 Pan Li1,2†Xueqin Liu1†Zhimin Hao1,2*Yanrong Jia1Xiangdong Zhao1Debao Xie1Jingao Dong1,2
Pan Li1,2†Xueqin Liu1†Zhimin Hao1,2*Yanrong Jia1Xiangdong Zhao1Debao Xie1Jingao Dong1,2 Fanli Zeng1,2*
Fanli Zeng1,2*- 1College of Life Sciences, Hebei Agricultural University, Baoding, China
- 2State Key Laboratory of North China Crop Improvement and Regulation, Baoding, China
Cip1, a newly identified yeast analog of p21, is a Cln3-CDK inhibitor that negatively regulates cell-cycle START. However, its function remains poorly understood. In this study, we found that deletion of CLN3 did not result in bypass of G1-phase arrest caused by Cip1 overexpression. Cip1 depletion in cln3-null mutants significantly advanced the timing of Cln2 expression, supporting the idea that Cip1 represses START in a Cln3-independent manner. We set to search for novel Cip1 interacting proteins and found that Ccr4, a known START regulator, and its associated factor Caf120, interact with Cip1. Ccr4-Caf120 acts redundantly with Cdk1-Cln3 to inhibit Whi5-mediated regulation of START. This interaction was conserved between human Ccr4 and p21. In addition, deletion of WHI5 robustly suppressed G1-phase arrest caused by Cip1 overexpression. We conclude that Cip1 negatively regulates START by acting as a dual repressor of Ccr4 in parallel with Cln3.
Introduction
In budding yeast, as well as in mammals, cell proliferation is primarily regulated at the G1/S-phase transition in response to a variety of environmental and internal signals (Bertoli et al., 2013). Regulation at this stage, which determines whether and when a cell will enter the division cycle, converges on a major conserved surveillance mechanism known as “START” in yeast and the “Restriction Point” in mammals (Johnson and Skotheim, 2013). START has evolved to be a critical decision-making point in late G1 phase that ensures a “no-return” cell-cycle entry. Loss of strict control of cell-cycle initiation results in uncontrolled proliferation and developmental diseases (Massague, 2004; Nojima, 2004).
START is promoted through activation of the cell size-dependent regulator Cln3-Cdk1, which relocalizes to the nucleus upon activation and phosphorylates the retinoblastoma (Rb) ortholog Whi5 (Wittenberg and Reed, 2005; Palumbo et al., 2016). Phosphorylated Whi5 is excluded from the nucleus, allowing activation of SBF. In turn, SBF is responsible for expression of late G1 genes, including cyclins encoded by CLN1 and CLN2, resulting in cell-cycle entry. This mechanism is analogous to the regulation of E2F transcription by the tumor suppressor Rb, which constitutes a key barrier to carcinogenesis in human cells (Bruin et al., 2004; Costanzo et al., 2004; Cooper, 2006).
In mammals, the tumor suppressor p21, a downstream target of p53, is the main factor responsible for negatively regulating the Cln3-Cdk1 ortholog CycD/E-CDK and suppressing unwanted cell division at the G1/S transition (Georgakilas et al., 2017). This strategy for regulating CDK activity is conserved from yeast to human, but previously no p21 or p53 homolog had been identified in yeast. Recently, the Saccharomyces cerevisiae protein Cip1 was shown to regulate G1/S transition in both the unperturbed and stressed cell cycle (Chang et al., 2017; Zhang Z. et al., 2017); accordingly, it was proposed as a functional analog of p21. Cip1 was originally identified as a G1-Cdk1 inhibitor that negatively regulates G1/S transition (Ren et al., 2016). Cip1 associates with G1-Cdk1s, including Cln3-Cdk1, and inhibits their activities both in vitro and in vivo (Ren et al., 2016; Chang et al., 2017). Targeting of Cln3-Cdk1 by Cip1 prevents Whi5 phosphorylation and thereby prevents the G1/S transition (Chang et al., 2017).
Cells have evolved strict and redundant pathways for crucial aspects of cell-cycle control. We sought to determine whether Cip1 regulates the G1/S transition in another manner, distinct from its previously identified regulation of Cln3-Cdk1. Cip1 overexpression blocks cell-cycle entry, arrests cells at G1 phase, and significantly increases cell size; all of these events are Cln3-dependent (Chang et al., 2017).
In this study, we found that CLN3 deletion could not suppress cell-cycle arrest caused by Cip1 overexpression, indicating that another target of Cip1, in parallel with Cln3-Cdk1, is involved in START control. Proteomic screening identified Ccr4-Caf120 complex as a novel downstream target of Cip1. The conserved Ccr4-Not complex, involved in mRNA biogenesis, fine-tunes the START program by modulating CLN1 and CLN2 expression through destabilization of WHI5 mRNAs (Manukyan et al., 2008). Consistent with this, the Ccr4-Not complex is essential for the G1/S transition in stressed cells (Westmoreland et al., 2004). However, the regulatory function of Ccr4-Not in cell-cycle control remains largely unknown. Our results demonstrate that Cip1 blocks the cell cycle at START by acting as a dual repressor of Ccr4-Caf120 and Cdk1-Cln3.
Results and Discussion
Cip1 Overexpression Causes Significant G1 Arrest in cln3 Mutants
Cip1 is an inhibitor of Cln3-Cdk1. To determine whether it might also play a Cln3-independent repressive role, we overexpressed Cip1 and monitored the effect on G1/S control. CIP1 was overexpressed under the GAL1 promoter. Upon galactose addition, Cip1 was functional largely induced as monitored at protein and mRNA levels shown in Figures 1A,B. In a wild-type CLN3 background, overexpression of Cip1 significantly inhibited cell growth, consistent with our previous observations (Ren et al., 2016). However in cln3-null cells, high levels of Cip1 resulted in a stronger growth defect (Figures 1C,D).
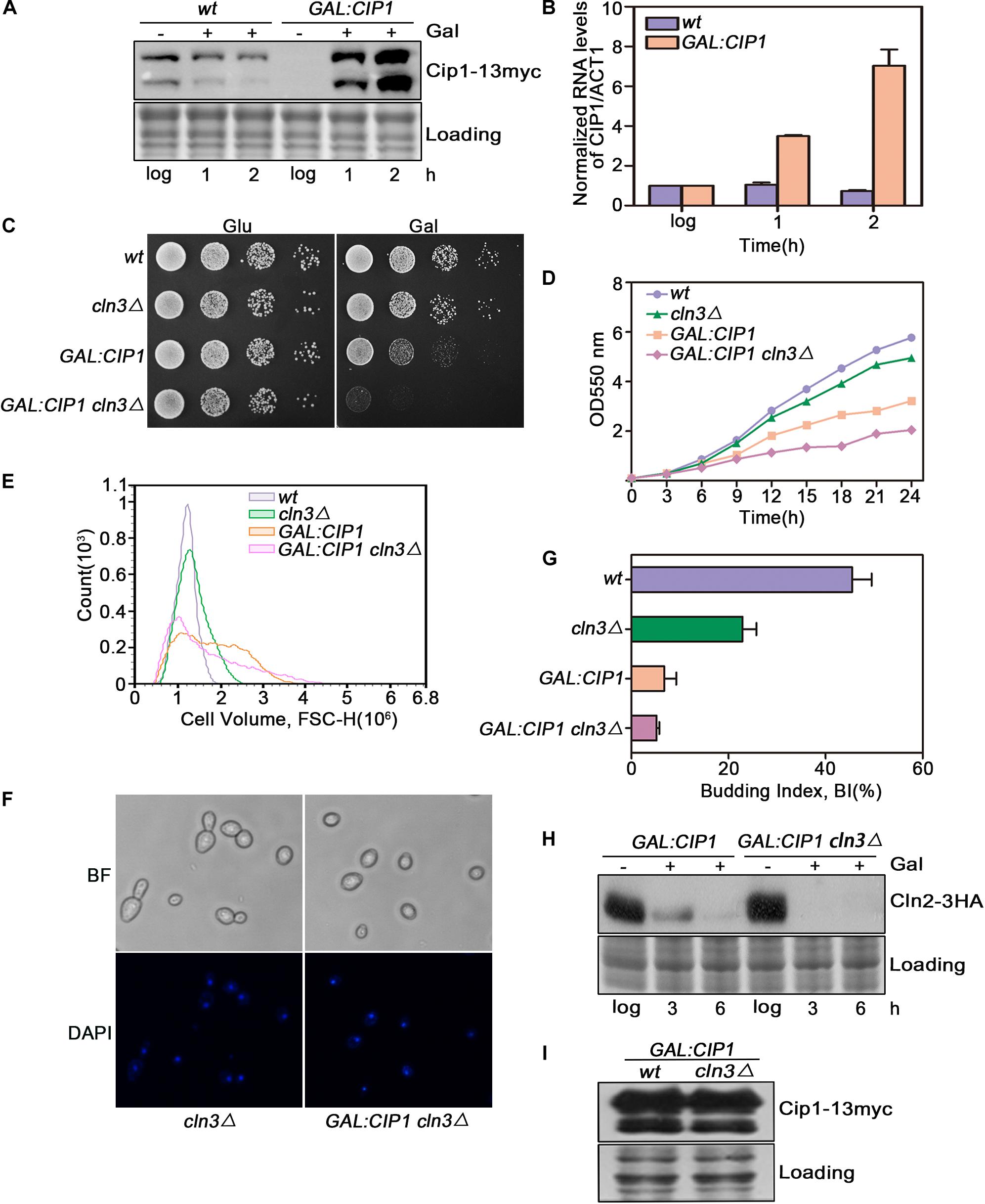
Figure 1. Cln3 depletion does not bypass G1 phase arrest caused by Cip1 overexpression. (A) Western blot analysis of Cip1 protein level in wild-type (YFL100) and galactose-induced Cip1 protein level in GAL:CIP1 (YFL97) cells. (B) Real-time quantitative PCR analysis of relative amount of CIP1 mRNA in wild-type (YFL100) and GAL:CIP1 (YFL97) cells after the addition of galactose. Mean values were from three independent replicates. Value was arbitrarily set at 1 for log samples. (C) 10-fold serial dilutions from cultures of wild-type (YFL3), cln3Δ (YXQ7), GAL:CIP1 (YFL97), and GAL:CIP1 cln3Δ (YXQ11) were spotted onto YPD and YPG, and incubated at 30°C for 3 days. (D) The same indicated strains were grown in YPR. Galactose (2%) was added to induce overexpression of Cip1, and growth curves were generated. (E) Cell volume distributions for the indicated strains are shown (20,000 cells analyzed/sample). Galactose (2%) was added to induce overexpression of Cip1, and samples were collected after 3h induction. (F,G) Cell-cycle distribution of cells after 3 h since the addition of galactose. Pictures show bright field images and DAPI staining of nuclei. Budded cells were counted and expressed as a percentage of total cells. (H) The levels of Cln2 protein were analyzed by western blotting at 3 and 6 h after addition of galactose. (I) Western blot analysis of Cip1 protein level in cells grown in YPG.
We found that this growth defect was due to a stronger inhibition of cell-cycle START in cln3-null mutants. In yeast overexpressing Cip1, more than 90% of cells arrested before START as large unbudded cells with a single nucleus, despite CLN3 deletion (Figures 1E–G). Expression of CLN2, a G1/S marker gene, was completely blocked in cells overexpressing Cip1 (Figure 1H). The overexpression extends of Cip1 in both wild-type and cln3-null cells is identical as controlled in Figure 1I. Therefore, START regulation by Cip1 might involve mechanisms independent of Cln3.
Cip1 Negatively Regulates START in a Cln3-Independent Manner
We observed a significant cell size decrease in cln3Δ cip1Δ double mutants compared to larger cln3Δ cells. We quantified this cell size alteration of each cell population by FACS analysis. As shown in Figure 2A, cln3Δ cip1Δ double mutants displayed a clear shift in the overall distribution toward small cell volumes compared to cln3Δ single mutants. We next asked whether deletion of CIP1 and CLN3 would advance START timing relative to single deletion of CLN3. For this purpose, we used Cln2 accumulation as the marker of START entry. Cln2 rapidly accumulated 20 min after wild-type cells were released from α-factor-induced pre-START arrest (Figures 2B,C). A similar expression pattern was observed in cip1Δ mutants. In cln3-mutant cells, which are defective in START activation, Cln2 expression was delayed for at least 10 min relative to wild-type cells. However, the delay in Cln2 expression and cell budding were rescued by double deletion of CLN3 and CIP1 (Figures 2B,C).
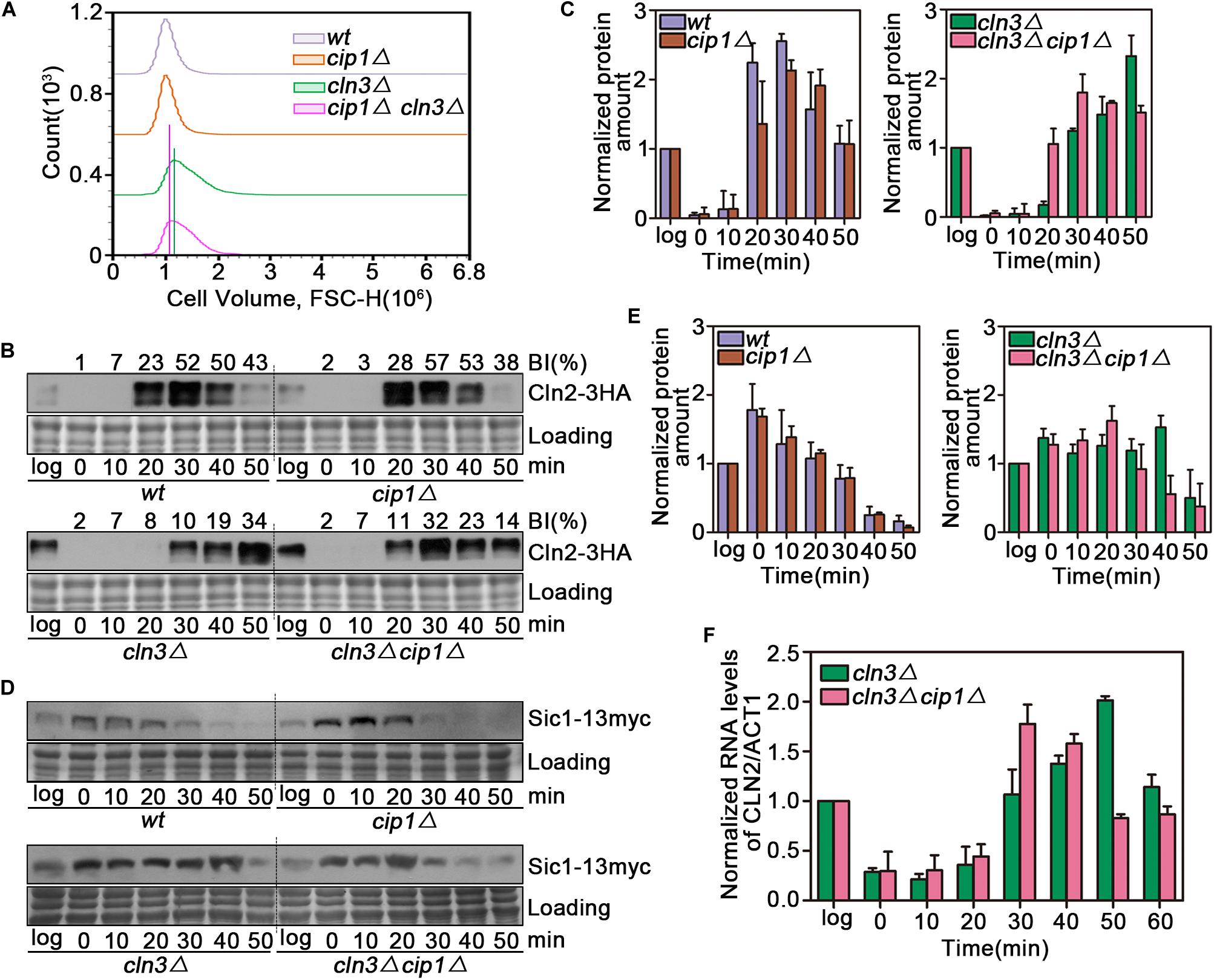
Figure 2. Effect of Cip1 depletion on START timing in a cln3 mutant strain. (A) Flow cytometry analysis of the cell volume distributions for the indicated strains (20,000 cells analyzed/sample). (B) Cultures of Cln2-3HA (YZZ6), cip1Δ Cln2-3HA (YZZ20), cln3Δ Cln2-3HA (YXQ8), and cln3Δ cip1Δ Cln2-3HA (YXQ9) were grown to mid-log phase in YPD and synchronized in pre-START G1. The cells were released from the α-factor arrest such that they entered the cell cycle synchronously. Samples were taken at the indicated time points (min). TCA whole-cell extracts were analyzed by immunoblotting with anti-HA (Cln2) antibodies. A Coomassie brilliant blue-stained region of the same membrane used for immunoblotting is shown as a loading control. The budding index (BI) monitoring the cell cycle progression were shown on the top of time course panels. (C) Graphs show quantification of Cln2, normalized against the loading control. Mean values were from three independent replicates. Value was arbitrarily set at 1 for log samples. (D) Cultures of Sic1-13myc (YLP139), cip1Δ Sic1-13myc (YLP149), cln3Δ Sic1-13myc (YLP155), and cln3Δ cip1Δ Sic1-13myc (YLP138) were grown to mid-log phase in YPD and synchronized in pre-START G1. The cells were released from the α-factor arrest such that they entered the cell cycle synchronously. Samples were taken at the indicated time points (min). TCA whole-cell extracts were analyzed by immunoblotting with anti-myc (Sic1) antibodies. A Coomassie brilliant blue-stained region of the same membrane used for immunoblotting is shown as a loading control. (E) Graphs show quantification of Sic1, normalized against the loading control. Mean values were from three independent replicates. Value was arbitrarily set at 1 for log samples. (F) Normalized relative amount of CLN2 mRNA vs. ACT1 control mRNA in cln3Δ (YXQ8) and cln3Δ cip1Δ cells (YXQ9). Mean values were from three independent replicates. Value was arbitrarily set at 1 for log samples.
To confirm this result, we monitored the stability of Sic1, whose disappearance is a marker of START execution. Sic1 began to be degraded at 20 min and disappeared 40 min after release from α-factor arrest in both wild-type and cip1Δ cells (Figures 2D,E). In cln3 mutants, Sic1 was stable for the duration of the experiment. By contrast, in double-mutant (cln3Δ cip1Δ) cells, START entry was advanced by more than 10 min.
To confirm that these observations were due to an effect on the START transcriptional program, we compared CLN2 mRNA levels between cln3Δ and cln3Δ cip1Δ mutants. G1/S gene expression, as reflected by CLN2 mRNA levels, was advanced by ∼20 min in cln3Δ cip1Δ double mutants relative to cln3Δ cells (Figure 2F). These observations strongly indicate that Cip1 has a Cln3-independent repressive function in START control.
Ccr4 Interacts With Cip1/p21
As shown above, CLN3 deletion could not suppress cell-cycle arrest induced by Cip1 overexpression, and cln3Δ cip1Δ double mutants exhibited accelerated entry into START. Hence, we performed proteomic assays to identify novel proteins associated with Cip1. The top nine hits are listed in Figure 3A. Most of the proteins in this list, such as Ccr4, Lge1 and Cln1 (Partridge et al., 1997; Westmoreland et al., 2003; Manukyan et al., 2008; Zhang W. et al., 2017), were previously implicated in G1/S cell-cycle regulation, confirming the specificity of our co-immunoprecipitation and mass spectrometry assays for Cip1-interacting proteins. Cln1 is a paralog of the G1/S cyclin Cln2, which binds to Cip1. Ccr4 is an essential component of the Ccr4-Not transcriptional complex, which negatively regulates the half-life of WHI5 mRNA and influences the timing of START (Manukyan et al., 2008).
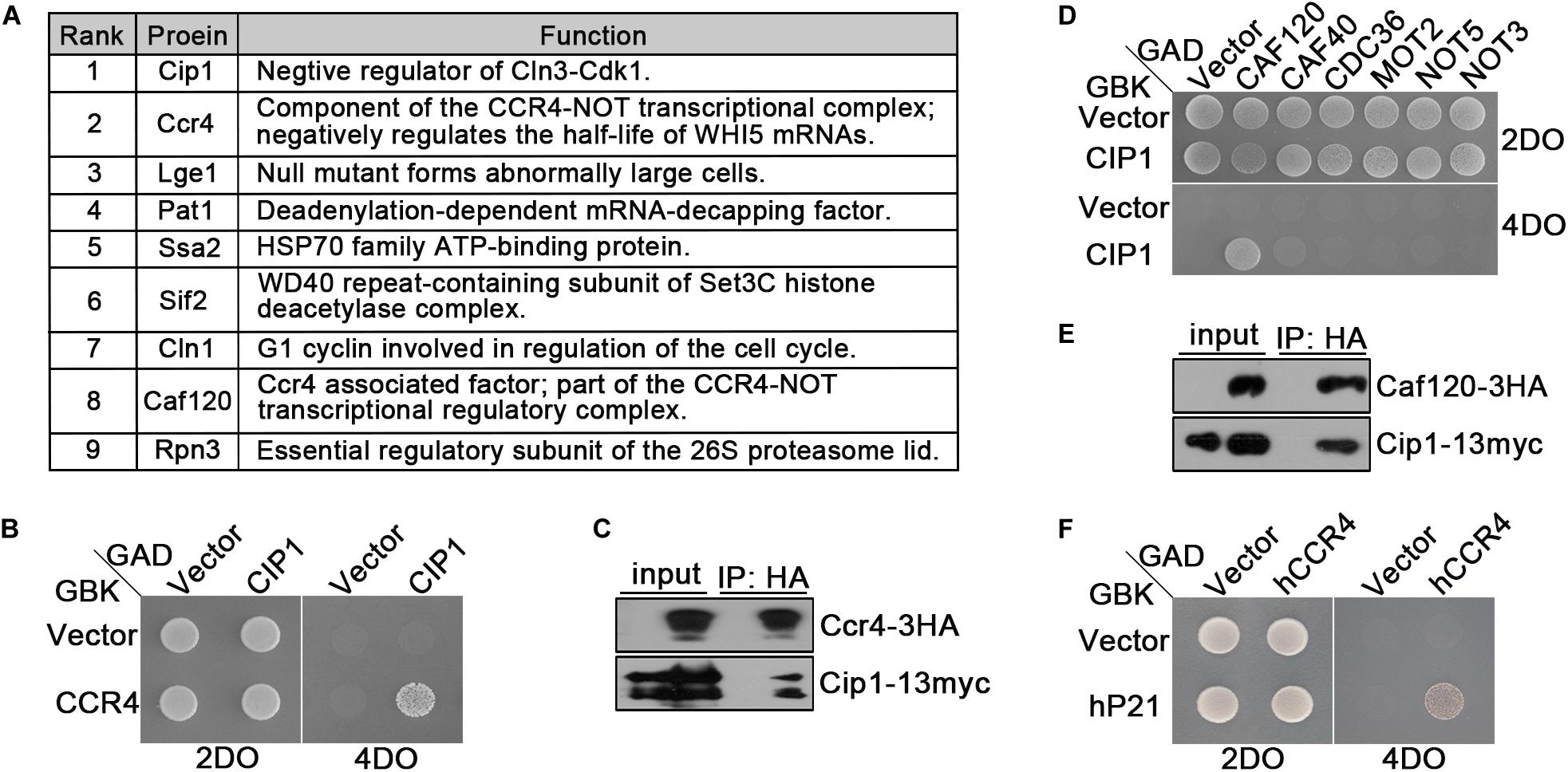
Figure 3. Ccr4 interacts with Cip1/p21. (A) Co-IP identified a range of Cip1 interacting proteins. Exponentially growing cells YFL100 (Cip1-13myc) and untagged control cells (YFL3) were harvested and extracted, and the extracts were immunoprecipitated using anti-myc antibodies. The precipitated proteins were separated by SDS-PAGE, and the differential bands were analyzed by mass spectrometry. In addition to Cip1 itself, the eight most abundant interacting proteins are shown in the table. The rank value is based on the frequency of peptide occurrence. (B) Cip1 interacted with Ccr4 in the yeast two-hybrid assay. Yeast cells transformed with the indicated plasmids were grown at 30°C on the control 2DO (SD-leucine and -tryptophan) and high-stringency 4DO (SD-adenine, -histidine, -leucine, and -tryptophan) plates. (C) Exponentially growing cultures of Cip1-13myc control cells (YFL100) and Cip1-13myc Ccr4-3HA cells (YLP41) were harvested. Ccr4-3HA was immunopurified from the indicated extracts using anti-HA antibodies. Copurified Cip1 was analyzed with anti-myc antibodies. (D) Yeast two-hybrid assay revealed that Cip1 robustly interacts with Caf120 but not with other Not subunits. (E) Co-IP assay showing that Cip1 interacts with Caf120. The procedure was similar to that described for Panel C. (F) Yeast two-hybrid assay showing that p21 interacts with human Ccr4.
Interestingly, our proteomic assays identified two subunits of the Ccr4-Not complex, Ccr4 and Caf120, as hits (Figure 3A). We verified the interaction between Cip1 and Ccr4 by using east two-hybrid assays (Figure 3B). We also carried out an experiment complementary to the one that led to the identification of Ccr4 as a Cip1-associated protein; we immunoprecipitated Ccr4-3HA from exponentially growing cells and probed for Cip1 among the associated proteins. Cip1 was reproducibly associated with Ccr4 (Figure 3C). Subsequent yeast two-hybrid assays and co-IP also confirmed the interaction between Cip1 and Caf120 (Figures 3D,E). Moreover, this interaction was specific to Caf120 and did not extent to other members of the Not complex, including Caf40, Cdc36, Mot2, Not5, and Not3 (Figure 3D).
Because Cip1 is functionally similar to human p21 (Ren et al., 2016; Chang et al., 2017), we speculated that the Ccr4 interaction might be conserved between the two proteins. Indeed, in a yeast two-hybrid assay, we identified a robust interaction between p21 and hCcr4 (Figure 3F). This result indicates that Ccr4 in both yeast and human cells is a bona fide target of Cip1/p21.
Ccr4 Is a Novel Target of Cip1 That Is Functionally Redundant With Cln3–Cdk1
Because Ccr4 exhibited a robust interaction with Cip1, we investigated whether Ccr4 is a component of Cip1-mediated START control, redundant with Cln3. Auxin-induced Ccr4 depletion could not bypass growth inhibition induced by Cip1 overexpression (Figure 4A). Hence, we generated ccr4-aid cln3Δ double mutants and found that Ccr4 and Cln3 are synthetically lethal, consistent with the previously reported inviability of ccr4Δ cln3Δ double mutants (Figures 4B,C; Manukyan et al., 2008). Moreover, this synthetically lethality was resulted from G1/S transition defects, as the double mutant cells died with larger cell volume and defected budding compared to each single mutants (Figures 4C–F). Ccr4 is a non-essential gene but Auxin mediated depletion of Ccr4 protein displayed slow growth rate with abnormally large cells (Figures 4C–E).
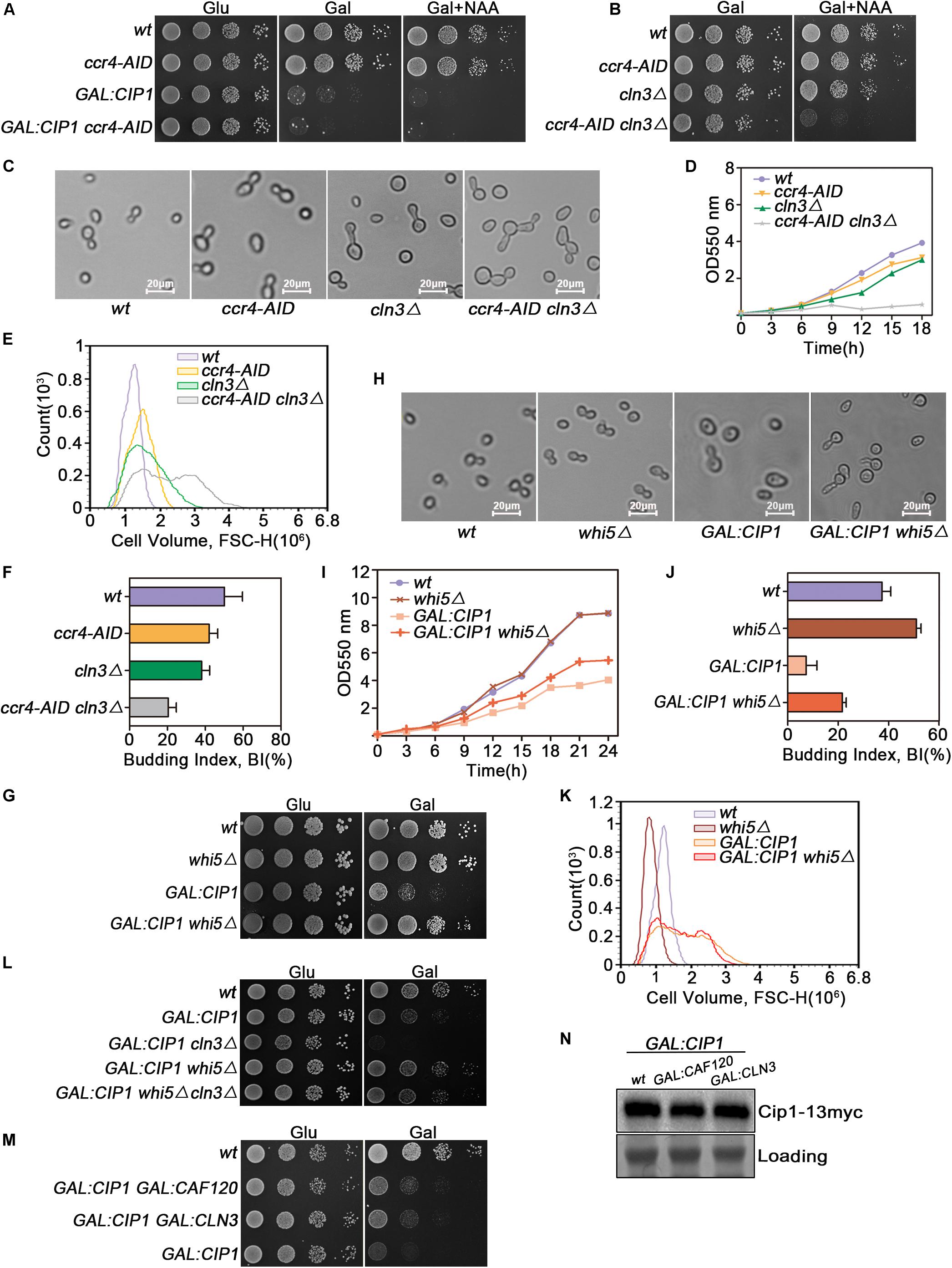
Figure 4. Cip1 acts as a dual suppressor of Cln3-Cdk1 and Ccr4-Caf120 to inhibit Whi5 function at START. (A) 10-fold serial dilutions from cultures of wild-type (YYJ138), ccr4-AID (YLP102), GAL:CIP1 (YLP105), and GAL:CIP1 ccr4-AID (YLP106) cells were spotted onto YPD and YPG with or without 1.0 mM NAA. The auxin-inducible degron (AID) system was turned on by addition of auxin (NAA) in the presence of galactose. (B) 10-fold serial dilutions from cultures of wild-type (YYJ138), ccr4-AID (YLP102), cln3Δ (YLP107), and ccr4-AID cln3Δ (YLP108) cells were spotted onto YPG with or without 1.0 mM NAA. (C) The same strains were grown to mid-log phase in YPR. Galactose was added for 1 h, and samples were taken at the indicated time points after addition of 0.5 μM NAA. Cell morphology was visualized by microscopy (scale bar, 20 μm). (D) Growth curves of the indicated strains. (E) Flow cytometry analysis of the cell volume distributions for the indicated strains (20,000 cells analyzed/sample). (F) Budded cells were counted and expressed as a percentage of total cells. (G) Deletion of WHI5 suppresses the cell-growth defect caused by Cip1 overexpression. 10-fold serial dilutions from cultures of wild-type (YFL3), whi5Δ (YLP23), GAL:CIP1 (YFL97), and GAL:CIP1 whi5Δ (YLP75) cells were spotted onto YPD and YPG medium and incubated at 30°C for 3 days. (H) The same strains were grown to mid-log phase at 30°C and then shifted to YPG medium for 18 h. Samples were collected, and cell morphology was visualized by microscopy (scale bar, 20 μm). (I) Growth curves of the indicated strains. The indicated strains were grown in YPR. Galactose (2%) was added to induce overexpression of Cip1, and growth curves were generated. (J) Budded cells were counted and expressed as a percentage of total cells. (K) Flow cytometry analysis of the cell volume distributions for the indicated strains (20,000 cells analyzed/sample). (L) 10-fold serial dilutions from cultures of wild-type (YFL3), GAL:CIP1 (YFL97), GAL:CIP1 cln3Δ (YXQ11), GAL:CIP1 whi5Δ (YLP75), and GAL:CIP1 whi5Δ cln3Δ (YLP78) cells were spotted onto YPD and YPG medium and incubated at 30°C for 3 days. (M) 10-fold serial dilutions from cultures of wild-type (YFL3), GAL:CIP1 (YFL97), GAL:CIP1 GAL:CLN3 (YLP174), and GAL:CIP1 GAL:CAF120 (YLP175) cells were spotted onto YPD and YPG medium and incubated at 30°C for 3 days. (N) Western blot analysis of Cip1 protein expression under galactose induction in the indicated strains grown in YPG.
Deletion of WHI5, Which Is Downstream of Cln3 and Ccr4, Suppresses G1-Phase Arrest Caused by Overexpression of Cip1
Ccr4 negatively regulates the half-life of WHI5 mRNA and hence influences the timing of G1/S cyclin transcription (Manukyan et al., 2008). Cln3-Cdk1, on the other hand, phosphorylates and inactivates Whi5 (Bruin et al., 2004). Hence, we asked whether deletion of WHI5, a common downstream target of both Ccr4 and Cln3, could restore growth to cells overexpressing Cip1. GAL-CIP1 whi5Δ cells had near-normal growth on plates (Figure 4G). Inhibition of budding and cell-cycle progression by Cip1 induction was significantly restored in the absence of Whi5 (Figures 4H–J). Intriguingly, cell buds elongated in GAL:CIP1 whi5Δ mutants (Figure 4H), which usually arises from a delayed switch from apical to isotropic bud growth, a process requiring M-CDK activity. Cip1 is an inhibitor of G1-CDK, however we could not rule out that Cip1 may exhibit inhibitory activity to Clb-Cdk1, specially in the absent of Whi5. This elongated cell buds in the mutants might be also explained by the defect of CLB5 and CLB6 expression when Whi5 is absent and G1/S transition is blocked by Cip1 overexpression.
The cell size distribution for GAL:CIP1 whi5Δ showed a shift to restored small cells compared to population of GAL:CIP1 cells (Figure 4K). The relative mean value of GAL:CIP1 whi5Δ cell size is 1.68 × 106, which is significantly smaller than 1.80 × 106 of GAL:CIP1 cells. We also generated GAL:CIP1 whi5Δ cln3Δ mutants and found that Whi5 deletion could also restore the growth defect in the GAL:CIP1 cln3Δ background (Figure 4L).
Previous study reported that induction of CLN3 in the Cip1 overexpressed cells partially restored the Cip1-dependent slow growth. To further support the idea that Ccr4-Caf120 works redundantly with Cln3-Cdk1, we generated GAL:CIP1 GAL:CAF120 cells and compared the growth status with GAL:CIP1 GAL:CLN3 cells. As shown in Figure 4M, simultaneous induction of CAF120 could partially rescue the Cip1-dependent slow growth as well as induction of CLN3. This suppression is not because different overexpression extends of Cip1 in the compared strains, as the amount of Cip1 induced is identical as controlled in Figure 4N. Therefore, these data indicate that Ccr4 and Cln3, two crucial proteins upstream of Whi5, are both negatively regulated by Cip1.
In this study, we identified a new target of Cip1 that acts in parallel with Cln3 to regulate START. Our results are summarized in Figure 5. Two different pathways downstream of Cip1, mediated by Cln3-Cdk1 and Ccr4-Caf120, down-regulate WHI5 through distinct mechanisms to inhibit the START transcriptional program. The previously reported interaction between Cip1 and the Cdk1-Cln3 complex inhibits G1-CDK kinase activity (Chang et al., 2017), whereas Ccr4 acts in parallel with Cln3-Cdk1 under the regulation of Cip1. Ccr4 regulates the size-dependent timing of CLN2 mRNA expression by modulating the half-life of WHI5 mRNA. Importantly, a previous study of the regulation of START also reported that Ccr4 is redundant with Cln3-Cdk1 (Manukyan et al., 2008). Thus, our results help to explain why Cln3-Cdk1 is dispensable for cell-growth inhibition induced by Cip1 overexpression. Nevertheless, further study is required to explore the molecular details of Ccr4 inhibition by Cip1.
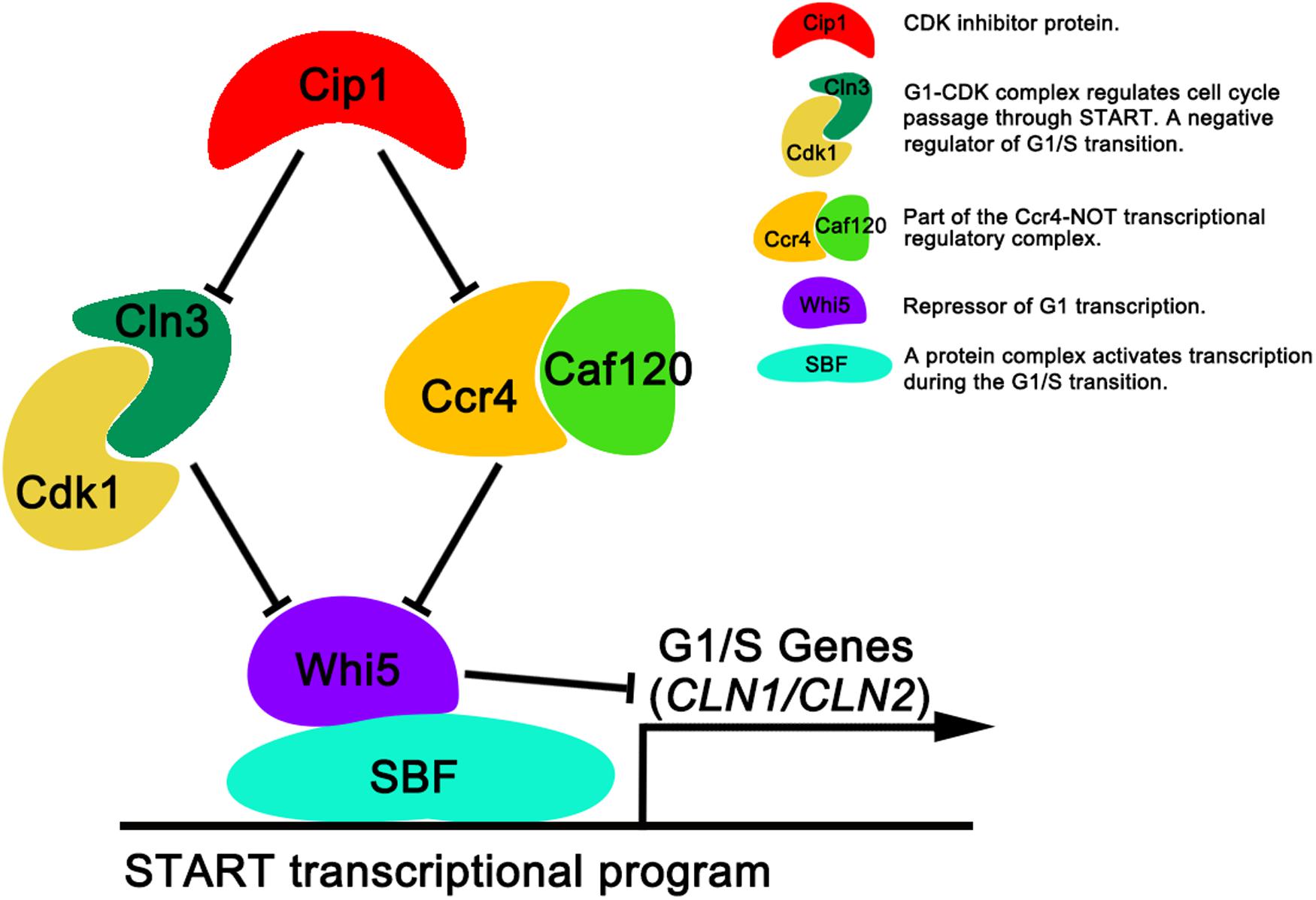
Figure 5. Dual repressive function of Cip1 on START timing via the redundant Cln3-Cdk1 and Ccr4-Caf120 pathways. The known interaction of Cip1 with the Cdk1-Cln3 complex inhibits its kinase activity. Ccr4 is a new target of Cip1, in parallel with Cln3-Cdk1. Dual inhibition by Cip1 down-regulates Whi5, preventing SBF from transcribing G1/S genes. The key description of each protein (protein complex) are shown.
In summary, our observations reveal that Cip1 acts as a dual repressor to negatively regulate both Cln3-Cdk1 and Ccr4 complex, with the final result of maintaining Whi5 active, thereby preventing SBF from transcribing G1/S genes. The situation in yeast may be conserved in other organisms, including humans. Notably in this regard, the Ccr4-Not complex in human cells was recently implicated in cancer development (Allison, 2017; Maolake et al., 2017; Vicente et al., 2018). Accordingly, it would be worthwhile to determine whether p21 is involved in a similar control mechanism in human cells.
Experimental Procedures
Constructs, Strains, Culture Conditions and Cell Size Analysis
All strains used in this work were derived from Saccharomyces cerevisiae W303-1a (Thomas and Rothstein, 1989) and listed in Table 1. Strains were generated following standard procedures by integration of PCR-generated cassettes as previously described (Zeng et al., 2018). Plasmids constructed for this study were validated by sequencing and are listed in Table 2. Cells were grown in standard media containing 1% yeast extract, 2% peptone, and 2% glucose. For induction experiments, cells were grown overnight in media containing 2% raffinose, and then inoculated into 2% galactose media or onto galactose plates with or without 1-naphthylacetic acid (NAA) as indicated. Fluorescent Activated Cell Sorting (FACS) was used in this study for the analysis of the cell size distribution in a given cell population. 20,000 cells were read for each profile on a NovoCyte D3130 (ACEA) instrument. The number of cells (y-axis) vs. values of the forward scatter signal band FSC-H (x-axis) was represented for cell size analysis.
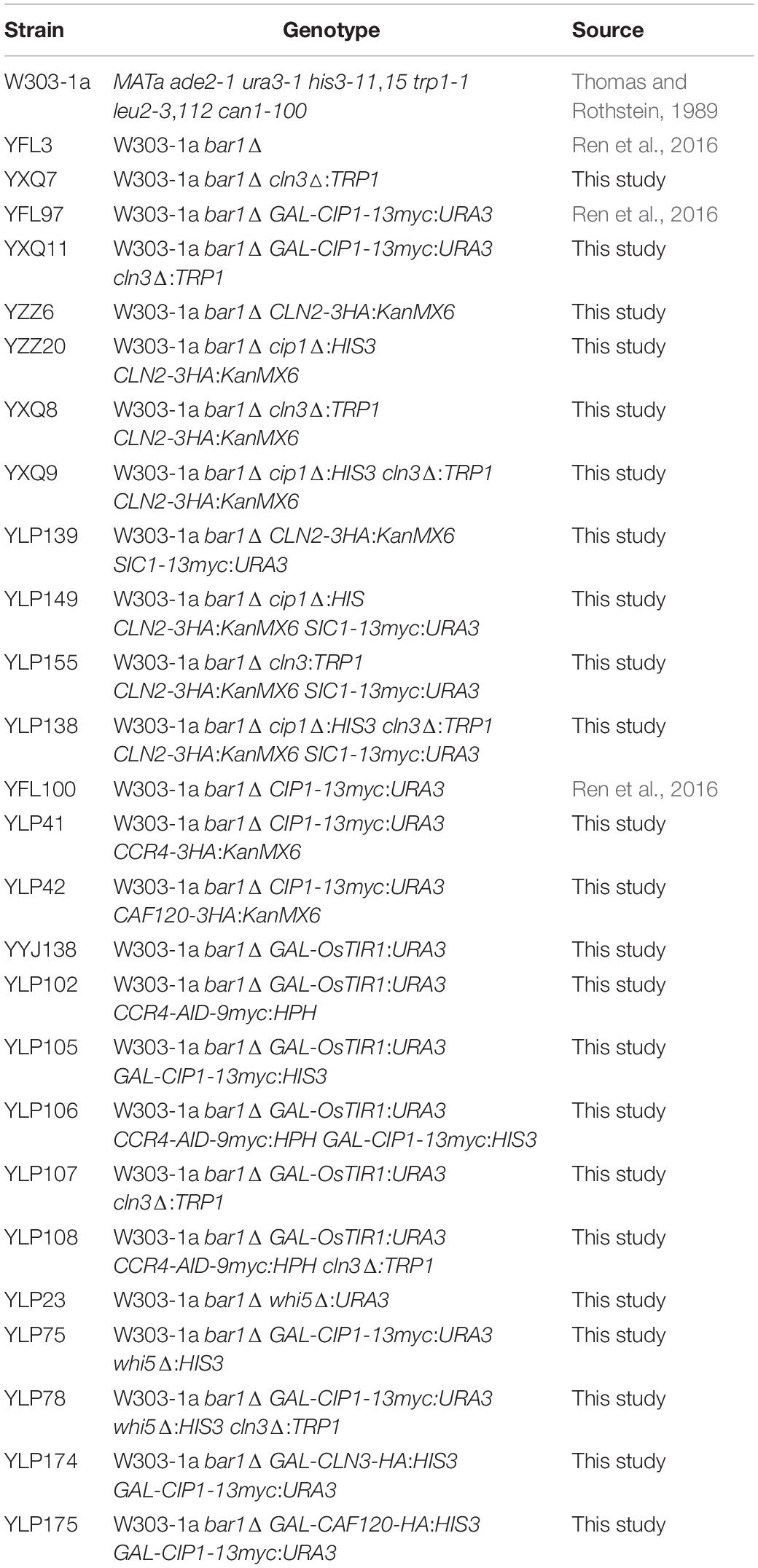
Table 1. Yeast strains in this study.
Table 2. Plasmids used in this study.
Co-IP and Western Blotting
For co-immunoprecipitation (Co-IP) and immunoprecipitation, 109 cells were collected and extracted by glass bead beating in lysis buffer [50 mM Tris–HCl, pH 7.8, 150 mM NaCl, 1 mM EDTA, 10% (v/v) glycerol, and 1% (v/v) Triton X-100] supplemented with protease inhibitors (1 mM PMSF, 0.15 mM aprotinin, 1 mM leupeptin, and 1 mM pepstatin) and phosphatase inhibitors (0.5 mM sodium pyrophosphate, 2 mM NaF, and 2 mM β-glycerophosphate). Twenty microliters of anti-HA HA-7 (Sigma A7470, mouse monoclonal purified IgG) or anti-myc HA-7 agarose matrix (Sigma A2095, mouse monoclonal purified IgG) was added to extracts to immunopurify proteins of interest C-terminally tagged with 13myc or 3HA, respectively. The samples were gently rotated for 1 h at 4°C, after which the beads were recovered by centrifugation at 500 × g for 1 min at 4°C, and the supernatants were discarded. The beads were then washed three times with 1 mL cold lysis buffer. Finally, the proteins were released by boiling the beads in Laemmli sample buffer. Enriched proteins were resolved by SDS-PAGE and subjected to western blotting.
Whole-cell extracts for western blotting were prepared by glass bead beating in trichloroacetic acid (TCA) and then resolved by SDS-PAGE as previously described. The primary antibodies used in this study were anti-HA (Sigma A7470, mouse monoclonal purified IgG) and anti-myc (Sigma A2095, mouse monoclonal purified IgG).
Large Scale Co-IP for Mass Spectroscopy
To identify the interacting proteins of Cip1 by mass spectroscopy proteomic analysis, a larger scale of co-immunoprecipitation procedure was followed. Around 10 g of cells expressing Cip1-13myc from its own promoter were collected and extracted. Upon extraction, samples were clarified for 1 h at 100,000 × g in a Thermo ultracentrifuge (Sorvall COMB PLUS). Extracts were then incubated with 100 μl of pre-equilibrated anti-myc HA-7 agarose matrix (Sigma A2095, mouse monoclonal purified IgG), and rotated gently for 1 h at 4°C. After that, beads were washed 6 times with lysis buffer and the proteins from the immunoprecipitate were released from the beads by mixing with 50 μl of room temperature 8M urea in 100 mM Tris pH 8.0 for 5 min. After releasing, the copurified proteins from the sample were separated by SDS-PAGE and stained with Coomassie Brilliant blue, followed by detection by mass spectrometry. As a control, a mock IP was performed using cells without myc-tagged Cip1 protein.
Yeast Two-Hybrid Assay
To investigate the interactions of Cip1 with yeast Ccr4–Not and p21 with human Ccr4, the Gal4-based Matchmaker Yeast Two-hybrid System 3 (Clontech Laboratories, Mountain View, CA, United States) was used. The protocol was essentially as previously described (Zeng et al., 2018). Briefly, the indicated genes, amplified from a budding yeast or a human cDNA library, were fused to the GAL4 activation domain (AD) in vector pGADT7 or the GAL4 DNA-binding domain (BD) in pGBKT7. Pairs of corresponding constructs were co-transformed into the tester strain AH109, in which the HIS3 and ADE2 reporter genes are under the control of three completely heterologous GAL4-responsive UAS, and promoter elements GAL1 and GAL2, respectively. Transformants were first grown on SD/-Leu/-Trp medium and subsequently plated on SD/-Ade/-His/-Leu/-Trp medium.
RNA Extraction and qRT-PCR
Around 109 cells were harvested for each sample and washed once with pre-chilled diethyl pyrocarbonate (DEPC)-treated water. Cell pellets were snap-frozen in liquid nitrogen and stored at −80°C prior to RNA extraction. Total RNA was obtained using the Trizol reagent (Invitrogen). RNA was then reverse transcribed using the PrimeScript RT kit (Takara, RR014A). The cDNA library was used as a template in real-time PCR using the Takara SYBR Premix Ex-Taq (Tli RNase H Plus) kit (Takara, RR420A). The primers for CLN2 and ACT1 were the same as in our previous study (Zhang Z. et al., 2017).
Statistics
All values were expressed as the mean ± SD, and statistical analyses were carried out using Student’s t-test.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
FZ, PL, XL, and ZH conceived and designed the experiments. PL, XL, ZH, and XZ performed the experiments. FZ, PL, ZH, and JD analyzed the data. FZ and PL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 31801039), Department of Human Resources and Social Security of Hebei Province (No. C201837), Youth Top Talent Project from Hebei Provincial Department of Education (No. BJ2020003), and Hebei Agricultural University (Nos. ZD201622 and 2018KYYJ01). Outstanding Youth Project from Natural Science Foundation of Hebei Province (No. C2020204109).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Zeyang Li and Prof. Jianguo Ji for their help with the mass spectrometry analysis and Prof. David G. Quintana for supplying strains and plasmids. We are grateful to all lab members for helpful discussions and suggestions in this work.
References
Allison, S. J. (2017). Kidney cancer: CCR4: a new target for RCC. Nat. Rev. Nephrol. 13:192. doi: 10.1038/nrneph.2017.14
Bertoli, C., Skotheim, J. M., and Bruin, R. A. M. D. (2013). Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518–528. doi: 10.1038/nrm3629
Bruin, R. A. M. D., McDonald, W. H., Kalashnikova, T. I., Yates, J., and Wittenberg, C. (2004). Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117, 887–898. doi: 10.1016/j.cell.2004.05.025
Chang, Y., Tseng, S., Huang, Y., Shen, Z., Hsu, P., Hsieh, M., et al. (2017). Yeast Cip1 is activated by environmental stress to inhibit Cdk1-G1 cyclins via Mcm1 and Msn2/4. Nat. Commun. 8:56. doi: 10.1038/s41467-017-00080-y
Cooper, K. (2006). Rb, whi it’s not just for metazoans anymore. Oncogene 25, 5228–5232. doi: 10.1038/sj.onc.1209630
Costanzo, M., Nishikawa, J. L., Tang, X., Millman, J. S., Schub, O., Breitkreuz, K., et al. (2004). CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117, 899–913. doi: 10.1016/j.cell.2004.05.024
Georgakilas, A. G., Martin, O. A., and Bonner, W. M. (2017). P21: a two-faced genome guardian. Trends Mol. Med. 23, 310–319. doi: 10.1016/j.molmed.2017.02.001
Johnson, A., and Skotheim, J. M. (2013). Start and the restriction point. Curr. Opin. Cell Biol. 25, 717–723. doi: 10.1016/j.ceb.2013.07.010
Manukyan, A., Zhang, J., Thippeswamy, U., Yang, J., Zavala, N., Mudannayake, M. P., et al. (2008). Ccr4 alters cell size in yeast by modulating the timing of CLN1 and CLN2 expression. Genetics 179, 345–357. doi: 10.1534/genetics.108.086744
Maolake, A., Izumi, K., Shigehara, K., Natsagdorj, A., Iwamoto, H., Kadomoto, S., et al. (2017). Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 8, 9739–9751. doi: 10.18632/oncotarget.14185
Massague, J. (2004). G1 cell-cycle control and cancer. Nature 432, 298–306. doi: 10.1038/nature03094
Nojima, H. (2004). G1 and S-phase checkpoints, chromosome instability, and cancer. Methods Mol. Biol. 3–49. doi: 10.1385/1-59259-788-2:003
Palumbo, P., Vanoni, M., Cusimano, V., Busti, S., Marano, F., Manes, C., et al. (2016). Whi5 phosphorylation embedded in the G1/S network dynamically controls critical cell size and cell fate. Nat. Commun. 7:11372. doi: 10.1038/ncomms11372
Partridge, J. F., Mikesell, G. E., and Breeden, L. L. (1997). Cell cycle-dependent transcription of CLN1 involves Swi4 binding to MCB-like elements. J. Biol. Chem. 272, 9071–9077. doi: 10.1074/jbc.272.14.9071
Ren, P., Malik, A., and Zeng, F. (2016). Identification of YPL014W (Cip1) as a novel negative regulator of cyclin-dependent kinase in Saccharomyces cerevisiae. Genes Cells 21, 543–552. doi: 10.1111/gtc.12361
Thomas, B. J., and Rothstein, R. (1989). Elevated recombination rates in transcriptionally active DNA. Cell 56, 619–630. doi: 10.1016/0092-8674(89)90584-9
Vicente, C., Stirparo, R., Demeyer, S., Bock, C. E. D., Gielen, O., Atkins, M., et al. (2018). The CCR4-NOT complex is a tumor suppressor in Drosophila melanogaster eye cancer models. J. Hematol. Oncol. 11:108. doi: 10.1186/s13045-018-0650-0
Westmoreland, T. J., Marks, J. R., Olson, J. A., Thompson, E. M., Resnick, M. A., and Bennett, C. B. (2004). Cell cycle progression in G1 and S phases is CCR4 dependent following ionizing radiation or replication stress in Saccharomyces cerevisiae. Eukaryot. Cell 3, 430–446. doi: 10.1128/EC.3.2.430-446.2004
Westmoreland, T. J., Olson, J. A., Saito, W. Y., Huper, G., Marks, J. R., and Bennett, C. B. (2003). DHH1 regulates the G1/S-checkpoint following DNA damage or BRCA1 expression in yeast. J. Surg. Res. 113, 62–73. doi: 10.1016/S0022-4804(03)00155-0
Wittenberg, C., and Reed, S. I. (2005). Cell cycle-dependent transcription in yeast: promoters, transcription factors, and transcriptomes. Oncogene 24, 2746–2755. doi: 10.1038/sj.onc.1208606
Zeng, F., Hua, Y., Liu, X., Liu, S., Lao, K., Zhang, Z., et al. (2018). Gpn2 and Rba50 directly participate in the assembly of the Rpb3 subcomplex in the biogenesis of RNA polymerase II. Mol. Cell. Biol. 38:e00091-18. doi: 10.1128/MCB.00091-18
Zhang, W., Yeung, C. H. L., Wu, L., and Yuen, K. W. Y. (2017). E3 ubiquitin ligase Bre1 couples sister chromatid cohesion establishment to DNA replication in Saccharomyces cerevisiae. eLife 6:e28231. doi: 10.7554/eLife.28231
Keywords: budding yeast, cell cycle, G1/S transition, Cip1, Ccr4, Caf120, p21
Citation: Li P, Liu X, Hao Z, Jia Y, Zhao X, Xie D, Dong J and Zeng F (2020) Dual Repressive Function by Cip1, a Budding Yeast Analog of p21, in Cell-Cycle START Regulation. Front. Microbiol. 11:1623. doi: 10.3389/fmicb.2020.01623
Received: 08 February 2020; Accepted: 22 June 2020;
Published: 09 July 2020.
Edited by:
Matteo Barberis, University of Surrey, United KingdomReviewed by:
Marti Aldea, Instituto de Biología Molecular de Barcelona (IBMB), SpainMarco Vanoni, University of Milano-Bicocca, Italy
Copyright © 2020 Li, Liu, Hao, Jia, Zhao, Xie, Dong and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fanli Zeng, ZmFubGkuemVuZ0Bwa3UuZWR1LmNu; Zhimin Hao, aGFvemhpbWluQGhlYmF1LmVkdS5jbg==
†These authors share first authorship