 Emilie Macke1
Emilie Macke1 Martijn Callens1,2
Martijn Callens1,2 Francois Massol3,4
Francois Massol3,4 Isabel Vanoverberghe1
Isabel Vanoverberghe1 Luc De Meester5
Luc De Meester5 Ellen Decaestecker1*
Ellen Decaestecker1*- 1Aquatic Biology, IRF Life Sciences, KU Leuven, Kortrijk, Belgium
- 2Centre d’Ecologie Fonctionnelle et Evolutive, UMR CNRS 5175, Montpellier, France
- 3CNRS, Lille–Sciences et Technologies, UMR 8198 Evo-Eco-Paleo, SPICI Group, Villeneuve-d’Ascq, France
- 4University of Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019–UMR 8204–CIIL–Center for Infection and Immunity of Lille, Lille, France
- 5Aquatic Ecology, Evolution and Conservation, KU Leuven, Leuven, Belgium
In spite of the growing interest in the role of the gut microbiome (GM) in host physiology and health, the mechanisms governing its assembly and its effects on the environment are poorly understood. In this article, we show that the host genotype and the GM of Daphnia influence the community structure of the surrounding bacterioplankton (BPK). When Daphnia genotypes were placed in an identical environment, both the GM and BPK showed a genotype and diet-dependent taxonomic composition. Overall, the GM strongly differed from the BPK in taxonomic composition and was characterized by a lower α-diversity, suggesting a selective rejecting of bacteria from the regional species pool. In a microbiome transplant experiment, the assembly of both the GM and BPK was strongly affected by the host genotype and the inoculum to which germ-free Daphnia were exposed. The combination of these results suggests a strong interaction between the host genotype, its GM and free-living microbial communities. Currently, it is generally assumed that an animal’s diet has a strong effect on the animal’s GM, but only a negligible (if any) effect on the surrounding environment. However, our results indicate that the diet/microbiome inocula have a small effect on the gut community and a large effect on the community in the surrounding environment. This structuring genotype × microbiome × environment effect is an essential prerequisite that could indicate that microbiomes play an important role in eco-evolutionary processes.
Introduction
The composition of the gut microbiome (GM) is characterized by a strong host-specific dominance of particular taxa, but with substantial variability over time within one host individual (Dabrowska and Witkiewicz, 2016; Adair and Douglas, 2017; Zhang et al., 2017). A key challenge is to decipher the factors shaping the host’s microbiome community and the relative contribution of different deterministic and stochastic processes to variation in its community structure (Alberdi et al., 2016; Adair and Douglas, 2017). So far, most studies on GM assembly have focused on within-host processes (Macke et al., 2017b; De Meester et al., 2019). Host-mediated control over the GM composition has been shown to occur in several taxa, most often due to antimicrobial defenses mounted by the host (Tasiemski et al., 2015) or developmentally regulated anatomical barriers to colonization (Ohbayashi et al., 2015; Lanan et al., 2016; Norman and Koskella, 2017). Behavioral traits such as selective feeding or sociality also contribute to shaping the GM by modifying the pool of potential colonizing microbes (Grossart et al., 2010; Koch and Schmid-Hempel, 2011; Mushegian et al., 2019). These deterministic processes partly depend on the host’s genetic background, suggesting an association between host genotype and GM composition (Macke et al., 2017a; Mushegian et al., 2019). On the other hand, microbe-microbe interactions, including competition and syntrophic interactions, have also been shown to affect the persistence of microbes within the gut (Adair and Douglas, 2017; Foster et al., 2017; Macke et al., 2017a). These interactions are strongly mediated by the host’s diet, which determines the available resources in the gut and, accordingly, what microbial species and types of metabolism can become dominant (Carmody et al., 2015; Foster et al., 2017).
The GM does not exist in isolation, but can contribute to a wider metacommunity maintained by the transmission between individual hosts and dispersal between host-associated and free-living microbial communities (Eckert et al., 2016; Murall et al., 2017). Dispersal of microbes is likely to be key in maintaining microbiome diversity and reducing variation in microbiome composition between individual hosts (Alberdi et al., 2016; Adair and Douglas, 2017; Macke et al., 2017b). Hence, processes involved in structuring the GM might often involve multiple spatial scales (Penczykowski et al., 2016; Norman and Koskella, 2017; Laine et al., 2019). In this way, the GM could be regarded as a local community colonized from a regional species pool, i.e., the microbial pool present in the environment (Shapira, 2016; Foster et al., 2017). The composition of this regional pool depends on environmental conditions, but might also be directly affected by hosts and their microbiomes (Wong et al., 2015; Callens et al., 2018). The interaction between hosts, their microbiomes and free-living communities is expected to play a crucial role in microbiome assembly. This metacommunity perspective on microbiome assembly might be particularly relevant for aquatic organisms and bacterioplankton (BPK), which are in close and continuous contact and are therefore expected to have strong reciprocal effects. If these interactions prove to be host genotype-dependent to a certain extent, then they may also be an important missing link in eco-evolutionary theory (Macke et al., 2017b; De Meester et al., 2019).
Combining metabarcoding with a GM transplant approach, we investigated the interplay between host genotype, GM, and BPK in the freshwater crustacean Daphnia magna. In a first experiment, we examined the effects of Daphnia genotype and diet on both the GM and BPK composition. Nine Daphnia genotypes were exposed to an environmental microbial source and were submitted to either a green algal or a cyanobacterial diet for approximately 58 generations. GM and BPK compositions were subsequently determined through next-generation sequencing and were found to be affected by host genotype and diet. In a second experiment, we examined the relative contribution of the microbial source versus Daphnia genotype-dependent sorting processes to GM and BPK assembly by performing a GM transplant. Germ-free Daphnia of two genotypes were exposed to different GM inocula. Two weeks after the transplant, GM and BPK compositions were assessed through next-generation sequencing, revealing a combined effect of microbial source and host genotype on the structuring of both communities.
MATERIALS AND METHODS
Daphnia magna Genotypes
Nine Daphnia genotypes (G1–G9) were used in Experiment 1. A subset of these genotypes (G2, G6, and G9) was used in Experiment 2. G1, G4, and G9 were originally isolated from an 8.7 ha shallow man-made pond located in Oud-Heverlee, Belgium. Clonal lineages were established from resting eggs sampled in three sediment core sections (18–21 cm, 11–14 cm, and top 3 cm depth, respectively), corresponding to three time periods (1970–1972, 1976–1979, and 1988, respectively). G3 was hatched from the top 3 cm sediment of a small, fishless and mesotrophic pond (350 m2) located near Knokke, Belgium (51°20′05.62″ N, 03°20′53.63″ E). G5–G8 were hatched from the top 3 cm sediment of a 3.7 ha eutrophic pond containing fish and located in Heverlee, Belgium (50°51′47.82″ N, 04°43′05.16″ E). G2 was isolated from Bysjön lake in Sweden. All genotypes were maintained in the laboratory under standardized stock conditions for several years prior to the experiment, in aged tap water, and for the two last years in re-constituted freshwater (ADaM medium) (Klüttgen et al., 1994), at a temperature of 19 ± 1°C and under a 16:8 h light:dark cycle, in 500 ml glass jars containing 20–30 individuals. The Daphnia were fed twice a week (0.8 mg C/L) with the green alga Scenedesmus obliquus; the medium was partly refreshed a few times a year and regularly replenished to compensate for evaporation. As all genotypes were maintained in the laboratory for several years prior to the experiment, we consider it unlikely that the microbiota of the stock cultures were dominated by bacteria from the pond of origin. However, we cannot exclude this possibility, given that recent research has indicated that some bacterial species are shared between adults and resting eggs (Mushegian et al., 2018).
Sterile Cultures of Green Algae and Cyanobacteria
The unicellular green alga S. obliquus (hereafter called Scenedesmus or abbreviated as “S”; strain CCAP 276/3A, provided by the Culture Collection of Algae and Protozoa, United Kingdom) and the unicellular cyanobacteria Microcystis aeruginosa (hereafter called Microcystis or abbreviated as “M”; strain PCC 7806, provided by the Pasteur Culture Collection, Institut Pasteur, Paris, France) were used to feed the Daphnia. Scenedesmus and Microcystis differ in their nutritional value and digestibility (von Elert et al., 2003). The Microcystis strain used in the present study produces toxins and bioactive compounds such as microcystins (Rohrlack et al., 2001). The Scenedesmus and Microcystis were grown in WC medium (Guillard and Lorenzen, 1972) and modified WC medium (WC medium without Tris), respectively, under sterile conditions at 20 ± 2°C and a light:dark cycle of 16:8 h in 2 L glass bottles with constant stirring and aeration. Filters (0.22 μm) were placed at the input and the output of the aeration system to avoid bacterial contamination. Algae were harvested weekly in their early stationary phase. Axenity was checked on LB medium agar plates. The ash-free dry weight of the cultures was determined according to Moheimani et al. (2013).
Experiment 1. Effects of Daphnia Genotype and Diet on Bacterioplankton and Gut Microbiome Composition
Previous research has already examined the GM composition of four genotypes (G1, G3, G7, and G8, see Macke et al., 2017a, Experiment 1) but in our experiment we extended the GM characterization from four to nine genotypes (G1–G9) and followed the population for a much longer time. We also included BPK analyses.
For each of the nine Daphnia genotypes, three maternal lines were cultured in ADaM medium (Klüttgen et al., 1994) in 2 L experimental glass jars, under standardized conditions (19 ± 1°C; 16:8 h light:dark cycle). They were fed daily with saturating amounts of Scenedesmus. Medium was refreshed once a week. When a sufficient number of individuals was reached, 120 juveniles were sampled from each maternal line and divided into two 2 L experimental jars (each containing 60 individuals). The first jar was fed a Scenedesmus diet, while the second was fed a Microcystis diet. In total, there were 54 populations (9 genotypes × 2 diets × 3 replicates). Algae were provided every other day with a final carbon concentration of approximately 1.5 mg C.L–1 according to (Macke et al., 2017a). Medium was refreshed every other week. Water from a pond situated in the ECOLAB on the campus (Kortrijk, Belgium, 50°48′30.3″ N, 3°17′38.0″ E) was added to the ADaM medium (15% of the final volume) every 2 weeks in order to provide a large diversity of bacteria and optimal growth conditions for the Daphnia, as detected in (Callens et al., 2016, 2018).
After 1.5 years (circa 58 generations) of exposure to the two types of diet, the composition and diversity of the BPK and GM community were assessed through next-generation sequencing of 16S rRNA. To obtain GM samples, 20 adult Daphnia were collected from each population and placed in a sterile (i.e., autoclaved) ADaM medium for 24 h to reduce the number of contaminating food particles within the gut (Callens et al., 2016). The Daphnia guts were subsequently extracted using sterilized dissecting needles under a stereomicroscope and jointly placed in 1.5 ml Eppendorf tubes containing 10 μL of deionized sterile water. For BPK characterization, 100 ml of medium was sampled from each population and filtered with a 0.22 μm syringe filter. The filter was subsequently placed in a 1.5 ml Eppendorf tube. GM and BPK samples were immediately placed at −20°C until further processing.
Experiment 2. Effects of Microbial Source and Host Genotype on Bacterioplankton and Gut Microbiome Assembly in a Microbiome Transplant Experiment
Previous research has already examined the GM composition of the recipient genotypes (Macke et al., 2017a, Experiment 3). Here, we extended the analyses to the surrounding BPK. Daphnia genotypes G2 and G9 were used as GM donors in the transplant experiment. For each genotype, six replicated populations were grown in 2 L jars and divided into two groups submitted to different experimental conditions in order to maximize variation in GM composition. In the first condition, the diet was composed of Microcystis, and filtered water from a pond situated on the university campus (ECOLAB, Kortrijk, Belgium, 50°48′30.3″ N, 3°17′38.0″ E) was added to the ADaM medium (5% of the final volume) every 2 to 3 weeks, to provide Daphnia with a diverse pool of microbes. In the second condition, the diet was composed of Scenedesmus and water from another pond on campus was added to provide Daphnia with another set of environmental bacteria. In this experiment, we made diverse inocula to examine whether Daphnia genotypes would consistently select for particular strains from these different inocula. However, by doing so the diet effect might be confounded by a pond effect, which is in line with our focus on microbiome effect in Experiment 2/shifting our focus from the diet effect to the microbiome inoculum effect in Experiment 2. Algae were provided every other day as in Macke et al. (2017a). After 8 weeks of exposure to the two conditions, 60 adult Daphnia individuals (i.e., 20 per replicate) were collected for each genotype and their GM was extracted. This way, we obtained four different microbial inocula, each one containing the GM of 60 Daphnia and representing the factorial combinations of donor genotypes (G2 and G9) and diets (Microcystis and Scenedesmus). These inocula are referred to as G2M, G2S, G9M, and G9S.
Twelve maternal lines were then created for each recipient genotype (G2 and G6) and grown separately in 500 ml jars for several generations on a diet of saturating amounts of Scenedesmus, under the conditions described above. Due to the loss of the G9 clone during the preparation of the transplant experiment, no full reciprocal transplant was possible, therefore G9 was replaced by G6. From each maternal line, germ-free individuals were obtained as described in Macke et al. (2017a). For each maternal line of each recipient clone, 40 germ-free juveniles were jointly placed in a 50 ml centrifuge tube containing 45 ml of sterile ADaM. The 12 tubes were divided into four groups, each one inoculated with either a “G2M,” “G2S,” “G9M,” or “G9S” microbial inoculum (on average one crushed gut for four germ-free recipient Daphnia) and 1 mg C.L–1 of Scenedesmus. Daphnia were left in these conditions for 2 days to ensure that they ingested enough bacteria and were subsequently transferred to a 500 ml jar containing fresh sterile ADaM medium. They were then submitted to an 80% Microcystis – 20% Scenedesmus diet with a final carbon concentration of 1 mg C.L–1. These algae were provided every day. Pond water was not added in this transplant experiment. In total, there were 24 recipient populations (2 recipient genotypes × 4 microbiome inocula × 3 replicates) with 40 Daphnia juveniles in each population. Two weeks after the transplant, a sample of medium (100 ml) and ten Daphnia guts were collected from each recipient population for BPK and GM characterization, using the protocols described in (Macke et al., 2017a).
Determination of Bacterioplankton and Gut Microbiome Community Composition and Diversity
DNA was extracted using a PowerSoil DNA Isolation Kit (MO BIO Laboratories) and dissolved in 20 μL MilliQ water. For Experiment 1, we obtained 108 DNA samples (9 genotypes × 2 diets × 3 replicates × 2 community types). For Experiment 2, we obtained 48 samples (2 recipient genotypes × 4 microbiome inocula × 3 replicates × 2 community types). DNA yield was determined using a Qubit dsDNA HS Assay Kit (Invitrogen) on 3 μL of the sample. Four samples from Experiment 1 (GM of G2–M, BPK of G5–M, and BPK of G5–S) and one sample from Experiment 2 (Recipient G2 – Inoculum G2S) did not contain sufficient DNA and were discarded. Because of the initially low bacterial DNA concentration in some samples, a nested PCR was applied to increase specificity and amplicon yield (Callens et al., 2016). The full length 16S rRNA gene was amplified with primers 27F and 1492R on 10 ng of template (94°C – 30 s; 50°C – 45 s; 68°C – 90 s; 30 cycles) by using a high-fidelity Pfx Polymerase (Life Technologies). PCR products were subsequently purified with the QIAquick PCR Purification Kit (Qiagen). To obtain dual-index amplicons of the V4 region, a second amplification was performed on 5 μL PCR product using primers 515F and 806R for 30 cycles (94°C – 30 s; 55°C – 30 s; 68°C – 60 s). Both primers contained an Illumina adapter and an 8-nt barcode at the 5′-end. For each sample, PCRs were performed in triplicate, pooled and gel purified using the QIAquick Gel Extraction Kit (Qiagen). An equimolar library was prepared by normalizing amplicon concentrations with a SequalPrep Normalization Plate Kit (Applied Biosystems) and subsequent pooling. Amplicons were sequenced using a v2 PE500 kit with custom primers (Kozich et al., 2013) on the Illumina MiSeq platform (KU Leuven Genomics Core) producing 2 × 250-nt paired-end reads.
Sequence reads were processed using R 3.3.2 (R Core Team, 2018) as previously reported by Callahan et al. (2016b). Sequences were trimmed (the first 10 nucleotides and all nucleotides from position 190 onward were removed) and filtered (maximum two expected errors per read) on paired ends jointly. Sequence variants were inferred using the high-resolution DADA2 method (Callahan et al., 2016a) and chimeras were removed. Taxonomy was assigned with a naive Bayesian classifier using the RDP v14 training set. Amplicon sequence variants (ASVs, hereafter called OTUs) which had no taxonomic assignment at phylum level or were assigned as “Chloroplast” or “Cyanobacteria” were removed from the dataset. For Experiment 1, the final dataset contained 2,887,669 reads, on average 27,766 reads per sample (minimum = 4,133 reads, maximum = 63,594 reads). For Experiment 2, the final dataset contained 1,500,800 reads, on average 29,427 reads per sample (min. = 5,804 reads, max. = 78,154 reads).
Statistical analyses and plots were performed using R 3.3.2 (R Core Team, 2018). To investigate differences in community composition among samples (β-diversity), Bray–Curtis dissimilarity indices were calculated and plotted using Principal Coordinates Analysis (PCoA) with the phyloseq package (McMurdie and Holmes, 2013). For Experiment 1, the effects of diet, genotype, community type (i.e., BPK and GM) and their interactions on β-diversity were assessed through a PERMANOVA, using the Adonis function of the vegan package (tested using 999 permutations of rows). For Experiment 2, we also used a PERMANOVA to examine how microbiome inoculum, recipient genotype, community type, and their interactions might affect community composition. For both experiments, analyses were carried out both for GMs and BPKs jointly and for each of them separately. Samples and taxa were clustered hierarchically based on Bray–Curtis dissimilarity indices, using the UPGMA (unweighted pair group method with arithmetic mean) agglomeration method, with the fastcluster package. A heat map of OTUs dominance in the different samples was obtained using the gplots package. To identify how bacterial classes differed among community types and diets, OTUs were grouped at class level, and differential abundance analyses were performed with the Bioconductor package DESeq2 (Love et al., 2014). Venn diagrams illustrating overlap of OTUs among community types and diets were produced using Venny 2.1.0 online freeware (Oliveros, 2007).
For both experiments, we investigated the correlation between variation in BPK composition on the one hand and variation in GM composition on the other hand by performing a Mantel test to compare the Bray–Curtis dissimilarity matrices of the two community types (permutations = 999; statistics based on Pearson’s correlation). Ordinations (i.e., PCoA) of BPK and GM were compared through a Procrustes analysis of the first two axes, and significance was assessed though permutation tests (999 permutations) with the protest function of the vegan package.
For α-diversity, OTU richness (total number of OTUs) was calculated using the vegan package. In Experiment 1, the effects of diet, genotype, community type, and their interactions on OTU richness were assessed through generalized linear models (GLM) with a Poisson distribution. p-values were adjusted for multiple comparisons through the control of the false discovery rate (FDR). When interactions with community type have proven to be significant, analyses were also performed separately for BPK and GM. Pairwise comparisons among genotypes and diets were performed by contrasting least-squares means. For Experiment 2, we also used GLM to test for the effects of microbiome inoculum, recipient genotype, community type, and their interactions on OTU richness. Similar analyses, which are presented in the Supplementary Material, were performed on the Shannon diversity index.
Results and Discussion
Our results show that Daphnia GM is not assembled randomly. There were strong differences between microbial communities in the Daphnia gut (GM) and in the surrounding water (BPK). In both experiments, the taxonomic composition of microbial communities (Figures 1, 2) differed between GM and BPK (Figures 3A, 4A and Tables 1a, 2a), mainly due to an overrepresentation of Actinobacteria (mostly Microbacteriaceae) and Flavobacteriia (Flavobacterium sp.) in BPK compared to GM (all p < 0.001 by Wald test; Supplementary Table S1). This suggests that these bacteria do not preferentially colonize the Daphnia gut environment. In Experiment 1, in which the cultures were regularly seeded with a diverse environmental sample of BPK, OTU richness was lower in GM than in BPK (p < 0.0001, GLM; Figure 3D and Supplementary Table S2a), suggesting a selection of particular BPK strains in the GM. β-proteobacteria (mostly Comamonadaceae) were over-represented in GM in Experiment 1 (p < 0.05 by Wald test; Supplementary Table S1a). The Comamonadaceae family is a major constituent of the microbiome and induces positive fitness effects in Daphnia (Eckert and Pernthaler, 2014; Peerakietkhajorn et al., 2015; Macke et al., 2017a; Callens et al., 2018; Mushegian et al., 2019). In Experiment 2, in which Daphnia were transferred into sterile water following the transplant so that BPK could only come from the leakage of Daphnia gut-associated microbes, OTU richness was higher in GM than in BPK (p < 0.0001, GLM; Figure 4D and Supplementary Table S3). This indicates that not all bacteria growing in Daphnia GM can thrive in an open environment. Important to note is that the GM is not the only potential host-derived source of microbiota. Daphnia contain microbiota on other structures as well, such as the carapace, the filter apparatus, etc. (Qi et al., 2009; Eckert and Pernthaler, 2014; Freese and Schink, 2011; Callens et al., 2016), which may explain the difference between GM and BPK. Taken together, our results suggest that under natural conditions with a diverse environmental microbial community, the gut microbiota will be a subset of the taxa occurring in the environment, while the taxa that leak from the GMs into the environment show differential capacities to contribute to the environmental microbial communities. Similar results were observed when considering the Shannon diversity index (Supplementary Tables S4, S5). Figures 3D, 4D suggest that α-diversity values were similar in both experiments for the GM, but not for the BPK, which was more diverse in the non-sterile medium of Experiment 1 than in the sterile medium of Experiment 2.
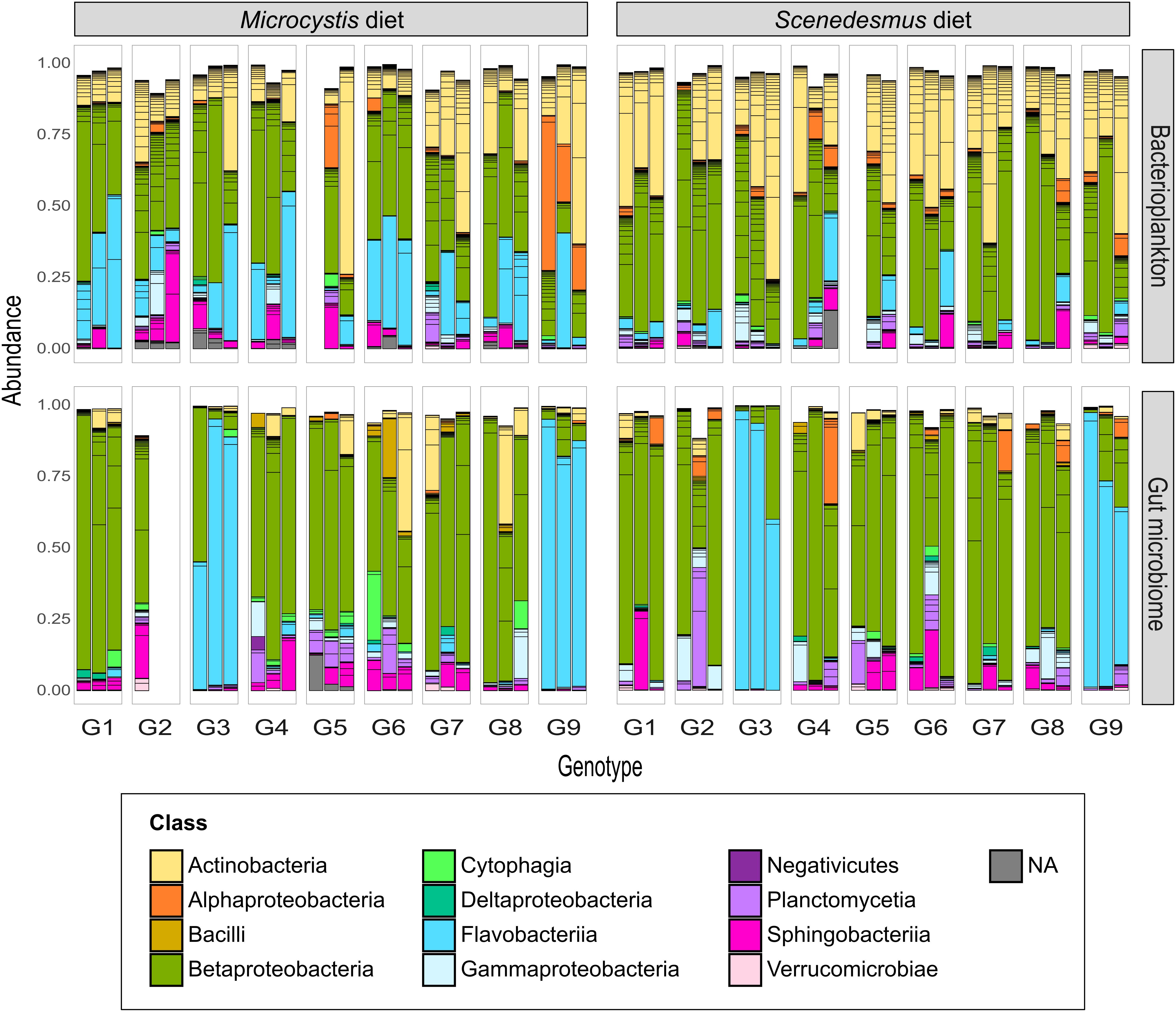
Figure 1. BPK and GM composition across Daphnia genotypes and diets (Experiment 1). Nine Daphnia genotypes (G1–G9) were exposed to either a Microcystis or a Scenedesmus diet. After 58 generations of exposure, both Daphnia GM and BPK were characterized through next generation sequencing (n = 3 replicated populations per genotype × diet combination). Bars indicate the relative abundance of OTUs. Colors indicate bacterial classes. OTUs with an occurrence lower than 1% are not represented.
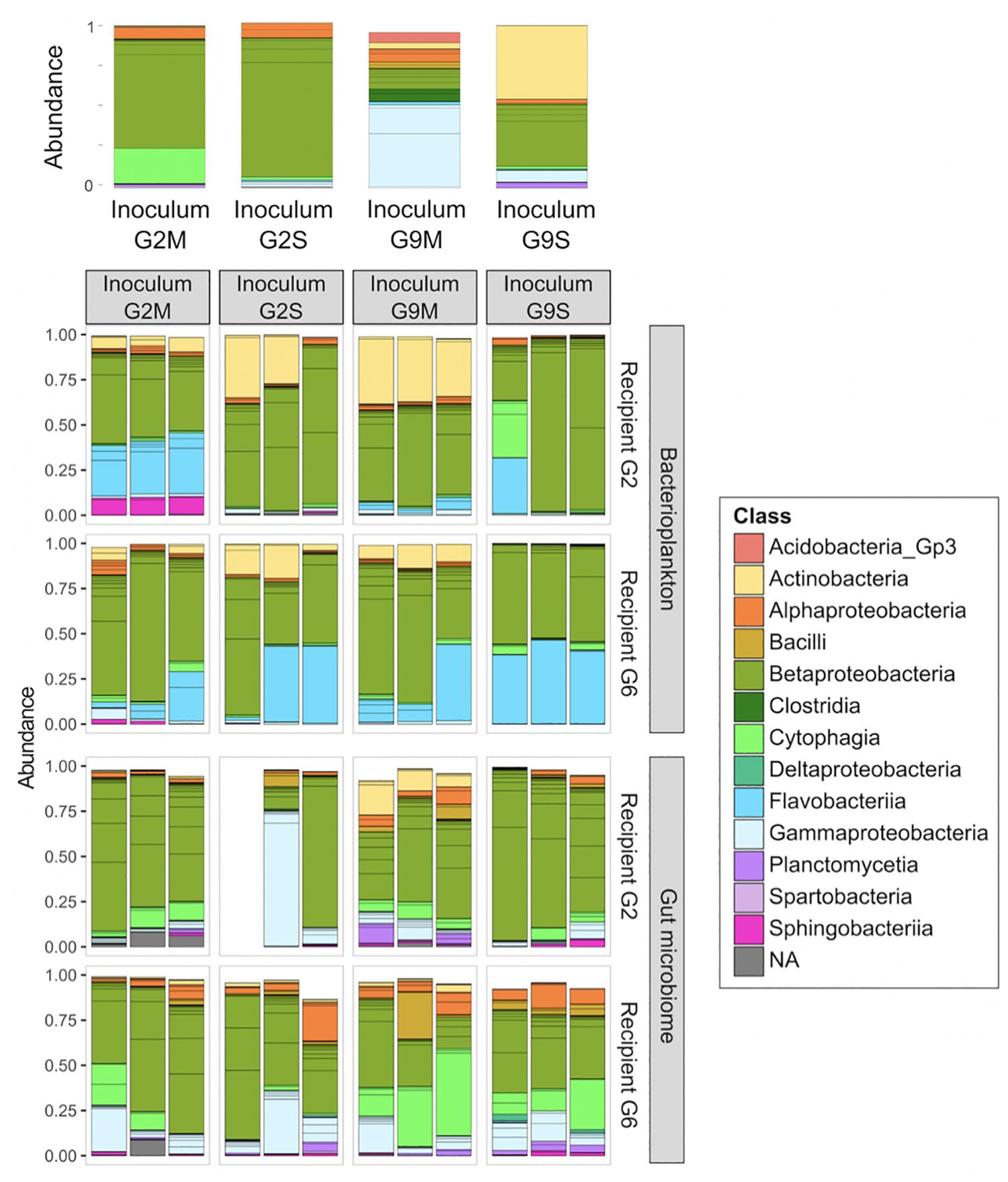
Figure 2. BPK and GM composition in recipient Daphnia genotypes following a GM transplant (Experiment 2). Two germ-free recipient Daphnia genotypes (G2 and G6) were inoculated with four types of microbial inocula (G2M, G2S, G9M, and G9S). Two weeks after the transplant, both Daphnia GM and BPK were characterized through next generation sequencing (n = 3 replicated populations per recipient genotype × inoculum combination). Bars indicate the relative abundance of OTUs. Colors indicate bacterial classes. OTUs with an occurrence lower than 1% are not represented. Upper panel is the GM composition of the inocula.
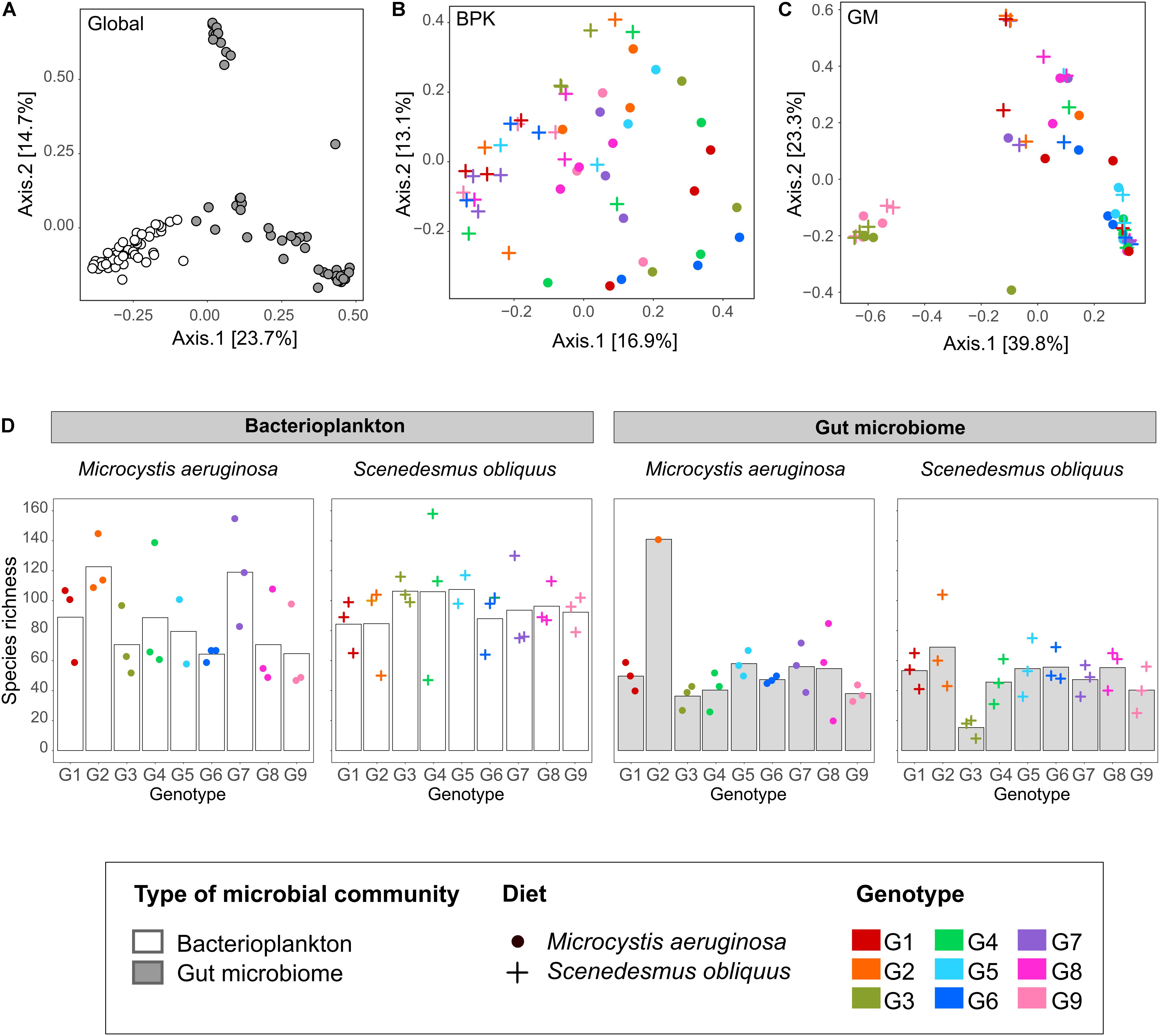
Figure 3. Daphnia genotype and diet effects on GM and BPK community structure (Experiment 1). (A–C) PCoA using Bray–Curtis dissimilarity indices considering all samples simultaneously (A), BPK samples only (B), and GM samples only (C). (D) α-diversity (OTU richness) in GM and BPK when Daphnia genotypes (G1–G9) were exposed to either a Microcystis or a Scenedesmus diet. Bars indicate mean values, points indicate specific values for each population.
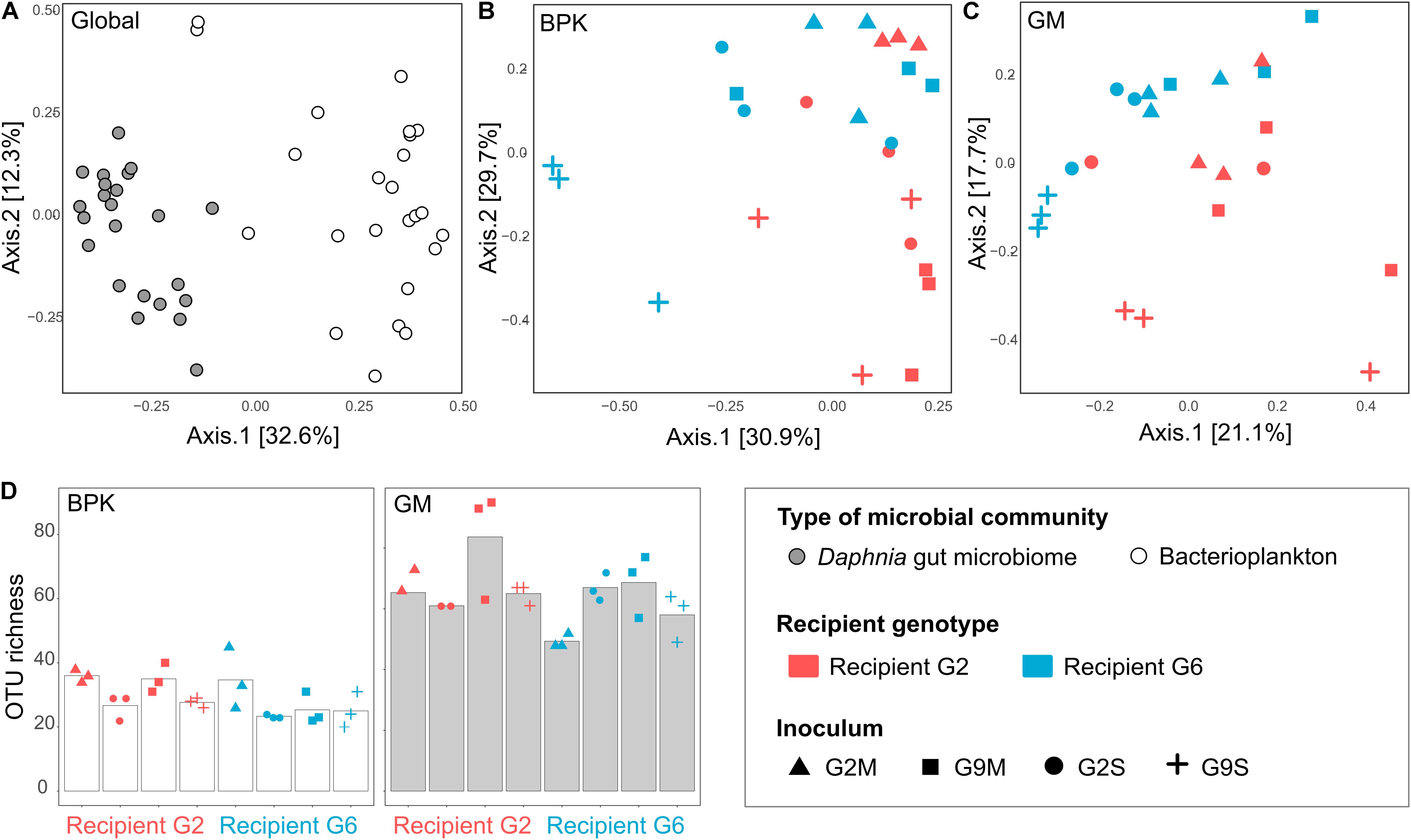
Figure 4. Contribution of recipient Daphnia genotype and microbial source to BPK and GM assembly in the transplant experiment (Experiment 2). (A–C) PCoA of microbial communities 2 weeks after the transplant, using Bray–Curtis dissimilarity indices, when considering all samples simultaneously (A), BPK samples only (B), and GM samples only (C). (D) α-diversity (OTU richness) in the BPK and GM of recipient Daphnia. Bars indicate mean values, points indicate specific values for each population.
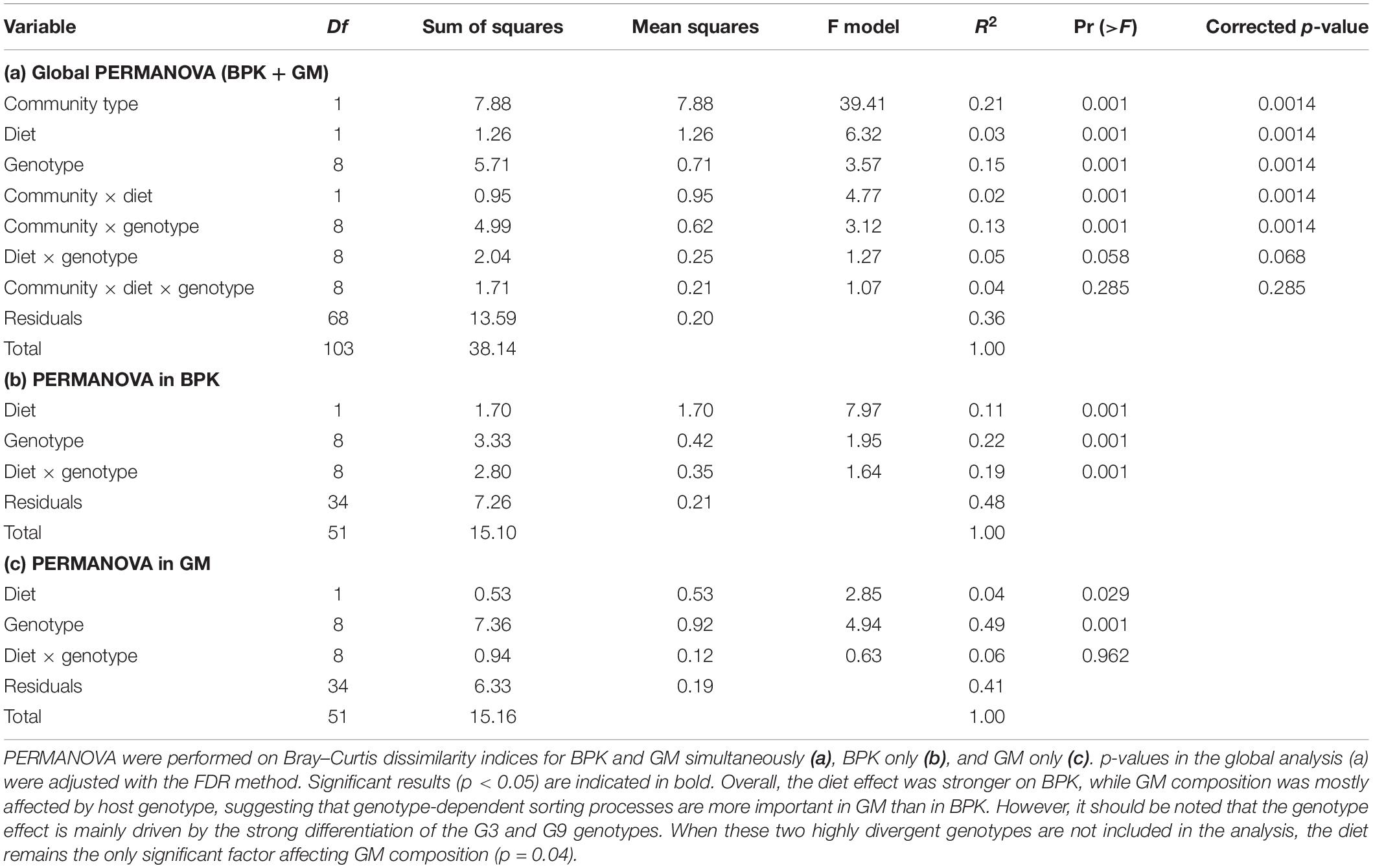
Table 1. Effects of community type, diet, and genotype on the taxonomic composition of microbial communities (Experiment 1).
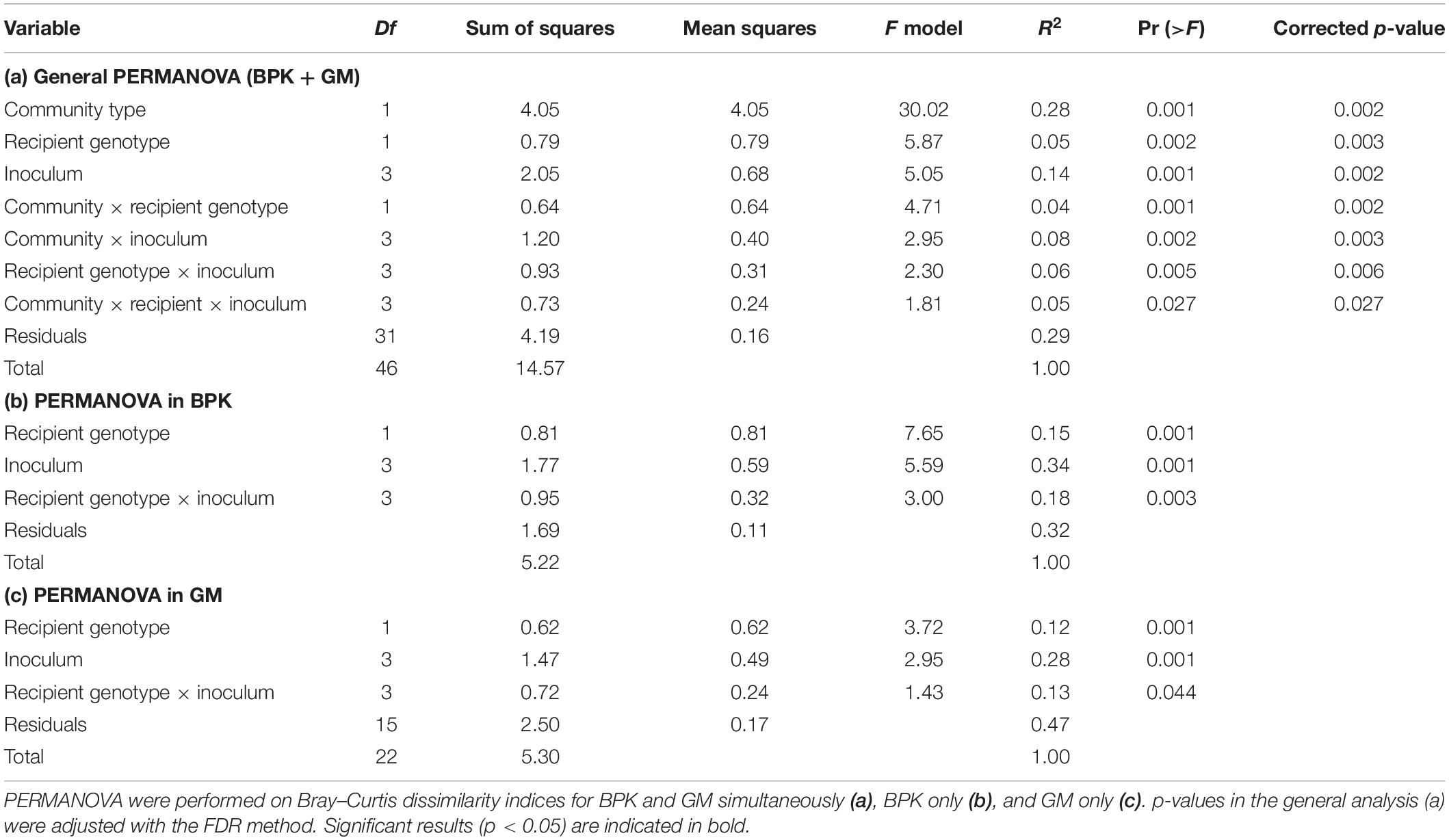
Table 2. Effects of community type, microbiome inoculum, and recipient genotype on the taxonomic composition of microbial communities (Experiment 2).
Another conclusion of our study is that the host genotype influences the composition of both the GM and BPK community. In Experiment 1, after ±58 generations of exposure of Daphnia to a shared environmental microbial pool, both GM and BPK compositions were influenced by Daphnia genotype. In Experiment 2, 2 weeks after a GM transplant, the two recipient genotypes differed in terms of both GM and BPK compositions. Host-mediated structuring of the external microbial environment has already been reported in Drosophila (Wong et al., 2015) and in Daphnia (Degans et al., 2002). Our study reveals that this impact can be genotype-dependent. In this way, our results indicate that the composition of the environmental microbial community is affected by the evolution of the host species (see also terhorst et al., 2014), providing a potentially important avenue for future research in the field of eco-evolutionary dynamics (Hendry, 2017; De Meester et al., 2019). Daphnia-mediated modification of the BPK might further influence the success of species in the freshwater community and induce ecosystem-level effects. In Experiment 1, the overall β-diversity of microbial communities was affected by host diet and genotype as well as by the interaction of these two factors (Table 1a). However, diet and genotype effects on BPK were different from those on GM (i.e., significant interactions with the community type; Table 1a). A Mantel test (R = −0.025, p = 0.68) and Procrustes analysis (Pr: m2 = 0.96, r = 0.19, p = 0.32; Figures 3B,C) revealed no association between community composition in GM and BPK. Focusing on BPK separately, both diet and host genotype had a substantial effect on the taxonomic composition, and as these two factors interacted (Figure 3B and Table 1b), this suggests that the effect of diet varied among genotypes. Figure 3B indicates that the genotype effect was stronger with the Microcystis than with the Scenedesmus diet. Overall, diet had the strongest impact on the proportion of Flavobacteriia and Sphingobacteriia, which were overrepresented in Daphnia on the Microcystis diet compared to those on the Scenedesmus diet (both p < 0.01, by Wald test; Supplementary Table S6). In GM, the taxonomic composition was mainly affected by the genotype, while the diet had a smaller effect (Figure 3C and Table 1c). Flavobacteriia, Cytophagia, Actinobacteria, and Bacilli were all more strongly associated with the Microcystis diet, whereas α- and γ-proteobacteria were more strongly associated with the Scenedesmus diet (all p < 0.05 by Wald test; Supplementary Table S6). The effects of genotype and diet on OTU richness were dependent on the community type (p < 0.0002 for all interactions involving the community type, GLM; Figure 3D and Supplementary Tables S2a, S7). In BPK, OTU richness differed among genotypes (p < 0.0001, GLM; Figure 3D and Supplementary Table S3b), and the diet had a genotype-dependent impact (Diet: p = 0.0042, diet × genotype: p < 0.0001, GLM; Figure 3D and Supplementary Table S2b). In GM, OTU richness was affected by genotype (p < 0.0001, GLM; Figure 3D and Supplementary Table S2c), diet (p = 0.048, GLM; Figure 3D and Supplementary Table S2c) and their interaction (p < 0.0001, GLM; Figure 3D and Supplementary Table S2c). The Shannon diversity index was influenced by host genotype in both BPK and GM (p = 0.005, GLM; Supplementary Tables S4, S8), but it was not influenced by the diet (p = 0.45, GLM; Supplementary Tables S4, S8).
The external microbial source interacts with the Daphnia genotype to shape GM and BPK. In addition to host-dependent processes, our results revealed that GM and BPK are affected by external factors including the diet (Experiment 1) and the external microbial source (Experiment 2). Both factors interacted with host genotype to shape the microbial communities. Overall, the diet effect was stronger in BPK, while GM composition was mostly affected by host genotype, suggesting that genotype-dependent sorting processes are more important in GM than in BPK. Exposure to Microcystis tended to decrease α-diversity in both types of communities and was associated with particular taxonomic groups, mainly Flavobacteria. Flavobacteria contribute to the lysis and degradation of Microcystis cells during cyanobacterial blooms (Maruyama et al., 2003; Kolmonen et al., 2004; Eiler and Bertilsson, 2007; Manage and Premetilake, 2011) and were found to be associated with Microcystis tolerance in Daphnia (Macke et al., 2017a). Interestingly, our results suggest that the structuring effect of host genotype on BPK is stronger with the Microcystis diet. Overall, the taxonomic composition of microbial communities was affected by both recipient genotype and microbiome inoculum (Figures 2, 4B,C and Table 2a). Differences among recipient genotypes and among microbiome inocula, however, varied depending on the community type (p < 0.03 for all interactions involving the community type, PERMANOVA; Table 2a). In both BPK and GM, both recipient genotype and microbiome inoculum affected the community structure, and both factors interacted (Figures 4B,C,5 and Tables 2b,c). The results of a Mantel test (R = 0.38, p = 0.002) and a Procrustes analysis (m2 = 0.59, r = 0.64, p = 0.001; Figures 4B,C) showed that variation in community composition among experimental treatments covaried between BPK and GM. OTU richness differed between the two recipient genotypes, clone G2 having a higher OTU richness than clone G6 for both BPK and GM (p = 0.003, GLM; Figure 4D and Supplementary Table S3). OTU richness was also affected by the microbiome inoculum (p = 0.009, GLM; Figure 4D and Supplementary Table S3), and this effect differed among community types (p = 0.002; Supplementary Table S3). The Shannon diversity index, however, was not affected by these factors (all p > 0.1, GLM; Supplementary Table S5).
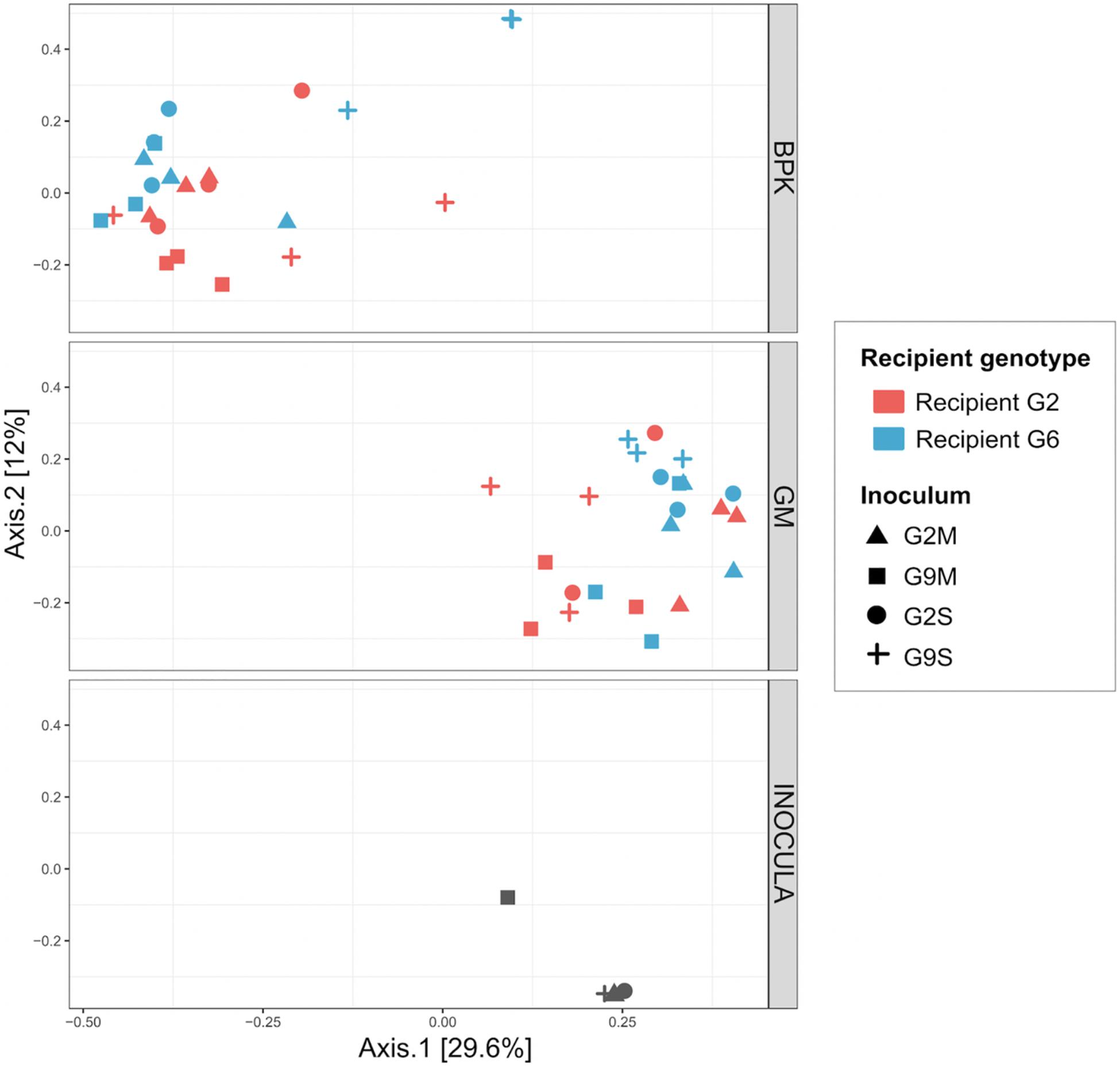
Figure 5. Principal coordinate analysis of microbial communities two weeks after the transplant, using Bray–Curtis dissimilarity indices (Experiment 2). Here, the microbiome inocula used to perform the transplant experiment were included in the analysis. BPK samples, GM samples and microbiome inocula were analyzed together, but are represented on different panels to facilitate interpretation.
Daphnia feeding behavior can play a role in structuring Daphnia-associated microbial communities (Degans et al., 2002; Mushegian et al., 2019). More specifically, by mediating the intake of microbes from the environment, Daphnia grazing activity might directly affect the relative abundance of certain bacterial strains both within the gut and in the BPK. Differential grazing might also influence microbial community composition indirectly, through its effect on phytoplankton. Algae produce exopolysaccharides and metabolites that are used by BPK and may influence their relative abundance (Taylor et al., 2014). By feeding on algae, Daphnia might affect the resources available for bacteria both in the BPK and in the GM and thus affect the community structure. Second, by shaping the intestinal environment, host metabolism and immunity, might contribute to drive GM assembly and favor the growth of specific microbes within the gut (Macke et al., 2017b). These effects may further extend to influence the external environment of Daphnia, for instance through the shedding of immune effectors (Otti et al., 2014), thus affecting the BPK community structure. Third, GM communities might influence environmental microbial communities by leakage of dominant taxa. If hosts are common in the environment, such leakage might be an important force in structuring free-living microbial communities. In Experiment 2, we found a positive correlation between variation in GM composition and variation in BPK composition. This either reflects a shared structuring effect of Daphnia on both GM and BPK, or a direct interaction between GM and BPK. This association was not present in Experiment 1, likely because it was masked in the presence of a more diverse environmental microbial community. Given the low congruence of GM and BPK community structure at the OTU level, it is, however, unlikely that GM contributed strongly to the structuring of BPK via the leakage of microbes, although some seeding cannot be excluded. Another explanation that does not discount the role of leakage of bacterial strains from the gut into the environment could be the different growth rate of bacteria in the gut versus the water. If some microbes have a higher growth rate in the gut than in the environment, this could lead to differences between GM and BPK, suggesting that some microbes, which are rare in the environment but common in the gut, may only be maintained by leakage from the host. Alternatively, GM may have contributed to structuring BPK indirectly via the expression of particular metabolites. Microbe–microbe interactions are indeed central in structuring microbial communities, more specifically through the production of antimicrobial molecules (Tasiemski et al., 2015; Adair and Douglas, 2017).
We can conclude by stating that host-dependent processes interact with external factors to shape both the GM and wider bacterial environment, suggesting that these two communities influence each other. Interactions between host genotypes, their GMs and free-living microbial communities might have important ecosystem level effects. This structuring effect is an essential prerequisite for the important role of the microbiome in eco-evolutionary processes and generates adaptive flexibility. An important avenue for future studies will be to determine how the microbiome can affect interactions among host species, e.g., by measuring the impact of microbial dispersal on the GMs, as well as on their fitness and competitive strength.
Data Availability Statement
The datasets generated for this study can be found in the NCBI, under accession numbers PRJNA498431 and PRJNA498417.
Author Contributions
EM, MC, ED, and LD conceived the ideas and designed the experiment approach. EM, MC, IV, and ED performed the experiments, did the sequencing and collected the data. EM, MC, and FM analyzed the data, with input from ED and LD. EM and ED then led the writing of the manuscript. All authors contributed to the revised versions.
Funding
Funding for this study was provided by the KU Leuven C16/2017/02, FWO G060216N, FWO G092619N projects, and an FWO postdoctoral fellowship (No. 12R4917N to EM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dries Bonte, Lore Bulteel, Steven Declerck, Shira Houwenhuyse, Lizanne Jansens, Gregory Maes, Koenraad Muylaert, Shinjini Mukherjee, Jeroen Raes, Robby Stoks, Caroline Souffreau, and the FWO EVENET network for stimulating discussions. We are also grateful to the KU Leuven Genomics Core for support with the sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00380/full#supplementary-material
References
Adair, K. L., and Douglas, A. E. (2017). Making a microbiome: the many determinants of host-associated microbial community composition. Curr. Opin. Microbiol. 35, 23–29. doi: 10.1016/j.mib.2016.11.002
Alberdi, A., Aizpurua, O., Bohmann, K., Zepeda-Mendoza, M. L., and Gilbert, M. T. P. (2016). Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol. Evol. 31, 689–699. doi: 10.1016/j.tree.2016.06.008
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016a). DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. doi: 10.1038/nmeth.3869
Callahan, B. J., Sankaran, K., Fukuyama, J. A., McMurdie, P. J., and Holmes, S. P. (2016b). Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Research 5:1492. doi: 10.12688/f1000research.8986.2
Callens, M., Macke, E., Muylaert, K., Bossier, P., Lievens, B., Waud, M., et al. (2016). Food availability affects the strength of mutualistic host–microbiota interactions in Daphnia magna. ISME J. 10, 911–920. doi: 10.1038/ismej.2015.166
Callens, M., Watanabe, H., Kato, Y., Jun, M., and Decaestecker, E. (2018). Microbiota inoculum composition affects holobiont assembly and host growth in Daphnia. Microbiome 6:56. doi: 10.1186/s40168-018-0444-1
Carmody, R. N., Gerber, G. K., Luevano, J. M., Gatti, D. M., Somes, L., Svenson, K. L., et al. (2015). Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84. doi: 10.1016/j.chom.2014.11.010
Dabrowska, K., and Witkiewicz, W. (2016). Correlations of host genetics and gut microbiome composition. Front. Microbiol. 7:1357. doi: 10.3389/fmicb.2016.01357
De Meester, L., Brans, K. I., Govaert, L., Souffreau, C., Mukherjee, S., Vanvelk, H., et al. (2019). Analysing eco-evolutionary dynamics—The challenging complexity of the real world. Funct. Ecol. 33, 43–59. doi: 10.1111/1365-2435.13261
Degans, H., Zöllner, E., De Gucht, K., De Meester, L., and Jürgens, K. (2002). Rapid Daphnia-mediated changes in microbial community structure: an experimental study. FEMS Micriobiol. Ecol. 2002, 137–149. doi: 10.1111/j.1574-6941.2002.tb01003.x
Eckert, E. M., Di Cesare, A., Stenzel, B., Fontaneto, D., and Corno, G. (2016). Daphnia as a refuge for an antibiotic resistance gene in an experimental freshwater community. Sci. Total Environ. 571, 77–81. doi: 10.1016/j.scitotenv.2016.07.141
Eckert, E. M., and Pernthaler, J. (2014). Bacterial epibionts of Daphnia: a potential route for the transfer of dissolved organic carbon in freshwater food webs. ISME J. 8, 1808–1819. doi: 10.1038/ismej.2014.39
Eiler, A., and Bertilsson, S. (2007). Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ. Microbiol. 9:838. doi: 10.1111/j.1462-2920.2006.01231.x
Foster, K. R., Schluter, J., Coyte, K. Z., and Rakoff-Nahoum, S. (2017). The evolution of the host microbiome as an ecosystem on a leash. Nature 548, 43–51. doi: 10.1038/nature23292
Freese, H., and Schink, B. (2011). Composition and stability of the microbial community inside the digestive tract of the aquatic crustacean Daphnia magna. Microb. Ecol. 62, 882–894. doi: 10.1007/s00248-011-9886-8
Grossart, H.-P., Dziallas, C., Leunert, F., and Tang, K. W. (2010). Bacterial dispersal by hitchiking on zooplankton. PNAS 107, 11959–11964. doi: 10.1073/pnas.1000668107
Guillard, R. R., and Lorenzen, C. J. (1972). Yellow-green algae with chlorophyllide C. J. Phycol. 8, 10–14. doi: 10.1111/j.0022-3646.1972.00010.x
Klüttgen, B., Dulmer, U., Engels, M., and Ratte, H. T. (1994). ADaM, an artificial freshwater for the culture of zooplankton. Water Res. 28, 743–746. doi: 10.1016/0043-1354(94)90157-0
Koch, H., and Schmid-Hempel, P. (2011). Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. U.S.A. 108, 19288–19292. doi: 10.1073/pnas.1110474108
Kolmonen, E., Sivonen, K., Rapala, J., and Haukka, K. (2004). Diversity of cyanobacteria and heterotrophic bacteria in cyanobacterial blooms in Lake Joutikas, Finland. Aquat. Microb. Ecol. 36, 201–211. doi: 10.3354/ame036201
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Laine, A. L., Barres, B., Numminen, E., and Siren, J. P. (2019). Variable opportunities for outcrossing result in hotspots of novel genetric variation in a pathogen metapopulation. eLife 8:e47091.
Lanan, M. C., Rodrigues, P. A. P., Agellon, A., Jansma, P., and Wheeler, D. E. (2016). A bacterial filter protects and structures the gut microbiome of an insect. ISME J. 10, 1866–1876. doi: 10.1038/ismej.2015.264
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Macke, E., Callens, M., De Meester, L., and Decaestecker, E. (2017a). Host-genotype dependent gut microbiota drives zooplankton tolerance to toxic cyanobacteria. Nat. Commun. 8:1608. doi: 10.1038/s41467-017-01714-x
Macke, E., Tasiemski, A., Massol, F., Callens, M., and Decaestecker, E. (2017b). Life history and eco-evolutionary dynamics in light of the gut microbiota. Oikos 126, 508–531. doi: 10.1111/oik.03900
Manage, P. M., and Premetilake, M. M. S. N. (2011). Occurrence of heterotrophic bacteria causing lysis of M. aeruginosa in Beira Lake, Sri Lanka. Vidyodaya J. Sci. 16, 31–56.
Maruyama, T., Kato, K., Yokoyama, A., Tanaka, T., Hiraishi, A., and Park, H. D. (2003). Dynamics of microcystin-degrading bacteria in mucilage of Microcystis. Microb. Ecol. 46, 279–288. doi: 10.1007/s00248-002-3007-7
McMurdie, P. J., and Holmes, S. P. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:11. doi: 10.1371/journal.pone.0061217
Moheimani, N., Borowitzka, M., Isdepsky, A., and Sing, F. (2013). “Standard methods for measuring growth of algae and their composition,” in Algae for Biofuels and Energy, eds N. R. Moheimani, M. A. Borowitzka, A. Isdepsky, and F. S. Sing (Dordrecht: Springer), 265–284. doi: 10.1007/978-94-007-5479-9_16
Murall, C. L., Abbate, J. L., Puelma Touzel, M., Allen-Vercoe, E., Alizon, S., Froissart, R., et al. (2017). “Invasions of host associated microbiome networks,” in Advances in Ecological Research, eds D. Bohan, A. Dumbrell, and F. Massol (Oxford: Elsevier), 201–281. doi: 10.1016/bs.aecr.2016.11.002
Mushegian, A., Arbore, R., Walser, J. C., and Ebert, D. (2019). Environmental sources of bacteria and genetic variation in behavior influence host-associated microbiota. Appl. Environ. Microbiol. 4:e1547-18.
Mushegian, A., Walser, J.-C., Sullam, K. E., and Ebert, D. (2018). The microbiota of diapause: how host-microbe associations are formed after dormancy in an aquatic crustacean. J. Anim. Ecol. 87, 400–413. doi: 10.1111/1365-2656.12709
Norman, N. M., and Koskella, B. (2017). The value of a comparative approach to understand the complex interplay between microbiota and host immunity. Front. Immunol. 8:1114. doi: 10.3389/fimmu.2017.01114
Ohbayashi, T., Takeshita, K., Kitagawa, W., Nikoh, N., Koga, R., Meng, X. Y., et al. (2015). Insect’s intestinal organ for symbiont sorting. Proc. Natl. Acad. Sci. U.S.A. 112, 5179–5188. doi: 10.1073/pnas.1511454112
Oliveros, J. C. (2007). VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online at: http://bioinfogp.cnb.csic.es/tools/venny?/index.html (accessed June 29, 2013).
Otti, O., Tragust, S., and Feldhaar, H. (2014). Unifying external and internal immune defences. Trends Ecol. Evol. 29, 625–634. doi: 10.1016/j.tree.2014.09.002
Peerakietkhajorn, S., Tsukada, K., Kato, Y., Matsuura, T., and Watanabe, H. (2015). Symbiotic bacteria contribute to increasing the population size of a freshwater crustacean, Daphnia magna. Environ. Microbiol. Rep. 7, 364–372. doi: 10.1111/1758-2229.12260
Penczykowski, R. M., Laine, A. L., and Koskella, B. (2016). Understanding the ecology and evolution of host-parasite interactions across scales. Evol. Appl. 9, 37–52. doi: 10.1111/eva.12294
Qi, W., Nong, G., Preston, J. F., Ben-Ami, F., and Ebert, D. (2009). Comparative metagenomics of Daphnia symbionts. BMC Genomics 10:172. doi: 10.1186/1471-2164-10-172
R Core Team (2018). R: A Language and Environment For Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: https://www.R-project.org/
Rohrlack, T., Dittmann, E., Borner, T., and Christoffersen, K. (2001). Effects of cell-bound microcystins on survival and feeding of Daphnia spp. Appl. Environ. Microbiol. 67, 3523–3529. doi: 10.1128/aem.67.8.3523-3529.2001
Shapira, M. (2016). Gut microbiotas and host evolution: scaling up symbiosis. Trends Ecol. Evol. 31, 539–549. doi: 10.1016/j.tree.2016.03.006
Tasiemski, A., Massol, F., Cuvillier-Hot, V., Boidin-Wichlacz, C., Roger, E., Rodet, F., et al. (2015). Reciprocal immune benefit based on complementary production of antibiotics by the leech Hirudo verbana and its gut symbiont Aeromonas veronii. Sci. Rep. 5:17498. doi: 10.1038/srep17498
Taylor, J. D., Cottingham, S. D., Billinge, J., and Cunliffe, M. (2014). Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J. 8, 245–248. doi: 10.1038/ismej.2013.178
terhorst, C. P., Lennon, J. T., and Lau, J. A. (2014). The relative importance of rapid evolution for plant-microbe interactions depends on ecological context. Proc. R. Soc. B Biol. Sci. 281:20140028. doi: 10.1098/rspb.2014.0028
von Elert, E., Martin-Creuzburg, D., and Le Coz, J. R. (2003). Absence of sterols constrains carbon transfer between cyanobacteria and a freshwater herbivore (Daphnia galeata). Proc. R. Soc. B Biol. Sci. 270, 1209–1214. doi: 10.1098/rspb.2003.2357
Wong, A. C. N., Luo, Y., Jing, X., Franzenburg, S., Bost, A., and Douglas, A. E. (2015). The host as the driver of the microbiota in the gut and external environment of Drosophila melanogaster. Appl. Environ. Microbiol. 81, 6232–6240. doi: 10.1128/AEM.01442-15
Keywords: microbiome, Daphnia, bacterioplankton, community structure, ecosystem level effects
Citation: Macke E, Callens M, Massol F, Vanoverberghe I, De Meester L and Decaestecker E (2020) Diet and Genotype of an Aquatic Invertebrate Affect the Composition of Free-Living Microbial Communities. Front. Microbiol. 11:380. doi: 10.3389/fmicb.2020.00380
Received: 08 October 2019; Accepted: 20 February 2020;
Published: 17 March 2020.
Edited by:
Pia H Moisander, University of Massachusetts Dartmouth, United StatesReviewed by:
Clay Cressler, University of Nebraska–Lincoln, United StatesAlexandra Mushegian, Georgetown University, United States
Copyright © 2020 Macke, Callens, Massol, Vanoverberghe, De Meester and Decaestecker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen Decaestecker, ZWxsZW4uZGVjYWVzdGVja2VyQGt1bGV1dmVuLmJl