
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 July 2020
Sec. Fungi and Their Interactions
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01535
 Luiza Souza Rodrigues1,2*†
Luiza Souza Rodrigues1,2*† Rajesh Kumar Gazara3,4,5†
Rajesh Kumar Gazara3,4,5† Hemanoel Passarelli-Araujo3,6
Hemanoel Passarelli-Araujo3,6 Andressa Eloisa Valengo2
Andressa Eloisa Valengo2 Paula Veronesi Marinho Pontes7
Paula Veronesi Marinho Pontes7 Rodrigo Nunes-da-Fonseca7
Rodrigo Nunes-da-Fonseca7 Robson Francisco de Souza8
Robson Francisco de Souza8 Thiago Motta Venancio3*†Libera Maria Dalla-Costa1,2*†
Thiago Motta Venancio3*†Libera Maria Dalla-Costa1,2*†Candida haemulonii is a complex formed by C. haemulonii sensu stricto, C. haemulonii var. vulnera, and C. duobushaemulonii. Members of this complex are opportunistic pathogens closely related to C. pseudohaemulonii, C. lusitaniae, and C. auris, all members of a multidrug-resistant clade. Complete genome sequences for all members of this group are available in the GenBank database, except for C. haemulonii var. vulnera. Here, we report the first draft genomes of two C. haemulonii var. vulnera (isolates K1 and K2) and comparative genome analysis of closely related fungal species. The isolates were biofilm producers and non-susceptible to amphotericin B and fluconazole. The draft genomes comprised 350 and 387 contigs and total genome sizes of 13.21 and 13.26 Mb, with 5,479 and 5,507 protein-coding genes, respectively, allowing the identification of virulence and resistance genes. Comparative analyses of orthologous genes within the multidrug-resistant clade showed a total of 4,015 core clusters, supporting the conservation of 24,654 proteins and 3,849 single-copy gene clusters. Candida haemulonii var. vulnera shared a larger number of clusters with C. haemulonii and C. auris; however, more singletons were identified in C. lusitaniae and C. auris. Additionally, a multiple sequence alignment of Erg11p proteins revealed variants likely involved in reduced susceptibility to azole and polyene antifungal agents. The data presented in this work will, therefore, be of utmost importance for researchers studying the biology of the C. haemulonii complex and related species.
Bloodstream infections caused by Candida spp. are associated with high healthcare costs and a mortality rate of 20–60% (Colombo et al., 2006; Nucci et al., 2013; Guinea, 2014; Doi et al., 2016). Candida albicans is the primary species involved in these infections. However, infections caused by non-albicans Candida, such as C. parapsilosis, C. glabrata, C. tropicalis, C. krusei, and C. guilliermondii, have been increasing, usually reported in critically ill patients, sometimes with overlapping C. albicans infections. In addition, other emerging non-albicans species are described as challenging to properly identify, causing hard to treat infections (Pfaller and Diekema, 2007; Wisplinghoff et al., 2014; Lamoth et al., 2018; Rodrigues et al., 2019; Ahangarkani et al., 2020).
In this context, Candida haemulonii is an uncommon and emerging yeast that has been detected in different geographic regions, and it is associated with superficial to deep infections, including chronic otitis media, peritonitis, candidemia, and osteitis, particularly in immunocompromised and neonatal patients (Gargeya et al., 1991; Kim et al., 2009; Hou et al., 2016; Ben-Ami et al., 2017; Arastehfar et al., 2018). In Brazil, species of the C. haemulonii complex have also been reported to cause candidemia, associated with infections in critically ill patients (Muro et al., 2012; Ramos et al., 2015; de Almeida et al., 2016).
Classical identification methods are unable to characterize this species, which can be accurately identified using molecular detection methods (Kim et al., 2009; Almeida et al., 2012; Muro et al., 2012; Kumar et al., 2016; Arastehfar et al., 2018). In 1993, C. haemulonii species were categorized in genetic groups I and II, which were later reclassified as C. haemulonii and C. haemulonii var. vulnera (group I) and C. duobushaemulonii (group II) (Lehmann et al., 1993; Cendejas-Bueno et al., 2012). Recently, two related species of the C. haemulonii complex, C. pseudohaemulonii and C. auris, were described and shown to form a multidrug-resistant (MDR) clade along with C. lusitaniae (Muñoz et al., 2018).
Candida haemulonii var. vulnera, as well as other related species, are commonly resistant to fluconazole and non-susceptible to amphotericin B (AMB). Mechanisms associated with reduced susceptibility to AMB and azoles are often associated with mutations in genes involved in ergosterol biosynthesis, stress responses, and in regulatory regions that promote the overexpression of efflux pumps (Kołaczkowska and Kołaczkowski, 2016; Whaley et al., 2016; Arendrup and Patterson, 2017).
The detection of MDR Candida species at the species level is required for better epidemiological surveillance and therapeutic interventions (Cendejas-Bueno et al., 2010; Lockhart et al., 2017b; Mizusawa et al., 2017; Sears and Schwartz, 2017; Spivak and Hanson, 2017; Jeffery-Smith et al., 2018; Taori et al., 2019). Although the molecular basis of the resistance of major Candida species to commonly used antifungal agents has been widely described, identifying the genetic determinants of virulence and antifungal resistance in emerging species remains a major challenge. For many of these microorganisms, there is a lack of whole-genome sequencing (WGS) data, leading researchers to use rRNA genes for species classification (Cowen and Steinbach, 2008; Khodadadi et al., 2017). WGS provides a first glimpse of the genomic basis of biological information about the microorganism and may allow further functional studies to elucidate the role of encoded proteins and their metabolic pathways, in order to expand the knowledge about the metabolism, virulence and resistance mechanisms. WGS also enables researchers to uncover phylogenetic relationships between isolates thoroughly (Piskur and Langkjaer, 2004; Binkley et al., 2014; Zoll et al., 2016).
Here we describe the first two genomes of C. haemulonii var. vulnera, isolated from bloodstream infections of two pediatric patients in a Brazilian hospital and compare them with other members of the MDR clade. Our data allowed the exploration of the phylogenetic context of these isolates, as well as the genomic data that enabled the identification and comparison of virulence and resistance genes. In addition, our orthologous clustering analysis represents a significant contribution to functional protein annotation and a resource for phylogenetic inference of the Candida MDR clade.
The studied clinical isolates were obtained from a tertiary children’s care hospital in the South of Brazil. The yeasts were isolated from blood culture sample of catheter of female patients under 10-years-old. The first case, in December 2009, was a patient with Down Syndrome, diagnosed with cardiopathy (total atrioventricular septal defect); and the second one, in April 2010, was hospitalized with febrile neutropenia after receiving chemotherapy for Ewing’s sarcoma. Patients were treated with a combination of liposomal amphotericin B and fluconazole; and amphotericin B deoxycholate, respectively, exhibiting good clinical recovery. Detailed clinical data sets were previously published (Muro et al., 2012).
Initially, blood cultures were performed using an automated BD BactecTM 9120 blood culture system (Becton, Dickinson, Franklin Lakes, NJ, United States) and Candida spp. were identified using phenotypic and molecular methods. An API 20C AUX system (BioMérieux, Marcy-l’Étoile, France) was used to identify the isolates as Kodamaea (Pichia) ohmeri, whereas the Vitek 2TM Compact, YST ID system (BioMérieux) showed inconclusive results for the first isolate (K1) and identified the other (K2) as C. haemulonii (Muro et al., 2012). The two strains were confirmed as C. haemulonii using matrix-assisted laser desorption ionization mass spectrometry (MALDI-TOF MS) using a MicroflexTM LT instrument (Bruker Daltonics, Billerica, MA, United States), and by sequence analysis of the ITS and D1/D2 regions of the rRNA genes (White et al., 1990; Leaw et al., 2006). The isolates were stored in skim milk and frozen at −80°C until processing for further study.
We reviewed the microbiology records including those of the two isolates identified as C. haemulonii. Before testing, each isolate was cultured on Sabouraud dextrose agar (Neogen Corporation, Lansing, MI, United States) and CHROMagar CandidaTM medium (BD Biosciences) for 48 h at 37 and 42°C to ensure their purity, viability, and thermotolerance. These organisms were identified with a Vitek 2TM Compact, YST ID system (BioMérieux) and MALDI-TOF MS, MicroflexTM LT instrument (Bruker Daltonics), using controls and the FlexControlTM version 3.4 software according to the manufacturer’s instructions.
Biofilm formation was evaluated as described by Branchini et al. (1994). A loop of microorganisms collected from Sabouraud dextrose agar was inoculated into a polystyrene tube (15 mL Falcon tube) containing 10 mL of Sabouraud dextrose broth supplemented with glucose (final concentration of 8%). The tubes were incubated at 35 ± 2°C for 24 h and the broth was gently aspirated. The tubes were washed thoroughly with phosphate-buffered saline (pH 7.2) and dried. Cells in the dried tubes were stained with 0.1% crystal violet (for 7 min), and excess dye was removed by washing with distilled water. After drying, the tubes were evaluated for biofilm formation. Biofilm formation was considered as positive when a visible film coated the wall and bottom of the tube. The experiments were performed in triplicate and biofilm production was evaluated independently by two different observers.
Antifungal susceptibility testing was performed with SensititreTM YeastOneTM (Thermo Fisher Scientific, Waltham, MA, United States) using the YO9 AST plate, containing serial drug dilutions of AMB, flucytosine, fluconazole, itraconazole, voriconazole, posaconazole, caspofungin, micafungin, and anidulafungin. Stock inoculum suspensions of the yeasts were obtained from 24-h cultures on Sabouraud dextrose agar at 35°C. The turbidity of each yeast suspension was adjusted to 0.5 McFarland standards and the isolates were tested according to the manufacturer’s instructions. The inoculum was evaluated by colony counting, with C. parapsilosis ATCC® 22019 used as the quality control strain. Plates were covered with adhesive seals and the colorimetric MIC endpoints were determined visually after 48 h of incubation at 35 ± 2°C in a non-CO2 atmosphere. The interpretative criteria for in vitro susceptibility testing of Candida spp., set by The Clinical & Laboratory Standards Institute (CLSI) M27-S3 guidelines, were followed, since CLSI M27-S4 contains species-specific clinical breakpoints, except rare species, such as C. haemulonii var. vulnera (CLSI, 2008, 2017).
Genomic DNA was extracted using the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, United States). The Nextera XT DNA system (Illumina, Inc., San Diego, CA, United States) was used to prepare sequencing-ready libraries, which were sequenced using an Illumina MiSeq instrument (paired-end mode, 250 × 2) using the MiSeq Reagent Kit v2 (Illumina, Inc.).
We evaluated read quality using fastqc (v.0.11.5)1. Trimmomatic (v.0.32) was used to remove adapters and trim and discard reads shorter than 50 base pairs (Bolger et al., 2014). Draft genomes were assembled de novo using SPAdes (v.3.5.0) (Bankevich et al., 2012). BUSCO (v.3.0) was used to assess genome assembly completeness (Simão et al., 2015). All genome assemblies were evaluated using QUAST (v.4.6.3), and repetitive sequences were masked using RepeatMasker (v.4.0.7) (Tarailo-Graovac and Chen, 2009; Gurevich et al., 2013). The rRNA and tRNA genes were predicted with RNAmmer (v.1.2.1) and ARAGORN, respectively (Laslett and Canback, 2004; Lagesen et al., 2007). Protein-coding genes were predicted using AUGUSTUS web (Stanke and Morgenstern, 2005). Proteins were annotated using BLAST2GO (v. 5.2.4) against the reference NCBI NR database, Eukaryota taxid: 2759 (minimal query coverage and maximum E-value of 40% and 1e-5, respectively) (Conesa et al., 2005). InterPro terms were obtained from InterProScan, which is available from EBI2, and converted and merged to Gene Ontology (GO) terms (Quevillon et al., 2005).
The ITS sequences of K1 and K2 were obtained from their respective genomes by identifying the scaffolds in which the ITS regions were located. We performed multiple sequence alignment between each genome and a randomly selected ITS sequence of a C. haemulonii species complex isolate with MUMmer (v.4.0.0beta2) (Kurtz et al., 2004). Each identified scaffold (K1 scaffold_270 for K1 and K2 scaffold_267 for K2) was aligned with ClustalW (Mega X) to a pair of previously described primers (ITS1 forward: 5’-TCCGTAGGTGAACCTGCGG-3’ and ITS4 reverse: 5’-TCCTCCGCTTATTGATATGC-3’) to identify the start and end of the ITS sequences (White et al., 1990; Kumar et al., 2018). After identifying the ITS sequences, they were extracted with the SAMtools faidx command (v.1.7) (Li et al., 2009).
Phylogenetic analyses were performed with the ITS sequences of the C. haemulonii species complex (Supplementary Table S1) and those from K1 and K2 using the unweighted pair group method with arithmetic mean with 1000 bootstrap replicates (Sneath and Sokal, 1973; Felsenstein, 1985). In addition to our two isolates, the analysis included 45 nucleotide sequences: 16 from C. haemulonii, 13 from C. duobushaemulonii, 12 from C. haemulonii var. vulnera, two from C. pseudohaemulonii, and two from C. auris (Chowdhary et al., 2014; Grenfell et al., 2016). The phylogenetic tree was visualized using Interactive Tree Of Life (v.4) (Letunic and Bork, 2016).
Average Nucleotide Identity (ANI) analysis was conducted with pyani (v.0.2.0) based on a whole-genome comparison among K1 and K2, C. haemulonii (B11899), C. pseudohaemulonii (B12108), C. duobushaemulonii (B09383), C. auris (6684 and B8441), and C. lusitaniae (ATCC 42720 and CBS6939) (Pritchard et al., 2016).
Single nucleotide variants (SNVs) and indel calling among K1, K2, and C. haemulonii (B11899) was performed according to GATK Best Practices recommendations (DePristo et al., 2011; Van der Auwera et al., 2013). The pipeline consists of aligning reads from one of the genomes to the other, which serves as a reference (Li, 2011). The alignment was performed using BWA-MEM, and the resulting alignment file was sorted using SAMtools (Li et al., 2009). Duplicated reads were marked using Picard’s MarkDuplicates and sorted with Picard’s SortSam. Variant calling was performed using GATK’s HaplotypeCaller, and the resulting set was normalized with vcflib’s vcfallelicprimitives and filtered (read depth >24 and quality >20) with vcflib’s vcffilter (McKenna et al., 2010). The pipeline was built using Snakemake (Köster and Rahmann, 2018).
We performed orthologous clustering analysis with the web server OrthoVenn2 (Xu et al., 2019). The protein FASTA file containing predicted protein sequences for C. haemulonii var. vulnera (K1), C. haemulonii (B11899), C. pseudohaemulonii (B12108), C. duobushaemulonii (B09383), C. auris (B8441), and C. lusitaniae (CBS6939) were used to predict the orthologous gene clusters among the MDR clade. Copy-number variation was assessed using predicted proteins against our database containing genes related to pathogenicity through BLASTp searches with 50 and 60% for similarity and query coverage thresholds, respectively.
Erg11p sequences from our isolates were aligned using MAFFT (v 7.271) (Katoh and Standley, 2013) with sequences from other related species: C. lusitaniae (ATCC 42720 and CBS 6936), C. auris (6684 and B8441), C. duobushaemulonii (B09383), C. pseudohaemulonii (B12108), C haemulonii (B11899), and C. albicans (SC5314).
The colonies of the two isolates, K1 and K2, when grown on Sabouraud dextrose agar, were white-to-cream-colored and smooth. They grew at 37°C, but not at 42°C, and showed a pink colony color on CHROMagar CandidaTM medium after incubation for 48 h. One of the isolates (K1) was identified as C. haemulonii by Vitek 2TM Compact with 97% certainty, whereas the other (K2) showed inconclusive (low discrimination) results. Considering that commercial phenotypic systems have a limited ability to discriminate C. haemulonii complex and related species, our two isolates had their identity confirmed as C. haemulonii using MALDI-TOF MS (Ramos et al., 2015). A previous study revealed similarity in the main spectrum profiles of C. haemulonii and C. haemulonii var. vulnera generated by this method, further demonstrating the difficulties of discriminating these microorganisms (Grenfell et al., 2016). Regarding the production of biofilm, both strains were in vitro biofilm producers by tube method.
The in vitro antifungal susceptibility data of C. haemulonii var. vulnera (n = 2) are presented in Table 1. C. haemulonii var. vulnera was described as a microorganism with high MICs for AMB and for non-susceptible to azoles (Cendejas-Bueno et al., 2012; de Almeida et al., 2016; Kumar et al., 2016). Although there are no interpretive criteria for AMB in the CLSI M27-S3 guidelines, both isolates presented high MIC values for this antifungal agent (≥2.0 μg/mL) (CLSI, 2008). Additionally, both isolates were classified as susceptible dose-dependent to itraconazole, K2 as susceptible dose-dependent to fluconazole, while K1 susceptible to fluconazole, and both as susceptible to echinocandins.
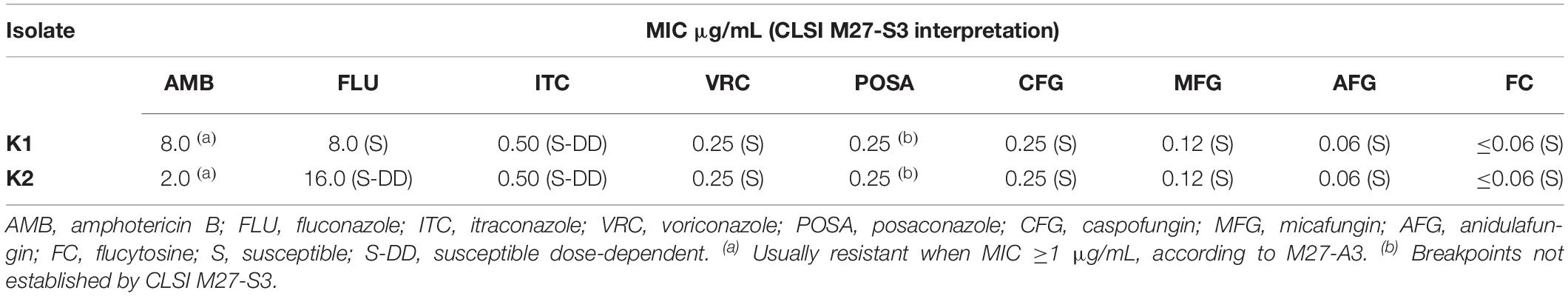
Table 1. SensititreTM YeastOneTM antifungal susceptibility profile of C. haemulonii var. vulnera (n = 2).
We sequenced the genomes of K1 and K2. The draft genomes comprised 350 and 387 contigs, total genome sizes of 13.21 and 13.26 Mb, and an average G + C content of 44%; BUSCO analysis indicated that the assemblies encompassed 94.6 and 94.5%, of the 1,771 genes from the Saccharomycetales reference dataset, respectively. A total of 5,484 and 5,508 protein-coding genes were predicted with AUGUSTUS and annotated with Blast2GO (Table 2). Although a major proportion of the C. haemulonii var. vulnera genome remains uncharacterized, as most of the other Candida genomes (Chatterjee et al., 2015); we annotated K1 and K2 genomes using GO. We assigned terms to 4,121 and 4,149 proteins and verified the presence of many catalytic proteins involved in cellular processes, particularly those with hydrolase (23%) and transferase (20%) activity.
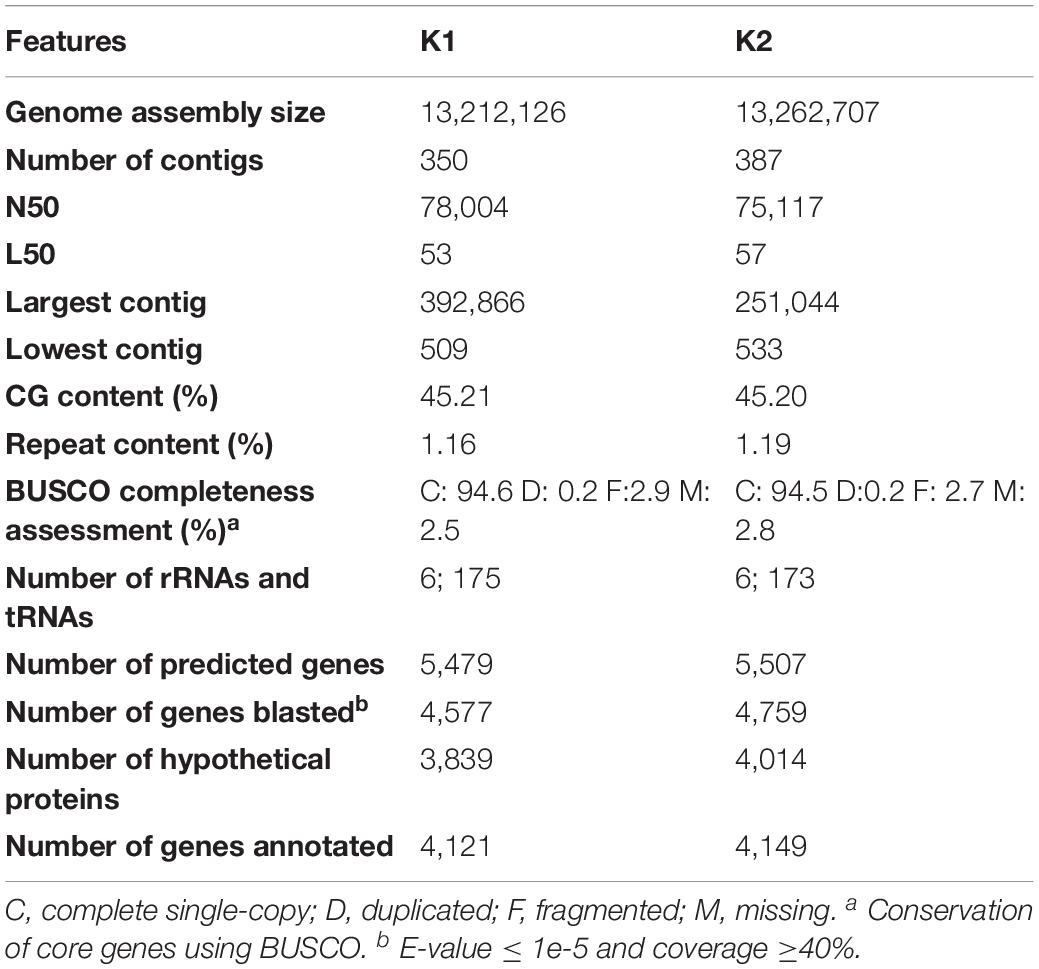
Table 2. Summary description of the C. haemulonii var. vulnera annotated genome assemblies.
The most frequent species among BLAST hits for K1, and K2 predicted proteins were C. haemulonii, C. pseudohaemulonii, and C. auris, when running against the NCBI nr database (Figure 1). We also annotated genes involved in biofilm formation, multidrug transport, and plasma membrane and cell wall biosynthesis, as well as genes involved in thermotolerance or environmental stress responses, such as MDR, ERG, SAP, ALS, MNN, LIP, HYR1, PMR1, VRG4, CDR, ARB1, FKS1, FUR1, ALG1, CHS1, CALS11, HSFC1B, and HSP1. This whole-genome shotgun project has been deposited at DDBJ/ENA/GenBank under the accession number PRJNA559198 (Biosamples: SAMN12529702 and SAMN12529703).
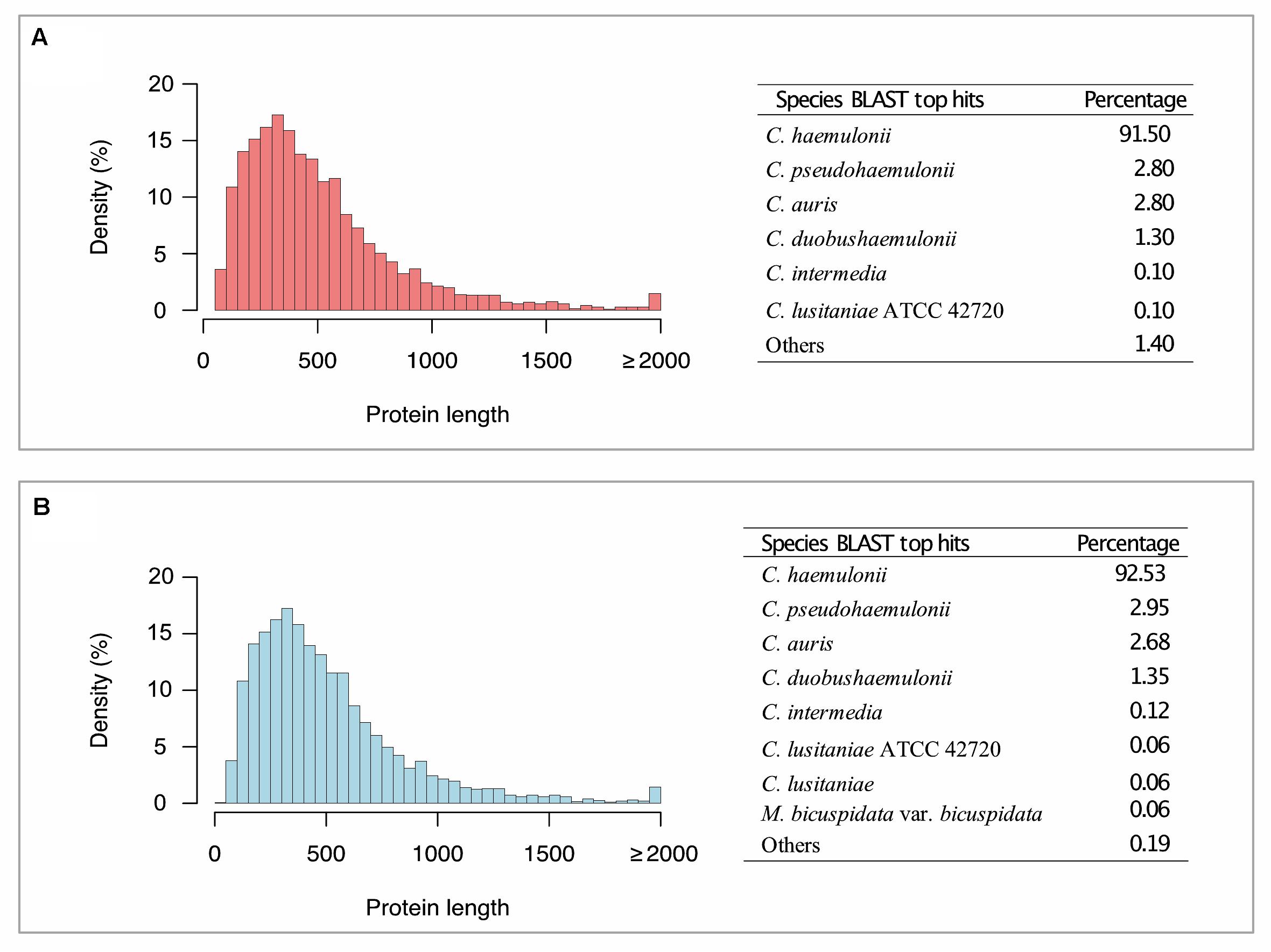
Figure 1. Histogram of coding genes predicted by AUGUSTUS and BLAST top hit species distribution summary. (A) Candida haemulonii var. vulnera K1. (B) Candida haemulonii var. vulnera K2. Candida haemulonii var. vulnera (K1 and K2) genes annotated against NR database (Eukaryota taxid: 2759), with an E-value ≤ 1e-5.
Based on ITS1 and ITS2 sequences from public repositories, we reconstructed the phylogeny of 47 isolates from five different Candida species (Supplementary Table S1), including K1 and K2 isolates. This analysis showed that K1 and K2 belong to the C. haemulonii var. vulnera clade, with 100% bootstrapped support (Figure 2), implying that they have been initially misidentified as C. haemulonii by MALDI-TOF MS.
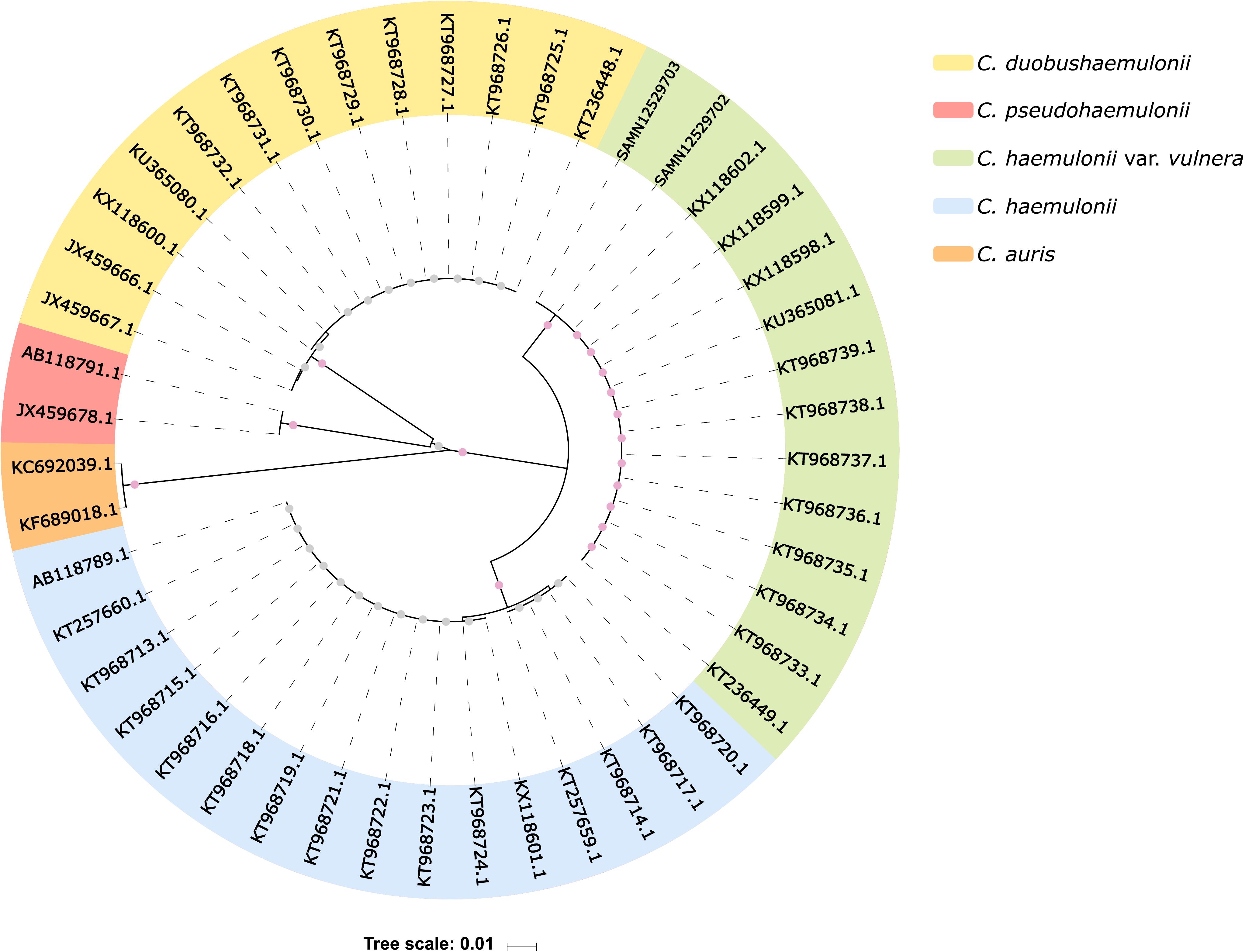
Figure 2. ITS sequence-based phylogenetic tree. Internal dots represent bootstrap values. Gray dots range from 0.60 to 0.70, and pink dots range from 0.99 to 1. SAMN12529703 and SAMN12529702 indicate our isolates (K1 and K2, respectively).
To further evaluate intraspecific variability among C. haemulonii complex and related species, we conducted ANI analysis among our isolates and the WGS of C. haemulonii, C. pseudohaemulonii, C. duobushaemulonii, C. auris, and C. lusitaniae deposited in NCBI. The results confirmed high levels of sequence identity between K1, K2, and C. haemulonii (Supplementary Figure S1).
Because this high genomic identity may be associated with repeated genomic regions in eukaryotes, we also searched for SNVs and indel differences among K1, K2, and C. haemulonii. Approximately 99.6% of the K2 raw reads were successfully aligned to the K1 genome, and the variant calling between these data resulted in a set of 1,033 SNVs and 154 indels. However, the variant calling of K2, using C. haemulonii as a reference resulted in a set of 1,422 SNVs and 1,510 indels, with 99.5% of K2 raw reads successfully aligned to the C. haemulonii genome.
Using the OrthoVenn2 web platform (Xu et al., 2019), we performed an orthologous clustering analysis of the predicted proteins in the MDR clade. Candida lusitaniae showed a smaller predicted proteome among the included isolates (C. haemulonii, C. haemulonii var. vulnera, C. duobushaemulonii, C. pseudohaemulonii, and C. auris). OrthoVenn2 clustering resulted in a total of 5,625 clusters, 3,849 of which were single-copy clusters.
A total of 4,015 core clusters were detected (e.g., those with at least one representative from all the six species included), with 24,654 proteins, supporting their conservation in the lineage (Figure 3). C. lusitaniae and C. auris were the species with the highest number of singleton protein sequences, orthologs for which could not be found in any of the other species. Besides, based on the similarity matrix, C. auris genomes formed a higher number of clusters with C. haemulonii var. vulnera (5,208) which, in turn, formed a higher number of clusters with C. haemulonii (5,344), as expected (Supplementary Figure S2).
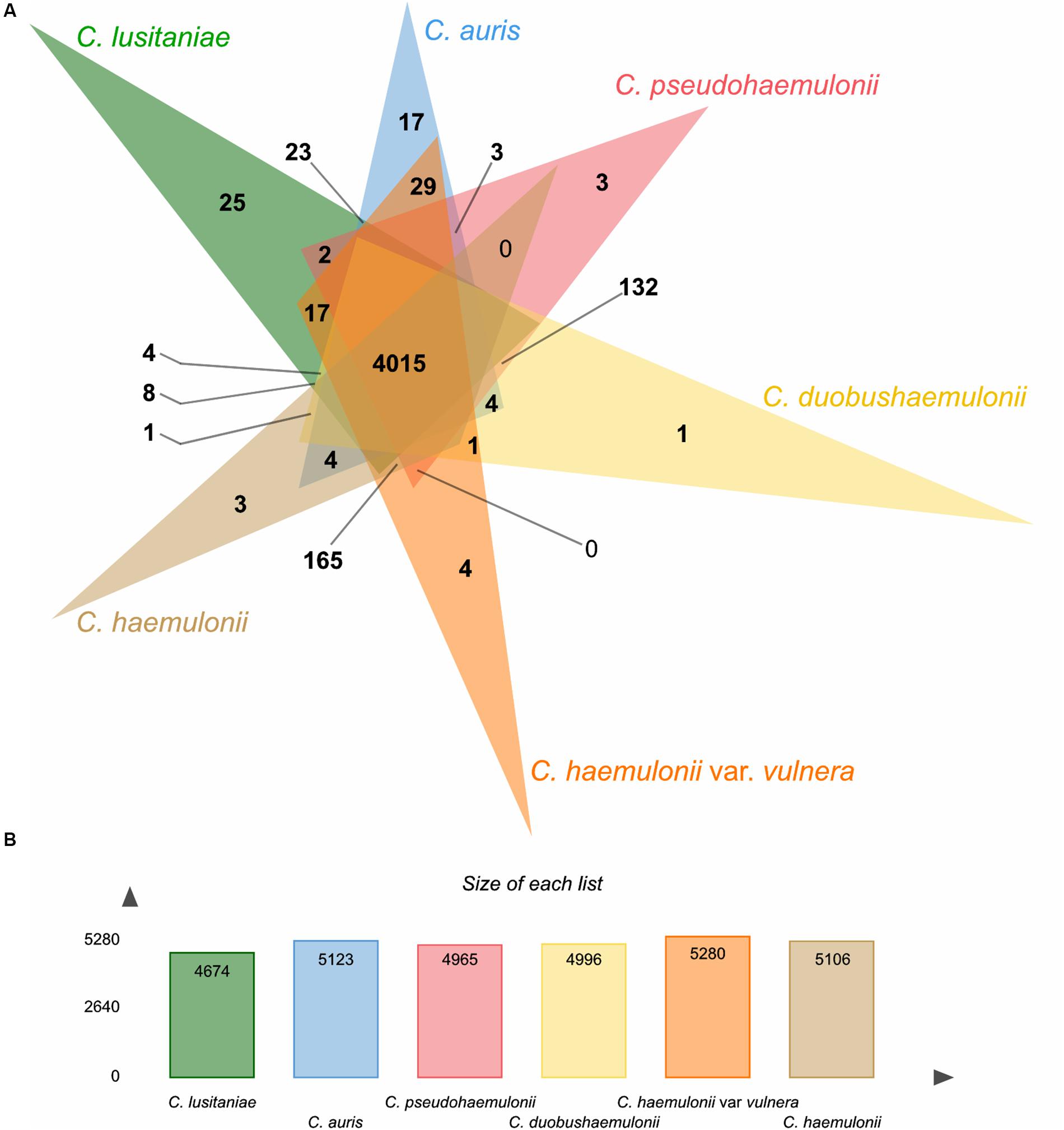
Figure 3. Summary of orthologs analysis using OrthoVenn2. (A) Venn diagram showing the distribution of orthologous clusters among C. haemulonii var. vulnera (K1) and related species. (B) Summary of protein data of each species.
In addition, 60 proteins from C. auris and C. haemulonii var. vulnera formed 29 clusters that did not include proteins from any other analyzed species. Most of these proteins were assigned the molecular function of ribonucleoprotein complex binding (GO: 0043021) in the nucleus cell region (GO: 0005634).
To characterize changes in gene content that may play a role in the evolution of MDR clade virulence and multidrug-resistance, we evaluated copy-number variation in genes among C. haemulonii var. vulnera and seven isolates of related species (Figure 4). Although many gene families involved in the pathogenesis of diseases caused by these species are present in similar numbers, some differences were observed, such as the expansion of lipases (particularly LIP1) shared by C. auris, C. pseudohaemulonii, C. duobushaemulonii, C haemulonii, and C. haemulonii var. vulnera, but not C. lusitaniae.
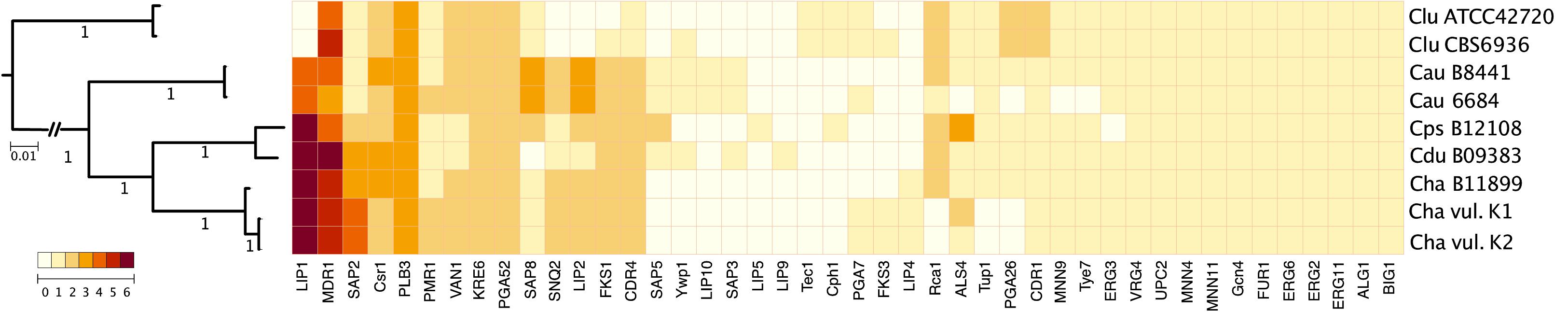
Figure 4. Gene number variation in MDR clade. Maximum likelihood phylogeny using 1737 single-copy core genes based on 1000 replicates among 9 annotated genome assemblies.
We identified, among species of the MDR clade, including our isolates, a greater number of ABC (ATP-binding cassette) transporter genes involved in clinical resistance to fluconazole and other toxic compounds, particularly the multidrug-resistant transporter 1 (MDR1). Additionally, C. lusitaniae was the only species that showed an increased copy-number of Candida drug resistance protein 1 (CDR1).
Alignment of the Erg11p amino acid sequences of K1 and K2 with MDR clade isolates and C. albicans (sensitive to polyene and azole) revealed ten amino acid substitutions previously reported as associated with azole resistance in Candida. All substitutions except E343S, V437S, A395V, and Y166V have been previously listed into three hotspot regions at amino acids 105–165, 266–287, and 405–488 of C. albicans Erg11p (Morio et al., 2010; Silva et al., 2016). However, the hotspot mutations Y132F and K143R, often found in resistant strains, were not identified in the two isolates of C. haemulonii var. vulnera described here (Figure 5; Healey et al., 2018).
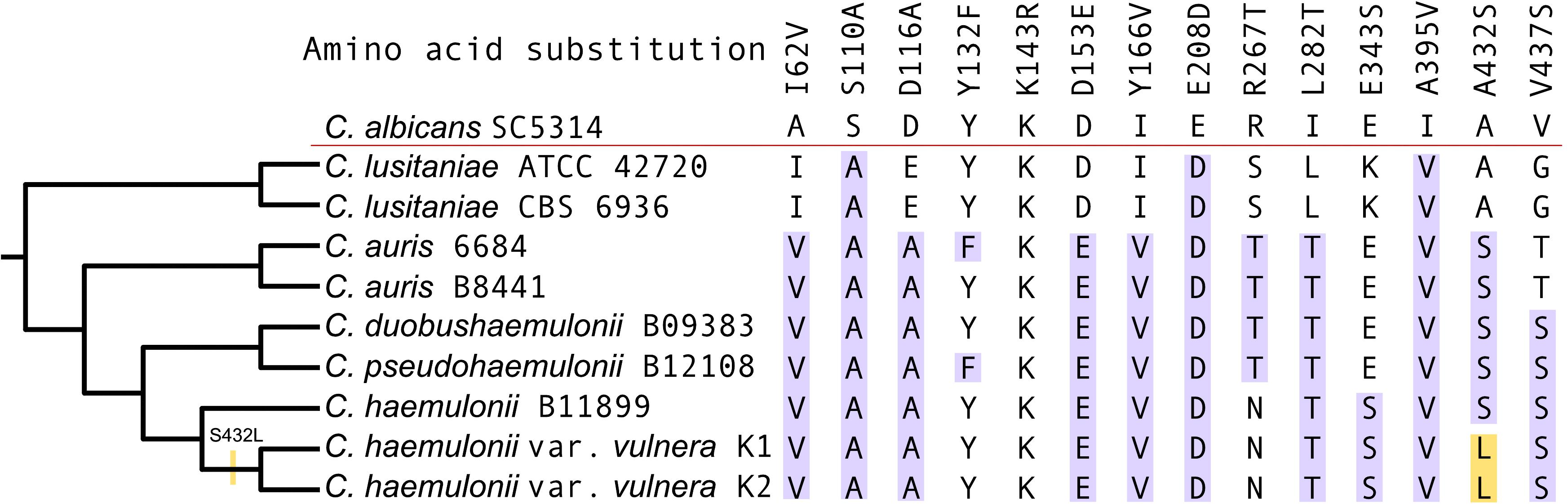
Figure 5. Multiple sequence alignment of the Erg11p from K1, K2, and other Candida species. Highlighted in blue are the previously described amino acid substitutions identified among the isolates and in yellow the apomorphy.
An apomorphy of the K1 and K2 isolates was observed at position 432 (A432L) in Erg11p, although serine in the position 432 was most likely the ancestral state of the MDR clade (Figure 5). The substitution of serine by leucine in the K1 and K2 isolates represented the reversal of e apolarity conferred by alanine in azole-sensitive members of Candida, such as C. albicans and C. lusitaniae.
The occurrence of candidiasis, caused by non-albicans species, is increasing. Although the reasons are still unclear, the use of fluconazole prophylaxis, the growing number of patients who are at risk of invasive fungal infections and advances in microbiological diagnosis, may have contributed (Sardi et al., 2013; Lee et al., 2018).
The emergence of non-albicans species has reinforced the importance of improving microbiological laboratory procedures, in particular concerning the adoption of molecular methods to overcome the limitation of classical methods in the identification of these opportunistic microorganisms (Lockhart et al., 2017a). Although MALDI-TOF MS is fast and accurate for yeast identification, it shows variable performance in the discrimination of C. haemulonii complex isolates and related species (Kathuria et al., 2015; Grenfell et al., 2016; Taverna et al., 2019). The two isolates reported here were identified as C. haemulonii sensu stricto using MALDI-TOF MS, while ITS1 and ITS2 analysis, obtained from the assembled genomes, identified the isolates as C. haemulonii var. vulnera (Supplementary Table S1).
Members of the C. haemulonii species complex are emerging yeasts associated with bloodstream and other invasive infections (Ruan et al., 2010). They are phylogenetically related to C. auris, the first global emerging yeast pathogen with potential for nosocomial transmission (Jeffery-Smith et al., 2018). Although C. auris is recognized as a virulent yeast, it exhibits high sequence divergence compared to Candida species commonly involved in human infections, such as C. albicans, C. tropicalis, C. parapsilosis, and C. glabrata. C. auris is more closely related to the C. haemulonii complex species, such as C. pseudohaemulonii and C. lusitaniae, which are less virulent but resistant to antifungals (Jeffery-Smith et al., 2018).
Although not yet reported in Brazil, C. auris has been identified in more than 30 countries. Complete genomic data revealed four distinct geographic clades: South Asian (I), East Asian (II), African (III), and South American (IV) (Lockhart et al., 2017b; Muñoz et al., 2018). More recently, a new clade, Iran (V), has also been proposed (Chow et al., 2019). Nevertheless, the origin of C. auris remains uncertain (Jackson et al., 2019). The generation and analysis of WGS data presented here can be used to explore the population structure and help to understand the species origin of MDR clade, and monitoring the global dissemination of them (Chybowska et al., 2020).
Here, we report the first WGS, assembly, and annotation of two C. haemulonii var. vulnera isolated from pediatric patients with candidemia in a tertiary pediatric hospital in the South of Brazil. The genomes are approximately 13.21 Mb and contain more than 5,400 predicted genes, similar to what has been previously described for other Candida species (Durrens et al., 2017; Chow et al., 2018a, b; Mohd Tap et al., 2018). ANI analysis showed high identity among K1 and K2 and C. haemulonii B11899 (99%). Comparison of C. haemulonii var. vulnera genomes to other related species showed an identity of 77% compared with C. duobushaemulonii or C. pseudohaemulonii, 75% with C. auris, and 72% with C. lusitaniae (Supplementary Figure S1).
A concatenated alignment of 1737 single-copy core genes from MDR clade members was used for the maximum likelihood tree inference. As expected, the data supported that C. haemulonii and C. haemulonii var. vulnera were closely related to each other, followed by C. duobushaemulonii and C. pseudohaemulonii that formed a sister group to C. haemulonii and C. auris as more divergent species. In turn, C. lusitaniae is a basally branching member of this group, consistent with previous phylogenetic analyses (Figure 4; Muñoz et al., 2018).
The pathogenicity of Candida spp. is attributed to virulence factors that allow evasion from the host’s defenses, enables adherence to surfaces, biofilm formation, alteration of cellular structure, and production of hydrolytic enzymes (Kumar and Menon, 2006; Ramage et al., 2009). A complex array of intracellular mechanisms and external factors influence biofilm formation and architecture in Candida species (Oh et al., 2011; Cavalheiro and Teixeira, 2018) as well as our two isolates, species from the C. haemulonii complex that have been reported to produce biofilm (Oh et al., 2011; Cendejas-Bueno et al., 2012; Muro et al., 2012; Ramos et al., 2017).
Detecting biofilm production is important, since biofilms provide greater resistance to antifungal agents and host’s defenses, contributing to the persistence of the infection. When detected on abiotic surfaces, such as catheters, the medical device should be removed (Tumbarello et al., 2012; Cavalheiro and Teixeira, 2018). Comparative genomics analysis revealed that, among the sequenced Candida species, C. haemulonii var. vulnera is closest to the previously characterized C. haemulonii and shares significant virulence attributes with the other members of the MDR clade, such as multiple drug transporter proteins, proteases and lipases.
Currently, invasive candidiasis is treated with three main classes of drugs: polyene antifungal agents, azoles, and echinocandins (Ben-Ami, 2018). Azole antifungals are important to treat Candida spp. infections, but the emergence of resistance to azoles and polyenes often culminates in treatment failure (Whaley et al., 2016; Lotfali et al., 2017). Voriconazole is associated with more adverse effects and drug interactions than fluconazole, and offers few advantages in candidiasis treatment, proving infection treatment by species intrinsically resistant to fluconazole (C. krusei) useful. Echinocandins are highly potent and safe antifungals for clinical use, against a wide variety of Candida species, including tolerant or azole-resistant ones (Ben-Ami, 2018).
Considering the few classes of antifungal agents available for the invasive candidiasis, the isolates described in this study were considered as MDR, because they were not susceptible to ≥1 agent (AMB, fluconazole and itraconazole) in ≥2 classes of drugs (polyene and azole) (Arendrup et al., 2017). Our results reinforced the importance of accurate identification and proper antifungal susceptibility testing to guide therapeutic strategies, instead of using AMB and azoles empirically.
Multiple mechanisms of resistance to azoles have been described for Candida species, including mutations in ergosterol biosynthesis genes (mainly resulting in increased expression of ERG11), overexpression of drug efflux pumps (e.g., CDR1, CDR2, and MDR1), and function-enhancing mutations in transcription factors (e.g., TAC1 and MRR1) that enhance their expression (Flowers et al., 2015; Whaley et al., 2016). Mechanisms of polyene resistance are less well studied than those of azoles resistance. However, the main mechanism of resistance to AMB involves ergosterol content reduction in the cell membrane (target abundance). Mutations in ergosterol biosynthesis genes (mainly ERG11 and ERG3) and biofilm formation can confer azole-polyene cross-resistance. Further, treatment with azoles that reduces cellular sterol concentrations can also confer polyene resistance (Lotfali et al., 2017; Perlin et al., 2017). Here we reported the presence of genes involved in biofilm formation, resistance genes and amino acid substitutions in Erg11p related to the increased MIC of azole and polyene antifungal agents in Candida spp.
The results presented here can be used to study fungal biology and virulence, as well as to understand the phylogenetic relationships between MDR Candida better, and to identify genomic regions associated with species-specific phenotypes.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA559198.
The studies involving human participants were reviewed and approved by HPP Institutional Review Board (IRB) number: 660-08/2008. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
LD-C, TV, LR, and HP-A conceived of the study. LD-C and TV supervised and managed the project. LR, RG, HP-A, AV, PP, RN, and RS performed research and data analysis. LR wrote the manuscript which was read, edited and approved by all authors. All authors contributed to the article and approved the submitted version.
The authors acknowledge the financial support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) (www.capes.gov.br) – Finance Code 001. TV was funded by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (www.faperj.br), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (www.cnpq.br). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Editage provided English editing of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01535/full#supplementary-material
Ahangarkani, F., Shokohi, T., Rezai, M. S., Ilkit, M., Mahmoodi Nesheli, H., Karami, H., et al. (2020). Epidemiological features of Nosocomial candidaemia in neonates, infants and children: a multicentre study in Iran. Mycoses 63, 382–394. doi: 10.1111/myc.13053
Almeida, J. N., Motta, A. L., Rossi, F., Abdala, E., Pierrotti, L. C., Kono, A. S., et al. (2012). First report of a clinical isolate of Candida haemulonii in Brazil. Clinics 67, 1229–1231. doi: 10.6061/clinics/2012(10)18
Arastehfar, A., Fang, W., Badali, H., Vaezi, A., Jiang, W., Liao, W., et al. (2018). Low-cost tetraplex PCR for the global spreading multi-drug resistant fungus. Front. Microbiol. 9:1119. doi: 10.3389/fmicb.2018.01119
Arendrup, M. C., and Patterson, T. F. (2017). Multidrug-resistant candida: epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 216(Suppl._3), S445–S451. doi: 10.1093/infdis/jix131
Arendrup, M. C., Prakash, A., Meletiadis, J., Sharma, C., and Chowdhary, A. (2017). Comparison of EUCAST and CLSI reference microdilution MICs of eight antifungal compounds for Candida auris and associated tentative epidemiological cutoff values. Antimicrob. Agents Chemother. 61:e00485-17. doi: 10.1128/AAC.00485-17
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Ben-Ami, R. (2018). Treatment of invasive Candidiasis: a narrative review. J. Fungi 4:97. doi: 10.3390/jof4030097
Ben-Ami, R., Berman, J., Novikov, A., Bash, E., Shachor-Meyouhas, Y., Zakin, S., et al. (2017). Multidrug-resistant Candida haemulonii and auris, C., Tel Aviv, Israel. Emerg. Infect. Dis. 23:486. doi: 10.3201/eid2302.161486
Binkley, J., Arnaud, M. B., Inglis, D. O., Skrzypek, M. S., Shah, P., Wymore, F., et al. (2014). The candida genome database: the new homology information page highlights protein similarity and phylogeny. Nucleic Acids Res. 42, D711–D716. doi: 10.1093/nar/gkt1046
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Branchini, M. L., Pfaller, M. A., Rhine-Chalberg, J., Frempong, T., and Isenberg, H. D. (1994). Genotypic variation and slime production among blood and catheter isolates of Candida parapsilosis. J. Clin. Microbiol. 32, 452–456. doi: 10.1128/jcm.32.2.452-456.1994
Cavalheiro, M., and Teixeira, M. C. (2018). Biofilms: threats, challenges, and promising strategies. Front. Med. 5:28. doi: 10.3389/fmed.2018.00028
Cendejas-Bueno, E., Gomez-Lopez, A., Mellado, E., Rodriguez-Tudela, J. L., and Cuenca-Estrella, M. (2010). Identification of pathogenic rare yeast species in clinical samples: comparison between phenotypical and molecular methods. J. Clin. Microbiol. 48, 1895–1899. doi: 10.1128/JCM.00336-10
Cendejas-Bueno, E., Kolecka, A., Alastruey-Izquierdo, A., Theelen, B., Groenewald, M., Kostrzewa, M., et al. (2012). Reclassification of the Candida haemulonii complex as Candida haemulonii (C. haemulonii group I), C. duobushaemulonii sp. nov (C. haemulonii group II), and C. haemulonii var. vulnera var. nov.: three multiresistant human pathogenic yeasts. J. Clin. Microbiol. 50, 3641–3651. doi: 10.1128/JCM.02248-12
Chatterjee, S., Alampalli, S. V., Nageshan, R. K., Chettiar, S. T., Joshi, S., and Tatu, U. S. (2015). Draft genome of a commonly misdiagnosed multidrug resistant pathogen Candida auris. BMC Genomics 16:686. doi: 10.1186/s12864-015-1863-z
Chow, N. A., de Groot, T., Badali, H., Abastabar, M., Chiller, T. M., and Meis, J. F. (2019). Potential Fifth Clade of Candida auris, Iran, 2018. Emerg. Infect. Dis. 25, 1780–1781. doi: 10.3201/eid2509.190686
Chow, N. A., Gade, L., Batra, D., Rowe, L. A., Juieng, P., Ben-Ami, R., et al. (2018a). Genome sequence of a multidrug-resistant Candida haemulonii isolate from a patient with chronic leg ulcers in Israel. Genome Announc. 6:e00176-18. doi: 10.1128/genomeA.00176-18
Chow, N. A., Gade, L., Batra, D., Rowe, L. A., Juieng, P., Loparev, V. N., et al. (2018b). Genome sequence of the amphotericin B-resistant Candida duobushaemulonii strain B09383. Genome Announc. 6:e00204-18. doi: 10.1128/genomeA.00204-18
Chowdhary, A., Anil Kumar, V., Sharma, C., Prakash, A., Agarwal, K., Babu, R., et al. (2014). Multidrug-resistant endemic clonal strain of Candida auris in India. Eur. J. Clin. Microbiol. Infect. Dis. 33, 919–926. doi: 10.1007/s10096-013-2027-1
Chybowska, A. D., Childers, D. S., and Farrer, R. A. (2020). Nine Things Genomics Can Tell Us About. Front. Genet. 11:351. doi: 10.3389/fgene.2020.00351
CLSI (2008). Reference Method For Broth Dilution Antifungal Susceptibility Testing Of Yeasts; Informational Supplement M27-S3. Wayne, PA: Clinical and Laboratory Standards Institute.
CLSI (2017). Reference Method For Broth Dilution Antifungal Susceptibility Testing Of Yeasts; Informational Supplement M27-S4. Wayne, PA: Clinical and Laboratory Standards Institute.
Colombo, A. L., Nucci, M., Park, B. J., Nouer, S. A., Arthington-Skaggs, B., da Matta, D. A., et al. (2006). Epidemiology of candidemia in Brazil: a nationwide sentinel surveillance of Candidemia in eleven medical centers. J. Clin. Microbiol. 44, 2816–2823. doi: 10.1128/JCM.00773-06
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Cowen, L. E., and Steinbach, W. J. (2008). Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukar. Cell 7, 747–764. doi: 10.1128/EC.00041-08
de Almeida, J. N., Assy, J. G., Levin, A. S., Del Negro, G. M., Giudice, M. C., Tringoni, M. P., et al. (2016). Candida haemulonii complex species, Brazil, January 2010-March 2015. Emerg. Infect. Dis. 22, 561–563. doi: 10.3201/eid2203.151610
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Doi, A. M., Pignatari, A. C., Edmond, M. B., Marra, A. R., Camargo, L. F., Siqueira, R. A., et al. (2016). Epidemiology and microbiologic characterization of nosocomial candidemia from a brazilian national surveillance program. PLoS One 11:e0146909. doi: 10.1371/journal.pone.0146909
Durrens, P., Klopp, C., Biteau, N., Fitton-Ouhabi, V., Dementhon, K., Accoceberry, I., et al. (2017). Genome sequence of the yeast Clavispora lusitaniae type strain CBS 6936. Genome Announc. 5:e00724-17. doi: 10.1128/genomeA.00724-17
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstra. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Flowers, S. A., Colón, B., Whaley, S. G., Schuler, M. A., and Rogers, P. D. (2015). Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob. Agents Chemother. 59, 450–460. doi: 10.1128/AAC.03470-14
Gargeya, I. B., Pruitt, W. R., Meyer, S. A., and Ahearn, D. G. (1991). Candida haemulonii from clinical specimens in the USA. J. Med. Vet. Mycol. 29, 335–338.
Grenfell, R. C., da Silva, Junior, A. R., Del Negro, G. M., Munhoz, R. B., Gimenes, V. M., et al. (2016). Identification of Candida haemulonii Complex Species: Use of ClinProTools(TM) to overcome limitations of the bruker biotyper(TM), VITEK MS(TM) IVD, and VITEK MS(TM) RUO databases. Front. Microbiol. 7:940. doi: 10.3389/fmicb.2016.00940
Guinea, J. (2014). Global trends in the distribution of Candida species causing candidemia. Clin. Microbiol. Infect. 20(Suppl. 6), 5–10. doi: 10.1111/1469-0691.12539
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Healey, K. R., Kordalewska, M., Jiménez Ortigosa, C., Singh, A., Berrío, I., Chowdhary, A., et al. (2018). Limited ERG11 mutations identified in isolates of Candida auris directly contribute to reduced azole susceptibility. Antimicrob. Agents Chemother. 62:e001427-18. doi: 10.1128/AAC.01427-18
Hou, X., Xiao, M., Chen, S. C., Wang, H., Cheng, J. W., Chen, X. X., et al. (2016). Identification and antifungal susceptibility profiles of Candida haemulonii species complex clinical isolates from a multicenter study in China. J. Clin. Microbiol. 54, 2676–2680. doi: 10.1128/JCM.01492-16
Jackson, B. R., Chow, N., Forsberg, K., Litvintseva, A. P., Lockhart, S. R., Welsh, R., et al. (2019). On the origins of a species: what might explain the rise of Candida auris? J. Fungi 5:58. doi: 10.3390/jof5030058
Jeffery-Smith, A., Taori, S. K., Schelenz, S., Jeffery, K., Johnson, E. M., Borman, A., et al. (2018). Candida auris: a review of the literature. Clin. Microbiol. Rev. 31:e0029-17. doi: 10.1128/CMR.00029-17
Kathuria, S., Singh, P. K., Sharma, C., Prakash, A., Masih, A., Kumar, A., et al. (2015). Multidrug-resistant Candida auris misidentified as Candida haemulonii: characterization by matrix-assisted laser desorption ionization-time of flight mass spectrometry and DNA sequencing and its antifungal susceptibility profile variability by vitek 2, CLSI broth microdilution, and ETEST method. J. Clin. Microbiol. 53, 1823–1830. doi: 10.1128/JCM.00367-15
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khodadadi, H., Karimi, L., Jalalizand, N., Adin, H., and Mirhendi, H. (2017). Utilization of size polymorphism in ITS1 and ITS2 regions for identification of pathogenic yeast species. J. Med. Microbiol. 66, 126–133. doi: 10.1099/jmm.0.000426
Kim, M. N., Shin, J. H., Sung, H., Lee, K., Kim, E. C., Ryoo, N., et al. (2009). Candida haemulonii and closely related species at 5 university hospitals in Korea: identification, antifungal susceptibility, and clinical features. Clin. Infect. Dis. 48, e57–e61. doi: 10.1086/597108
Kołaczkowska, A., and Kołaczkowski, M. (2016). Drug resistance mechanisms and their regulation in non-albicans Candida species. J. Antimicrob. Chemother. 71, 1438–1450. doi: 10.1093/jac/dkv445
Köster, J., and Rahmann, S. (2018). Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 34:3600. doi: 10.1093/bioinformatics/bty350
Kumar, A., Prakash, A., Singh, A., Kumar, H., Hagen, F., Meis, J. F., et al. (2016). Candida haemulonii species complex: an emerging species in India and its genetic diversity assessed with multilocus sequence and amplified fragment-length polymorphism analyses. Emerg. Microb. Infect. 5:e49. doi: 10.1038/emi.2016.49
Kumar, C. P., and Menon, T. (2006). Biofilm production by clinical isolates of Candida species. Med. Mycol. 44, 99–101.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lamoth, F., Lockhart, S. R., Berkow, E. L., and Calandra, T. (2018). Changes in the epidemiological landscape of invasive candidiasis. J. Antimicrob. Chemother. 73(Suppl._1), i4–i13. doi: 10.1093/jac/dkx444
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Leaw, S. N., Chang, H. C., Sun, H. F., Barton, R., Bouchara, J. P., and Chang, T. C. (2006). Identification of medically important yeast species by sequence analysis of the internal transcribed spacer regions. J. Clin. Microbiol. 44, 693–699. doi: 10.1128/JCM.44.3.693-699.2006
Lee, W. J., Hsu, J. F., Lai, M. Y., Chiang, M. C., Lin, H. C., Huang, H. R., et al. (2018). Factors and outcomes associated with candidemia caused by non-albicans Candida spp versus Candida albicans in children. Am. J. Infect. Control 46, 1387–1393. doi: 10.1016/j.ajic.2018.05.015
Lehmann, P. F., Wu, L. C., Pruitt, W. R., Meyer, S. A., and Ahearn, D. G. (1993). Unrelatedness of groups of yeasts within the Candida haemulonii complex. J. Clin. Microbiol. 31, 1683–1687. doi: 10.1128/jcm.31.7.1683-1687.1993
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lockhart, S. R., Berkow, E. L., Chow, N., and Welsh, R. M. (2017a). for the clinical microbiology laboratory: not your grandfather’s. Clin. Microbiol. Newsl. 39, 99–103. doi: 10.1016/j.clinmicnews.2017.06.003
Lockhart, S. R., Etienne, K. A., Vallabhaneni, S., Farooqi, J., Chowdhary, A., Govender, N. P., et al. (2017b). Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin. Infect. Dis. 64, 134–140. doi: 10.1093/cid/ciw691
Lotfali, E., Ghajari, A., Kordbacheh, P., Zaini, F., Mirhendi, H., Mohammadi, R., et al. (2017). Regulation of ERG3, ERG6, and ERG11 genes in antifungal-resistant isolates of Candida parapsilosis. Iran. Biomed. J. 21, 275–281.
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mizusawa, M., Miller, H., Green, R., Lee, R., Durante, M., Perkins, R., et al. (2017). Can multidrug-resistant Candida auris be reliably identified in clinical microbiology laboratories? J. Clin. Microbiol. 55, 638–640. doi: 10.1128/JCM.02202-16
Mohd Tap, R., Kamarudin, N. A., Ginsapu, S. J., Ahmed, A. R., Bakri Ahmad, N., Amran, F., et al. (2018). Draft genome sequence of Candida pseudohaemulonii isolated from the blood of a neutropenic patient. Genome Announc. 6:e00166-18. doi: 10.1128/genomeA.00166-18
Morio, F., Loge, C., Besse, B., Hennequin, C., and Le Pape, P. (2010). Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn. Microbiol. Infect. Dis. 66, 373–384. doi: 10.1016/j.diagmicrobio.2009.11.006
Muñoz, J. F., Gade, L., Chow, N. A., Loparev, V. N., Juieng, P., Berkow, E. L., et al. (2018). Genomic insights into multidrug-resistance, mating and virulence in Candida auris and related emerging species. Nat. Commun. 9:5346. doi: 10.1038/s41467-018-07779-6
Muro, M. D., Motta, F. E. A., Burger, M., Melo, A. S., and Dalla-Costa, L. M. (2012). Echinocandin resistance in two Candida haemulonii isolates from pediatric patients. J. Clin. Microbiol. 50, 3783–3785. doi: 10.1128/JCM.01136-12
Nucci, M., Queiroz-Telles, F., Alvarado-Matute, T., Tiraboschi, I. N., Cortes, J., Zurita, J., et al. (2013). Epidemiology of candidemia in Latin America: a laboratory-based survey. PLoS One 8:e59373. doi: 10.1371/journal.pone.0059373
Oh, B. J., Shin, J. H., Kim, M. N., Sung, H., Lee, K., Joo, M. Y., et al. (2011). Biofilm formation and genotyping of Candida haemulonii, Candida pseudohaemulonii, and a proposed new species (Candida auris) isolates from Korea. Med. Mycol. 49, 98–102. doi: 10.3109/13693786.2010.493563
Perlin, D. S., Rautemaa-Richardson, R., and Alastruey-Izquierdo, A. (2017). The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect. Dis. 17, e383–e392. doi: 10.1016/S1473-3099(17)30316-X
Pfaller, M. A., and Diekema, D. J. (2007). Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20, 133–163. doi: 10.1128/CMR.00029-06
Piskur, J., and Langkjaer, R. B. (2004). Yeast genome sequencing: the power of comparative genomics. Mol. Microbiol. 53, 381–389. doi: 10.1111/j.1365-2958.2004.04182.x
Pritchard, L., Glover, H. R., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 10–24. doi: 10.1039/C5AY02550H
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Ramage, G., Mowat, E., Jones, B., Williams, C., and Lopez-Ribot, J. (2009). Our current understanding of fungal biofilms. Crit. Rev. Microbiol. 35, 340–355. doi: 10.3109/10408410903241436
Ramos, L. S., Figueiredo-Carvalho, M. H., Barbedo, L. S., Ziccardi, M., Chaves, A. L., Zancopé-Oliveira, R. M., et al. (2015). Candida haemulonii complex: species identification and antifungal susceptibility profiles of clinical isolates from Brazil. J. Antimicrob. Chemother. 70, 111–115. doi: 10.1093/jac/dku321
Ramos, L. S., Oliveira, S. S. C., Souto, X. M., Branquinha, M. H., and Santos, A. L. S. (2017). Planktonic growth and biofilm formation profiles in Candida haemulonii species complex. Med. Mycol. 55, 785–789. doi: 10.1093/mmy/myx005
Rodrigues, L. S., Motta, F. A., Picharski, G. L., Vasconcelos, T. M., Riccieri, M. C., and Dalla-Costa, L. M. (2019). Invasive candidiasis: risk factor for mortality in a pediatric tertiary care hospital in south of Brazil. Medicine 98:e15933. doi: 10.1097/MD.0000000000015933
Ruan, S. Y., Kuo, Y. W., Huang, C. T., Hsiue, H. C., and Hsueh, P. R. (2010). Infections due to Candida haemulonii: species identification, antifungal susceptibility and outcomes. Int. J. Antimicrob. Agents 35, 85–88. doi: 10.1016/j.ijantimicag.2009.08.009
Sardi, J. C., Scorzoni, L., Bernardi, T., Fusco-Almeida, A. M., and Mendes Giannini, M. J. (2013). Candida species: current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J. Med. Microbiol. 62(Pt 1), 10–24. doi: 10.1099/jmm.0.045054-0
Sears, D., and Schwartz, B. S. (2017). Candida auris: an emerging multidrug-resistant pathogen. Int. J. Infect. Dis. 63, 95–98. doi: 10.1016/j.ijid.2017.08.017
Silva, D. B., Rodrigues, L. M., Almeida, A. A., Oliveira, K. M., and Grisolia, A. B. (2016). Novel point mutations in the ERG11 gene in clinical isolates of azole resistant Candida species. Mem. Inst Oswaldo. Cruz. 111, 192–199. doi: 10.1590/0074-02760150400
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sneath, P. H. A., and Sokal, R. R. (1973). Numerical Taxonomy: The Principles and Practice of Numerical Classification. New York, NY: W H Freeman & Company.
Spivak, E. S., and Hanson, K. E. (2017). Candida auris: an emerging fungal pathogen. J. Clin. Microbiol. 15:e1007638. doi: 10.1128/JCM.01588-17
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. doi: 10.1093/nar/gki458
Taori, S. K., Khonyongwa, K., Hayden, I., Athukorala, G. D. A., Letters, A., Fife, A., et al. (2019). Candida auris outbreak: mortality, interventions and cost of sustaining control. J. Infect. 79, 601–611. doi: 10.1016/j.jinf.2019.09.007
Tarailo-Graovac, M., and Chen, N. (2009). Using repeatmasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 4:Unit4.10. doi: 10.1002/0471250953.bi0410s25
Taverna, C. G., Mazza, M., Bueno, N. S., Alvarez, C., Amigot, S., Andreani, M., et al. (2019). Development and validation of an extended database for yeast identification by MALDI-TOF MS in argentina. Med. Mycol. 57, 215–225. doi: 10.1093/mmy/myy021
Tumbarello, M., Fiori, B., Trecarichi, E. M., Posteraro, P., Losito, A. R., De Luca, A., et al. (2012). Risk factors and outcomes of candidemia caused by biofilm-forming isolates in a tertiary care hospital. PLoS One 7:e33705. doi: 10.1371/journal.pone.0033705
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11–33. doi: 10.1002/0471250953.bi1110s43
Whaley, S. G., Berkow, E. L., Rybak, J. M., Nishimoto, A. T., Barker, K. S., and Rogers, P. D. (2016). Azole antifungal resistance in Candida albicans and emerging non-albicans candida species. Front. Microbiol. 7:2173. doi: 10.3389/fmicb.2016.02173
White, T. J., Bruns, T. D., Lee, S. B., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfang, J. J. Snisky, and T. J. White (Cambridge, MA: Academic Press).
Wisplinghoff, H., Ebbers, J., Geurtz, L., Stefanik, D., Major, Y., Edmond, M. B., et al. (2014). Nosocomial bloodstream infections due to Candida spp. in the USA: species distribution, clinical features and antifungal susceptibilities. Int. J. Antimicrob. Agents 43, 78–81. doi: 10.1016/j.ijantimicag.2013.09.005
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Keywords: Candida haemulonii var. vulnera, candidemia, whole-genome sequencing, multidrug-resistance clade, fungal infection
Citation: Rodrigues LS, Gazara RK, Passarelli-Araujo H, Valengo AE, Pontes PVM, Nunes-da-Fonseca R, de Souza RF, Venancio TM and Dalla-Costa LM (2020) First Genome Sequences of Two Multidrug-Resistant Candida haemulonii var. vulnera Isolates From Pediatric Patients With Candidemia. Front. Microbiol. 11:1535. doi: 10.3389/fmicb.2020.01535
Received: 01 April 2020; Accepted: 12 June 2020;
Published: 03 July 2020.
Edited by:
Attila Gacser, University of Szeged, HungaryReviewed by:
Hamid Badali, The University of Texas Health Science Center at San Antonio, United StatesCopyright © 2020 Rodrigues, Gazara, Passarelli-Araujo, Valengo, Pontes, Nunes-da-Fonseca, de Souza, Venancio and Dalla-Costa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luiza Souza Rodrigues, bHVpemFiaW9tZWRAaG90bWFpbC5jb20=; Thiago Motta Venancio, dGhpYWdvLnZlbmFuY2lvQGdtYWlsLmNvbQ==; dG12ZW5hbmNpb0B1ZW5mLmJy; Libera Maria Dalla-Costa, bG1kYWxsYWNvc3RhQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.