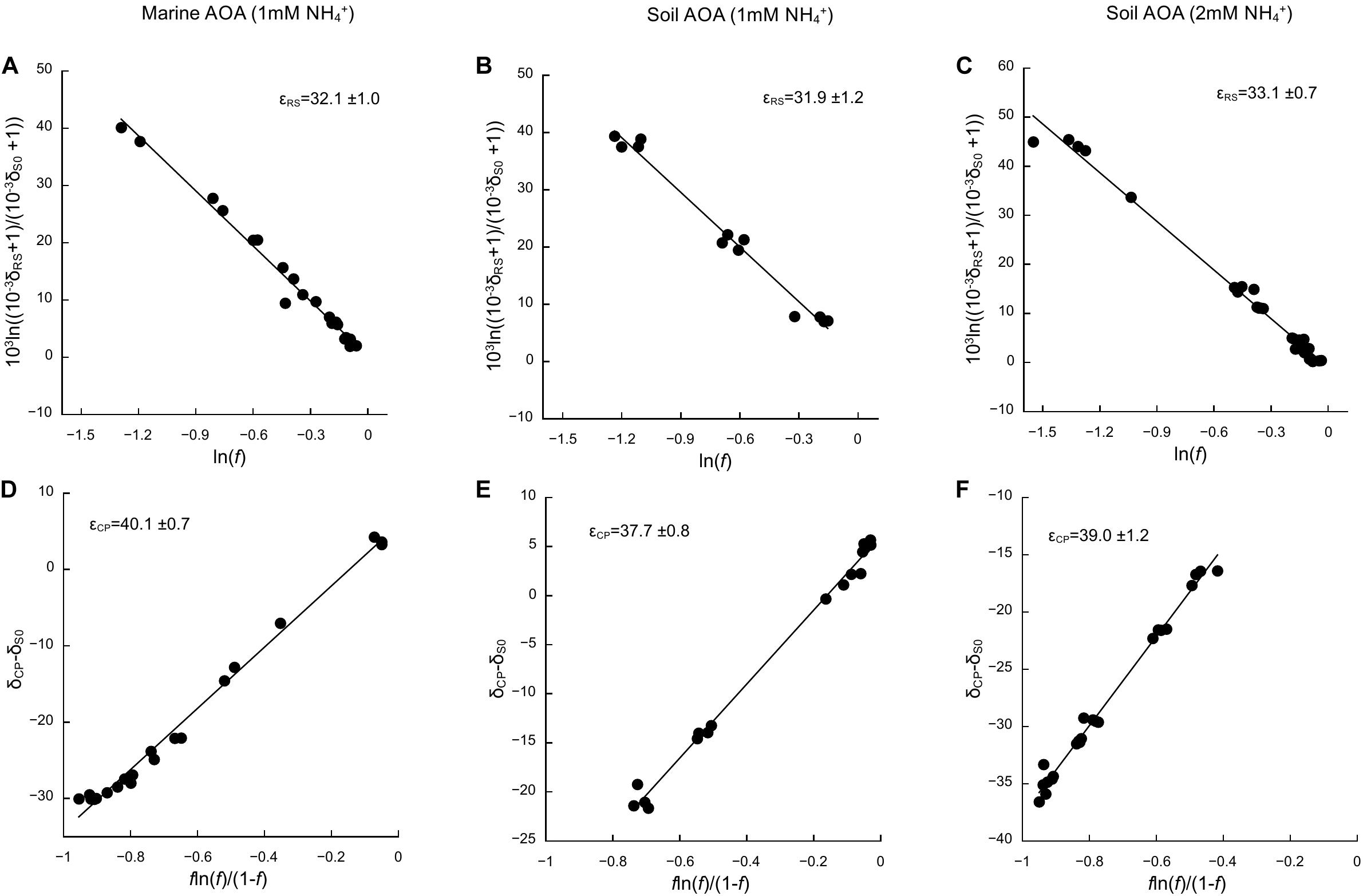
Figure 2. Kinetic isotope effects of the marine AOA N. adriaticus and the soil AOA N. viennensis. Isotope fractionation factors (εRS and εCP) were calculated based on changes in (A–C) δ15N-NH4+ (δRS) and (D–F) δ15N-NO2– (δCP), using linear regressions of versus ln(f) and δCP−δS0 versus , respectively, as described in Mariotti et al. (1981). δS0 is δ15N of initial NH4+ and f is the fraction of the initial [NH4+] remaining in the culture. Uncertainties of εRS and εCP are expressed as SE of the slope. We used multiple-point estimates because they have lower uncertainty than single-point analyses.
Nitrogen isotope fractionation has been studied in several AOB strains, but only in three marine and one thermophilic AOA enrichment cultures. These AOA enrichment cultures showed average N isotope fractionation factors between 22 and 25‰ at low substrate concentrations, and up to 32.0‰ at higher ammonium concentrations (Santoro and Casciotti, 2011; Nishizawa et al., 2016, measured via the isotopic composition of the product nitrite; see Table 1). These estimates are in the same range as the reported average isotope effects for different AOB strains, i.e., 14–38‰ (Delwiche and Steyn, 1970; Mariotti et al., 1981; Casciotti et al., 2003). 15N isotope fractionation factors of N. viennensis and N. adriaticus are in the upper range, or higher, than those previously reported for AOA, which might be due to the higher ammonia concentrations applied in our study (1–2 mM in our study vs. 200 μM in Nishizawa et al., 2016; 10–75 μM in Santoro and Casciotti, 2011). Previous studies have indicated that higher initial ammonia concentrations lead to more stable and higher 15N isotope fractionation (Casciotti et al., 2003; Santoro and Casciotti, 2011).
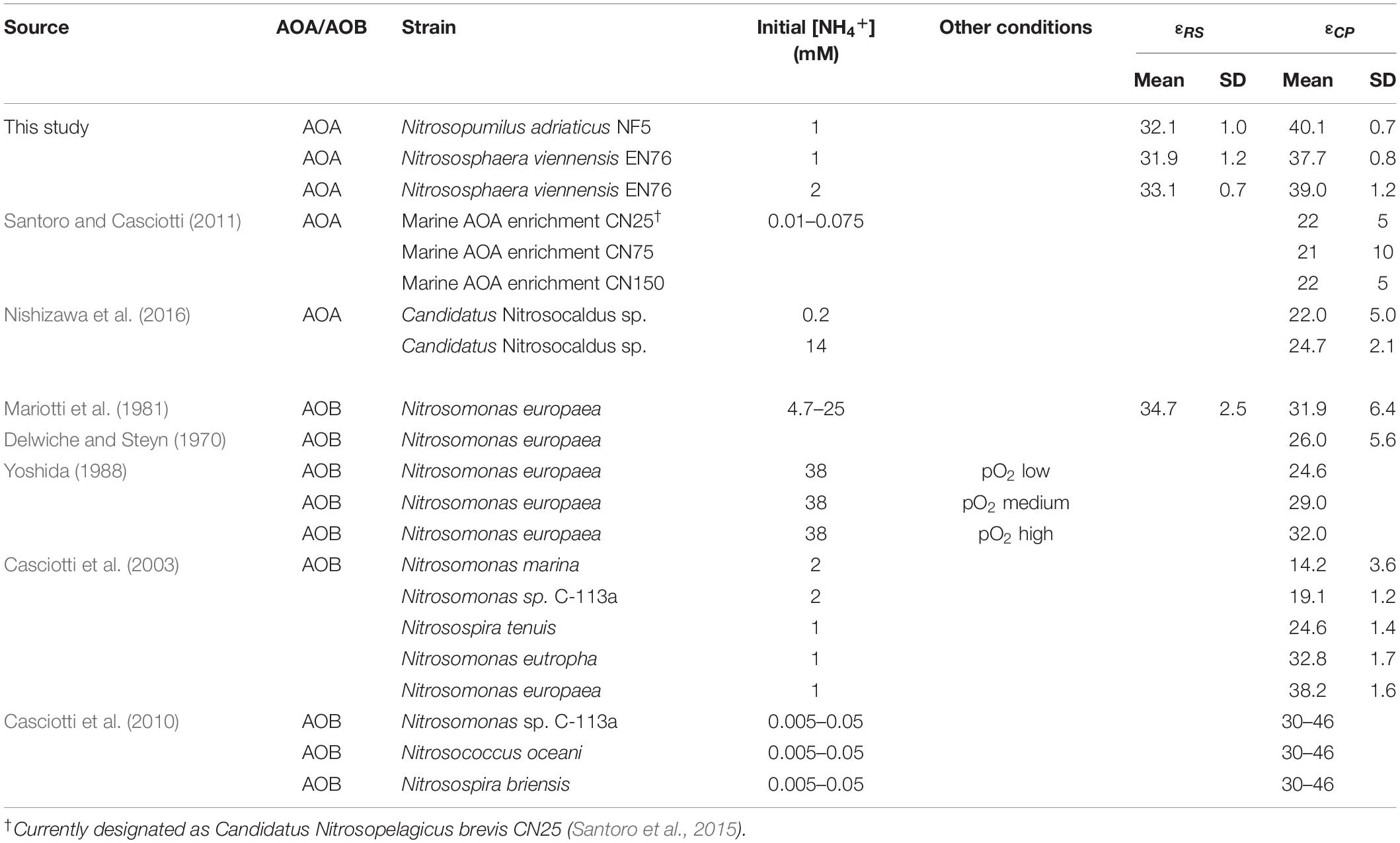
Table 1. Compilation of published kinetic isotope effects of AOA and AOB.
We also measured εAMO based on changes in δ15NH4+ (i.e., the residual substrate) to both circumvent and assess potential biases associated with estimates based on δ15NO2– (i.e., the cumulative product). It should be noted, however, that different apparent isotope effects in whole cells may also be observed in the NH4+ pool, despite constant AMO enzyme-level isotope effects, depending, for example, on the balance between ammonia oxidation rates and ammonia diffusion across the S-layer (i.e., outermost cell envelope component in AOA) (Casciotti et al., 2003; Li et al., 2018). Published models of AOA and AOB metabolism favor the hypothesis of a (pseudo-)periplasmic location of the ammonia oxidation process (Arp and Stein, 2003; Walker et al., 2010; Simon and Klotz, 2013). However, AOA and AOB harbor very distinct NH3/NH4+ transport systems (Offre et al., 2014), whose role in ammonia oxidation and contribution to observed differences in 15N isotope fractionation remain unclear (Arp and Stein, 2003). At low ammonia concentrations, ammonia oxidation rates are expected to become limited by NH4+ transport/NH3 diffusion, resulting in the convergence of the isotope effect toward that of NH4+/NH3 equilibrium (if NH3 is mainly taken up by the cells) or NH4+/NH3 transport. The NH4+/NH3 equilibrium isotope effect has been estimated to be 19.2‰ in aqueous solution (Hermes et al., 1985), whereas secondary active ammonium (AMT) transporters, which are highly expressed in AOA (Carini et al., 2017), have been shown to exert isotope fractionation of around 13–15‰, due to deprotonation of NH4+ during transport (Ariz et al., 2018). It is unlikely that ammonia oxidation has been limited by NH3 availability in our study, because of the high substrate concentrations used, which are well above the Km of the AMO of N. viennensis (5.4 μM NH3 + NH4+; Kits et al., 2017) and that of the marine strain Nitrosopumilus maritimus strain SCM1 (0.13 μM NH3 + NH4+; Martens-Habbena et al., 2009), which is closely related to N. adriaticus. Furthermore, Nishizawa et al. (2016) estimated that, when NH3 concentrations in the pseudo-periplasm are lower than in the medium under laboratory conditions, cell-specific NH3 diffusion rates into the pseudo-periplasm are higher than cell-specific ammonia oxidation rates. It has also been proposed that the charged S-layer proteins of AOA enhance the diffusion of charged solutes, such as NH4+, which concentrates NH4+ in the pseudo-periplasmic space near the active site of the AMO (Li et al., 2018), where then the equilibrium reaction between NH4+ and NH3 is relatively fast and considered not to be rate-limiting.
Even if ammonia oxidation was not limited by periplasmatic NH3 availability, the apparent isotope effect of the AMO can also be underestimated due to concurrent NH4+ assimilation, which has a smaller isotope effect. This process would alter observed εRS estimates in proportion to the amount of NH4+ assimilated and the isotope effect for NH4+ assimilation (4–27‰; Hoch et al., 1992). Therefore, we also measured δ15N of the cell biomass at the end of incubation of N. viennensis grown on 2 mM NH4+ (Figures 1F, 3). Although it is impossible to infer directly the contribution of N assimilation to εRS from just one end-point measurement, we propose that N assimilation substantially contributed to the decrease of εRS relative to εCP in our study, as biomass was 15N-enriched by ∼10‰ compared to initial NH4+. Biomass N represented 3.1% (±0.3 SE) of ammonia oxidized by N. viennensis grown on 2 mM NH4+. Although dissolved inorganic N (DIN) concentrations (sum of [NH4+] and [NO2–]) were relatively constant over the course of ammonia oxidation, we recovered only 81.9% (±1.5 SE) of the initial DIN by the end of incubation of N. adriaticus, and 94.7% (±3.4 SE) and 90.7% (±1.1 SE) of N. viennensis grown on 1 mM NH4+ or 2 mM NH4+, respectively. In N. adriaticus cultures, assimilation of N by contaminant bacteria likely did not contribute substantially to the lower εRS relative to εCP, due to the high enrichment level of the culture (95%) at the time of the experiment, and the fact that εRS of N. adriaticus was similar to that of N. viennensis in pure culture. In addition, the 15N-enrichment of N. viennensis’ biomass shows that AMO preferentially, and primarily, reacts on pseudo-periplasmatic NH3, causing 15N-enrichment of the residual ammonia, which is subsequently assimilated into biomass. We thus propose that under substrate replete conditions, the observed isotope effects of εRS of 31.9–33.1‰ primarily reflect the kinetic isotope effect of the AMO-catalyzed reaction, modified by the NH4+/NH3 equilibrium isotope effect (19.2‰; Hermes et al., 1985) and decreased by the contribution of the lower kinetic isotope effect of NH4+ assimilation for anabolic purposes (4–27‰; Hoch et al., 1992). Moreover, it should be noted that some ammonia oxidizers use distinct pathways of NH4+ assimilation, even among just AOA, which may contribute to different kinetic isotope effects. For instance, some members of the AOA genus Candidatus Nitrosocosmicus appear to assimilate NH4+ via glutamate synthase (GOGAT), whereas all other known AOA use the glutamate dehydrogenase (GDH) pathway (Alves et al., 2019).
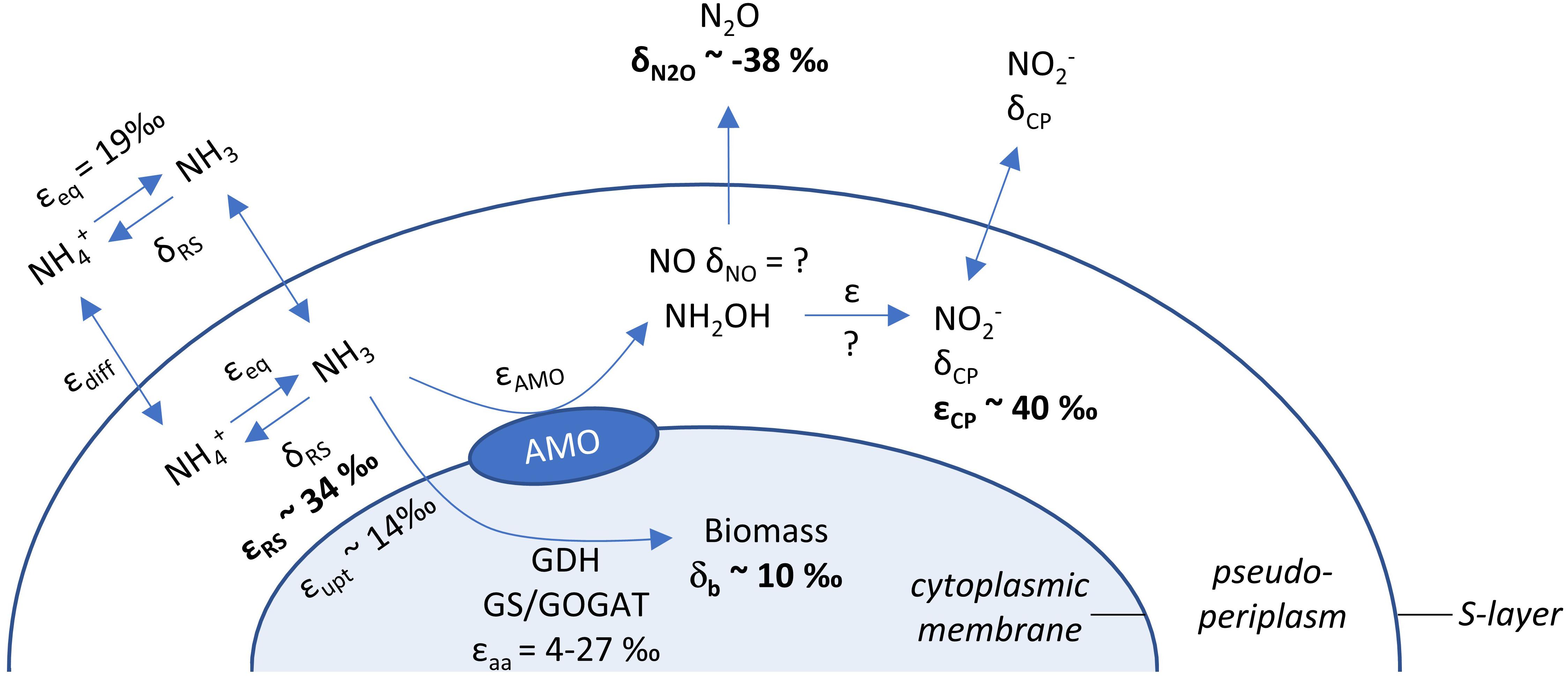
Figure 3. Schematic overview of processes and isotope fractionation effects involved in ammonia oxidation, growth and intermediate formation of the soil AOA N. viennensis. δ15N values are given for endpoint measurements of N2O and biomass, while average kinetic isotope effects of ammonia oxidation are presented for substrate (NH4+, εRS) and product (NO2–, εCP). Literature values for isotope fractionation of NH3/NH4+ equilibration (εeq; causing 15N depletion of NH3), for secondary active NH4+ uptake (εupt) and ammonia assimilation (εaa) are presented as well. The identity of the enzyme oxidizing NH2OH to NO2– and its inherent isotope fractionation are currently unknown for AOA.
Despite these potential isotope fractionation effects on the RS level, a higher εCP relative to εRS may also result from accumulation of metabolic intermediates, allowing for at least a second 15N isotope fractionation step to be observed. First, accumulation of NH2OH, or any other intermediate, has not been observed in AOA cells, and the coupled activities of AMO and hydroxylamine oxidoreductase (yet unknown in AOA) are assumed to maintain NH2OH at low steady-state concentrations. Therefore, isotope effects associated with NH2OH oxidation should have a limited impact on δ15NO2–. Second, any process that adds an additional isotope fractionation step, either prior, or subsequent, to NO2– formation, such as the production of the gases NO and N2O, may result in an under- or overestimation of the kinetic isotope fractionation factor. The effect of N2O and NO production on δ15NO2– will depend on their formation pathways, and respective isotope effects and by-product yields. In the N. viennensis culture grown on 2 mM NH4+, we measured cumulative N2O at the end of the incubation, which represented 0.5% (±0.01 SE) of the NH4+ oxidized, or 3.5% (±0.4 SE) of the “missing” N pool. N2O yields of AOA are generally low. For example, N. viennensis has been shown to produce N2O at rates of about 0.1% of those of ammonia oxidation when grown on 1 mM NH4+/NH3 (Stieglmeier et al., 2014b), whereas the marine AOA N. maritimus SCM1 produces even less (0.002–0.03%; Löscher et al., 2012). Here, the cumulative N2O of the N. viennensis culture had a δ15N of −38.1‰ (±0.3 SE) (Figures 1F, 3), which was more 15N-depleted than previously observed for AOA enrichment cultures. AOA have been shown to produce N2O with δ15N signatures ranging between −35 and −13‰ in soil enrichment cultures and −9‰ in marine enrichment cultures (Santoro et al., 2011; Jung et al., 2014), while N2O produced by AOB tends to have lower δ15N, ranging between −66‰ (Nitrosomonas europaea; Yoshida, 1988) and −10‰ (Nitrosomonas marina; Frame and Casciotti, 2010). Furthermore, by-products that are more 15N-depleted than the main product of ammonia oxidation (i.e., NO2–) would decrease the apparent kinetic isotope effect of AMO (εCP), instead of increasing it. Using an isotopic mass balance approach, we calculated that the missing N pool (i.e., the NH4+ taken up which was not oxidized to NO2–), would need to have a δ15N of −18.5‰ (±1.7 SE) in order to account for the difference in isotope fractionation between εRS and εCP (Table 2). Therefore, the δ15N2O signature of −38.1‰ cannot explain the observed large isotope fractionation based on δ15NO2–, since the N2O produced would need to be a larger contributor to the “missing” N pool, as well as to be 15N-enriched relative to NO2–.
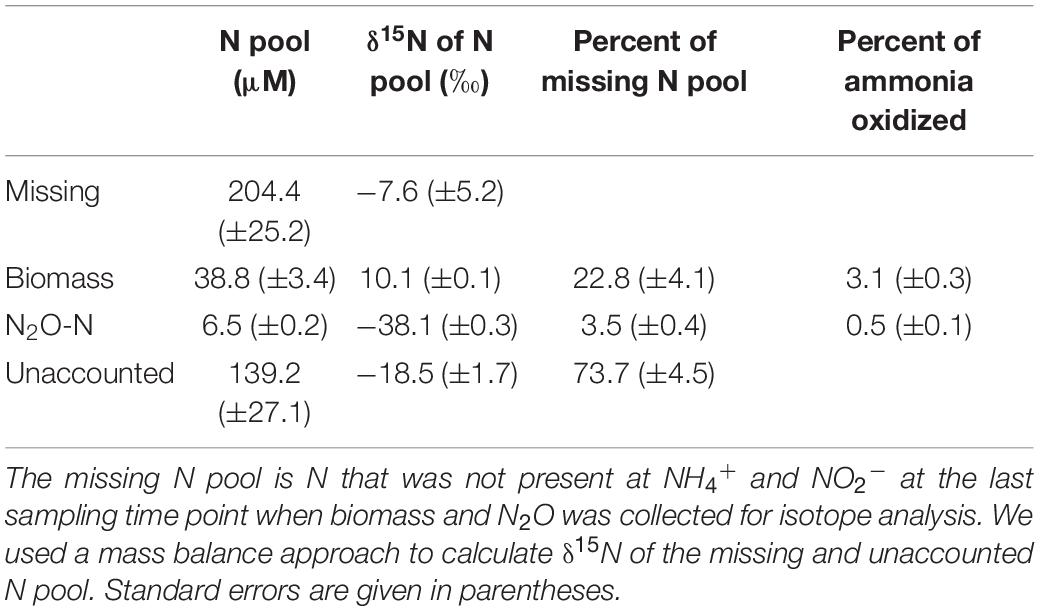
Table 2. Nitrogen pools for N. viennensis culture grown on 2 mM NH4+.
Nitric oxide is an important intermediate in the ammonia oxidation pathway, particularly in that of AOA. Unlike in AOB, NO is a necessary co-reactant for the oxidation of NH2OH to NO2– in AOA, despite being produced in relatively small amounts (Kozlowski et al., 2016). Although the δ15N signature of NO produced by AOA has not yet been determined, Yoshida (1988) found that NO produced during nitrification by N. europaea had a δ15N between 0 and +20‰. The production of such 15N-enriched NO could significantly contribute to the observed overestimation of εCP in AOA.
Conclusion
In conclusion, our results show that, under non-limiting substrate conditions, the εAMO of two phylogenetically and ecologically distinct AOA strains was 31.9–33.1‰ based on δ15NH4+, whereas the more commonly estimated εAMO based on δ15NO2– was higher (37.7–40.1‰). Thus, NH4+ assimilation, but not N2O production, significantly affected the isotope fractionation factor of AMO estimated for N. viennensis (Figure 3). Although the potential role of NO in this context remains to be tested, isotopic analysis of this molecule is difficult and therefore future measurements of εAMO may rely on coupled estimates from δ15NH4+ and δ15NO2–.
Data Availability Statement
The raw data supporting the conclusion of this article have been deposited at DRYAD (doi: 10.5061/dryad.0gb5mkkz1).
Author Contributions
WW designed the study. MMo, RA, BB, MMe, MS, LJ, SR, and MW performed the experiments. MMo, RA, BB, MS, and LJ analyzed the data. GH and CS provided the resources and strains. MMo, RA, and WW wrote the manuscript with contributions from all co-authors.
Funding
This study was funded by the Austrian Science Fund (FWF; project P28037-B22). Open access funding was provided by University of Vienna.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the students of the course “Stable Isotopes in Ecology” of the years 2012–2014 at the University of Vienna, who contributed to the sample analyses and discussion of the data.
References
Alves, R. J. E., Kerou, M., Zappe, A., Bittner, R., Abby, S. S., Schmidt, H., et al. (2019). Ammonia oxidation by the arctic terrestrial thaumarchaeote Ca. Nitrosocosmicus arcticus is stimulated by increasing temperatures. Front. Microbiol. 10:1571. doi: 10.3389/fmicb.2019.01571
Ariz, I., Boeckstaens, M., Gouveia, C., Martins, A. P., Sanz-Luque, E., Fernández, E., et al. (2018). Nitrogen isotope signature evidences ammonium deprotonation as a common transport mechanism for the AMT-Mep-Rh protein superfamily. Sci. Adv. 4:eaar3599. doi: 10.1126/sciadv.aar3599
Arp, D. J., and Stein, L. Y. (2003). Metabolism of inorganic N compounds by ammonia-oxidizing bacteria. Crit. Rev. Biochem. Mol. Biol. 38, 471–495. doi: 10.1080/10409230390267446
Bayer, B., Pelikan, C., Bittner, M. J., Reinthaler, T., Könneke, M., Herndl, G. J., et al. (2019). Proteomic response of three marine ammonia-oxidizing archaea to hydrogen peroxide and their metabolic interactions with a heterotrophic alphaproteobacterium. mSystems 4:e00181-19. doi: 10.1128/mSystems.00181-19
Bayer, B., Vojvoda, J., Offre, P., Alves, R. J. E., Elisabeth, N. H., Garcia, J. A. L., et al. (2016). Physiological and genomic characterization of two novel marine thaumarchaeal strains indicates niche differentiation. ISME J. 10, 1051–1063. doi: 10.1038/ismej.2015.200
Buchwald, C., and Casciotti, K. L. (2013). Isotopic ratios of nitrite as tracers of the sources and age of oceanic nitrite. Nat. Geosci. 6, 308–313. doi: 10.1038/ngeo1745
Caranto, J. D., and Lancaster, K. M. (2017). Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 114, 8217–8222. doi: 10.1073/pnas.1704504114
Carini, P., Dupont, C., and Santoro, A. E. (2017). Correlated expression of archaeal ammonia oxidation machinery across disparate environmental and culture conditions. BioRxiv [Preprint]. doi: 10.1101/175141
Casciotti, K. L. (2016). Nitrogen and oxygen isotopic studies of the marine nitrogen cycle. Ann. Rev. Mar. Sci. 8, 379–407. doi: 10.1146/annurev-marine-010213-135052
Casciotti, K. L., and Buchwald, C. (2012). Insights on the marine microbial nitrogen cycle from isotopic approaches to nitrification. Front. Microbiol. 3:356. doi: 10.3389/fmicb.2012.00356
Casciotti, K. L., McIlvin, M., and Buchwald, C. (2010). Oxygen isotopic exchange and fractionation during bacterial ammonia oxidation. Limnol. Oceanogr. 55, 753–762. doi: 10.4319/lo.2010.55.2.0753
Casciotti, K. L., Sigman, D. M., and Ward, B. B. (2003). Linking diversity and stable isotope fractionation in ammonia-oxidizing bacteria. Geomicrobiol. J. 20, 335–353. doi: 10.1080/01490450303895
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by Nitrospira bacteria. Nature 528:504. doi: 10.1038/nature16461
Delwiche, C. C., and Steyn, P. L. (1970). Nitrogen isotope fractionation in soils and microbial reactions. Environ. Sci. Technol. 4, 929–935. doi: 10.1021/es60046a004
Frame, C. H., and Casciotti, K. L. (2010). Biogeochemical controls and isotopic signatures of nitrous oxide production by a marine ammonia-oxidizing bacterium. Biogeosciences 7, 2695–2709. doi: 10.5194/bg-7-2695-2010
Goyal, S. S., Rains, D. W., and Huffaker, R. C. (1988). Determination of ammonium ion by fluorometry or spectrophotometry after on-line derivatization with o-phthalaldehyde. Anal. Chem. 60, 175–179. doi: 10.1021/ac00153a016
Hermes, J. D., Weiss, P. M., and Cleland, W. W. (1985). Use of nitrogen-15 and deuterium isotope effects to determine the chemical mechanism of phenylalanine ammonia-lyase. Biochemistry 24, 2959–2967. doi: 10.1021/bi00333a023
Hoch, M. P., Fogel, M. L., and Kirchman, D. L. (1992). Isotope fractionation associated with ammonium uptake by a marine bacterium. Limnol. Oceanogr. 37, 1447–1459. doi: 10.4319/lo.1992.37.7.1447
Hood-Nowotny, R., Umana, N. H.-N., Inselbacher, E., Oswald- Lachouani, P., and Wanek, W. (2010). Alternative methods for measuring inorganic, organic, and total dissolved nitrogen in soil. Soil Sci. Soc. Am. J. 74:1018. doi: 10.2136/sssaj2009.0389
Houlton, B. Z., and Bai, E. (2009). Imprint of denitrifying bacteria on the global terrestrial biosphere. Proc. Natl. Acad. Sci. U.S.A. 106, 21713–21716. doi: 10.1073/pnas.0912111106
Jung, M.-Y., Well, R., Min, D., Giesemann, A., Park, S.-J., Kim, J.-G., et al. (2014). Isotopic signatures of N 2 O produced by ammonia-oxidizing archaea from soils. ISME J. 8, 1115–1125. doi: 10.1038/ismej.2013.205
Kerou, M., Offre, P., Valledor, L., Abby, S. S., Melcher, M., Nagler, M., et al. (2016). Proteomics and comparative genomics of Nitrososphaera viennensis reveal the core genome and adaptations of archaeal ammonia oxidizers. Proc. Natl. Acad. Sci. U.S.A. 113, E7937–E7946. doi: 10.1073/pnas.1601212113
Kits, K. D., Sedlacek, C. J., Lebedeva, E. V., Han, P., Bulaev, A., Pjevac, P., et al. (2017). Kinetic analysis of a complete nitrifier reveals an oligotrophic lifestyle. Nature 549:269. doi: 10.1038/nature23679
Kozlowski, J. A., Stieglmeier, M., Schleper, C., Klotz, M. G., and Stein, L. Y. (2016). Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 10, 1836–1845. doi: 10.1038/ismej.2016.2
Lachouani, P., Frank, A. H., and Wanek, W. (2010). A suite of sensitive chemical methods to determine the δ15N of ammonium, nitrate and total dissolved N in soil extracts. Rapid Commun. Mass Spectrom. 24, 3615–3623. doi: 10.1002/rcm.4798
Li, P.-N., Herrmann, J., Tolar, B. B., Poitevin, F., Ramdasi, R., Bargar, J. R., et al. (2018). Nutrient transport suggests an evolutionary basis for charged archaeal surface layer proteins. ISME J. 12, 2389–2402. doi: 10.1038/s41396-018-0191-0
Löscher, C. R., Kock, A., Koenneke, M., LaRoche, J., Bange, H. W., and Schmitz, R. A. (2012). Production of oceanic nitrous oxide by ammonia-oxidizing archaea. Biogeosciences 9, 2419–2429. doi: 10.5194/bg-9-2419-2012
Mariotti, A., Germon, J. C., Hubert, P., Kaiser, P., Letolle, R., Tardieux, A., et al. (1981). Experimental determination of nitrogen kinetic isotope fractionation: some principles; illustration for the denitrification and nitrification processes. Plant Soil 62, 413–430. doi: 10.1007/bf02374138
Martens-Habbena, W., Berube, P. M., Urakawa, H., José, R., and Stahl, D. A. (2009). Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature 461:976. doi: 10.1038/nature08465
Nishizawa, M., Sakai, S., Konno, U., Nakahara, N., Takaki, Y., Saito, Y., et al. (2016). Nitrogen and oxygen isotope effects of ammonia oxidation by thermophilic Thaumarchaeota from a geothermal water stream. Appl. Environ. Microbiol. 82, 4492–4504. doi: 10.1128/aem.00250-16
Offre, P., Kerou, M., Spang, A., and Schleper, C. (2014). Variability of the transporter gene complement in ammonia-oxidizing archaea. Trends Microbiol. 22, 665–675. doi: 10.1016/j.tim.2014.07.007
Prosser, J. I, Hink, L., Gubry-Rangin, C., and Nicol, G. W. (2019). Nitrous oxide production by ammonia oxidisers: physiological diversity, niche differentiation and potential mitigation strategies. Glob. Chang. Biol. 26, 103–118. doi: 10.1111/gcb.14877
Prosser, J. I., and Nicol, G. W. (2012). Archaeal and bacterial ammonia-oxidisers in soil: the quest for niche specialisation and differentiation. Trends Microbiol. 20, 523–531. doi: 10.1016/J.TIM.2012.08.001
R Development Core Team (2012). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Santoro, A. E., Buchwald, C., McIlvin, M. R., and Casciotti, K. L. (2011). Isotopic signature of N2O produced by marine ammonia-oxidizing archaea. Science 333, 1282–1285. doi: 10.1126/science.1208239
Santoro, A. E., and Casciotti, K. L. (2011). Enrichment and characterization of ammonia-oxidizing archaea from the open ocean: phylogeny, physiology and stable isotope fractionation. ISME J. 5:1796. doi: 10.1038/ismej.2011.58
Santoro, A. E., Dupont, C. L., Richter, R. A., Craig, M. T., Carini, P., McIlvin, M. R., et al. (2015). Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc. Natl. Acad. Sci. U.S.A. 112, 1173–1178. doi: 10.1073/pnas.1416223112
Shen, T., Stieglmeier, M., Dai, J., Urich, T., and Schleper, C. (2013). Responses of the terrestrial ammonia-oxidizing archaeon Ca. Nitrososphaera viennensis and the ammonia-oxidizing bacterium Nitrosospira multiformis to nitrification inhibitors. FEMS Microbiol. Lett. 344, 121–129. doi: 10.1111/1574-6968.12164
Sigman, D. M., and Casciotti, K. L. (2001). “Nitrogen isotopes in the ocean,” in Encyclopedia of Ocean Sciences, ed. J. H. Steele (Cambridge, MA: Academic Press), 1884–1894. doi: 10.1006/rwos.2001.0172
Simon, J., and Klotz, M. G. (2013). Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta Bioenerget. 1827, 114–135. doi: 10.1016/j.bbabio.2012.07.005
Sørensen, P., and Jensen, E. S. (1991). Sequential diffusion of ammonium and nitrate from soil extracts to a polytetrafluoroethylene trap for 15N determination. Anal. Chim. Acta 252, 201–203. doi: 10.1016/0003-2670(91)87215-s
Stieglmeier, M., Klingl, A., Alves, R. J. E., Rittmann, S. K.-M. R., Melcher, M., Leisch, N., et al. (2014a). Nitrososphaera viennensis gen. nov., sp. nov., an aerobic and mesophilic, ammonia-oxidizing archaeon from soil and a member of the archaeal phylum Thaumarchaeota. Int. J. Syst. Evol. Microbiol. 64, 2738–2752. doi: 10.1099/ijs.0.063172-0
Stieglmeier, M., Mooshammer, M., Kitzler, B., Wanek, W., Zechmeister-Boltenstern, S., Richter, A., et al. (2014b). Aerobic nitrous oxide production through N-nitrosating hybrid formation in ammonia-oxidizing archaea. ISME J. 8, 1135–1146. doi: 10.1038/ismej.2013.220
Tourna, M., Stieglmeier, M., Spang, A., Könneke, M., Schintlmeister, A., Urich, T., et al. (2011). Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc. Natl. Acad. Sci. U.S.A. 108, 8420–8425. doi: 10.1073/pnas.1013488108
van Kessel, M. A. H. J., Speth, D. R., Albertsen, M., Nielsen, P. H., den Camp, H. J. M. O., Kartal, B., et al. (2015). Complete nitrification by a single microorganism. Nature 528, 555–559. doi: 10.1038/nature16459
Vitousek, P. M., Menge, D. N. L., Reed, S. C., and Cleveland, C. C. (2013). Biological nitrogen fixation: rates, patterns and ecological controls in terrestrial ecosystems. Philos. Trans. R. Soc. B Biol. Sci. 368:20130119. doi: 10.1098/rstb.2013.0119
Walker, C. B., De La Torre, J. R., Klotz, M. G., Urakawa, H., Pinel, N., Arp, D. J., et al. (2010). Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. U.S.A. 107, 8818–8823. doi: 10.1073/pnas.0913533107
Yoshida, N. (1988). 15N-depleted N2O as a product of nitrification. Nature 335:528. doi: 10.1038/335528a0
Keywords: ammonia oxidation, nitrification, nitrous oxide, stable isotope fractionation, Thaumarchaeota
Citation: Mooshammer M, Alves RJE, Bayer B, Melcher M, Stieglmeier M, Jochum L, Rittmann SK-MR, Watzka M, Schleper C, Herndl GJ and Wanek W (2020) Nitrogen Isotope Fractionation During Archaeal Ammonia Oxidation: Coupled Estimates From Measurements of Residual Ammonium and Accumulated Nitrite. Front. Microbiol. 11:1710. doi: 10.3389/fmicb.2020.01710
Received: 18 February 2020; Accepted: 29 June 2020;
Published: 28 July 2020.
Edited by:
Laura E. Lehtovirta-Morley, University of East Anglia, United KingdomReviewed by:
Soo-Je Park, Jeju National University, South KoreaAnne E. Taylor, Oregon State University, United States
Karen L. Casciotti, Stanford University, United States
Copyright © 2020 Mooshammer, Alves, Bayer, Melcher, Stieglmeier, Jochum, Rittmann, Watzka, Schleper, Herndl and Wanek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Wanek, d29sZmdhbmcud2FuZWtAdW5pdmllLmFjLmF0
†Present address: Maria Mooshammer, Department of Environmental Science, Policy, and Management, University of California, Berkeley, Berkeley, CA, United States; Ricardo J. E. Alves, Lawrence Berkeley National Laboratory, Climate and Ecosystem Sciences Division, Earth and Environmental Sciences, Berkeley, CA, United States; Barbara Bayer, Department of Ecology, Evolution and Marine Biology, University of California, Santa Barbara, Santa Barbara, CA, United States