 Natalia M. Vior
Natalia M. Vior Eva Cea-Torrescassana
Eva Cea-Torrescassana Tom H. Eyles
Tom H. Eyles Govind Chandra
Govind Chandra Andrew W. Truman
Andrew W. Truman- Department of Molecular Microbiology, John Innes Centre, Norwich, United Kingdom
Bottromycin is a ribosomally synthesized and post-translationally modified peptide (RiPP) produced by several streptomycetes, including the plant pathogen Streptomyces scabies. There is significant interest in this molecule as it possesses strong antibacterial activity against clinically relevant multidrug resistant pathogens and is structurally distinct from all other antibiotics. However, studies into its efficacy are hampered by poor yields. An understanding of how bottromycin biosynthesis is regulated could aid the development of strategies to increase titres. Here, we use 5′-tag-RNA-seq to identify the transcriptional organization of the gene cluster, which includes an internal transcriptional start site that precedes btmD, the gene that encodes the bottromycin precursor peptide. We show that the gene cluster does not encode a master regulator that controls pathway expression and instead encodes a regulatory gene, btmL, which functions as a modulator that specifically affects the expression of btmD but not genes up- or downstream of btmD. In order to identify non-cluster associated proteins involved in regulation, proteins were identified that bind to the main promoter of the pathway, which precedes btmC. This study provides insights into how this deceptively complex pathway is regulated in the absence of a pathway specific master regulator, and how it might coordinate with the central metabolism of the cell.
Introduction
Infectious diseases are responsible for one-fifth of deaths worldwide, and antimicrobial resistance is becoming an increasingly serious threat to global public health, which makes the development of new antibiotics a pressing need (Laxminarayan et al., 2016; Martens and Demain, 2017). The effective treatment of multidrug resistant bacteria requires the identification of compounds with either unusual chemical scaffolds or novel molecular targets, capable of overcoming multiple and widespread resistance mechanisms (Laxminarayan, 2014; Newman and Cragg, 2016; Martens and Demain, 2017). One antimicrobial compound of great interest is bottromycin, a peptide antibiotic with promising activity against multi-drug resistant Gram-positive bacterial pathogens (Shimamura et al., 2009; Tacconelli and Magrini, 2017). Bottromycin was first isolated in 1957 from cultures of Streptomyces bottropensis (Waisvisz et al., 1957), and then more recently as a product of the plant pathogen Streptomyces scabies (Crone et al., 2012) and other Streptomyces species (Gomez-Escribano et al., 2012; Hou et al., 2012; Huo et al., 2012). Bottromycin is a ribosomally synthesized and post-translationally modified peptide (RiPP). In its biosynthetic pathway, a precursor peptide (BtmD) undergoes a complex and unprecedented series of modifications catalyzed by enzymes encoded in the bottromycin (btm) gene cluster (Figure 1) (Crone et al., 2012, 2016; Gomez-Escribano et al., 2012; Huo et al., 2012; Franz et al., 2017; Schwalen et al., 2017; Sikandar et al., 2019).
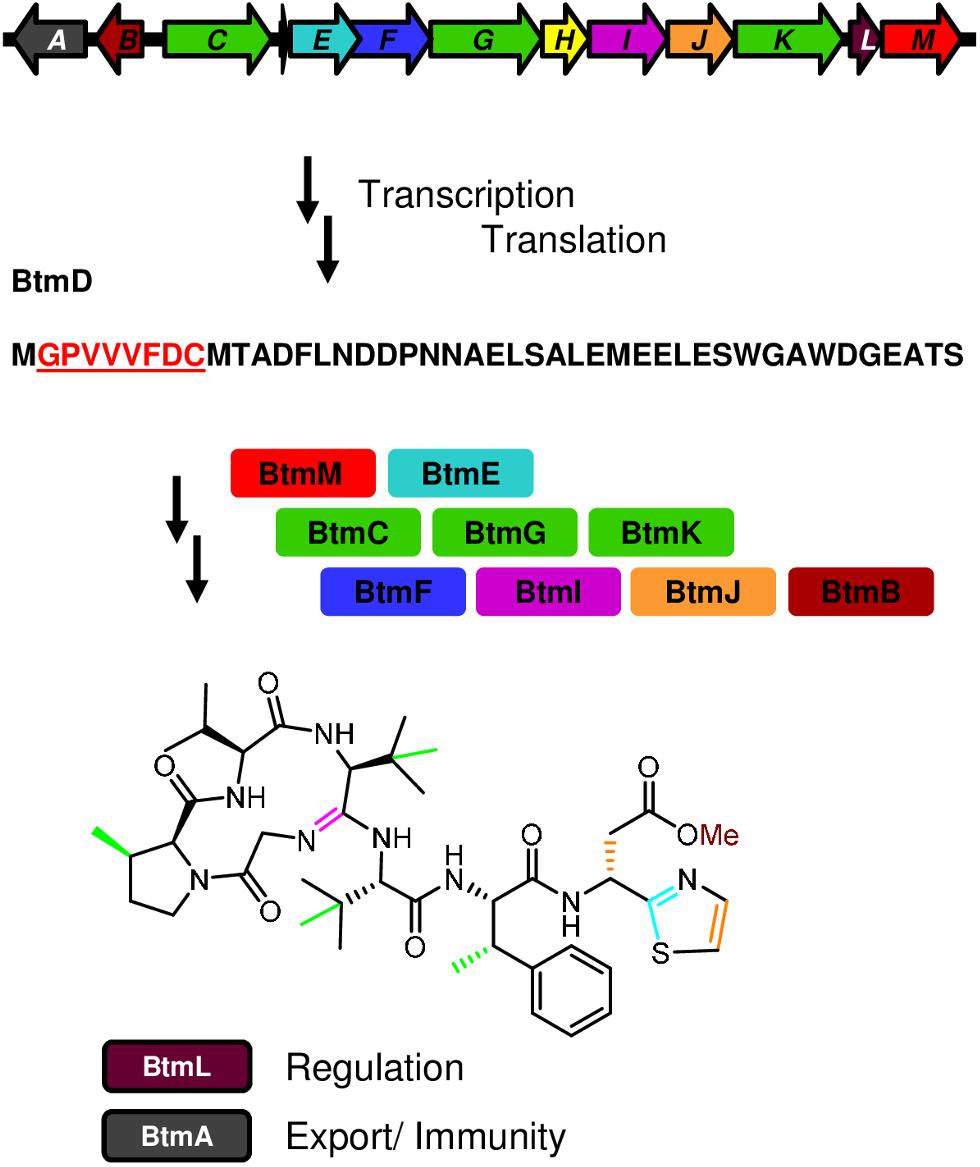
Figure 1. Biosynthesis of bottromycin. The diagram shows the genetic organization of the bottromycin (btm) cluster and summarizes its biosynthetic pathway, from the transcription and translation of the precursor peptide gene btmD, to the posttranslational modifications leading to the final compound.
Bottromycin is a structurally novel compound that selectively blocks the binding of aminoacyl t-RNAs to the A-site in the 50S subunit of bacterial ribosomes, ultimately leading to protein synthesis inhibition (Otaka and Kaji, 1976, 1981, 1983). This makes bottromycin an interesting lead for development, as it attacks a molecular target that is currently unexploited in the clinic. However, bottromycin is produced in low yields, where titres in wild type producers are below 1 mg/L (Huo et al., 2012). These low production levels complicate both drug development and further biosynthetic studies of these molecules (Huo et al., 2012; Crone et al., 2016). An understanding of how bottromycin biosynthesis is regulated may enable the rational improvement of yield by modifying pathway regulation, especially as the pathway is inefficient in laboratory conditions (Crone et al., 2016; Eyles et al., 2018).
It is very well documented that altering the regulatory networks that control secondary metabolite biosynthesis often leads to significant yield increases, either by overexpressing positive regulators, inactivating repressors, or altering higher levels of regulation in the producer strain (Makitrynskyy et al., 2013; Vior et al., 2014; Yoo et al., 2015; Huang et al., 2016). RiPP biosynthesis, especially in the case of lantibiotics, is often regulated by classic two-component systems or orphan response regulators (Bierbaum and Sahl, 2009; Cooper et al., 2010), but this is not a mechanism common to all RiPP pathways. Systems involving sigma-anti-sigma factor pairs, SARP, LuxR, or TetR regulators have also been documented (Izawa et al., 2013; Flinspach et al., 2014; Zhang et al., 2014; Fernández-Martínez et al., 2015), and more diverse regulatory systems are likely to be found in the future, especially because many RiPP gene clusters do not encode any obvious pathway specific regulators (Bartholomae et al., 2017). The btm gene cluster in S. scabies encodes one potential regulatory protein, BtmL (Figure 1) (Crone et al., 2012). This protein is conserved across all characterized bottromycin gene clusters (Crone et al., 2012; Gomez-Escribano et al., 2012; Huo et al., 2012), but nothing is known about how this putative regulator controls bottromycin biosynthesis, nor whether additional regulators have critical roles in btm cluster regulation.
In this work, we apply transcriptomic, proteomic and metabolomic techniques in combination with qRT-PCR and reporter activity experiments in order to obtain key details on the regulation of bottromycin biosynthesis. We show that BtmL is not a master regulator of biosynthesis and instead specifically enhances expression levels of the precursor peptide gene btmD. This occurs in conjunction with a transcriptional start site for btmD that is internal to the preceding gene in the cluster. We show that this pathway is surprisingly complex and provide evidence into how it is regulated in the absence of a pathway specific master regulator.
Results
BtmL Specifically Modulates the Expression of the Precursor Peptide Gene btmD and Is Independent of Cobalt Levels in the Medium
The btm gene cluster in S. scabies encodes a single putative regulator, BtmL, a 20.5 kDa protein that contains a C-terminal domain of unknown function (DUF2087 or PF09860), which has been associated with putative transcriptional regulators and is proposed to bind nucleic acids (Rigden, 2011). To date (July 2019), over 6,500 DUF2087-containing proteins have been sequenced and deposited in Genbank, of which more than 2,000 have additional DNA-binding domains. In silico analysis of BtmL using both Phyre2 (Crone et al., 2012; Kelley et al., 2015) and I-TASSER (Yang et al., 2015) predicted that the N-terminus of the protein has structural homology with SmtB-ArsR-like repressors, and therefore would feature a winged helix-turn-helix (wHTH) domain characteristic of this family of transcriptional regulators (Busenlehner et al., 2003; Osman and Cavet, 2010; Chakravorty and Merz, 2014). However, no canonical wHTH domain was detected by sequence analysis and the protein appears to lack the conserved residues characteristically involved in ArsR-family metal binding (Osman and Cavet, 2010; Crone et al., 2012). Despite this, previous empirical evidence showed that addition of cobalt(II) to the production medium increased bottromycin yields (Crone et al., 2012), inferring that the ArsR-like structure of BtmL may control cluster expression via a metal-binding mechanism. This hypothesis was especially compelling given that several genes in the btm cluster encode class B radical SAM methyltransferases, which are cobalamin-dependent enzymes (Bauerle et al., 2015).
To investigate the role of BtmL in bottromycin biosynthesis, a mutant S. scabies strain carrying an in-frame deletion of btmL was generated (Crone et al., 2012) and its ability to produce bottromycin was assessed using liquid chromatography - mass spectrometry (LC-MS). Inactivation of btmL did not abolish bottromycin biosynthesis, but we could observe a moderate and consistent decrease in production levels to approximately 40% of wild type (WT) levels (Figure 2A). This result suggests that btmL acts as a positive modulator of bottromycin biosynthesis, but is not the master activator of the pathway. WT levels of bottromycin production were restored in ΔbtmL upon in trans complementation with a copy of btmL under the control of the constitutive promoter ermE∗p (Bibb et al., 1985), confirming that the phenotype was due to the deletion of btmL. Surprisingly, when that same btmL expression construct was introduced in the WT strain (generating strain WT + L), there was no increase in bottromycin levels (Figure 2A).
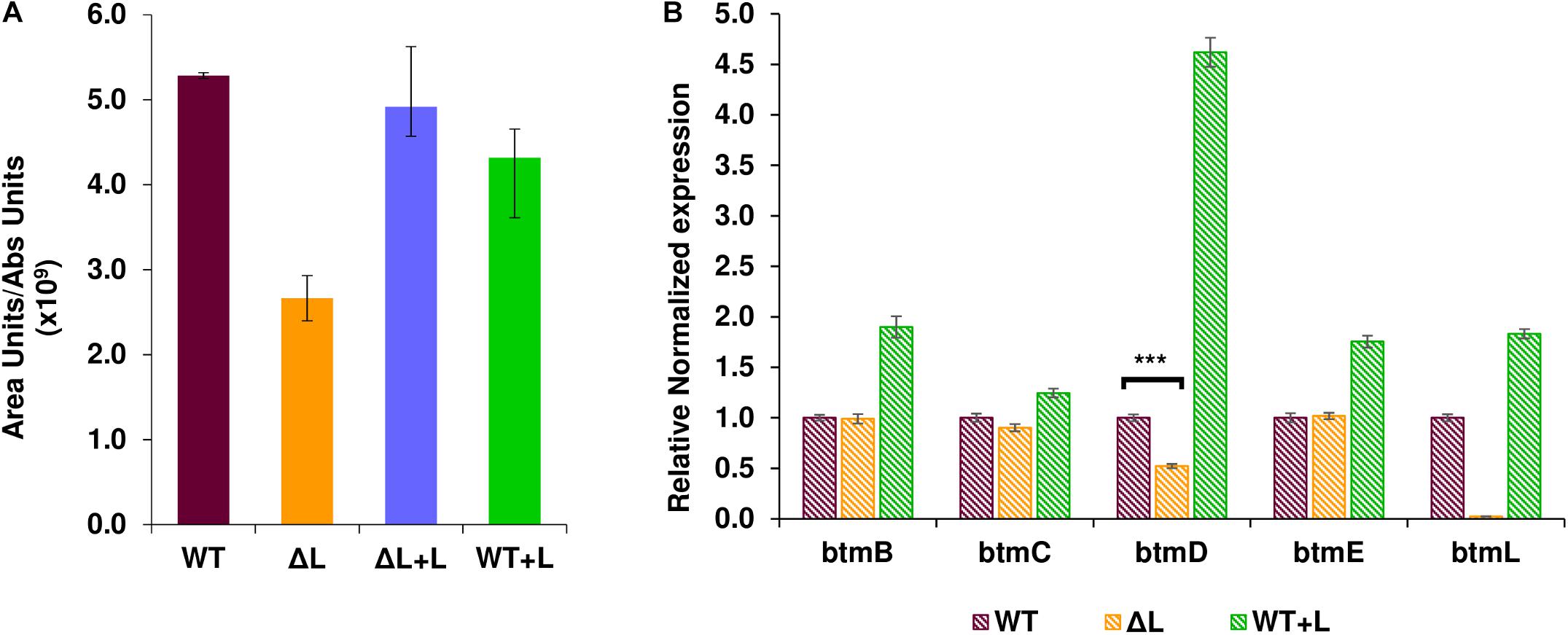
Figure 2. Effect of btmL on bottromycin production and btm gene transcription. (A) LC-MS analysis of bottromycin production in S. scabies WT, ΔbtmL (ΔL), ΔL complemented with a copy of btmL (ΔL + L) and the WT strain overexpressing btmL (WT + L). Error bars represent the standard deviation of the average production values in biological triplicates, which are normalized by culture growth. (B) qRT-PCR analysis of the transcription of representative btm genes in the WT (purple bars), ΔbtmL (yellow bars) and WT + L (green bars) at 72 h of growth in BPM. Expression values are relative to the expression of the target gene in the WT, which was set at 1. Transcription of hrdB was used to normalize the expression levels across samples. Error bars represent standard error of the mean from triplicate experiments, and the asterisk represents a statistically significant difference in btmD expression between wt and the mutant ΔbtmL (p-value < 0.001).
To evaluate quantitatively whether btmL could be acting as a transcriptional regulator, qRT-PCR experiments were carried out with RNA samples from both the WT and ΔbtmL strains (Figure 2B). The btm gene cluster is organized in two divergent groups of genes: btmA and btmB on one strand and btmC-btmM on the other strand (Figure 1). We therefore measured the expression of btmB from the smaller block, and btmC, btmD (the gene encoding the precursor peptide), btmE and btmL from the rest of the cluster. Expression values of these genes were normalized using the expression of hrdB, which encodes the principal sigma factor of RNA polymerase. A time course experiment determined that the optimal time to measure btm gene expression was at 72 h post-inoculation of bottromycin production medium (Supplementary Figure S1). At this time point, it could be observed that deletion of btmL has no significant effect on most of the genes tested, with one notable exception: btmD, the gene encoding the bottromycin precursor peptide. Transcription of btmD is reduced to approximately 40% of WT levels, which correlates with the reduction in bottromycin production (Figure 2B). Furthermore, qRT-PCR analysis of the WT + L strain revealed that transcription of btmD is significantly increased in this strain, confirming that BtmL is a specific and positive regulator of btmD. Interestingly, upstream and downstream genes (btmB, btmC, and btmE) are only slightly overexpressed or not overexpressed at all, suggesting the presence of an alternative operon encompassing only btmD.
After confirming that btmL is involved in bottromycin regulation, we assessed whether cobalt(II) levels influence pathway productivity, and whether this effect was mediated by btmL. Bottromycin production was measured in S. scabies WT and ΔbtmL cultured in bottromycin production medium (BPM) supplemented with different concentrations of CoCl2 (Figure 3A). In absolute terms, bottromycin production increased upon addition of cobalt(II), reaching a maximum at 15–25 μg/mL CoCl2 after which production steadily decreased until almost disappearing at 200 μg/mL CoCl2. However, the increase in production simply corresponded to a proportional increase in mycelial growth upon addition of up to 25 μg/mL of cobalt(II) in the medium (Figure 3A). At higher cobalt(II) concentrations mycelial growth was sustained, so the drop in production was probably due to a general inhibitory effect of excess metal in the medium, a phenomenon reported for the biosynthesis of other antibiotics (Rogers and Birnbaum, 1974; Abbas and Edwards, 1990). The growth-dependent production increase was also noticeable in ΔbtmL, which followed a similar pattern, albeit at a reduced level of production. Moreover, a comparison of qRT-PCR data for the expression levels of several pathway genes in the presence or absence of added cobalt(II) (Figure 3B) showed that these are nearly identical in either condition for both the WT and ΔbtmL strains, discarding any specific regulatory effect of this metal. This result is in accordance with the observation that btmL lacks the conserved metal-binding residues characteristic of ArsR-SmtB regulators (Osman and Cavet, 2010; Crone et al., 2012).
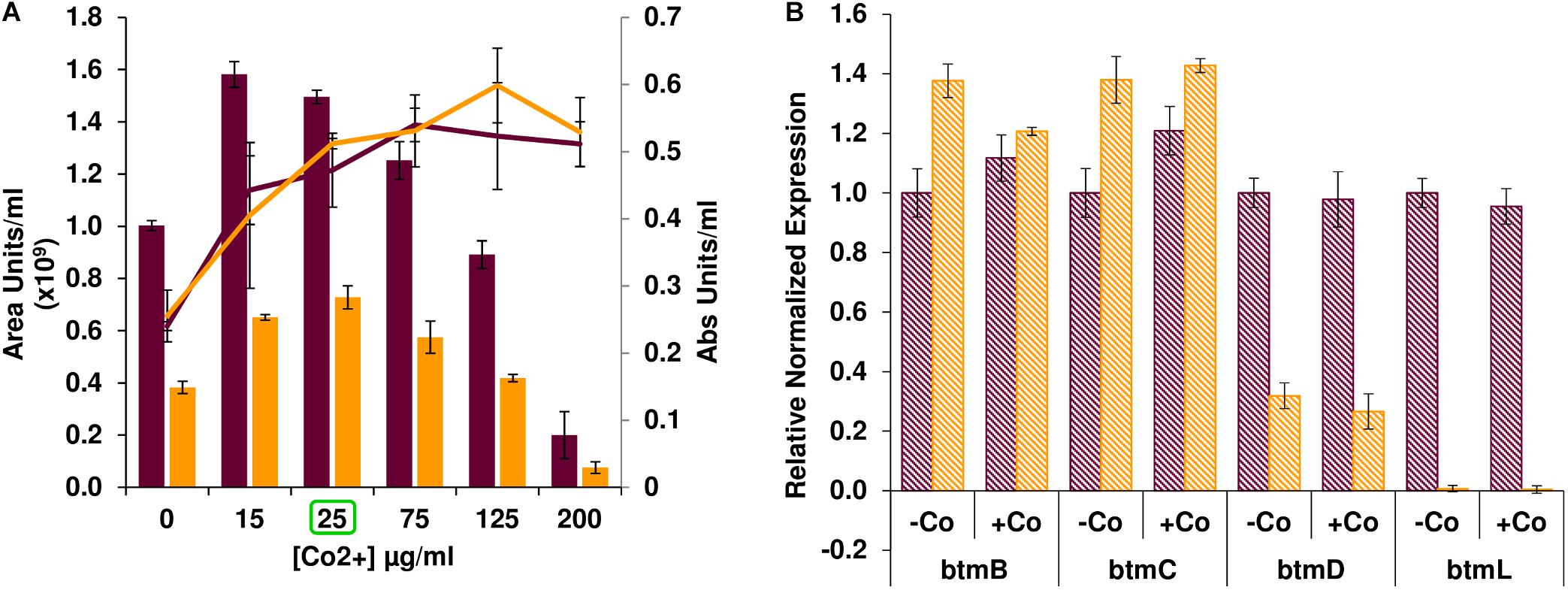
Figure 3. Effect of cobalt(II) on bottromycin biosynthesis. (A) Bottromycin production in S. scabies WT (purple bars) and ΔbtmL (yellow bars) in BPM supplemented with increasing concentrations of CoCl2. Culture growth (calculated from a DNA quantification assay, see section “Materials and Methods”) is overlaid and represented on the secondary axis. Error bars represent the standard deviation of biological triplicate data. The concentration of cobalt used for the qRT-PCR experiment in (B) appears highlighted with a green square. (B) qRT-PCR analysis of the expression of btm genes in the WT (purple bars) and ΔbtmL mutant (yellow bars) at 72 h of growth in BPM with and without added cobalt. Expression values are relative to the expression of the target gene in the WT without added cobalt, which was set at 1. Transcription of hrdB was used to normalize the expression levels across samples and error bars represent standard error of the mean from triplicate experiments.
These results ruled out cobalt(II)-dependent pathway regulation by BtmL, which is instead a cobalt-independent transcriptional modulator that selectively increases btmD transcript levels in order to generate high levels of the precursor peptide in a feed-forward loop. Other positive loop systems have been reported in the biosynthesis of several RiPPs like microbisporicin and planosporicin, where the autoinduction mechanism relies of the detection of small quantities of precursor peptide by a sigma-antisigma complex (Sherwood and Bibb, 2013; Fernández-Martínez et al., 2015), or cinnamycin, where production is launched after onset of immunity to the compound in the producer strain (O’Rourke et al., 2017).
Overexpression of Exporter BtmA Has a Moderate Positive Effect on Bottromycin Production
The discordance between the increase in btmD expression and the lack of change in bottromycin production in the WT + L strain (Figure 2A) led us to postulate that the potential increase in available BtmD might not be effectively channeled by the pathway and exported out of the cell, leading to increased degradation of pathway intermediates and generation of shunt metabolites. Supporting this hypothesis, previous work showed that overexpression of botT, the major facilitator superfamily (MFS) transporter gene (Kumar et al., 2016; Quistgaard et al., 2016) in the bottromycin cluster from Streptomyces sp. BC16019 increased bottromycin production in a heterologous host, although it still did not reach wild type production levels (Huo et al., 2012). We therefore assessed whether overexpressing the homolog of this gene (btmA, 88% identity to botT) in the S. scabies btm gene cluster might increase the efficiency of the pathway and self-resistance to potentially toxic levels of the antibiotic.
Expression of a second copy of btmA under the control of the constitutive promoter ermE∗p provided a 25% increase in bottromycin production with respect to the WT strain (Figure 4A), an effect that was maintained, but not improved, when it was expressed in WT + L (generating WT + L + A). When the quantification was extended to other bottromycin related metabolites described previously (Crone et al., 2016; Eyles et al., 2018) this pattern was still observed for the total set of mature bottromycins (those including the main posttranslational modifications) (Figure 4A). However, in the case of the pathway
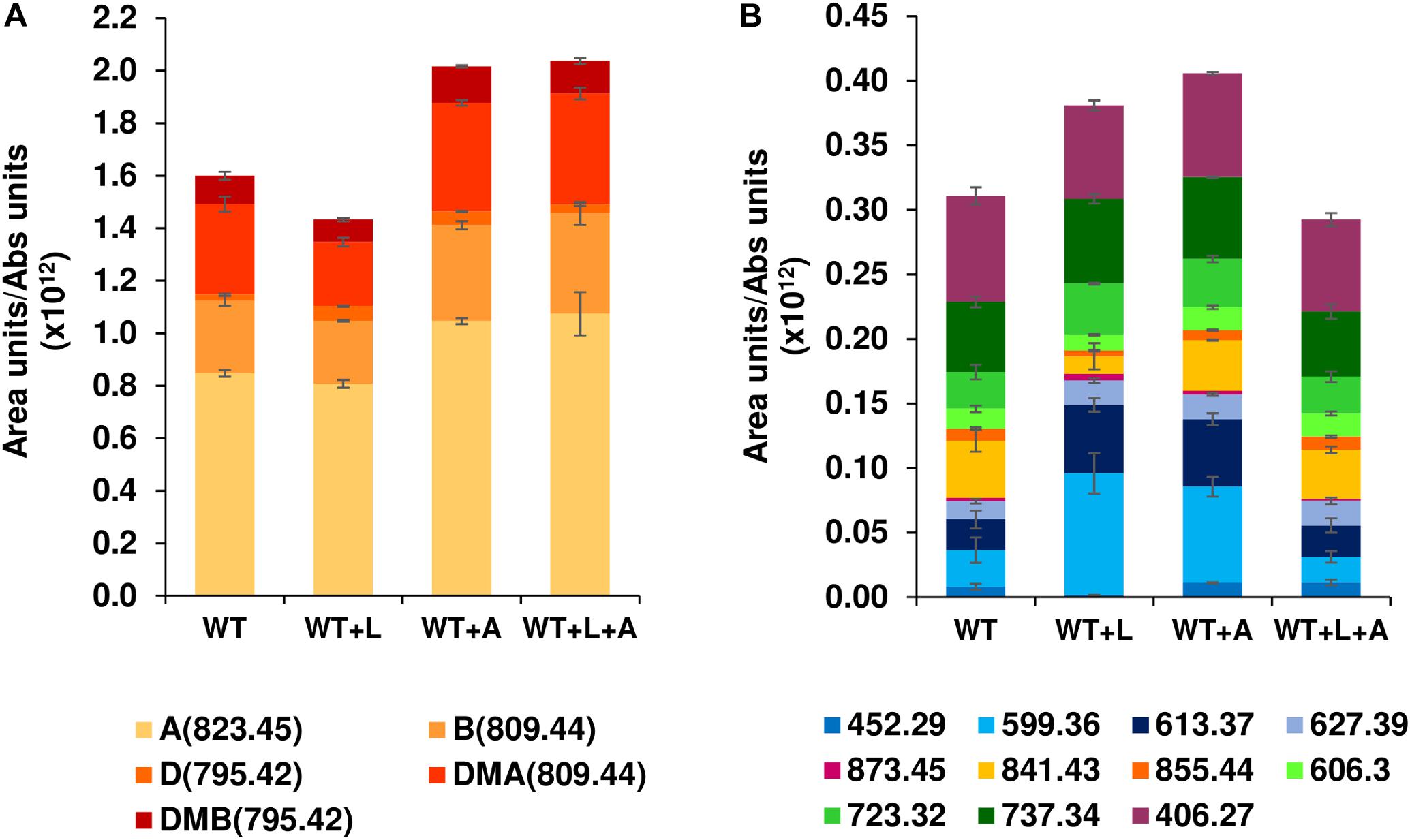
Figure 4. Effect of the MFS transporter gene btmA on bottromycin production. Comparison of WT and WT + L strains with strains overexpressing btmA (WT + A and WT + L + A). (A) Production of mature bottromycins. (B) Production of intermediates and shunt products of the bottromycin pathway. Legends show all the compounds quantified in the experiment, and each compound is labeled with its observed mass as described in Crone et al. (2016) and Eyles et al. (2018). Mature bottromycins are also labeled with letters (A: bottromycin A2, B: bottromycin B, D: bottromycin D, DMA: desmethyl bottromycin A2 and DMB: desmethyl bottromycin B). Error bars represent the standard deviation of biological triplicate data, and production values are normalized by culture growth.
intermediates and shunt metabolites the trend was not present, with WT + L and WT + A producing more of them than the WT and the double overexpression strain WT + L + A (Figure 4B). This result indicates that efficiency can be increased to some extent by improving export of the molecule, but this is not the major bottleneck for bottromycin production. The main limiting factor for bottromycin production is likely to be the post-translational maturation of the precursor peptide instead, as we previously showed that the bottromycin pathway inefficiently stalls at numerous biosynthetic steps, even in the WT strain (Crone et al., 2016).
Mapping of the Transcriptional Organization of the btm Cluster Identifies a Secondary Transcriptional Start Site for btmD
Given that btmD appears to be situated within a larger operon, the specific increase in btmD transcription following btmL overexpression was unexpected and prompted us to investigate the transcriptional organization of the btm gene cluster. This cluster contains 13 genes, where 11 (btmC-M) are arranged in the same orientation and appear tightly clustered, with btmE to btmJ physically overlapping and btmK starting 8 bp after the btmJ stop codon. To assess the possibility of co-transcription of the remaining genes, RT-PCR experiments were performed on 72 h cultures of both WT and ΔbtmL strains with specific primer pairs for the intergenic regions btmA-B, btmC-D, btmD-E, and btmK-L (Figure 5A). These showed co-transcription of all of the tested regions except btmA-B, indicating that genes btmC-M co-transcribe and can behave as a single 14.5 kb operon, whereas btmA and btmB are transcribed separately. The gene cluster was analyzed for putative terminators using WebGeSTer (Mitra et al., 2011). Consistent with the RT-PCR results, terminators were predicted spanning up to 90 bp away from the stop codon of btmA (ΔG = −19.5), between btmA and btmB (ΔG = −19.43) and 149 bp away from the stop codon of btmM (ΔG = −19.27) (Figure 5A).
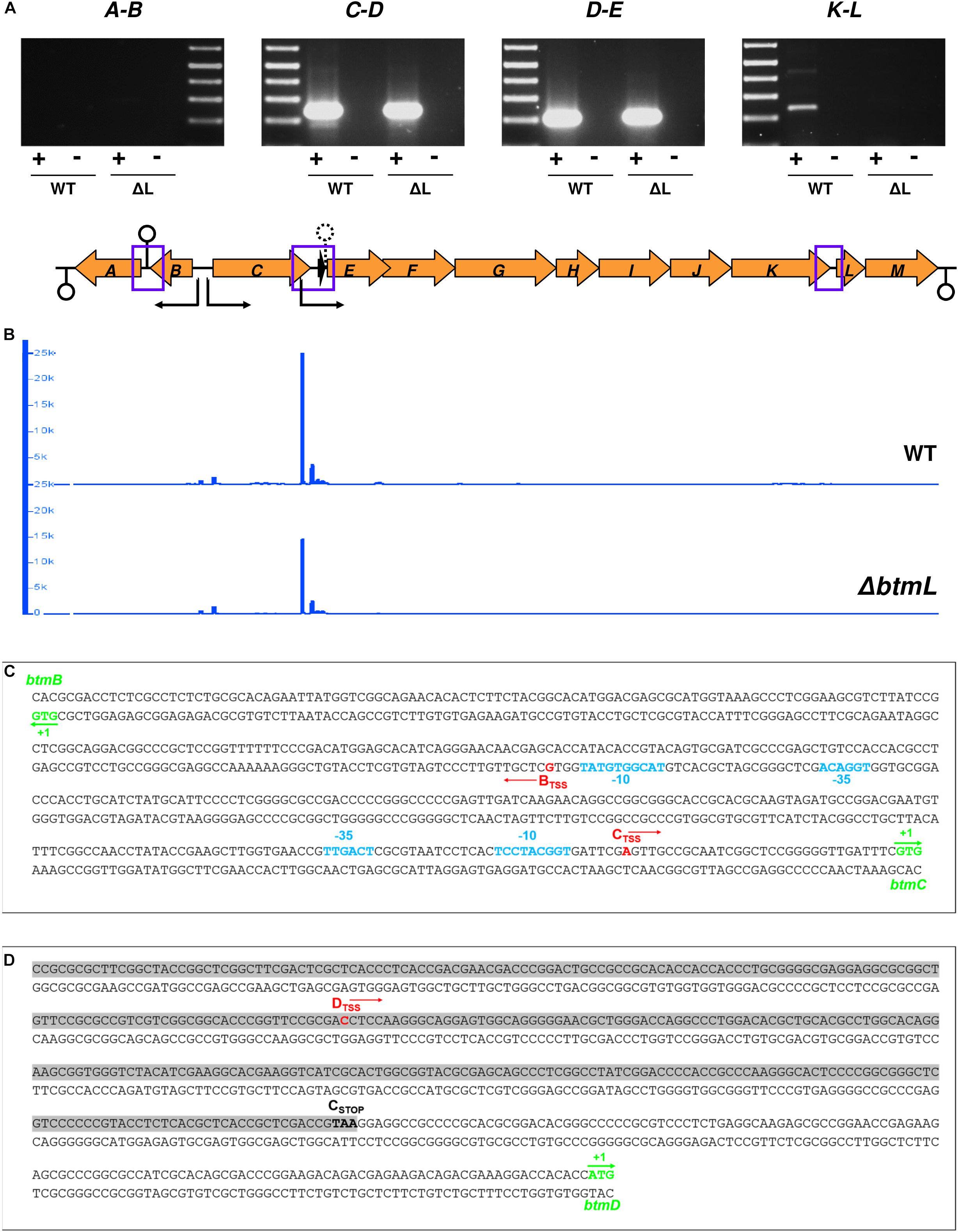
Figure 5. Transcriptional organization of the btm gene cluster. (A) RT-PCR cotranscription analysis of the intergenic regions with no overlapping genes in the btm cluster (highlighted with blue squares in the schematic representation of the btm gene cluster below the gel images; loop symbols in this diagram indicate the location of predicted transcription terminators -solid line- and attenuators -dashed line- in the cluster). (B) Transcription start site mapping of the btm cluster using 5′-tag-RNA-seq. The plots (generated in IGB) show the mapping of reads with TSS tags in the btm cluster in the WT (top) and ΔbtmL (bottom) strains. Sharp peaks corresponding to mapped reads precede btmB, btmC, and btmD, along with some non-specific transcription initiation in the btmD region. The vertical axis in the plots represents read counts and the horizontal axis maps to the gene cluster shown in (A). The direction of transcription from the identified TSSs is represented with black arrows in the cluster. (C) Precise mapping of TSS and potential promoter regions in the btmB-C intergenic region. btmB and btmC start codons are marked in green, and the TSSs identified for btmB and btmC using both 5′tag-RNA-seq and 5′-RACE are labeled in red. −10 and −35 promoter regions for btmC (predicted with BPROM) and btmB (annotated manually) are represented in light blue. (D) Mapping of btmD TSS (red) within the coding sequence of btmC (shadowed in gray, with its stop codon highlighted in bold).
This evidence for btmC-M co-transcription did not exclude the possibility of alternative operons, and the specific increase in btmD transcription following btmL overexpression hinted toward an alternative transcription start site (TSS) for btmD that would explain how it is differentially regulated from its surrounding genes. In order to map the TSSs for the whole cluster we employed 5′-tag-RNA-seq (also called tagRNA-seq) (Innocenti et al., 2015) that enables the identification of TSSs in an untargeted, genome-wide fashion. This powerful technique has the advantage of differentially distinguishing primary transcripts (those generated by an RNA polymerase and therefore coming from true TSSs) from processed transcripts, which arise upon degradation or RNase mediated cleavage of the original transcripts at specific processing sites (PSs). In this particular case, with an operon spanning over 10 kb, degradation or post-transcriptional modification of the resulting transcript could also account for the difference between btmD expression and the remainder of the btmC-M operon.
5′-tag-RNA-seq reads mapped to the S. scabies 87-22 genome clearly showed the presence of three TSSs in the btm cluster: one preceding btmB, one before btmC, and an extra one preceding btmD and within the coding region of the upstream gene, btmC (Figure 5B). Unexpectedly, no TSS was found before btmA, in contrast with what was expected from the intergenic RT-PCR result (Figure 5A). To independently confirm the existence of these TSSs, we performed 5′-RACE (Rapid amplification of cDNA ends) experiments for btmBTSS, btmCTSS, and btmDTSS, which yielded identical results to the 5′tag-RNA-seq (Figures 5C,D). btmCTSS is in position -32 with respect to the btmC start codon, and a prediction of putative σ70 promoters in the btmB-btmC intergenic region using BPROM (Solovyev and Salamov, 2011) identified a nearly canonical −35 sequence (TTGACT) and a poorly conserved −10 region (TCCTACGGT) that are consistent with the location of the btmCTSS (Figure 5C). It was not possible to confidently identify −10 and −35 boxes for the btmBTSS using BPROM or other prediction tools, so potential sequences were identified manually (Figure 5C). The btmDTSS is located 348 bp away from the start of btmD and 215 bp before the end of btmC coding sequence (Figure 5D), but we could not identify any suitable −10 or −35 regions for this TSS, either manually or with predictive tools. Unexpectedly, btmDTSS appears both in the WT and in ΔbtmL, albeit with lower intensity in ΔbtmL (Figure 5B), which indicates that although btmL does affect the transcription of btmD, it is not essential for the expression of this alternative transcript.
Interestingly, the 5′tagRNA-seq results showed that at 72 h of growth there is an extremely high level of transcription of some btm genes, in particular btmD. Transcription initiation preceding this gene seems rather promiscuous with some background noise (Figure 5B). Strikingly, an analysis of the full transcriptome to assess levels of gene expression as reads per kilobase per million reads (RPKM) and transcripts per million (TPM) (Wagner et al., 2012), showed that btmD and btmC are the 4th and 9th most highly transcribed genes in the genome at this time point (Supplementary Datasheet S1). Transcription after btmD drops considerably, showing that even upon overexpression of btmL, expression of btmE did not increase in a comparable way to btmD, in accordance with the qRT-PCR results (Figure 2B). The transcriptomic data also shows that all downstream genes in the operon are transcribed at lower levels than their preceding genes. This may be explained by a transcription attenuator downstream of btmD that likely serves to dampen transcription of downstream genes in the cluster. Accordingly, a putative terminator sequence between btmD and btmE (ΔG = −21.22) is predicted by WebGeSTer (Mitra et al., 2011).
Genome-Wide Analysis of Transcriptional Start Sites in S. scabies
In RiPP biosynthesis, it appears to be beneficial to evolve mechanisms to selectively increase precursor peptide production in relation to catalytic proteins, given that precursor peptides are structural rather than catalytic. The mechanisms by which this happens are poorly understood, especially in pathways where the precursor peptide is apparently co-transcribed with upstream genes encoding catalytic proteins. The secondary transcriptional start site for btmD provides a previously unobserved mechanism by which this can occur. To assess whether this strategy was apparent in other RiPP gene clusters in S. scabies, we assessed their transcriptional organization using the 5′tagRNA-seq dataset. This showed that for all of the putative RiPP clusters identified in the S. scabies genome, the precursor peptide gene always has its own TSS (Supplementary Figures S2, S3). This happens even when the gene is preceded by genes predicted to encode tailoring enzymes (Supplementary Figure S2). Notably, in the case of SCAB_32021, which is tightly clustered with a preceding gene encoding a putative methyltransferase, its dedicated TSS overlaps with the end of this gene, in a similar scenario to btmC and btmD (Supplementary Figure S2). This suggests that having a dedicated TSS could be a widespread strategy for obtaining the appropriate stoichiometry of precursor peptide and tailoring enzymes in those cases where the precursor peptide gene is not at the beginning of an operon.
Reporter Assays Show Strong Transcription From the btmC Promoter Yet Negligible Transcription From the Putative btmD Promoter
To further characterize the main promoter regions in the pathway, promoter activity was measured using the β-glucuronidase reporter gene gusA (Myronovskyi et al., 2011). A 414 bp fragment containing the whole intergenic region btmB-btmC, including btmCTSS but not its RBS, was cloned in vector pIJ10742 (Feeney et al., 2017) to create a transcriptional fusion with gusA. This construct was introduced in S. scabies WT, ΔbtmL, and WT + L to test whether the different genetic backgrounds would affect promoter activity. As a positive control, vector pIJ10741 carrying gusA under the control of the constitutive promoter ermE∗p (Feeney et al., 2017) was used, while empty pIJ10742 with no promoter was employed as a negative control. btmCp proved to be an exceptionally strong promoter in both in solid and in liquid culture (Figure 6), and at 72 h of growth its activity was comparable to ermE∗p. This result correlates with the high levels of expression of btmC observed in the transcriptomic analysis. The same gusA reporter experiment was performed with a 545 bp region that was predicted to contain the promoter preceding btmDTSS, but no reporter activity could be detected in any of the genetic backgrounds tested (Figure 6).
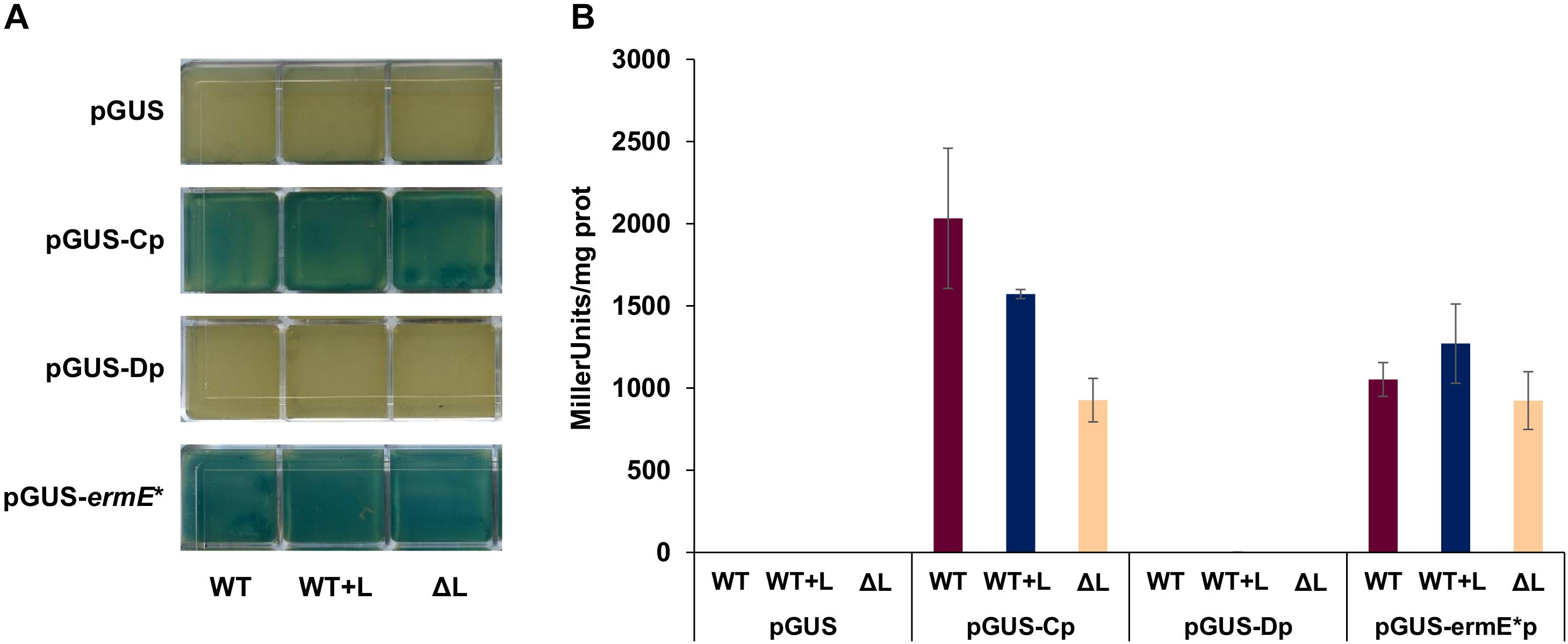
Figure 6. Promoter activity using β-glucuronidase (GUS) reporter assays. The putative promoter regions for bmtC and btmD were inserted before the gusA gene in plasmid pIJ10740 to create transcriptional fusions. The promoterless vector was used as a negative control, and pIJ10741 (containing gusA under the control of ermE*p) was used as a positive control. (A) Qualitative GUS assay in solid medium. Each of the squares represents one independent culture, with squares in the same row corresponding to strains containing the construct listed to their left, and the annotation at the bottom of each column indicating the genetic background. (B) Quantitative GUS assay in liquid medium, with reporter activity measured as Miller units/mg protein.
Therefore, further reporter constructs were made that progressively extended the region tested until it included the whole coding sequence of btmC (but not btmCp), but these provided identical negative results (Supplementary Figures S4, S5). This result pointed to the possibility that transcription from btmDTSS is not driven from its own promoter, but rather from btmCp, where some sort of secondary structure might help enhance transcription of this particular gene. To test this, we assessed the promoter activity of the whole region (btmCp, btmC, and the intergenic region before btmD) and compared its promoter activity to that of the original btmCp reporter. Two promoter versions were generated, one containing the WT sequence, and another one where several synonymous point mutations were introduced in the region of btmDTSS via yeast refactoring (Supplementary Figure S4) in order to disrupt possible DNA secondary structures in this region. Although these new transcriptional fusions were more active than all the other sequences lacking btmCp, they showed drastically reduced activity when compared to the original btmCp reporter, indicating that the additional DNA sequence dampened the promoter activity of btmCp. The mutated btmDTSS region did not significantly affect this phenotype (Supplementary Figures S4, S5).
Identification of Potential Pathway Regulators Using DNA Affinity Capture Assays
Our transcriptional analysis combined with the data from S. scabies ΔbtmL and WT + L show that the master regulator(s) of the btm cluster are not encoded by the btm cluster and are therefore unlikely to be pathway-specific. Identification of such regulators could aid in improving the yield of bottromycin. Given the difficulty in predicting binding sequences for transcriptional activators in streptomycetes without extensive transcriptomic and metabolic datasets (Iqbal et al., 2012; Hwang et al., 2019), the intergenic region btmB-C was used as a probe in a DNA-binding protein capture experiment. This region was amplified using biotinylated primers and the resulting fragment was immobilized on streptavidin coated magnetic beads. The beads were then incubated with lysates of 72 h cultures of S. scabies WT grown in BPM. As a negative control, the same promoter region was incubated with lysates of cultures grown in GYM medium, where the cluster is not expressed and therefore there is no bottromycin production (Supplementary Figure S6). After incubation and several washes with competitor DNA to eliminate proteins binding non-specifically to the probe, the remaining proteins were eluted, washed again and analyzed using quantitative proteomics. A total of 321 proteins were identified in the 2 sets of samples, 120 of which appeared in both conditions, whereas 177 where present exclusively in the BPM samples and only 24 were specific to GYM samples. These results were filtered to identify annotated regulatory proteins or uncharacterized proteins binding to btmCp specifically in bottromycin production conditions. This reduced the candidates to 21 proteins (Supplementary Table S3), of which three were annotated as regulators, while the remainder were uncharacterized proteins.
Based on the intensity of the signal obtained or the biological relevance of the hit, we selected four promising candidates for further genetic analysis to assess their involvement in bottromycin biosynthesis: MtrA (SCAB_55281), GlnK (SCAB_61751), a putative regulatory protein (SCAB_ 85931) and an uncharacterized protein that binds specifically to btmCp under production conditions (SCAB_51451). MtrA forms a three-component system with the histidine kinase MtrB and LpqB, a lipoprotein that is involved in signal transduction (Hoskisson and Hutchings, 2006; Nguyen et al., 2010). This system is widely conserved in Actinobacteria, where it has been shown to regulate both development and antibiotic production in Streptomyces venezuelae and in Streptomyces coelicolor (Som et al., 2017; Zhang et al., 2017). SCAB_61751 encodes a homolog of GlnK, a signal transduction protein with a central role in nitrogen metabolism (Thomas et al., 2000). SCAB_ 85931 is annotated as a putative regulatory protein and contains domains characteristic of RsbR-like anti-anti-sigma factors. Rsb proteins form part of the “stressosome,” which responds to bacterial stress and ultimately leads to the activation of the alternative sigma factor σB (Lee et al., 2004). SCAB_51451 contains no conserved domains. In order to test the role of these genes in bottromycin biosynthesis, each of them was constitutively overexpressed under the control of ermE∗p in S. scabies WT using the integrative plasmid pIB139-RBS, a derivative of pIB139 (Wilkinson et al., 2002). The potential involvement of the MtrAB two-component system prompted us to also express mtrB, which encodes the cognate histidine kinase. Despite the evidence provided by DNA binding, the bottromycin titres were comparable to the WT under the conditions tested (Supplementary Figure S7).
Discussion
An understanding of genetic regulation is required to inform rational approaches to overproduce or engineer the biosynthesis of medicinally promising specialized metabolites. In the case of bottromycin and other RiPPs, their ribosomal origin enables the rapid generation of analogs by precursor peptide mutagenesis, which can lead to the rapid generation of libraries of bioactive analogs (Yang et al., 2018). A distinct challenge for the regulation of RiPP biosynthesis is to enable the expression of a sufficiently large amount of the precursor peptide to support RiPP production, given that this is the substrate for the pathway, while producing other pathway proteins in catalytic quantities. Numerous genetic strategies have evolved that enable this differential production of the precursor peptide. Most commonly, precursor peptide genes are encoded at the beginning of RiPP operons, where they can be followed by imperfect transcriptional terminators (sometimes called “attenuators”) that dampen transcript levels of the following genes (Severinov et al., 2007; Foulston and Bibb, 2010; O’Rourke et al., 2017). Alternatively, some RiPP gene clusters contain multiple precursor peptide genes (Li et al., 2010), while others feature a single precursor peptide containing multiple core peptides (Leikoski et al., 2013; Santos-Aberturas et al., 2019).
There is substantial interest in the RiPP antibiotic bottromycin due to its novel structure, unique molecular target and its activity toward multi-drug resistant pathogens, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) (Shimamura et al., 2009; Tacconelli and Magrini, 2017). Identification of the bottromycin biosynthetic gene cluster (Crone et al., 2012; Gomez-Escribano et al., 2012; Huo et al., 2012) showed that the precursor peptide gene, btmD, appeared to be encoded in the middle of a larger operon. In this study, we used 5′-tag-RNA-seq and 5′-RACE to reveal that the bottromycin gene cluster features an internal transcription start site that enhances transcription of btmD in relation to the remainder of the gene cluster (Figure 5B). The use of 5′-tag-RNA-seq provides confidence that this is a primary transcript and not simply the result of degradation of a longer transcript. In bottromycin production conditions, btmD is one of the most highly expressed genes in the genome. The presence of an internal TSS (btmDTSS) to boost precursor peptide transcript levels has not previously been reported but our data indicate that it may be a common regulatory feature in RiPP biosynthesis. Evidence for this was provided by an analysis of other RiPP gene clusters in the S. scabies genome, which showed that their precursor peptides also have dedicated TSSs, in some cases overlapping with the coding sequence of the preceding genes, as with btmD. The occurrence of intragenic TSS is not a rare phenomenon, and recent TSS mapping work in Streptomyces clavuligerus has revealed the presence of 155 intragenic TSSs in its genome out of a total of 2659 identified (Hwang et al., 2019) and even higher numbers have been reported for other bacteria (Wade, 2015; Brito et al., 2017).
The gene cluster encodes one putative transcriptional regulator, BtmL (Figure 1), but deletion and over-expression experiments showed that this does not function as a master regulator of biosynthesis (Figure 2) and instead functions to increase transcript levels of btmD. Notably, btmDTSS is still present in S. scabies ΔbtmL (Figure 5B), indicating that BtmL does not solely control this regulatory feature. Unexpectedly, a gusA reporter experiment did not reveal any promoter activity associated to btmDTSS, indicating that transcription from this TSS might instead be driven from btmCp. This could also explain why mobility shift assays with purified BtmL using this region as probe were unsuccessful (data not shown). However, a transcriptional fusion containing the entire region from btmCp to the region before btmD provided lower GusA reporter activity than only btmCp. This unexpected result indicates that a complex and unusual system is functioning to regulate btmD gene expression, and further supports the theory that btmDTSS does not have its own dedicated promoter. These data highlight the need for further studies to elucidate the precise role of internal TSSs in RiPP biosynthesis.
Despite these null results, the importance of btmDTSS is supported by our prior work that showed that altering the region preceding btmD practically abolishes bottromycin production (Eyles et al., 2018), either by deletion of btmC or by swapping the positions of btmC and btmD in the gene cluster. Similarly, DNA affinity capture with the btmB-C region did not pull down BtmL (Supplementary Table S3). Therefore, the mechanism by which BtmL modulates btmD transcription remains cryptic. One possible hypothesis would be the interaction with a cis-regulatory region in the 5′ untranslated region (UTR) of btmD mRNA. Transcripts with UTRs bigger than 100–150 bp are usually considered “extended-leadered mRNAs” and are likely candidates to contain such a regulatory feature (Brito et al., 2017; Hwang et al., 2019). The btmD 5’UTR (347 bp) is within this category, but searches in this sequence against the Rfam database for any known RNA structures did not yield a clear result.
We sought to use our knowledge of pathway expression to increase bottromycin titre, firstly by over-expressing btmL either alone or along with btmA, which encodes a MFS transporter. While this increased btmD transcription (Figure 2B), it did not lead to higher bottromycin yields, indicating that btmD levels are not the rate limiting factor for pathway expression. Similarly, in S. scabies, export by BtmA is not a bottleneck in this pathway. We had previously shown that the bottromycin pathway produces a significant amount of side-products in both S. scabies and a heterologous expression system (Crone et al., 2016; Eyles et al., 2018). Pathway refactoring in this heterologous system showed that constitutive high expression of the btm cluster leads to high yields of shunt metabolites and low titres of mature products, while controlled expression using a riboswitch increases overall production with higher ratios of mature bottromycins versus shunt metabolites. This is consistent with a study that refactored the bottromycin gene cluster from Streptomyces sp. BC16019 (Horbal et al., 2018), which also concluded that high levels of transcription did not fully correlate with increases in production. Promoter choice is a key factor for optimizing production of RiPPs, and can also depend on the timing of gene expression, such as in the heterologous production of telomestatin in Streptomyces avermitilis (Amagai et al., 2017).
There is surprisingly little known about the regulation of specialized metabolites that do not contain master regulators in their gene clusters. A rare example is erythromycin biosynthesis in Saccharopolyspora erythraea, which is regulated by BldD, a key regulator of actinomycete development, as well as two other regulatory genes which are not located within its biosynthetic cluster (Chng et al., 2008; Kirm et al., 2013; Wu et al., 2014). We sought to identify the master regulators of bottromycin biosynthesis using a DNA affinity capture experiment with the intergenic region between btmB and btmC, which contains the major promoters that control the pathway. This provided a series of promising candidate proteins, including the well-characterized regulatory proteins MtrA and GlnK. However, over-expression of these genes also did not lead to increased bottromycin production in S. scabies. This negative result does not disprove the involvement of these regulators, in particular in the case of MtrA and GlnK. As a member of classical two-component system the activity of the response regulator MtrA is dependent on its phosphorylation state (Kenney, 2002; Desai and Kenney, 2017). Overexpression of the gene, if it is not appropriately phosphorylated, might not be enough to reveal a phenotype. GlnK is a PII family signal transducer protein that modulates the activity of multiple biosynthetic and regulatory proteins in nitrogen metabolism (Arcondéguy et al., 2001; Gerhardt et al., 2015; Shimizu, 2016). It would make sense that nitrogen levels would exert an effect on bottromycin biosynthesis, due to the peptidic nature of bottromycin and in fact a paralog of this protein has been shown to affect secondary metabolite production in S. coelicolor (Waldvogel et al., 2011). These proteins also rely on post-translational modifications to properly function, which adds a layer of complication in their regulatory functions that may dampen the effect of their overexpression. Further studies will be necessary to fully characterize the role of these regulators, as well as the uncharacterized proteins identified in this work, on bottromycin production.
In summary, we have revealed that the regulation of bottromycin biosynthesis is surprisingly complex and features interplay between global regulatory proteins and a cluster-situated modulator, BtmL. Precursor peptide transcription is enhanced in relation to surrounding genes by BtmL and a cryptic internal transcriptional start site. This single-gene internal transcript makes the precursor peptide gene one of the most highly transcribed genes in the S. scabies genome. A wider assessment of the S. scabies genome shows that this strategy could represent a widespread regulatory mechanism for the expression of RiPP precursor peptide genes.
Materials and Methods
Chemicals and Molecular Biology Reagents
Unless otherwise specified, antibiotics, media components and chemical reagents used in this work were purchased from Sigma-Aldrich (United Kingdom) with the exception of soy flour, which was purchased from Holland & Barret (United Kingdom). Enzymes and molecular biology kits were purchased from New England Biolabs and Promega Healthcare, respectively.
Bacterial Strains and Culture Conditions
The following strains were used in this work: S. scabies DSM 41658, both the wild-type (WT) and the mutant strain ΔbtmL (ΔL), whose construction was described previously (Crone et al., 2012). Additionally, Escherichia coli K-12 strain DH5α (Invitrogen) was used for plasmid propagation and DNA manipulation, and the methylation deficient strain E. coli ET12567 containing pUZ8002 (Paget et al., 1999) was used for intergeneric conjugal transfer of genetic material to S. scabies, which was performed following standard procedures (Kieser et al., 2000). E. coli culture media used in this work are described in Sambrook et al. (1989). Several Streptomyces culture media were used: mannitol soya flour medium (SFM) (Kieser et al., 2000) was used for Streptomyces propagation and conjugations, and instant potato mash agar [2% Smash (Premier Foods), 2% agar] was used to grow S. scabies for spore harvesting. GYM medium (0.4% glucose, 0.4% yeast extract, and 1.0% malt extract, in Milli-Q water) and bottromycin production medium (BPM: 1% glucose, 1.5% soluble starch, 0.5% yeast extract, 1.0% soy flour, 0.5% NaCl, and 0.3% CaCO3, in Milli-Q water) were used for bottromycin production. Production experiments were performed as follows: 30 μL of concentrated spores were used to inoculate 10 mL of GYM medium in 50 mL flasks and were incubated for 48 h at 30°C and 250 rpm. 250 μL of seed culture were used to inoculate 10 mL BPM in 50 mL Falcon tubes covered with foam bungs instead of caps. Alternatively, 1 mL of seed culture was used to inoculate 50 mL of BPM in 250 mL flasks containing a spring. When necessary, cultures were supplemented with appropriate concentrations of CoCl2 (25 μg/mL unless otherwise stated). Triplicate production cultures were incubated for 5–6 days at 28°C and 230 rpm, at which point samples were collected and processed immediately, or frozen at −20°C until further processing and analysis. When antibiotic selection was necessary, culture media were supplemented with the appropriate antibiotics at the following final concentrations: kanamycin at 50 μg/mL, apramycin at 50 μg/mL, hygromycin at 50 μg/mL, chloramphenicol at 25 μg/mL, and nalidixic acid at 25 μg/mL.
Gene Expression Plasmids Construction
All the primers used for gene amplification and generation of the following constructs are listed in Supplementary Table S1. The gene expression vectors used in this work were pIB139-RBS-btmD (Crone et al., 2016) and pIJ10257 (Hong et al., 2005), which are both integrative plasmids containing the constitutive ermE∗ promoter. pIB139-RBS-btmD is a derivative of pIB139 (Wilkinson et al., 2002) that carries the btmD gene preceded by a ribosome binding site (RBS) and an NdeI restriction site installed to facilitate cloning and improve gene expression (Crone et al., 2016). This plasmid was linearized using restriction enzymes NdeI and EcoRI, releasing btmD and allowing the introduction of the gene of interest, either via ligation of an EcoRI/NdeI treated PCR fragment or by Gibson assembly, following published protocols (Crone et al., 2012). The ligation method was used to introduce btmL and generate pIB139-RBS-btmL, which was used to complement the mutant ΔbtmL and to overexpress this gene in the WT strain (WT + L). The Gibson assembly method was used to introduce mtrA, mtrB, glnB, SCAB_85931, and SCAB_51451, into pIB139-RBS for gene overexpression the WT strain. An empty version of pIB139-RBS was generated to use as a control in production experiments, via Gibson assembly, using the NdeI/EcoRI linearized vector and primers pIB-RBS_fw and pIB-RBS_rv, carrying the RBS sequence. pIJ10257 was used for the expression of btmA. This gene was amplified using primers btmA-start and btmA-end, digested with NdeI and HindIII and ligated into NdeI/HindIII digested pIJ10257. Correct construction of all expression plasmids was confirmed using colony PCR and sequencing, after which they were transferred to E. coli ET12567/pUZ8002 cells for conjugation into S. scabies (WT or ΔbtmL as appropriate). pIB139 integrates in the ϕC31 phage integration site, and pIJ10257 integrates in the ϕBT1 site, which allowed for the simultaneous overexpression of two genes when necessary. Correct integration of the plasmids was verified by colony PCR using ermEp_chk1 in combination with the reverse amplification primer for each of the genes. As a control for the production experiments, a strain carrying the corresponding empty vector was generated in each case.
LC-MS Analysis of Bottromycin Production
Bottromycin production culture samples (1 mL) were extracted with an equal volume of methanol, mixed with shaking for a minimum of 10 min. The mixtures were then centrifuged for 4 min at 13,000 rpm to pellet cellular material and other particulate contaminants. 2 μL of the resulting supernatants were injected onto a Phenomenex Kinetex 2.6 μm XB-C18 column (50 mm × 2.1 mm, 100 Å) attached to a Shimadzu Nexera X2 UHPLC and eluted with a linear gradient of 5 to 95% acetonitrile (ACN) in water + 0.1% formic acid (FA) over 6 min, with a flow-rate of 0.6 mL/min. MS data were obtained in positive mode using a Shimadzu IT-TOF mass spectrometer coupled to the UHPLC and analyzed using LabSolutions software (Shimadzu). Bottromycin production was plotted in peak area units. To normalize production values across samples, culture growth was quantified by measuring DNA concentration with an adaptation of the Burton diphenylamine colorimetric assay (Zhao et al., 2013).
Isolation of Total RNA
Two milliliter samples of S. scabies liquid cultures were harvested after 72 h incubation in conditions as described above. Samples were washed with an equal volume of RNAlater (Thermo Fisher Scientific) and stored at −80°C until further processing. RNA was then extracted following previous protocols (Crone et al., 2016), resuspending the mycelium in 1 mL of RLT buffer from the RNeasy Kit (Qiagen) and homogenizing the sample in lysing matrix B tubes (MP Biomedicals) using a FastPrep instrument and a program of 3 × 30 s pulses at 6 m/s with 1 min cooling intervals on ice. The lysates were then centrifuged at 13,000 rpm and 700 μL of supernatant from each sample were then transferred to spin tubes from the RNeasy Kit to undergo purification following manufacturer instructions. Chromosomal DNA contamination was eliminated with on-column DNase I treatment (Qiagen) and a further cleanup step using TURBO DNA-free Kit (Ambion, Invitrogen). RNA concentration in the samples was quantified measuring A260 using a Nanodrop. In the case of the samples for 5′tag-RNAseq, further quantity and integrity measurements were performed with RNA ScreenTape (Agilent).
RT and qRT-PCR Analyses
Both RT-PCR and qRT-PCR analyses were carried out using 250 ng of total RNA as template in a two-step protocol. The first step consisted of cDNA synthesis with the QuantiTect Reverse Transcription Kit (Qiagen), as detailed in the manufacturer’s instructions. For RT-PCR analyses the cDNA was then used as a template for PCR reactions using Taq polymerase and specific primers for the regions to test (Supplementary Table S1). The amplification conditions were as follows: initial denaturation at 95°C for 3 min followed by 33 cycles of 95°C for 30 s, 58–62°C for 30 s and 72°C for 40–60 s, with a final extension step at 72°C for 5 min. The resulting RT-PCR products were separated in 2% agarose gels and stained with ethidium bromide for visualization. For qRT-PCR analyses the aforementioned cDNA was used as template in quantitative reactions with the SensiFAST SYBR No-ROX Kit (Bioline) following the manufacturer’s instructions. The reactions were run in a Bio-Rad CFX96 thermocycler and the amplification protocol was a 2-step cycling PCR program: 1 cycle at 95°C for 2 min followed by 40 cycles of 5 s at 95°C and 30 s at 60°C. An additional melting curve step was used at the end of the reaction to assess the specificity of the amplified products. The qRT-PCR results were analyzed with CFX Manager software (Bio-Rad). In both cases, negative control samples of the cDNA synthesis step with no retrotranscriptase were included, in order to control for the presence of contaminating chromosomal DNA in the RNA samples. Primers used in these analyses (Supplementary Table S1) were designed with a preference for 17–23 mers and Tm ∼ 65°C with the help of Vector NTI Advance 11.5 (Invitrogen) and Primer3 software (Untergasser et al., 2012) and validated used the online tool NetPrimer (Premier Biosoft). In the specific case of qRT-PCR primers, these were designed to amplify fragments of ∼100 bp and their efficiency was tested using serial dilutions of chromosomic DNA as template. For both RT-PCR and qRT-PCR, primers for hrdB, encoding a housekeeping sigma factor, were used as an internal control to assess the quality of RNA and in the latter case to normalize gene expression levels.
5′tag-RNA-Seq (tagRNA-Seq)
Library Construction and Sequencing
Total RNA samples of S. scabies WT and ΔbtmL were extracted as previously described and, following quality control assays to ensure their integrity, they were submitted to Vertis Biotechnologie AG (Germany) for the construction and sequencing of tagRNA-seq libraries in a protocol adapted from the technique described in Innocenti et al. (2015). Prior to library construction, rRNA was depleted in the samples using the Ribo-Zero rRNA Removal Kit for bacteria (Epicenter). The remaining material was labeled sequentially as follows: sequence tag CTGAAGCT was ligated to transcripts presenting 5′-monophosphate groups (processed transcripts). The samples were then treated with RNA 5′ polyphosphatase (5′PP; Epicenter) to convert the 5′-triphosphate groups of primary transcripts into 5′-monophosphate ends amenable for ligation with the alternative tag sequence TAATGCGC. Once labeled, the samples were used as template for first-strand cDNA synthesis using random hexameric primers. After fragmentation and RNA clean up, Illumina TruSeq sequencing adapters were ligated in a strand specific manner to the 5′ and 3′ ends of the cDNA fragments. The cDNA was then amplified with a proof-reading enzyme to enrich the samples. At this point, it is possible to specifically PCR amplify the 5′-ends which carried the two tag sequences, but that would mean losing the information relating to the rest of the transcriptome. Therefore, a full transcriptome enrichment (for fragments that had Illumina adapters on both ends) was carried out in order to preserve the full coverage of the transcriptome in our libraries. The resulting material was purified with the Agencourt AMPure XP Kit (Beckman Coulter Genomics) and analyzed by capillary electrophoresis. The cDNA preparations were pooled in equimolar amounts and size selected (240–450 bp range) and the pooled libraries were sequenced on an Illumina NextSeq 500 system using 75 bp read lengths. RNA-seq data has been deposited in the ArrayExpress database (accession number E-MTAB-8236).
Data Analysis
Sequencing reads were first sorted according to their tags in order to generate independent fastq files for each of the datasets (see Supplementary Table S2 for a summary of the sequencing results). Once sorted, the reads were trimmed to remove the tag sequences and aligned to S. scabies 87-22 genome (RefSeq NC_013929.1) using Bowtie2 (Langmead and Salzberg, 2012), which yielded SAM alignment files for each of the fastq files. Downstream processing of the SAM alignment files was performed using a series of Perl scripts supported by the Bioperl (Stajich et al., 2002) toolkit. Using the SAM alignments as input, the number of reads mapping to each nucleotide position of the S. scabies genome was calculated and saved in a coverage file. The coverage information was then integrated with a feature table including the coordinates of all of the gene coding sequences in the genome, in order to calculate the number of reads per gene, or gene counts. These gene counts were used to make an estimation of gene expression levels by calculating RPKM and TPM values for each gene (Wagner et al., 2012) (Supplementary Datasheet S1). For mapping and visualization of the transcription start sites (TSSs) in the bottromycin cluster, the SAM files containing the sequences labeled with 5′ tags were transformed into wig files suitable for visualization in the Integrated Genome Browser (IGB) software (Freese et al., 2016) plotted against the S. scabies genome. These files were normalized to eliminate biases due to difference in sequencing depth using the normalizeQuantiles function in the limma package (Ritchie et al., 2015) of R (R Core Team, 2014). Given the high amount of processed transcripts present in the btmC-D region of the bottromycin cluster, a “wig minus wig” file was generated in which processed transcript reads were subtracted from the primary transcript reads. The regions mapped in this filtered file were considered to be true TSSs.
5′-RACE Experiments
The TSSs identified in the 5′tag-RNA-seq experiment were validated using a 5′-RACE system for rapid amplification of cDNA ends (Invitrogen), using the manufacturer’s instructions (version 2.0). 1 μg of total RNA from S. scabies WT and ΔbtmL harvested after 72 h of growth at 28°C and 230 rpm was used to carry out cDNA synthesis with specific primers for each of the promoters tested (Supplementary Table S1). The cDNA was purified and treated with terminal deoxynucleotidyl transferase (TdT) to add poly(dC) tails to its 3′ends. After an initial PCR amplification of the tailed fragments with the 5′-RACE abridged anchor primer and subsequent amplifications with the universal amplification primer (both provided by the kit) and specific nested primers (Supplementary Table S1), defined amplification products were observed. These products were gel purified and submitted for Sanger sequencing to confirm the position of the TSSs.
gusA Transcriptional Fusions Construction and β-Glucuronidase Reporter Activity Assays
Putative promoter regions of btmC (btmCp, 414 bp) and btmD (btmDp, 545 bp) and extended promoter regions spanning different lengths of btmC were amplified from S. scabies genomic DNA or from a refactored bottromycin cluster containing 33 synonymous point mutations in the btmDTTS region (Supplementary Figure S4) with primers containing NdeI and XhoI restriction sites (Supplementary Table S1). These were verified by sequencing, and then ligated or assembled into NdeI/XhoI digested pIJ10742, which contains a promoterless copy of the reporter gene gusA (Feeney et al., 2017). These plasmids were introduced via intergeneric conjugation into S. scabies (WT, ΔbtmL, and WT + L) using E. coli ET12567/pUZ8002, where they integrated in the ϕBT1 phage integration site. Hygromycin resistant exconjugants were analyzed by colony PCR with primers pGUS_chk_fw and pGUS_chk_fw. β-glucuronidase assays in solid and liquid medium were carried out as described previously (Sherwood and Bibb, 2013). In the case of the liquid assays, reporter activity was represented as Miller units/mg protein. In both cases, pIJ10742 carrying no promoter was used as a negative control, and pIJ10741, carrying gusA under the control of ermE∗p (Feeney et al., 2017), was included as a positive control of promoter activity.
DNA Affinity Protein Capture Assay
Sample Preparation
The intergenic region btmB-btmC (414 bp) was PCR amplified using primers probe_BC_fw2_b and probe_BC_rv2 to generate a 5′ biotinylated probe. 40 μg of this probe were inmobilised onto 10 mg of streptavidin magnetic beads (Dynabeads® MyOneTM Streptavidin T1, Invitrogen, United Kingdom), following the manufacturer’s instructions. Protein extracts from S. scabies wild type were obtained from 500 mL cultures in either BPM or GYM incubated for 72 h at 28°C and 250 rpm. Cell pellets were harvested by centrifugation for 15 min at 7,000 rpm, resuspended in binding buffer (20 mM TrisHCl pH7.5, 1 mM EDTA, 100 mM NaCl, 10% glycerol, and 1 mM DTT) supplemented with protease inhibitors (cOmpleteTM Protease Inhibitor Cocktail, Roche, United Kingdom) and lysed by sonication with a Vibra-cell sonicator (Sonics & Materials Inc., United States) using 150 × 2 s pulses at 40% amplitude alternated by 5 s rest on ice. Binding assays were performed using a modification of the protocol reported by Bekiesch et al. (2016) using 60 mg of total protein in a 10 mL final volume of the aforementioned binding buffer supplemented with 0.1 mg/mL of salmon sperm DNA (Invitrogen, Germany). Proteins were eluted twice in 250 μL binding buffer containing 2 M NaCl and the binding assay was repeated twice for each sample, generating 1 mL of final eluate per sample. These eluates were then acidified to pH3 with trifluoroacetic acid (TFA) and applied to an C4 SPE column (OMIX C4, Agilent). The samples were washed twice with 0.1% TFA in water and three times with 0.1% FA before eluting in three fractions (200 μL 30% ACN and 0.1% FA, 200 μL 30% ACN and 200 μL 70% ACN) that were pooled and kept frozen on dry ice until analysis.
Sample Analysis
After purification and clean up, duplicate samples for each condition were analyzed by LC-MS/MS on an Orbitrap-FusionTM mass spectrometer (Thermo Fisher, United Kingdom) equipped with an UltiMateTM 3000 RSLCnano System using an Acclaim PepMap C18 column (2 μm, 75 μm × 500 mm, Thermo). Samples were loaded and trapped using a pre-column which was then switched in-line to the analytical column for separation. Peptides were eluted with a gradient of 5–40% ACN in water/0.1% FA at a rate of 0.5% min–1. The column was connected to a 10 μm SilicaTipTM nanospray emitter (New Objective, United States) for infusion into the mass spectrometer. Data dependent analysis was performed using a CID/HCD fragmentation method with the following parameters: positive ion mode, orbitrap MS resolution = 60 k, mass range (quadrupole) = 300–1500 m/z, AGC target 2e5, MS2 in ion trap, threshold 1e4, isolation window 1.6 Da (quadrupole), charge states 2–6, MS2 top20, AGC target 1.5e4, max inject time 200 ms, dynamic exclusion 1 count, 60 s exclusion, exclusion mass window ±7 ppm. MS scans were saved in profile mode while MS2 scans were saved in centroid mode. Raw files were processed with MaxQuant (version 1.5.3.30) (Tyanova et al., 2016). The peak lists were searched against a S. scabies protein database downloaded from Uniprot.org (25.05.2016) with 16846 entries together with the MaxQuant contaminants database (249 entries) using an in-house Mascot Server (2.4.1, Matrix Science, United Kingdom) with trypsin with 2 missed cleavages, carbamidomethylation (C) as fixed and oxidation (M), acetylation (protein N-terminus), and deamidation (N,Q) as variable modifications. Mass tolerances were 6 ppm for precursor ions and 0.6 Da for fragment ions. Mascot search results were imported into the Scaffold software (Proteome Software Inc., United States) to probabilistically validate protein identifications derived from the MS/MS sequencing results using the X!Tandem (Craig and Beavis, 2003) and ProteinProphet algorithms (Nesvizhskii et al., 2003). Validation parameters were set to 95% protein probability and 95% peptide probability.
Data Availability Statement
The datasets generated for this study can be found in the ArrayExpress Archive of Functional Genomics Data (accession number E-MTAB-8236).
Author Contributions
NV performed genetic manipulation experiments, LC-MS analyses, RNA extraction, and gene expression analyses. NV, EC-T, and TE performed promoter activity assays. GC and NV performed RNA-seq data processing and analysis. AT devised and supervised this work. NV and AT wrote the manuscript.
Funding
Financial support was provided by a Royal Society University Research Fellowship (AT), Biotechnology and Biological Sciences Research Council (BBSRC) MET and MfN Institute Strategic Programme grants (BB/J004596/1 and BBS/E/J/000PR9790) to the John Innes Centre (AT and NV), BBSRC grant BB/M003140/1 (AT and NV), a European Commission Erasmus+ traineeship (EC-T), and a Ph.D. studentship (BBSRC Doctoral Training Program grant BB/M011216/1) (TE). Open access publication fees are paid by a United Kingdom Research Council grant to the John Innes Centre.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Gerhard Saalbach (John Innes Centre Proteomics Facility) for his help with the proteomic analysis of the DNA binding protein capture samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00495/full#supplementary-material
References
Abbas, A. S., and Edwards, C. (1990). Effects of metals on Streptomyces coelicolor growth and actinorhodin production. Appl. Environ. Microbiol. 56, 675–680. doi: 10.1128/aem.56.3.675-680.1990
Amagai, K., Ikeda, H., Hashimoto, J., Kozone, I., Izumikawa, M., Kudo, F., et al. (2017). Identification of a gene cluster for telomestatin biosynthesis and heterologous expression using a specific promoter in a clean host. Sci. Rep. 7:3382. doi: 10.1038/s41598-017-03308-5
Arcondéguy, T., Jack, R., and Merrick, M. (2001). P(II) signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol. Mol. Biol. Rev. 65, 80–105. doi: 10.1128/MMBR.65.1.80-105.2001
Bartholomae, M., Buivydas, A., Viel, J. H., Montalbán-López, M., and Kuipers, O. P. (2017). Major gene-regulatory mechanisms operating in ribosomally synthesized and post-translationally modified peptide (RiPP) biosynthesis. Mol. Microbiol. 106, 186–206. doi: 10.1111/mmi.13764
Bauerle, M. R., Schwalm, E. L., and Booker, S. J. (2015). Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J. Biol. Chem. 290, 3995–4002. doi: 10.1074/jbc.R114.607044
Bekiesch, P., Franz-Wachtel, M., Kulik, A., Brocker, M., Forchhammer, K., Gust, B., et al. (2016). DNA affinity capturing identifies new regulators of the heterologously expressed novobiocin gene cluster in Streptomyces coelicolor M512. Appl. Microbiol. Biotechnol. 100, 4495–4509. doi: 10.1007/s00253-016-7306-1
Bibb, M. J., Janssen, G. R., and Ward, J. M. (1985). Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. Gene 38, 215–226. doi: 10.1016/0378-1119(85)90220-3
Bierbaum, G., and Sahl, H.-G. (2009). Lantibiotics: mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 10, 2–18. doi: 10.2174/138920109787048616
Brito, L. F., Irla, M., Kalinowski, J., and Wendisch, V. F. (2017). Detailed transcriptome analysis of the plant growth promoting Paenibacillus riograndensis SBR5 by using RNA-seq technology. BMC Genomics 18:846. doi: 10.1186/s12864-017-4235-z
Busenlehner, L. S., Pennella, M. A., and Giedroc, D. P. (2003). The SmtB/ArsR family of metalloregulatory transcriptional repressors: structural insights into prokaryotic metal resistance. FEMS Microbiol. Rev. 27, 131–143. doi: 10.1016/S0168-6445(03)00054-8
Chakravorty, D. K., and Merz, K. M. (2014). Studying allosteric regulation in metal sensor proteins using computational methods. Adv. Protein Chem. Struct. Biol. 96, 181–214. doi: 10.1016/bs.apcsb.2014.06.009
Chng, C., Lum, A. M., Vroom, J. A., and Kao, C. M. (2008). A key developmental regulator controls the synthesis of the antibiotic erythromycin in Saccharopolyspora erythraea. Proc. Natl. Acad. Sci. U.S.A. 105, 11346–11351. doi: 10.1073/pnas.0803622105
Cooper, L. E., Li, B., and van der Donk, W. A. (2010). 5. 08 biosynthesis and mode of action of lantibiotics. Compr. Nat. Prod. II 1971, 217–256. doi: 10.1016/B978-008045382-8.00116-7
Craig, R., and Beavis, R. C. (2003). A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17, 2310–2316. doi: 10.1002/rcm.1198
Crone, W. J. K., Leeper, F. J., and Truman, A. W. (2012). Identification and characterisation of the gene cluster for the anti-MRSA antibiotic bottromycin: expanding the biosynthetic diversity of ribosomal peptides. Chem. Sci. 3, 3516–3521. doi: 10.1039/c2sc21190d
Crone, W. J. K., Vior, N. M., Santos-Aberturas, J., Schmitz, L. G., Leeper, F. J., and Truman, A. W. (2016). Dissecting bottromycin biosynthesis using comparative untargeted metabolomics. Angew. Chemie Int. Ed. 55, 9639–9643. doi: 10.1002/anie.201604304
Desai, S. K., and Kenney, L. J. (2017). To P or Not to P? Non-canonical activation by two-component response regulators. Mol. Microbiol. 103, 203–213. doi: 10.1111/mmi.13532
Eyles, T. H., Vior, N. M., and Truman, A. W. (2018). Rapid and robust yeast-mediated pathway refactoring generates multiple new bottromycin-related metabolites. ACS Synth. Biol. 7, 1211–1218. doi: 10.1021/acssynbio.8b00038
Feeney, M. A., Chandra, G., Findlay, K. C., Paget, M. S. B., and Buttner, M. J. (2017). Translational control of the SigR-directed oxidative stress response in Streptomyces via IF3-mediated repression of a noncanonical GTC start codon. mBio 8, e815–e817. doi: 10.1128/mBio.00815-17
Fernández-Martínez, L. T., Gomez-Escribano, J. P., and Bibb, M. J. (2015). A relA-dependent regulatory cascade for auto-induction of microbisporicin production in Microbispora corallina. Mol. Microbiol. 97, 502–514. doi: 10.1111/mmi.13046
Flinspach, K., Kapitzke, C., Tocchetti, A., Sosio, M., and Apel, A. K. (2014). Heterologous expression of the thiopeptide antibiotic GE2270 from planobispora rosea ATCC 53733 in Streptomyces coelicolor requires deletion of ribosomal genes from the expression construct. PLoS One 9:e90499. doi: 10.1371/journal.pone.0090499
Foulston, L. C., and Bibb, M. J. (2010). Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. U.S.A. 107, 13461–13466. doi: 10.1073/pnas.1008285107
Franz, L., Adam, S., Santos-Aberturas, J., Truman, A. W., and Koehnke, J. (2017). Macroamidine formation in bottromycins is catalyzed by a divergent YcaO enzyme. J. Am. Chem. Soc. 139, 18158–18161. doi: 10.1021/jacs.7b09898
Freese, N. H., Norris, D. C., and Loraine, A. E. (2016). Integrated genome browser: visual analytics platform for genomics. Bioinformatics 32, 2089–2095. doi: 10.1093/bioinformatics/btw069
Gerhardt, E. C. M., Rodrigues, T. E., Müller-Santos, M., Pedrosa, F. O., Souza, E. M., Forchhammer, K., et al. (2015). The Bacterial signal transduction protein GlnB regulates the committed step in fatty acid biosynthesis by acting as a dissociable regulatory subunit of acetyl-CoA carboxylase. Mol. Microbiol. 95, 1025–1035. doi: 10.1111/mmi.12912
Gomez-Escribano, J. P., Song, L., Bibb, M. J., and Challis, G. L. (2012). Posttranslational ß-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics. Chem. Sci. 3, 3522–3525. doi: 10.1039/C2SC21183A
Hong, H.-J., Hutchings, M. I., Hill, L. M., and Buttner, M. J. (2005). The role of the novel Fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J. Biol. Chem. 280, 13055–13061. doi: 10.1074/jbc.M413801200
Horbal, L., Marques, F., Nadmid, S., Mendes, M. V., and Luzhetskyy, A. (2018). Secondary metabolites overproduction through transcriptional gene cluster refactoring. Metab. Eng. 49, 299–315. doi: 10.1016/J.YMBEN.2018.09.010
Hoskisson, P. A., and Hutchings, M. I. (2006). MtrAB-LpqB: a conserved three-component system in actinobacteria? Trends Microbiol. 14, 444–449. doi: 10.1016/j.tim.2006.08.005
Hou, Y., Tianero, M. D. B., Kwan, J. C., Wyche, T. P., Michel, C. R., Ellis, G. A., et al. (2012). Structure and biosynthesis of the antibiotic bottromycin D. Org. Lett. 14, 5050–5053. doi: 10.1021/ol3022758
Huang, Y., Yang, D., Pan, G., Tang, G.-L., and Shen, B. (2016). Characterization of LnmO as a pathway-specific Crp/Fnr-type positive regulator for leinamycin biosynthesis in Streptomyces atroolivaceus and its application for titer improvement. Appl. Microbiol. Biotechnol. 100, 10555–10562. doi: 10.1007/s00253-016-7864-7862
Huo, L., Rachid, S., Stadler, M., Wenzel, S. C., and Müller, R. (2012). Synthetic biotechnology to study and engineer ribosomal bottromycin biosynthesis. Chem. Biol. 19, 1278–1287. doi: 10.1016/j.chembiol.2012.08.013
Hwang, S., Lee, N., Jeong, Y., Lee, Y., Kim, W., Cho, S., et al. (2019). Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 47, 6114–6129. doi: 10.1093/nar/gkz471
Innocenti, N., Golumbeanu, M., Fouquier, d’Hérouël, A., Lacoux, C., Bonnin, R. A., et al. (2015). Whole-genome mapping of 5’ RNA ends in bacteria by tagged sequencing: a comprehensive view in Enterococcus faecalis. RNA 21, 1018–1030. doi: 10.1261/rna.048470.114
Iqbal, M., Mast, Y., Amin, R., Hodgson, D. A., Wohlleben, W., and Burroughs, N. J. (2012). Extracting regulator activity profiles by integration of de novo motifs and expression data: characterizing key regulators of nutrient depletion responses in Streptomyces coelicolor. Nucleic Acids Res. 40, 5227–5239. doi: 10.1093/nar/gks205
Izawa, M., Kawasaki, T., and Hayakawa, Y. (2013). Cloning and heterologous expression of the thioviridamide biosynthesis gene cluster from Streptomyces olivoviridis. Appl. Environ. Microbiol. 79, 7110–7113. doi: 10.1128/AEM.01978-13
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kenney, L. J. (2002). Structure/function relationships in OmpR and other winged-helix transcription factors. Curr. Opin. Microbiol. 5, 135–141. doi: 10.1016/S1369-5274(02)00310-7
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F., and Hopwood, D. A. (2000). Practical Streptomyces Genetics. Norwich: John Innes Foundation.
Kirm, B., Magdevska, V., Tome, M., Horvat, M., Karničar, K., Petek, M., et al. (2013). SACE_5599, a putative regulatory protein, is involved in morphological differentiation and erythromycin production in Saccharopolyspora erythraea. Microb. Cell Fact. 12:126. doi: 10.1186/1475-2859-12-126
Kumar, S., He, G., Kakarla, P., Shrestha, U., Ranjana, K. C., Ranaweera, I., et al. (2016). Bacterial multidrug efflux pumps of the major facilitator superfamily as targets for modulation. Infect Disord. Drug Targets 16, 28–43. doi: 10.2174/1871526516666160407113848
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Laxminarayan, R. (2014). Antibiotic effectiveness: balancing conservation against innovation. Science 345, 1299–1301. doi: 10.1126/science.1254163
Laxminarayan, R., Amábile-Cuevas, C. F., Cars, O., Evans, T., Heymann, D. L., Hoffman, S., et al. (2016). UN high-level meeting on antimicrobials—what do we need? Lancet 388, 218–220. doi: 10.1016/S0140-6736(16)31079-0
Lee, E.-J., Cho, Y.-H., Kim, H.-S., Ahn, B.-E., and Roe, J.-H. (2004). Regulation of sigmaB by an anti- and an anti-anti-sigma factor in Streptomyces coelicolor in response to osmotic stress. J. Bacteriol. 186, 8490–8498. doi: 10.1128/JB.186.24.8490-8498.2004
Leikoski, N., Liu, L., Jokela, J., Wahlsten, M., Gugger, M., Calteau, A., et al. (2013). Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem. Biol. 20, 1033–1043. doi: 10.1016/J.CHEMBIOL.2013.06.015
Li, B., Sher, D., Kelly, L., Shi, Y., Huang, K., Knerr, P. J., et al. (2010). Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl. Acad. Sci. 107, 10430–10435. doi: 10.1073/PNAS.0913677107
Makitrynskyy, R., Ostash, B., Tsypik, O., Rebets, Y., Doud, E., Meredith, T., et al. (2013). Pleiotropic regulatory genes bldA, adpA and absB are implicated in production of phosphoglycolipid antibiotic moenomycin. Open Biol. 3:130121. doi: 10.1098/rsob.130121
Martens, E., and Demain, A. L. (2017). The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 70, 520–526. doi: 10.1038/ja.2017.30
Mitra, A., Kesarwani, A. K., Pal, D., and Nagaraja, V. (2011). WebGeSTer DB—a transcription terminator database. Nucleic Acids Res. 39, D129–D135. doi: 10.1093/nar/gkq971
Myronovskyi, M., Welle, E., Fedorenko, V., and Luzhetskyy, A. (2011). ß-Glucuronidase as a sensitive and versatile reporter in Actinomycetes. Appl. Environ. Microbiol. 77, 5370–5383. doi: 10.1128/AEM.00434-11
Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R. (2003). A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal Chem 75, 4646–4658. doi: 10.1021/ac0341261
Newman, D. J., and Cragg, G. M. (2016). Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. doi: 10.1021/acs.jnatprod.5b01055
Nguyen, H. T., Wolff, K. A., Cartabuke, R. H., Ogwang, S., and Nguyen, L. (2010). A lipoprotein modulates activity of the MtrAB two-component system to provide intrinsic multidrug resistance, cytokinetic control and cell wall homeostasis in Mycobacterium. Mol. Microbiol. 76, 348–364. doi: 10.1111/j.1365-2958.2010.07110.x
O’Rourke, S., Widdick, D., and Bibb, M. (2017). A novel mechanism of immunity controls the onset of cinnamycin biosynthesis in Streptomyces cinnamoneus DSM 40646. J. Ind. Microbiol. Biotechnol. 44, 563–572. doi: 10.1007/s10295-016-1869-9
Osman, D., and Cavet, J. S. (2010). Bacterial metal-sensing proteins exemplified by ArsR-SmtB family repressors. Nat. Prod. Rep. 27, 668–680. doi: 10.1039/b906682a
Otaka, T., and Kaji, A. (1976). Mode of Action of Bottromycin A2. Release of aminoacyl- or peptidyl-tRNA from ribosomes. J. Biol. Chem. 251, 2299–2306.
Otaka, T., and Kaji, A. (1981). Mode of action of bottromycin A2: effect on peptide bond formation. FEBS Lett. 123, 173–176. doi: 10.1016/0014-5793(81)80280-3
Otaka, T., and Kaji, A. (1983). Mode of action of bottromycin A2: effect of bottromycin A2 on polysomes. FEBS Lett. 153, 53–59. doi: 10.1016/0014-5793(83)80118-5
Paget, M. S., Chamberlin, L., Atrih, A., Foster, S. J., and Buttner, M. J. (1999). Evidence that the extracytoplasmic function sigma factor sigmaE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J. Bacteriol. 181, 204–211. doi: 10.1128/jb.181.1.204-211.1999
Quistgaard, E. M., Low, C., Guettou, F., and Nordlund, P. (2016). Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132. doi: 10.1038/nrm.2015.25
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rigden, D. J. (2011). Ab initio modeling led annotation suggests nucleic acid binding function for many DUFs. Omi. A J. Integr. Biol. 15, 431–438. doi: 10.1089/omi.2010.0122
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rogers, T. O., and Birnbaum, J. (1974). Biosynthesis of fosfomycin by Streptomyces fradiae. Antimicrob. Agents Chemother. 5, 121–132. doi: 10.1128/aac.5.2.121
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual, 2nd editio Edn. New York, NY: Cold Spring Harbor Laboratory Press.
Santos-Aberturas, J., Chandra, G., Frattaruolo, L., Lacret, R., Pham, T. H., Vior, N. M., et al. (2019). Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucleic Acids Res. 47, 4624–4637. doi: 10.1093/nar/gkz192
Schwalen, C. J., Hudson, G. A., Kosol, S., Mahanta, N., Challis, G. L., and Mitchell, D. A. (2017). In vitro biosynthetic studies of bottromycin expand the enzymatic capabilities of the YcaO superfamily. J. Am. Chem. Soc. 139, 18154–18157. doi: 10.1021/jacs.7b09899
Severinov, K., Semenova, E., Kazakov, A., Kazakov, T., and Gelfand, M. S. (2007). Low-molecular-weight post-translationally modified microcins. Mol. Microbiol. 65, 1380–1394. doi: 10.1111/j.1365-2958.2007.05874.x
Sherwood, E. J., and Bibb, M. J. (2013). The antibiotic planosporicin coordinates its own production in the actinomycete Planomonospora alba. Proc. Natl. Acad. Sci. U.S.A. 110, E2500–E2509. doi: 10.1073/pnas.1305392110
Shimamura, H., Gouda, H., Nagai, K., Hirose, T., Ichioka, M., Furuya, Y., et al. (2009). Structure determination and total synthesis of bottromycin A2: a potent antibiotic against MRSA and VRE. Angew. Chem Int. Ed. 48, 914–917. doi: 10.1002/anie.200804138
Shimizu, K. (2016). “Metabolic regulation and coordination of the metabolism in bacteria in response to a variety of growth conditions,” in Bioreactor Engineering Research and Industrial Applications I: Cell Factories, eds Q. Ye, J. Bao, and J.-J. Zhong (Berlin: Springer), 1–54. doi: 10.1007/10_2015_320
Sikandar, A., Franz, L., Melse, O., Antes, I., and Koehnke, J. (2019). Thiazoline-specific amidohydrolase PurAH is the gatekeeper of bottromycin biosynthesis. J. Am. Chem. Soc. 141:9748. doi: 10.1021/jacs.8b12231
Solovyev, V., and Salamov, A. (2011). “Automatic annotation of microbial genomes and metagenomic sequences,” in Metagenomics & its Applications in Agriculture, Biomedicine & Environmental Studies, ed. R. W. Li (New York, NY: Nova Science Publishers Inc), 61–78.
Som, N. F., Heine, D., Holmes, N. A., Munnoch, J. T., Chandra, G., Seipke, R. F., et al. (2017). The conserved actinobacterial two-component system MtrAB coordinates chloramphenicol production with sporulation in Streptomyces venezuelae NRRL B-65442. Front. Microbiol. 8:1145. doi: 10.3389/fmicb.2017.01145
Stajich, J. E., Block, D., Boulez, K., Brenner, S. E., Chervitz, S. A., Dagdigian, C., et al. (2002). The Bioperl toolkit: perl modules for the life sciences. Genome Res. 12, 1611–1618. doi: 10.1101/gr.361602
Tacconelli, E., and Magrini, N. (2017). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Geneva: World Health Organization.
Thomas, G., Coutts, G., and Merrick, M. (2000). The glnKamtB operon. A conserved gene pair in prokaryotes. Trends Genet. 16, 11–14. doi: 10.1016/S0168-9525(99)01887-9
Tyanova, S., Temu, T., and Cox, J. (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319. doi: 10.1038/nprot.2016.136
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3–new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Vior, N. M., Olano, C., García, I., Méndez, C., and Salas, J. A. (2014). Collismycin a biosynthesis in Streptomyces sp. CS40 is regulated by iron levels through two pathway-specific regulators. Microbiology 160, 467–478. doi: 10.1099/mic.0.075218-0
Wade, J. T. (2015). Where to begin? Mapping transcription start sites genome-wide in Escherichia coli. J. Bacteriol. 197, 4–6. doi: 10.1128/JB.02410-14
Wagner, G. P., Kin, K., and Lynch, V. J. (2012). Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285. doi: 10.1007/s12064-012-0162-3
Waisvisz, J. M., van der Hoeven, M. G., van Peppen, J., and Zwennis, W. C. M. (1957). Bottromycin. I. A new sulfur-containing antibiotic. J. Am. Chem. Soc. 79, 4520–4521. doi: 10.1021/ja01573a072
Waldvogel, E., Herbig, A., Battke, F., Amin, R., Nentwich, M., Nieselt, K., et al. (2011). The PII protein GlnK is a pleiotropic regulator for morphological differentiation and secondary metabolism in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 92, 1219–1236. doi: 10.1007/s00253-011-3644-1
Wilkinson, C. J., Hughes-Thomaz, Z. A., Martin, C. J., Bohm, I., Mironenko, T., Deacon, M., et al. (2002). Increasing the efficiency of heterologous promoter in actinomycetes. J. Mol. Microbiol. Biotechnol. 4, 417–426.
Wu, P., Pan, H., Zhang, C., Wu, H., Yuan, L., Huang, X., et al. (2014). SACE-3986, a TetR family transcriptional regulator, negatively controls erythromycin biosynthesis in Saccharopolyspora erythraea. J. Ind. Microbiol. Biotechnol. 41, 1159–1167. doi: 10.1007/s10295-014-1449-9
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yang, X., Lennard, K. R., He, C., Walker, M. C., Ball, A. T., Doigneaux, C., et al. (2018). A lanthipeptide library used to identify a protein–protein interaction inhibitor. Nat. Chem. Biol. 14, 375–380. doi: 10.1038/s41589-018-0008-5
Yoo, Y. J., Hwang, J., Shin, H., Cui, H., Lee, J., and Yoon, Y. J. (2015). Characterization of negative regulatory genes for the biosynthesis of rapamycin in Streptomyces rapamycinicus and its application for improved production. J. Ind. Microbiol. Biotechnol. 42, 125–135. doi: 10.1007/s10295-014-1546-9
Zhang, P., Wu, H., Chen, X. L., Deng, Z., Bai, L., and Pang, X. (2014). Regulation of the biosynthesis of thiopeptide antibiotic cyclothiazomycin by the transcriptional regulator SHJG8833 in Streptomyces hygroscopicus 5008. Microbiology 160, 1379–1392. doi: 10.1099/mic.0.076901-0
Zhang, P., Wu, L., Zhu, Y., Liu, M., Wang, Y., Cao, G., et al. (2017). Deletion of MtrA inhibits cellular development of Streptomyces coelicolor and alters expression of developmental regulatory genes. Front. Microbiol. 8:2013. doi: 10.3389/fmicb.2017.02013
Keywords: Streptomyces, bottromycin, RiPP, antibiotic, regulation, transcription, RNA-seq, natural product
Citation: Vior NM, Cea-Torrescassana E, Eyles TH, Chandra G and Truman AW (2020) Regulation of Bottromycin Biosynthesis Involves an Internal Transcriptional Start Site and a Cluster-Situated Modulator. Front. Microbiol. 11:495. doi: 10.3389/fmicb.2020.00495
Received: 07 August 2019; Accepted: 06 March 2020;
Published: 26 March 2020.
Edited by:
Yinhua Lu, Shanghai Normal University, ChinaReviewed by:
Weishan Wang, Institute of Microbiology (CAS), ChinaDi Sun, Jiangsu Normal University, China
Copyright © 2020 Vior, Cea-Torrescassana, Eyles, Chandra and Truman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew W. Truman, YW5kcmV3LnRydW1hbkBqaWMuYWMudWs=