
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 May 2020
Sec. Infectious Agents and Disease
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00965
This article is part of the Research Topic Factors Affecting Host Selection by Mosquitoes: Implications for the Transmission of Vector-Borne Pathogens View all 12 articles
 Krajana Tainchum1,2†
Krajana Tainchum1,2† Chloé Dupont3,4†
Chloé Dupont3,4† Theeraphap Chareonviriyaphap2
Theeraphap Chareonviriyaphap2 Estelle Jumas-Bilak3,4
Estelle Jumas-Bilak3,4 Michael J. Bangs2,5
Michael J. Bangs2,5 Sylvie Manguin3*
Sylvie Manguin3*Among the complex microbial community living in the mosquito midgut, some bacteria (e.g., Enterobacter spp.) can deliver effector molecules with anti-Plasmodium effects suppressing the development of malaria parasites (Plasmodium falciparum) before the öokinete can penetrate the mosquito midgut epithelium. Despite knowledge of this phenomenon, only a few studies have defined the diversity of microbiota in wild-caught adult Anopheles species. The objective of this study was to analyze and compare the bacterial microbiota in different Anopheles species, including representatives of the primary malaria vectors in western Thailand. Wild female Anopheles species were sampled from malaria-endemic areas in Tak and Mae Hong Son provinces near the Thai-Myanmar border. Midgut/abdominal bacterial diversity was assessed by examining the 16S rRNA gene, V3 hypervariable region, using PCR-Temporal Temperature Gel Electrophoresis (PCR-TTGE) profiling and sequence analysis. A total of 24 bacterial genera were identified from eight Anopheles species. Five bacterial genera were newly reported in Anopheles mosquitoes (Ferrimonas, Megasphaera, Pectobacterium, Shimwellia, and Trabulsiella). Five genera, including Megasphaera, were detected exclusively in a single-malaria (Plasmodium vivax) infected Anopheles minimus and not observed in other non-infected mosquitoes. The use of PCR-TTGE provides the first characterization of the midgut bacterial microbiome present in wild adult Anopheles in Thailand. Evidence that microbiota might impact pathogen development (suppression) in Anopheles and thereby reduce the risk of pathogen transmission deserves more studies to describe the presence and better understand the biological role of bacteria in natural mosquito populations.
Despite significant progress in the control of malaria throughout the country, Thailand remains malaria-endemic, particularly along the international borders with Cambodia, Lao PDR, Malaysia, and Myanmar (DDC, 2018). The vast majority of recorded malaria cases, primarily Plasmodium vivax (73%) and Plasmodium falciparum (18%), occur along the Thai-Myanmar border (70–80%), especially in Tak and Mae Hong Son provinces (DDC, 2018). Of the 79 recognized Anopheles species present in Thailand, the most important malaria vectors include two sibling species in the Dirus Complex (Anopheles baimaii Sallum & Peyton, and Anopheles dirus Peyton & Harrison), Anopheles minimus Theobald, Anopheles aconitus Dönitz, and three members of the Maculatus Group (Anopheles maculatus Theobald, Anopheles pseudowillmori [Theobald], and Anopheles sawadwongporni Rattanarithikul & Green) (Tananchai et al., 2019).
The malaria sporogonic cycle begins when a female mosquito ingests infective-stage (gametocytes) Plasmodium parasites from the blood of an infected human host. The ingested parasites undergo syngamy to create motile stage öokinetes that penetrate the mosquito midgut to become oocysts. The oocyst gradually enlarges as parasites multiply to produce many sporozoites. Once released from the oocyst, sporozoites migrate to the salivary glands to await transmission to another host via a mosquito bite. Although many Plasmodium gametocytes can be ingested during a single blood-feeding, only a small fraction typically develop to form oocysts (Leroy et al., 2014). The reason for this sharp decrease in potential infection density is multifactorial (genetic and non-genetic) and in large part influenced by the parasite-vector species relationship and individual mosquito susceptibility (competence) to Plasmodium infection allowing successful sporogonic development of the parasite while minimizing significant adverse effects on mosquito fitness (survival, fecundity, etc.) (Black and Severson, 2005; Lefevre et al., 2013). One area of research on this relationship has been the modulating effects linked to certain naturally occurring microbiota in the midgut and abdomen of mosquitoes that can suppress or prevent Plasmodium development (Cirimotich et al., 2011b). In particular, the role of enterobacteria that influences parasite development and transmission has been investigated in Anopheles mosquitoes. Additional investigations on this phenomenon may help to develop novel methods involving bacterial symbionts to arrest malaria from vector to host.
Symbiotic bacteria, such as Pantoea agglomerans and Asaia spp., have been successfully transformed to express anti-malaria molecules (anti-plasmodia effector proteins) that render host mosquitoes refractory to malaria infection (Favia et al., 2008; Damiani et al., 2010; Wang et al., 2012); in effect, becoming a paratransgenic means for preventing malaria transmission (i.e., transmission-blocking strategy) (Doumbo et al., 2018). Engineered P. agglomerans strains can inhibit up to 98% of P. falciparum development in infected mosquitoes (Riehle et al., 2007). Enterobacter (Esp_Z) is shown to inhibit öokinete, oocyst, and sporozoite formation of P. falciparum in Anopheles gambiae by up to 99% (Cirimotich et al., 2011b). Co-infections with Serratia marcescens and P. vivax in Anopheles albimanus have resulted in only 1% of mosquitoes being able to develop oocysts and complete sporogonic development (Minard et al., 2013). In another study, Serratia marcescens Y1 strain isolated from field-collected female Anopheles, induced anti-plasmodia factors that activated the immune system in Anopheles stephensi effectively rendering the mosquito resistant to Plasmodium berghei infection (Bai et al., 2019).
The bacterial biodiversity in nine species of field-collected Anopheles in Thailand and Vietnam demonstrated complex microbiota in the mosquito midgut and abdomen, primarily Gram-negative bacterial rods, including Serratia marcescens, Klebsiella ozaenae, Pseudomonas aeruginosa, Escherichia coli, and Enterobacter spp. (Manguin et al., 2013). Other studies have reported the majority of adult mosquito midgut microbiota were Gram-negative species in the phylum Proteobacteria (Tandina et al., 2016; Zoure et al., 2020). At least three mosquito-specific bacterial species have been isolated from the midgut of African malaria vectors in the Gambiae Complex, including Thorsellia anophelis, Janibacter anophelis (Kampfer et al., 2006) and Elizabethkingia anophelis (Kampfer et al., 2011). Despite a large amount of work done on malaria vectors over many decades, few studies have examined the natural diversity of microbiota in adult mosquitoes (Manguin et al., 2013; Bassene et al., 2018). Therefore, the objective of this study is to use a sensitive molecular method to evaluate the natural bacterial diversity in wild-caught adult Anopheles in a malaria-endemic region of western Thailand.
Human use protocol (human-landing collections) for this study was reviewed and approved by the Ethical Research Committee, Chulalongkorn University, Bangkok, Thailand (No. 0961/56).
In 2011, adult Anopheles mosquitoes were collected from two hypoendemic malaria locations along the Thai-Myanmar border in Tak and Mae Hong Son provinces, respectively (Figure 1), using a standard human-landing collection technique (Tainchum et al., 2014). Table 1 provides geographic locations, prevailing climatic factors, environment biotype, and crude malaria incidence rate (2010–2012). The abundance of the primary malaria vectors, An. minimus, An. dirus complex, and An. maculatus group, by month of collection (from February to November 2011) and location, is presented in Figure 2. Live mosquitoes were initially sorted using discriminating morphological criteria for species, complex or group level identification (Rattanarithikul et al., 2006). For further mosquito identification, Anopheles specimens were assayed using the appropriate allele-specific PCR technique by species complex or group (Tainchum et al., 2014). Each mosquito was divided in two parts, head-thorax for mosquito species identification and detection of malaria (Plasmodium) infection, and abdomen retained for bacteriological analysis. Abdomens were stored at −80°C until further processing.
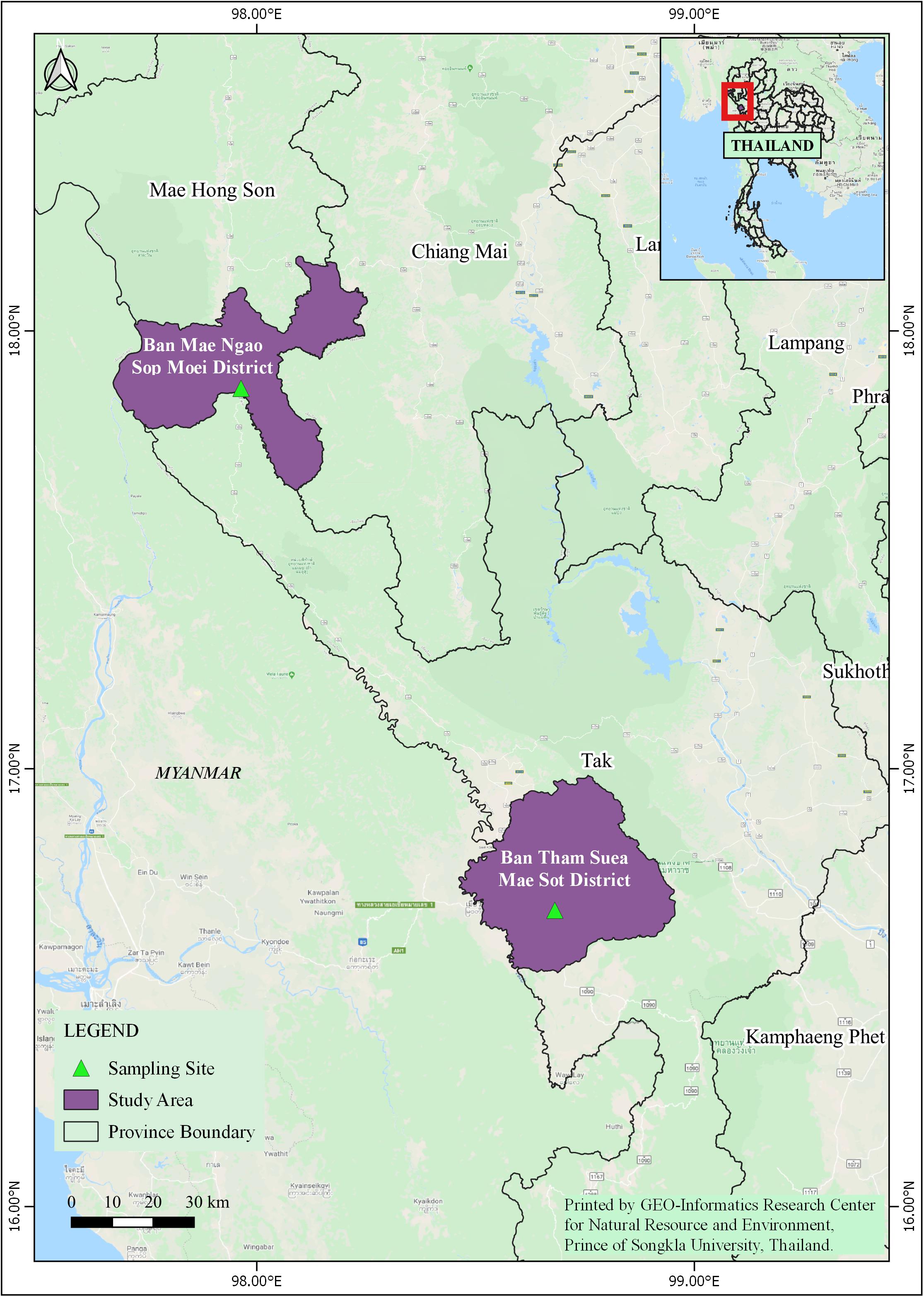
Figure 1. Study site locations in Mae Sot District (Tak Province) and Sop Moei District (Mae Hong Son Province), western Thailand.
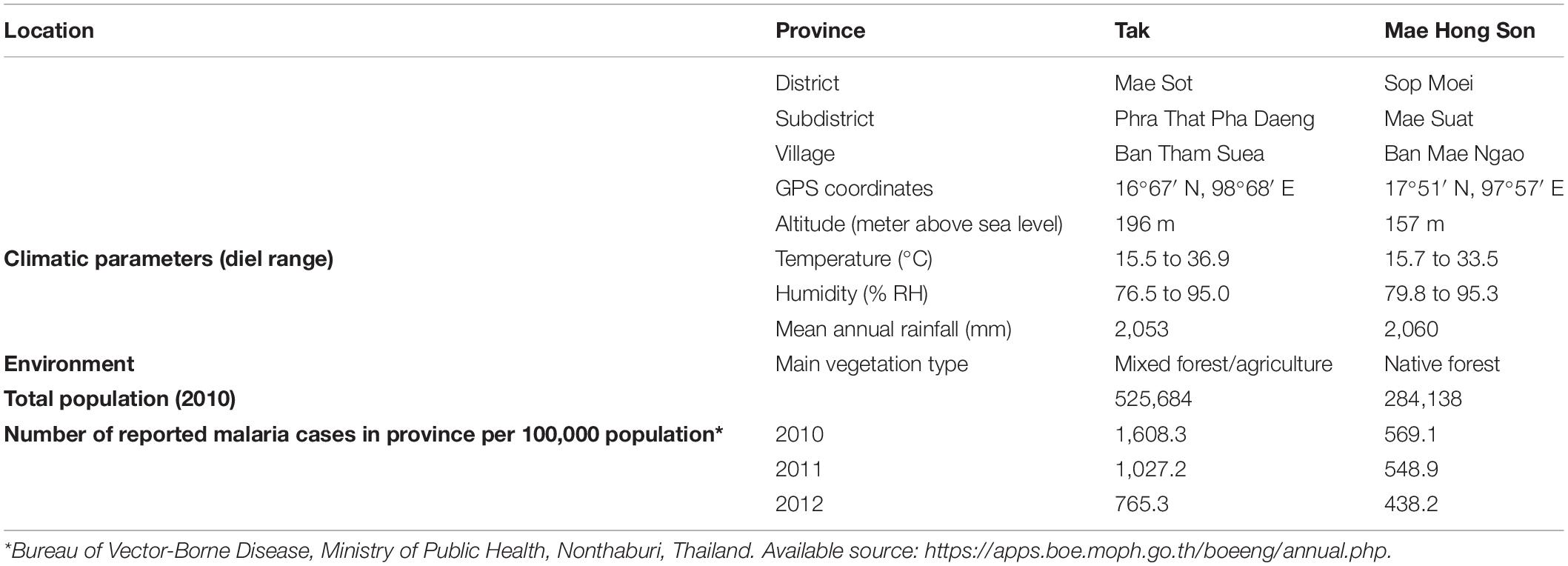
Table 1. Background information on sampled locations with malaria transmission.
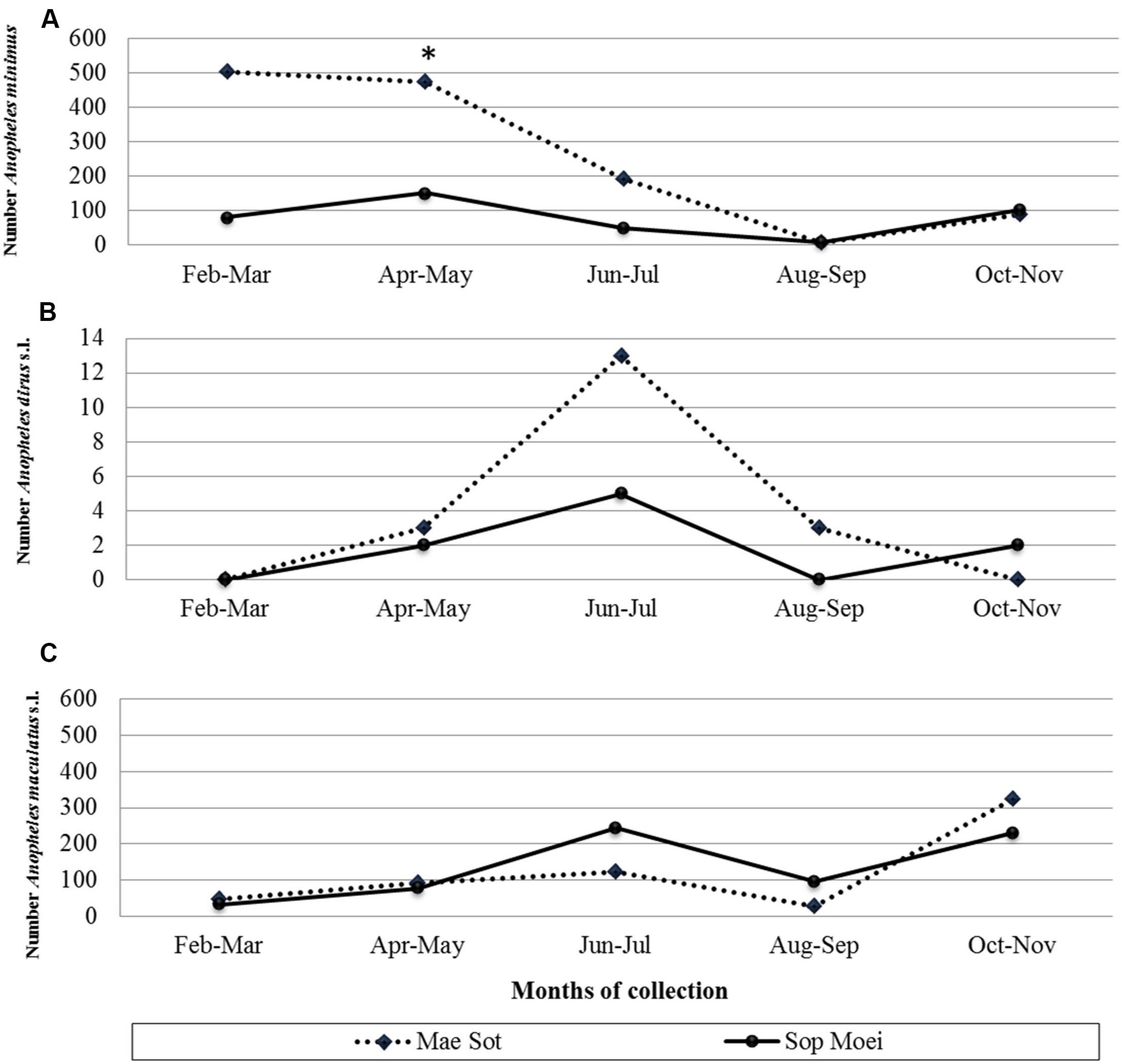
Figure 2. Number of (A) Anopheles minimus (scale from 0 to 600 specimens), (B) An. dirus complex (scale from 0 to 14 specimens), and (C) An. maculatus group (scale from 0 to 600 specimens) mosquitoes collected by location and month (February to November 2011). *, month (April) and location (Mae Sot) where the vivax-infected An. minimus was collected.
Plasmodium detection used an aliquot of the head-thorax DNA extraction used for Anopheles species identification. A Roche LightCycler480 (Software Version LCS480 1.5.0.39) was performed for real-time PCR with TaqMan reagents and hydrolysis probes for the detection of P. falciparum, P. vivax, and P. knowlesi, following a slightly modified methodology of Divis et al. (2010).
Each abdomen was rinsed twice in purified water (from sealed injectable solution) before complete disruption using a tissue crusher device in 150 μL of TE buffer. DNA was extracted using the Master Pure Gram Positive DNA purification kit following supplier instructions (Epicentre Biotechnologies, Madison WI, United States).
For each sample, the V2–V3 region of the 16S rRNA bacteria gene was amplified using the primers HDA1/HDA2 (Roudière et al., 2009); HDA1: 5′-ACTC CTA CGG GAG GCA GCA GT-3′, HDA2: 5′-GTA TTA CCG CGG CTG CTG GCA-3′. A 40-bp clamp, named GC (5′-CGC CCG GGG CGC GCC CCG GGC GGG GCG GGG GCA CGG GGG G-3′) flanked the 5′ extremity of HDA1 (Ogier et al., 2002) to form HDA1-GC. PCR was performed using an Eppendorf thermal cycler® (Eppendorf, Le Pecq, France) in sterile 0.5 ml tubes. The reaction mixture (50 μL) contained 2.5 units of Taq DNA Polymerase (FastStart High Fidelity PCR System, Roche, Meylan, France), 0.2 mM of each primer and 1 μl of DNA in the appropriate reaction buffer. The amplification cycle was 95°C for 2 min, 35 cycles at 95°C for 1 min, 62°C for 30 s, 72°C for 1 min, and 7 min at 72°C for the final extension step. Solutions were prepared using sterile DNA-free water with the addition of template DNA and gel electrophoresis of PCR products carried out in separate rooms to avoid any possible contamination. PCR amplification of DNA was detected by 2% agarose gel electrophoresis containing ethidium bromide and visualized under ultraviolet light.
Temporal temperature gel electrophoresis was performed using the DCode universal mutation detection system (Bio-Rad Laboratories, Marne-la-Coquette, France) with 16 cm × 16 cm × 1 mm gels (Roudière et al., 2009). The 60 ml gels were composed of 8% (w/v) bisacrylamide (37.5:1), 7 M urea, 60 μl of N,N,N′,N′-tetramethylethylenediamine (TEMED), and 0.1% (w/v) ammonium persulfate. Gels were run with 1X Tris-acetate-EDTA buffer at pH 8.4. A 5 μl PCR product was loaded on the gel with 5 μl in-house dye marker (50% saccharose 50%, 0.1% bromophenol blue) using capillary tips. Denaturing electrophoresis was performed at 46 V with a temperature ramp from 63 to 70°C during 16 h (increment 0.4°C/h) following a pre-migration step of 15 min at 20 V and 63°C. Gels were stained with ethidium bromide solution (5 μg/ml) for 20 min, washed with de-ionized water, and viewed using a UV transillumination system (Vilber Lourmat Sté, France) and photographed.
On each TTGE gel, approximately 50% of the bands were sequenced, while other bands were assigned to an affiliated operational taxonomic unit (OTU) by comparing their migration distance with that of sequenced bands. TTGE bands were excised and DNA eluted with 50 μl of elution buffer using Qiaquick PCR purification kit (Qiagen, Courtaboeuf, France) overnight at 37°C before PCR amplification with HDA1/HDA2 without GC clamp. The reaction conditions were identical to those described above. PCR products were sequenced using an ABI 3730xl sequencer (Cogenics, Meylan, France). Each sequencing chromatograph was visually inspected and corrected as appropriate.
The sequences were quality-checked using the SEQMATCH program in the 16S rDNA-specialized database, Ribosomal Database Project (RDP1). The 16S rDNA sequences were analyzed using the Basic Local Alignment Search Tool (BLAST) from the GenBank database2 and/or the RDP (Cole et al., 2005), for initial sample identifications. The reference sequence with the highest percentage was used for OTU affiliation. Clustered OTUs are based on 97% DNA sequence identity threshold of the 16S gene sequences to distinguish between bacteria at the genus level. A sequence was affiliated to a species-level OTU when the percent of sequence similarity was > 99% (as proposed by Drancourt et al., 2000). This value is above the recognized cut-off value standard for the delineation of species (Stackebrandt and Goebel, 1994), but warrants high stringency for species-level OTU affiliation. Below 99% similarity, the sequence is affiliated to the genus of the reference sequence having the highest percentage. When different species reference sequences match (or near equal), affiliation was done to the genus level. For example, 99.5% sequence similarity between species Aeromonas caviae and Aeromonas hydrophila was only assigned to the genus Aeromonas. Low cut-off is not defined for the genus delineation as affiliation to a higher taxonomic rank, e.g., family or order, was to be done considering the taxonomic frame of the clade using Greengenes database (McDonald et al., 2012).
The OTU means between the two different regions were compared using a Mann–Whitney U-test for the non-parametric test with the statistical significance level set at p < 0.05 using SPSS for Windows version 16 (Chicago, IL, United States).
A total of 190 Anopheles specimens, representing eight Anopheles species, were collected from Mae Sot (Tak Province) and Sop Moei (Mae Hong Son Province) (Table 1 and Figure 1). The peak collection numbers of mosquitoes (An. minimus, An. dirus complex, and An. maculatus group) occurred during the same periods in both locations but differed by species (Figure 2). For instance, in Mae Sot An. minimus had higher captures during February and April, while a more modest increase was seen in Sop Moei in May. Anopheles dirus s.l. peaked in June and July in Mae Sot and Sop Moei, respectively. For An. maculatus s.l., between both locations greater numbers were captured in June-July and October-November timeframes compared to other months (Figure 2). Within the Minimus Complex, only An. minimus was identified in association with very few specimens (n = 2) of a closely related species, An. aconitus (Funestus Group) (Table 2). Two species in the Dirus Complex, An. dirus and An. baimaii, and four Maculatus Group species, An. maculatus, An. sawadwongporni, An. pseudowillmori and An. dravidicus were molecularly identified (Table 2).
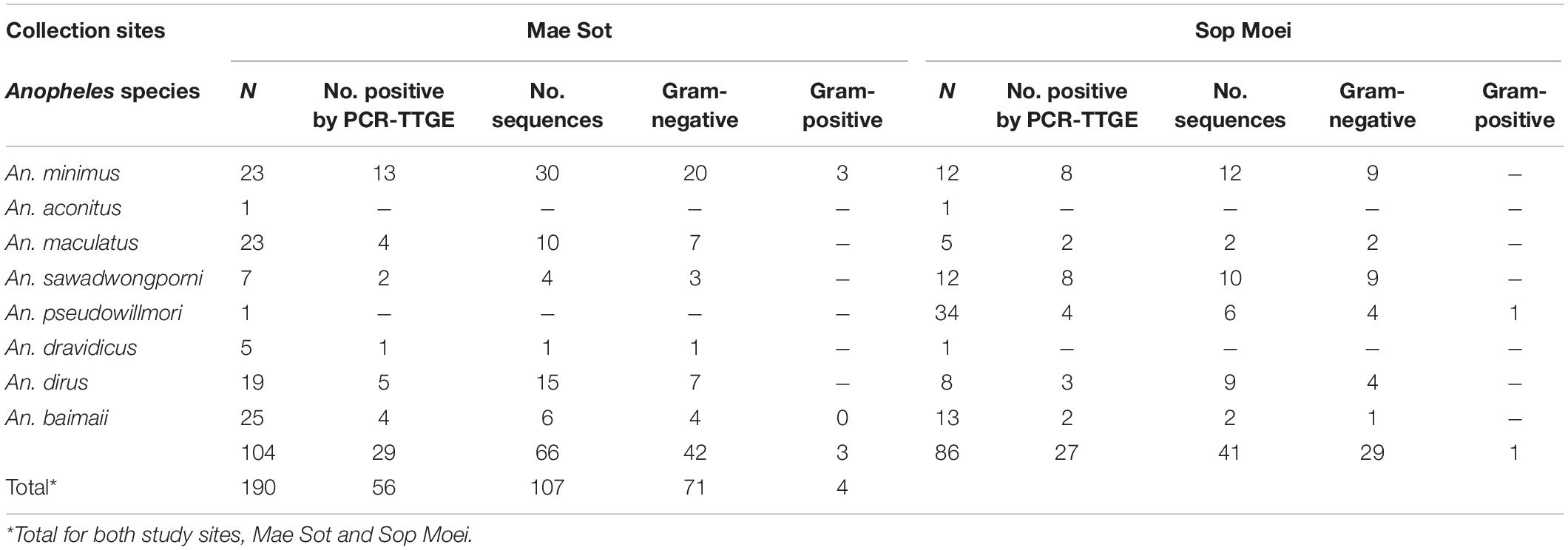
Table 2. Sample number and species of Anopheles assayed for abdominal bacteria, those found PCR-TGGE bacteria positive, and bacteria designated either Gram-negative or Gram-positive.
From 104 Anopheles assayed from Mae Sot and 86 from Sop Moei, only one (0.53%) mosquito, An. minimus, was found with malaria (P. vivax) sporozoites. This specimen was collected in Mae Sot (Tak Province) in April 2011 during the typical warm-dry season (Figure 2). The abdominal microbiota findings of all 190 assayed specimens, including the malaria-infected sample (Figure 3), was based on isolating the 16S rRNA gene using PCR-TTGE. In total, 107 sequences were obtained from 56 mosquitoes (30% of total sample) (Table 2).

Figure 3. PCR-TTGE banding patterns for bacterial genera detected in the abdomen of one naturally Plasmodium vivax infected An. minimus.
A raw diversity index that globally reflects the bacterial diversity in a sample is typically reflected by counting the resulting bands in TTGE profiles. The number of bands ranged from one to 10, suggesting that the bacterial diversity per mosquito also ranged from one to 10 OTUs. However, subsequent sequencing showed that bands with different migration distances could belong to the same OTU. This atypical phenomenon was observed for bacteria displaying sequence heterogeneity among the 16S rRNA gene copies. For example, most members of genera in the large family Enterobacteriaceae displayed a high level of 16S rRNA gene heterogeneity, thus producing complex banding patterns. Considering that Enterobacteriaceae were relatively common in our samples, the raw diversity index overestimated the actual bacterial diversity. Therefore, a refined diversity index was calculated after affiliation of each band to an OTU by sequencing or by a comparative analysis approach. The resulting index showed a bacterial diversity with an average of 1.7 OTU per specimen. The number of OTUs per specimen did not differ between mosquitoes in the two locations, with an average OTU of 1.69 and 1.7 per specimen in Mae Sot and Sop Moei, respectively (p = 0.345). Anopheles minimus hosted the majority of OTU identified in this study, 50% and 29% of OTU identified in Mae Sot and Sop Moei, respectively; however, it was also the most abundant Anopheles species captured (Figure 4).
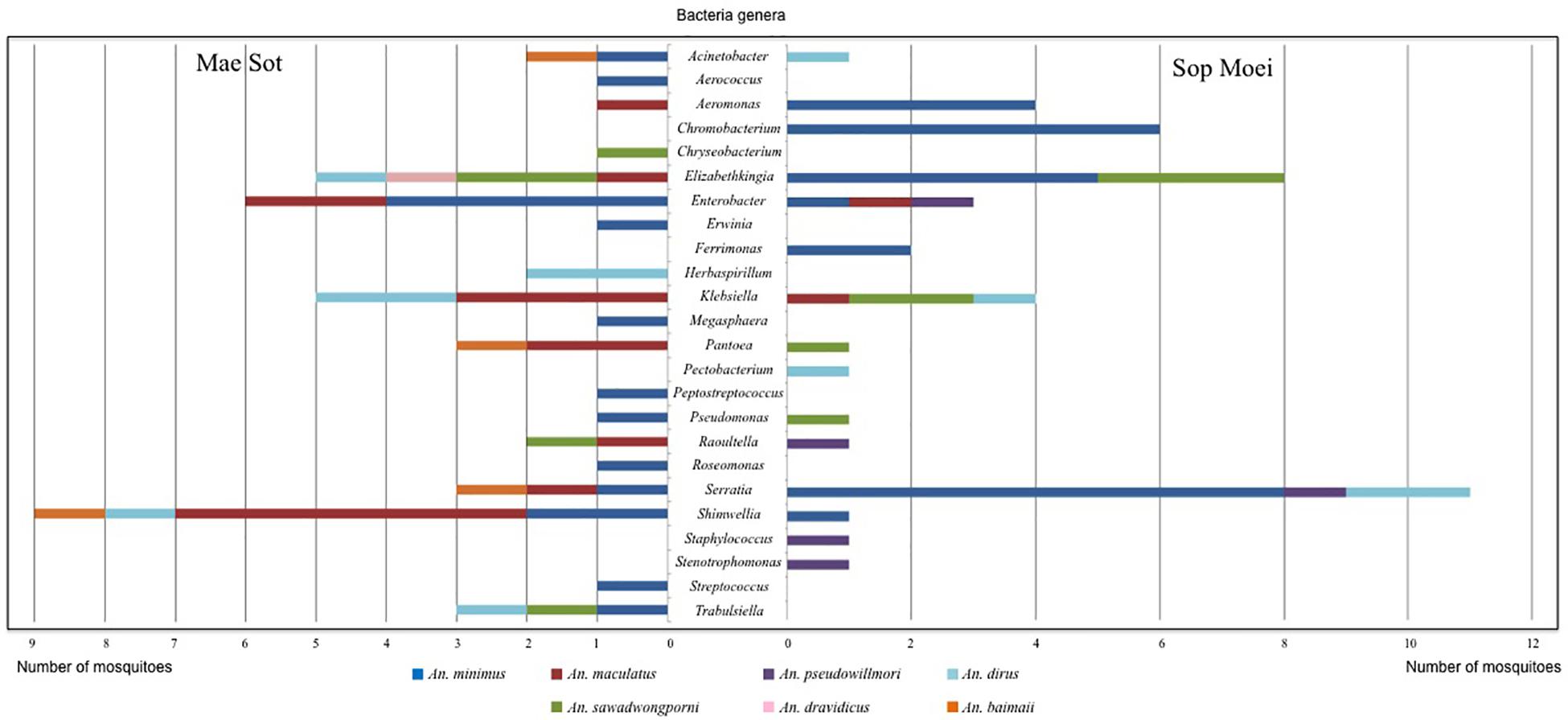
Figure 4. Bacterial genera identified in Anopheles species by PCR-TTGE.
The 16S rRNA gene PCR-TTGE that focused on the hypervariable V3 region produced sequences of approximately 200 bp, which are generally of size not informative enough for species-specific affiliation. Consequently, this study presents the bacterial diversity at the genus level. However, probable species affiliation was proposed for several genera when the phylogenetic signal of the V3 region was regarded significant. Contrasting with the low diversity per individual (average 1.7), collectively the OTU diversity in the entire sampling was high with 24 different bacterial genera distributed among three phyla; Proteobacteria (71%), Firmicutes (21%), and Bacteroidetes (8%) (Table 3). Proteobacteria, the predominant microbiota, encompassed the Alpha, Beta, and Gamma superclasses.
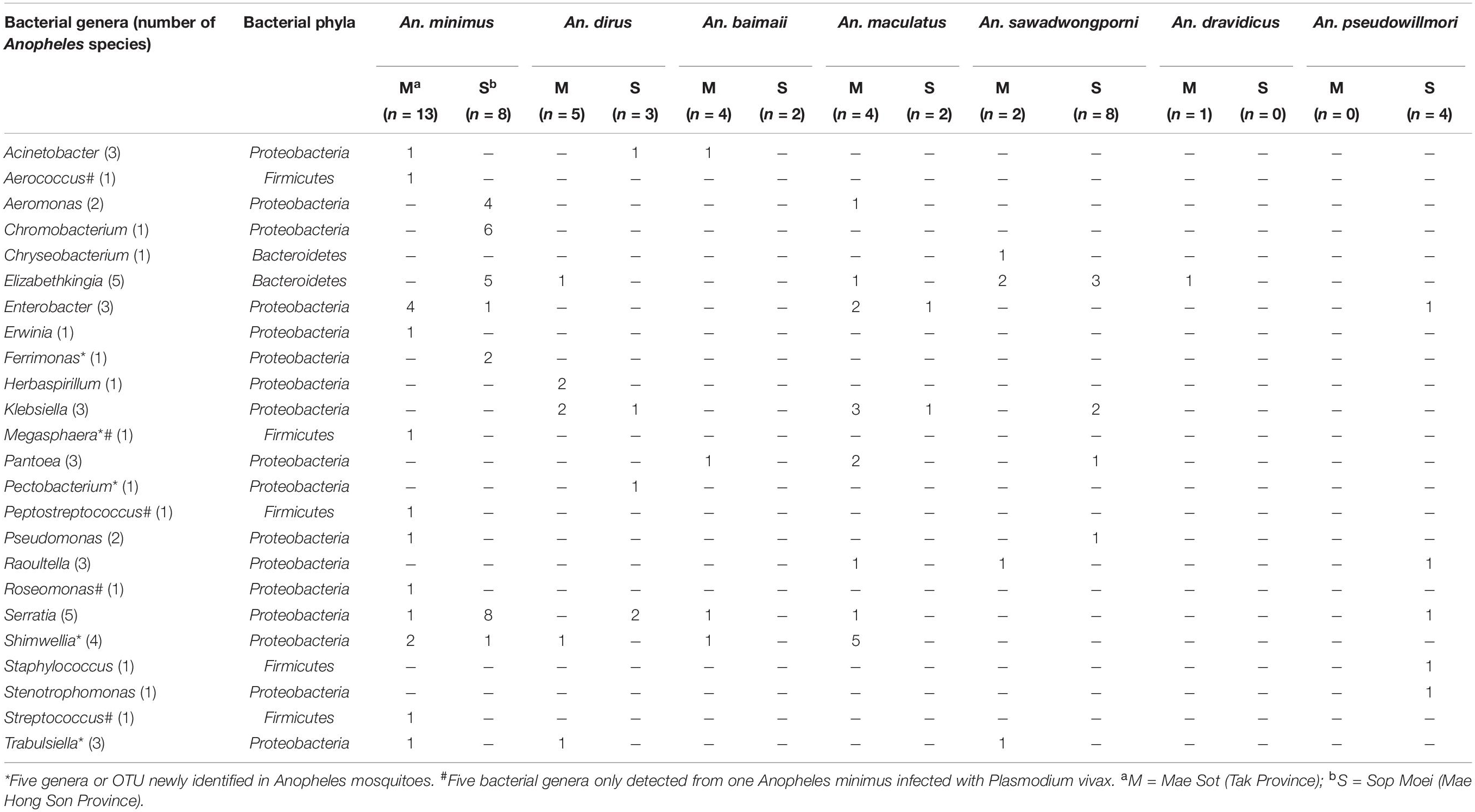
Table 3. Bacterial genera detected in adult Anopheles abdomens collected in Mae Sot and Sop Moei districts, western Thailand.
Within the seven Anopheles species analyzed (An. aconitus excluded), An. minimus was more microbiota diverse with 16 bacterial genera identified, followed by An. dirus and An. maculatus, with half the number of genera (8) detected per species. However, An. minimus was a dominant species captured (n = 35) and analyzed based on PCR-TTGE (21 specimens out of 56) (Figure 4 and Table 3). Nineteen and 15 genera were identified in Mae Sot and Sop Moei respectively, with 10 shared genera (Figure 4). The comparison of bacterial diversity between the four Anopheles species collected at both locations (An. minimus, An. dirus, An. maculatus, and An. sawadwongporni) revealed in three instances the number of genera was higher in Mae Sot than Sop Moei.
Among the 24 bacterial genera identified, 14 (58%) were shared by at least two specimens and 10 were identified in only one mosquito (Table 3). Among these 14 genera, two (Elizabethkingia and Serratia) were shared by five Anopheles species out of seven, one (Shimwellia) by four species, six (Acinetobacter, Enterobacter, Klebsiella, Pantoea, Raoultella, Trabulsiella) in three species, two (Aeromonas, Pseudomonas) in two species, and three (Chromobacterium, Ferrimonas, Herbaspirillum) identified in at least two specimens of the same species (Table 3 and Figure 4).
Nine of the 24 genera (37.5%) belonged to Enterobacteriaceae, including species within the genus Enterobacter, Erwinia, Klebsiella, Pantoea, Pectobacterium, Raoultella, Serratia, Shimwellia, and Trabulsiella (Table 3). The Anopheles microbiota included four primary species, Serratia marcescens (n = 14), Shimwellia blattae (n = 10), Enterobacter cloacae (n = 9), and Klebsiella pneumoniae (n = 9). Shimwellia was the dominant genus identified in Mae Sot (n = 9 specimens), while Serratia was dominant in Sop Moei (n = 11 specimens) (Figure 4). Elizabethkingia, which belongs to the Bacteroidetes phylum, was the second most common genus identified, present in the abdomen of 13 Anopheles specimens among five species (71.5%) – An. minimus, An. dirus, An. maculatus, An. sawadwongporni, and An. dravidicus. Sequences affiliated with the genus Elizabethkingia could not be definitively assigned to either E. anophelis or Elizabethkingia meningoseptica as the V3 region cannot discriminate between the Anopheles-specific bacterium and the bacterium as human pathogen. Among the 10 genera identified in only one Anopheles adult, six were found in An. minimus (Aerococcus, Erwinia, Megasphaera, Peptostreptococcus, Roseomonas, and Streptococcus). The other four genera were Staphylococcus and Stenotrophomonas in An. pseudowillmori, Pectobacterium in An. dirus, and Chryseobacterium in An. sawadwongporni.
In all, 24 bacterial genera were identified in this study, of which 14 have been reported previously in another study (Manguin et al., 2013). Five genera are newly identified in wild-caught adult Anopheles, including Megasphaera found only in a malaria-infected An. minimus in Mae Sot (Figures 3, 4 and Table 3). In addition, this single malaria-infected mosquito hosted five genera not identified in the remaining non-infected Anopheles assayed, including four genera within the phylum Firmicutes (Aerococcus, Megasphaera, Peptostreptococcus, Streptococcus) and one genus, Roseomonas, in the phylum Proteobacteria (Figure 3 and Table 3). Interestingly, three of these five genera are Gram-positive cocci, out of the total four cocci genera identified in this study. Of the 10 genera that were observed only once, half were identified in the only Plasmodium-infected mosquito from the 190 sampled (Table 3).
Native adult Anopheles species were sampled from malaria-endemic areas in Tak and Mae Hong Son provinces near the Thai-Myanmar border. Bacterial diversity was assessed by the 16S rRNA gene, V3 hypervariable region, PCR-Temporal Temperature Gel Electrophoresis (PCR-TTGE) profiling and sequence analysis. The method provides the first estimation of the abdominal bacterial biodiversity present in field-collected Anopheles mosquitoes in Thailand. A total of 24 bacterial genera were identified from eight Anopheles species. Anopheles minimus presented a higher bacterial diversity than the other sampled Anopheles species identified in this study. Five bacterial genera were detected from a single malaria (P. vivax) infected An. minimus. Five genera are newly reported from field-collected Anopheles mosquitoes (Ferrimonas, Megasphaera, Pectobacterium, Shimwellia, and Trabulsiella), suggesting that the diversity of bacteria in Anopheles remains largely underestimated. Elizabethkingia spp., a relatively common midgut bacterium in Anopheles was also detected. This genus appears to have a role in increasing iron metabolism necessary for bacterial growth and Plasmodium development (Clark et al., 2014; Sharma et al., 2019). Bacterial microbiota in Anopheles and their effects on the development of Plasmodium parasites as a potential means of controlling malaria transmission have been reported in various Anopheles species, but the precise mechanisms of activity remain poorly understood (Dong et al., 2009; Meister et al., 2009; Abdul-Ghani et al., 2012; Gendrin and Christophides, 2013; Minard et al., 2013; Hughes et al., 2014; Sharma et al., 2014; Villegas and Pimenta, 2014; Romoli and Gendrin, 2018). The PCR-TTGE described bacteria for 30% of Anopheles (56/190) analyzed and appears an efficient tool to investigate bacterial diversity in large samplings of mosquitoes. This method is appropriate to detect bacterial communities with low to moderate diversities as seen in wild-caught Anopheles samples in this study and other investigations (Manguin et al., 2013; Ngo et al., 2015, 2016). However, the method presents some limitations due to the restricted number of bands that can be separated within the length of the gel. Optimization of TTGE conditions allows the separation of bands with a minimum distance of 0.1 mm. Therefore, the TTGE procedure would present difficulties to interpret bacterial diversity that exceeds 25 to 30 OTUs per mosquito sample (Roudière et al., 2009).
In this study, the anopheline microbiota displayed TTGE profiles that did not exceed 10 bands; however, the profiles have been interpreted with some difficulty due to inherent heterogeneities in rRNA genes for many bacterial species present in the mosquito ecosystem. At the genomic level, rRNA genes are generally organized in multigene families (Acinas et al., 2004) and sequences show low variability within species, subspecies or genome level (Liao, 2000). That aside, TTGE remains useful by providing an accurate ‘snapshot’ of microbiota in different populations of hosts. Results obtained with TTGE fingerprinting compared to pyrosequencing or Next-Generation Sequencing have demonstrated good correlation for the detection of the majority of OTUs in complex microbiotic communities (Manguin et al., 2013; Li et al., 2014).
Nineteen of 24 bacterial genera detected have been reported from field-collected Anopheles (Abdul-Ghani et al., 2012; Gendrin and Christophides, 2013; Minard et al., 2013; Hughes et al., 2014; Sharma et al., 2014; Villegas and Pimenta, 2014). From western Thailand, the abdominal microbiota presented large inter-specimen variability but was dominated by Serratia, Elizabethkingia, Shimwellia, Enterobacter and Klebsiella, particularly members in the family Enterobacteriaceae. These results demonstrate that PCR-TTGE has fairly low detectability of the minority and/or low-density bacteria populations. This low resolution is a limitation but alternatively can be beneficial for this type of work as the majority of taxa detected by TTGE probably corresponds to true symbiotic endo-colonizers in Anopheles and are not likely to be transient or incidental contaminant bacteria.
The presence of enterobacteria is of particular interest because mosquitoes harboring abundant commensal Enterobacteriaceae appear more likely to be infected by P. falciparum suggesting a possible protective role of these bacteria for natural Plasmodium infection (Boissière et al., 2012). On the contrary, some species of Enterobacteriaceae, particularly S. marcescens and Enterobacter (Esp_Z), are able to inhibit Plasmodium development in Anopheles midguts (Gonzalez-Ceron et al., 2003; Cirimotich et al., 2011a; Bai et al., 2019). For instance, S. marcescens in An. albimanus is associated with inhibition of P. vivax oocyst development (Gonzalez-Ceron et al., 2003). In the same phylum (Proteobacteria, family Neisseriaceae), the presence of Chromobacterium Csp_P is associated with a significant reduction in susceptibility of An. gambiae to P. falciparum (Ramirez et al., 2014).
Members of the genus Elizabethkingia detected in assayed mosquitoes from Thailand could not be definitively identified as E. anophelis given its close relatedness in the 16S rRNA gene sequence with E. meningoseptica. This latter bacteria species is generally widely dispersed in the environment and recognized as an occasional serious bacterial pathogen in humans giving rise to meningitis and pneumonia (Xie et al., 2009). Previous studies reported that E. anophelis is a dominant midgut bacterium of laboratory-reared An. stephensi and An. gambiae (Boissière et al., 2012; Ngwa et al., 2013). Sharma et al. (2019) reported an interesting microbial interaction and immune modulation, whereby the presence of P. vivax in An. stephensi provoked a metabolic alteration of the availability of iron and nutritional physiology required for ideal bacterial growth resulting in a reduction of the microbiota present in the midgut, particularly the predominant bacteria Elizabethkingia and Pseudomonas. In addition to the parasite suppressing gut immunity in the mosquito host, by retarding bacterial growth, a greater amount of time is then allowed for the parasite to successfully cross the midgut wall and develop into the oocyst stage.
Within the genus Staphylococcus, S. sciuri was detected in An. pseudowillmori, a bacterium also isolated in the abdomen of other Anopheles species in Vietnam, such as Anopheles crawfordi and Anopheles barbumbrosus (Ngo et al., 2015). Enterobacter and Staphylococcus are among the genera showing trans-stadial maintenance in Anopheles and have been found in An. albimanus (Enterobacter) and mainly in male An. stephensi (Staphylococcus) (Rani et al., 2009; Minard et al., 2013; Galeano-Castaneda et al., 2020). Two other genera, Aerococcus and Peptostreptococcus, were found in An. minimus from Thailand, similar to findings from Vietnam (Ngo et al., 2016).
In this study, one An. minimus from Mae Sot was found naturally infected with P. vivax (Tainchum et al., 2014). This mosquito had at least five genera (Aerococcus, Megasphaera, Peptostreptococcus, Roseomonas, Streptococcus), bacteria not present in the malaria-free Anopheles. Moreover, the malaria-infected mosquito was devoid of Enterobacteriaceae and had three of the Gram-positive cocci out of the four total cocci genera detected in the study. Cocci bacteria are commonly present in larval habitats, which might explain their presence in Anopheles mosquitoes (Rejmankova et al., 2000; Piyaratne et al., 2005). Although no reasonable extrapolation or conclusion can be drawn from the findings in a single infected mosquito, the larger microbiota diversity found in this specimen is concordant with observations on microbiota detected in African An. gambiae s.l. and Anopheles funestus in which microbial diversity was greater in P. falciparum-infected samples than in non-infected ones (Bassene et al., 2018). Some bacterial symbionts were absent in the infected An. minimus specimen, notably Elizabethkingia and Serratia, which were commonly present in non-infected mosquitoes. Again, the single malaria-infected Anopheles was insufficient to allow a comparative analysis of bacterial species biodiversity between malaria-infected and non-infected mosquitoes. One possible limitation of the study design was only focusing on detection of infectious stage sporozoites (post-oocyst) present in the head-thorax portion of the mosquito – the final stage of the Plasmodium sporogony. We acknowledge a potential methodology flaw in which additional mosquito infections present as developing oocysts, would have escaped detection. However, having detected Plasmodium in the head-thorax indicates that we found sporozoites and the infected mosquito was a malaria vector, while detecting parasite stages in the abdomen is not a guaranty that the mosquito is a vector.
Although, the infected An. minimus was collected during a peak abundance period characteristic for this species, malaria-infected mosquitoes are becoming relatively rare findings due to a substantial decrease of malaria transmission in Thailand. A recent study in western Thailand showed P. vivax-infected An. minimus during an April period with an overall 0.76% sporozoite rate for the Minimus Complex (Tainchum et al., 2014; Sriwichai et al., 2016), comparable to this study (0.53%) and higher than the 0.092% reported by Tainchum et al. (2014). Therefore, in order to investigate and compare the natural microbiota between Plasmodium-infected and non-infected Anopheles, further collections and analyses are required. The mosquitoes found without Plasmodium displayed relatively high enterobacteria diversity, especially the genus Serratia. Identification of enterobacteria species in more samples will be the next step in the search for Enterobacter (Esp_Z) and Chromobacterium (Csp_P), both known to inhibit P. falciparum development (Cirimotich et al., 2011a).
The analysis of the microbiota detected in the abdomens of eight field-caught Anopheles species from Thailand resulted in 24 bacterial genera, among which five were only detected from one P. vivax-infected An. minimus specimen. A total of five genera were newly reported in Anopheles mosquitoes of which Megasphaera was only found in the malaria-infected mosquito. Serratia and Elizabethkingia were the most frequent bacteria found in the anopheline abdomens. Findings of low bacterial diversity, ranging from one to five genera per Anopheles, contrasted with a high overall OTU diversity in the entire sampling of species from both localities, which included three major bacterial phyla, Proteobacteria, Firmicutes, and Bacteroidetes. An analysis of the bacterial biodiversity in mosquitoes infected by malaria parasites compared with non-infected specimens was not possible due to the insufficient number of Plasmodium infections detected. Five genera identified in the infected specimen were not detected in other specimens, including three Gram-positive cocci. The PCR-TTGE method with bacterial 16S rRNA provided the first estimation of bacterial biodiversity present in Anopheles in Thailand. These findings suggest that bacterial diversity in Anopheles remains underestimated and requires further investigation. As some microbiota can suppress or block human pathogen development in Anopheles vectors, thus reducing the risk of transmission, more studies are needed to better understand the role of naturally occurring bacteria in wild mosquito populations as a potential method of disease control.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
KT collected the specimens, made the mosquito identification, participated in the bacterial analyses and result’s interpretation, and wrote the draft. CD made the bacterial analyses, their interpretation and improved substantially the manuscript. EJ-B supervised the study design and was involved in the bacterial sequence analyses. TC supervised the mosquito collections and analyses done in Thailand. MB improved the manuscript and participated in the data analyses. SM was at the origin of the study design, was highly involved in data analyses and writing the manuscript. All the authors contributed to the manuscript redaction.
This work was supported by the Center for Advanced Studies for Agriculture and Food (CASAF PD003), Institute for Advanced Studies, Kasetsart University under the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, Ministry of Education, Thailand, as well as the French Government with the PHC Siam Franco-Thai program (Grant No. 20627SD), and the French Institut de Recherche pour le Développement, Montpellier, France, for additional financial support, molecular training, and associated laboratory expenses.
MB was employed by PT Freeport Indonesia/International SOS.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Isabelle Zorgniotti (Equipe Pathogènes Hydriques, Santé et Environnements, UMR 5569 HSM, UM, Faculté de Pharmacie, Montpellier) for the DNA extraction and sequencing of cultured isolates.
Abdul-Ghani, R., Al-Mekhlafi, A. M., and Alabsi, M. S. (2012). Microbial control of malaria: biological warfare against the parasite and its vector. Acta Trop. 121, 71–84. doi: 10.1016/j.actatropica.2011.11.001
Acinas, S. G., Marcelino, L. A., Klepac-Ceraj, V., and Polz, M. F. (2004). Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186, 2629–2635. doi: 10.1128/jb.186.9.2629-2635.2004
Bai, L., Wang, L., Vega-Rodriguez, J., Wang, G., and Wang, S. (2019). A gut symbiotic bacterium Serratia marcescens renders mosquito resistance to Plasmodium infection through activation of mosquito immune responses. Front. Microbiol. 10:1580. doi: 10.3389/fmicb.2019.01580
Bassene, H., Niang, E. H. A., Fenollar, F., Dipankar, B., Doucoure, S., Ali, E., et al. (2018). 16S metagenomic comparison of Plasmodium falciparum-infected and noninfected Anopheles gambiae and Anopheles funestus microbiota from Senegal. Am. J. Trop. Med. Hyg. 99, 1489–1498. doi: 10.4269/ajtmh.18-0263
Black, W. C., and Severson, D. W. (2005). “Genetics of vector competence,” in Biology of Disease Vectors, ed. W. C. Marquardt (San Diego, CA: Academic Press), 415–448.
Boissière, A., Tchioffo, M. T., Bachar, D., Abate, L., Marie, A., Nsango, S. E., et al. (2012). Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 8:e1002742. doi: 10.1371/journal.ppat.1002742
Cirimotich, C. M., Dong, Y., Clayton, A. M., Sandiford, S. L., Souza-Neto, J. A., Mulenga, M., et al. (2011a). Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 332, 855–858. doi: 10.1126/science.1201618
Cirimotich, C. M., Ramirez, J. L., and Dimopoulos, G. (2011b). Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 10, 307–310. doi: 10.1016/j.chom.2011.09.006
Clark, M. A., Goheen, M. M., and Cerami, C. (2014). Influence of host iron status on Plasmodium falciparum infection. Front. Pharmacol. 5:84. doi: 10.3389/fphar.2014.00084
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam, S. A., McGarrell, D. M., et al. (2005). The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33, D294–D296. doi: 10.1093/nar/gki038
Damiani, C., Ricci, I., Crotti, E., Rossi, P., Rizzi, A., Scuppa, P., et al. (2010). Mosquito-bacteria symbiosis: the case of Anopheles gambiae and Asaia. Microb. Ecol. 60, 644–654. doi: 10.1007/s00248-010-9704-8
DDC (2018). Annual Epidemiological Surveillance Report 2018. Nonthaburi: Ministry of Public Health, Department of Disease Control (DDC).
Divis, P. C., Shokoples, S. E., Singh, B., and Yanow, S. K. (2010). A TaqMan real-time PCR assay for the detection and quantitation of Plasmodium knowlesi. Malar. J. 9:344. doi: 10.1186/1475-2875-9-344
Dong, Y., Manfredini, F., and Dimopoulos, G. (2009). Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 5:e1000423. doi: 10.1371/journal.ppat.1000423
Doumbo, O. K., Niaré, K., Healy, S. A., Sagara, I., and Duffy, P. E. (2018). “Malaria transmission-blocking vaccines: present status and future perspectives,” in Towards Malaria Elimination: A Leap Forward, eds S. Manguin and V. Dev (London: IntechOpen), 363–386.
Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J. P., and Raoult, D. (2000). 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 38, 3623–3630.
Favia, G., Ricci, I., Marzorati, M., Negri, I., Alma, A., Sacchi, L., et al. (2008). Bacteria of the genus Asaia: a potential paratransgenic weapon against malaria. Adv. Exp. Med. Biol. 627, 49–59. doi: 10.1007/978-0-387-78225-6_4
Galeano-Castaneda, Y., Bascunan, P., Serre, D., and Correa, M. M. (2020). Trans-stadial fate of the gut bacterial microbiota in Anopheles albimanus. Acta Trop. 201:105204. doi: 10.1016/j.actatropica.2019.105204
Gendrin, M., and Christophides, G. K. (2013). “The Anopheles mosquito microbiota and their impact on pathogen transmission,” in Anopheles Mosquitoes - New Insights into Malaria Vectors, ed. S. Manguin (Rijeka: InTech Open Access), 525–548.
Gonzalez-Ceron, L., Santillan, F., Rodriguez, M. H., Mendez, D., and Hernandez-Avila, J. E. (2003). Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J. Med. Entomol. 40, 371–374. doi: 10.1603/0022-2585-40.3.371
Hughes, G. L., Dodson, B. L., Johnson, R. M., Murdock, C. C., Tsujimoto, H., Suzuki, Y., et al. (2014). Native microbiome impedes vertical transmission of Wolbachia in Anopheles mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 111, 12498–12503. doi: 10.1073/pnas.1408888111
Kampfer, P., Matthews, H., Glaeser, S. P., Martin, K., Lodders, N., and Faye, I. (2011). Elizabethkingia anophelis sp. nov., isolated from the midgut of the mosquito Anopheles gambiae. Int. J. Syst. Evol. Microbiol. 61, 2670–2675. doi: 10.1099/ijs.0.026393-0
Kampfer, P., Terenius, O., Lindh, J. M., and Faye, I. (2006). Janibacter anophelis sp. nov., isolated from the midgut of Anopheles arabiensis. Int. J. Syst. Evol. Microbiol. 56, 389–392. doi: 10.1099/ijs.0.63905-0
Lefevre, T., Vantaux, A., Dabire, K. R., Mouline, K., and Cohuet, A. (2013). Non-genetic determinants of mosquito competence for malaria parasites. PLoS Pathog. 9:e1003365. doi: 10.1371/journal.ppat.1003365
Leroy, D., Campo, B., Ding, X. C., Burrows, J. N., and Cherbuin, S. (2014). Defining the biology component of the drug discovery strategy for malaria eradication. Trends Parasitol. 30, 478–490. doi: 10.1016/j.pt.2014.07.004
Li, W., Han, L., Yu, P., Ma, C., Wu, X., Moore, J. E., et al. (2014). Molecular characterization of skin microbiota between cancer cachexia patients and healthy volunteers. Microb. Ecol. 67, 679–689. doi: 10.1007/s00248-013-0345-6
Liao, D. (2000). Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J. Mol. Evol. 51, 305–317. doi: 10.1007/s002390010093
Manguin, S., Ngo, C. T., Tainchum, K., Juntarajumnong, W., Chareonviriyaphap, T., Michon, A. L., et al. (2013). “Bacterial biodiversity in midguts of Anopheles mosquitoes, malaria vectors in Southeast Asia,” in Anopheles Mosquitoes - New Insights into Malaria Vectors, ed. S. Manguin (Rijeka: InTechOpen Access), 549–576.
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Meister, S., Agianian, B., Turlure, F., Relogio, A., Morlais, I., Kafatos, F. C., et al. (2009). Anopheles gambiae PGRPLC-mediated defense against bacteria modulates infections with malaria parasites. PLoS Pathog. 5:e1000542. doi: 10.1371/journal.ppat.1000542
Minard, G., Mavingui, P., and Moro, C. V. (2013). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vect. 6:146. doi: 10.1186/1756-3305-6-146
Ngo, C. T., Aujoulat, F., Veas, F., Jumas-Bilak, E., and Manguin, S. (2015). Bacterial diversity associated with wild caught Anopheles mosquitoes from Dak Nong province, vietnam using culture and DNA fingerprint. PLoS One 10:e0118634. doi: 10.1371/journal.pone.0118634
Ngo, C. T., Romano-Bertrand, S., Manguin, S., and Jumas-Bilak, E. (2016). Diversity of the bacterial microbiota of Anopheles mosquitoes from Binh Phuoc Province, Vietnam. Front. Microbiol. 7:2095. doi: 10.3389/fmicb.2016.02095
Ngwa, C. J., Glockner, V., Abdelmohsen, U. R., Scheuermayer, M., Fischer, R., Hentschel, U., et al. (2013). 16S rRNA gene-based identification of Elizabethkingia meningoseptica (Flavobacteriales: Flavobacteriaceae) as a dominant midgut bacterium of the Asian malaria vector Anopheles stephensi (Diptera: Culicidae) with antimicrobial activities. J. Med. Entomol. 50, 404–414. doi: 10.1603/me12180
Ogier, J. C., Son, O., Gruss, A., Tailliez, P., and Delacroix-Buchet, A. (2002). Identification of the bacterial microflora in dairy products by temporal temperature gradient gel electrophoresis. Appl. Environ. Microbiol. 68, 3691–3701. doi: 10.1128/aem.68.8.3691-3701.2002
Piyaratne, M. K., Amerasinghe, P. H., Amerasinghe, F. P., and Konradsen, F. (2005). Food of larval Anopheles culicifacies and Anopheles varuna in a stream habitat in Sri Lanka. J. Am. Mosq. Control Assoc. 21, 387–394. doi: 10.2987/8756-971X(2006)21[387:FOLACA]2.0.CO;2
Ramirez, J. L., Short, S. M., Bahia, A. C., Saraiva, R. G., Dong, Y., Kang, S., et al. (2014). Chromobacterium Csp_P reduces malaria and dengue infection in vector mosquitoes and has entomopathogenic and in vitro anti-pathogen activities. PLoS Pathog. 10:e1004398. doi: 10.1371/journal.ppat.1004398
Rani, A., Sharma, A., Rajagopal, R., Adak, T., and Bhatnagar, R. K. (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi -an Asian malarial vector. BMC Microbiol. 9:96. doi: 10.1186/1471-2180-9-96
Rattanarithikul, R., Harrison, B. A., Harbach, R. E., Panthusiri, P., Coleman, R. E., and Panthusiri, P. (2006). Illustrated keys to the mosquitoes of Thailand. IV. Anopheles. Southeast Asian J. Trop. Med. Public Health 37(Suppl. 2), 1–128.
Rejmankova, E., Harbin-Ireland, A., and Lege, M. (2000). Bacterial abundance in larval habitats of four species of Anopheles (Diptera: Culicidae) in Belize, Central America. J. Vector Ecol. 25, 229–239.
Riehle, M. A., Moreira, C. K., Lampe, D., Lauzon, C., and Jacobs-Lorena, M. (2007). Using bacteria to express and display anti- Plasmodium molecules in the mosquito midgut. Int. J. Parasitol. 37, 595–603. doi: 10.1016/j.ijpara.2006.12.002
Romoli, O., and Gendrin, M. (2018). The tripartite interactions between the mosquito, its microbiota and Plasmodium. Parasit. Vect. 11:200. doi: 10.1186/s13071-018-2784-x
Roudière, L., Jacquot, A., Marchandin, H., Aujoulat, F., Devine, R., Zorgniotti, I., et al. (2009). Optimized PCR-temporal temperature gel electrophoresis compared to cultivation to assess diversity of gut microbiota in neonates. J. Microbiol. Methods 79, 156–165. doi: 10.1016/j.mimet.2009.08.005
Sharma, P., Rani, J., Chauhan, C., Kumari, S., Tevatiya, S., De, T D., et al. (2019). Altered gut microbiota and immunity defines Plasmodium vivax survival in Anopheles stephensi. BioRxiv [preprint]. doi: 10.1101/774075
Sharma, P., Sharma, S., Maurya, R. K., Das, D. T., Thomas, T., Lata, S., et al. (2014). Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasit. Vect. 7:235. doi: 10.1186/1756-3305-7-235
Sriwichai, P., Samung, Y., Sumruayphol, S., Kiattibutr, K., Kumpitak, C., Payakkapol, A., et al. (2016). Natural human Plasmodium infections in major Anopheles mosquitoes in western Thailand. Parasit. Vect. 9:17. doi: 10.1186/s13071-016-1295-x
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44, 846–849.
Tainchum, K., Ritthison, W., Chuaycharoensuk, T., Bangs, M. J., Manguin, S., and Chareonviriyaphap, T. (2014). Diversity of Anopheles species and trophic behavior of putative malaria vectors in two malaria endemic areas of northwestern Thailand. J. Vector Ecol. 39, 424–436. doi: 10.1111/jvec.12118
Tananchai, C., Manguin, S., Bangs, M. J., and Chareonviriyaphap, T. (2019). Malaria vectors and species complexes in Thailand: implications for vector control. Trends Parasitol. 35, 544–558. doi: 10.1016/j.pt.2019.04.013
Tandina, F., Almeras, L., Kone, A. K., Doumbo, O. K., Raoult, D., and Parola, P. (2016). Use of MALDI-TOF MS and culturomics to identify mosquitoes and their midgut microbiota. Parasit. Vect. 9:495. doi: 10.1186/s13071-016-1776-y
Villegas, L. M., and Pimenta, P. F. (2014). Metagenomics, paratransgenesis and the Anopheles microbiome: a portrait of the geographical distribution of the anopheline microbiota based on a meta-analysis of reported taxa. Mem. Inst. Oswaldo Cruz 109, 672–684. doi: 10.1590/0074-0276140194
Wang, S., Ghosh, A. K., Bongio, N., Stebbings, K. A., Lampe, D. J., and Jacobs-Lorena, M. (2012). Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 109, 12734–12739. doi: 10.1073/pnas.1204158109
Xie, Z. Y., Zhou, Y. C., Wang, S. F., Mei, B., Xu, X. D., Wen, W. Y., et al. (2009). First isolation and identification of Elizabethkingia meningoseptica from cultured tiger frog, Rana tigerina rugulosa. Vet. Microbiol. 138, 140–144. doi: 10.1016/j.vetmic.2009.02.011
Keywords: Anopheles mosquitoes, malaria, bacterial microbiota, biodiversity, Thailand
Citation: Tainchum K, Dupont C, Chareonviriyaphap T, Jumas-Bilak E, Bangs MJ and Manguin S (2020) Bacterial Microbiome in Wild-Caught Anopheles Mosquitoes in Western Thailand. Front. Microbiol. 11:965. doi: 10.3389/fmicb.2020.00965
Received: 20 December 2019; Accepted: 22 April 2020;
Published: 21 May 2020.
Edited by:
Josué Martínez-de la Puente, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Guido Favia, University of Camerino, ItalyCopyright © 2020 Tainchum, Dupont, Chareonviriyaphap, Jumas-Bilak, Bangs and Manguin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvie Manguin, c3lsdmllLm1hbmd1aW5AaXJkLmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.