
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 May 2020
Sec. Food Microbiology
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00636
 Natalie Witt1,2
Natalie Witt1,2 Sandro Andreotti3
Sandro Andreotti3 Anne Busch4,5
Anne Busch4,5 Kerstin Neubert6
Kerstin Neubert6 Knut Reinert6
Knut Reinert6 Herbert Tomaso4
Herbert Tomaso4 David Meierhofer1*
David Meierhofer1*Zoonotic pathogens that can be transmitted via food to humans have a high potential for large-scale emergencies, comprising severe effects on public health, critical infrastructures, and the economy. In this context, the development of laboratory methods to rapidly detect zoonotic bacteria in the food supply chain, including high-resolution mass spectrometry proteotyping are needed. In this work, an optimized sample preparation method for liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based proteome profiling was established for Francisella isolates, and a cluster analysis, as well as a phylogenetic tree, was generated to shed light on evolutionary relationships. Furthermore, this method was applied to tissues of infected hare carcasses from Germany. Even though the non-informative data outnumbered by a manifold the information of the zoonotic pathogen in the resulting proteome profiles, the standardized evaluation of MS data within an established automated analysis pipeline identified Francisella (F.) tularensis and, thus, could be, in principle, an applicable method to monitor food supply chains.
Zoonotic diseases are a biological threat to humans, and some have even the potential to be misused as biological weapons. In order to assess the epidemiological situation, knowledge about the origin and the distribution of an outbreak by fast and reliable methods is essential. Cultivation of bacteria in biosafety levels 2 and 3 laboratories is the gold standard for diagnosing these pathogens and allows subsequent typing of bacteria, e.g., with PCR assays and other DNA-based techniques. Currently, matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS) approaches are commonly used to identify Francisella in an automated way at low costs in a very short time (Hubalek et al., 2004; Kilmury and Twine, 2010; Seibold et al., 2010; Durighello et al., 2014; Karatuna et al., 2016; Kasap et al., 2017). However, only pure isolates can be identified, adequate reference databases are required, and some species cannot be discriminated against due to their close phylogenetic relationship. MALDI-based proteome profiling is, thus, not capable of identifying pathogens in complex biological matrices, as only the most abundant proteins can be detected.
In contrast, high-resolution electrospray ionization (ESI) LC-MS/MS has progressed tremendously in recent years (Dworzanski et al., 2004; Cheng et al., 2016). Shotgun proteomics, in combination with bioinformatics, has enabled proteomics-based microbial identifications, even to the strain level (Dworzanski et al., 2006; Karlsson et al., 2015, 2018; Hayoun et al., 2019). In parallel with the introduction of proteomics for analyzing microorganisms, this resulted in the development of an analytical methodology called proteotyping. Proteotyping is a technique, which uses high-resolution MS and proteomic analysis to comprehensively characterize, classify, and identify microorganisms. This can, for example, include taxonomic features for identification, features important for clinical responses (e.g., biomarkers of antibiotic resistance and virulence), as well as markers for biotechnological and environmental applications (e.g., biomarkers of catabolic and anabolic pathways in cell metabolism; Karlsson et al., 2015). Proteotyping, therefore, uses an amino acid sequence list of all isoforms, which are seen as mass shifts in LC-MS/MS spectra, that have arisen from non-synonymous mutations in the genes between the species. The advantage of proteotyping over whole spectrum clustering approaches is that only mass changes associated with a particular set of allelic isoforms of the same protein are considered for phylogeny derivation (Emele et al., 2019). Other methods instead also take the presence or absence of individual masses as well as peak intensity into account, which delivers less accurate results (Zautner et al., 2015).
F. tularensis is a highly infectious Gram-negative bacterium causing the zoonotic disease tularemia. The Centers for Disease Control (CDC) considers this bacterium as a potential biological agent of category A, which may have a major impact on an exposed human population (Rotz et al., 2002). In Germany, most humans get infected due to contact with infected hare (Lepus europaeus). The clinical signs and symptoms of the disease vary depending on the route of transmission and can present as ulceroglandular, oculoglandular, oropharyngeal, or pneumonic tularemia. Cultivation of the slow-growing bacterium F. tularensis takes several days, and optimal growth conditions require nutrient media, such as the Cystine Heart Agar. Routine laboratories usually do not offer specific diagnostic tests for this relatively rare pathogen. Serological assays can help to establish the diagnosis in human cases and have been used to screen indicator animals, such as foxes and wild boars (Sharma et al., 2013, 2014), but food samples still require a classical approach.
Here, we demonstrate – as proof of principle – the use of shotgun proteome profiling by high-resolution ESI LC-MS/MS for direct evidence of Francisella ssp., as an exemplary representative of zoonotic bacteria, in tissues of infected hare carcasses without prior cultivation of the pathogen. Furthermore, the lower limit of detection (LLOD) of this method was determined by a spike-in of known concentrations of a Francisella isolate into the hare matrix. A phylogenetic tree was generated to show evolutionary relationships among Francisella ssp. isolates based on similarities and differences in their genetic characteristics. In addition, a cluster analysis, in which proteome data from 45 Francisella strains were grouped (clustered) in a way that strains in the same cluster are more similar to each other on a proteomic level than those in other groups, was applied. Therefore, an optimized LC-MS/MS sample preparation protocol was established to generate the proteome profiles of the Francisella isolates, along with strain-specific whole-genome sequencing (WGS) data. A scheme of the methodological strategy is outlined in Figure 1. ESI LC-MS/MS may help to analyze suspicious samples in outbreak scenarios and can be applied to inactivated specimens. This approach has the potential to screen simultaneously for a vast variety of pathogens in complex food matrices.
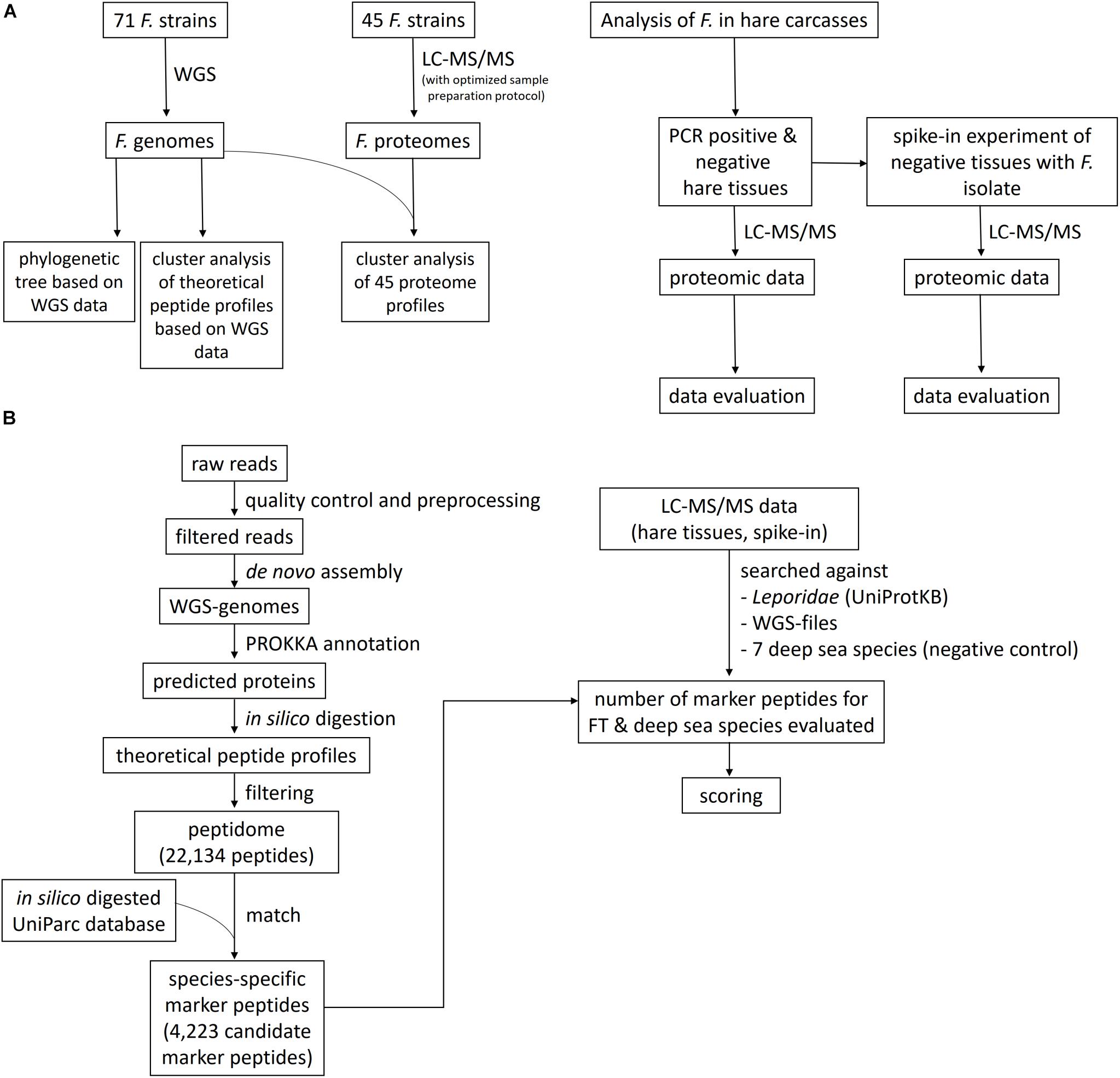
Figure 1. Flowchart of the study design and the bioinformatics data evaluation pipeline. (A) Francisella isolates were sequenced using whole-genome sequencing (WGS) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) proteome profiling, applying an optimized sample preparation protocol for MS. A phylogenetic tree and a cluster analysis of the theoretical peptide profiles based on the WGS data were established, together with a cluster analysis of the proteome profiles. The proteome profiling method was applied to tissues of with F. tularensis-infected hare carcasses and to an artificial spike-in experiment, where the lower limit of detection was determined. (B) The bioinformatics data evaluation comprises the WGS pipeline, containing the read preprocessing, the assembly of the genomes, and the Prokka annotation. The predicted proteins were in silico digested, and the peptidome was matched against an in silico digested UniParc database. The number of these species-specific peptides was determined in the LC-MS/MS data of the hare tissues and the spike-in experiment, together with peptide hits for seven deep-sea species, which served as a negative control to determine the specificity of the applied method. A scoring function was created to evaluate the significance of the found matches.
Francisella strains (71) were selected for WGS. Institutional handling of tularemia-positive samples or cultures were done as described before (Busch et al., 2018), following the code of conduct to minimize dual-use risks (dual-use potential of life sciences research)1. The isolates were obtained from liver and spleen samples of hare carcasses collected by hunters in Germany between 2012 and 2018. The samples were identified and characterized at the Friedrich-Loeffler-Institut (Jena, Germany), using cultivation, MALDI-TOF MS of isolates, conventional PCR assays targeting the gene tul4 (recognizing all three Francisella biogroups type A tularensis, type B holarctica, and Novicida), and WGS, as reported previously (Busch et al., 2018). The assemblies were deposited in public repositories at www.ncbi.nlm.nih.gov/bioproject (PRJNA560345, PRJNA353900, and PRJNA575140), as referenced in Supplementary Table S1.
Two primary spleen and two liver tissues derived from a total of three individual infected hare carcasses were investigated (Table 1). All four samples (16T0017, 18T0118, 18T0123, 18T0124) were autoclaved at 121°C for 30 min to allow safe handling of the material in the laboratory. In addition, spleen (n = 3) and liver tissue (n = 3) of hare carcasses that were negative for F. tularensis (as determined by cultivation and PCR assays) were used as negative controls (16T1200, 16T1202, 16T1188, 16T1203, 16T1205, 16T1215).
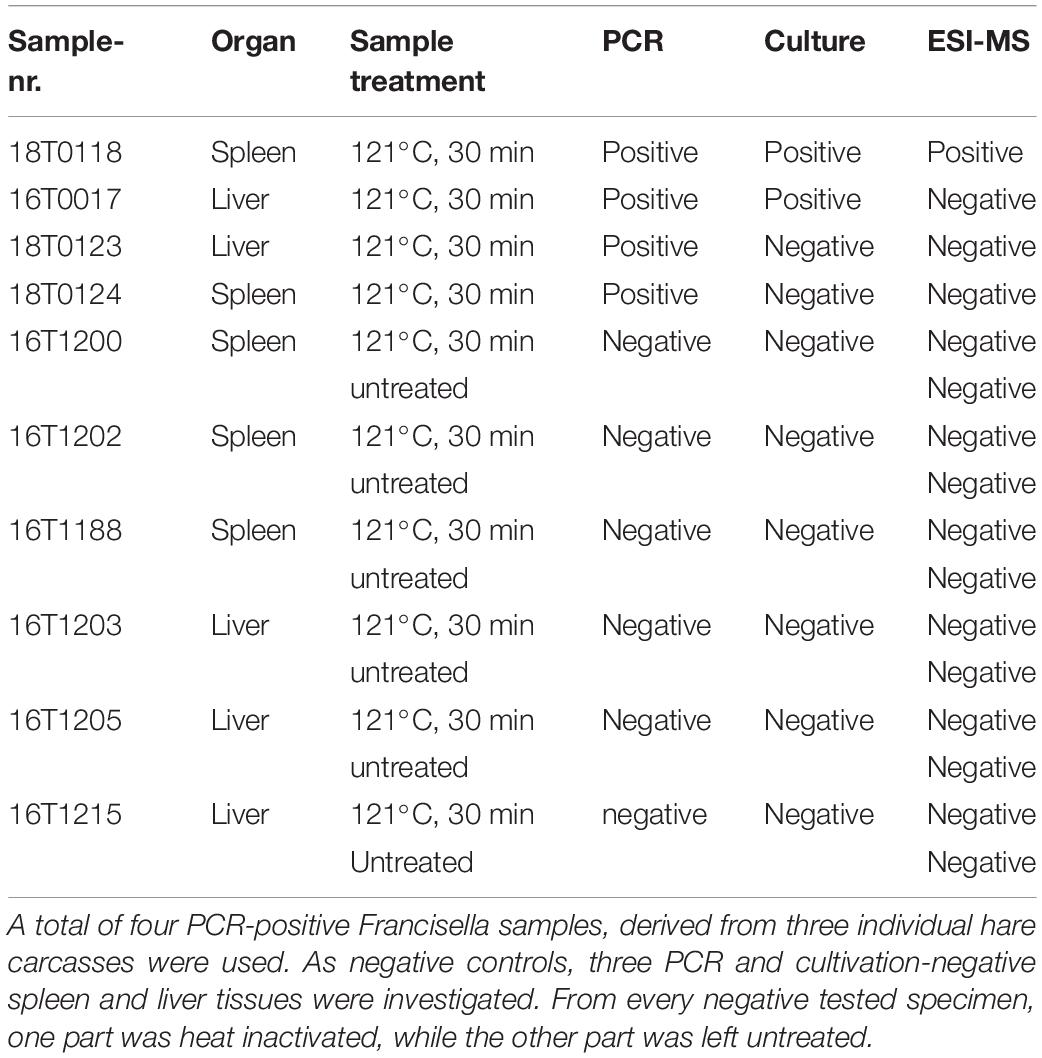
Table 1. Sample and method overview to directly identify Francisella in tissues collected from hare carcasses.
WGS was performed as described previously (Busch et al., 2018). In brief, the DNA was prepared from a 10 mL culture in brain heart infusion broth (Brain, Heart Infusion Broth, Sifin, Berlin, Germany). Bacterial cells were harvested after 72 h by centrifugation, and the DNA was purified using QIAGEN Genomic-tip 20/G and a QIAGEN Genomic DNA buffer set kit (Qiagen, Hilden, Germany). The DNA quality was examined using a Qubit 2.0 fluorometer (Life Technologies, Germany) and agarose gel electrophoresis. All strains were subjected only to Illumina HiSeq and/or MiSeq sequencing using the Nextera XT DNA protocol for library preparation (in house; GATC, Konstanz, and BfR, Berlin, Germany). The raw reads for data deposited in PRJNA575140 were filtered using Flexbar (version 3.0.3) to remove adapters and perform quality trimming (BWA trimming mode with a quality offset of 25). Overlapping paired-end reads were merged using FLASH2 (version 2.2) with a minimum required overlap length between reads of 50 bp. The resulting extended fragments and non-overlapping reads were assembled using SPAdes (version 3.10.0) with the options “careful” and “cov-cutoff auto”. Data were filtered excluding data with a < 5 coverage and < 500 bp in length. Prokka (version 1.14-dev) was used to identify genomic features with the inclusion of a genus-specific BLAST database of Francisella and subsequent translation of CDS sequences.
Two sample preparation protocols, namely, the trichloroacetic acid (TCA) extraction protocol and the iST kit (PreOmics, Martinsried, Germany) were applied to the two F. tularensis test strains, 08T0073 and FSC237, that were inactivated by either autoclavation (121°C, 30 min) or inactivated in 75% ethanol, performed in technical duplicates.
The samples were prepared according to the TCA protocol as reported previously (Deatherage Kaiser et al., 2015). In brief, 40 μg of protein of each sample was pelleted for 10 min at maximum speed (21,130 × g) and resuspended in 0.5 mL of 20% TCA (Sigma-Aldrich, St. Louis, MO, United States) solution in water. After incubation for 24 h at −20°C, the samples were thawed and pelleted for 10 min at 21,130 × g at 4°C. The pellets were washed twice in 200 μl of ice-cold (−20°C) acetone (VWR, Radnor, PA, United States), the supernatant was discarded, and the pellets were dried under the fume hood. The dried pellets were resuspended for 1 h at 60°C and 400 rpm in 100 μl of lysis buffer containing 6 M urea (GE Healthcare, Chicago, IL, United States), 14.3 mM 2-mercaptoethanol (Roth, Karlsruhe, Germany) in 50 mM Tris/HCl (pH 8.5), followed by an ultrasonic treatment until complete resuspension of the pellets. Of each protein lysate, 20 μl was digested in a total volume of 150 μl – containing 40 mM ammonium bicarbonate (Sigma-Aldrich) in water, 400 ng of trypsin (Roche, Basel, Switzerland), and 400 ng of Lys C (Thermo Fisher Scientific, Waltham, MA, United State) – for 4 h at 37°C and 300 rpm. The peptides were desalted by C18 tips (Thermo Scientific) for subsequent LC-MS/MS analysis.
The iST Kit (PreOmics) was used according to the manufacturer’s protocol (pelleted cells and precipitated protein, v. 2.5). For this purpose, 8 μg of each sample was pelleted for 10 min at maximum speed (21,130 × g), and the digestion was performed using the provided trypsin-Lys-C mixture for 4 h.
Desalted peptides from both protocols were dried in a vacuum concentrator and dissolved in either 12 μl of LC Load (iST kit) or 5% acetonitrile (ACN, VWR) and 2% formic acid (FA; TCA protocol, Fisher Chemicals, Waltham, MA, United States), briefly vortexed and sonicated in a water bath for 2 min. After centrifugation at 16,000 × g for 5 min, 6 μl was injected for nano-LC-MS/MS analysis. To avoid any carry-over of peptides, one wash was always run in between all individual samples in all experiments. In order to benchmark the two sample preparation methods, the RAW MS data were processed with the MaxQuant software (v.1.5.3.30) (Cox and Mann, 2008) to search against an F. tularensis database obtained from UniProtKB with 19,197 entries, released in 10/2017. Mass spectra were searched by MaxQuant default settings for the iST protocol and no fixed modifications for the TCA protocol.
Forty-five selected Francisella isolates were used for proteome profiling (Supplementary Table S2) to establish a cluster analysis on the proteome level. For this high-resolution MS analysis, a randomized block design was applied (Oberg and Vitek, 2009), which uses a randomized sample cohort and technical replicates to ensure the reliability of the conclusions and to assess whether the observed differences in a measurement are likely to occur by random chance and reducing bias and variance due to known sources of experimental variation. This strategy employed technical duplicates for 42 and octuplets for three strains, a total of 108 LC-MS/MS runs (see schema in Supplementary Table S3). For every strain, 30 μg of protein was processed according to the above-mentioned iST kit, and about 1 μg of the digest was injected for nano-LC-MS/MS analysis.
The sample preparation workflow for complex matrices was adapted as follows: The four inactivated positive hare tissues were prepared in quintuplicates, whereas the six negative tissues were split—one half was subjected to heat-inactivation at 121°C for 30 min and the other half was left untreated, prepared in triplicates.
Ten milligrams of each tissue was homogenized under denaturing conditions with a FastPrep (twice for 60 s; 6.5 m × s–1; MP Biomedicals, Santa Ana, CA, United States) in a buffer containing 1% (w/v) sodium deoxycholate, 10 mM Tris(2-carboxyethyl)phosphine hydrochloride, and 40 mM 2-chloroacetamide in 100 mM Tris/HCl (pH 8.5) (Kulak et al., 2014) (all purchased from Sigma-Aldrich, St. Louis, MO, United States). Following homogenization, the samples were centrifuged for 1 min (4°C, 150 × g) and subsequently boiled for 10 min at 95°C and 1,000 rpm in a ThermoMixer (Eppendorf, Hamburg, Germany). The samples were transferred into new tubes and sonicated for 2 min twice (UP200St, Hielscher Ultrasonics, Teltow, Germany). For each replicate, a total protein amount of 30 μg was digested for 18 h with the iST kit (PreOmics) according to the manufacturer’s protocol with the modification that the lysis was performed using the FastPrep. In order to reduce sample complexity, a three-step fractionation was carried out. The first fraction was eluted with 100 μl of SDB-RPSx1 [100 mM ammonium formate, (AF, Santa Cruz Biotechnology, Dallas, TX, United States), 40% ACN, and 0.5% FA in water], the second fraction with 100 μl of SDB-RPSx2 (150 mM AF, 60% ACN, and 0.5% FA in water; Kulak et al., 2014), and the third fraction was eluted with the elution buffer from the iST kit for 3 min at 3,800 × g each. All eluates were dried in a vacuum concentrator and dissolved in 60 μl of 5% ACN, 2% FA, briefly vortexed and sonicated in a water bath for 2 min. After centrifugation for 5 min at 16,000 × g, 6 μl per fraction was injected for nano-LC-MS/MS analysis.
The samples for the positive hare tissues were measured using complete sample randomization, whereby the fractions belonging to one sample were measured together. The negative hare samples were measured applying a randomized block design, in which the spleen and liver samples form alternating randomized blocks.
The capability of the LC-MS/MS method, the bioinformatics pipeline, and the LLOD of F. tularensis in a complex matrix were tested by spike-ins of F. tularensis into F. tularensis-negative liver (16T1215) and spleen (16T1200) tissues from autoclaved hare carcasses. Therefore, 30 μg of protein lysates from both tissues were spiked with a dilution series of the F. tularensis strain 08T0075. Between 100 genome equivalents (GE) to 1E10 GE in the highest concentration were used for the dilution series in 10-fold steps, processed according to the iST protocol. One third was finally injected for LC-MS/MS analysis in three fractions.
LC-MS/MS analysis was performed by nano-flow reverse-phase liquid chromatography (Dionex Ultimate 3000, Thermo Scientific) coupled online to a Q-Exactive HF Orbitrap mass spectrometer (Thermo Scientific) via a nano-electrospray ion source, as described previously (Kurschner et al., 2017). In brief, about 1 μg of desalted peptides were injected for every sample. Peptides were first loaded on a trapping column (μ-precolumn; 300 μm ID × 5 mm; C18 PepMap100; 5 μm; 100 Å; Thermo Scientific) and washed for 5 min (2% ACN, 0.1% FA, in water), before separation on a PicoFrit analytical column (75 μm ID × 55 cm long, 15 μm Tip ID (New Objectives, Woburn, MA, United States), in-house packed with 3 μm of C18 resins (Reprosil-AQ Pur, Dr. Maisch, Ammerbuch-Entringen, Germany) at a controlled temperature of 50°C. Peptides were eluted applying a non-linear 121 min gradient with a flow rate of 0.266 μl/min as follows: Peptides were first eluted for 1 min using 12% solvent B in solvent A, before increasing the concentration over 100 min to 38% solvent B. After stepping up the concentration of solvent B for 3 min to 95%, it was reduced to the starting concentration of 3.8% B. Solvent A contained 0.1% FA and solvent B 80% ACN, 0.1% FA in water. An electrospray was generated by applying 3.5 kV. The Orbitrap was operated in a data-dependent manner (m/z range of 300 to 1,750 m/z, resolution of 60,000 at m/z 200, AGC target 1E6), followed by 12 data-dependent MS/MS scans (resolution of 30,000 with a normalized collision energy of 25 eV, AGC target 5E5). In order to avoid repeated sequencing of the same peptides, a dynamic exclusion window of 30 s was used. In addition, only peptide charge states between two to eight were sequenced.
For the 45 Francisella isolates, 108 LC-MS/MS RAW files were processed using tools of the open-source library OpenMS (v. 2.4) (Röst et al., 2016) and searched individually against our Francisella WGS files. RAW files were converted to the mzML format, and as a first preprocessing step, a mass calibration based on identified peptides was conducted. Calibrated spectra were then searched with the MS-GF+ search engine (Kim and Pevzner, 2014) and filtered at a 1% false discovery rate. For hierarchical cluster analysis, peptides belonging to the 25% highest abundant in at least three samples were selected. For this subset, the peptide incidence vectors for each sample were generated and used to compute a hierarchical clustering using Hamming distance and average linkage.
Proteins predicted for assembled genomes were in silico digested, and the resulting theoretical peptide profiles were analyzed and screened for species-specific peptides, as outlined in Figure 1. In silico digestion was performed with trypsin and without missed cleavages keeping all peptides of length 8–30 amino acids. To reduce the effect of erroneous genome assemblies or annotation failures, all peptides not occurring in at least three samples were removed. From this set of 22,134 peptides (peptidome), a subset of species-specific peptides was extracted. For this purpose, the complete UniParc database (31/07/2019) with 281.8 million proteins was in silico digested, and the resulting peptides were matched against the set of F. tularensis peptides. To ensure specificity, a conservative approach was chosen by treating the isomeric amino acids leucine and isoleucine and isobaric amino acids glutamine and lysine as equivalent during the search. Peptides not matching any other species than F. tularensis were retained as the final set of 4,223 species-specific peptides (Supplementary Table S4).
To assess the specificity of the method to truly identify F. tularensis within hare tissues, we performed similar steps to obtain species-specific peptides for seven other species (alpha proteobacterium HIMB5, Geobacillus sp. 12AMOR1, Lutibacter profundi, Vallitalea guaymasensis, Caloranaerobacter sp. TR13, Deferribacter desulfuricans, Hydrogenovibrio crunogenus), where it is ensured that they cannot occur in hare carcasses since their natural environment is deep-sea hydrothermal vents. The peptidome for these species was generated from all available proteins in the UniprotKB and NCBI protein databases.
For the identification of Francisella in hare carcasses, a total of 168 RAW files were processed using the same pipeline as described above and searched against all proteins of family Leporidae (UniProtKB with 27,234 entries, released in 02/2019), our Francisella specific WGS files, and the proteins for seven additional deep-sea species, impossible to occur in hare and used to assess the specificity of the method. From the resulting high-confidence peptide identifications, the number of specific peptides for F. tularensis and the other seven species was evaluated for each sample. To decide whether a certain number of identified species-specific peptides for a sample is significant or due to false peptide hits, we computed a score based on a simple probabilistic model that accounts for the total number of peptide matches and also for the number of species-specific peptides relative to the size of the searched database (Supplementary Definition 1). For complete preprocessing, identification, and downstream analysis, several KNIME pipelines (Berthold et al., 2008) were created to ensure portability and reproducibility, which are available upon request. The processed MS raw and KNIME output files have been deposited to the ProteomeXchange Consortium2 and can be found in the PRIDE depository PXD13979.
PhyloPhlAn, a method to assign microbial phylogeny and putative taxonomy to measure the sequence diversity of clades and subspecies (Segata et al., 2013; Seemann, 2014), was used in combination with annotation files derived from Prokka 1.14-dev, performed with standard settings to create a dendroscope (Huson et al., 2007). PhyloPhlAn includes a non-redundant database of 400 proteins generated from 3,737 genomes of all microbial taxa to assign microbial phylogeny and putative taxonomy. The software builds phylogenetic trees based on >4,600 aligned amino acid positions, mirroring more the changes in the protein sequence and, thus, functionality than on nucleotide changes that might be silent. PhyloPhlAn was able to measure the sequence diversity of all sequenced Francisella strains allowing even the resolution of the different clades (Figure 2). A similar clustering analysis based on all predicted tryptic peptides for the 71 isolates gave similar results (Supplementary Figure S1). It could be shown that predicted core proteome analyses lead to accurate classification of clades of Francisella tularensis. This raised the question if the same was true for the analysis of 45 LC-MS/MS data sets of Francisella. Therefore, we employed systematic and MS-based proteome profiling based on random block design, and case-specific WGS data were used to map peptides derived from the Francisella strains. The cluster analysis, based on the 45 proteomic profiles, assigned the isolates to the respective genetic clades as well (Figure 3) and is, thus, a versatile tool for this task.
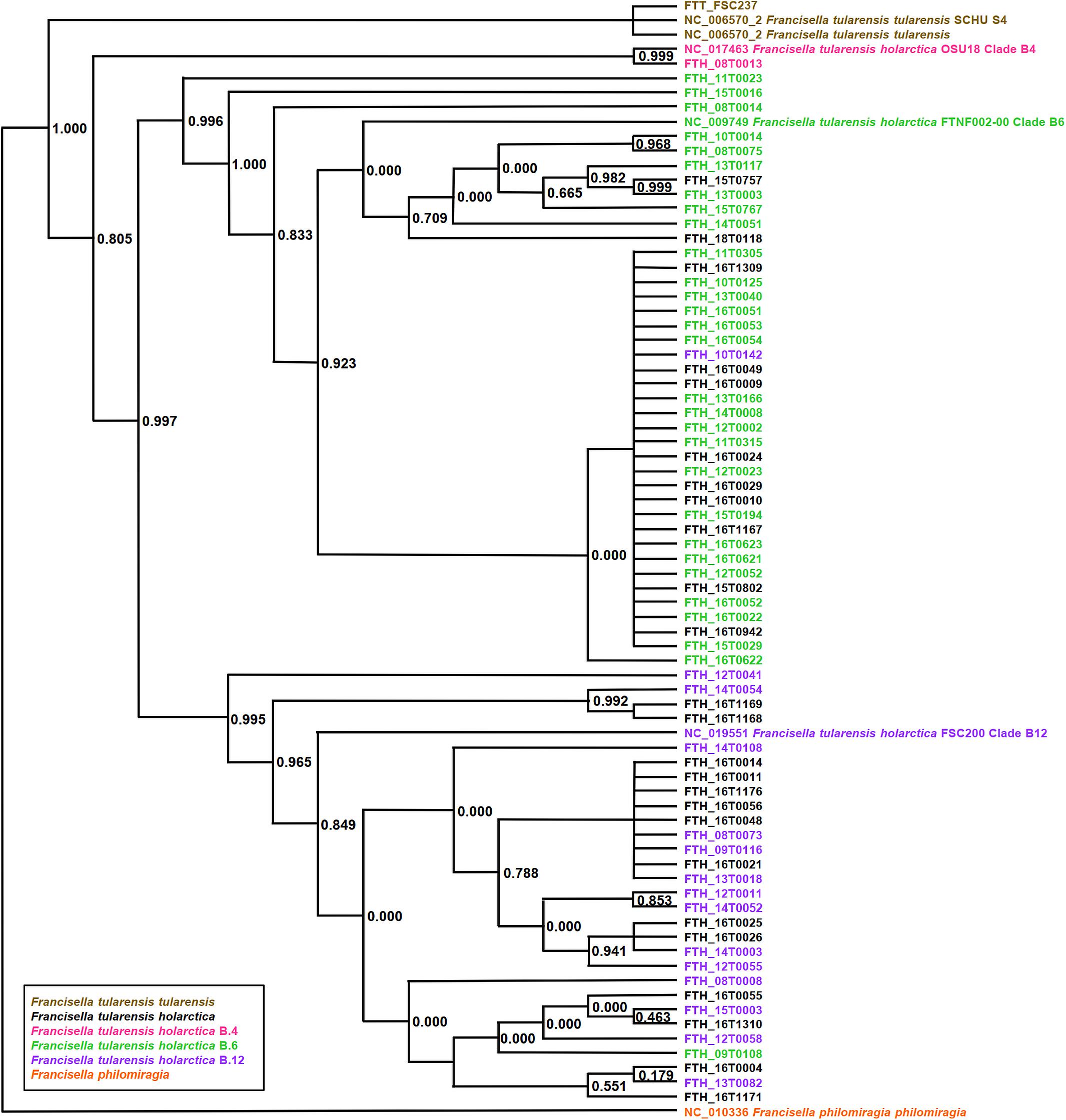
Figure 2. Phylogeny-based on whole-genome sequencing data from 71 Francisella (F.) isolates. SNP detection and phylogenetic analysis of genomes were carried out without genome alignment or reference genome by kSNP3.0. Assignment to clade B.4, B.6, and clade B.12 was done according to the results of real-time PCR targeting the respective loci. Colors indicate assignment to F. tularensis holarctica (pink to clade B.4, green to clade B.6, purple to clade B.12), F. tularensis tularensis (brown), and F. philomiragia (orange). The graphic scale equals 0.1 amino acids.
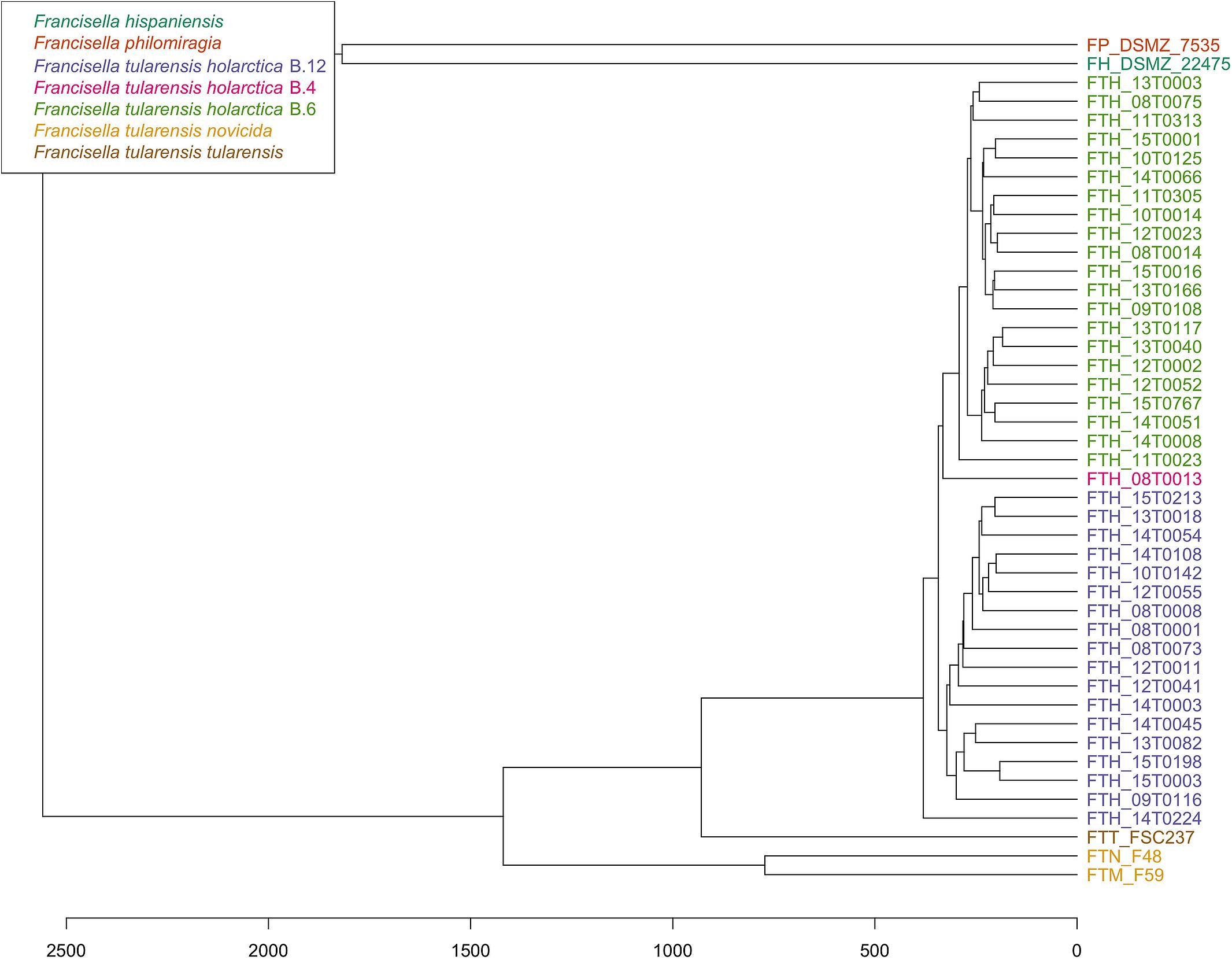
Figure 3. Cluster analysis of 45 Francisella proteome profiles. For strains with octuplets, two runs were randomly selected, and for the resulting 45 duplicates, the peptide lists were combined. Only peptides that were among the 25% highest abundant in at least three runs were considered for hierarchical clustering using Hamming distance (x-axis) and average linkage.
Bacterial pathogens have to be inactivated before they can be discharged from biosafety levels 2 and 3 laboratories. We asked whether the common heat inactivation at 121°C affects the subsequent LC-MS/MS analysis of proteins and, therefore, compared this method with an inactivation method in ethanol. The focus was to benchmark the number of identified unique peptides and protein groups under the premise that the number of enzymatic missed cleavages should be as low as possible. Furthermore, we aimed to identify a robust, reliable, and fast MS sample preparation protocol for F. tularensis that can also be easily handled in unexperienced laboratories. Ethanol inactivation of both F. tularensis strains resulted independent of the used sample preparation protocol in a higher average number of protein groups (Figure 4A) and unique peptides (Figure 4B) compared with heat inactivation. Even though the ethanol-inactivated samples resulted in higher proteome identification rates, the heat-inactivated samples are also applicable for MS sample preparation. This is specifically of relevance when working with highly infectious pathogens, such as F. tularensis, where safe handling has a high priority. Hence, inactivation by autoclavation was chosen as a default method for all further work in our study.
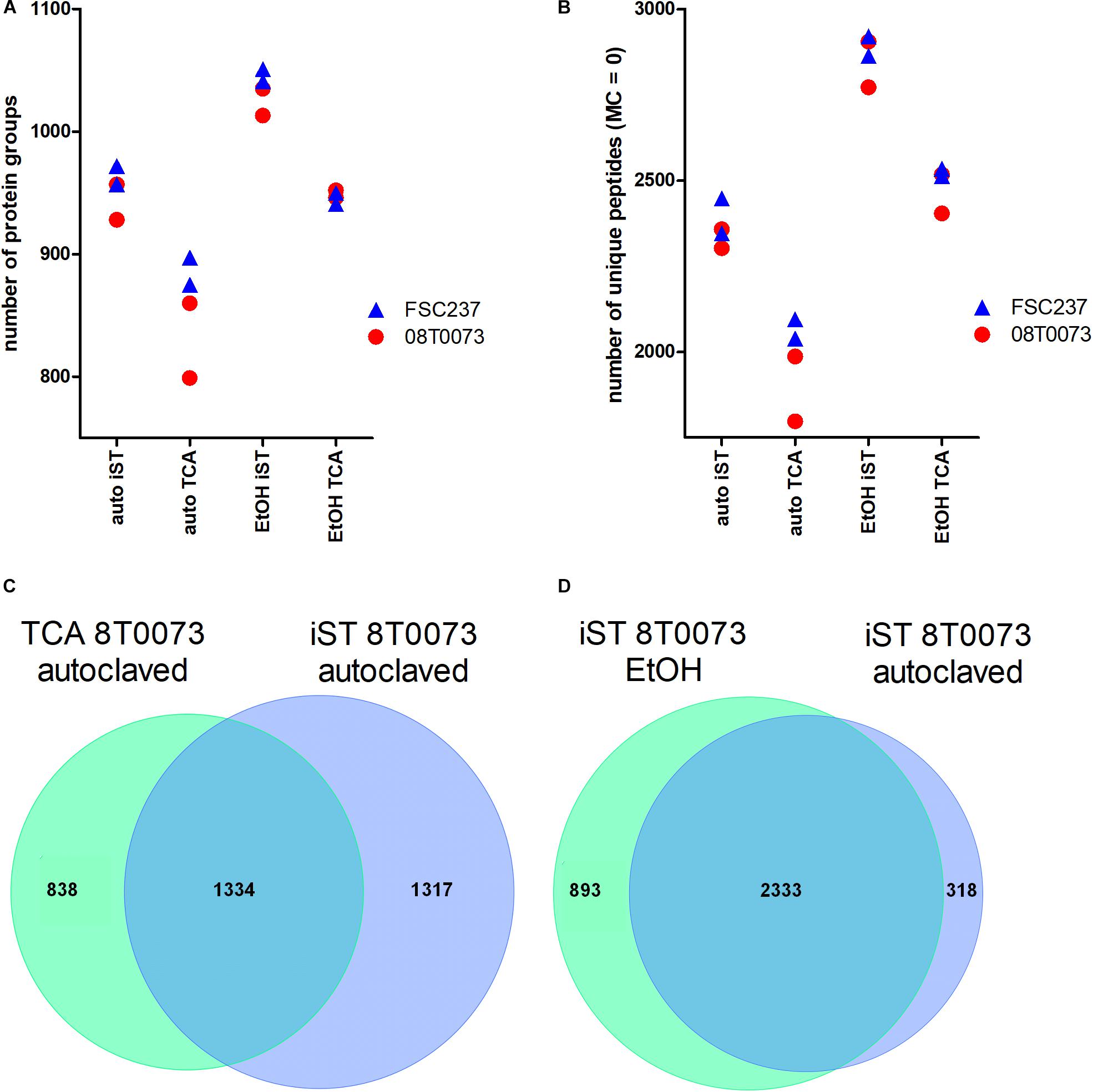
Figure 4. Protocol comparisons between iST and TCA sample preparation methods and autoclavation vs. ethanol inactivation to detect F. tularensis strains. (A) The identified protein groups and the unique peptides without missed cleavages (MC, B) were plotted for two indicated strains in technical duplicates. Furthermore, the overlap of unique peptides between both protocols for the same sample and inactivation method (C) and the overlap of unique peptides for the iST protocol for both inactivation methods within the same sample (D) are shown.
The comparison of the iST kit with the TCA sample preparation protocol revealed considerably better results for the iST kit in terms of the number of identified protein groups and unique peptides without tryptic missed cleavages, independent of the inactivation method (Figures 4A,B). Furthermore, the overlap of unique peptides for the TCA and the iST protocol was evaluated for the different samples and inactivation methods. The results showed that within the same inactivation method, ∼50% of all unique peptides were shared between the iST and the TCA protocol (Figure 4C). Moreover, the overlap of the unique peptides for different inactivation methods within the same digestion protocol was compared and showed that over 80% of the unique peptides were shared (Figure 4D). Thus, it is of importance to apply the same methodology for a given set of species-specific peptides. Next, we compared the ease and time of handling and the robustness of these two methods. Albeit both protocols resulted in a robust outcome, the iST kit can be performed within one working day without the need of experienced skills, whereas the TCA protocol takes twice as long and requires higher experimental acquirements. For these reasons, the iST kit was applied for all subsequent sample preparations.
To answer the open question, whether a direct identification of the pathogen F. tularensis in a complex biological matrix, such as hare tissue, is possible, we employed systematic and MS-based proteome profiling in two infected and three non-infected spleen and liver tissue samples of the hare. In one sample (18T0118) that was positive for F. tularensis, verified by cultivation and further characterization of the isolate (Table 1), 52 specific peptides were found in total, ranging between 32 and 36 peptides for individual replicates, and the diagnosis could be established (Supplementary Tables S5, S6A,B). In two samples that were only PCR positive (18T0123 and 18T0124), the number of bacteria in the tissue was seemingly too low, as even cultivation was not possible (Table 1). However, for sample 18T0124, five specific peptides were matched in one replicate producing a signal above noise level but did not reach significance (score < 50) and was also not consistent across the replicates. Hence, without further investigation, no unequivocal diagnosis could be established for this case.
As expected for the seven deep-sea species, only a few species-specific peptides could be matched. Although the absolute number of matched specific peptides for V. guaymasensis was up to 15 hits for some samples, this did not produce a significant score as the number of specific peptides for this species is more than 14 times higher than for F. tularensis (Supplementary Table S7). The same holds for the negative samples where neither F. tularensis nor any of the other species had a significant number of species-specific peptide hits (Supplementary Tables S8A,B), demonstrating the specificity of the method.
Furthermore, to resolve the influence of the inactivation procedure on the proteome data quality, e.g., protein degradation and modification at high temperatures, one part of each negative tested specimen was investigated after inactivation at 121°C for 30 min, while the other part was untreated.
This analysis was solely based on Leporidae and Francisella specific databases. The results showed that the number of identified unique peptides was higher for the untreated [26,674 ± 2,856 standard deviation (SD)], compared to the heat-inactivated (20,578 ± 3,980 SD) tissues (Supplementary Table S9). As a result, protein degradation and modifications at high temperatures do not appear to play a major role in the integrity of peptides in LC-MS/MS analysis.
To evaluate the power of the ESI proteome profiling approach for detecting F. tularensis in complex matrices, an artificial F. tularensis spike-in was performed. A similar number of identified peptides mostly derived by the matrix were identified in all samples for liver (mean 9,501 ± 872 SD) and spleen (10,531 ± 535 SD), whereas the number of F. tularensis-specific peptides correlated to the number of spiked-in GE (Figure 5). The LLOD of F. tularensis-specific peptides was for both tissues 3E8 GE, since the computed scores for the identified species-specific peptides in these samples (Supplementary Table S10) were above the significance level ( >50). The scores were not significant for the other seven deep-sea species.
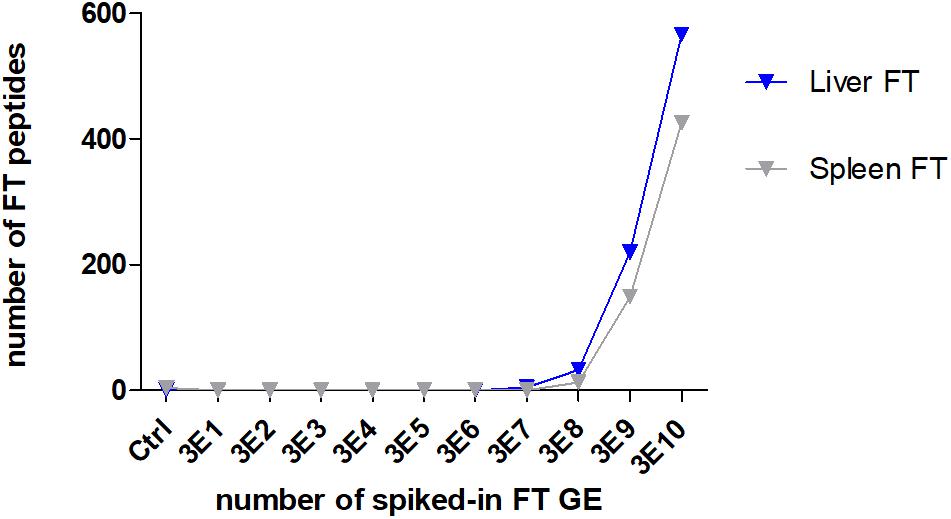
Figure 5. Lower limit of detection (LLOD) for an F. tularensis strain spiked into lysates of a liver and a spleen sample. The numbers of identified specific F. tularensis peptides vs. the numbers of spiked-in GE per measured sample are displayed.
Tularemia is a rather rare disease, but nevertheless, the causative agent F. tularensis is of major concern because it can be used as a biological agent. Therefore, it is prudent to constantly improve the diagnostic repertoire for this pathogen. F. tularensis can be transmitted by contaminated aerosols, water, food, as well as via contact with infected animals and skin lesions. Contaminated hares are the most important source of human cases of tularemia in Germany, but also other food or water can be contaminated and lead to clinical presentation such as oropharyngeal tularemia. Most human cases are diagnosed using serological assays, but seroconversion takes time, which results in a diagnostic window of ∼2 weeks (Chaignat et al., 2014). In outbreak scenarios, it is important to screen potential sources as soon as the index case has been diagnosed. Fast assessment of the contaminated food items will help to identify exposed people and to offer adequate prophylactic or therapeutic antibiotic treatment.
The currently most common mass spectrometry approach to identify and classify bacteria was coined proteotyping and was originally based on MALDI-TOF instrumentation. Proteotyping achieves several tasks, e.g., the identification and quantification of expression patterns of proteins, which can be further used to identify active metabolic pathways or pathogenic patterns. In combination with genotyping, proteotyping offers a holistic characterization of microorganisms and is, as such, heavily dependent on bioinformatic analysis pipelines. The fast-growing genome reference sequence databases, as well as streamlined bioinformatics pipelines, are a major driver for the development and use of high-resolution tandem mass spectrometry and proteotyping, which will refine the microbial phylogeny and creates a basis for proteomic identification.
A main advantage of proteotyping over classical methods (e.g., serotyping, fatty acid and lipid profiling, SDS-PAGE, etc.) is that the phenotypic characteristics of the entire taxonomic range from family, genus, species, and strain are covered, as it is the case for next-generation sequencing for genotyping. There are several distinguishable bioinformatic concepts on how to analyze proteome profiles for phylogenic purposes: The abundance and presence of marker peptides can either be used alone or in addition aligned to genome reference data for the establishment of a dendrogram. A single amino acid substitution in a peptide is already efficient enough to discriminate between two closely related strains. It should be noted that the number of species-specific peptides is always based on a current reference dataset. Changes to the reference set will consequently alter the set and number of species-specific peptides.
Direct identification of bacteria may facilitate targeted antimicrobial therapy and may help to analyze outbreaks within a short time. ESI LC-MS/MS has been shown previously to be useful for identifying clinical isolates such as Acinetobacter baumannii and a variety of other respiratory pathogens even in clinical samples (Wang et al., 2016, 2017).
We tested a collection of Francisella strains from Germany and compared the proteome results with the proteome prediction based on whole-genome sequencing data (PhyloPhlAn). As described before, a distinct differentiation of F. tularensis ssp. holarctica was possible. This could be confirmed with Francisella and F. tularensis-specific peptide profiles that could be established in silico and could be measured in ESI LC-MS/MS. In our study, we showed that the cluster analysis of proteome profiles can also be used as a substitute for genotyping by canonical single nucleotide polymorphisms (canSNP) and core genome multilocus sequence typing (cgMLST). Even though the F. tularensis subsp. holarctica is genetically closely related, we were able to separate the clades also on the proteome level. Interestingly, the F. tularensis strains 09T0108 and 10T0142 clustered on the proteome level according to their clades, while the phylogenetic tree showed the contrary. This fact requires closer inspection, but resequencing excluded a sample mix up.
The method optimization for the F. tularensis proteome profiling revealed that the iST kit showed robust results, together with an easy and fast handling routine, even possible in unexperienced labs. Furthermore, it could be shown that heat inactivation is a suitable method for MS sample preparation, and protein degradation at high temperatures does not appear to play a significant role on the proteome data quality. Especially while working with highly infectious pathogens like F. tularensis, autoclavation should – for security reasons – be the method of choice for sample inactivation. The comparison between sample preparation protocols and inactivation methods for detecting F. tularensis revealed further that the overlap between peptides is low. Consequently, species-specific peptides have to be established by the same workflow in order to get acceptable numbers of identifiable markers.
No similar LC-MS/MS studies could be found for F. tularensis in the literature, where an identification of bacteria direct out of naturally infected or spiked-in tissues were identified. LC-MS/MS approaches, which use additional immunoprecipitation steps, can detect even lower numbers of colony-forming units (cfu), such as for Yersinia pseudotuberculosis (2 × 104/mL) (Chaignat et al., 2014), or phage amplification combined with immunoprecipitation, as shown for E. coli (1 cfu) (Wang et al., 2016).
In our study, fast identification of the pathogen F. tularensis of infected hare tissues by ESI LC-MS/MS without prior cultivation was shown to be possible, and we could demonstrate that the influence of heat inactivation did not preclude identification of the pathogen, which is very important with regard to laboratory safety. From a scientific point of view, it would be desirable to further differentiate on the subspecies level, although this is not necessary for patient treatment – the prophylactic and therapeutic measures for F. tularensis exposures and infections are identical.
Even though the limitation of this study is the relatively small number of primary samples that were available, we could show, as proof of principle, that it is possible to identify F. tularensis by direct LC-MS/MS analysis of infected hare tissue without prior cultivation. This direct ESI LC-MS/MS-based bioinformatics strategy could pave the way for future analyses to characterize microorganisms.
In summary, high-resolution LC-MS/MS proteome profiling has the potential to be used as a fast and versatile tool for the identification of pathogens in complex biological matrices. This method can be used to screen suspicious samples in order to determine the origin and spread of an outbreak quickly. The preliminary identification of the pathogen using ESI LC-MS/MS will especially help to choose adequate cultivation conditions to obtain bacterial isolates that can be further characterized.
Future studies with an increased number of positive samples for different pathogens will be necessary to investigate the sensitivity and generalizability of the data analysis. This also involves more elaborate scoring functions incorporating peptide ID scores, signal intensity, and corrections for strong deviations in species-specific peptide numbers or peptide compositions that affect the peptide identification.
The datasets generated for this study can be found in the www.ncbi.nlm.nih.gov/bioproject: PRJNA560345, PRJNA353900, and PRJNA575140 Proteomics Data: http://proteomecentral.proteomexchange.org) via the PRIDE partner repository PXD013979.
No ethical review process was necessary, as only hare carcasses found in nature and collected by hunters were used for this study.
HT, DM, NW, and SA contributed to the conception and design of the study. NW performed the MS experiments with exception of the spike-in experiment, which was done by DM. SA, KN, AB, and KR performed the data processing and the statistical analysis. DM wrote the first draft of the manuscript. DM, NW, SA, AB, and HT wrote the sections of the manuscript. All authors contributed to the manuscript revision, and read and approved the submitted version.
This work was funded by grants of the German Federal Ministry of Education and Research within the framework of the project Ess-B.A.R (FKZ 13N13983, FKZ 13N13984, FKZ 13N13985) and the Max Planck Society. AB was currently funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2051 – Project-ID 390713860.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Kerstin Cerncic and Anja Hackbart for their skillful technical assistance. Last but not least, we are immensely grateful to all people that provided samples and isolates.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00636/full#supplementary-material
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berthold, M. R., Cebron, N., Dill, F., Gabriel, T. R., Kötter, T., Meinl, T., et al. (2008). KNIME: The Konstanz Information Miner. Berlin: Springer.
Busch, A., Thomas, P., Zuchantke, E., Brendebach, H., Neubert, K., Gruetzke, J., et al. (2018). Revisiting Francisella tularensis subsp. holarctica, causative agent of tularemia in Germany With bioinformatics: new insights in genome structure, DNA methylation and comparative phylogenetic analysis. Front. Microbiol. 9:344. doi: 10.3389/fmicb.2018.00344
Chaignat, V., Djordjevic-Spasic, M., Ruettger, A., Otto, P., Klimpel, D., Muller, W., et al. (2014). Performance of seven serological assays for diagnosing tularemia. BMC Infect Dis. 14:234. doi: 10.1186/1471-2334-14-234
Cheng, K., Chui, H., Domish, L., Hernandez, D., and Wang, G. (2016). Recent development of mass spectrometry and proteomics applications in identification and typing of bacteria. Proteomics Clin. Appl. 10, 346–357. doi: 10.1002/prca.201500086
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367.
Deatherage Kaiser, B. L., Wunschel, D. S., Sydor, M. A., Warner, M. G., Wahl, K. L., and Hutchison, J. R. (2015). Improved proteomic analysis following trichloroacetic acid extraction of Bacillus anthracis spore proteins. J. Microbiol. Methods 118, 18–24. doi: 10.1016/j.mimet.2015.08.008
Durighello, E., Bellanger, L., Ezan, E., and Armengaud, J. (2014). Proteogenomic biomarkers for identification of Francisella species and subspecies by matrix-assisted laser desorption ionization-time-of-flight mass spectrometry. Anal. Chem. 86, 9394–9398. doi: 10.1021/ac501840g
Dworzanski, J. P., Deshpande, S. V., Chen, R., Jabbour, R. E., Snyder, A. P., Wick, C. H., et al. (2006). Mass spectrometry-based proteomics combined with bioinformatic tools for bacterial classification. J. Proteome Res. 5, 76–87.
Dworzanski, J. P., Snyder, A. P., Chen, R., Zhang, H., Wishart, D., and Li, L. (2004). Identification of bacteria using tandem mass spectrometry combined with a proteome database and statistical scoring. Anal. Chem. 76, 2355–2366.
Emele, M. F., Mozina, S. S., Lugert, R., Bohne, W., Masanta, W. O., Riedel, T., et al. (2019). Proteotyping as alternate typing method to differentiate Campylobacter coli clades. Sci. Rep. 9:4244. doi: 10.1038/s41598-019-40842-w
Hayoun, K., Gouveia, D., Grenga, L., Pible, O., Armengaud, J., and Alpha-Bazin, B. (2019). Evaluation of sample preparation methods for fast proteotyping of microorganisms by tandem mass spectrometry. Front. Microbiol. 10:1985. doi: 10.3389/fmicb.2019.01985
Hubalek, M., Hernychova, L., Brychta, M., Lenco, J., Zechovska, J., and Stulik, J. (2004). Comparative proteome analysis of cellular proteins extracted from highly virulent Francisella tularensis ssp. tularensis and less virulent F. tularensis ssp. holarctica and F. tularensis ssp. mediaasiatica. Proteomics 4, 3048–3060.
Huson, D. H., Richter, D. C., Rausch, C., Dezulian, T., Franz, M., and Rupp, R. (2007). Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. doi: 10.1186/1471-2105-8-460
Karatuna, O., Celebi, B., Can, S., Akyar, I., and Kilic, S. (2016). The use of Matrix-assisted laser desorption ionization-time of flight mass spectrometry in the identification of Francisella tularensis. Bosn J. Basic Med. Sci. 16, 132–138. doi: 10.17305/bjbms.2016.894
Karlsson, R., Gonzales-Siles, L., Boulund, F., Svensson-Stadler, L., Skovbjerg, S., Karlsson, A., et al. (2015). Proteotyping: proteomic characterization, classification and identification of microorganisms–A prospectus. Syst. Appl. Microbiol. 38, 246–257.
Karlsson, R., Gonzales-Siles, L., Gomila, M., Busquets, A., Salva-Serra, F., Jaen-Luchoro, D., et al. (2018). Proteotyping bacteria: characterization, differentiation and identification of pneumococcus and other species within the Mitis Group of the genus Streptococcus by tandem mass spectrometry proteomics. PLoS One 13:e0208804. doi: 10.1371/journal.pone.0208804
Kasap, M., Karadenizli, A., Akpinar, G., Uzuner, H., Ayimugu, A., Karaosmanoglu, K., et al. (2017). Comparative analysis of proteome patterns of Francisella tularensis isolates from patients and the environment. Curr. Microbiol. 74, 230–238. doi: 10.1007/s00284-016-1178-6
Kilmury, S. L., and Twine, S. M. (2010). The francisella tularensis proteome and its recognition by antibodies. Front. Microbiol. 1:143. doi: 10.3389/fmicb.2010.00143
Kim, S., and Pevzner, P. A. (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5:5277. doi: 10.1038/ncomms6277
Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., and Mann, M. (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Meth. 11, 319–324. doi: 10.1038/nmeth.2834
Kurschner, G., Zhang, Q., Clima, R., Xiao, Y., Busch, J. F., Kilic, E., et al. (2017). Renal oncocytoma characterized by the defective complex I of the respiratory chain boosts the synthesis of the ROS scavenger glutathione. Oncotarget 8, 105882–105904. doi: 10.18632/oncotarget.22413
Oberg, A. L., and Vitek, O. (2009). Statistical design of quantitative mass spectrometry-based proteomic experiments. J Proteome Res. 8, 2144–2156.
Röst, H. L., Sachsenberg, T., Aiche, S., Bielow, C., Weisser, H., Aicheler, F., et al. (2016). OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748. doi: 10.1038/nmeth.3959
Rotz, L. D., Khan, A. S., Lillibridge, S. R., Ostroff, S. M., and Hughes, J. M. (2002). Public health assessment of potential biological terrorism agents. Emerg. Infect. Dis. 8, 225–230.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Bornigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/ncomms3304
Seibold, E., Maier, T., Kostrzewa, M., Zeman, E., and Splettstoesser, W. (2010). Identification of Francisella tularensis by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry: fast, reliable, robust, and cost-effective differentiation on species and subspecies levels. J. Clin. Microbiol. 48, 1061–1069. doi: 10.1128/JCM.01953-09
Sharma, N., Hotta, A., Yamamoto, Y., Fujita, O., Uda, A., Morikawa, S., et al. (2013). Detection of Francisella tularensis-specific antibodies in patients with tularemia by a novel competitive enzyme-linked immunosorbent assay. Clin. Vaccine Immunol. 20, 9–16. doi: 10.1128/CVI.00516-12
Sharma, N., Hotta, A., Yamamoto, Y., Uda, A., Fujita, O., Mizoguchi, T., et al. (2014). Serosurveillance for Francisella tularensis among wild animals in Japan using a newly developed competitive enzyme-linked immunosorbent assay. Vec. Borne Zoonotic Dis. 14, 234–239. doi: 10.1089/vbz.2013.1349
Wang, H., Drake, S. K., Yong, C., Gucek, M., Lyes, M. A., Rosenberg, A. Z., et al. (2017). A genoproteomic approach to detect peptide markers of bacterial respiratory pathogens. Clin. Chem. 63, 1398–1408. doi: 10.1373/clinchem.2016.269647
Wang, H., Drake, S. K., Yong, C., Gucek, M., Tropea, M., Rosenberg, A. Z., et al. (2016). A novel peptidomic approach to strain typing of clinical Acinetobacter baumannii isolates using mass spectrometry. Clin. Chem. 62, 866–875. doi: 10.1373/clinchem.2015.253468
Keywords: Francisella tularensis, high-resolution tandem mass spectrometry proteotyping, proteome profiling, detection of pathogens, phylogenetic analysis
Citation: Witt N, Andreotti S, Busch A, Neubert K, Reinert K, Tomaso H and Meierhofer D (2020) Rapid and Culture Free Identification of Francisella in Hare Carcasses by High-Resolution Tandem Mass Spectrometry Proteotyping. Front. Microbiol. 11:636. doi: 10.3389/fmicb.2020.00636
Received: 08 October 2019; Accepted: 20 March 2020;
Published: 08 May 2020.
Edited by:
David Rodriguez-Lazaro, University of Burgos, SpainReviewed by:
Jean Armengaud, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), FranceCopyright © 2020 Witt, Andreotti, Busch, Neubert, Reinert, Tomaso and Meierhofer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Meierhofer, TWVpZXJob2ZlckBtb2xnZW4ubXBnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.