
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 July 2020
Sec. Virology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.01472
This article is part of the Research Topic Shiga Toxin-Converting Bacteriophages View all 8 articles
 Michele Zuppi1
Michele Zuppi1 Rosangela Tozzoli1*
Rosangela Tozzoli1* Paola Chiani1Pablo Quiros2Adan Martinez-Velazquez2
Paola Chiani1Pablo Quiros2Adan Martinez-Velazquez2 Valeria Michelacci1
Valeria Michelacci1 Maite Muniesa2
Maite Muniesa2 Stefano Morabito1
Stefano Morabito1Bacteriophages are pivotal elements in the dissemination of virulence genes. The main virulence determinants of Shiga Toxin producing E. coli, Shiga Toxins (Stx), are encoded by genes localized in the genome of lambdoid bacteriophages. Stx comprise two antigenically different types, Stx1 and Stx2, further divided into subtypes. Among these, certain Stx2 subtypes appear to be more commonly occurring in the most severe forms of the STEC disease, haemorrhagic colitis and haemolytic uremic syndrome (HUS). This study aimed at obtaining insights on the evolution of Stx2 bacteriophages, due to their relevance in public health, and we report here on the analysis of the genomic structure of Stx2 converting phages in relation with the known reservoir of the E. coli strains harboring them. Stx2-converting phages conveying the genes encoding different stx2 subtypes have been isolated from STEC strains and their whole genomes have been sequenced, analyzed and compared to those of other Stx2 phages available in the public domain. The phages’ regions containing the stx2 genes have been analyzed in depth allowing to make inference on the possible mechanisms of selection and maintenance of certain Stx2 phages in the reservoir. The “stx regions” of different stx2 gene subtypes grouped into three different evolutionary lines in the comparative analysis, reflecting the frequency with which these subtypes are found in different animal niches, suggesting that the colonization of specific reservoir by STEC strains could be influenced by the Stx phage that they carry. Noteworthy, we could identify the presence of nanS-p gene exclusively in the “stx regions” of the phages identified in STEC strains commonly found in cattle. As a matter of fact, this gene encodes an esterase capable of metabolizing sialic acids produced by submaxillary glands of bovines and present in great quantities in their gastrointestinal tract.
Shiga Toxin-producing E. coli (STEC) are pathogens of public health concern, capable of inducing severe disease in humans, either at the gastrointestinal or systemic level (Caprioli et al., 2005). STEC disease includes different symptoms, ranging from non-complicated diarrhea to haemorrhagic colitis and the Haemolytic Uremic Syndrome (HUS), which represents the most severe form of STEC infection.
The STEC pathotype consists in a heterogeneous group of intestinal pathogenic E. coli and owes the name to the ability of elaborating one or more toxins known as Shiga toxins (Stx), whose activity can block the protein synthesis in target eukaryotic cells, causing their death (Melton-Celsa, 2014). Stx-coding genes are carried by lambdoid prophages, referred to as Stx-converting phages or Stx phages (O’Brien et al., 1983; Huang et al., 1987; Bryan et al., 2015) and encode two main antigenically distinct toxin types, Stx1 and Stx2. Each of these types is further divided into subtypes, three for Stx1 (Stx1a, Stx1c and Stx1d) and at least seven for Stx2 (Stx2a, Stx2b, Stx2c, Stx2d, Stx2e, Stx2f and Stx2g) (Scheutz et al., 2012), although new Stx2 subtypes are continuously described, including Stx2h to k (Lacher et al., 2016; Bai et al., 2018; Yang et al., 2020). The Stx and their subtypes have been associated with different frequency to the onset of severe diseases (Fuller et al., 2011; Melton-Celsa, 2014; Krüger and Lucchesi, 2015). In fact, Stx2 is more frequently associated, compared to Stx1, to HUS and haemorrhagic colitis and even more striking is the association between HUS and the subtypes Stx2a, Stx2c and Stx2d (Fuller et al., 2011; Melton-Celsa, 2014; Krüger and Lucchesi, 2015).
STEC are zoonotic pathogens and, although cattle have been recognized as the main asymptomatic animal reservoir (Karmali, 2018), these bacteria can be found in the intestinal tract of many animals, including other ruminants, pigs and pigeons (Mughini-Gras et al., 2018). It is important to note that STEC strains producing different Stx2 subtypes can be associated with different animal reservoirs. The E. coli strains producing Stx2a, Stx2c and to a lesser extent Stx2d, are mainly isolated from the bovine reservoir (Januszkiewicz and Rastawicki, 2016; Akiyama et al., 2017; Jajarmi et al., 2017; Tostes et al., 2017; Cha et al., 2018; Rahman et al., 2018; Dehdashti et al., 2019). Other subtypes, such as the Stx2b and Stx2g, can be also found in STEC isolated from ruminants but with a lower prevalence (Januszkiewicz and Rastawicki, 2016; Akiyama et al., 2017; Jajarmi et al., 2017; Dehdashti et al., 2019). The Stx2e subtype is produced by STEC found almost exclusively in pigs (Krüger et al., 2011; Cha et al., 2018), while strains producing the Stx2f subtype seem to preferentially or exclusively colonize pigeons (Krüger and Lucchesi, 2015; Van Hoek et al., 2019). An explanation for such observations has not been proposed yet. In general, it is conceivable that STEC strains producing distinctive Stx2 subtypes may have acquired advantages facilitating the survival in the specific reservoir.
The role of prophages is crucial in the microbial evolution (Javadi et al., 2017), as they may increase the evolutionary fitness of their host by favoring the rearrangement of genomic regions and the acquisition of foreign DNA providing new features through Horizontal Gene Transfer (Feiner et al., 2015). In particular, the spreading of Stx phages contributed to the emergence of different Shiga toxin-producing E. coli types (e.g., Stx-producing Enteroaggregative E. coli (Navarro-Garcia, 2014) and Stx producing Enterotoxigenic E. coli (Michelacci et al., 2018), among others), by providing the ability of producing the Stx to different diarrheagenic E. coli.
In this study, we focused on the evolution of Stx2 phages conveying the genes encoding different Stx2 subtypes based on whole genome sequencing data. We isolated and sequenced a set of Stx2 phages and analyzed their genomes in comparison to a panel of Stx2 phages’ genomes available in the public domain in the attempt to shed light on their evolution and correlating this analysis with the Stx subtype conveyed.
The STEC strains used for the induction of the phages were part of the culture collections of the European Union Reference Laboratory for E. coli and the University of Barcelona and are listed in Table 1. The E. coli K12 strain XL1-Blue (Bullock et al., 1987) was used as propagator strain for the amplification of the induced phages.
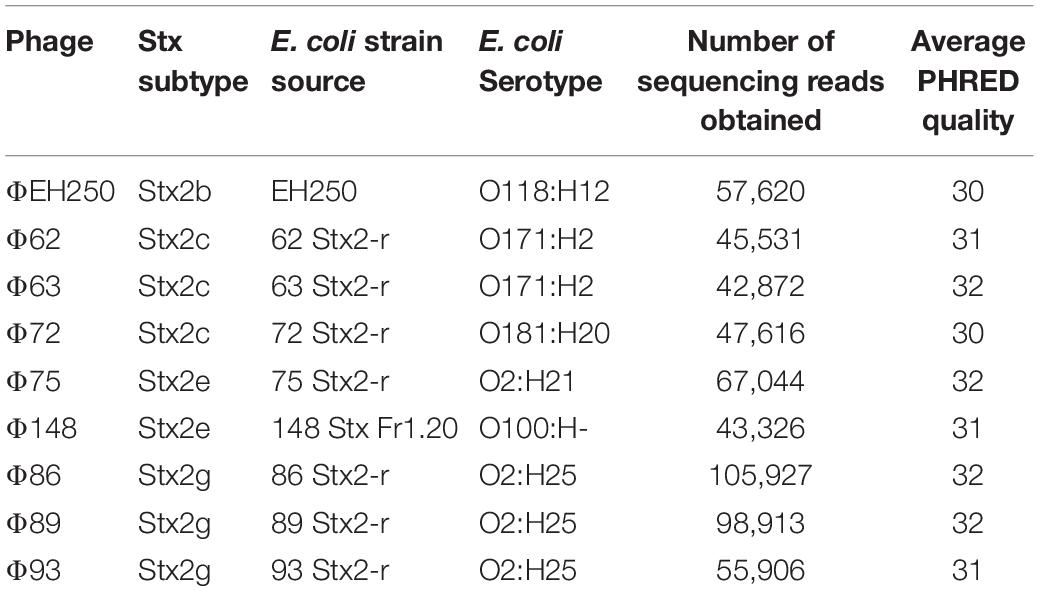
Table 1. Bacteriophages isolated and sequenced in this study together with sequence quality controls.
The STEC strains were treated with Mitomycin C to promote the induction of the Stx bacteriophages. In detail, the bacterial strains were grown in Luria-Bertani (LB) broth overnight at 37°C, diluted 1:100 into 10 ml of LB broth supplemented with 5 mM CaCl2 and grown with vigorous shaking (200 rpm) at 37°C, until an OD600 of 0.3 was reached. The cultures were then added with Mitomycin C (final concentration 0.5 μg/ml) and further incubated overnight in the dark. The treated cultures were finally filtered through sterile 0.22 μm Millex-GP filters (Merck KGaA, Darmstadt, Germany) to prepare the phage suspensions for the following amplification and purification. One hundred microliters of the phage suspensions were added to 100 μl of culture of the propagator strain XL1-Blue, grown in LB broth to 0.3 OD600, and incubated in a thermostatic bath at 37°C for 20 min. The cultures were added to 3.5 ml of LB modified soft agar (LB broth with 0.001% thiamine V/V and 5 mM CaCl2 with agar 7 g/l), kept at 46°C, and instantly poured on LB agar plates, which were then incubated overnight at 37°C.
Five milliliters of SM buffer (5.8 g NaCl, 2 g MgSO4⋅7 H2O, 50 ml Tris-HCl 1 M pH 7.5, 0.1 g gelatin, 950 ml deionized H2O) were dispensed to each plate to recover the phage particles that underwent lytic cycle and kept at 4°C overnight. The phage suspensions in SM buffer were recovered, added with chloroform at 5% final concentration and centrifuged twice at 4500 × g at 4°C for 20 min, in order to recover the phage particles from the supernatant while eliminating bacterial and agar remains. The phage suspensions were then used to re-infect the propagator strain XL1-Blue, as described above, in order to increase the phage titre, recovered with SM buffer and concentrated with Amicon® Ultra-15 Centrifugal Filter Units with Ultracel-30 tubes (Merck KGaA, Darmstadt, Germany) with a cut-off of 30 kDa.
The phage suspensions were purified by CsCl density gradients, following the protocol described by Sambrook and Russel (2001). Briefly, 2 ml of sucrose 20% (w/v) was first added to the tubes to ultra-Clear BECKMAN® tubes (Life Sciences Division Headquarters, Indianapolis, IN, United States) before adding 1 ml of CsCl solutions of density of 1.7, 1.5, and 1.3 g/ml, respectively. The different solutions of CsCl were sequentially added for the creation of the gradient. The phage suspensions were added with 0.5 g/ml of solid CsCl, incubated until the CsCl salts had completely dissolved and eventually 1 ml of the phage suspension containing CsCl was deposited above the gradient. The tubes containing the phages on the CsCl gradient were centrifuged at 80,000 × g for 2 h at 4°C in a Swinging-Bucket Rotor (Beckman, rotor SW41 Ti) (Life Sciences Division Headquarters, Indianapolis, IN, United States). The bands resulting from the centrifugation of the samples containing the phage particles were collected with a sterile syringe by puncturing the tube, added to a tube filled with a CsCl solution (density of 1.5 g/ml) and centrifuged again at 160,000 × g for 24 h at 4°C. Collection of the band was repeated, and the final suspension was dialyzed against 10 mM NaCl, 50 mM Tris-HCl pH 8.0, 10 mM MgCl2.
Each suspension was added with 100 units/ml of Sigma-Aldrich® DNase I RNase-free (Merck KGaA, Darmstadt, Germany) and incubated at 37°C for 1 h for the removal of free DNA. The phages’ suspensions were treated with 50 μg/ml of Proteinase K Roche® (Merck KGaA, Darmstadt, Germany) for 1 h at 56°C for the disruption of the phage capsid, and the DNA was extracted with phenol-chloroform-isoamyl alcohol, as reported by Sambrook and Russel (2001).
When more than one band was visualized, they were punctured separately and PCR targeting stx genes was carried out as described below to verify which band corresponded to the Stx phage.
End-point PCR amplifications were performed with a GeneAmp PCR system 2400 (Perkin-Elmer, PE Applied Biosystem, Waltham, MA, United States). A 378 bp fragment of the stx2A subunit was amplified with primers UP378/LP378 (Muniesa and Jofre, 1998) that detected all stx2 subtypes, except stx2f. The stx genes subtypes carried by the different phages were confirmed by PCR using the primers and amplification conditions described by Scheutz et al. (2012).
Phage suspensions were 100-fold concentrated using Amicon Ultra 15 ml tubes (100 kDa Amicon Ultra centrifugal filter units, Millipore, Bedford, MA, United States) by centrifuging at 3,000 × g for 10 min. Ten microliter of each phage suspension were dropped onto copper grids with carbon coated Formvar films, negatively stained with 2% ammonium molybdate (pH 6.8) and examined under a Jeol 1010 transmission electron microscope (TEM) (JEOL Inc. Peabody, MA, United States) operating at 80 kV.
The DNA libraries were prepared using the NEBNext® Fast DNA Fragmentation & Library Prep Set for Ion Torrent (New England Biolabs, Ipswich, MA, United States). The template preparation and sequencing run were performed with an Ion One Touch 2 System and an Ion Torrent PGM sequencer (Life Technologies, Carlsbad, CA, United States) following the manufacturer’s instructions for 400 bp DNA libraries on an Ion 314TM chip v2 (Life Technologies, Carlsbad, CA, United States).
The reads resulting from the sequencing of the phage genomes (Table 1) were uploaded to the public server ARIES1 for the bioinformatics analyses (Knijn et al., 2020). The quality of the reads was assessed using the tool FASTQC (Galaxy v0.63)2. The reads were then trimmed with “FASTQ positional and quality trimming” (Galaxy v0.0.1) (Cuccuru et al., 2014), in order to remove the segments corresponding to the Ion Torrent adapters and the regions presenting a Phred Quality Score for each base < 26. The reads were assembled through the software “SPAdes genome assembler for regular and single-cell projects” (Galaxy v3.11.1) (Bankevich et al., 2012). The contigs shorter than 1000 bp were filtered out [“Filter SPAdes output” (Galaxy v0.1)]. The filtered contigs were annotated with the tool “Prokka” (Galaxy v1.12.0) (Seemann, 2014), using both the bacterial and viral databases, together with the genomes recovered from the public domain (Table 2). The annotation of these latter genomes, already annotated, was repeated to avoid possible differences due to the use of different annotation tools.
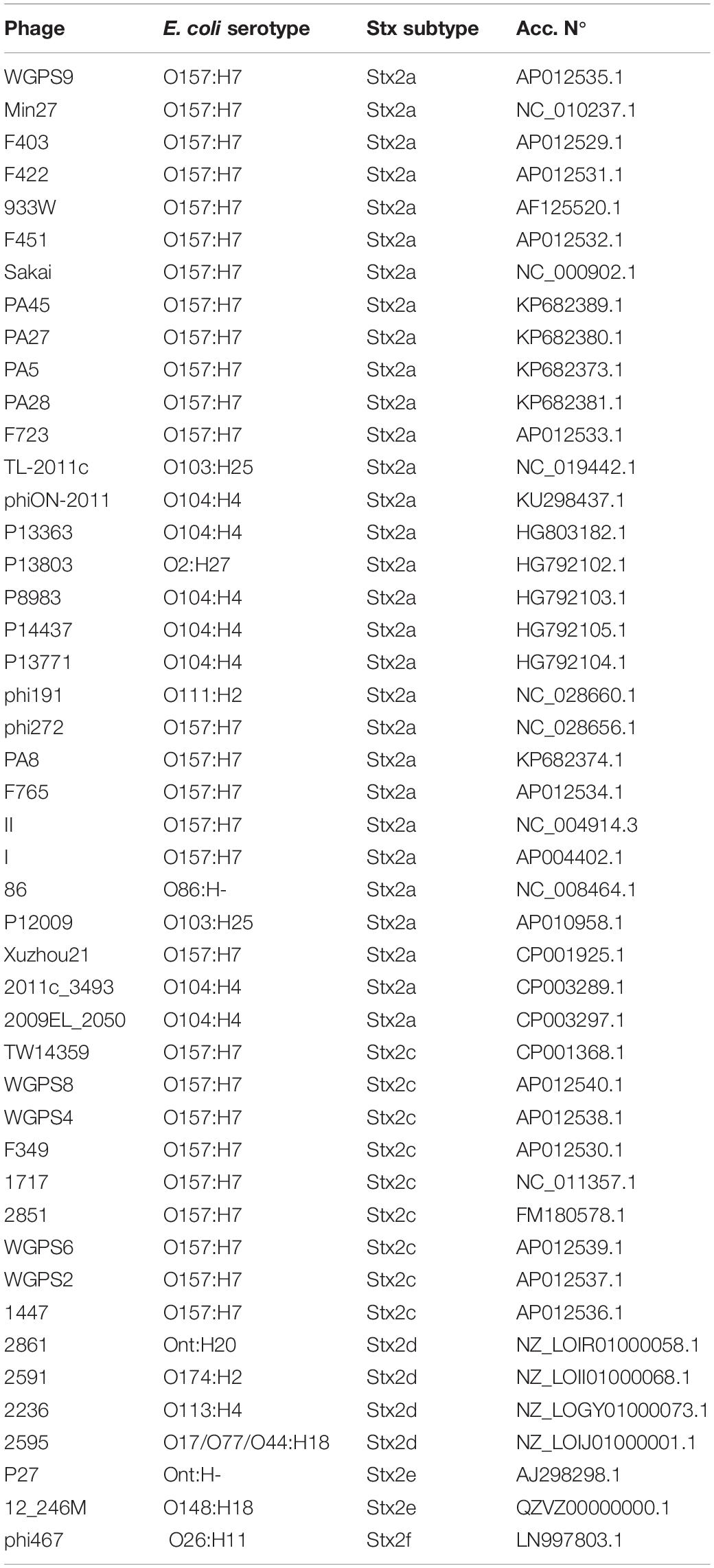
Table 2. Bacteriophages genomes retrieved from GenBank.
The presence of the stx genes in all the genomes was assessed through the web application “virulence finder”3. The contigs that did not display the stx genes in the output were further manually screened using the NCBI BLAST+ blastn algorithm.
The annotated sequences were visualized with the software Snapgene® (v4.1.9) (GSL, Biotech), through which it was possible to localize, isolate and characterize the gene composition of the “stx regions,” defined as the phage’s genome region encompassed by the genes q and s and containing the Stx-coding genes.
All the genomes of the phages and the respective “stx regions” were compared through the software for multiple sequences alignment “MAFFT Multiple alignment program for amino acid or nucleotide sequences” (Galaxy Version 7.221.3) (Katoh and Standley, 2013). The alignment algorithm included the following parameters: L-INS-i, most accurate; recommended for < 200 sequences; with the iterative refinement method (max. 1000 iterations) incorporating local pairwise alignment information. We have used 0 as “Gap extend penalty” value, in order to eliminate the differences due to the alignment of sequences of different lengths. The alignments were used to produce a maximum likelihood tree with the ultrafast bootstrap based on 1000 replicates, visualized through the online software iTOL (v 4.3.2) (Letunic and Bork, 2016). The comparative analysis based on the structure of the phage genomes was carried out by computing the distance matrix based on the presence or absence of each ORF with an annotated function and using the GC content of each of the ORF as an additional criterion (Supplementary Table S1). In detail, each ORF with the same annotation was considered as being a different allele when displaying a different GC content. The presence, absence and the different allelic types were all used to compute the Hamming distance between pairs of samples by using the function “Distance matrix” (ver. 0.1.9) of the MentaLIST software (Feijao et al., 2018) (galaxy version 0.2.3). The matrix obtained was eventually used to produce a Neighbor Joining tree with the function “MentaLIST Tree” of the same software. The comparison of the phage structures was carried out using the BLAST algorithm embedded into the Circoletto online tool4 using the ultra-strict parameter (E-value-180) (Darzentas, 2010). For the pictogram construction, bit-score values have been used to describe the quality of the alignment at a given point, displayed in different colors (blue ≤ 50%; green ≤ 75%, orange ≤ 99%, and red = 100% identity). The bit-score is a normalized version of the score value returned by the BLAST searches, expressed in bits (Altschul et al., 1990).
The purification of the Stx phages from the majority of the STEC strains allowed the observation of one single band after the CsCl density gradients, indicative of the presence of one single prophage in enough density to be visualized (Figure 1A). The band was collected and the presence of the stx in phage DNA was confirmed by PCR (data not shown). Strains EH250 and 86 Stx2-r (Table 1) showed two bands which, after collection and DNA extraction, were tested by PCR to determine which of the two corresponded to Stx2b phage ΦEH250 and Stx2g phage Φ86, respectively. The second band in these strains plausibly corresponded to another non-Stx inducible phage present in the chromosome that was not used in this study.
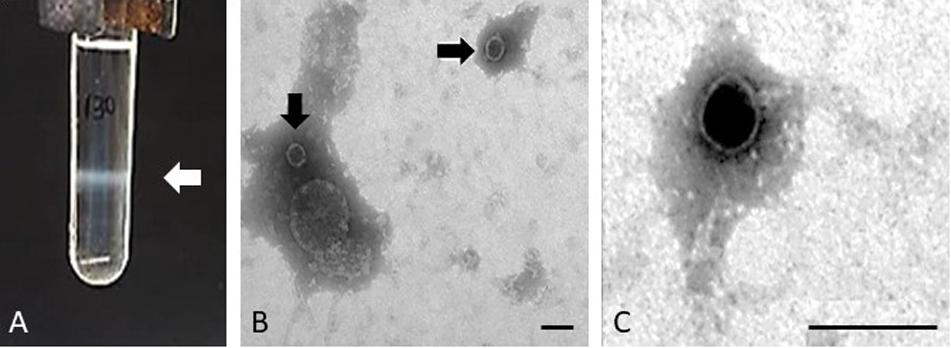
Figure 1. Phage purification. Example of the band comprising the Stx2 phage of isolate 62 and corresponding to a density of 1.5 g/ml in gradient of CsCl (A) and electron micrographs of Stx phage Φ62 (B,C). Bar 100 nm.
Since some strains induce Stx prophages at low rates and in order to increase the volume of each Stx phage suspension to guarantee enough DNA for whole genome sequencing, several bands from different CsCl tubes were collected for each phage, purified and pooled together prior to DNA extraction. Some Stx phages were isolated in enough concentration to be visualized by electron microscopy. Phages ΦEH250, Φ62, Φ63, and Φ72 showed the same morphological type corresponding to phages of icosahedral capsids of 50 ± 3 nm of diameter with very small tails compatible with phages of the Podoviridae, a family of viruses in the order Caudovirales (King et al., 2011; Figures 1B,C).
The whole genome sequencing of the phages produced a number of reads ranging from 42,872 to 105,927 (Table 1). All the sequences produced were of good quality with an average PHRED value per base of 30–32 (Table 1). When the sequences were assembled, most of them yielded one or two contigs of total length > 39,000 bp, approximating the reported dimensions of the Stx phages’ genomes (Krüger and Lucchesi, 2015). The only exception was the genome of the phage ΦEH250, which was assembled into five different short contigs (Table 3). All the contigs produced were characterized by a high coverage values (145X-496X), with the exception of the contigs from the phage ΦEH250 sequence, whose coverage values ranged from 4.5X to 45.09X (Table 3).
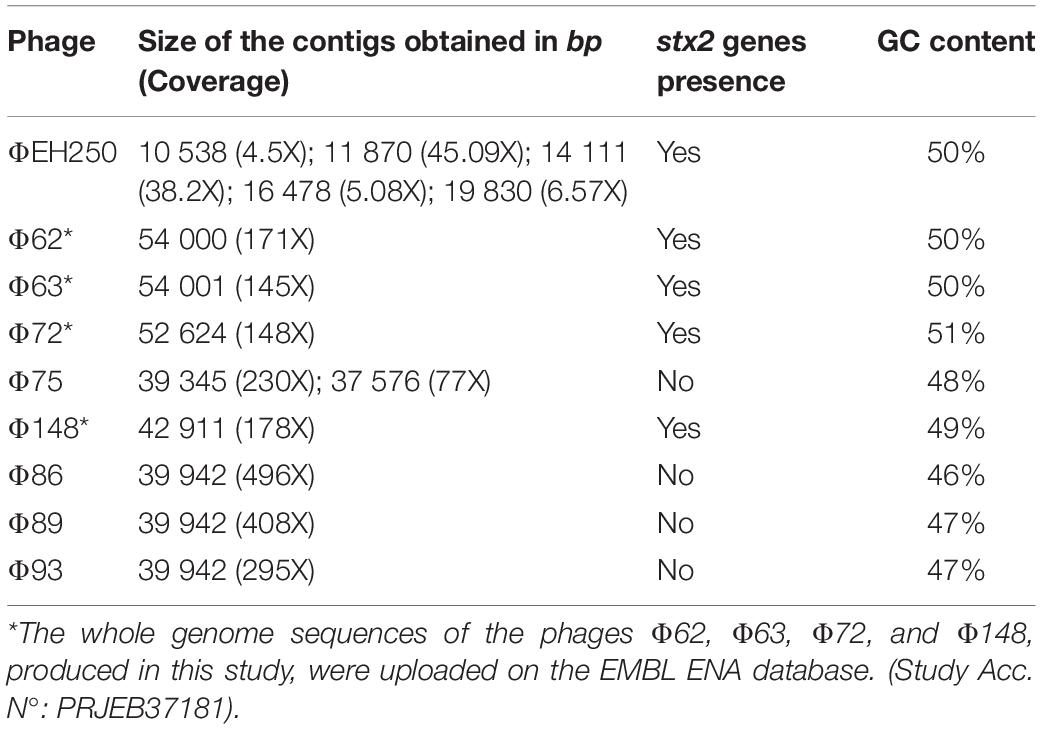
Table 3. Characteristics of the assemblies of the phages’genomes sequenced in this study and identification of the presence of stx2 genes.
The stx gene subtypes carried by the different phages, identified by PCR on the DNA from the STEC strains or on the purified phage DNA (data not shown), were confirmed through the analysis of the draft genomes using the web application “virulence finder”5 for five out of the nine phages studied. The stx genes couldn’t be found in the sequenced genomes of the phages Φ75, Φ86, Φ89, and Φ93 (Table 3), either using the virulence finder application or by manually searching the contigs through the use of the NCBI BLAST+ blastn algorithm. This was probably caused by the sequencing of a phage, which did not possess the Stx2-coding genes that was present in the same CsCl band together with the one encoding the Stx. It is possible that the non-Stx phage was preferentially selected during the propagation of the phages. Interestingly, the phages that did not display the presence of the Stx-coding genes had on average a lower GC content of 46–48% with respect to the phages with the genes coding for the different Stx subtypes, which were all characterized by a GC content comprised between 49 and 51% (Table 3).
The whole genome sequences of the phages Φ62, Φ63, Φ72, and Φ148 were used for the following analyses and uploaded on the database EMBL ENA (Study Accession No. PRJEB37181, Table 3).
The regions of the draft phage genomes containing the Stx-coding genes, located between the phage genes q and s and named hereafter the “stx regions,” were identified and extracted from all the annotated genomes and visualized using the software Snapgene® (v4.1.9) (GSL, Biotech). The genomic structure of these regions was analyzed in more detail (Figure 2). The “stx region” identified in the phage ΦEH250 was not complete, lacking the part comprised between the Stx-coding genes and the s gene, probably due to the problems encountered during the assembly of this phage’s genome (Figure 2B).
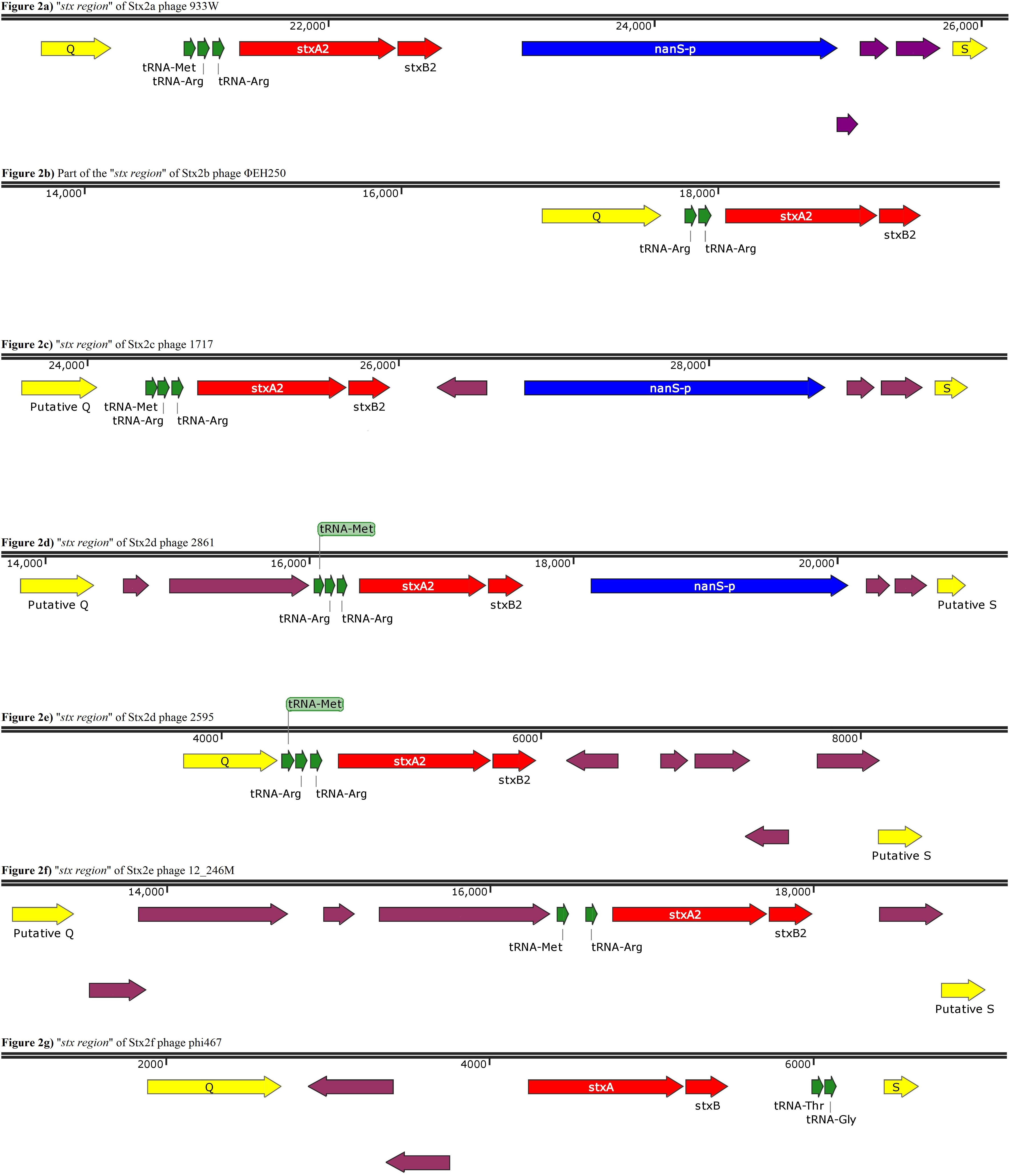
Figure 2. Panels (a) to (g) show the gene organization of gene organization of the “stx region” of Stx2 phages encoding different Stx2 subtypes. Panel (b) shows a contig displaying a partial “stx region” as from this phage an entire region was not obtained. The purple arrows indicate ORFs without an annotated function.
This analysis showed that the structure of the “stx regions” presented a common arrangement of features, which seemed to be associated with the subtype of Stx-coding genes conveyed. In particular, all the “stx regions” investigated presented a set of tRNA-coding genes that varied among the subtypes (Figure 2). These genes were always positioned between the late-phase regulator gene q and the stx genes, except for the Stx2f phage phi467, in which the “stx region” showed an opposite orientation regarding the q and s genes, when compared to the regions of the other phages analyzed, with the tRNA-coding genes placed close to the s gene (Figure 2G). In detail, the “stx region” of the phages encoding the Stx2a, Stx2c, and Stx2d all presented three genes for tRNA methionine, arginine and arginine (Figure 2), except for the phage TL-2011c (Table 2), encoding the Stx2a subtype, which presented the tRNA triplet methionine, proline and arginine (data not shown). The Stx2e phages all presented only two genes coding for the tRNA, encoding the tRNA methionine and arginine (Figure 2F), while the Stx2f phage also presented two of these genes, coding for the tRNA threonine and glycine (Figure 2G). Finally, the Stx2b phage analyzed presented two genes coding both for tRNA arginine (Figure 2B).
The “stx region” of all the Stx2a, Stx2c, and Stx2d phages differed from the regions of the phages encoding the other Stx2 subtypes also for the presence of the nanS-p gene (Figure 2), positioned downstream of the stx genes, with the exception of the Stx2d phage, 2595, which lacked this open reading frame (Figure 2E).
The gene nanS-p is the phage homolog of the bacterial gene nanS, which codes for an esterase involved in the metabolism of the 5-N-acetyl-9-O-acetyl neuraminic acid (Neu5,9Ac2) (Saile et al., 2018).
The multiple sequence alignment analysis of the whole genomes and of the “stx regions” of the phages didn’t include the phages Φ75, Φ86, Φ89, and Φ93 as they didn’t show the presence of the stx genes (Table 3).
The phage ΦEH250 (Table 1) was excluded from the analysis as well. As matter of fact, the assembly of the draft genome of this phage did not yield contigs of sufficient length to be representative of the whole genome and the “stx region” was fragmented into different contigs.
The phylogenetic analyses were conducted by comparing the whole genome sequences of the phages (Figure 3) and their “stx regions” (Figure 4). An additional analysis was carried out considering the presence and the absence of the annotated ORFs together with their GC content (Figure 5). All the dendrograms obtained analyzing the phage genomes showed that the Stx2a and Stx2c phages constituted homogenous populations descending from single ancestors. On the other hand, the Stx2d and Stx2e phages displayed a topology suggesting a relationship between these two populations with both the approaches (Figures 3, 5), although additional analyses of more sequence data would be needed to strengthen this result. Interestingly, both the dendrograms obtained with the whole genome sequences and the presence of the annotated ORFs produced the same topology (Figures 3, 5), with the phage 2236 (Stx2d) interspersed in the clade containing the phages encoding Stx2a and Stx2c, which seemed to originate from a single ancestor (Figures 3, 5). Noteworthy, all the phylogenetic analyses were concordant in placing the Stx2f phage into a single clade distant from all the other phages (Figures 3–5). The comparative analysis of the phage genomes confirmed the phylogenetic analysis and showed extensive variability within the same populations of phages encoding the different Stx2 subtypes, either in term of sequence homology or in the presence of phage’s regions (Figures 6, 7). Interestingly, the phage 2236, encoding Stx2d, was largely different from the other three phages carrying the genes coding for the same Stx2 subtype (Figure 7). This observation is in agreement with what observed with the phylogenetic analyses based either on the identification of differences in the phage sequences through multiple alignment (Figure 3) or on the presence of specific phage genes (Figure 5). This phage was also different from the other Stx2d phages and more similar to the Stx2a phages (Supplementary Figure S1).
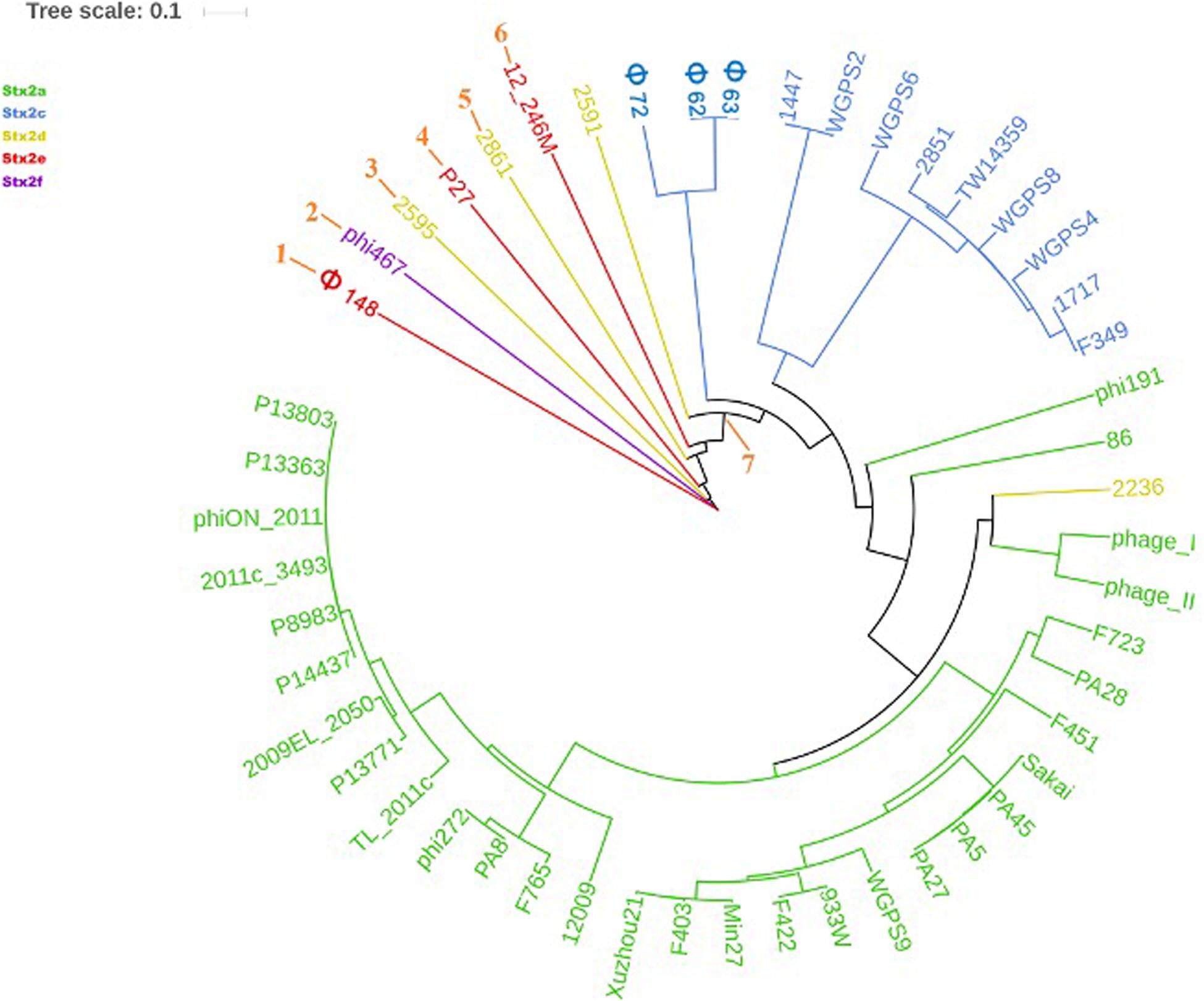
Figure 3. Dendrogram generated from the multiple sequence alignment of the whole genomes of the Stx2 phages with the MAFFT software. The different clades are numbered in orange at the node or the tip of the branch. The Maximum Likelihood tree has been built to display the topology with 95% bootstrap coverage estimated from 1000 repetitions and annotated using the iTOL software.
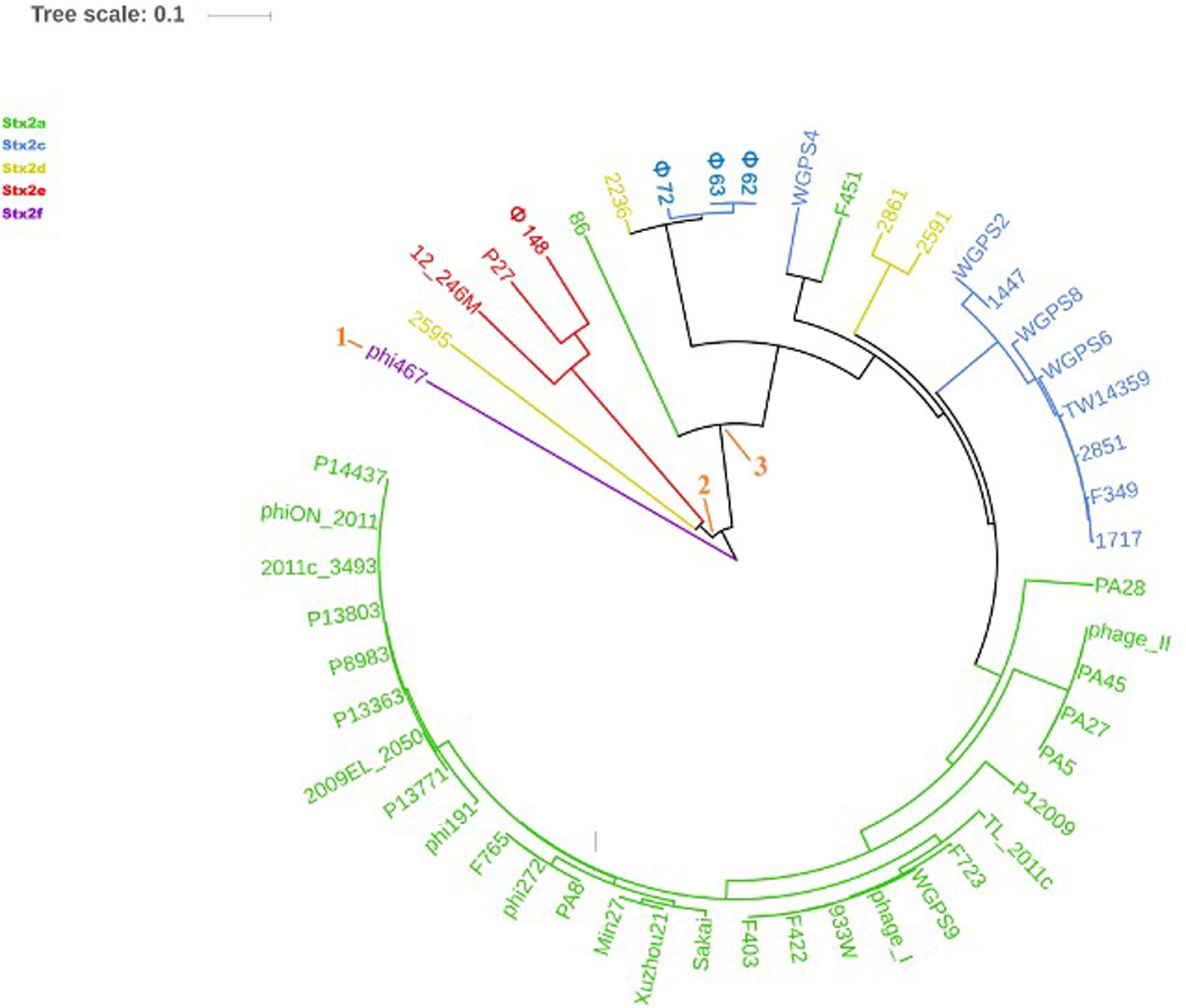
Figure 4. Dendrogram generated from the multiple sequence alignment of the “stx regions” of the Stx2 phages with the MAFFT software. The different clades are numbered in orange at the node or the tip of the branch. The Maximum Likelihood tree has been built to display the topology with 95% bootstrap coverage estimated from 1000 repetitions and annotated using the iTOL software.
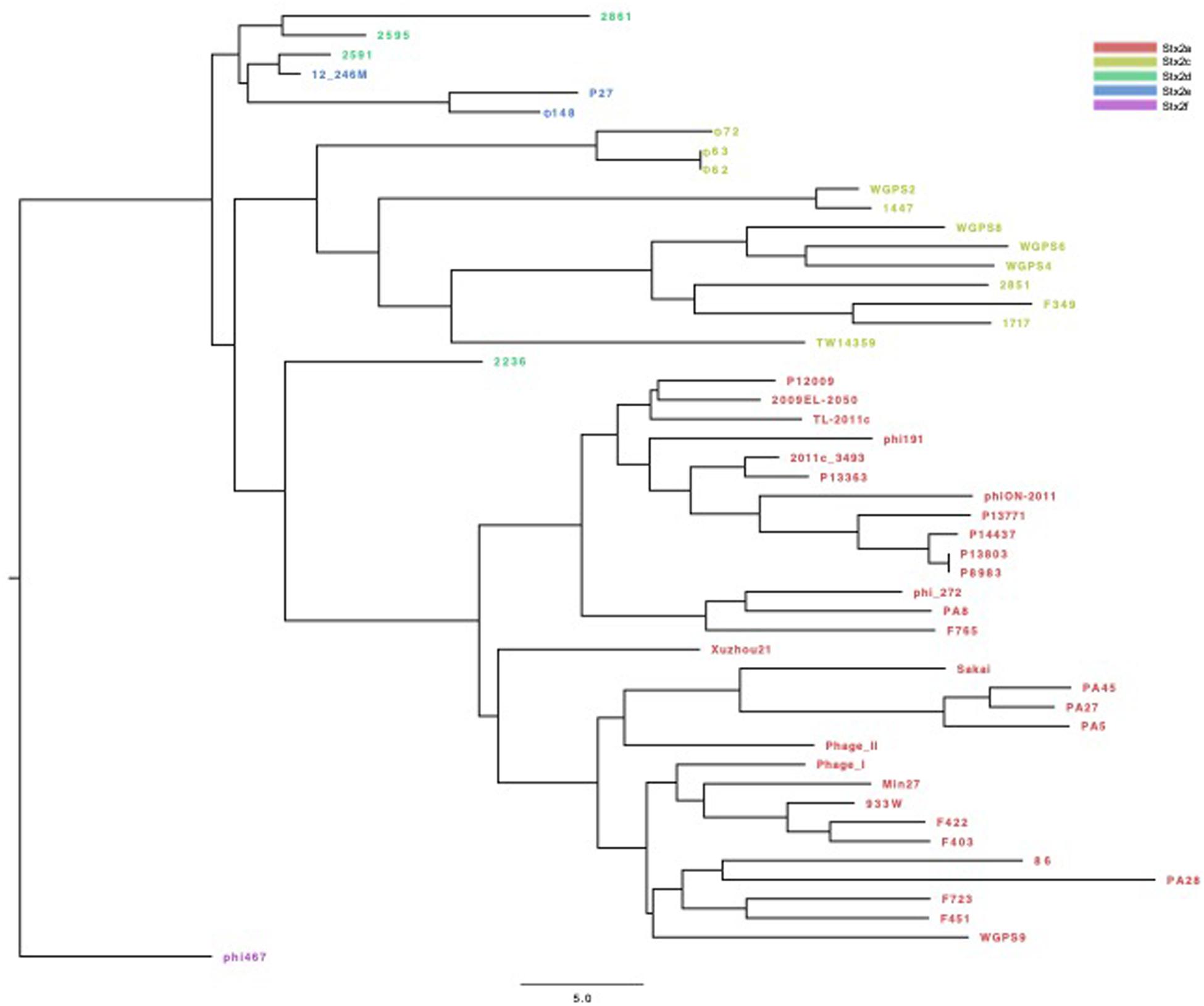
Figure 5. Dendrogram of relationships between the Stx phages’ genomes excluding the “stx regions.” The Neighbor Joining tree was constructed by comparing the presence and the GC content of the annotated ORFs in the different phages. Each ORF with the same annotation was considered as being a different allele when displaying a different GC content. The presence, absence and the different allelic types were all used to compute the Hamming distance. The phages are colored differently according to the Stx2 gene subtypes.
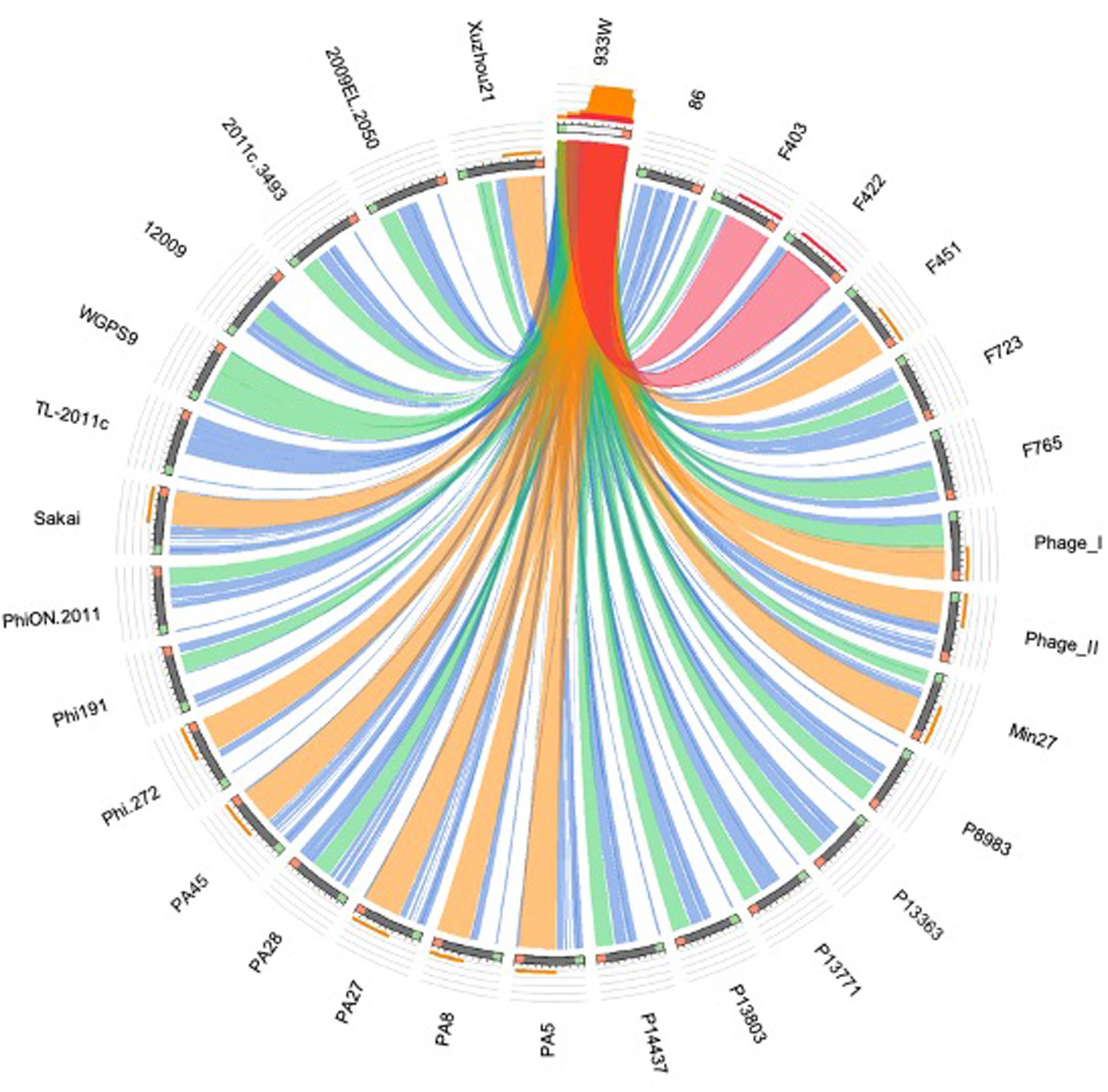
Figure 6. Sequence similarities between the stx-phages carrying the genes encoding the Stx2a subtype. The picture shows the results of the BLAST local alignments using BP933W as a query against all the Stx2a-phage. The colors codes blue, green, orange and red represent the overall quality of the aligned segments along the phage sequences, evaluated on the basis of the bit-score values in the worst-to-the-best order (blue to red, blue ≤ 50%; green ≤ 75%, orange ≤ 99%, and red = 100% identity). The bit-score is a normalized version of the score value returned by the BLAST searches, expressed in bits. The height of the colored bars in the histogram on the top of the BP933W ideogram shows how many times each color hits a specific fragment of the other phage sequences. A twist in a ribbon indicates that the local alignment is inverted (query and database sequence on opposite strands).
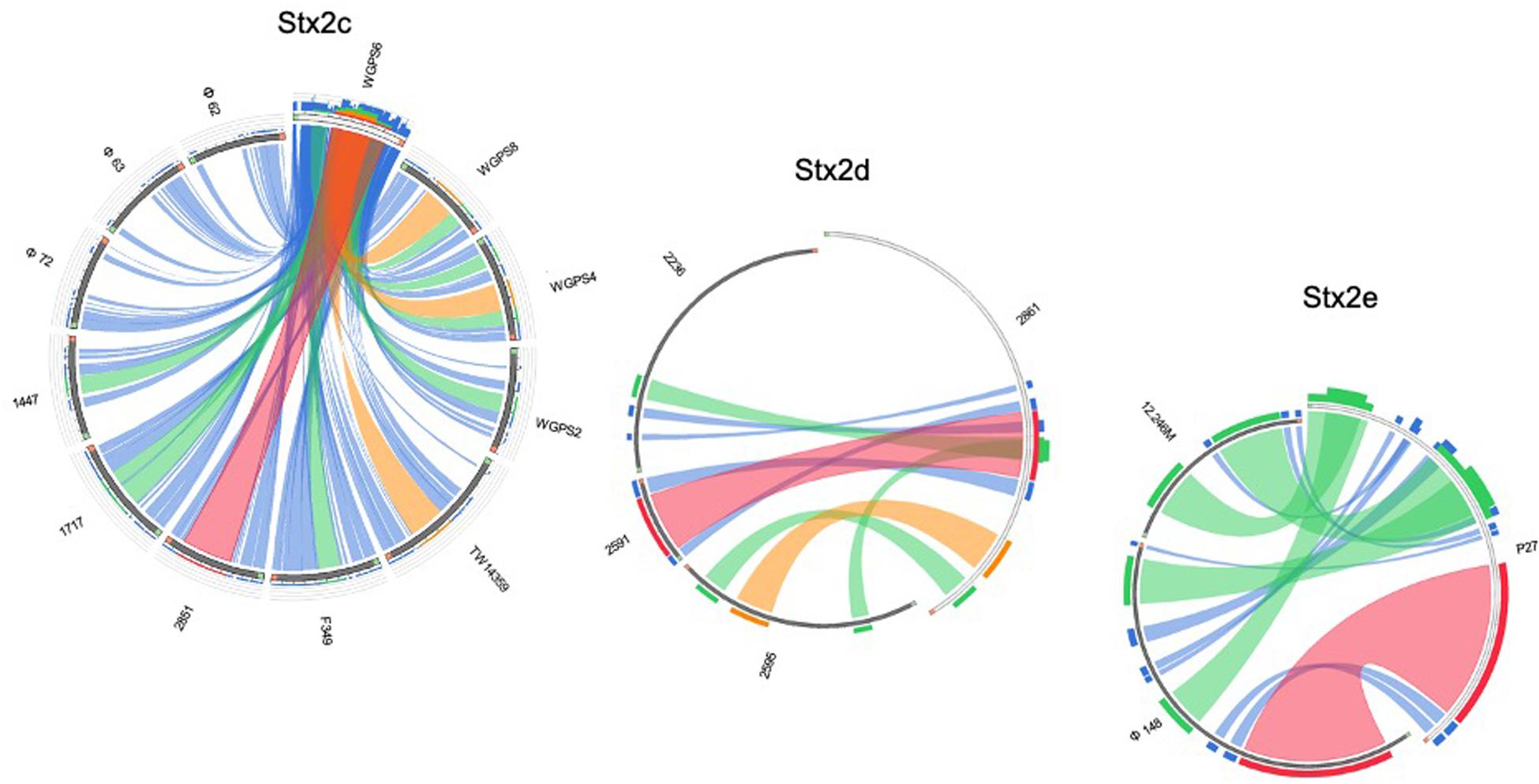
Figure 7. Sequence similarities between the stx-phages carrying the genes encoding the Stx2c, Stx2d, and Stx2e subtypes. The picture shows the results of the BLAST local alignments using WGPS6, 2861, and P27, respectively, as queries against all the Stx2-phages belonging to the related Stx2 subtypes. The colors codes blue, green, orange and red represent the overall quality of the aligned segments along the phage sequences, evaluated on the basis of the bit-score values in the worst-to-the-best order (blue to red, blue ≤ 50%; green ≤ 75%, orange ≤ 99%, and red = 100% identity). The bit-score is a normalized version of the score value returned by the BLAST searches, expressed in bits. The height of the colored bars in the histogram on the top of the query ideograms shows how many times each color hits a specific fragment of the other phage sequences. A twist in a ribbon indicates that the local alignment is inverted (query and database sequence on opposite strands).
The analysis of the “stx regions” carried out in the attempt to obtain information on the source of this DNA, which represents the transduced region of the phage’s genome, showed that the “stx regions” fell into three major clades (Figure 4), one composed, again, by the Stx2f phage phi467 only, one comprising the Stx2e phages and the Stx2d phage 2595, and the last clade including the Stx2a, Stx2c and the remaining Stx2d phages.
The bacteriophages encoding the Shiga toxins (Stx) are a heterogeneous group of lambdoid phages whose members can present different morphologies and variability in the host infectivity range (Krüger and Lucchesi, 2015).
While sharing the presence of the Shiga toxin genes and a few other characteristics, these phages show a considerable genomic diversity (Krüger and Lucchesi, 2015), which has not been thoroughly described, yet. Most of the studies present in the literature were performed with a limited number of phages (Recktenwald and Schmidt, 2002; Smith et al., 2012; Steyert et al., 2012; Tozzoli et al., 2014; Zhang et al., 2018), didn’t distinguish between the stx subtypes (Smith et al., 2007) or investigated the phage population present in STEC strains with respect to other features, such as the effect of stress factors on the induction of the prophages (Zhang et al., 2020).
In this study we meant to investigate the evolution of the phages encoding Stx2 belonging to different subtypes through whole genome sequencing. The choice to restrict this study to the phages encoding the Stx2 was due to the observation that this toxin type is associated with the most severe forms of the infections in humans (Krüger and Lucchesi, 2015). In order to do so, the genomes of four Stx2 phages, purified from STEC strains and sequenced in this study, were compared through multiple sequence alignment to the genomes of 46 other Stx2 phages found in NCBI GenBank. The same approach was used to investigate the structure of the phage regions conveying the Stx2-coding genes, the “stx region,” of all the bacteriophages analyzed.
Alongside other mobile genetic elements, phages are one of the most important engines of bacterial evolution, contributing enormously to their genetic variability through Horizontal Gene Transfer (Javadi et al., 2017). The phages genomes are known to be highly mosaic, being composed by segments with different evolutionary histories, which were acquired by phages through a variety of mechanisms, such as homologous recombination, transposition and site-specific recombination (Hatfull and Hendrix, 2011). Events of genomic rearrangement such as recombination involving regions of the host, as well as regions of other phages, active or cryptic, have been proposed to drive the evolution of the Stx phages, representing an important factor for their diversity (Smith et al., 2012).
The phylogenetic analysis of the whole genomes of the phages investigated in this study confirmed the significant diversity already described among the Stx phages (Zhang et al., 2020), even within the same toxin subtype (Figures 3, 5–7 and Supplementary Figure S1), suggesting that they could, in certain cases, have descended from different ancestors that acquired the Shiga toxin genes in independent transduction events. The phages encoding the Stx2a and Stx2c subtypes seemed to be part of a single lineage deriving from a common ancestor (Figures 3, 5). Interestingly, the same lineage seemed to have originated also at least one of the Stx2d phages, 2236, which shares a higher degree of homology in the sequence and gene content with the Stx2a phages than with those encoding the Stx2d subtype (Figures 3, 5 and Supplementary Figure S1). This observation may indicate that the Stx2d phages population may be the result of multiple events of transduction of the Stx genes operated by different phages. The remaining Stx2d phages seemed to be closer to those carrying the genes encoding the subtype Stx2e. STEC producing this latter Stx2 subtype are almost exclusively found in swine and the genomes of the Stx2e phages shared the same clade with the Stx2d phages 2861, 2595, and 2591 (Figures 3, 5). This finding is not surprising, since the Stx2d phages can be isolated from a variety of different niches besides bovines, including pigs (Baranzoni et al., 2016).
The Stx2f phages are found in E. coli strains whose natural reservoir corresponds to pigeons. These animals have a gastric physiology different from ruminants and it is conceivable that the E. coli part of the pigeon intestinal microbiota may have evolved differently from those found in ruminants. Accordingly, the phylogenetic and structural analyses showed the phage phi467, carrying the Stx2f-coding genes, falling into a clade separated from the others (Figures 3–5). We did not include many Stx2f-encoding phages in this study as they are not available in the public databases for downloading, but previous evidences showed that these phages share a very high degree of similarity (Grande et al., 2016). It is thus conceivable that they could have descended from a common ancestor and possibly remained confined to the pigeon host.
The “stx regions” of the phages investigated showed lower variability compared to the whole genomes and were divided into three clades (Figure 4). These overlapped only partially the topology of the dendrogram obtained through the whole genome analyses, reinforcing the hypothesis of the occurrence of multiple transduction events, with some of the “stx regions” carrying the same Stx2 subtype-coding genes present in phages belonging to different clades, as observed for the Stx2d and, to a lesser extent, Stx2e bacteriophages. This hypothesis may be also supported by the presence of the same tRNA genes in the stx region of phages carrying the same Stx2 subtype. In this respect, the tRNA gene sequences may be considered as a marker for the region captured by the ancestor phages during the excision from the bacterial chromosome and may have been involved in the transduction event.
Interestingly, the three clades observed in the phylogenetic analysis of the “stx regions” reflected, to a certain level, the frequency with which the Stx2 subtypes are encoded by E. coli strains isolated from the animal reservoirs. As a matter of fact, the “stx regions” of the Stx2a, Stx2c and most of the Stx2d phages were included into a single line (line 3, Figure 4). As reported previously, these are the subtypes which are commonly produced by E. coli strains isolated from ruminants, especially bovines.
It is known that prophages can improve the fitness of their bacterial host by carrying genes that can provide an evolutionary advantage, in a phenomenon known as “lysogenic conversion” (Feiner et al., 2015), of which the Shiga toxin could be an example. As a matter of fact, the Stx phages have been proposed to augment the fitness of certain E. coli K12 and clinical isolates of E. coli O157 by conferring protection against grazing protozoan (Steinberg and Levin, 2007). However, this observation has not been extended to other STEC serogroups and other authors have reported the lack of a protective effect provided by the Stx on protozoan predation of E. coli (Schmidt et al., 2016). As a matter of fact, at least in the laboratory conditions, they could not observe a differential decline of the STEC population with respect to other E. coli. Additionally, the same was observed when the grazing experiments were conducted using a wild type STEC and its lysogenic mutant, lacking the Stx phage (Schmidt et al., 2016). These observations make this aspect a gap yet to be filled in the knowledge of the evolution of the STEC strains and their Stx phages. In this respect, the nanS-p gene present in the “stx regions” of certain phages could be an example illustrating the lysogenic conversion. This gene has been identified in this study only in the phages conveying the genes encoding Stx2a, Stx2c and Stx2d subtypes, which are mainly present in E. coli strains isolated from ruminants. As reported previously, the nanS-p gene is a homologous of the gene nanS, located in the E. coli sialoregulon, which encodes an esterase able to mediate the release of 5-acetyl-neuroamminic acid (Neu5Ac) from Neu5,9Ac2 and from bovine submaxillary gland mucin (Saile et al., 2018). Neu5Ac, by disabling the repressor NanR, activates the E. coli sialoregulon, from which it is then utilized to produce pyruvate, used for the tricarboxylic acid cycle (Saile et al., 2018). Saile et al. (2018) suggested that the activity of additional NanS esterase could provide an evolutionary advantage to the bacteria by facilitating the utilization of sialic acids as an ATP source. Therefore, this gene could promote the colonization of the bovine reservoir through the degradation and utilization of bovine submaxillary mucin’s sialic acids.
Interestingly, the STEC strains that carry the phages conveying the subtypes Stx2a, Stx2c and Stx2d are also those most often linked to severe disease, during human infection (Koutsoumanis et al., 2020). Additional to the advantage offered to the E. coli in the ruminants’ gastro-intestinal (GI) tract, the nanS-p could also cause, as a side effect, an improved colonization of the human GI tract, rich of Neu-5,9-Ac2 (Saile et al., 2018), facilitating the insurgence of the most severe forms of the infections in humans. The only exception was represented by the Stx2d phage 2595, which did not show the presence of this gene and whose “stx region” wasn’t included in the same clade as the other Stx2d phages (Figure 4). Stx2d-producing E. coli have been isolated from ruminants (Tasara et al., 2008) and have also been reported in other animal species (Baranzoni et al., 2016). This may account for a wider ecological niche of this group of Stx phages, also explaining the variability observed in the sequence and gene content of these phages and of their “stx regions.”
In conclusion, we showed that the Stx2 phages analyzed in this study presented a considerable variability and that some of them could have originated from different ancestors that acquired the stx genes in multiple transduction events. We make the hypotheses that Stx2a, Stx2c and some Stx2d phages may have derived from the same ancestor and that the “stx regions” could present genetic determinants promoting or facilitating the colonization of specific reservoirs and environments by the bacterial host. As it has been proposed by Saile et al. (2018), nanS-p could be one of these, and this reinforces the hypothesis of a role for the Stx phages in conferring an advantage to the host bacterial strain. However, we cannot exclude that also other determinants present on the phage’s genome outside the “stx region” could have contributed to the adaptation of their bacterial host to specific niches. As a matter of fact, most of the genes present in the different phages studied could not be annotated resulting as hypothetical proteins whose function could not be defined. Therefore, it is certainly possible that the adaptation to the host reservoir could be a multifactorial process implying both genes present on the phage’s genome and the transduced DNA and that nanS-p gene could be part of a more complex asset of determinants influencing the fitness of the bacterial host in the reservoir.
While the comparative approach presented here has limitations linked to the scarce availability of genomes of phages encoding some of the Stx subtypes, at the same time it gives indications that may direct further investigations. Further analyses involving an enlarged panel of Stx phage genomes, including those encoding the more recent Stx2 subtypes and the Stx1 phages, would be beneficial for a better comprehension of the mechanisms underlying the evolution of the Stx phages.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB37181.
MZ performed most of the sequencing and bioinformatic analyses of data and drafted the manuscript. RT contributed to the scientific discussion and particularly in the analysis of the genomic data and to the critical revision of the manuscript. PC prepared and checked the bacterial cultures and performed the Real Time PCR experiments. PQ and AM-V performed the phages’ induction, amplification, and purification. VM contributed to the bioinformatic analyses and to the critical revision of the manuscript. MM supervised all the phages purification activities and actively contributed to the scientific discussion and to the critical revision of the manuscript. SM conceived the study, contributed to the scientific discussion and thoroughly revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was partially supported by funds from the Spanish Ministerio de Innovación y Ciencia (AGL2016-75536-P), the Agencia Estatal de Investigación (AEI), the European Regional Fund (ERF), and intramural research funds from the Istituto Superiore di Sanità.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Dr. Marco Crescenzi, Fiorella Ciaffoni, and Manuela Marra from the ISS Core Facilities Technical-Scientific Service for the Next Generation Sequencing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01472/full#supplementary-material
FIGURE S1 | Illustrates the relationships between the Stx2d and Stx2a phages through the Circoletto software alignment of the respective phage sequences.
TABLE S1 | Includes the analysis on the presence or absence of the genes with an annotated function found in all the phages’ genomes investigated.
Akiyama, Y., Futai, H., Saito, E., Ogita, K., Sakae, H., Fukunaga, M., et al. (2017). Shiga toxin subtypes and virulence genes in Escherichia coli isolated from cattle. Jpn. J. Infect. Dis. 70, 181–185. doi: 10.7883/yoken.JJID.2016.100
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bai, X., Fu, S., Zhang, J., Fan, R., Xu, Y., Sun, H., et al. (2018). Identification and pathogenomic analysis of an Escherichia coli strain producing a novel Shiga toxin 2 subtype. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-25233-x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baranzoni, G. M., Fratamico, P. M., Gangiredla, J., Patel, I., Bagi, L. K., Delannoy, S., et al. (2016). Characterization of shiga toxin subtypes and virulence genes in porcine shiga toxin-producing Escherichia coli. Science 7, 1–10. doi: 10.3389/fmicb.2016.00574
Bryan, A., Youngster, I., and McAdam, A. J. (2015). Shiga toxin producing Escherichia coli. Clin. Lab. Med. 35, 247–272. doi: 10.1016/j.cll.2015.02.004
Bullock, J. M., Fernandez, J. M., and Short, W. O. (1987). XL1Blue, a high efficiency\rplasmid transforming recA Escherichia coli Strain with b-Galactosidase selection. Biotechniques 5, 376–378.
Caprioli, A., Morabito, S., and Brugère, H. O. E. (2005). Enterohaemorrhagic Escherichia coli: emerging issues on virulence and modes of transmission. Vet. Res. 36, 289–311. doi: 10.1051/vetres:2005002
Cha, W., Fratamico, P. M., Ruth, L. E., Bowman, A. S., Nolting, J. M., Manning, S. D., et al. (2018). Prevalence and characteristics of Shiga toxin-producing Escherichia coli in finishing pigs: implications on public health. Int. J. Food Microbiol. 264, 8–15. doi: 10.1016/j.ijfoodmicro.2017.10.017
Cuccuru, G., Orsini, M., Pinna, A., Sbardellati, A., Soranzo, N., Travaglione, A., et al. (2014). Orione, a web-based framework for NGS analysis in microbiology. Bioinformatics 30, 1928–1929. doi: 10.1093/bioinformatics/btu135
Darzentas, N. (2010). Circoletto: visualizing sequence similarity with circos. Bioinformatics 26, 2620–2621. doi: 10.1093/bioinformatics/btq484
Dehdashti, S., Ghanbarpour, R., and Hajikolaei, M. R. H. (2019). Molecular detection of Shiga toxin-producing and antibiotic-resistant Escherichia coli isolates from buffaloes in southwest of Iran. Trop. Anim. Health Prod. 51, 1725–1736. doi: 10.1007/s11250-019-01869-3
Feijao, P., Yao, H. T., Fornika, D., Gardy, J., Hsiao, W., Chauve, C., et al. (2018). MentaLiST - A fast MLST caller for large MLST schemes. Microb. Genomi. 4, 1–8. doi: 10.1099/mgen.0.000146
Feiner, R., Argov, T., Rabinovich, L., Sigal, N., Borovok, I., and Herskovits, A. A. (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 13, 641–650. doi: 10.1038/nrmicro3527
Fuller, C. A., Pellino, C. A., Flagler, M. J., Strasser, J. E., and Weiss, A. A. (2011). Shiga toxin subtypes display dramatic differences in potency. Infect. Immun. 79, 1329–1337. doi: 10.1128/IAI.01182-10
Grande, L., Michelacci, V., Bondì, R., Gigliucci, F., Franz, E., Badouei, M. A., et al. (2016). Whole-genome characterization and strain comparison of VT2f- producing Escherichia coli causing hemolytic uremic syndrome. Emerg. Infect. Dis. 22. 2078–2086. doi: 10.3201/eid2212.160017
Hatfull, G. F., and Hendrix, R. W. (2011). Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303. doi: 10.1016/j.coviro.2011.06.009
Huang, A., Friesen, J., and Brunton, J. L. (1987). Characterization of a bacteriophage that carries the genes for production of Shiga-like toxin 1 in Escherichia coli. J. Bacteriol. 169, 4308–4312. doi: 10.1128/jb.169.9.4308-4312.1987
Jajarmi, M., Imani Fooladi, A. A., Badouei, M. A., and Ahmadi, A. (2017). Virulence genes, Shiga toxin subtypes, major O-serogroups, and phylogenetic background of Shiga toxin-producing Escherichia coli strains isolated from cattle in Iran. Microb. Pathog. 109, 274–279. doi: 10.1016/j.micpath.2017.05.041
Januszkiewicz, A., and Rastawicki, W. (2016). Molecular characterization of Shiga toxin-producing Escherichia coli strains isolated in Poland. Polish J. Microbiol. 65, 261–269. doi: 10.5604/17331331.1215601
Javadi, M., Bouzari, S., and Oloomi, M. (2017). “Horizontal gene transfer and the diversity of Escherichia coli,” in Recent Advances on Physiology, Pathogenesis and Biotechnological Applications, ed. A. Samie (London: IntechOpen), doi: 10.5772/intechopen.68307
Karmali, M. A. (2018). Factors in the emergence of serious human infections associated with highly pathogenic strains of shiga toxin-producing Escherichia coli. Int. J. Med. Microbiol. 308, 1067–1072. doi: 10.1016/j.ijmm.2018.08.005
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
King, A. M. Q., Lefkowitz, E., Adams, M. J., and Carstens, E. B. (2011). Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. Amsterdam: Elsevier.
Knijn, A., Michelacci, V., Orsini, M., and Morabito, S. (2020). Advanced research infrastructure for experimentation in genomicS (ARIES): a lustrum of Galaxy experience. bioRxiv [Preprint], doi: 10.1101/2020.05.14.095901
Koutsoumanis, K., Allende, A., Alvarez-Ordóñez, A., Bover-Cid, S., Chemaly, M., Davies, R., et al. (2020). Pathogenicity assessment of Shiga toxin-producing Escherichia coli (STEC) and the public health risk posed by contamination of food with STEC. EFSA J. 18:e05967. doi: 10.2903/j.efsa.2020.5967
Krüger, A., and Lucchesi, P. M. A. (2015). Shiga toxins and stx phages: highly diverse entities. Microbiology 161, 1–12. doi: 10.1099/mic.0.000003
Krüger, A., Lucchesi, P. M. A., and Parma, A. E. (2011). Verotoxins in bovine and meat verotoxin-producing Escherichia coli isolates: type, number of variants, and relationship to cytotoxicity. Appl. Environ. Microbiol. 77, 73–79. doi: 10.1128/AEM.01445-10
Lacher, D. W., Gangiredla, J., Patel, I., Elkins, C. A., and Feng, P. C. H. (2016). Use of the Escherichia coli identification microarray for characterizing the health risks of Shiga toxin-producing Escherichia coli isolated from foods. J. Food Prot. 79, 1656–1662. doi: 10.4315/0362-028X.JFP-16-176
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Melton-Celsa, A. R. (2014). Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2, 1–21. doi: 10.1128/microbiolspec.ehec-0024-2013
Michelacci, V., Maugliani, A., Tozzoli, R., Corteselli, G., Chiani, P., Minelli, F., et al. (2018). Characterization of a novel plasmid encoding F4-like fimbriae present in a Shiga-toxin producing enterotoxigenic Escherichia coli isolated during the investigation on a case of hemolytic-uremic syndrome. Int. J. Med. Microbiol. 308, 947–955. doi: 10.1016/j.ijmm.2018.07.002
Mughini-Gras, L., van Pelt, W., van der Voort, M., Heck, M., Friesema, I., and Franz, E. (2018). Attribution of human infections with Shiga toxin-producing Escherichia coli (STEC) to livestock sources and identification of source-specific risk factors, The Netherlands (2010-2014). Zoon. Public Health 65, e8–e22. doi: 10.1111/zph.12403
Muniesa, M., and Jofre, J. (1998). Abundance in sewage of bacteriophages that infect Escherichia coli O157:H7 and that carry the Shiga toxin 2 gene. Appl. Environ. Microbiol. 64, 2443–2448. doi: 10.1128/aem.64.7.2443-2448.1998
Navarro-Garcia, F. (2014). Escherichia coli O104: H4 pathogenesis: an enteroaggregative E. coli / Shiga toxin-producing E. coli explosive cocktail of high virulence. Microbiol Spectr 2, 1–19.
O’Brien, A. D., Newland, J. W., Holmes, R. K., Smith, H. W., and Formal, S. B. (1983). Shiga-like toxin-converting phages from Eschenrchia coli. Science 226, 694–696. doi: 10.1126/science.6387911
Rahman, M., Nabi, A., Asadulghani, M., Faruque, S. M., and Islam, M. A. (2018). Toxigenic properties and stx phage characterization of Escherichia coli O157 isolated from animal sources in a developing country setting. BMC Microbiol. 18:98. doi: 10.1186/s12866-018-1235-3
Recktenwald, J., and Schmidt, H. (2002). The nucleotide sequence of Shiga toxin (Stx) 2e-encoding phage φP27 is not related to other Stx phage genomes, but the modular genetic structure is conserved. Infect. Immun. 70, 1896–1908. doi: 10.1128/IAI.70.4.1896-1908.2002
Saile, N., Schwarz, L., Eißenberger, K., Klumpp, J., Fricke, F. W., and Schmidt, H. (2018). Growth advantage of Escherichia coli O104:H4 strains on 5-N-acetyl-9-O-acetyl neuraminic acid as a carbon source is dependent on heterogeneous phage-Borne nanS-p esterases. Int. J. Med. Microbiol. 308, 459–468. doi: 10.1016/j.ijmm.2018.03.006
Sambrook, J., and Russel, D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd Edn, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Schmidt, C. E., Shringi, S., and Besser, T. E. (2016). Protozoan predation of Escherichia coli O157: H7 is unaffected by the carriage of shiga toxin-encoding bacteriophages. PLoS One 11:e0147270. doi: 10.1371/journal.pone.0147270
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Smith, D. L., Rooks, D. J., Fogg, P. C. M., Darby, A. C., Thomson, N. R., Mccarthy, A. J., et al. (2012). Comparative genomics of shiga toxin encoding bacteriophages. BMC Genomics 13:311. doi: 10.1186/1471-2164-13-311
Smith, D. L., Wareing, B. M., Fogg, P. C. M., Riley, L. M., Spencer, M., Cox, M. J., et al. (2007). Multilocus characterization scheme for shiga toxin-encoding bacteriophages. Appl. Environ. Microbiol. 73, 8032–8040. doi: 10.1128/AEM.01278-07
Steinberg, K. M., and Levin, B. R. (2007). Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc. R. Soc. B Biol. Sci. 274, 1921–1929. doi: 10.1098/rspb.2007.0245
Steyert, S. R., Sahl, J. W., Fraser, C. M., Teel, L. D., Scheutz, F., and Rasko, D. A. (2012). Comparative genomics and stx phage characterization of LEE-negative Shiga toxin-producing Escherichia coli. Front. Cell Infect. Microbiol. 2:133. doi: 10.3389/fcimb.2012.00133
Tasara, T., Bielaszewska, M., Nitzsche, S., Karch, H., Zweifel, C., and Stephan, R. (2008). Activatable Shiga toxin 2d (Stx2d) in STEC strains isolated from cattle and sheep at slaughter. Vet. Microbiol. 131, 199–204. doi: 10.1016/j.vetmic.2008.03.001
Tostes, R., Goji, N., Amoako, K., Chui, L., Kastelic, J., Devinney, R., et al. (2017). Subtyping Escherichia coli virulence genes isolated from feces of beef cattle and clinical cases in alberta. Foodborne Pathog. Dis. 14, 35–42. doi: 10.1089/fpd.2016.2199
Tozzoli, R., Grande, L., Michelacci, V., Fioravanti, R., Gally, D., Xu, X., et al. (2014). Identification and characterization of a peculiar vtx2-converting phage frequently present in verocytotoxin-producing Escherichia coli O157 isolated from human infections. Infect. Immun. 82, 3023–3032. doi: 10.1128/IAI.01836-14
Van Hoek, A. H. A. M., Van Veldhuizen, J. N. J., Friesema, I., Coipan, C., Rossen, J. W. A., Bergval, I. L., et al. (2019). Comparative genomics reveals a lack of evidence for pigeons as a main source of stx 2f -carrying Escherichia coli causing disease in humans and the common existence of hybrid Shiga toxin-producing and enteropathogenic E. coli pathotypes. BMC Genomics 20:35. doi: 10.1186/s12864-019-5635-z
Yang, X., Bai, X., Zhang, J., Sun, H., Fu, S., Fan, R., et al. (2020). Escherichia coli strains producing a novel Shiga toxin 2 subtype circulate in China. Int. J. Med. Microbiol. 310:151377. doi: 10.1016/j.ijmm.2019.151377
Zhang, L. X., Simpson, D. J., McMullen, L. M., and Gänzle, M. G. (2018). Comparative genomics and characterization of the late promoter pR’ from shiga toxin prophages in Escherichia coli. Viruses 10:595. doi: 10.3390/v10110595
Keywords: Shiga toxin, phages, Stx2, ecology, evolution, WGS
Citation: Zuppi M, Tozzoli R, Chiani P, Quiros P, Martinez-Velazquez A, Michelacci V, Muniesa M and Morabito S (2020) Investigation on the Evolution of Shiga Toxin-Converting Phages Based on Whole Genome Sequencing. Front. Microbiol. 11:1472. doi: 10.3389/fmicb.2020.01472
Received: 27 March 2020; Accepted: 05 June 2020;
Published: 10 July 2020.
Edited by:
Andrew S. Lang, Memorial University of Newfoundland, CanadaReviewed by:
Paul Fogg, University of York, United KingdomCopyright © 2020 Zuppi, Tozzoli, Chiani, Quiros, Martinez-Velazquez, Michelacci, Muniesa and Morabito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosangela Tozzoli, cm9zYW5nZWxhLnRvenpvbGlAaXNzLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.