 Roland T. Ashford
Roland T. Ashford Jakub Muchowski
Jakub Muchowski Mark Koylass
Mark Koylass Holger C. Scholz
Holger C. Scholz Adrian M. Whatmore
Adrian M. Whatmore- 1Department of Bacteriology, Animal and Plant Health Agency, Weybridge, United Kingdom
- 2Department of Bacteriology and Toxinology, Bundeswehr Institute of Microbiology, Munich, Germany
The bacterial family Brucellaceae is currently composed of seven genera, including species of the genus Brucella, a number of which are significant veterinary and zoonotic pathogens. The bacteriological identification of pathogenic Brucella spp. may be hindered by their close phenotypic similarity to other members of the Brucellaceae, particularly of the genus Ochrobactrum. Additionally, a number of novel atypical Brucella taxa have recently been identified, which exhibit greater genetic diversity than observed within the previously described species, and which share genomic features with organisms outside of the genus. Furthermore, previous work has indicated that the genus Ochrobactrum is polyphyletic, raising further questions regarding the relationship between the genus Brucella and wider Brucellaceae. We have applied whole genome sequencing (WGS) and pan-family multi-locus sequence analysis (MLSA) approaches to a comprehensive panel of Brucellaceae type strains, in order to characterize relationships within the family. Phylogenies based on WGS core genome alignments were able to resolve phylogenetic relationships of 31 non-Brucella spp. type strains from within the family, alongside type strains of twelve Brucella species. A phylogeny based on concatenated pan-family MLSA data was largely consistent with WGS based analyses. Notably, recently described atypical Brucella isolates were consistently placed in a single clade with existing species, clearly distinct from all members of the genus Ochrobactrum and wider family. Both WGS and MLSA methods closely grouped Brucella spp. with a sub-set of Ochrobactrum species. However, results also confirmed that the genus Ochrobactrum is polyphyletic, with seven species forming a separate grouping. The pan-family MLSA scheme was subsequently applied to a panel of 50 field strains of the family Brucellaceae, isolated from a wide variety of sources. This analysis confirmed the utility of the pan-Brucellaceae MLSA scheme in placing field isolates in relation to recognized type strains. However, a significant number of these isolates did not cluster with currently identified type strains, suggesting the existence of additional taxonomic diversity within some members of the Brucellaceae. The WGS and pan-family MLSA approaches applied here provide valuable tools for resolving the identity and phylogenetic relationships of isolates from an expanding bacterial family containing a number of important pathogens.
Introduction
The bacterial family Brucellaceae (class Alphaproteobacteria, order Rhizobiales) is currently comprised of seven genera; Brucella, Daeguia, Falsochrobactrum, Mycoplana, Ochrobactrum, Paenochrobactrum, and Pseudochrobactrum (Kämpfer and Glaeser, 2019). The family contains species with a wide range of habitat or host preferences, encompassing obligate intracellular pathogens of animals (e.g., Brucella melitensis), opportunistic pathogens often associated with nosocomial infections (e.g., Ochrobactrum anthropi), plant associated pathogens and symbionts (e.g., O. lupini) and organisms isolated from the natural and anthropogenic environment (e.g., Paenochrobactrum glaciei and Pseudochrobactrum lubricantis, respectively).
The type genus Brucella (Scholz et al., 2018) contains the causative agents of brucellosis, which remains one of the most important zoonotic diseases globally, with more than 500,000 new cases reported each year (Pappas et al., 2006). For several decades the genus Brucella was described as consisting of six “classical” species (B. abortus, B. melitensis, B. ovis, B. suis, B. canis, and B. neotomae), with well characterized mammalian host preferences (Corbel, 2006). Subsequently, additional species have been described, with the genus expanding to include B. pinnipedialis and B. ceti isolated from marine mammals (Foster et al., 2007), B. microti from voles (Scholz et al., 2008b), B. inopinata isolated from a human infection (Scholz et al., 2010), B. papionis from baboons (Whatmore et al., 2014) and B. vulpis, isolated from foxes (Scholz et al., 2016a). Additionally, a number of novel atypical strains from human and other mammalian hosts have been described, but await formal classification (Tiller et al., 2010a, b; Guzmán-Verri et al., 2019). These species and strains reflect an ongoing expansion of the known host range and genetic diversity of the genus. In particular, B. microti, B. inopinata and B. vulpis have been described as “atypical” Brucella species, exhibiting either atypical phenotypic traits (B. microti), or greater genetic diversity (B. inopinata and B. vulpis).
In addition to the novel atypical Brucella species and strains identified from mammalian hosts there is an expanding body of literature describing the isolation of Brucella sp. organisms from poikilothermic hosts, namely amphibians (Eisenberg et al., 2012; Whatmore et al., 2015; Scholz et al., 2016b; Soler-Lloréns et al., 2016) and cartilaginous fish (Eisenberg et al., 2017). In many cases, these isolates have initially been misidentified as Ochrobactrum anthropi on the basis of automated phenotyping or mass spectrometry (Eisenberg et al., 2017). However, further genetic investigation has identified the isolates as representing distinct Brucella lineages (Scholz et al., 2016b). Such isolates fall outside of the core Brucella, exhibiting greatest similarity to previously described atypical isolates from mammals (B. inopinata and B. vulpis). Analyses based on whole genome sequencing (WGS) have indicated that, whilst such amphibian isolates maintain a high degree of genetic homology with core Brucella species, they exhibit a degree of horizontal gene transfer, with the incorporation of genomic regions exhibiting sequence identity to soil living or facultatively pathogenic Alphaproteobacteria, most notably from the genus Ochrobactrum (Scholz et al., 2016b; Al Dahouk et al., 2017).
The genus Ochrobactrum, is the largest within the family Brucellaceae, and has expanded significantly in the past decade (Kämpfer et al., 2018). The genus currently consists of 18 validly published species, plus a number of others awaiting valid publication. It has previously been noted that the genus Ochrobactrum appears to be polyphyletic (Kämpfer et al., 2013). Furthermore, studies based on sequencing of single locus targets (most commonly 16S ribosomal RNA or recA), have generated conflicting results on phylogenetic relationships within the family.
These issues highlight the well described problems of inferring phylogenetic relationships from single genetic loci, including lack of resolution, stochastic variation and the influence of recombination or horizontal gene transfer (Gevers et al., 2005). Previously, few studies had attempted to apply a multi-locus analysis approach to interrogate phylogenetic relationships within the family Brucellaceae. Aujoulat et al. (2014) applied an existing multi-locus sequence typing scheme for Ochrobactrum anthropi (Romano et al., 2009) to a wider panel of type strains from 14 Ochrobactrum species and two Brucella species. These authors again identified the genus Ochrobactrum as polyphyletic, with a robust clade containing O. anthropi and O. intermedium and five other species grouping distinctly from a less well supported clade containing the remaining Ochrobactrum type strains and the two Brucella species included. However, this analysis did not incorporate the full diversity of either Ochrobactrum or Brucella, or the remaining five genera which make up the family.
More recently, a number of studies have combined multi-locus sequencing and whole genome sequencing (WGS) based approaches to address specific taxonomic issues within the genus Ochrobactrum. Gazolla Volpiano et al. (2019) for example, identified O. lupini as a heterotypic synonym of O. anthropi. Similarly, Krzyzanowska et al. (2019) used a combination of these approaches to investigate the relationship of a proposed novel Ochrobactrum species (O. quorumnocens) to previously described members of the genus.
Very recently, Leclercq et al. (2019) used WGS data to investigate relationships within the family Brucellaceae, as well as its position within the wider Rhizobiales. These authors confirmed the genus Brucella to be a monophyletic grouping within the genus Ochrobactrum, and that Ochrobactrum spp. divide into two distinct clades. However, Leclercq et al. (2019) did not incorporate type strains reflecting the full taxonomic diversity of either Ochrobactrum or Brucella. Additionally, the remaining genera within the Brucellaceae were either under-represented (Pseudochrobactrum and Mycoplana) or absent (Paenochrobactrum and Daeguia).
The continuing expansion of the recognized genetic diversity of the genus Brucella, and the uncertainty regarding phylogenetic relationships amongst members of related genera, highlight the need for a comprehensive multi-gene phylogeny of the Brucellaceae. Here, we describe the parallel application of WGS and multi-locus sequence analysis (MLSA) approaches to investigate phylogenetic relationships within the family. The pan-family MLSA method developed incorporates loci identical those used in existing schemes for Brucella species (Whatmore et al., 2007, 2016) thereby permitting the comparison of data generated under both schemes. Subsequently, we have applied the pan-family MLSA scheme to panels of field isolates, to assess its utility in establishing relationships with extant species.
Materials and Methods
Type Strains
The taxonomic status of species within the Brucellaceae was retrieved via the online resource List of Prokaryotic Names with Standing in Nomenclature (Euzeby, 1997; Parte, 2014 accessed 14/11/2019). The type strains of all Brucella species were available from the collection of the APHA Brucella Reference Laboratory. The type strains of other species identified as belonging to the family Brucellaceae were purchased from established culture collections (Table 1). Strains were grown on solid media at optimal conditions as described by the supplier. A single pure colony from each agar plate was transferred to 100 μl of molecular grade water and cells were lysed by heating at 100°C for 10 min. Type strain 16S rRNA sequences were retrieved from the National Centre for Biotechnology Information (NCBI) database.
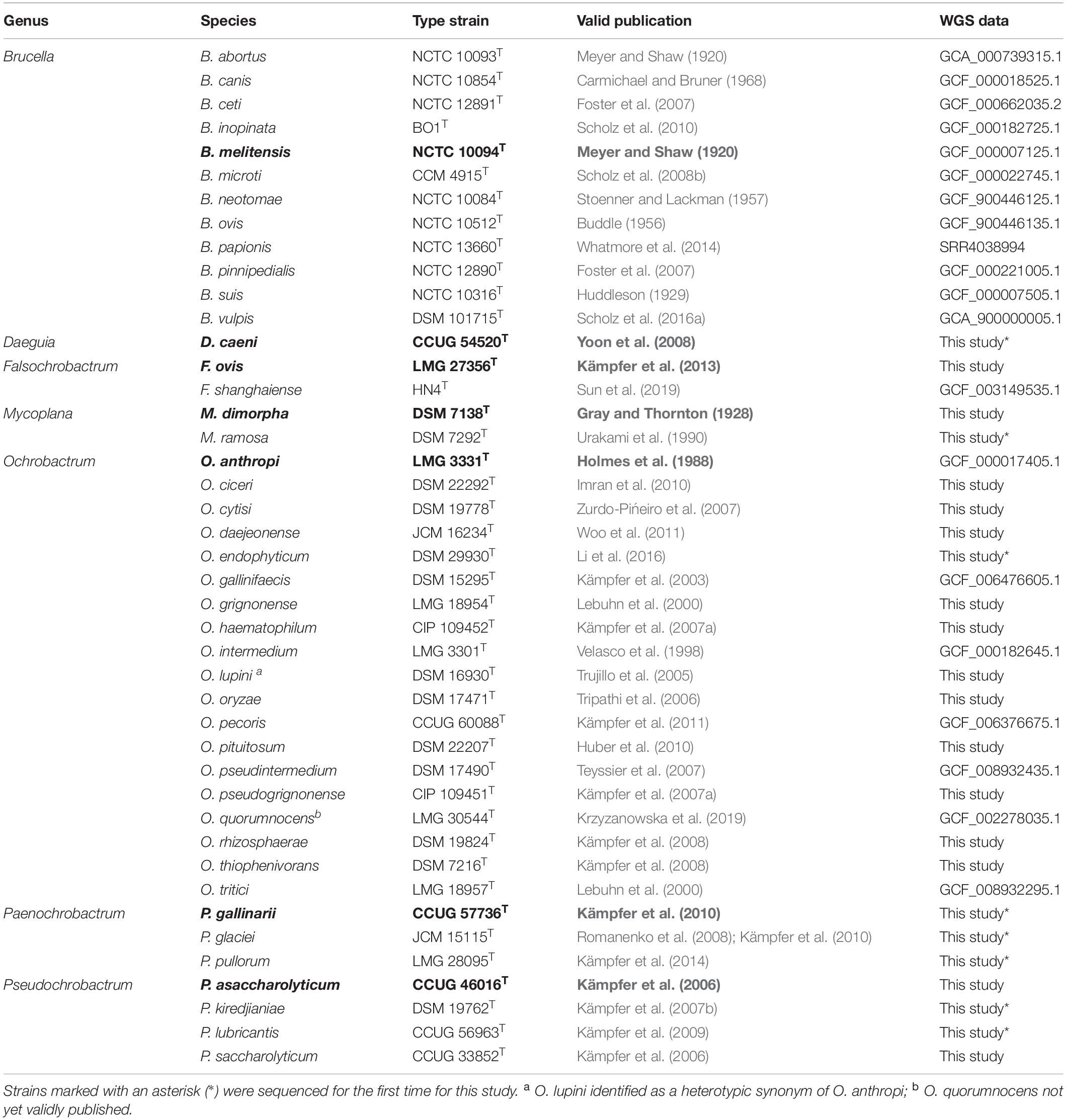
Table 1. Type strains of species belonging to the Brucellaceae, used for whole genome sequence analysis and pan-family multi-locus sequence analysis (genus type species shown in bold).
Additional Atypical Brucella sp. Strains
In addition to Brucellaceae type strains, atypical Brucella sp. from both mammalian and cold-blooded hosts were included in phylogenetic analyses (Table 2). These included previously described atypical isolates from a human patient (BO2: Tiller et al., 2010b) and from Australian rodents (Tiller et al., 2010a). These latter strains (referred to here as 83/13 and 83/4) were originally isolated as part of the same cohort as the Australian rodent strain NF2653 (Cook et al., 1966). Additionally, a number of more recently described atypical isolates from amphibians were analyzed. Isolate UK8/14 was recovered from a White’s tree frog (Whatmore et al., 2015). Six African bullfrog strains from the 21 previously analyzed by Scholz et al. (2016b) were also selected, on the basis of representing all sequence types from this panel in the existing Brucella MLSA scheme (09RB8471: ST63; 09RB8910: ST64; 09RB8913; ST65; 10RB9205: ST66; 10RB9215: ST67 and 10RB9213: ST68 (Al Dahouk et al., 2017).
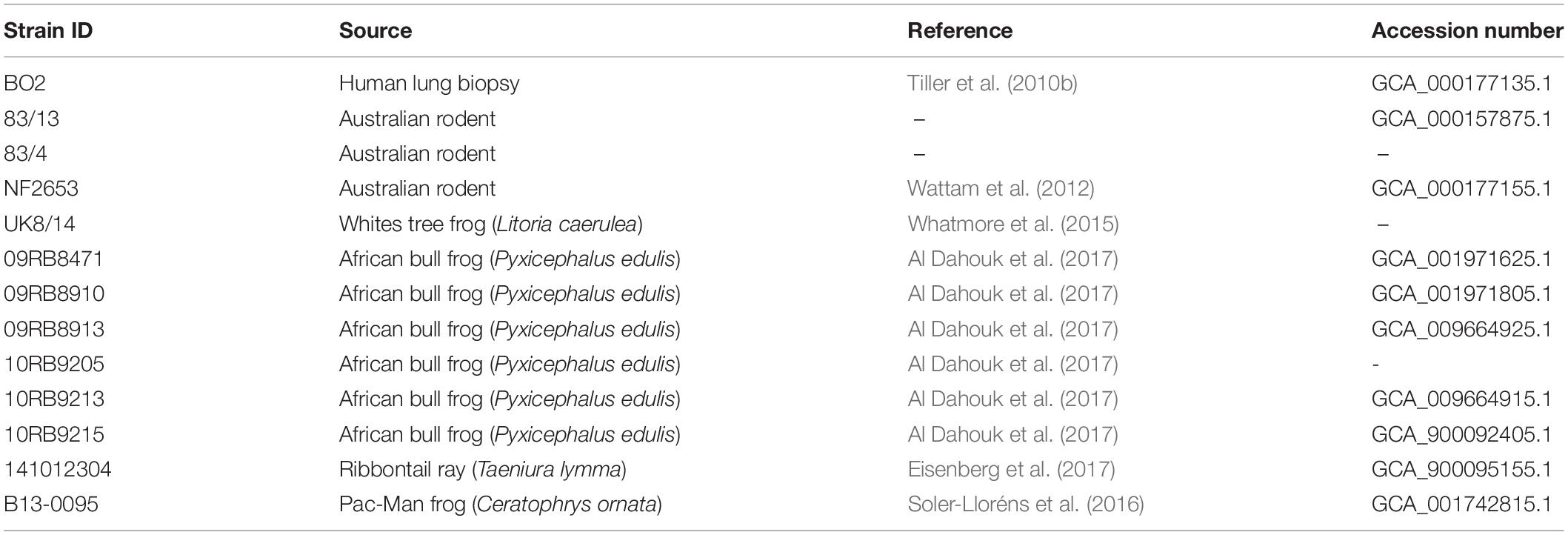
Table 2. Atypical Brucella sp. strains used for whole genome sequence analysis and pan-family multi-locus sequence analysis.
Field Strains
The performance of the MLSA scheme was further evaluated using field isolates provided by the Bundeswehr Institute of Microbiology (Munich, Germany) and obtained by the Animal and Plant Health Agency (Weybridge, United Kingdom) via brucellosis surveillance activities. In the former case, these represented a diverse panel of Ochrobactrum spp. from a variety of sources and geographical locations, confirmed as Ochrobactrum sp. by both 16S rRNA and recA sequencing (see Scholz et al., 2008a for full details). A panel of 37 isolates was examined (Table 3), representing major clades identified by recA sequence analysis (Scholz et al., 2008a). DNA was prepared as previously described (Scholz et al., 2008a). In the case of Animal and Plant Health Agency (APHA) isolates (n = 13), these were submitted as suspect Brucella sp. of veterinary origin but subsequently identified as non-Brucella members of the Brucellaceae by 16S rRNA sequencing (Wragg et al., 2014) (Table 3). Pure cultures were stored frozen in either Luria Broth with 15% glycerol or using proprietary MicroBank tubes (ProLab Diagnostics, United Kingdom). Frozen strains were revived by plating onto sheep blood and/or nutrient agar and incubating at 28°C for 24–48 h. Crude thermo-lysates from single colonies were prepared as described above.
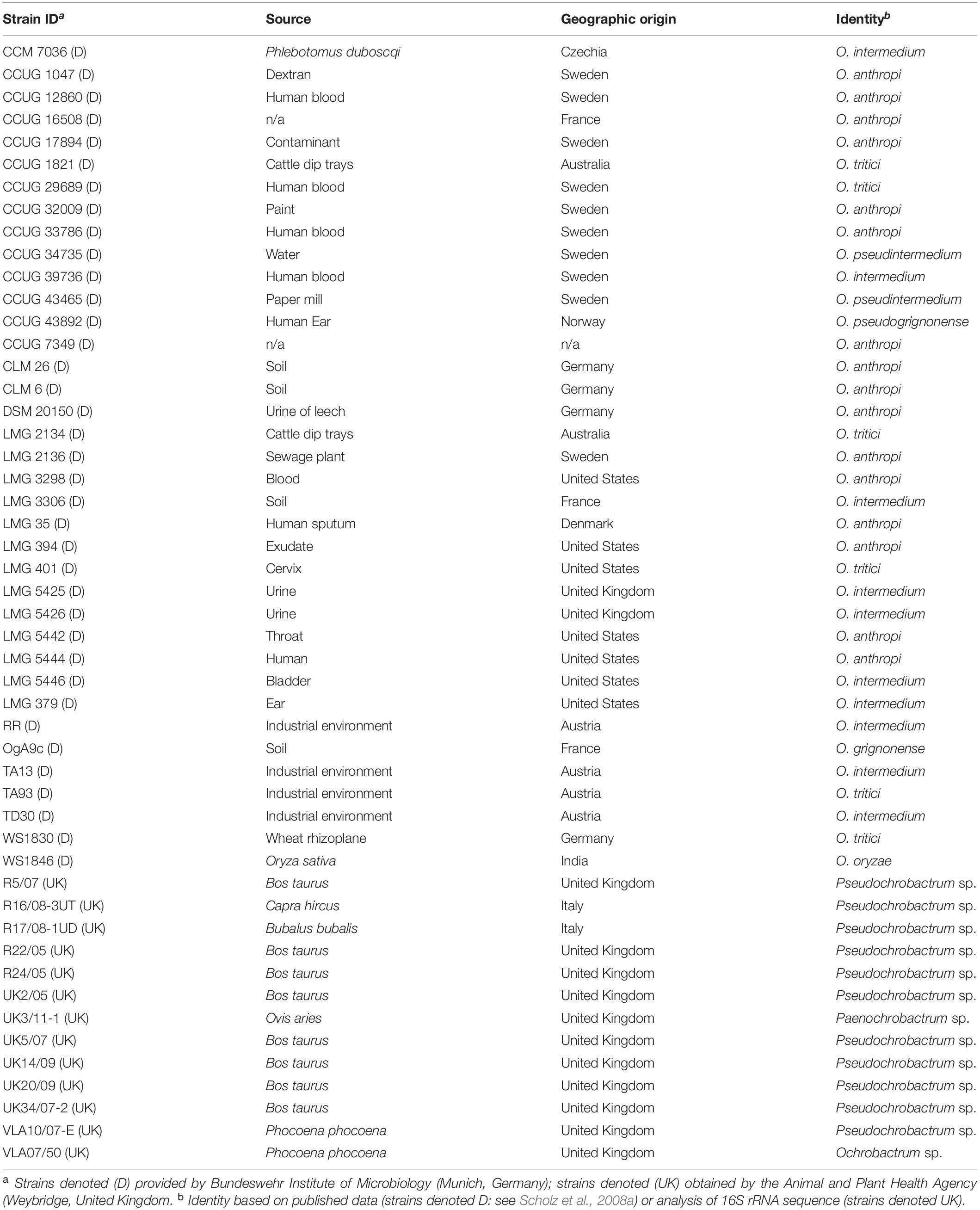
Table 3. Field strains used for evaluation of the pan-Brucellaceae multi-locus sequence analysis scheme.
Multi-Locus Sequence Analysis (MLSA)
Genomic targets for the pan-Brucellaceae MLSA scheme were selected from a larger panel of 21 loci used in an existing scheme for the Brucella genus (Whatmore et al., 2016). Alignments of sequences retrieved from NCBI were constructed for all 21 Brucella genus MLSA loci using CLUSTALW, implemented in the Lasergene software suite (version 12, DNASTAR Inc., WI, United States), for processing by the degenerate primer design program HYDEN (Linhart and Shamir, 2007) using default settings (25 bp in length, up to 1 mis-match in the 5′ and 3′ region allowed). Further iterations of primers were generated as alternatives, by modifying these default settings (primer length, number of mis-matches allowed and the level of degeneracy allowed across the primer).
Thermo-lysates from Brucellaceae type strains (generated as described above) were used to evaluate MLSA targets and optimize PCR conditions, to identify a panel of loci which amplified reliably using a standard thermal profile and reaction conditions. Optimal reaction conditions were identified as 5.0 μl of Roche 10x PCR buffer, 200 μM final concentration of dNTPs, 1.25 units of FastStart Taq polymerase, 2 mM of MgCl2, 0.4 μM of forward and reverse primers and 1 μl of template DNA (thermo-lysate), in a total reaction volume of 50 μl. Amplification was performed using an Eppendorf MasterCyclerPro (Eppendorf, Germany), with the following thermal cycle: 94°C for 5 min. followed by 35 cycles of 94°C for 1 min., 55°C for 1 min. and 72°C for 1 min. and a final extension step of 72°C for 7 min. Products were separated by agarose gel electrophoresis to confirm that only a single product of the expected size was present. PCR products were then purified by passage through QIAquick PCR purification columns (Qiagen, United Kingdom) and sequenced in both directions using the same forward and reverse primers as used for initial PCR amplification. The Big Dye Terminator Cycle Sequencing Kit (version 3.1, Applied Biosystems, Carlsbad, CA, United States) was used according to manufacturer’s instructions, with capillary electrophoresis on an ABI 3130xl DNA Analyzer (Applied Biosystems, Carlsbad, CA, United States). In a small number of cases an amplified product of the correct size was extracted from an agarose gel following electrophoresis and purified using the MinElute Gel Extraction Kit (Qiagen, United Kingdom), according to manufacturer’s instructions. Purified PCR products were then sequenced as described.
Sanger sequence data were manipulated using the Lasergene software suite (version 12, DNASTAR Inc., WI, United States), to produce a trimmed consensus sequence file for each strain and locus. Consensus sequences were used to perform phylogenetic analyses, for both individual loci and concatenated sequences (in the following order: gpd, dnaK, trpE, csdB, leuA, and acnA). The Lasergene MegAlign module was used to estimate percentage similarity between sequences. Other summary statistics were calculated using MEGA5 (version 5.2; Tamura et al., 2011). Three type strains were not available when MLSA work was undertaken (O. endophyticum DSM 29930T; O. quorumnocens LMG 30544T; F. shanghaiense HN4T), and therefore MLSA sequences were retrieved from WGS data as described below. Similarly, MLSA data for three atypical Brucella sp. isolates (141012304, B13-0095 and NF2653) were extracted from WGS data available on NCBI. Phylogenetic analyses for MLSA data were performed using MEGA5. Maximum likelihood trees were constructed using a general time reversible substitution model (gamma distribution plus invariant sites), with percentage bootstrap confidence levels of internal branches calculated from 1000 re-samplings of the original data. Sequence data from Rhizobium etli CFN42T (Rhizobiales; Rhizobiaceae; assembly accession GCF_000092045.1) were included and defined as an out-group in all analyses.
Whole Genome Sequencing (WGS)
Whole genome sequencing data from type strains were either retrieved from publically available databases (NCBI), or generated for this study (Table 1). For this purpose DNA was extracted from crude thermo-lysates, using the Qiagen DNEasy Blood and Tissue Kit (Qiagen, Manchester, United Kingdom). Library preparation was performed using the Nextera XT Library Preparation Kit (Illumina Inc., San Diego, CA, United States) according to the manufacturer’s protocol. Libraries were sequenced on the Illumina MiSeq platform, using the MiSeq v2 Reagent Kit (Illumina Inc., San Diego, CA, United States), producing 150 bp paired-end reads.
Raw sequence data were processed using the Nullarbor pipeline (version 2.01), which integrates a number of previously published bioinformatic tools. Briefly, quality assessment and trimming of raw read data was performed using Trimmomatic (version 0.39; Bolger et al., 2014). De novo assembly of raw reads was performed using the SPAdes assembler (version 3.13.1; Bankevich et al., 2012). De novo assembled genomes were then annotated using Prokka (version 1.13.1; Seemann, 2014). Genomes retrieved from publically available databases (either as finished genomes or whole genome shotgun assembly contigs) were likewise annotated using Prokka. Where an assembled genome was not available via NCBI then short-read data were downloaded from the NCBI SRA database and assembled as described. Genomic average nucleotide identity (ANI) estimates were calculated from fasta formatted genome assemblies, using FastANI (version 1.3; Jain et al., 2018).
In order to generate pan-genomes from WGS data, Roary (version 3.13.0; Page et al., 2015) was used, with a minimum sequence identity of 70% and gene presence in ≥99% of isolates to be identified as a core gene. Core genome sequence alignments were generated using MAFFT (Katoh et al., 2002). Core genome alignments were used to produce maximum likelihood phylogenies with RAxML-NG (Kozlov et al., 2019) applying a general time reversible model (gamma distribution plus invariant sites), with 100 bootstraps. The resulting phylogenies were viewed and further annotated using MEGA5. Rhizobium etli CFN42T (Rhizobiales; Rhizobiaceae; assembly accession GCF_000092045.1) was included and defined as an out-group. Additional outgroup strains were incorporated in some analyses to investigate the placement of Mycoplana spp. (Agrobacterium tumefaciens Ach5: GCF_000971565.1; Aquamicrobium aerolatum DSM 21857T: GCF_900113935.1; Ensifer adhaerens ATCC 33212T: GCF_000697965.2; Ensifer sojae DSM 26426T: GCF_000261485.1; Mesorhizobium ciceri: WSM 1271: GCF_000185905.1; Mesorhizobium loti NZP 2037T: GCF_001676765.1; Rhizobium etli CFN42T: GCF_000092045.1; Sinorhizobium meliloti DSM 30135T: GCF_000006965.1).
Results
WGS Core Genome Analyses and Selection of MLSA Loci
Summary statistics for de novo genome assemblies are provided in Supplementary Table S1. Of those type strain genomes sequenced for this study (see Table 1) the genome size ranged from 3.2 to 5.8 Mb, corresponding to between 3030 and 5542 annotated coding sequences (Supplementary Table S1). The mean GC content of these genomes ranged from 48.6% to 63.5%. Pan genome analysis of annotated assemblies identified a core genome of 450 genes amongst the 43 type strains included in the analysis (plus R. etli CFN42T). The concatenated core genome alignment of these genes comprised 449,688 bp. Inclusion of additional outgroup strains reduced the core genome to 410 genes (419,690 bp), whilst inclusion of atypical Brucella strains resulted in a core genome of 400 genes (395,037 bp).
Six loci were identified which amplified reliably in all type species of the Brucellaceae, using the standard thermal profile and reaction conditions described, and were therefore incorporated into the pan-family MLSA scheme (Supplementary Table S2). All loci represent informative housekeeping genes of the type conventionally used in MLSA. Mean GC content in the six loci ranged from 54.5% to 59.4%, with an average of 57.0% across all loci. The proportion of polymorphic sites in each of the six loci, across all Brucellaceae type species, ranged from 42.34% (dnaK) to 53.50% (trpE), with an average of 48.37%. Nucleotide diversity (π) in the six loci ranged from 0.1256 (dnaK) to 0.1975 (trpE), whilst nucleotide diversity across the entire 3,004 bp concatenated sequence was 0.1532 (Supplementary Table S2).
Sequence Diversity Indices From WGS and MLSA
The level of genomic ANI and MLSA nucleotide identity observed within and between genera of the Brucellaceae varied considerably (Table 4 and Supplementary Table S3). As anticipated, the highest intra-generic ANI values were observed within the Brucella (average 99.1%) whilst the lowest average ANI value was seen amongst Falsochrobactrum (average 77.9%). The largest intra-generic range (highest to lowest) in ANI values was observed within the Ochrobactrum genus (75.4–98.5%). Between genera, the highest ANI was observed between Brucella and Ochrobactrum (80.9%), whilst the lowest was between Mycoplana and Paenochrobactrum or Pseudochrobactrum (both 74.3%). The same pattern was observed in nucleotide identity estimates derived from MLSA data (Table 4). Again, nucleotide identity within the genus Brucella was very high (average 99.3%). Average nucleotide identity within the other genera of the Brucellaceae ranged from 79.4% (Falsochrobactrum) to 93.7% (Pseudochrobactrum). The largest intra-generic range (highest to lowest) in nucleotide identity was observed within the Ochrobactrum (78.2–99.7%). Between genera, the highest level of average nucleotide identity was observed between Brucella and Ochrobactrum (86.9%), whilst the lowest was between Mycoplana and Paenochrobactrum (70.7%).
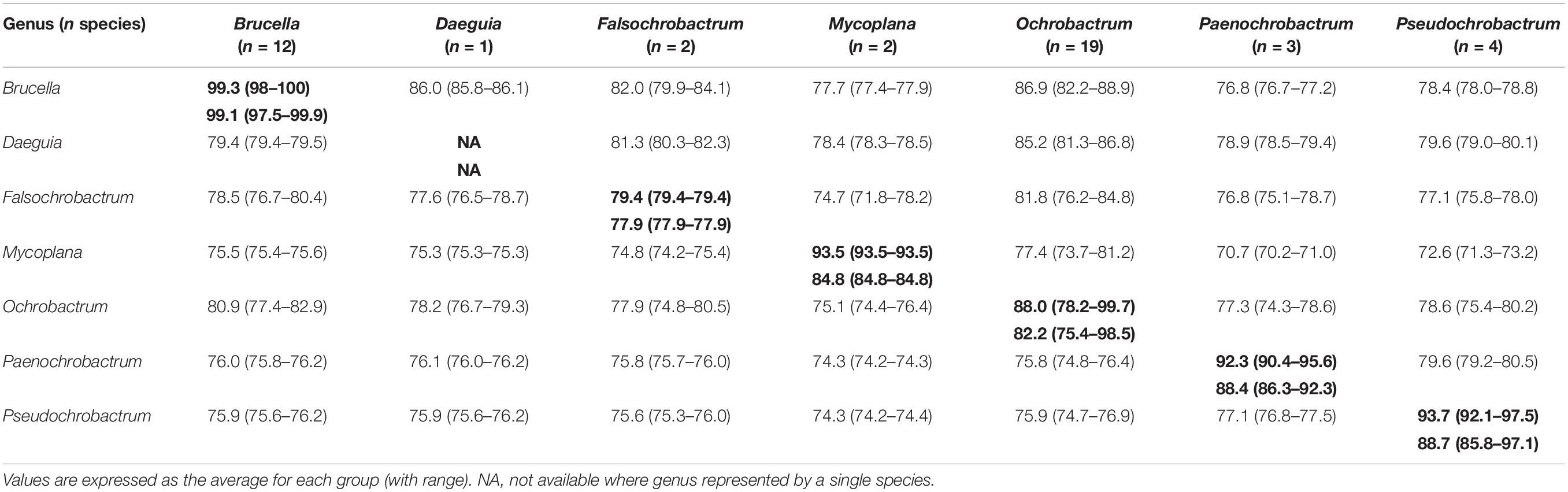
Table 4. Intra-generic (bold) and inter-generic similarity of seven genera of the family Brucellaceae, measured by genomic ANI values from WGS data (bottom left matrix) and nucleotide identity from concatenated MLSA data (upper right matrix).
As anticipated, measures of sequence similarity for WGS (percentage ANI) and MLSA (percentage nucleotide identity) showed very high levels of agreement [Figure 1A; semi-logarithmic regression: Y = −283.2 + 193∗log(X); R2: 0.89]. The relationship between genomic ANI and MLSA nucleotide identity was further investigated by comparing their values for all species, relative to the Brucellaceae type species B. melitensis NCTC 10094T (Figure 1B). Most notable was the very large dispersion of Ochrobactrum species, which were split into distinct groups by both metrics (Groups A and B as defined in section “Phylogenetic Analyses Based on WGS”), with one outlier (O. endophyticum) highly divergent from other members of the genus. Similarly, the two Falsochrobactrum species were characterized by very different levels of diversity relative to B. melitensis.
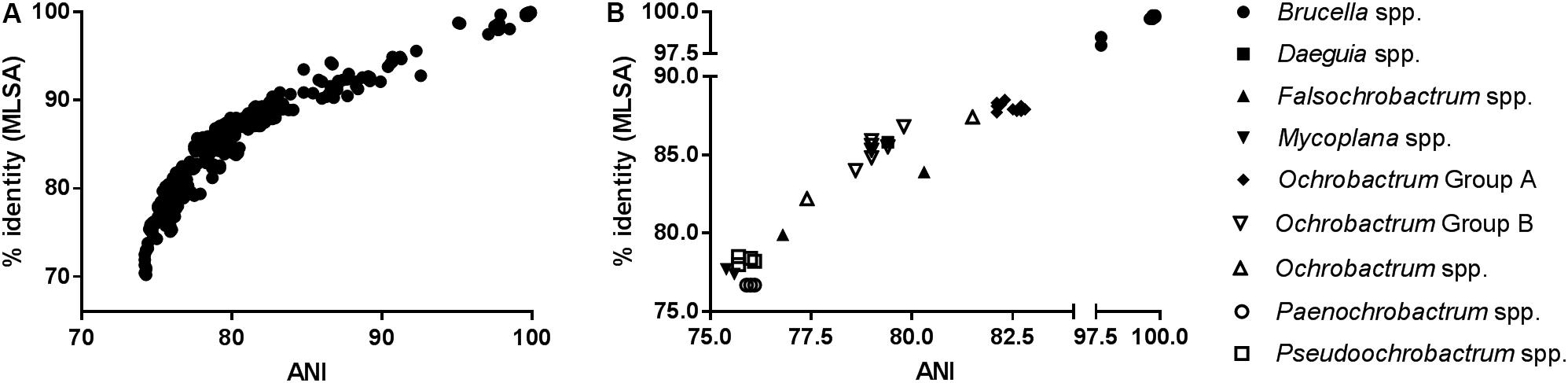
Figure 1. Association between sequence similarity indices from WGS (average nucleotide identity, ANI) and MLSA (percentage nucleotide identify of concatenated sequence) for Brucellaceae type strains. (A) The distribution of values for both indices, from all pairwise comparisons between members of the family (n = 903 comparisons). (B) Values for members of each genus, relative to the Brucellaceae type species (B. melitensis NCTC 10094T).
Further investigation of genomic ANI values (Figure 2) demonstrated that all pairwise comparisons between species within the genus Brucella exceeded 97.5%. Furthermore, a number of other intra-generic pairings with very high ANI values were identified. Specifically, within the genus Ochrobactrum, O. ciceri and O. intermedium were identified as sharing 98.5% genomic ANI, whilst O. anthropi and O. lupini (the latter already identified as a homotypic synonym of the former) shared 97.9% ANI. Outside of the Brucella and Ochrobactrum genera, the highest ANI value observed was between Pseudochrobactrum lubricantis and P. sacchrolyticum (97.1%).
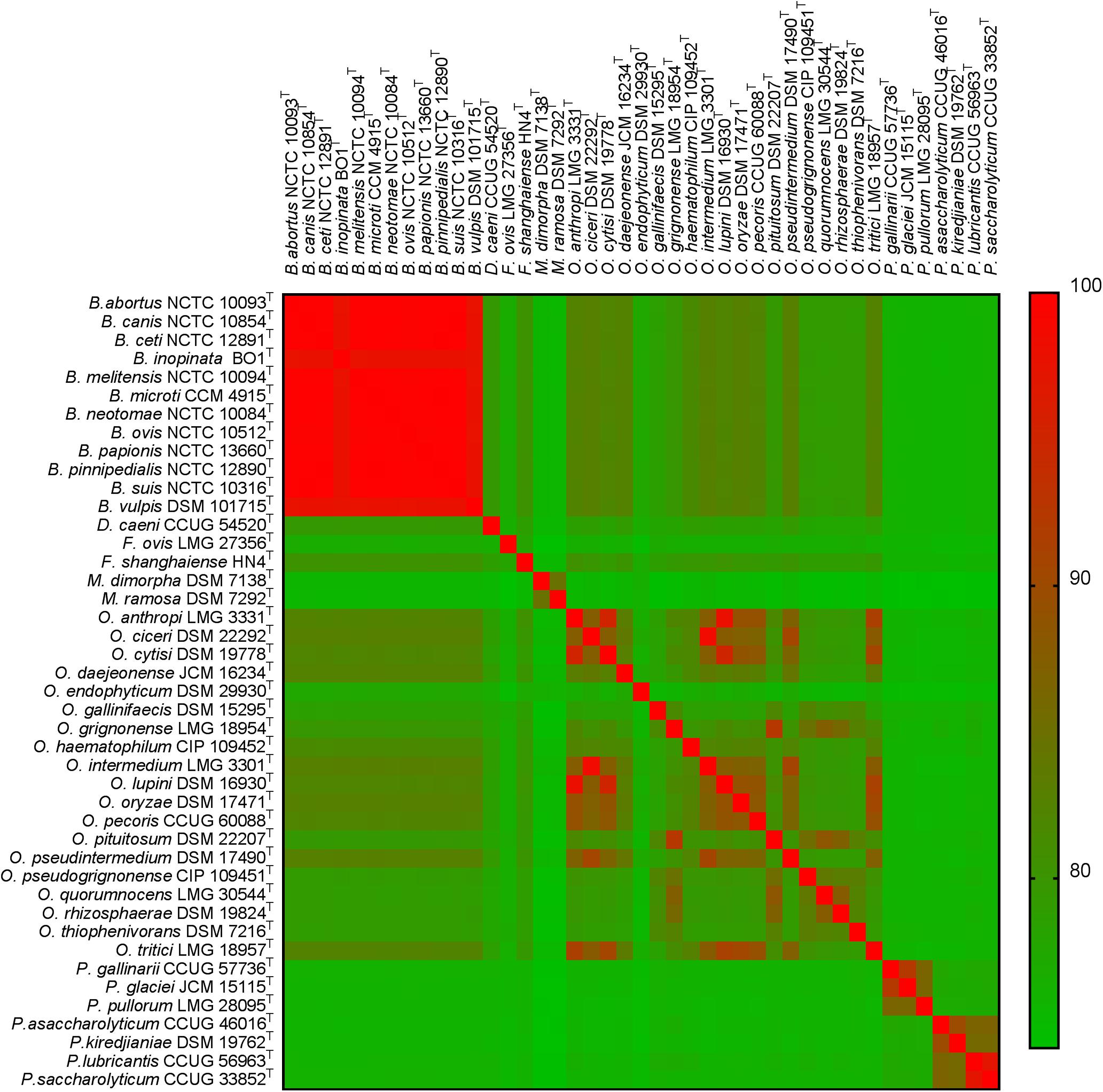
Figure 2. Heatmap of genomic average nucleotide identity (ANI) values for pairwise comparisons between all type strains of the family Brucellaceae (n = 43).
Phylogenetic Analysis of Brucellaceae Type Strains
Phylogenetic Analyses Based on WGS
Maximum likelihood analysis based on the core genome alignment from Brucellaceae type strains resulted in an almost fully resolved phylogeny (Figure 3A), with the only nodes lacking full bootstrap support sitting within the genus Brucella. Nonetheless, the monophyly of this genus was highly supported. The genus Ochrobactrum was identified as polyphyletic. A sister group to the Brucella consisted of 10 Ochrobactrum species (Figure 3A: Group A), containing O. anthropi – O. lupini – O. cytisi – O. tritici – O. pecoris – O. oryzae – O. ciceri – O. intermedium – O. pseudintermedium – O. daejeonense. A single species (O. haematophilum), branched basally, to form a sister group to the clade containing these two taxa (Ochrobactrum Group A and Brucella spp.). A second group of seven Ochrobactrum species (Figure 3A: Group B) formed a sister group to the Brucella and all other Ochrobactrum species (with the exception of O. endophyticum). This clade was comprised of O. grignonense – O. pituitosum – O. quorumnocens – O. rhizosphaerae – O. pseudogrignonense – O. thiophenivorans – O. gallinifaecis.
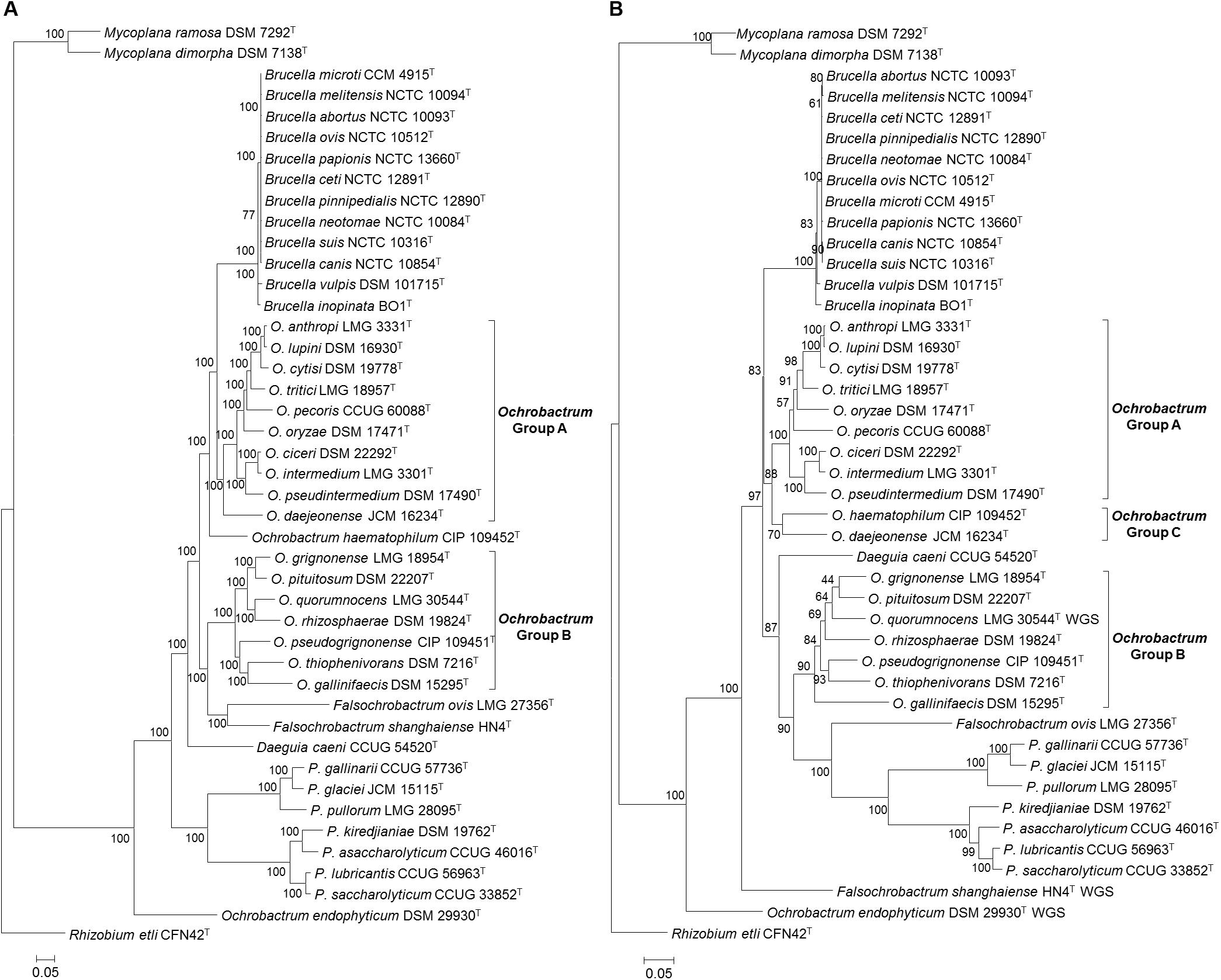
Figure 3. Phylogenetic relationship between type strains of the family Brucellaceae (n = 43), based on WGS (A) and MLSA (B). In both cases the evolutionary history was inferred using the maximum likelihood method, based on the general time reversible model. Node labels give percentage bootstrap support. The scale bar shows the number of base substitutions per site. Rhizobium etli (CFN42T) was used to root the phylogenetic tree.
Maximum likelihood phylogeny based on WGS data indicated that Falsochrobactrum spp. formed a sister clade to Ochrobactrum Group B. Further phylogenies were estimated using the WGS dataset (both with and without additional outgroups), removing each species in turn (F. ovis and F. shanghaiense). The phylogenies produced were identical in their placement of the two Falsochrobactrum species, which were individually placed as a sister taxon to Ochrobactrum Group B (Supplementary Figure S1). The single Daeguia species (D. caeni) branched basally to Brucella spp., Ochrobactrum Groups A and B and Falsochrobactrum spp. Both Paenochrobactrum spp. and Pseudochrobactrum spp. formed well-supported sister taxa in a single clade. A single Ochrobactrum species (O. endophyticum) branched basally to all members of the family, other than Mycoplana spp. (Figure 3A). Maximum likelihood phylogenetic analysis incorporating eight outgroup strains from the wider Rhizobiales indicated that Mycoplana spp. were placed with type strains of Sinorhizobium meliloti DSM 30135T and Ensifer spp. (E. sojae DSM 26426T and E. adhaerens ATCC 33212T), of the family Rhizobiaceae, rather than with members of the Brucellaceae (Supplementary Figure S1).
Phylogenetic Analysis Based on MLSA
Phylogenetic analyses based on concatenated sequences (3,004 bp) for all Brucellaceae type strains demonstrated that the pan-family MLSA scheme was able to distinguish all members of the taxon, with reasonable levels of bootstrap support in the majority of cases (Figure 3B). A single strongly supported clade containing all currently recognized Brucella species was evident. The species of Ochrobactrum included in the analysis of concatenated sequences formed three distinct clades. The first of these clades (Figure 3B: Group A) consisted of nine species, containing the grouping of O. anthropi – O. lupini O. cytisi – O. tritici – O. oryzae – O. pecoris – O. ciceri – O. intermedium – O. pseudintermedium. However, the node separating O. pecoris from other members of this clade was weakly supported. A second Ochrobactrum clade (Figure 3B: Group B) contained seven species (O. grignonense – O. pituitosum – O. quorumnocens – O. rhizosphaerae – O. pseudogrignonense – O. thiophenivorans – O. gallinifaecis) and grouped more closely with a clade containing Pseudochrobactrum spp., Paenochrobactrum spp. and Falsochrobactrum ovis than with the Ochrobactrum Group A clade. Bootstrap support for nodes within this grouping of seven Ochrobactrum species was generally low, with the exception of the nodes dividing O. gallinifaecis from all others, and dividing O. thiophenivorans from O. pseudogrignonense. A third clade of two Ochrobactrum species (Figure 3B: Group C: O. daejeonense and O. haematophilum) formed a distinct sister clade to Group A, though with only moderate bootstrap support. A well-supported clade contained Pseudochrobactrum spp., Paenochrobactrum spp. and Falsochrobactrum ovis, with F. ovis branching basally to form a sister taxon to the other two genera. The recently described F. shanghaiense, however, branched basally to all other Brucellaceae type strains, with the exception of a single Ochrobactrum species (O. endophyticum) and Mycoplana spp. Daeguia caeni was placed as a sister taxon to Ochrobactrum Group B and Pseudochrobactrum spp., Paenochrobactrum spp. and F. ovis.
Phylogenetic Analyses Based on Individual MLSA Loci and 16S rRNA Sequence
Maximum-likelihood phylogenies for each locus are shown in Supplementary Figure S2. In all cases, a single clade containing Brucella type species, including divergent atypical species (B. inopinata and B. vulpis) was identified, with a high level of support (bootstrap values of 100% for all loci). Only one locus (trpE) indicated the existence of a monophyletic Ochrobactrum clade. All other loci indicated that the genus was polyphyletic. A group of species, corresponding to Ochrobactrum Group A (as identified in analysis of both WGS and MLSA datasets), was evident for all loci except acnA. Within Ochrobactrum Group A, O. anthropi and O. lupini were consistently shown to be very closely related, with O. cytisi also consistently grouping with the former species, but somewhat divergent. Similarly, O. ciceri and O. intermedium were consistently identified as highly similar, with O. pseudintermedium forming a slightly divergent sister taxon. The relative position of the seven species within Ochrobactrum Group B (as identified in analysis of both WGS and MLSA datasets) was not consistent between loci, and generally had low levels of bootstrap support. Both O. daejeonense and O. haematophilum were inconsistently placed, and often weakly supported, in analyses of data from individual loci. Analysis of individual MLSA loci consistently identified well supported Pseudochrobactrum and Paenochrobactrum groups, as sister taxa. The position of the two Falsochrobactrum species was variable, however, and they were not placed in a unified taxon in any single locus analysis. The placement of the single Daeguia species was highly variable in analyses based on individual loci, and frequently had low levels of bootstrap support. In the majority of cases O. endophyticum branched basally to all members of the family except Mycoplana spp., and clearly distinct from other Ochrobactrum species. Finally, data from individual loci other than gpd identified the two Mycoplana species as branching basally to all other members of the family.
As observed in previous studies, analysis of almost full-length 16S rRNA sequences from Brucellaceae type strains did not provide sufficient resolution to correctly place species, even at the genus level (Supplementary Figure S2).
Atypical Brucella Isolates
Inclusion of atypical Brucella isolates with type strains of the family Brucellaceae clearly identified them as belonging within the genus Brucella, with maximum bootstrap support for a monophyletic genus (Figures 4A,C). Additionally, both WGS and MLSA based approaches identified the six classical Brucella species, plus the more recently described B. ceti, B. pinnipedialis, B. microti and B. papionis as forming a well-supported group of ten core species, with limited diversity between them (Figures 4B,D). Outside of these core species, the basal branching order of atypical Brucella species and strains within the genus was relatively weakly supported by both WGS and MLSA datasets, with low bootstrap values for several nodes (Figures 4B,D). Amongst these isolates there were only a small number of groupings well supported by bootstrap values using MLSA data. Strains from Australian rodents (83/4, 83/13 and NF2653) formed a well-supported clade (Figure 4D), as did two strains from African bullfrogs (Brucella sp. 09RB8471 and 10RB9205). Based on MLSA data the human-associated strain B. inopinata formed a moderately well-supported group with a novel isolate from an amphibian (Brucella sp. B13-095), and distinct from the other atypical human isolate Brucella sp. BO2. In contrast, WGS based analysis identified these human associated isolates as sister taxa, with the amphibian isolate Brucella sp. B13-095 branching basally (Figure 4B). Branch lengths based on MLSA data indicated that Brucella sp. 141012304, isolated from a cartilaginous fish, was highly divergent from other atypical strains. Branch lengths of atypical Brucella strains calculated from WGS data, however, indicated a more consistent level of divergence from core species.
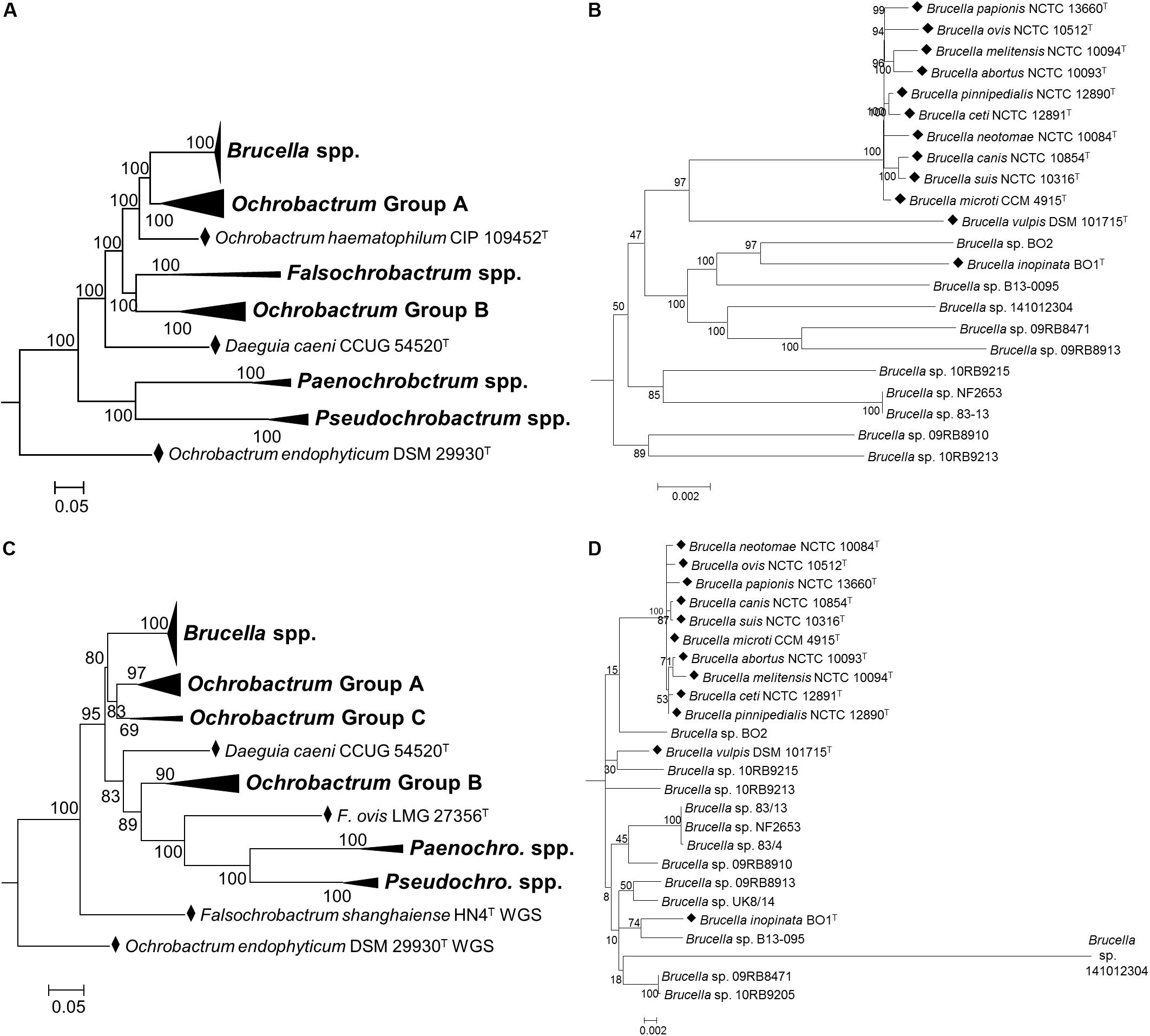
Figure 4. Phylogenetic relationship between type strains of the family Brucellaceae (and selected atypical Brucella sp. strains), based on WGS (A,B) and MLSA (C,D). Panels (A,C) show the phylogeny inferred for the entire family, whilst panels (B,D) show only the Brucella spp. clade. Type strains are denoted by a black diamond (◆). In both cases the evolutionary history was inferred using the maximum likelihood method, based on the general time reversible model. Node labels give percentage bootstrap support. The scale bar shows the number of base substitutions per site. Rhizobium etli (CFN42T) was used to root the phylogenetic tree (not shown).
Field Isolates
In order to assess the value of the pan-family MLSA scheme in placing field isolates, subsequent analysis incorporated panels of field isolates, alongside type strains of all species within the Brucellaceae. This demonstrated that the MLSA scheme assigned field isolates to phylogenetic locations largely consistent with their identities assigned on the basis of phenotypic, recA and/or 16S rRNA sequence analysis (Figure 5).
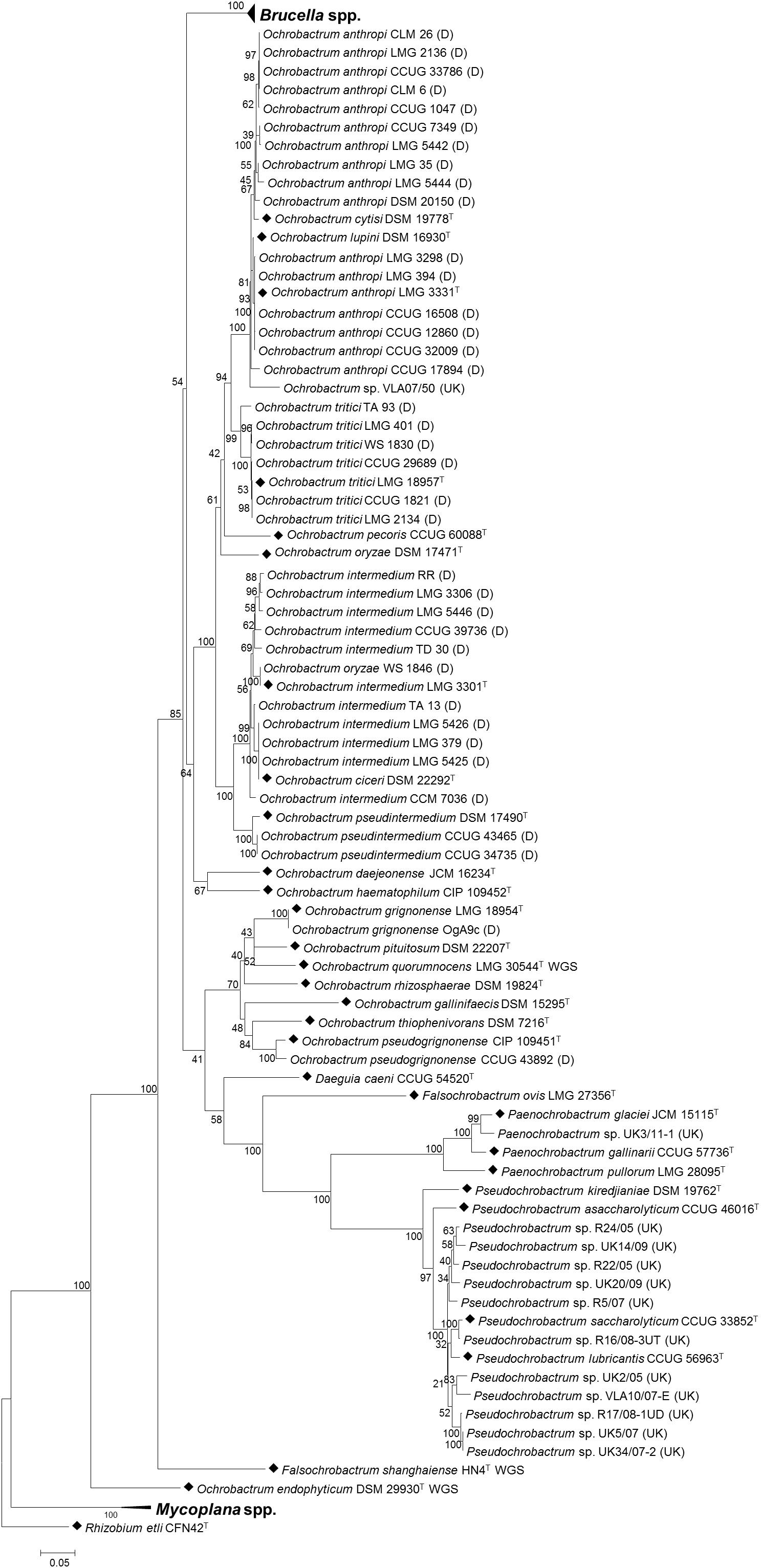
Figure 5. Phylogenetic relationship between type strains of the family Brucellaceae (n = 43) and field strains (n = 50), based on multi-locus sequence analysis. Type strains are denoted by a black diamond (◆), whilst strains from Bundeswehr Institute of Microbiology, Germany are denoted (D) and field strains from the Animal and Plant Health Agency, United Kingdom are denoted (UK). The evolutionary history was inferred using the maximum likelihood method, based on the general time reversible model. Node labels give percentage bootstrap support. The scale bar shows the number of base substitutions per site. Rhizobium etli (CFN42T) was used to root the phylogenetic tree.
Isolates previously identified as Ochrobactrum anthropi formed a well-supported clade containing the type strains of O. anthropi – O. cytisi and O. lupini. However, isolates did not fall into three separate clusters corresponding to the three species but rather represented a continuum of diversity. Only five O. anthropi field strains clustered with the type strain for the species. A further ten O. anthropi field strains grouped more closely with O. cytisi although with low levels of bootstrap support for the node separating them from the well supported O. anthropi – O. lupini cluster. A single field isolate (VLA07/50) grouped within the O. anthropi – O. cytisi and O. lupini clade with a high degree of bootstrap support, but appeared to be sufficiently divergent from other strains within the cluster to potentially represent a novel species. Interestingly, this isolate originated from a harbor porpoise (Phocoena phocoena) and hence may represent a marine or marine-mammal associated strain.
A sister clade to the O. anthropi – O. cytisi and O. lupini grouping contained the type strain of O. tritici and all field isolates identified as belonging to this species. A sister clade to the grouping of O. anthropi – O. cytisi – O. lupini and O. tritici contained the type species of O. pseudintermedium – O. ciceri and O. intermedium and associated field isolates. All field isolates of O. pseudintermedium grouped with their respective type strain (DSM 17490T). In the case of O. intermedium, five of the ten field isolates grouped most closely with the type strain (LMG 3301T), though with a continuum of diversity from the parent species. Four O. intermedium field strains clustered more closely with the Ochrobactrum ciceri type strain (DSM 22292T). A single O. intermedium field strain (CCM 7036) branched basally to the cluster containing type species of O. intermedium – O. ciceri and associated field isolates, indicating additional diversity within this group that may merit description of further species. A single field isolate originally identified as O. oryzae (WS 1846), clustered with the O. intermedium type strain and field isolates. The location of the type strain for this species (DSM 17471T) was relatively poorly supported.
A single field isolate grouped with the type strains of both O. grignonense and O. pseudogrignonense (field isolates OgA9c and CCUG 43892, respectively). In the case of the former (OgA9c), this strain was isolated simultaneously to the type strain, and from the same material (Lebuhn et al., 2000) which is reflected in their sequence homology. Strain CCUG 43892 was isolated independently from the type strain of O. pseudogrignonense (CIP 109451T), but formally described at the same time (Kämpfer et al., 2007a) due to their phenotypic and genetic similarity. This is reflected in the high level of bootstrap support for their placement together, though branch lengths indicate a degree of sequence divergence in the six MLSA loci described here.
With the exception of a single strain, United Kingdom field isolates (obtained via surveillance, for exclusion as Brucella), were identified as either Pseudochrobactrum sp. or Paenochrobactrum sp. by 16S sequencing (Table 3). Pseudochrobactrum sp. field strains formed a diverse cluster with the type strains of P. lubricantis and P. saccharolyticum suggesting these species poorly reflect actual diversity of these bacterial groups. In particular, a group of five field isolates within this grouping formed two distinct and well-supported clusters of two and three isolates, respectively. The single Paenochrobactrum sp. field strain (UK3/11-1) grouped in a single well-supported clade with type strains of the three described Paenochrobactrum species, and was identified as most closely resembling P. glaciei.
Discussion
In this study, we have investigated phylogenetic relationships within the Brucellaceae, using a comprehensive WGS dataset incorporating all currently described species and a pan-family MLSA approach developed to be compatible with existing Brucella schemes.
Increasingly, WGS approaches are being used to generate data for phylogenetic placement of novel bacterial isolates. Nonetheless, the pan-family MLSA approach developed here remains valuable, in providing a defined list of genomic targets for future phylogenetic analysis and a comprehensive database of both reference and field isolates for comparison. Additionally, whilst the generation of WGS data for bacterial isolates is increasing, such data is absent for a large number of strains described before the approach became widespread. Again, the data generated in the current work will permit their integration into future multi-gene analyses incorporating results derived from both whole genome and “first-generation” sequencing methods. As an example of this approach, Krzyzanowska et al. (2019) recently described a putative novel Ochrobactrum species (Ochrobactrum quorumnocens sp. nov.) isolated from the potato rhizosphere. These authors applied a comparative genomic analysis with related Ochrobactrum type strains, using WGS data from the novel isolate and six existing species. These data were used to apply an MLSA based approach using concatenated nucleotide sequences of three genes: 16S rRNA gene, groEL and gyrB. Retrieval of data for the genes adopted in the current work would permit the comparison of this novel strain to a wider diversity of species within the Brucellaceae.
Our work provides an additional tool to understand strains where conventional diagnostic approaches are confounded. Recently described “atypical” Brucella sp. isolates can be confused with Ochrobactrum sp. by diagnostic approaches such as API 20NE tests (Scholz et al., 2010; Tiller et al., 2010b; Soler-Lloréns et al., 2016). These isolates can also confound identification by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS), an increasingly widely used front-line tool in bacterial diagnostics (Cunningham and Patel, 2013). Thus there are examples where atypical Brucella either return no reliable identification or are misidentified as B. melitensis (Whatmore et al., 2015; Mühldorfer et al., 2017). The pan-family MLSA approach applied here clearly identified that all atypical strains incorporated into the analysis are much more closely related to Brucella than to Ochrobactrum.
The WGS and MLSA approaches applied here are of value in informing the ongoing debate concerning Brucella taxonomy and evolution, and allow accurate placement and understanding of the relationships of these organisms with extant isolates. Both methods identified a monophyletic Brucella clade, with a highly conserved group of ten core species and greater diversity in recently identified atypical strains. Basal branches in this clade were, however, relatively weakly supported by both methods. Nonetheless, based on currently accepted practices for defining species based on genomic ANI values (Varghese et al., 2015) all Brucella strains incorporated into our analyses would represent a single species. Brucella taxonomy continues to be controversial both at the intra-genus level and in terms of relationships with closely related species. The previous re-designation of the then six original “core” Brucella species as a single species based on genetic homogeneity (Verger et al., 1985) was reversed, based on community preference for a nomenclature that reflected clear differences in epidemiology and pathogenic potential of the nomenspecies (Osterman and Moriyón, 2006). The current study emphasizes the genetic homogeneity of the expanded core group, in contrast to the greater diversity seen in the recently emerged atypical Brucella. Attempts to describe emerging amphibian isolates as new species failed because of the heterogeneity of the isolates (Al Dahouk et al., 2017) and their placement here confirms that there are currently few distinct clusters of isolates among the atypical Brucella that would readily facilitate description of any well-defined new species. A more pragmatic, though not cladistic, solution may be to update the species description for B. inopinata, originally based on only a single isolate (BO1), taking into account the large number of recently described strains, and potentially classify all atypical Brucella as B. inopinata. Arguments that these isolates should not be members of the genus Brucella, as currently defined, are not supported by the genetic relationships observed here, as they clearly remain well separated from the closest Ochrobactrum isolates.
The current work supports previous findings, based on single-gene, multi-locus and whole genome analyses, which indicated that the genus Ochrobactrum represents a polyphyletic grouping (Kämpfer et al., 2013; Aujoulat et al., 2014; Leclercq et al., 2019). Our study provides the most comprehensive analysis to date of the composition of these groupings, and their relationships to other members of the family. Both WGS and MLSA based analyses identified two main clusters, with Ochrobactrum Group A (O. anthropi; O. lupini, O. cytisi; O. tritici; O. pecoris; O. oryzae; O. ciceri, O. intermedium, O. pseudintermedium) forming a well-supported sister taxa to all Brucella species. The position of two further species (O. daejeonense and O. haematophilum) diverged slightly between the two methods, though both were placed with the Brucella-Ochrobactrum Group A clade (with WGS indicating O. haematophilum to branch basally to this group). A further Ochrobactrum clade (Group B) contained seven species (O. gallinifaecis – O. quorumnocens – O. rhizosphaerae – O. grignonense – O. pituitosum – O. pseudogrignonense – O. thiophenivorans) and formed a sister taxon to the Brucella – Ochrobactrum Group A clade in WGS based analyses (An Ochrobactrum group of the same composition was identified in MLSA based analysis, but in this case it was placed as a sister taxon to the clade containing Pseudochrobactrum spp., Paenochrobactrum spp. and Falsochrobactrum ovis). Recently, Leclercq et al. (2019) applied a phylogenomic approach to identify similar polyphyletic Ochrobactrum clades to those identified here. These authors suggest that there is a separation in the ecological niche of species belonging to Ochrobactrum groups A and B, with the former characterized by human and animal derived isolates, and the latter mainly composed of environmental and plant-associated isolates. Grouping type strains by their source of isolation did not reveal any association between Ochrobactrum groups and human/animal origin of isolation in the current study (Fisher’s exact test, P > 0.05 for both WGS and MLSA data).
Two Ochrobactrum species (O. daejeonense and O. haematophilum) were inconsistently placed, and often weakly supported, in analyses of data from individual loci. Examples of incongruence in placement between single gene trees of Ochrobactrum species are supportive of previous suggestions that horizontal gene transfer may be a driving force in the evolution of both atypical Brucella (Scholz et al., 2016b) and other free-living members of the Brucellaeae (Aujoulat et al., 2014). This is in contrast to the situation in the core Brucella which have been considered largely clonal with only rare horizontal gene transfer events contributing to speciation (Whatmore et al., 2007; Wattam et al., 2014).
Analyses in the current work (WGS, MLSA, individual gene and 16S rRNA based) indicated that O. endophyticum (DSM 29930T) is not located within the Ochrobactrum groupings described above, but instead forms a distinct taxon, basal to other members of the family (with the exception of Mycoplana spp., though see below). Analysis of WGS data incorporating additional outgroup species (Supplementary Figure S1) also indicated that O. endophyticum remained basal to other members of the Brucellaceae, but was more closely related to them than the nearest taxonomic neighbors included in the analysis (Mesorhizobium spp. and Aquamicrobium aerolatum). Clarification of the true taxonomic position of O. endophyticum will require the incorporation of additional strains outside of the Brucellaceae, but it appears that it may require amendment.
Analysis of WGS and MLSA data from Brucellaceae type strains identified O. anthropi and O. lupini to be closely related, consistent with a recent analysis based on WGS data, which proposed that O. lupini should be re-classified as a later heterotypic synonym of O. anthropi (Gazolla Volpiano et al., 2019). These authors also highlighted the high level of similarity observed between O. anthropi/O. lupini and O. cytisi, and recommended WGS of the type strain for the latter species, to clarify its position. Incorporation of WGS data for O. cytisi DSM 19778T in the current study indicated that it is correctly identified as an independent species (adopting the threshold of 96.5% proposed by Varghese et al., 2015) with genomic ANI values of 95.2% and 95.1% relative to O. anthropi/O. lupini, respectively. Leclercq et al. (2019) drew the same conclusion based on sequencing of a non-type strain of O. cytisi (IPA7.2). High levels of similarity, consistent with possible re-classification as a single species were observed between type strains of O. ciceri and O. intermedium (genomic ANI of 98.5%) and P. lubricantis and P. saccharolyticum (genomic ANI of 97.1%).
Furthermore, it is also clear from the inclusion of field isolates into multi-locus analysis of Brucellaceae that there is much previously unrecognized diversity that is not reflected in currently identified species. Notable diversity was evident within the O. anthropi – O. cytisi – O. lupini, O. intermedium – O. ciceri and P. saccharolyticum – P. lubricantis groupings, resulting in a lack of clear monophyletic lineages for any of these taxa once field isolates were incorporated.
Basal branching orders within type strain phylogenies were largely consistent between WGS and MLSA based approaches (Figure 3). One instance where this was not the case, however, was in the placement of Falsochrobactrum. Both F. ovis and F. shanghaiense were placed as a sister taxon to Ochrobactrum Group B in analyses based on WGS data. Conversely, in analyses using MLSA, the taxon was split, with F. ovis placed as a sister taxon to the Paenochrobactrum spp., and Pseudochrobactrum spp. group, and F. shanghaiense placed basal to all member of the family other than O. endophyticum and Mycoplana spp. Leclercq et al. (2019) reported inconsistent placement of Falsochrobactrum spp. using a whole genome dataset, with the location differing when both species were included relative to when either was included independently. However, neither placement was fully supported by bootstrap values in their analyses. The inclusion of a more comprehensive panel of related species and, potentially, the larger number of loci employed, provides greater confidence in the placement of these species in WGS analyses from the present study, as evidenced by the level of bootstrap support achieved. Nonetheless, it is clear from levels of sequence similarity observed within this genus (both genomic ANI and MLSA nucleotide identity) that these two species are highly divergent (Figure 1 and Table 4).
Finally, incorporation of additional outgroup strains in analyses of WGS data in the current work has demonstrated that Mycoplana spp. were placed with maximum bootstrap support within the family Rhizobiaceae rather than within the Brucellaceae. This is consistent with the findings of Leclercq et al. (2019) who incorporated a dataset encompassing the wider order Rhizobiales and demonstrated the same placement of Mycoplana spp. with Ensifer and Sinorhizobium species.
Conclusion
The work presented here provides a comprehensive multi-gene analysis of the phylogeny of an expanding bacterial family, which includes a number of significant human and veterinary pathogens. It further demonstrates the importance of a multi-gene approach, be it WGS or MLSA based, in establishing robust relationships within this group. This work has demonstrated that the genus Brucella as currently described forms a well-separated monophyletic group, despite its on-going expansion to incorporate genetically diverse strains from a wide range of host species. Furthermore, the work confirms Ochrobactrum to be polyphyletic. The analysis of non-type strains of some members of Brucellaceae reveals that there remains significant genetic diversity not captured within existing species. The tools and databases developed in this study provide a valuable resource against which to place novel isolates and begin to identify new groups.
Data Availability Statement
WGS data generated for this study can be found in the NCBI SRA database under BioProject PRJNA638390. MLSA data can be found in either the Brucella PubMLST database (https://pubmlst.org/brucella/) or in the NCBI GenBank database (accession numbers given in Supplementary Table S4).
Author Contributions
RA analyzed the data and drafted the manuscript. JM and MK undertook experimental work and data analysis. HS provided strains and contributed to the manuscript. AW conceived of and designed the study and contributed to the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Brucellosis research at APHA is supported by the United Kingdom Department for Environment, Food and Rural Affairs (Defra). Development and implementation of the pan-family MLSA scheme was specifically funded as part of project SE0313.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01329/full#supplementary-material
Footnotes
References
Al Dahouk, S., Köhler, S., Occhialini, A., Jiménez de Bagüés, M. P., Hammerl, J. A., Eisenberg, T., et al. (2017). Brucella spp. of amphibians comprise genomically diverse motile strains competent for replication in macrophages and survival in mammalian hosts. ı Sci. Rep. 7:44420. doi: 10.1038/srep44420
Aujoulat, F., Romano-Bertrand, S., Masnou, A., Marchandin, H., and Jumas-Bilak, E. (2014). Niches, population structure and genome reduction in Ochrobactrum intermedium: clues to technology-driven emergence of pathogens. PLoS One 9:e83376. doi: 10.1371/journal.pone.0083376
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buddle, M. B. (1956). Studies on Brucella ovis (n.sp.), a cause of genital disease of sheep in New Zealand and Australia. J. Hyg. (Lond) 54, 351–364. doi: 10.1017/s0022172400044612
Carmichael, L. E., and Bruner, D. W. (1968). Characteristics of a newly-recognized species of Brucella responsible for infectious canine abortions. Cornell Vet. 48, 579–592.
Cook, I., Campbell, R. W., and Barrow, G. (1966). Brucellosis in North Queensland rodents. Aust. Vet. J. 42, 5–8. doi: 10.1111/j.1751-0813.1966.tb04603.x
Cunningham, S. A., and Patel, R. (2013). Importance of using Bruker’s security-relevant library for Biotyper identification of Burkholderia pseudomallei, Brucella species, and Francisella tularensis. J. Clin. Microbiol. 51, 1639–1640. doi: 10.1128/JCM.00267-13
Eisenberg, T., Hamann, H.-P., Kaim, U., Schlez, K., Seeger, H., Schauerte, N., et al. (2012). Isolation of potentially novel Brucella spp. from frogs. Appl. Environ. Microbiol. 78, 3753–3755. doi: 10.1128/aem.07509-11
Eisenberg, T., Riße, K., Schauerte, N., Geiger, C., Blom, J., and Scholz, H. C. (2017). Isolation of a novel ‘atypical’ Brucella strain from a bluespotted ribbontail ray (Taeniura lymma). Antonie Van Leeuwenhoek 110, 221–234. doi: 10.1007/s10482-016-0792-4
Euzeby, J. P. (1997). List of bacterial names with standing in nomenclature: a folder available on the Internet. Int. J. Syst. Bacteriol. 47, 590–592. doi: 10.1099/00207713-47-2-590
Foster, G., Osterman, B. S., Godfroid, J., Jacques, I., and Cloeckaert, A. (2007). Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int. J. Syst. Evol. Microbiol. 57, 2688–2693. doi: 10.1099/ijs.0.65269-0
Gazolla Volpiano, C., Sant, H., Anna, F., Ambrosini, A., Brito Lisboa, B., et al. (2019). Reclassification of Ochrobactrum lupini as a later heterotypic synonym of Ochrobactrum anthropi based on whole-genome sequence analysis. Int. J. Syst. Evol. Microbiol. 69, 2312–2314. doi: 10.1099/ijsem.0.003465
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., et al. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Gray, P. H. H., and Thornton, H. G. (1928). Soil bacteria that decompose certain aromatic compounds. Zentralbl. Bakteriol. Parasitenkd. Infektionskr. Hyg. Abteilung II. 73, 74–96.
Guzmán-Verri, C., Suárez-Esquivel, M., Ruíz-Villalobos, N., Zygmunt, M. S., Gonnet, M., Campos, E., et al. (2019). Genetic and phenotypic characterization of the etiological agent of canine orchiepididymitis smooth Brucella sp. BCCN84.3. Front. Vet. Sci. 6:175. doi: 10.3389/fvets.2019.00175
Holmes, B., Popoff, M., Kiredjian, M., and Kersters, K. (1988). Ochrobactrum anthropi gen. nov., sp. nov. from human clinical specimens and previously known as group Vd. Int. J. Syst. Evol. Microbiol. 38, 406–416. doi: 10.1099/00207713-38-4-406
Huber, B., Scholz, H. C., Kämpfer, P., Falsen, E., Langer, S., and Busse, H. J. (2010). Ochrobactrum pituitosum sp. nov., isolated from an industrial environment. Int. J. Syst. Evol. Microbiol. 60, 321–326. doi: 10.1099/ijs.0.011668-0
Huddleson, I. F. (1929). The differentiation of the species of genus Brucella. Michigan State Coll. Agric. Exp. Station Tech. Bull. 100, 1–16.
Imran, A., Hafeez, F. Y., Frühling, A., Schumann, P., Malik, K. A., and Stackebrandt, E. (2010). Ochrobactrum ciceri sp. nov., isolated from nodules of Cicer arietinum. Int. J. Syst. Evol. Microbiol. 60, 1548–1553. doi: 10.1099/ijs.0.013987-0
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Kämpfer, P., Buczolits, S., Albrecht, A., Busse, H.-J., and Stackebrandt, E. (2003). Towards a standardized format for the description of a novel species (of an established genus): Ochrobactrum gallinifaecis sp. nov. Int. J. Syst. Evol. Microbiol. 53, 893–896. doi: 10.1099/ijs.0.02710-0
Kämpfer, P., Glaeser, S., Busse, H.-J., Eisenberg, T., and Scholz, H. (2013). Falsochrobactrum ovis gen. nov., sp. nov., isolated from a sheep. Int. J. Syst. Evol. Microbiol. 63, 3841–3847. doi: 10.1099/ijs.0.049627-0
Kämpfer, P., and Glaeser, S. P. (2019). “Brucellaceae,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, et al. (Hoboken, NJ: John Wiley & Sons, Inc). doi: 10.1002/9781118960608.fbm00166.pub2
Kämpfer, P., Glaeser, S. P., and Holmes, B. (2018). “Ochrobactrum,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, et al. (Hoboken, NJ: John Wiley & Sons, Inc). doi: 10.1002/9781118960608.gbm00809.pub2
Kämpfer, P., Huber, B., Busse, H. J., Scholz, H. C., Tomaso, H., Hotzel, H., et al. (2011). Ochrobactrum pecoris sp. nov., isolated from farm animals. Int. J. Syst. Evol. Microbiol. 61, 2278–2283. doi: 10.1099/ijs.0.027631-0
Kämpfer, P., Huber, B., Lodders, N., Warfolomeow, I., Busse, H.-J., and Scholz, H. C. (2009). Pseudochrobactrum lubricantis sp. nov., isolated from a metal-working fluid. Int. J. Syst. Evol. Microbiol. 59, 2464–2467. doi: 10.1099/ijs.0.008540-0
Kämpfer, P., Martin, E., Lodders, N., Jackel, U., Huber, B. E., Schumann, P., et al. (2010). Paenochrobactrum gallinarii gen. nov., sp. nov., isolated from air of a duck barn, and reclassification of Pseudochrobactrum glaciei as Paenochrobactrum glaciei comb. nov. Int. J. Syst. Evol. Microbiol. 60, 1493–1498. doi: 10.1099/ijs.0.015842-0
Kämpfer, P., Poppel, M. T., Wilharm, G., Glaeser, S. P., and Busse, H. J. (2014). Paenochrobactrum pullorum sp. nov. isolated from a chicken. Int. J. Syst. Evol. Microbiol. 64, 1724–1728. doi: 10.1099/ijs.0.061101-0
Kämpfer, P., Rosselló-Mora, R., Scholz, H. C., Welinder-Olsson, C., Falsen, E., and Busse, H.-J. (2006). Description of Pseudochrobactrum gen. nov., with the two species Pseudochrobactrum asaccharolyticum sp. nov. and Pseudochrobactrum saccharolyticum sp. nov. Int. J. Syst. Evol. Microbiol. 56, 1823–1829. doi: 10.1099/ijs.0.64256-0
Kämpfer, P., Scholz, H. C., Huber, B., Falsen, E., and Busse, H.-J. (2007a). Ochrobactrum haematophilum sp. nov. and Ochrobactrum pseudogrignonense sp. nov., isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 57, 2513–2518. doi: 10.1099/ijs.0.65066-0
Kämpfer, P., Scholz, H., Huber, B., Thummes, K., Busse, H.-J., Maas, E. W., et al. (2007b). Description of Pseudochrobactrum kiredjianiae sp. nov. Int. J. Syst. Evol. Microbiol. 57, 755–760. doi: 10.1099/ijs.0.64714-0
Kämpfer, P., Sessitsch, A., Schloter, M., Huber, B., Busse, H. J., and Scholz, H. C. (2008). Ochrobactrum rhizosphaerae sp. nov. and Ochrobactrum thiophenivorans sp. nov., isolated from the environment. Int. J. Syst. Evol. Microbiol. 58, 1426–1431. doi: 10.1099/ijs.0.65407-0
Katoh, K., Misawa, K., Kuma, K.-I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Krzyzanowska, D. M., Maciąg, T., Ossowicki, A., Rajewska, M., Kaczyński, Z., Czerwicka, M., et al. (2019). Ochrobactrum quorumnocens sp. nov., a quorum quenching bacterium from the potato rhizosphere, and comparative genome analysis with related type strains. PLoS One 14:e0210874. doi: 10.1371/journal.pone.0210874
Lebuhn, M., Achouak, W., Schloter, M., Berge, O., Meier, H., Barakat, M., et al. (2000). Taxonomic characterization of Ochrobactrum sp. isolates from soil samples and wheat roots, and description of Ochrobactrum tritici sp. nov. and Ochrobactrum grignonense sp. nov. Int. J. Syst. Evol. Microbiol. 50, 2207–2223. doi: 10.1099/00207713-50-6-2207
Leclercq, S. O., Cloeckaert, A., and Zygmunt, M. S. (2019). Taxonomic organization of the family Brucellaceae based on a phylogenomic approach. Front. Microbiol. 10:3083. doi: 10.3389/fmicb.2019.03083
Li, L., Li, Y. Q., Jiang, Z., Gao, R., Nimaichand, S., Duan, Y. Q., et al. (2016). Ochrobactrum endophyticum sp. nov., isolated from roots of Glycyrrhiza uralensis. Arch. Microbiol. 198, 171–179. doi: 10.1007/s00203-015-1170-8
Linhart, C., and Shamir, R. (2007). Degenerate primer design: theoretical analysis and the HYDEN program. Methods Mol. Biol. 402, 221–244. doi: 10.1007/978-1-59745-528-2_11
Meyer, K. F., and Shaw, E. B. (1920). A comparison of the morphological, cultural and biochemical characteristics of B. abortus and B. melitensis, Studies on the genus Brucella nov. gen. I. J. Infect. Dis. 27, 173–184.
Mühldorfer, K., Wibbelt, G., Szentiks, C. A., Fischer, D., Scholz, H. C., Zschöck, M., et al. (2017). The role of ‘atypical’ Brucella in amphibians: are we facing novel emerging pathogens? J. Appl. Microbiol. 122, 40–53. doi: 10.1111/jam.13326
Osterman, B., and Moriyón, I. (2006). International committee on systematics of prokaryotes; subcommittee on the taxonomy of Brucella. Int. J. Syst. Evol. Microbiol. 56, 1173–1175. doi: 10.1099/ijs.0.64349-0
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pappas, G., Papadimitriou, P., Akritidis, N., Christou, L., and Tsianos, E. V. (2006). The new global map of human brucellosis. Lancet Infect. Dis. 6, 91–99. doi: 10.1016/S1473-3099(06)70382-6
Parte, A. C. (2014). LPSN–list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, D613–D616. doi: 10.1093/nar/gkt1111
Romanenko, L. A., Tanaka, N., Frolova, G. M., and Mikhailov, V. V. (2008). Pseudochrobactrum glaciei sp. nov., isolated from sea ice collected from Peter the Great Bay of the Sea of Japan. Int. J. Syst. Evol. Microbiol. 58, 2454–2458. doi: 10.1099/ijs.0.65828-0
Romano, S., Aujoulat, F., Jumas-Bilak, E., Masnou, A., Jeannot, J.-L., Falsen, E., et al. (2009). Multilocus sequence typing supports the hypothesis that Ochrobactrum anthropi displays a human-associated subpopulation. BMC Microbiol. 9:267. doi: 10.1186/1471-2180-9-267
Scholz, H. C., Banai, M., Cloeckaert, A., Kämpfer, P., and Whatmore, A. M. (2018). “Brucella,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, P. DeVos, et al. (Hoboken, NJ: John Wiley & Sons, Inc). doi: 10.1002/9781118960608.gbm00807.pub2
Scholz, H. C., Al Dahouk, S., Tomaso, H., Neubauer, H., Witte, A., Schloter, M., et al. (2008a). Genetic diversity and phylogenetic relationships of bacteria belonging to the Ochrobactrum-Brucella group by recA and 16S rRNA gene-based comparative sequence analysis. Syst. Appl. Microbiol. 31, 1–16. doi: 10.1016/j.syapm.2007.10.004
Scholz, H. C., Hubalek, Z., Sedláèek, I., Vergnaud, G., Tomaso, H., Al Dahouk, S., et al. (2008b). Brucella microti sp. nov., isolated from the common vole Microtus arvalis. Int. J. Syst. Evol. Microbiol. 58, 375–382. doi: 10.1099/ijs.0.65356-0
Scholz, H. C., Nöckler, K., Göllner, C., Bahn, P., Vergnaud, G., Tomaso, H., et al. (2010). Brucella inopinata sp. nov., isolated from a breast implant infection. Int. J. Syst. Evol. Microbiol. 60, 801–808. doi: 10.1099/ijs.0.011148-0
Scholz, H. C., Revilla-Fernandez, S., Al Dahouk, S., Hammerl, J. A., Zygmunt, M. S., Cloeckaert, A., et al. (2016a). Brucella vulpis sp nov., isolated from mandibular lymph nodes of red foxes (Vulpes vulpes). Int. J. Syst. Evol. Microbiol. 66, 2090–2098. doi: 10.1099/ijsem.0.000998
Scholz, H. C., Muehldorter, K., Shilton, C., Benedict, S., Whatmore, A. M., Blom, J., et al. (2016b). The change of a medically important genus: worldwide occurrence of genetically diverse novel Brucella species in exotic frogs. PLoS One 11:e0168872. doi: 10.1371/journal.pone.0168872
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Soler-Lloréns, P. F., Quance, C. R., Lawhon, S. D., Stuber, T. P., Edwards, J. F., Ficht, T. A., et al. (2016). A Brucella spp. isolate from a Pac-Man Frog (Ceratophrys ornata) reveals characteristics departing from classical Brucellae. Front. Cell. Infect. Microbiol. 6:116. doi: 10.3389/fcimb.2016.00116
Stoenner, H. G., and Lackman, D. B. (1957). A new species of Brucella isolated from the desert wood rat, Neotoma lepida Thomas. Am. J. Vet. Res. 18, 947–951.
Sun, L., Yao, L., Gao, X., Huang, K., Bai, N., Lyu, W., et al. (2019). Falsochrobactrum shanghaiense sp. nov., isolated from paddy soil and emended description of the genus Falsochrobactrum. Int. J. Syst. Evol. Microbiol. 69, 778–782. doi: 10.1099/ijsem.0.003236
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Teyssier, C., Marchandin, H., Jean-Pierre, H., Masnou, A., Dusart, G., and Jumas-Bilak, E. (2007). Ochrobactrum pseudintermedium sp. nov., a novel member of the family Brucellaceae, isolated from human clinical samples. Int. J. Syst. Evol. Microbiol. 57, 1007–1013. doi: 10.1099/ijs.0.64416-0
Tiller, R. V., Gee, J. E., Frace, M. A., Taylor, T. K., Setubal, J. C., Hoffmaster, A. R., et al. (2010a). Characterization of novel Brucella strains originating from wild native rodent species in North Queensland, Australia. Appl. Environ. Microbiol. 76, 5837–5845. doi: 10.1128/aem.00620-10
Tiller, R. V., Gee, J. E., Lonsway, D. R., Gribble, S., Bell, S. C., Jennison, A. V., et al. (2010b). Identification of an unusual Brucella strain (BO2) from a lung biopsy in a 52 year-old patient with chronic destructive pneumonia. BMC Microbiol. 10:23. doi: 10.1186/1471-2180-10-23
Tripathi, A. K., Verma, S. C., Chowdhury, S. P., Lebuhn, M., Gattinger, A., and Schloter, M. (2006). Ochrobactrum oryzae sp. nov., an endophytic bacterial species isolated from deep-water rice in India. Int. J. Syst. Evol. Microbiol. 56, 1677–1680. doi: 10.1099/ijs.0.63934-0
Trujillo, M. E., Willems, A., Abril, A., Planchuelo, A.-M., Rivas, R., Ludeńa, D., et al. (2005). Nodulation of Lupinus albus by strains of Ochrobactrum lupini sp. nov. Appl. Environ. Microbiol. 71, 1318–1327. doi: 10.1128/AEM.71.3.1318-1327.2005
Urakami, T., Oyanagi, H., Araki, H., Suzuki, K.-I., and Komagata, K. (1990). Recharacterization and emended description of the genus Mycoplana and description of two new species, Mycoplana ramosa and Mycoplana segnis. Int. J. Syst. Evol. Microbiol. 40, 434–442. doi: 10.1099/00207713-40-4-434
Varghese, N. J., Mukherjee, S., Ivanova, N., Konstantinidis, K. T., Mavrommatis, K., Kyrpides, N. C., et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771. doi: 10.1093/nar/gkv657
Velasco, J., Romero, C., Lopez-Goni, I., Leiva, J., Diaz, R., and Moriyon, I. (1998). Evaluation of the relatedness of Brucella spp. and Ochrobactrum anthropi and description of Ochrobactrum intermedium sp. nov., a new species with a closer relationship to Brucella spp. Int. J. Syst. Bacteriol. 48, 759–768. doi: 10.1099/00207713-48-3-759
Verger, J.-M., Grimont, F., Grimont, P. A. D., and Grayon, M. (1985). Brucella, a monospecific genus as shown by deoxyribonucleic acid hybridization. Int. J. Syst. Evol. Microbiol. 35, 292–295. doi: 10.1099/00207713-35-3-292
Wattam, A. R., Foster, J. T., Mane, S. P., Beckstrom-Sternberg, S. M., Beckstrom-Sternberg, J. M., Dickerman, A. W., et al. (2014). Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J. Bacteriol. 196, 920–930. doi: 10.1128/jb.01091-13
Wattam, A. R., Inzana, T. J., Williams, K. P., Mane, S. P., Shukla, M., Almeida, N. F., et al. (2012). Comparative genomics of early-diverging Brucella strains reveals a novel lipopolysaccharide biosynthesis pathway. mBio 3:e00246-12. doi: 10.1128/mBio.00388-12
Whatmore, A. M., Dale, E. J., Stubberfield, E., Muchowski, J., Koylass, M., Dawson, C., et al. (2015). Isolation of Brucella from a White’s tree frog (Litoria caerulea). JMM Case Rep. 2015:2. doi: 10.1099/jmmcr.0.000017
Whatmore, A. M., Davison, N., Cloeckaert, A., Al Dahouk, S., Zygmunt, M. S., Brew, S. D., et al. (2014). Brucella papionis sp. nov., isolated from baboons (Papio spp.). Int. J. Syst. Evol. Microbiol. 64, 4120–4128. doi: 10.1099/ijs.0.065482-0
Whatmore, A. M., Koylass, M. S., Muchowski, J., Edwards-Smallbone, J., Gopaul, K. K., and Perrett, L. L. (2016). Extended multilocus sequence analysis to describe the global population structure of the genus Brucella: phylogeography and relationship to biovars. Front. Microbiol. 7:2049. doi: 10.3389/fmicb.2016.02049
Whatmore, A. M., Perrett, L. L., and MacMillan, A. P. (2007). Characterisation of the genetic diversity of Brucella by multilocus sequencing. BMC Microbiol. 7:34. doi: 10.1186/1471-2180-7-34
Woo, S. G., Ten, L. N., Park, J., and Lee, M. (2011). Ochrobactrum daejeonense sp. nov., a nitrate-reducing bacterium isolated from sludge of a leachate treatment plant. Int. J. Syst. Evol. Microbiol. 61, 2690–2696. doi: 10.1099/ijs.0.025510-0
Wragg, P., Randall, L., and Whatmore, A. M. (2014). Comparison of Biolog GEN III MicroStation semi-automated bacterial identification system with matrix-assisted laser desorption ionization-time of flight mass spectrometry and 16S ribosomal RNA gene sequencing for the identification of bacteria of veterinary interest. J. Microbiol. Methods 105, 16–21. doi: 10.1016/j.mimet.2014.07.003
Yoon, J. H., Kang, S. J., Park, S., and Oh, T. K. (2008). Daeguia caeni gen. nov., sp. nov., isolated from sludge of a textile dye works. Int. J. Syst. Evol. Microbiol. 58, 168–172. doi: 10.1099/ijs.0.65483-0
Keywords: Brucella, Brucellaceae, multi-locus locus sequence analysis, pan-family, phylogeny, Ochrobactrum
Citation: Ashford RT, Muchowski J, Koylass M, Scholz HC and Whatmore AM (2020) Application of Whole Genome Sequencing and Pan-Family Multi-Locus Sequence Analysis to Characterize Relationships Within the Family Brucellaceae. Front. Microbiol. 11:1329. doi: 10.3389/fmicb.2020.01329
Received: 22 November 2019; Accepted: 25 May 2020;
Published: 14 July 2020.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
David O’Callaghan, Université de Montpellier, FranceMartin W. Hahn, University of Innsbruck, Austria
Copyright © 2020 Ashford, Muchowski, Koylass, Scholz and Whatmore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland T. Ashford, Um9sYW5kLkFzaGZvcmRAYXBoYS5nb3YudWs=
†Present address: Mark Koylass, AstraZeneca, Cambridge, United Kingdom