 Xiaoting Hua
Xiaoting Hua Linyue Zhang1,2
Linyue Zhang1,2 Sebastian Leptihn
Sebastian Leptihn Yunsong Yu
Yunsong Yu- 1Department of Infectious Diseases, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Key Laboratory of Microbial Technology and Bioinformatics of Zhejiang Province, Hangzhou, China
- 3Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, Hangzhou, China
Recently, discussions arose about the accuracy of the two Acinetobacter baumannii multilocus sequence typing (MLST) Schemes (Hamidian et al., 2017; Castillo-Ramirez and Grana-Miraglia, 2019; Gaiarsa et al., 2019). Santiago et al. showed that neither the Oxford scheme nor the Pasteur scheme could reflect the relationships among A. baumannii isolates accurately (Castillo-Ramirez and Grana-Miraglia, 2019). Stefano Gaiarsa et al. reported issues of the Oxford scheme regarding gdhB paralogy, recombination, primer sequences, and position of the genes on the genome (Gaiarsa et al., 2019). The importance of using more powerful genotyping strategies is highlighted when analyzing bacteria with highly dynamic genomes like Acinetobacter baumannii. The decrease in costs of whole genome sequencing would make the technology an ideal tool for genotyping of bacterial species. Here, we provide experimental data obtained from the two methods, allowing a direct comparison between the core genome MSLT (cgMLST) and core SNP (cgSNP) with the MLST.
First, we revisited the genomic sequences of 172 previously described A. baumannii isolates that were obtained from three hospitals in Hangzhou, China (Hua et al., 2017). Six isolates that did not belong to A. baumannii were excluded. The aim was to construct robust phylogenetic trees of the 166 genome sequences by using MLST, cgMLST and cgSNP data individually, and to then determine the differences and similarities between the two methods. To avoid confusion, we used SToxf and STpas to distinguish Oxford scheme and Pasteur scheme in A. baumannii MLST. The MSLT results showed that our datasets covered 20 SToxfs and 10 STpass. Compared to the Oxford scheme (SToxf), there were fewer STs in the Pasteur scheme (STpas) (Figure 1A). The diversity of the two MLST schemes were assessed using the Simpson's Diversity Index with a 95% confidence interval (http://www.comparingpartitions.info/). For the Pasteur scheme, the Simpson's Diversity Index is 0.291 (0.202–0.380) while it is 0.823 (0.781–0.866) for the Oxford scheme. The Oxford scheme was more discriminatory than the Pasteur scheme. The result of the Oxford scheme, which displayed a higher Simpson's Diversity Index, is confirmed by Gaiarsa et al. who based their calculation on the genomes of the three International Clones using 730 genomes (Gaiarsa et al., 2019). We used the genome dataset used in Bautype which contained all the available genome assemblies from Genbank (3519 as of May, 2019) to confirm the comparative result of the Oxford scheme and the Pasteur scheme (Hua et al., 2020). For the Pasteur scheme, the Simpson's Diversity Index is 0.645 (0.626–0.664) while it is 0.934 (0.929–0.940) for the Oxford scheme.
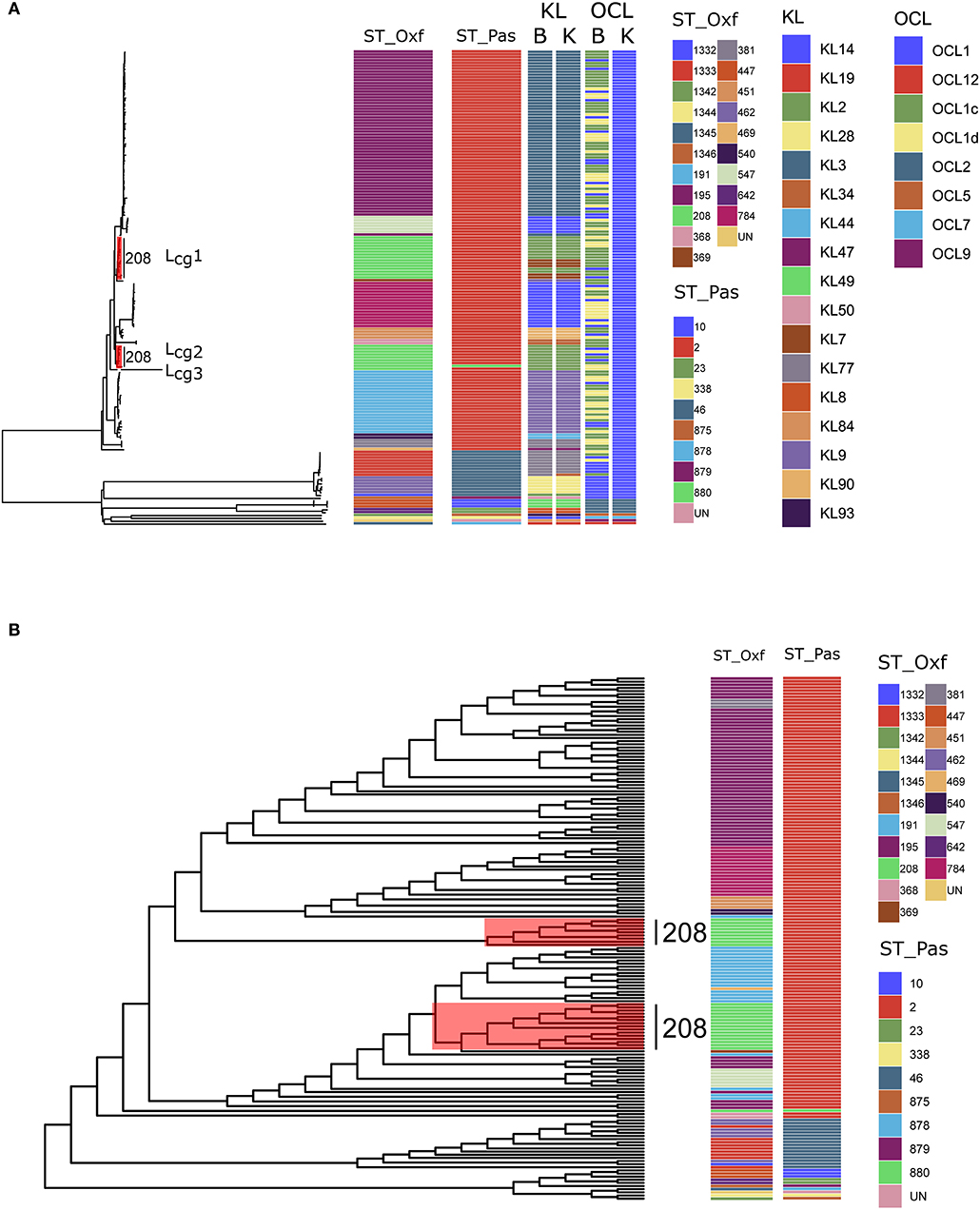
Figure 1. (A) Maximum-likelihood phylogeny reflecting the relationships among Acinetobacter baumannii isolates via cgMLST. The corresponding MLST types, KL types, and OCL types are shown on the right. Three distinct lineages (Lcg1, 2, 3) from SToxf208 were marked with red. ST_Oxf: types defined by the Oxford scheme; ST_Pas: types defined by the Pasteur scheme; B: types defined by Bautype; K: types defined by Kaptive; (B) Maximum-likelihood phylogeny via cgSNP. The corresponding MLST types, are shown on the right. Three distinct lineages (Lcg1, 2, 3) from SToxf208 were marked with red.
In the first step of our work, among 166 isolates, 145 isolates were assigned to 11 different STs by the Oxford scheme but only one ST (STpas 2) when using the Pasteur scheme. This observation provides further support for the opinion that was put forward by Santiago et al. who elaborates that the two schemes show different levels of resolution with the Pasteur scheme, revealing considerably less detail than the Oxford scheme when distinguishing between A. baumannii isolates (Castillo-Ramirez and Grana-Miraglia, 2019). This observation also confirmed the result of Gaiarsa et al. that the Oxford MLST scheme has higher discriminatory power and higher concordance than Pasteur MLST (Gaiarsa et al., 2019).
Second, we performed a genome-wide gene-by-gene comparison by employing the cgMLST target definer function of the software Ridom SeqSphere+, version 5.1.0 (Ridom, Münster, Germany) using default parameters (Junemann et al., 2013). We constructed a maximum-likelihood phylogeny via 2390 core genes to determine the differences between the isolates. The cgMLST method showed higher Simpson's Diversity Index (0.964, CI: 0.951–0.976) than the Pasteur scheme and the Oxford scheme. Based on our cgMLST profiles, all STs in either scheme formed coherent lineages except for SToxf208. Surprisingly, we found that SToxf208 strains could be divided into three distinct lineages (named as Lcg1, 2, 3) rather than subdivided internally (Figure 1A). This phenomenon indicated that the internal differences determined by cgMLST of these 24 strains were greater than the differences determined by the seven housekeeping genes applied in the Oxford scheme. The capsular polysaccharide (KL) and lipooligosaccharide outer core (OCL) were detected by Kaptive (Wyres et al., 2020) and Bautype (Hua et al., 2020). For the KL characterization, results from Kaptive and Bautype correlate, except for XH662. The KL type of XH662 in Kaptive is KL34, while it is KL77 in Bautype. The different predicted KL type was caused by the difference among reference sequences of KL77 used in Kaptive and Bautype. The sequence of KL77 in Kaptive has one more A at the position 21925 compared to that in Bautype. KL recombination was observed in Lcg1 (Figure 1A). There were two KLs, KL2 and KL7 in Lcg1. For OCL, all ST208 belonged to OCL1 when performing the analysis using Kaptive. The Bautype result showed that OCL1, OCLc, and OCLd appeared in ST208 strains. The difference in OCL results indicated a different OCL database curator strategy in Kaptive vs. Bautype.
Next, we constructed phylogenetic tree base core genome SNPs. Core genome SNPs were called for each isolate using Snippy v4.4.5 (https://github.com/tseemann/snippy), and each isolate was mapped to the reference genome XH386 (CP010779.1). Alignments were filtered for recombinations using Gubbins v2.3.4 (Croucher et al., 2015). A maximum likelihood tree was inferred with RAxML v8.2.12 under the GTRCAT model (Stamatakis, 2014). The phylogenetic trees were visualized using ggtree v1.16 (Yu, 2020). The result of cgSNP also support the finding of cgMLST that SToxf208 strains could be divided into three distinct lineages (Figure 1B).
Last, in order to investigate the reasons that might cause this phenomenon, we analyzed the variable allelic genes in cgMLST within SToxf208 strains and defined them as two gene sets. The first gene set contained 11 adjacent genes and formed two different allelic variation combinations, based on which SToxf 208 strains could be roughly divided into 2 groups (Lcg2 as a group1, Lcg1 and Lcg3 as group2). The second gene set contained 14 genes distributed discretely on the chromosome and formed three different allelic variation combinations that could further distinguish Lcg1 and Lcg3 from the previously described “group 2.” We also found the same allelic variation combinations in the first gene set appearing in other non-SToxf208 strains as well (Table 1). For example, SToxf208 Lcg2 strains and a SToxf381 strain (XH663) shared the same allelic variation combination. Similarly, SToxf208 Lcg1, Lcg3 strains and a SToxf191 strain (XH697) exhibited the same phenomenon. Considering the involved genes were adjacent, we speculate that this is due to homologous recombination, a mechanism which was proposed previously (Castillo-Ramirez and Grana-Miraglia, 2019). A previously published report describes that the gpi gene, which is used in the Oxford scheme, is located near the capsule biosynthesis gene cluster, with recombination replacements of this cluster region being common, again possibly due to homologous recombination. As a result, a different gpi sequences were introduced, leading to problems for typing (Hamidian et al., 2017). This illustrates that a more suitable MLST scheme, including more reliable housekeeping genes, needs to be developed.
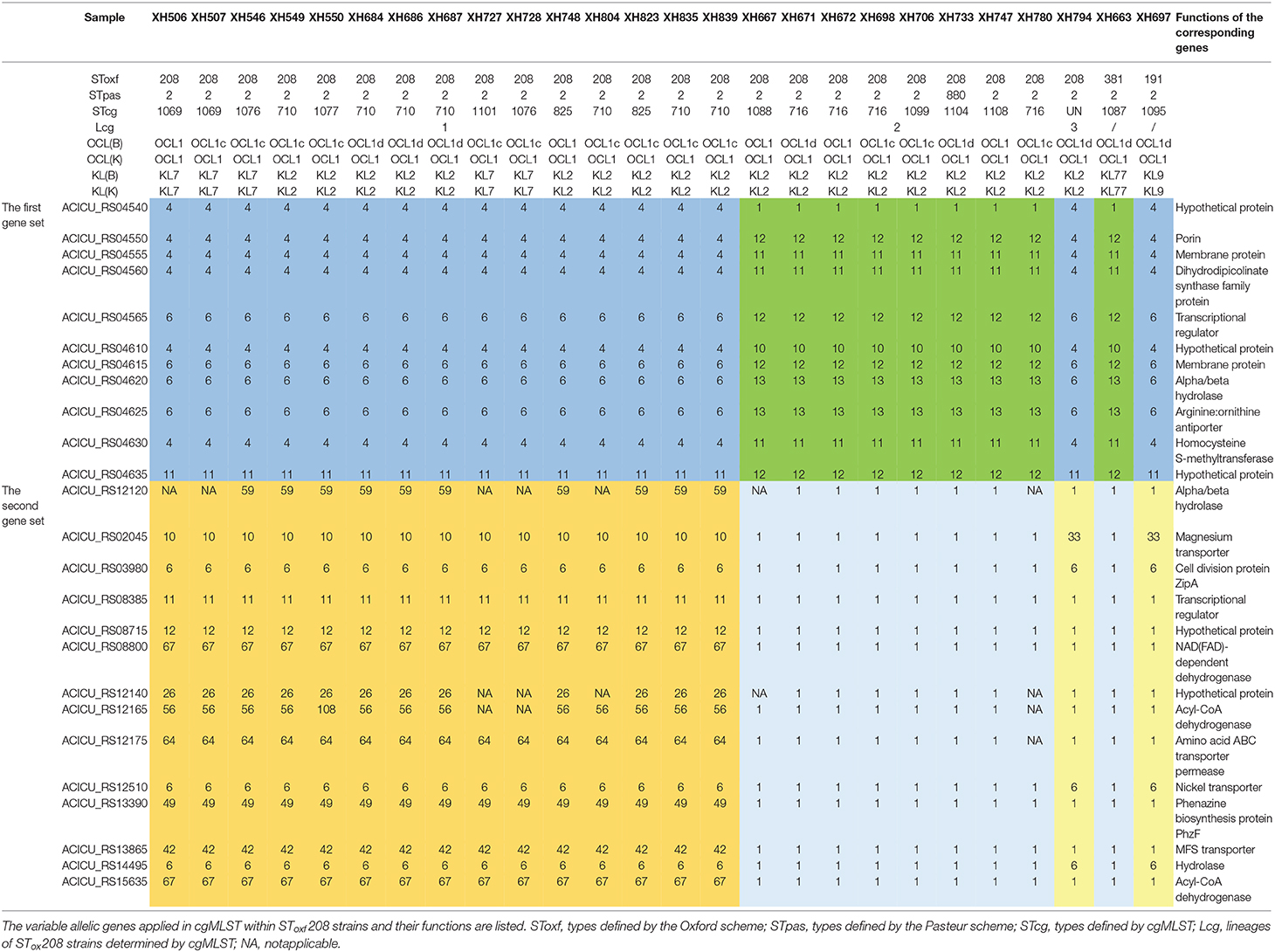
Table 1. The cgMLST and MSLT results of all SToxf208 strains and two non-SToxf208 strains.
A. baumannii has emerged as a critical pathogen of nosocomial infections worldwide, particularly in intensive care (Wong et al., 2017). SToxf208 was identified as predominant ST of carbapenem resistant A. baumannii in China (Deng et al., 2014). The MLST scheme has been used for infectious disease epidemiology, revealing the relationships within and between bacterial lineages. Although MLST has many advantages over other molecular typing methods, it cannot reflect the true relationships of isolates for A baumannii using only a small number of chromosomal segments due to the high levels of recombination events in the bacterial genome (Castillo-Ramirez and Grana-Miraglia, 2019). Our analysis demonstrated that strains of the predominant type SToxf208 could be divided into three distinct lineages when employing cgMLST and cgSNP, which showed a superior discriminatory ability compared with the conventional MLST technique, where all strains are clustered together as the same type. By using the information from cgMLST and/or cgSNP, we might be able to gain a better perspective to understand the epidemiology of A. baumannii infections in China, but also globally.
Author Contributions
XH and YY designed the study. LZ and XH analyzed the bioinformatics data. LZ, JH, and SL wrote the manuscript.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (31670135, 31770142, 81861138054, and 31970128) and the Zhejiang Province Medical Platform (2020RC075).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Bartual and coworkers for the introduction of the Oxford scheme, and Diancourt and coworkers for the introduction of the Pasteur scheme.
References
Castillo-Ramirez, S., and Grana-Miraglia, L. (2019). Inaccurate Multilocus Sequence Typing of Acinetobacter baumannii. Emerg. Infect. Dis. 25, 186–187. doi: 10.3201/eid2501.180374
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Deng, M., Zhu, M. H., Li, J. J., Bi, S., Sheng, Z. K., Hu, F. S., et al. (2014). Molecular epidemiology and mechanisms of tigecycline resistance in clinical isolates of Acinetobacter baumannii from a Chinese university hospital. Antimicrob Agents Chemother. 58, 297–303. doi: 10.1128/AAC.01727-13
Gaiarsa, S., Batisti Biffignandi, G., Esposito, E. P., Castelli, M., Jolley, K. A., Brisse, S., et al. (2019). Comparative analysis of the two Acinetobacter baumannii Multilocus Sequence Typing (MLST) schemes. Front. Microbiol. 10:930. doi: 10.3389/fmicb.2019.00930
Hamidian, M., Nigro, S. J., and Hall, R. M. (2017). Problems with the oxford multilocus sequence typing scheme for acinetobacter baumannii: do sequence Type 92 (ST92) and ST109 Exist? J. Clin. Microbiol. 55, 2287–2289. doi: 10.1128/JCM.00533-17
Hua, X., Liang, Q., Fang, L., He, J., Wang, M., Hong, W., et al. (2020). Bautype: capsule and lipopolysaccharide serotype prediction for: Acinetobacter baumannii: genome. Infect. Microbes Dis. 2, 18–25. doi: 10.1097/im9.0000000000000019
Hua, X., Zhou, Z., Yang, Q., Shi, Q., Xu, Q., Wang, J., et al. (2017). Evolution of Acinetobacter baumannii in vivo: international Clone II, more resistance to ceftazidime, mutation in ptk. Front. Microbiol. 8:1256. doi: 10.3389/fmicb.2017.01256
Junemann, S., Sedlazeck, F. J., Prior, K., Albersmeier, A., John, U., Kalinowski, J., et al. (2013). Updating benchtop sequencing performance comparison. Nat. Biotechnol. 31, 294–296. doi: 10.1038/nbt.2522
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Wong, D., Nielsen, T. B., Bonomo, R. A., Pantapalangkoor, P., Luna, B., and Spellberg, B. (2017). Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin. Microbiol. Rev. 30, 409–447. doi: 10.1128/CMR.00058-16
Wyres, K. L., Cahill, S. M., Holt, K. E., Hall, R. M., and Kenyon, J. J. (2020). Identification of Acinetobacter baumannii loci for capsular polysaccharide (KL) and lipooligosaccharide outer core (OCL) synthesis in genome assemblies using curated reference databases compatible with Kaptive. Microb Genom. 6. doi: 10.1099/mgen.0.000339
Keywords: Acinetobacter baumannii, whole genome sequencing, cgMLST, mlst, genotyping
Citation: Hua X, Zhang L, He J, Leptihn S and Yu Y (2020) Population Biology and Epidemiological Studies of Acinetobacter baumannii in the Era of Whole Genome Sequencing: Is the Oxford Scheme Still Appropriate? Front. Microbiol. 11:775. doi: 10.3389/fmicb.2020.00775
Received: 05 March 2020; Accepted: 31 March 2020;
Published: 29 April 2020.
Edited by:
Jörg Linde, Friedrich Loeffler Institute, GermanyReviewed by:
Raffaele Zarrilli, University of Naples Federico II, ItalyCopyright © 2020 Hua, Zhang, He, Leptihn and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunsong Yu, eXZ5czExOUB6anUuZWR1LmNu