
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 07 May 2020
Sec. Infectious Agents and Disease
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00778
 Xiong Zhu1Hai Chen1
Xiong Zhu1Hai Chen1 Sha Li1Li-cheng Wang1Duo-rong Wu2Xu-ming Wang3Ru-shou Chen4
Sha Li1Li-cheng Wang1Duo-rong Wu2Xu-ming Wang3Ru-shou Chen4 Zhen-jun li5
Zhen-jun li5 Zhi-guo Liu1,5*
Zhi-guo Liu1,5*Melioidosis is a common infectious disease in Southeast Asia and Northern Australia. In Hainan, several cases have been reported, but no systematic study has yet been done on the molecular epidemiology profiles of the organism. An investigation of the molecular epidemiology links and population structure of Burkholderia pseudomallei would help to better understand the clonally of the isolates and differences among them. In this study, multilocus variable-number tandem repeat analysis (MLVA), and multilocus sequence typing (MLST) were applied to examine the epidemiological relatedness and population structure of 166 B. pseudomallei isolates obtained during 2002–2014 in Hainan, China. Both the MLVA_4 and MLST approaches had high discriminatory power for this population, with diversity indices of 0.9899 and 0.9457, respectively. However, the MLVA_4 assay showed a higher discriminatory power than the MLST approach, and a variable-number tandem repeat (VNTR3 933) found by the MLVA approach was the most useful in discriminating strains from this province. A total of 166 strains yielded 99 MLVA_4 genotypes, of which 34 genotypes were shared by 101 isolates, for a clustering rate of 60.8% (101/166), which suggested that some cases may have a common source. Additionally, 65 isolates showed distinct genotypes, indicating that more than 39.2% (65/166) of melioidosis cases in Hainan had epidemiologically unrelated or sporadic characteristics. The 166 isolates were resolved into 48 STs, of which five STs (ST55, -70, -46, -50, and -58) were here found to be predominant. Phylogenetic analysis of 116 isolates conducted using the eBURST v3 segregated the 48 STs into eight groups with ST50 as predicted founder, and 21 STs were found to be singletons, which suggest that the strains in the Hainan region represent a high diversity of ST clones, indicating that many B. pseudomallei clone groups are endemic to this region. Moreover, ST50 had 5 SLV, 7 DLV, 6 TLV, and 29 satellite STs and formed a radial expansion pattern, suggesting that the melioidosis epidemic in this study was mainly caused by the clonal expansion of ST 50. Phylogenetic analysis on global scale suggests that China’s isolates are closely related to isolates from Southeast Asia, particularly from Thailand and Malaysia.
Melioidosis, a disease hyperendemic in Northern Australia and Southeast Asia, is caused by the environmental bacterium Burkholderia pseudomallei (Wiersinga et al., 2012), and is considered a potential emerging infectious disease in many tropical developing countries (Currie et al., 2008). Melioidosis has a wide variety of symptoms, many of which are shared with other infections, including pyogenic bacterial infection and tuberculosis. Thus, the lack of defining clinical symptoms (Le Tohic et al., 2019) makes melioidosis challenging to diagnose (Shrestha et al., 2019). Most patients are infected through contact with contaminated soil or water. Diabetes, alcoholism, renal insufficiency, and chronic steroid use are important risk factors for the infection by B. pseudomallei (Kronsteiner et al., 2019). Melioidosis can have an acute or chronic presentation, and relapse may occur if there is inadequate adherence to treatment or an occult focus of the infection (Currie et al., 2000; Koshy et al., 2019). Based on a geographic information system modeling prediction, the annual incidence of human melioidosis is up to 165,000 cases worldwide, resulting in approximately 89,000 deaths annually (Limmathurotsakul et al., 2016). However, the true incidence of this disease remains difficult to determine. Moreover, due to misdiagnosis and underreporting, it appears likely that there has been a severe underestimation of the incidence of the disease in tropical areas of the world.
Melioidosis has become a significant public health issue in tropical and sub-tropical areas (Kong et al., 2016), including in Hainan province, one of the few tropical areas in China. Although B. pseudomallei was detected in the environment in the 1970s, the first human case was not identified until 1989 (Yang, 2000). More than 200 culture-confirmed cases were reported during 2002–2014, and there is evidence that the trend is increasing each year. Hainan is a well-known and important open international island. With a continually developing economy and rising international influence, it is expected to become a major international tourist destination area, a new business center, and an important stage for international exchange. Thus, many individuals are at risk of contracting B. pseudomallei, which represents a significant public health concern.
An investigation of the comprehensive molecular epidemiological characteristics of B. pseudomallei from clinical data is needed. Multilocus variable-number tandem repeat analysis (MLVA) enables the estimation of epidemiological relatedness among isolated strains, as well as the tracking of pathogens such as B. pseudomallei in epidemiological outbreaks at the phylogenetic scale (Pearson et al., 2007; Currie et al., 2009). In addition, multilocus sequence typing (MLST) offers the ability to explore the population structure and evolutionary characteristics. In the present study, a molecular investigation of B. pseudomallei strains from clinical samples collected from 2002 to 2014 was performed to estimate the epidemiological relationship and population structure of isolates in Hainan, China.
This study is a retrospective investigation of historical strain collections (2002–2014) using molecular typing methods. The study protocol was approved by the Ethics Committees of the National Institute for Communicable Disease Control and Prevention and the Chinese Center for Disease Control and Prevention. Informed consent was obtained from all patients before testing. Isolated B. pseudomallei strains were used to confirm the diagnosis.
Clinical samples from suspected patients were cultured on Columbia blood agar (P0188, Sigma, United States) and incubated at 37°C for 1 week. Colonies of suspected isolates were selected and identified using the Vitek 2 Compact system (Vitek 2 27220, BioMerieux, France), phenotypic features, and 16S rRNA PCR amplification, as previously described (Lowe et al., 2006). A total of 166 strains were characterized in this study, of which 163 were obtained from patients; the remaining three strains were isolated from well water samples. Moreover, there were 14 strains obtained from six infections events (IDs 1–6), five of which (ID 1–5) contained two strains each that were isolated from the same patient, from different clinical samples, or at different point times. ID1 (HNBP040 and HNBP041) and ID4 (HNBP115 HNBP116) each contained two strains that were obtained from different time points. ID2 (HNBP052 and HNBP082), ID3 (HNBP055 and HNBP129), and ID5 (HNBP114 and HNBP135) each contained two strains that were isolated from different clinical samples. However, ID 6 included four strains (HNBP163–HNBP166) from a traceback investigation of one infection event. HNBP163 was obtained from the blood of a patient, and the remaining three (HNBP164–HNBP166) were recovered from well water samples. HNBP164 and HNBP165 were isolated from water from a well located in a patient’s house, and HNBP166 was isolated from the well water of the patient’s neighbor. HNBP134 was recovered from the blood of a patient who was a journalist from Inner Mongolia and conducted news reporting in Sanya City for 2 weeks. DNA was extracted from all strains using a Nucleic Acid Automatic Extraction System (LLXBIO China Ltd., China) with a single loop of fresh bacterial cells according to the manufacturer’s instructions. DNA concentrations were measured by UV spectrophotometry (NanoDrop 2000, Thermo Fisher, United States). The DNA extracted from all isolates was stored at −20°C.
The MLVA_4 genotyping assay was performed as previously described (Currie et al., 2009). Briefly, the four higher-variation variable-number tandem repeat (VNTR) loci 2341, 389, 1788, and 933 were chosen for MLVA genotyping (U’Ren et al., 2007). A 20-μL amplification system was applied, and all PCR involved an initial denaturation at 95°C for 5 min, followed by 30 cycles at 94°C for 30 s, 68°C for 30 s, and 72°C for 30 s, with a final extension step at 72°C for 5 min. Next, 5 μL of each PCR product was separated by gel electrophoresis, and the bands of the expected amplicons from the four loci were denatured and resolved by capillary electrophoresis using an ABI Prism 3130 automated fluorescent capillary DNA sequencer (Applied Biosystems, United States). The fragments were sized following comparison with a ROX (carboxy-X-rhodamine)-labeled molecular ladder (MapMaker 1000; BioVentures Inc., Murfreesboro, TN, United States) and Gene Mapper software version 4.0 (Applied Biosystems). The fragment sizes were subsequently converted to repeat unit numbers using a published allele numbering system. The MLVA_4 data were imported into BioNumerics 7.6 software (Applied Maths, Sint-Martens-Latem, Belgium) for cluster analysis (Supplementary Table S1). The molecular epidemiological relatedness of isolates was evaluated using a matrix of the pairwise differences for the 4 VNTR loci, with a dendrogram produced using the unweighted pair group method with arithmetic averages (UPGMA).
Multilocus sequence typing assays were performed as previously described (Price et al., 2016a). Each allele was assigned a different number, and the allelic profile (a string of seven integers) was used to define the sequence type (ST) for that isolate (Supplementary Table S1). The allelic profiles of the isolates were imported into BioNumerics version 7.6, and the relatedness of the isolates was displayed as a dendrogram using the matrix of pairwise differences in the allelic profiles and UPGMA clustering. The genetic diversity and discriminatory power of each typing method were calculated based on the Hunter-Gaston diversity index (HGDI), according to a previously published method (Hunter, 1990).
The similarity of MLST profiles of isolates identified in this study or elsewhere in China (Supplementary Table S2) in B. pseudomallei MLST database was assessed using eBURST software as described previously (Kamthan et al., 2018) STs (Supplementary Table S3). The relationship of China STs to the global collection of STs was assessed using the eBURST algorithm with PHYLOViZ 2.0 (Nascimento et al., 2017) available at MLST site1. All MLST profiles have been submitted to the MLST DB2.
A total of 166 strains were investigated in this study; three strains were from environmental samples (well water), and 163 B. pseudomallei isolates were obtained from clinical samples (158 patients). Ten of the strains were obtained from five patients, each harboring two strains. Regarding the 158 B. pseudomallei isolates obtained from 158 individual patients, 59% (94/158) of the patients were engaged in livestock farming and related work. The mean age of these 158 patients was 48.6 years (range: 1–82 years), and the ratio of males (n = 132) to females (n = 26) was 5.1 (Supplementary Table S4). The patients had wide-ranging symptoms: 61 (38.6%) had sepsis, 58 (36.7%) had pulmonary infections, and 34 (21.5%) had local abscesses. Moreover, 73 (46.2%) patients had known diabetes risk factors. A total of 117 (75.3%) patients were cured, 39 (24.7%) died, and the other two had unknown outcomes. Among the 39 patients who died, 18 (46.2%) had diabetes, 27 (69.2%) had septicemia, and 19 (48.7%) had pulmonary infections. The age range of the patients who died was 1–77 years, and the predominant age range was 40–50 years.
A total of 163 strains were collected from clinical patient samples, including blood, secretions, and pus, and three strains were isolated from well water samples. All the strains were identified as B. pseudomallei, which is a gram-negative, bipolar, aerobic, motile, and rod-shaped bacterium. Colonies of the strains were wrinkled, had a robust metallic appearance, and emitted an acrid, earthy smell. Of the 166 strains collected from 2002 to 2014, 65.7% (109/166) were obtained between 2010 and 2012 (Supplementary Table S5). Five strains were isolated from the blood of patients from Fujian (n = 2), Inner Mongolia (n = 1), Hunan (n = 1), and Russia (n = 1); these patients lived in Hainan for a period before they became sick. Other strains were obtained from 14 counties in Hainan Province (Figure 1).
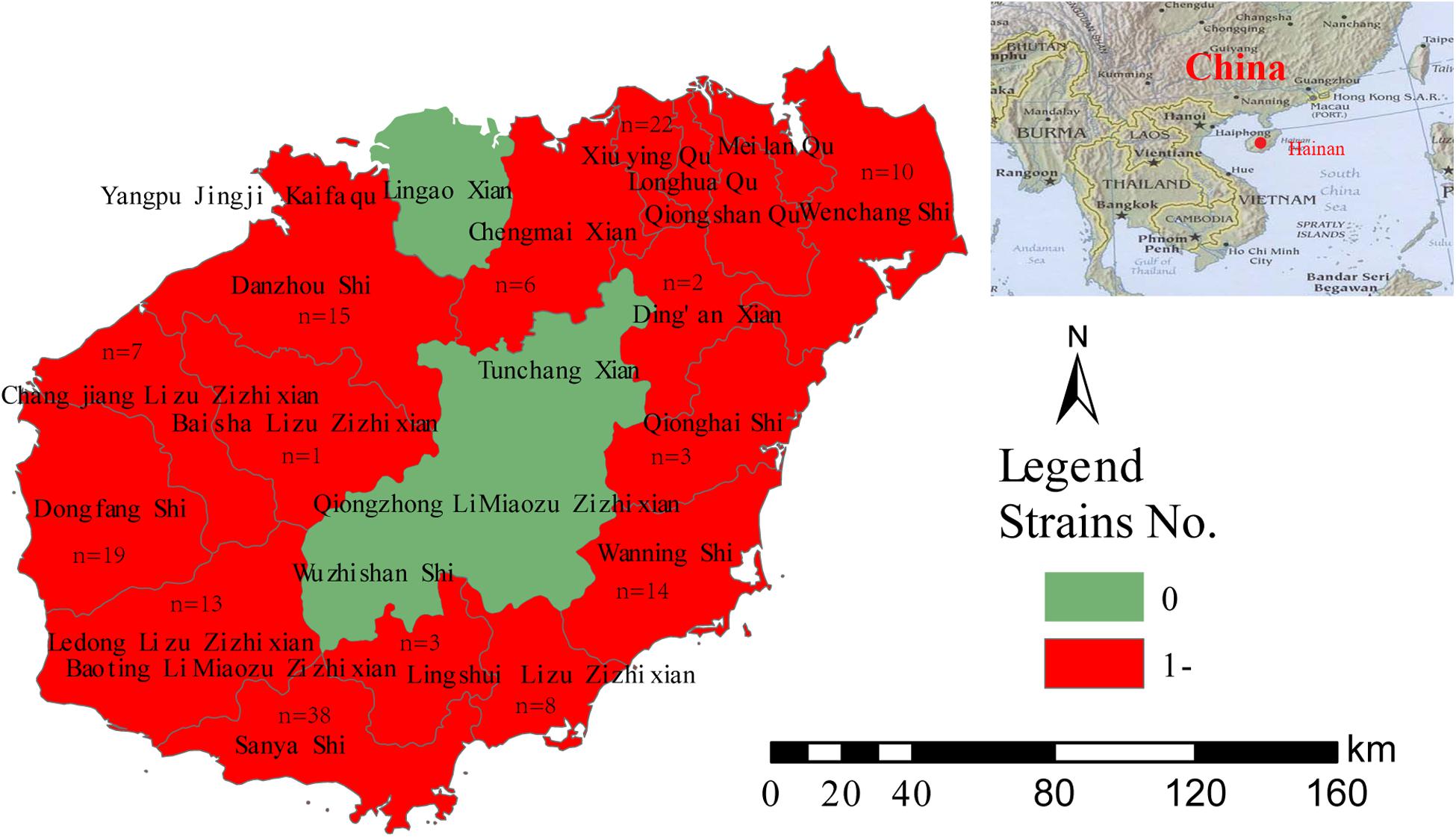
Figure 1. Geographic distribution of 161 clinical B. pseudomallei strains.
In this study, the results of B. pseudomallei isolate diversity analysis confirmed that both the MLVA_4 and MLST approaches offer high discriminatory power for this population, with diversity indices of 0.9899 and 0.9457, respectively, although the former displayed higher discriminatory power than the latter (Table 1). We sorted 166 strains into 48 STs in the MLST assay and 99 different MLVA genotypes in the MLVA_4 assay. The HGDI for MLVA was the highest (0.9025) for VNTR3 (933), which had 15 allele types. The allele numbers for VNTR1 (2341), VNTR2 (1788), and VNTR4 (389) by MLVA were 11, 9, and 14, respectively, and the polymorphism levels of all loci were ≥0.8207. In MLST, the highest number of alleles was found for the gmhD locus (10), which also had the highest HGDI (0.7925), followed by narK with eight alleles, gltB, and lepA with five alleles, and the remaining loci with three alleles (Table 1).
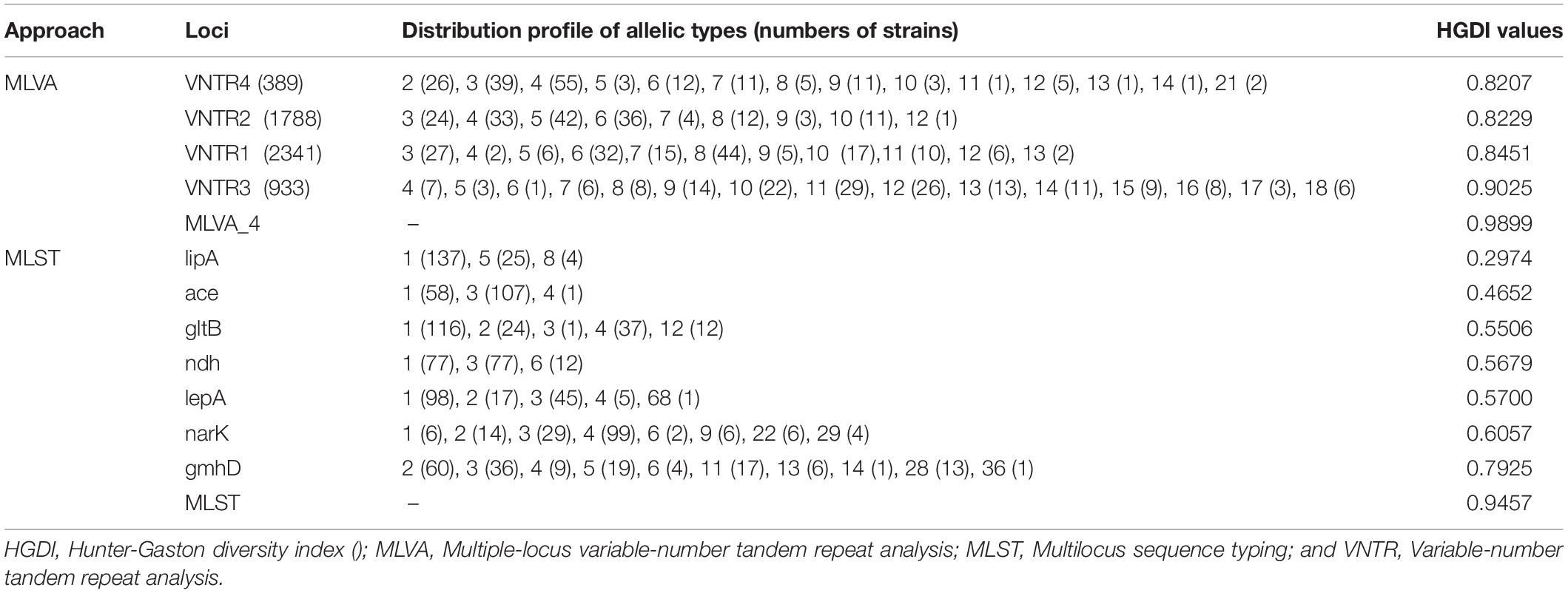
Table 1. Allelic types and Hunter-Gaston diversity index (HGDI) of B. pseudomallei for 11 typing loci in this study.
A total of 166 strains were sorted into 99 different MLVA_4 genotypes; 34 genotypes were found to be shared by 101 strains, accounting for 61% (Figures 2, 3). Seven shared MLVA_4 genotypes (GT11, 38, 42, 54, 63, 77, and 89), and each one of them was comprised of two strains from identical locations and similar times (Table 3). Five shared MLVA_4 genotypes (GT 21, 38, 39, 53, and 63), including strains with identical STs and from two to five different regions over a long period (Table 4). Notably, MLVA_4 genotype 21 is shared by eight strains obtained from five areas and isolated 10 years apart. The other 65 strains harbor unique genotypes, with each genotype representing a single strain. Moreover, the MLVA and MLST data for six individual infection events (IDs 1–6) showed that the strains with IDs 1 and 4 have identical MLVA_4 genotypes and the same ST; the strains with IDs 2, 3, and 5 exhibit different MLVA_4 profiles and STs (Table 2). A total of four strains were isolated from the ID 6 infection event on a farm in Saya. The HNBP163 strain, derived from the blood of one patient, has an MLVA_4 genotype (GT9) and ST (ST667) identical to those of HNBP164, which was isolated from the well water of the same patient. However, there was a significant difference between the strains (HNBP165 and HNBP166) collected from the two other well water samples (Table 2). HNBP134 has an MLVA_4 genotype and ST identical to those of strains from five different regions in Hainan Province. Two strains from Fujian Province exhibit different MLVA_4 genotypes and STs. GT60 and GT97 each represent unique strains obtained from the blood of patients from Russia and Hunan, respectively.
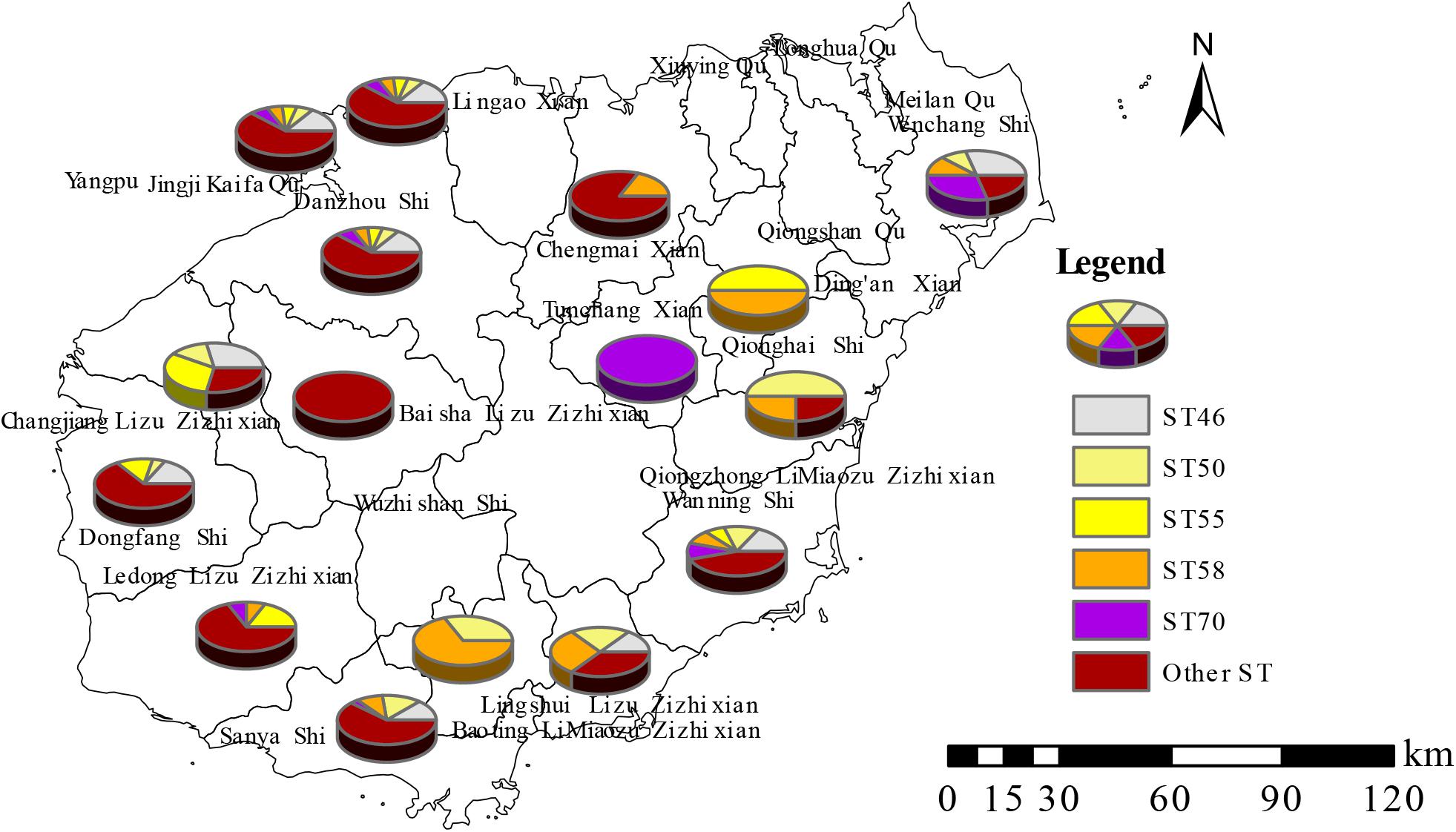
Figure 2. Distribution characteristics of five predominant STs in Hainan Province.

Figure 3. A dendrogram of the 166 B. pseudomallei strains showing the strain identification features, MLVA_4 and MLST characters’ values, their geographical origins, and the year of isolation.
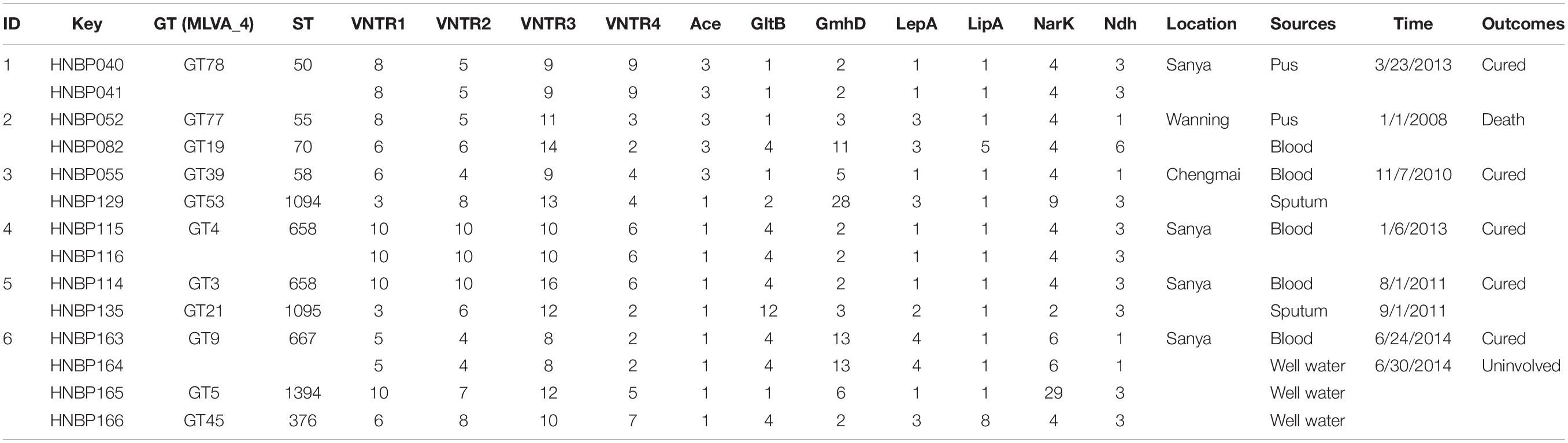
Table 2. Information on scene epidemiology, MLVA and MLST for six infection events (ID1–6).
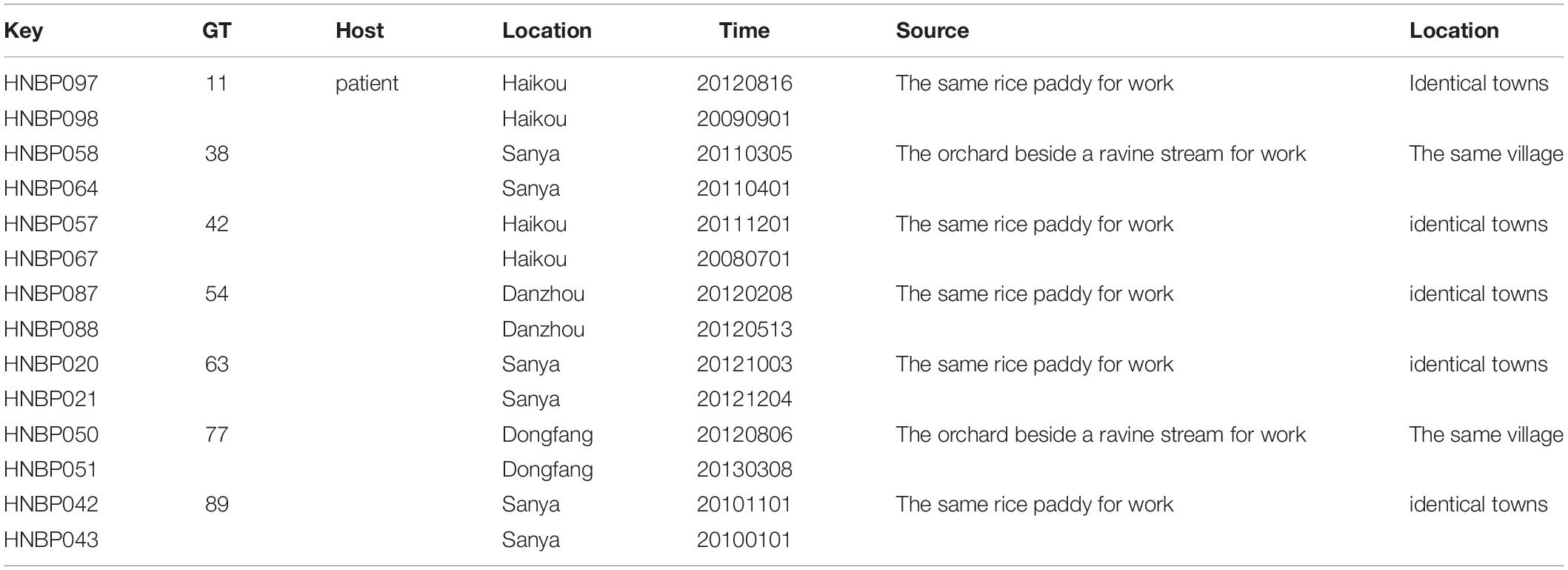
Table 3. Strains with shared genotypes and the same source of infection.
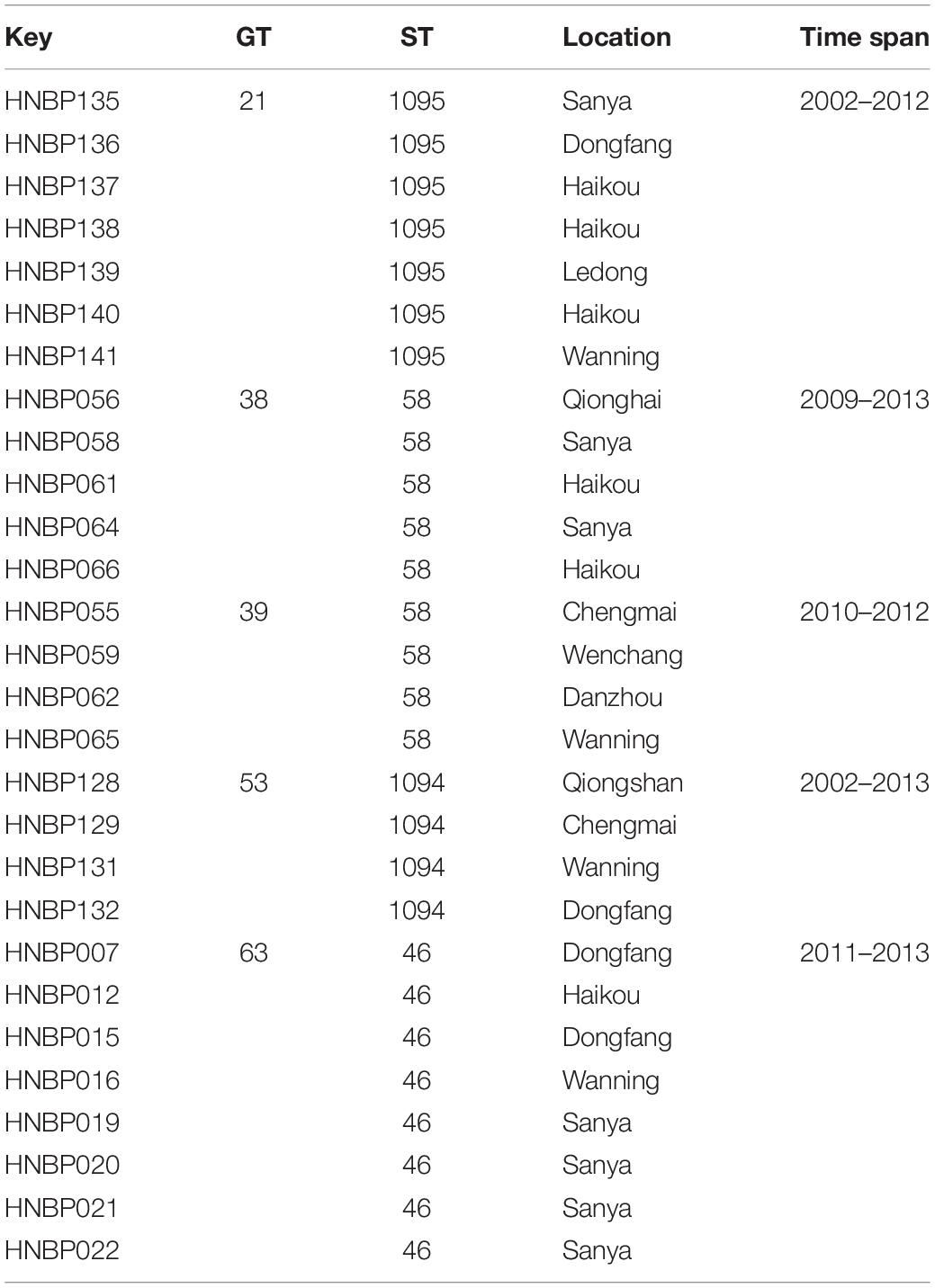
Table 4. Strains with an identical MLVA_4 genotype and ST from distinct regions.
All 166 strains were resolved into 48 STs, three of which (ST167, −168, and −389) were singleton STs that had not been previously reported from Hainan. The other STs are identical to those previously reported. Nineteen STs were observed in Sanya, 13 in Dongfang and Haikou, 12 in Danzhou, 11 in Ledong and Wanning, and six in Lingshui and Wenchang. Other regions had one to five different STs (Supplementary Table S6). Five STs (ST55, −70, −46, −50, and −58) were predominant in the population in this study (Figure 4). The dominant STs, which occurred in ≥11 cases, were ST46 (twenty cases; 12.0%), ST50 and ST58 (nineteen; 11.4%), ST70 (twelve; 7.2%), and ST55 (eleven; 6.6%). These five STs accounted for 48.8% of all cases, and the remainder were associated with one to eight cases. Two STs, ST168, and -389, are single-locus variations of ST48. ST167 is a single-locus variation of ST562. Moreover, twelve different STs, including four cases of ST50 and two cases of both ST55 and −58, were responsible for 18 deaths. Eight STs (ST46, −50, −55, −58, −70, −658, −1094, and −1095) each included more than five strains, and these STs were found to have one to thirteen similar or identical MLVA_4 genotypes (Supplementary Table S7).
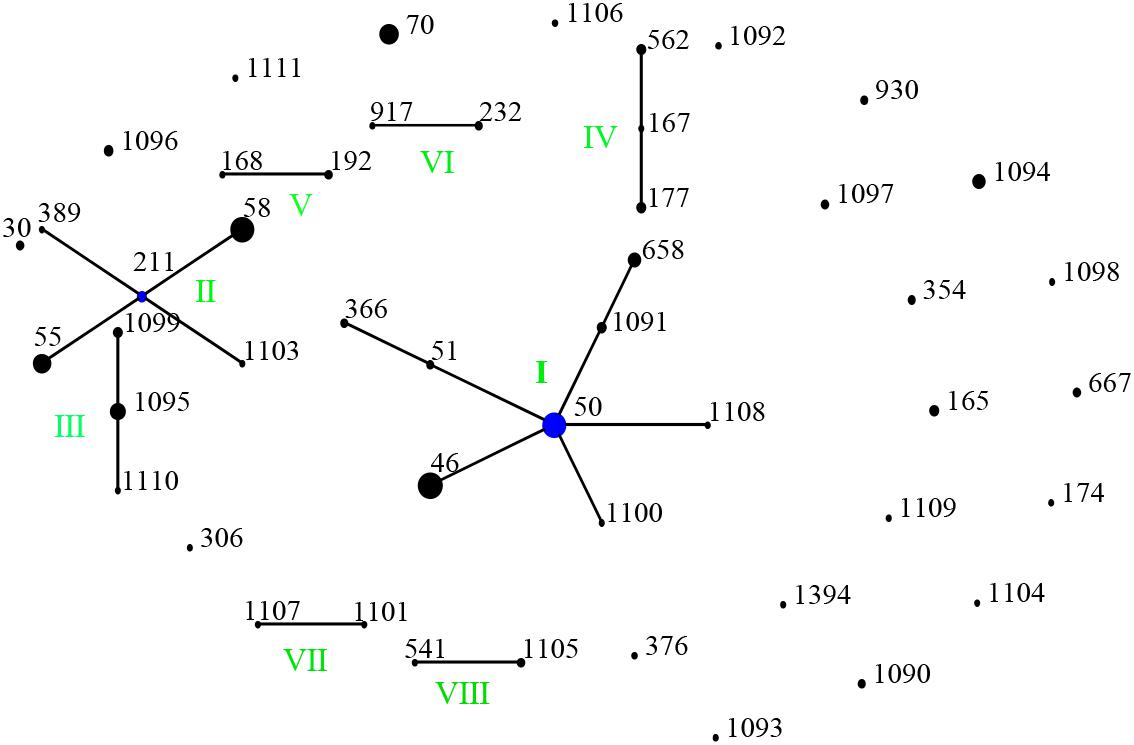
Figure 4. Genetic relationship of 166 B. pseudomallei isolates of this study using eBURST. (Each black dot represents single genotype, and blue dot refers to group founder). Strains from this study formed eight (I-VII) ST groups.
When analyzed using eBURST, the 48 STs evaluated in this study formed eight groups (I–VIII) with ST50 as predicted founder, and 21 STs were found to be singletons (Figure 4). These two groups (I and II) form a radial expansion pattern. ST50 had 5 SLV, 7 DLV, 6 TLV, and 29 satellite STs. Based on the eBURST analysis, 479 isolates from China were clustered into 95 STs, and the 62 STs were clustered in six groups (a–f) with the remaining 33 STs being singletons. ST50, the predicted founder ST in this population (Figure 5), had a frequency of 42 with 8 SLV, 13 DLV, 15 TLV, and 58 satellite STs. ST46 and ST51 were subgroup founders. The present study expanded the clonal cluster of China isolates by adding more branching STs. When Chinese STs were analyzed using PHYLOViZ against the global collection of 6046 isolates in the B. pseudomallei MLST isolate database (as on 20th May 2020), the majority of Chinese isolates grouped into four groups (A–D; Figure 6) and were almost exclusively clustered in the Southeast Asia clade. It appears that there are a few outliers (e.g., ST37, −660, −1099, −1101, and −1107) that are distantly related to the majority of Hainan STs. China’s isolates appeared to be different from the Oceania cluster (Australia) and grouped closer to isolates from Southeast Asia, e.g., Thailand, Malaysia, and Vietnam (Figure 6).
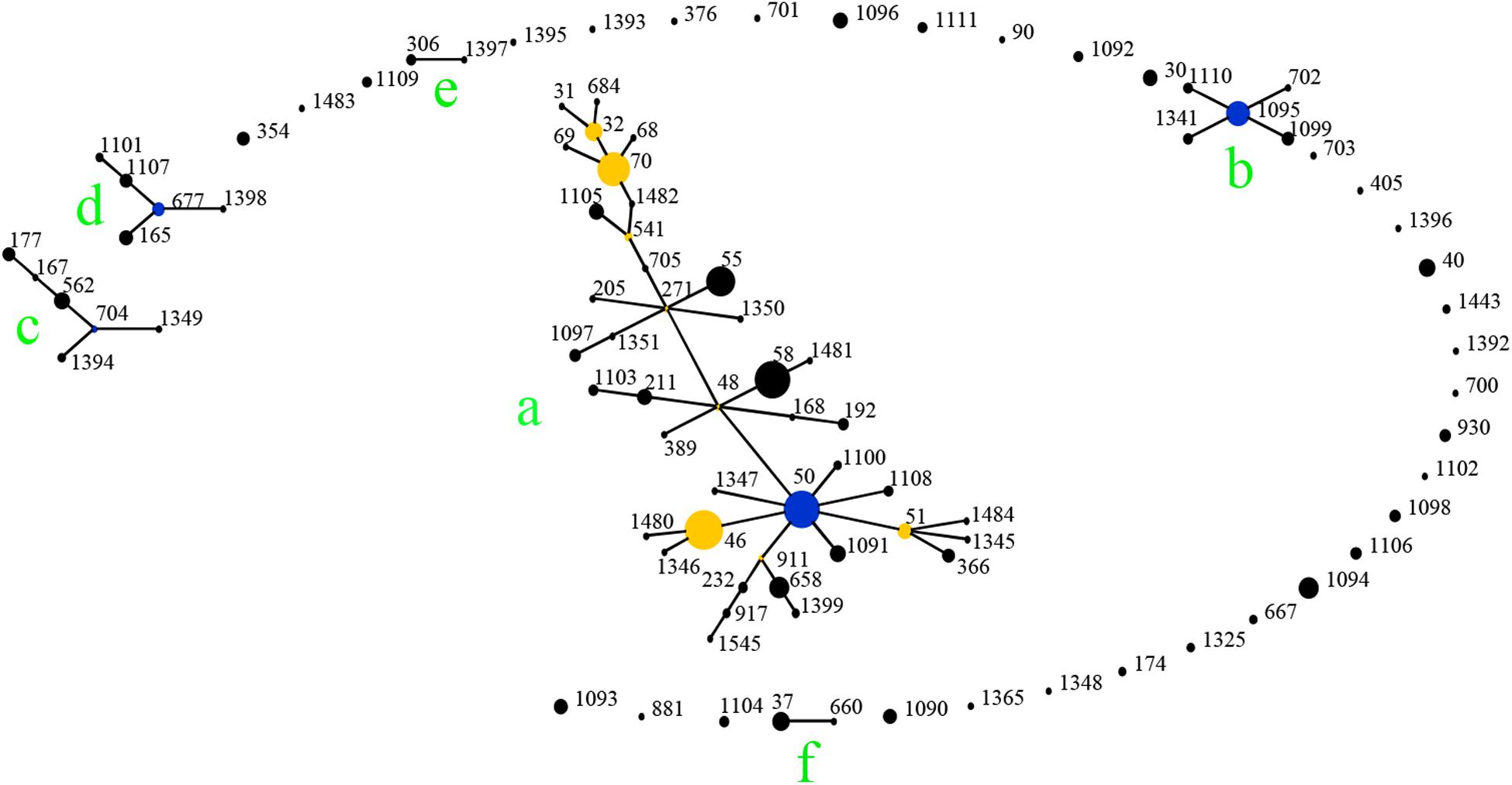
Figure 5. Genetic relationship of all China B. pseudomallei isolates (n = 479) using eBURST. (Blue dot refers to group founder, and yellow dot refers to sub-group founder. Each black dot represents single genotype. Re-sampling for bootstrapping = 10, 000; minimum number of identical loci for group definition = 6; minimum number of SLV for subgroup definition = 3). 479 isolates from China were clustered into 95 STs, and the 62 STs were clustered in six groups (a–f).
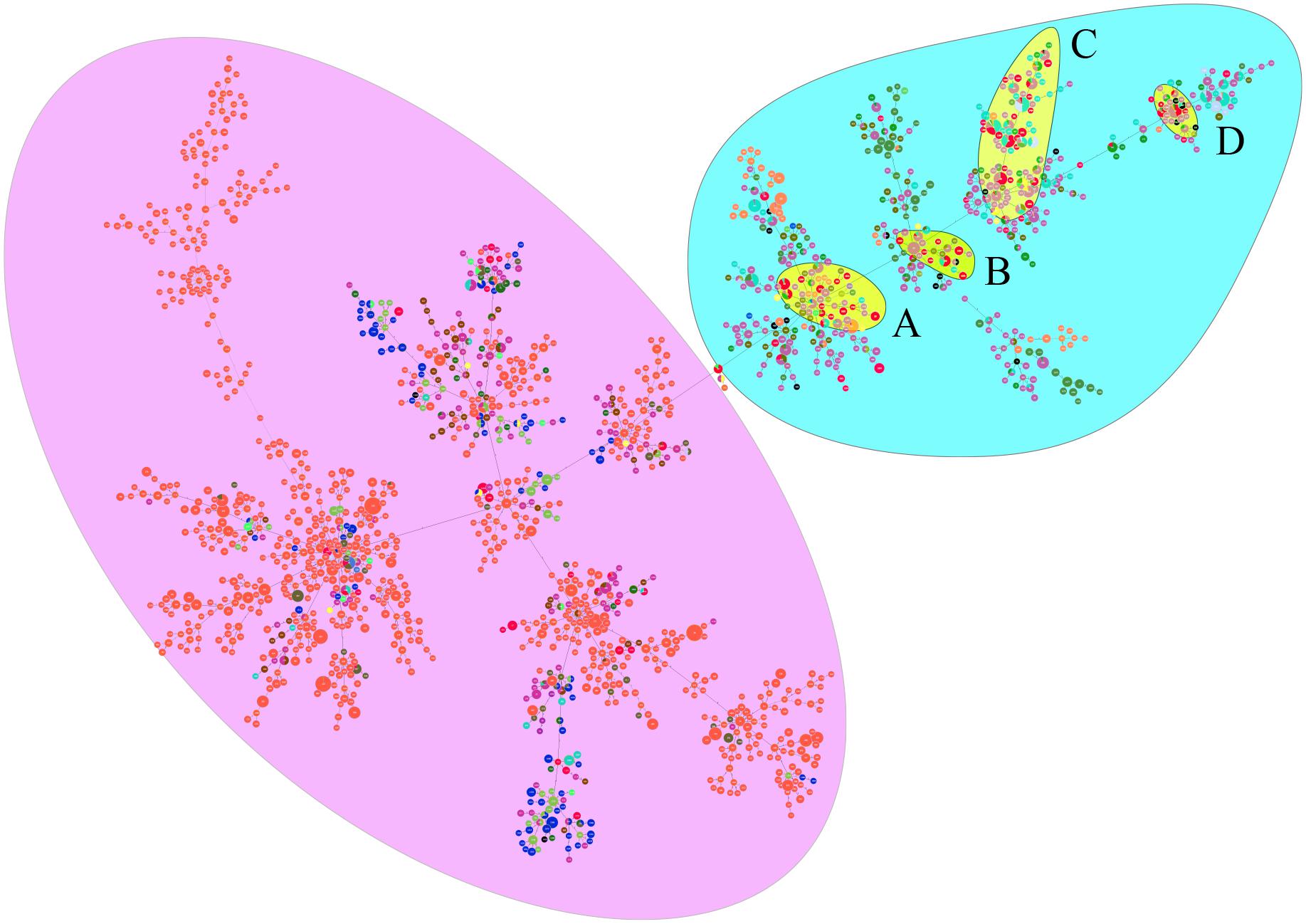
Figure 6. PHYLOViZ analysis showing the genetic relationship among global collection of sequence types (STs) of 6161 B. pseudomallei. Each dot represents a distinct ST. Oceania and Southeast Asian dominant STs are shaded in purple and light blue, respectively. China STs (shaded in yellow) cluster in four groups—All four groups cluster with STs from Southeast Asia. STs from China are shown in red. Different colored dots represent STs from Australia (orange), Thailand (light violet), Malaysia (sky blue), other countries (deep olive green), India (blue), Cambodia (brown), Vietnam (mint green), Sri Lanka (grass green), Singapore (gray), Bangladesh (yellow), Burma (light green), Laos (black), Turkey (indigo), Philippines (purple), and Japan (chartreuse). The majority of Chinese isolates grouped into four groups (A–D).
In this study, pneumonia and sepsis were the most common clinical symptoms of melioidosis patients; 46.2% of patients had known diabetes risk factors, and those with diabetes mellitus have a more than 10-fold higher susceptibility to melioidosis (Currie et al., 2004; Kasantikul et al., 2016). A high death rate (25.0%) was observed, and 69.2% (27/39) of the deceased patients were septic. Septic shock cases have a greater than 90% mortality rate (Cheng and Currie, 2005; Kingsley et al., 2016). Thus, early diagnosis and timely treatment are crucial to obtaining satisfactory therapeutic effects. A total of 65.7% (109/166) of the strains in this study were obtained between 2010 and 2012. These data suggest that this disease exhibited an increasing trend after 2010, in part due to greater awareness and improved diagnostics.
Moreover, these strains were found in 14 areas (of a total of 22 administrative areas) of Hainan Province, indicating a wide distribution for these organisms, although the majority of the strains were detected in coastal areas, where the population density is higher than in center counties (cities). Moreover, due to poor economic resources, many patients from central regions had few chances to be treated. The highest isolation rate from soil samples was observed in the southern coastal region (Dong et al., 2018). The positivity rate for B. pseudomallei in coastal areas or wet rice fields is higher than in mountainous regions in many parts of the globe, rendering the former areas serious epidemic areas for melioidosis (Hsun-Pi et al., 2007; Archana et al., 2014).
MLVA_4 and MLST have higher discrimination power than do ribotyping and RAPD based on PCR amplification (Currie et al., 2009). The MLVA_4 assay showed a higher discriminatory power than MLST (Pearson et al., 2007). The HGDI for MLVA was the highest (0.9025) for VNTR3, with fifteen allele types, suggesting that VNTR3 in the MLVA approach is most useful for discriminating among strains from this province. Furthermore, the most variable locus in MLST was gmhD, with ten alleles. This locus may play a dominant role in the population diversity of B. pseudomallei in this region (Wang et al., 2016). Moreover, B. pseudomallei is a highly recombinogenic species, and recombination events are a key factor for genetic differentiation (Price et al., 2015). The strains from many STs corresponded to three to thirteen similar or related MLVA_4 genotypes, suggesting that MLVA_4 can be used to discriminate closely related clones of B. pseudomallei.
More than 60.0% (101/166) of the strains were in clusters, suggesting that some cases may have a common source. Currie et al. (2009) found that MLVA_4 was able to distinguish epidemiologically unlinked strains that were identical by MLST and PFGE, although the isolates from confirmed point-source outbreaks were either identical or clustered closely. Patients carrying B. pseudomallei with seven shared MLVA_4 genotypes (GT11, −38, −42, −54, −63, −77, and −89) represent identical sources of infection, of which five patients (GT11, −42, −54, −63, and −89) shared the same rice paddy for work. The patients carrying the remaining two genotypes (GT38 and -77) shared an orchard beside a ravine stream, suggesting that these strains are epidemiologically related. In addition, five shared MLVA_4 genotypes (GT21, −38, −39, −53, and −63) included strains with identical STs from different regions over a long time span; these data show an ongoing spread of melioidosis not only within a specific region but also among different regions of Hainan. Nonetheless, without WGS, it is difficult to define how closely related these identical genotypes are without shared epidemiology. Additionally, 65 isolates showed distinct genotypes, indicating that more than 39.2% (65/166) of the melioidosis cases in Hainan had epidemiologically unrelated sporadic characteristics. Two strains from the same patient (IDs 1 and 4) have identical MLVA_4 genotypes and STs, suggesting the occurrence of a single B. pseudomallei infection. Different MLVA_4 profiles and STs were observed in pairs of strains from single infection events (IDs 2, 3, and 5), suggesting that these cases may have been infected by strains from two different B. pseudomallei colony populations or that variation occurred within the strains (Pearson et al., 2007).
Moreover, previous research confirmed that a single unchlorinated water source harboring multiple B. pseudomallei strains was linked to an outbreak (Sarovich et al., 2017). In this study, HNBP163, which was isolated from a patient, has an MLVA_4 genotype (GT9) and ST (667) identical to that of a strain (HNBP164) from a water well located in the patient’s house, suggesting that the source of infection, in this case, was the well water. In addition, strains with two different STs (ST1394 and ST376) were isolated from the same well water sample, and five loci differences were found between ST1394 and ST376. These data provide strong evidence that two strains with distinct STs can be isolated from the same well. Moreover, strains from patients from Inner Mongolia, Fujian, and Hunan had an MLVA_4 genotype and ST identical to those of strains from Hainan. These patients presented to the hospital after traveling to Hainan. Combined field epidemiology suggests that these patients may have had travel-associated infections. HNBP033 (GT60) was obtained from Russia, presenting unique MLVA_4 genotypes, and a further survey of these isolates by WGS may help better trace the sources of infection (Currie et al., 2015; McRobb et al., 2015).
A total of 166 strains were divided into 48 STs, 5 STs accounted for 48.8% of all cases, suggesting that the most common STs are overrepresented in the isolate population associated with disease (Vesaratchavest et al., 2006). When the STs were analyzed using eBURST, the 48 STs were divided into 8 groups and 21 singletons, suggesting that the strains in the Hainan region represent a high diversity of ST clones (Wang et al., 2016). ST50, the predicted founder ST in this study, connected to two dominant ST58 and ST46 and most of the STs by SLV, DLV, or TLV. ST50 is also the predicted founder ST in the Chinese population, and it had a frequency of 42 with 8 SLV, 13 DLV, 15 TLV, and 58 satellite STs. These data suggest that the melioidosis epidemic in China was mainly due to the clonal expansion of ST 50. ST 50 is common in Malaysia and Thailand. Malaysian STs were clustered into a single group with ST50 as the predicted founder (Zueter et al., 2018). ST167 is a single-locus variation of the ST562 type. It was likely imported into Australia from somewhere in Asia (Price et al., 2016b). The other two newly detected STs, 168 and −389, are single or (double)-locus variations of ST48, which is of Thai origin (McCombie et al., 2006). These regions are geographically close, suggesting a potential molecular epidemiology connection between strains from the ST50 clone complex in these regions. The majority of China isolates clustered in Southeast Asia clade suggest the possible dissemination of melioidosis across these Asian countries. There appear to be a few outliers that are distantly related to the majority of Hainan STs and group in the Oceania lineage. There are also three predominant STs (ST46, −58, and −70). These included both strains from this study and strains from Australia, suggesting the probable travel of these predominant STs over time in a global context. The communication and commerce activities between countries may promote the spread of B. pseudomallei strains with different genetic backgrounds. Exploring the geographical expansion and spread of STs among countries and regions is essential to better understand the epidemiology of melioidosis at the global level (Cheng et al., 2008). Another possible explanation is the possibility of ST homoplasy among strains from distinct regions, whereby isolates have the same ST, but do not have shared ancestry and may be distantly related at the whole-genome level. Thus, future work includes performing whole-genome sequencing on all isolates, which is much higher resolution compared to MLST and MLVA (Gee et al., 2017).
Our study has some limitations. First, the data used were collected from passive diagnoses that might have been influenced by case definitions, laboratory tests, or each physician’s understanding of the disease. Second, due to variability in the number of strains collected among the different counties and years, further research with additional strains is essential. Third, a limited number of environmental samples were included (n = 3), and thus extensive environmental sampling is needed to accurately determine the distribution of the STs. This will be vital for source attribution, in order to determine where patients are gaining infection, as well as for guiding public health initiatives and remediation activities on patient property in other regions around Hainan.
In conclusion, a molecular investigation of B. pseudomallei during 2002–2014 was performed in this study. An MLVA_4 assay confirmed that a significant proportion of melioidosis in this province was due to multiple contaminations from a limited number of sources. Moreover, the melioidosis cases in Hainan showed epidemiologically unrelated or sporadic characteristics. Our results demonstrate high diversity was observed in the Hainan strain population, and extensive ST sharing between the strains from this study and those from Thailand, Malaysia, and Vietnam. Determining the homoplasy between the strains of the same ST and MLVA_4 genotype in different geographical locations using WGS is essential to better understand the epidemiology of melioidosis at the global level.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
This study is a retrospective investigation of historical strain collections using modern typing methods. The study protocol was approved by the Ethics Committees of the National Institute for Communicable Disease Control and Prevention and the Chinese Center for Disease Control and Prevention. Informed consent was obtained from all the patients prior to testing. B. pseudomallei strains isolated were using for confirmation diagnosis.
XZ performed most of the strain isolation and MLVA typing. ZGL performed the MLVA cluster analysis and MLST typing and drafted the manuscript. HC, SL, and LCW performed the strain biotyping. DRW and XMW prepared the DNA samples. RSC and ZJL participated in the design of the study and critically reviewed the manuscript. ZGL and ZJL participated in the design of the study and managed the project. All authors read and approved the final manuscript.
This study was supported by the Key R&D Programs of Hainan Province (Grant Nos. ZDYF2019149 and ZDYF2017163), Science and Technology Innovation Project of Medical and Health of Sanya city (2019YW20), Natural Science Fund of the Hainan province (Nos. 814389 and 817319), and National Natural Science Foundation of China (No. 81573208). The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to all the staff in the laboratory and Sanya People’s Hospital, Hainan, for assistance in patient screening and patient epidemiological data collection. We are grateful to Lian-xu Xia and Xiao Zheng from the China CDC for experimental guidance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00778/full#supplementary-material
TABLE S1 | Strain identification codes (Key), GT (MLVA_4), MLST characteristics, location, sources, year of isolation, and outcomes for 166 B. pseudomallei isolates.
TABLE S2 | The 479 Chinese B. pseudomallei strains used to eBURST analysis.
TABLE S3 | The 6161 B. pseudomallei strains used to phylogenetic analysis on a global scale.
TABLE S4 | Patient demographic features.
TABLE S5 | Distribution of 166 B. pseudomallei strains from 2002 to 2014.
TABLE S6 | Distribution characteristics of 48 STs of B. pseudomallei in Hainan Province.
TABLE S7 | Links between MLST and MLVA_4 genotyping approaches.
DLV, Double-locus variants; MLST, Multilocus sequence typing; MLVA, Multiple-locus variable-number tandem repeat analysis; MST, Minimum spanning tree; PCR, Polymerase chain reaction; RAPD, Random amplified polymorphic DNA; SLV, Single-locus variants; ST, Sequence type; VNTR, Variable-number tandem repeat analysis; WGS, Whole-genome sequencing; TLV, Triple-locus variants.
Archana, P., Duraipandian, T., Ashu, K., Ajith, K., Sonia, A., Sapana, T., et al. (2014). Isolation, identification and characterization of Burkholderia pseudomallei from soil of coastal region of India. Springerplus 3:438. doi: 10.1186/2193-1801-3-438
Cheng, A. C., and Currie, B. J. (2005). Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18, 383–416. doi: 10.1128/cmr.18.2.383-416.2005
Cheng, A. C., Ward, L., Godoy, D., Norton, R., Mayo, M., Gal, D., et al. (2008). Genetic diversity of Burkholderia pseudomallei isolates in Australia. J. Clin. Microbiol. 46, 249–254. doi: 10.1128/jcm.01725-07
Currie, B. J., Dance, D. A., and Cheng, A. C. (2008). The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans. R. Soc. Trop Med. Hyg. 102(Suppl. 1), S1–S4.
Currie, B. J., Fisher, D. A., Anstey, N. M., and Jacups, S. P. (2000). Melioidosis: acute and chronic disease, relapse and re-activation. Trans. R. Soc. Trop. Med. Hyg. 94, 301–304. doi: 10.1016/s0035-9203(00)90333-x
Currie, B. J., Haslem, A., Pearson, T., Hornstra, H., Leadem, B., Mayo, M., et al. (2009). Identification of melioidosis outbreak by multilocus variable number tandem repeat analysis. Emerg. Infect. Dis. 15, 169–174. doi: 10.3201/eid1502.081036
Currie, B. J., Jacups, S. P., Cheng, A. C., Fisher, D. A., Anstey, N. M., Huffam, S. E., et al. (2004). Melioidosis epidemiology and risk factors from a prospective whole-population study in northern Australia. Trop Med. Int. Health 9, 1167–1174. doi: 10.1111/j.1365-3156.2004.01328.x
Currie, B. J., Price, E. P., Mayo, M., Kaestli, M., Theobald, V., Harrington, I., et al. (2015). Use of whole-genome sequencing to link burkholderia pseudomallei from air sampling to mediastinal melioidosis. Austr. Emerg. Infect. Dis. 21, 2052–2054. doi: 10.3201/eid2111.141802
Dong, S., Wu, L., Long, F., Wu, Q., Liu, X., Pei, H., et al. (2018). The prevalence and distribution of Burkholderia pseudomallei in rice paddy within Hainan. China. Acta Trop. 187, 165–168. doi: 10.1016/j.actatropica.2018.08.007
Gee, J. E., Gulvik, C. A., Elrod, M. G., Batra, D., Rowe, L. A., Sheth, M., et al. (2017). Phylogeography of Burkholderia pseudomallei Isolates, Western Hemisphere. Emerg. Infect. Dis. 23, 1133–1138.
Hsun-Pi, S., Hsiao-Wei, Y., Ya-Lei, C., Tien-Lin, F., Yu-Ling, C., Tung-Ching, C., et al. (2007). Prevalence of melioidosis in the Er-ren river basin, taiwan: implications for transmission. J. Clin. Microbiol. 45:2599. doi: 10.1128/jcm.00228-07
Hunter, P. R. (1990). Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol. 28, 1903–1905. doi: 10.1128/jcm.28.9.1903-1905.1990
Kamthan, A., Shaw, T., Mukhopadhyay, C., and Kumar, S. (2018). Molecular analysis of clinical Burkholderia pseudomallei isolates from southwestern coastal region of India, using multi-locus sequence typing. PLoS Negl. Trop. Dis. 12:e0006915. doi: 10.1371/journal.pntd.0006915
Kasantikul, T., Sommanustweechai, A., Polsrila, K., Kongkham, W., Chaisongkram, C., Sanannu, S., et al. (2016). Retrospective study on fatal melioidosis in captive zoo animals in thailand. Transbound Emerg. Dis. 63, e389–e394. doi: 10.1111/tbed.12315
Kingsley, P. V., Leader, M., Nagodawithana, N. S., Tipre, M., and Sathiakumar, N. (2016). Melioidosis in malaysia: a review of case reports. PLoS Negl. Trop. Dis. 10:e0005182. doi: 10.1371/journal.pntd.0005182
Kong, Z., Fang, Y., Zhang, M., Hong, J., Tan, Z., Yuan, Z., et al. (2016). Melioidosis acquired by a traveler from Papua New Guinea. Travel. Med. Infect. Dis. 14, 267–270.
Koshy, M., Jagannati, M., Ralph, R., Victor, P., David, T., Sathyendra, S., et al. (2019). Clinical manifestations, antimicrobial drug susceptibility patterns, and outcomes in melioidosis cases, India. Emerg. Infect. Dis. 25, 316–320.
Kronsteiner, B., Chaichana, P., Sumonwiriya, M., Jenjaroen, K., Chowdhury, F. R., Chumseng, S., et al. (2019). Diabetes alters immune response patterns to acute melioidosis in humans. Eur. J. Immunol. 49, 1092–1106.
Le Tohic, S., Montana, M., Koch, L., Curti, C., and Vanelle, P. (2019). A review of melioidosis cases imported into Europe. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1395–1408.
Limmathurotsakul, D., Golding, N., Dance, D. A., Messina, J. P., Pigott, D. M., Moyes, C. L., et al. (2016). Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis. Nat. Microbiol. 1:15008.
Lowe, P., Haswell, H., and Lewis, K. (2006). Use of various common isolation media to evaluate the new VITEK 2 colorimetric GN Card for identification of Burkholderia pseudomallei. J. Clin. Microbiol. 44, 854–856.
McCombie, R. L., Finkelstein, R. A., and Woods, D. E. (2006). Multilocus sequence typing of historical Burkholderia pseudomallei Isolates collected in Southeast Asia from 1964 to 1967 provides insight into the epidemiology of melioidosis. J. Clin. Microbiol. 44, 2951–2962.
McRobb, E., Sarovich, D. S., Price, E. P., Kaestli, M., Mayo, M., Keim, P., et al. (2015). Tracing melioidosis back to the source: using whole-genome sequencing to investigate an outbreak originating from a contaminated domestic water supply. J. Clin. Microbiol. 53, 1144–1148.
Nascimento, M., Sousa, A., Ramirez, M., Francisco, A. P., Carrico, J. A., and Vaz, C. (2017). PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129.
Pearson, T., U’ren, J. M., Schupp, J. M., Allan, G. J., Foster, P. G., Mayo, M. J., et al. (2007). VNTR analysis of selected outbreaks of Burkholderia pseudomallei in Australia. Infect. Genet. 7, 416–423.
Price, E. P., Machunter, B., Spratt, B. G., Wagner, D. M., Currie, B. J., and Sarovich, D. S. (2016a). Improved multilocus sequence typing of Burkholderia pseudomallei and closely related species. J. Med. Microbiol. 65, 992–997.
Price, E. P., Sarovich, D. S., Smith, E. J., Machunter, B., Harrington, G., Theobald, V., et al. (2016b). Unprecedented melioidosis cases in northern australia caused by an asian burkholderia pseudomallei strain identified by using large-scale comparative genomics. Appl. Environ. Microbiol. 82, 954–963.
Price, E. P., Sarovich, D. S., Viberg, L., Mayo, M., Kaestli, M., Tuanyok, A., et al. (2015). Whole-genome sequencing of Burkholderia pseudomallei isolates from an unusual melioidosis case identifies a polyclonal infection with the same multilocus sequence type. J. Clin. Microbiol. 53, 282–286.
Sarovich, D. S., Chapple, S. N. J., Price, E. P., Mayo, M., Holden, M. T. G., Peacock, S. J., et al. (2017). Whole-genome sequencing to investigate a non-clonal melioidosis cluster on a remote Australian island. Microb. Genom. 3:e000117.
Shrestha, N., Adhikari, M., Pant, V., Baral, S., Shrestha, A., Basnyat, B., et al. (2019). Melioidosis: misdiagnosed in Nepal. BMC Infect. Dis. 19:176. doi: 10.1186/s12879-019-3793-x
U’Ren, J. M., Schupp, J. M., Pearson, T., Hornstra, H., Friedman, C. L., Smith, K. L., et al. (2007). Tandem repeat regions within the Burkholderia pseudomallei genome and their application for high resolution genotyping. BMC Microbiol. 7:23. doi: 10.1186/s12879-019-23-x
Vesaratchavest, M., Tumapa, S., Day, N. P., Wuthiekanun, V., Chierakul, W., Holden, M. T., et al. (2006). Nonrandom distribution of Burkholderia pseudomallei clones in relation to geographical location and virulence. J. Clin. Microbiol. 44, 2553–2557.
Wang, X. M., Zheng, X., Wu, H., Zhou, X. J., Kuang, H. H., Guo, H. L., et al. (2016). Multilocus sequence typing of clinical isolates of Burkholderia pseudomallei collected in hainan, a tropical island of southern China. Am. J. Trop. Med. Hyg. 95, 760–764.
Wiersinga, W. J., Currie, B. J., and Peacock, S. J. (2012). Melioidosis. N. Engl. J. Med. 367, 1035–1044.
Keywords: melioidosis, Burkholderia pseudomallei, molecular characteristics, MLVA, MLST, Hainan
Citation: Zhu X, Chen H, Li S, Wang L, Wu D, Wang X, Chen R, li Z and Liu Z (2020) Molecular Characteristics of Burkholderia pseudomallei Collected From Humans in Hainan, China. Front. Microbiol. 11:778. doi: 10.3389/fmicb.2020.00778
Received: 21 October 2019; Accepted: 01 April 2020;
Published: 07 May 2020.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
Ella Meumann, Charles Darwin University, AustraliaCopyright © 2020 Zhu, Chen, Li, Wang, Wu, Wang, Chen, li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-guo Liu, bGl1emhpZ3VvQGljZGMuY24=; d2xjYmx6Z0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.