
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 February 2020
Sec. Microbial Symbioses
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00311
 Camille Clerissi1,2*
Camille Clerissi1,2* Julien de Lorgeril1
Julien de Lorgeril1 Bruno Petton3
Bruno Petton3 Aude Lucasson1
Aude Lucasson1 Jean-Michel Escoubas1
Jean-Michel Escoubas1 Yannick Gueguen1
Yannick Gueguen1 Lionel Dégremont4
Lionel Dégremont4 Guillaume Mitta1
Guillaume Mitta1 Eve Toulza1*
Eve Toulza1*Pacific Oyster Mortality Syndrome (POMS) affects Crassostrea gigas oysters worldwide and causes important economic losses. Disease dynamic was recently deciphered and revealed a multiple and progressive infection caused by the Ostreid herpesvirus OsHV-1 μVar, triggering an immunosuppression followed by microbiota destabilization and bacteraemia by opportunistic bacterial pathogens. However, it remains unknown if microbiota might participate to protect oysters against POMS, and if microbiota characteristics might be predictive of oyster mortalities. To tackle this issue, we transferred full-sib progenies of resistant and susceptible oyster families from hatchery to the field during a period in favor of POMS. After 5 days of transplantation, oysters from each family were either sampled for individual microbiota analyses using 16S rRNA gene-metabarcoding or transferred into facilities to record their survival using controlled condition. As expected, all oysters from susceptible families died, and all oysters from the resistant family survived. Quantification of OsHV-1 and bacteria showed that 5 days of transplantation were long enough to contaminate oysters by POMS, but not for entering the pathogenesis process. Thus, it was possible to compare microbiota characteristics between resistant and susceptible oysters families at the early steps of infection. Strikingly, we found that microbiota evenness and abundances of Cyanobacteria (Subsection III, family I), Mycoplasmataceae, Rhodobacteraceae, and Rhodospirillaceae were significantly different between resistant and susceptible oyster families. We concluded that these microbiota characteristics might predict oyster mortalities.
The farmed oyster Crassostrea gigas is heavily affected by the Pacific Oyster Mortality Syndrome (POMS) targeting juveniles (Barbosa Solomieu et al., 2015; Pernet et al., 2016). This disease is multifactorial and depends on water temperature (Petton et al., 2015), development stage (Azéma et al., 2017), and oyster diet (Pernet et al., 2019). It is also polymicrobial due to the combined development of viral and bacterial infections (de Lorgeril et al., 2018). Different susceptibility levels were previously associated with oyster physiological status, genetic backgrounds (Dégremont et al., 2005; Samain et al., 2007; Wendling et al., 2017), in association with microbiota dysbiosis (Lokmer and Wegner, 2015).
Recently, holistic molecular approaches revealed the mechanism of POMS (de Lorgeril et al., 2018; Rubio et al., 2019). These studies showed that an infection by the Ostreid herpesvirus (OsHV-1 μVar) is the critical step in the infectious process leading to an immune-compromised state by altering hemocyte physiology. This first process is followed by a microbiota destabilization which “opens the door” to bacterial pathogens (e.g., vibrios) that target hemocytes to induce their lysis. The infectious process is completed with subsequent bacteraemia, which is the ultimate step inducing oyster death.
So far, it is still unknown whether oyster microbial associates might influence disease development, and if microbiota characteristics might predict oyster mortalities. However, microbiota can play a role of physical barriers against pathogens. For example, it was suggested that part of the resident hemolymph bacteria may contribute to oysters protection by producing antimicrobial peptides (Desriac et al., 2014). Other studies highlighted protective effects of bacteria, such as secondary endosymbionts of aphids against parasitoid wasps (Oliver et al., 2003), or symbionts of frogs against pathogenic fungi (Woodhams et al., 2007). Furthermore, microbial associates might also stimulate immunity of their hosts (Wang et al., 2019), and thus indirectly limit pathogen development.
To tackle this issue, we used one resistant oyster family (RF21) and two susceptible families (SF15 and SF32) from a previous study (de Lorgeril et al., 2018). Pathogen-free oysters (reproduced and grown in bio-secured conditions) from these three full-sib families were placed for 5 days in the field during an infectious period (Figure 1). According to previous observations, 16°C was a relevant threshold to define the infectious period, as high mortality rates were observed above this temperature (Pernet et al., 2012; Petton et al., 2013; Dégremont et al., 2015). Moreover, 5 days were considered sufficient for oyster contamination by the causal agents of the disease (Petton et al., 2015), and allowed microbiota analyses before disease development and animal death. In both the hatchery (control) and after 5 days of transplantation in the field, oysters were sampled, and we analyzed oyster-associated bacterial communities using 16S rRNA gene-metabarcoding.
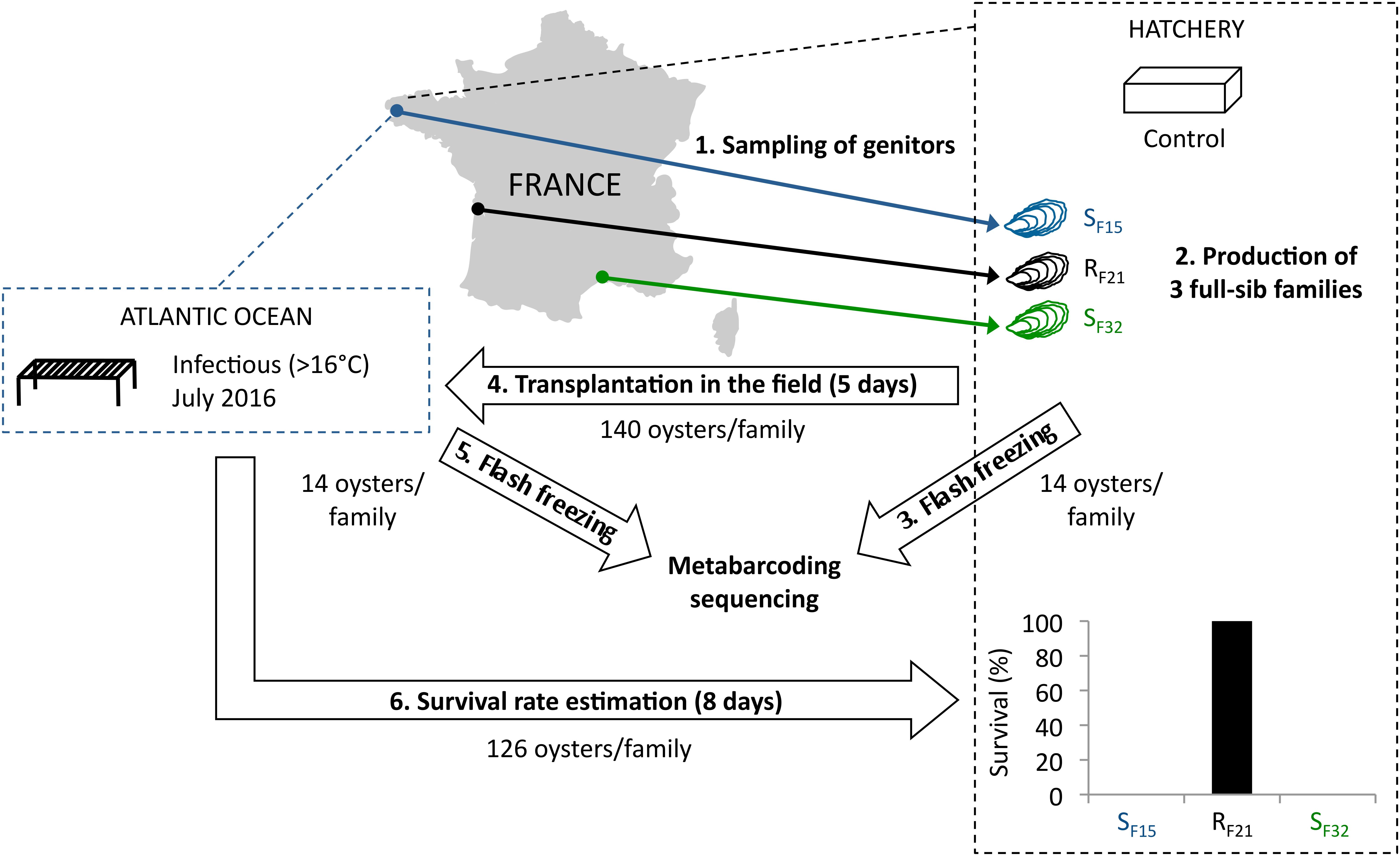
Figure 1. Experimental design. Three oyster full-sib families were produced in controlled condition (hatchery), and placed for 5 days in the environment (Atlantic Ocean, latitude: 48.335263; longitude: –4.317922) during an infectious period (July 2016). Then, oysters were flash frozen, DNA were extracted, and microbiota were sequenced using 16S rRNA gene-metabarcoding. Oysters deployed in the field during 5 days were followed during 8 days in the hatchery. Survival rates were recorded at endpoint.
This study aimed at comparing bacteria content of resistant and susceptible families in order to possibly identify microbiota features associated with oyster mortality and/or resistance. According to the previous holistic study that used the resistant family RF21 and another susceptible family (SF11) (de Lorgeril et al., 2018), we expected that susceptible families had (i) early microbiota destabilization (increase of species richness and microbiota dispersions, decrease of evenness), and (ii) possibly contained the opportunistic and/or pathogenic bacteria that participated to bacteraemia in the holistic study (e.g., vibrios).
In 2015, full-sib C. gigas oyster families were produced using a methodology that allowed the production of pathogen-free juveniles (de Lorgeril et al., 2018). Two oyster families (SF15 and SF32) were produced using one female and one male sampled from wild populations in the Atlantic Ocean and the Mediterranean Sea, respectively (Figure 1). In addition, family RF21 was produced using genitors from a mass selective breeding program aiming to increase the resistance of C. gigas oysters against OsHV-1. It was performed by breeding disease survivors throughout four generations of selection (Dégremont et al., 2015). Thus, each family (cohort) corresponded to the offspring of a biparental reproduction (full-sib progenies). Conditioning, reproduction and larval breeding were performed as described previously (de Lorgeril et al., 2018). At the larval and post-larval stages, oysters were fed with the same diet as the genitors at a concentration between 1500–2000 μm3.μl–1 (Rico-Villa et al., 2009). Before experiments, all oyster families were maintained in controlled condition at the laboratory (Argenton-sur-Creuse, France) using seawater treated with UV, filtered through 1 μm mesh, and enriched with a bi-specific phytoplankton diet made of Tisochrysis lutea (CCAP 927/14) and Chaetoceros muelleri (CCAP 1010/3) (in equal biomass proportion) at a ratio equivalent to 6% of the oyster dry mass (Rico-Villa et al., 2009). Finally, all oysters remained free of any abnormal mortality.
About 140 juveniles per oyster family were either kept in the controlled condition or placed in the field for 5 days. The field site was located within a oyster farm in the Atlantic Ocean during an infectious period (Figure 1). This infectious period was selected according to seawater temperatures (above 16°C). It was confirmed by the observed mortality rates, and the presence within some oysters of pathogens and/or opportunists previously described in the same region (Atlantic Ocean) (de Lorgeril et al., 2018). After 5 days of transplantation (July 2016), no mortality occurred and 14 individuals per family were flash frozen in liquid nitrogen and stored at −80°C. The remaining oysters were then transferred into the hatchery under controlled conditions to monitor the survival rates of the three families. The number of dead oysters was recorded at day 13 (i.e., 8 days after the end of transplantation). Similarly, 14 individuals per family (except 13 for SF32, because DNA extraction failed for one sample) kept in the controlled condition were flash frozen in liquid nitrogen and stored at −80°C.
Frozen oysters were ground in liquid nitrogen in 50 ml stainless steel bowls using 20 mm diameter grinding balls (Retsch MM400 mill). The powders were stored at −80°C, and were then used for DNA extractions using the DNA from tissue Macherey-Nagel kit (reference 740952.250) according to the manufacturer’s protocol. In order to improve DNA extractions, we added a crushing step, which consisted in an additional 12 min mechanical lysis using zirconium beads before the 90 min enzymatic lysis in the presence of proteinase K. DNA concentration and quality were checked with Epoch microplate spectrophotometer (BioTek Instruments, Inc).
Then, the 16S rRNA gene of bacterial communities was amplified and sequenced using the variable V3V4 loops (341F: 5′-CCTACGGGNGGCWGCAG-3′; 805R: 5′-GACTACHVGGGTATCTAATCC-3′) (Klindworth et al., 2013). Paired-end sequencing (250 bp read length) was performed at the McGill University (Génome Québec Innovation Centre, Montréal, Canada) on the MiSeq system (Illumina) using the v2 chemistry according to the manufacturer’s protocol. Raw sequence data are available in the SRA database (BioProject ID PRJNA419907).
Quantification of OsHV-1, total bacteria 16S rDNA, and total Vibrio 16S rDNA was performed using quantitative PCR (qPCR). All amplification reactions were analyzed using a Roche LightCycler 480 Real-Time thermocycler (qPHD-Montpellier GenomiX platform, Montpellier University, France). A Labcyte acoustic automated liquid handling platform (ECHO) was used for pipetting into the 384-well plate (Roche). The total qPCR reaction volume was 1.5 μl and consisted of 0.5 μl DNA (40 ng.μl–1) and 1 μl LightCycler 480 SYBR Green I Master mix (Roche) containing 0.5 μM PCR primer (Eurogentec SA). Virus-specific primer pairs targeted a DNA polymerase catalytic subunit (DP, ORF100, AY509253): 5′-ATTGATGATGTGGATAATCTGTG-3′ and 5′-GGTAAATACCATTGGTCTTGTTCC-3′ (Davison et al., 2005). Total bacteria specific primer pairs were the 341F-805R primers targeting the variable V3V4 loops for bacterial communities (see above). Total Vibrio specific primer pairs were 5′-GGCGTAAAGCGCATGCAGGT-3′ and 5′-GAAATTCTACCCCCCTCTACAG-3′ (Mansergh and Zehr, 2014). qPCR reactions were performed with the following program: 95°C for 10 min, followed by 40 cycles of denaturation (95°C, 10 s), hybridization (60°C, 20 s) and elongation (72°C, 25 s) (de Lorgeril et al., 2018). After these PCR cycles a melting temperature curve of the amplicon was generated to verify the specificity of the amplification. Absolute quantification of OsHV-1 copies was calculated by comparing the observed Cq values to a standard curve generated from the DNA polymerase catalytic subunit amplification product cloned into the pCR4-TOPO vector. For total bacteria and total Vibrio 16S rDNA, we used the relative quantification calculated by the 2–ΔΔCq method (Pfaffl, 2001) with the mean of the measured threshold cycle values of two reference genes: Cg-BPI (GenBank: AY165040, Cg-BPI-F: 5′-ACGGTACAGAACGGATCTACG-3′; Cg-BPI-R: 5′-AATCGTG GCTGACATCGTAGC-3′), and Cg-actin (GenBank: AF026063, Cg-actin-F: 5′-TCATTGCTCCACCTGAGAGG-3′; Cg-actin-R: 5′-AGCATTTCCTGTGGACAATGG-3′).
The FROGS pipeline (Find Rapidly OTU with Galaxy Solution) implemented into a galaxy instance was used to define Operational Taxonomic Units (OTU), and computed taxonomic affiliations (Escudié et al., 2017). Briefly, paired reads were merged using FLASH (Magoc and Salzberg, 2011). After denoising and primer/adapters removal with cutadapt (Martin, 2011), de novo clustering was performed using SWARM that uses a local clustering threshold, with aggregation distance d = 3 (Mahé et al., 2015). Chimera were removed using VSEARCH (de novo chimera detection) (Rognes et al., 2016). Particularly, this method divided each sequence into four fragments, and then looked for similarity with putative parents in the whole set of OTUs. We filtered the dataset for singletons and we annotated OTUs using Blast + against the Silva database (release 123, September 2015) to produce an OTU and affiliation table in standard BIOM format. Rarefaction curves of species richness were produced using the {phyloseq} R package, and the ggrare function (McMurdie and Holmes, 2013). In order to compare samples for alpha and beta diversity, we used the rarefy_even_depth function to subsample dataset to 5148 reads per sample excluding chloroplasts. The alpha diversity metrics (Chao1 and Shannon) were estimated at the OTU level with the estimate_richness function. Moreover, Pielou’s measure of species evenness was computed using the diversity function in {vegan}. We also used phyloseq to obtain abundances at differents taxonomic ranks (for genus and family) (tax_glom function). Because agglomerate of multi-affiliation and unknown taxa does not make sense at higher taxonomic ranks, we only kept taxa having a true annotation for each corresponding taxonomic rank. We computed Bray–Curtis dissimilarities to study beta diversity, i.e., distances between samples for OTU compositions (vegdist function, {vegan}).
All statistical analyses were done using R v3.3.1 (R Development Core Team, 2008).
Principal coordinate analyses (pcoa, {vegan}) were computed to describe compositions of microbial communities between samples using Bray–Curtis dissimilarities (vegdist, {vegan}). Multivariate homogeneity of group dispersions was tested between microbial assemblages of resistant and susceptible oyster families using 999 permutations (permutest, {vegan}). We used DESeq2 (Love et al., 2014) (DESeq {DESeq2}) to identify OTUs having differential abundances between resistant and susceptible oyster families. Heatmaps of significant bacterial genera and families were computed using relative abundances and the heatmap.2 function ({gplots}).
We performed one-way ANOVA or non-parametric Kruskal–Wallis tests [when normality of residuals was rejected (Shapiro test)] to compare alpha diversity metrics of microbiota. When ANOVA or Kruskal–Wallis tests were significant, we then computed pairwise comparisons between group levels (post hoc analyses) using pairwise-t-tests or Dunn tests, respectively.
For all analyses, the threshold significance level was set at 0.05. P-values were corrected for multiple comparisons using Benjamini and Hochberg’s method (Benjamini and Hochberg, 1995) (p.adjust, {stats}).
Full-sib progenies from one resistant (RF21) and two susceptible (SF15 and SF32) oyster families were produced and reared in bio-secured conditions (Argenton-sur-Creuse, France). About 140 pathogen-free oysters per family were transferred to the infectious environment for 5 days (Brest, Atlantic Ocean, temperature above 16°C) (Figure 1). 14 oysters per family were then flash frozen for individual bacterial microbiota analyses. The other 126 oysters per family were placed back in hatchery to monitor survival rates. As expected, all oysters from the two susceptible families died, whereas all oysters from the resistant family survived (Figure 1). This observation showed that oysters were contaminated during transplantation, and then developed the disease. Quantification of OsHV-1 and bacteria at day 5 post-transplantation showed that only three oysters from susceptible families displayed moderate to important viral infection, and that only one individual displayed both high viral infection and bacteraemia (SF15.I.R14) (Figure 2). These results suggested that 5 days of transplantation in the field during an infectious period were long enough for oyster contamination, but not for entering the pathogenesis process (except for three oysters).
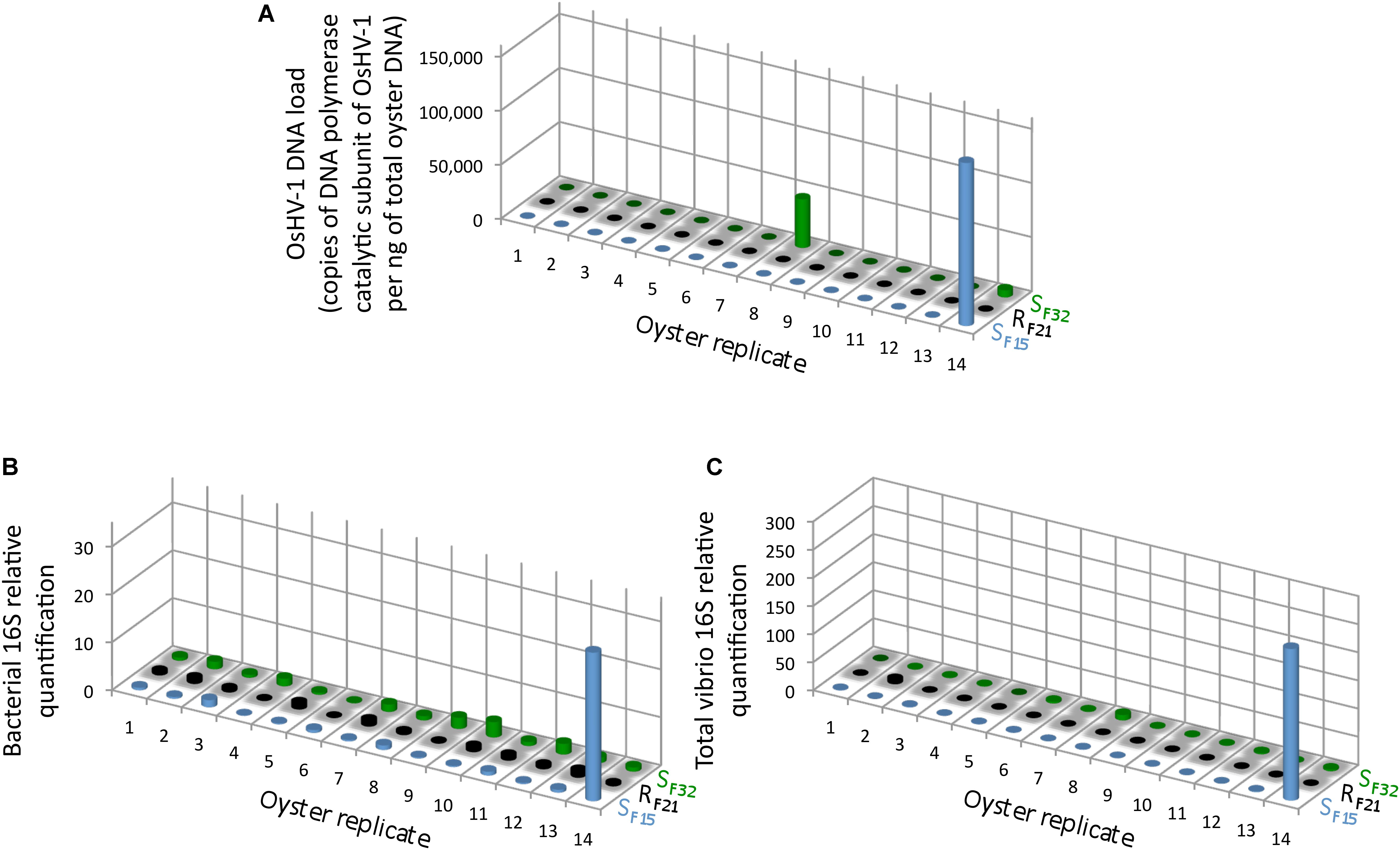
Figure 2. OsHV-1 and bacterial colonization in oysters at day 5 post-transplantation in field condition during an infectious period. (A) The OsHV-1 load was quantified by qPCR and expressed as copies of DNA polymerase catalytic subunit of OsHV-1 per ng of total oyster DNA. Relative quantification of total bacteria (B), and total vibrios (C) abundance were measured by qPCR (n = 42).
The whole bacterial communities were sequenced using the 16S rRNA gene from 14 oysters per family and per condition (except 13 oysters for SF32 in hatchery) (i.e., 83 oyster-associated microbiota for control and infectious conditions). In average, each sample contained 21,423 sequences representing 1,075 OTUs (Supplementary Figure S1 and Supplementary Tables S1, S2). First, we searched for known opportunistic genera within microbiota of diseased oysters, i.e., already identified when the disease dynamic was described using RF21 and another susceptible family (SF11) in the same region (Atlantic Ocean) (de Lorgeril et al., 2018). Nine out of ten previously identified genera were present in our dataset (Figure 3). Most were abundant for the oyster highly infected by OsHV-1 and displaying bacteraemia (SF15.I.R14) (Figures 2, 3A). Both Vibrio and Psychromonas genera were found within the three diseased oysters (Figure 3A). However, Vibrio occurred in the majority of healthy individuals as well (Figure 3B).
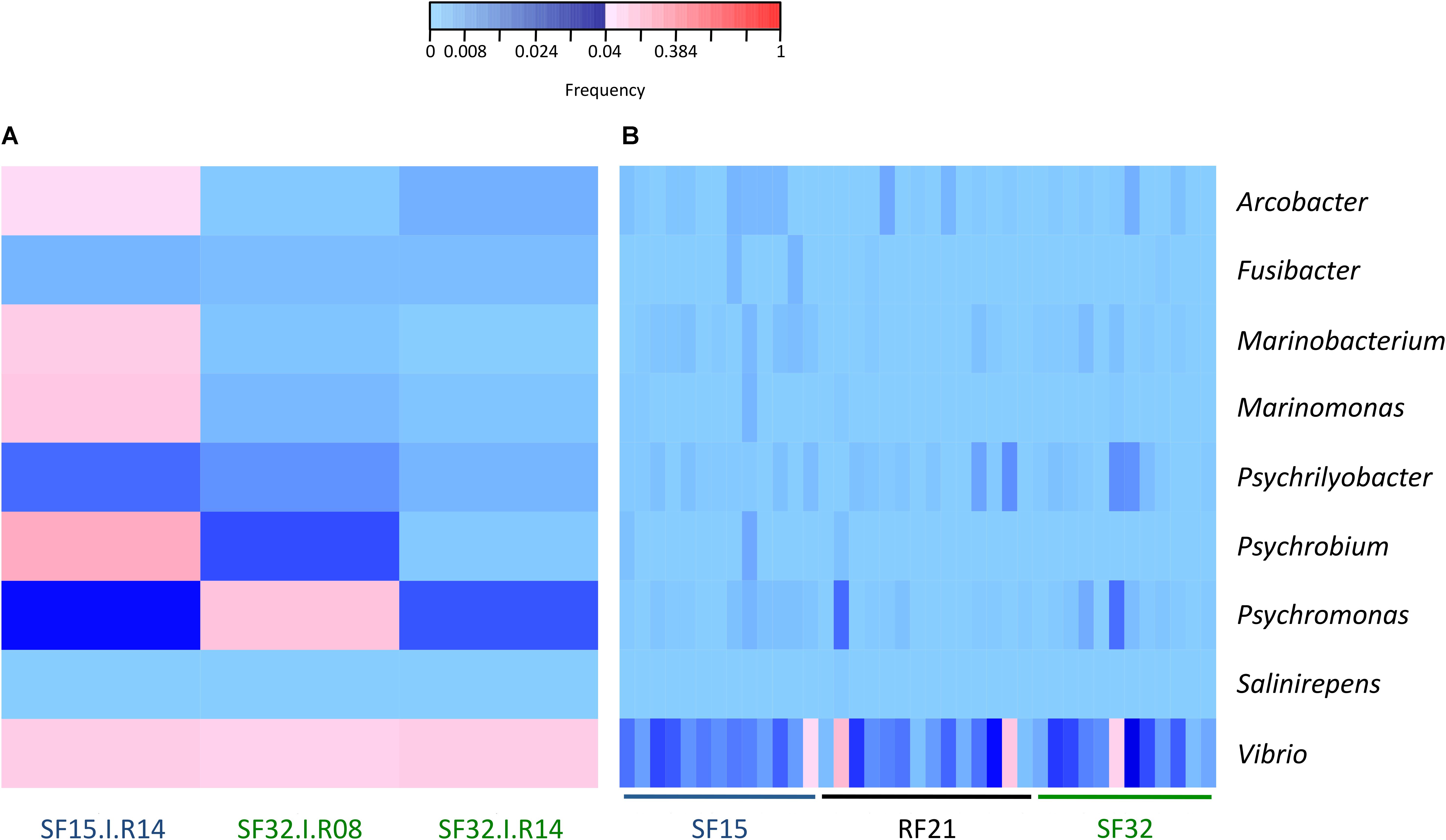
Figure 3. Frequencies of known opportunistic bacterial genera associated with POMS. (A) Positive oysters for OsHV-1 detection (SF15.I.R14, SF32.I.R08, and SF32.I.R14). (B) Negative oysters for OsHV-1 detection for each family. These opportunistic genera were identified in a previous experimental infection (de Lorgeril et al., 2018). Frequencies above and below 4% are displayed in red and blue, respectively (n = 42).
Although 5 days of transplantation were long enough for oyster contamination, only three individuals entered the pathogenesis process as indicated by virus load. Thus, after discarding these three individuals, it was possible to compare microbiota characteristics of the other oysters between resistant and susceptible families before the onset of disease development.
First, we compared alpha diversity indices (Chao1, evenness and Shannon) between oyster families (Figure 4 and Supplementary Table S3). This analysis showed that Chao1 increased for all families between hatchery and the infectious environment. Moreover, evenness and Shannon indices increased only for the resistant family RF21. Notably, RF21 had higher evenness than SF15 and SF32 during the infectious condition. Secondly, we computed a principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarities to describe microbiota compositions (Figure 5A). This ordination highlighted that oyster microbiota changed between hatchery and the infectious environment for the three oyster families. Unexpectedly, microbiota dispersion of RF21 was not lower than the susceptible families, and even showed higher dispersions for the infectious condition than in hatchery (Figure 5B).
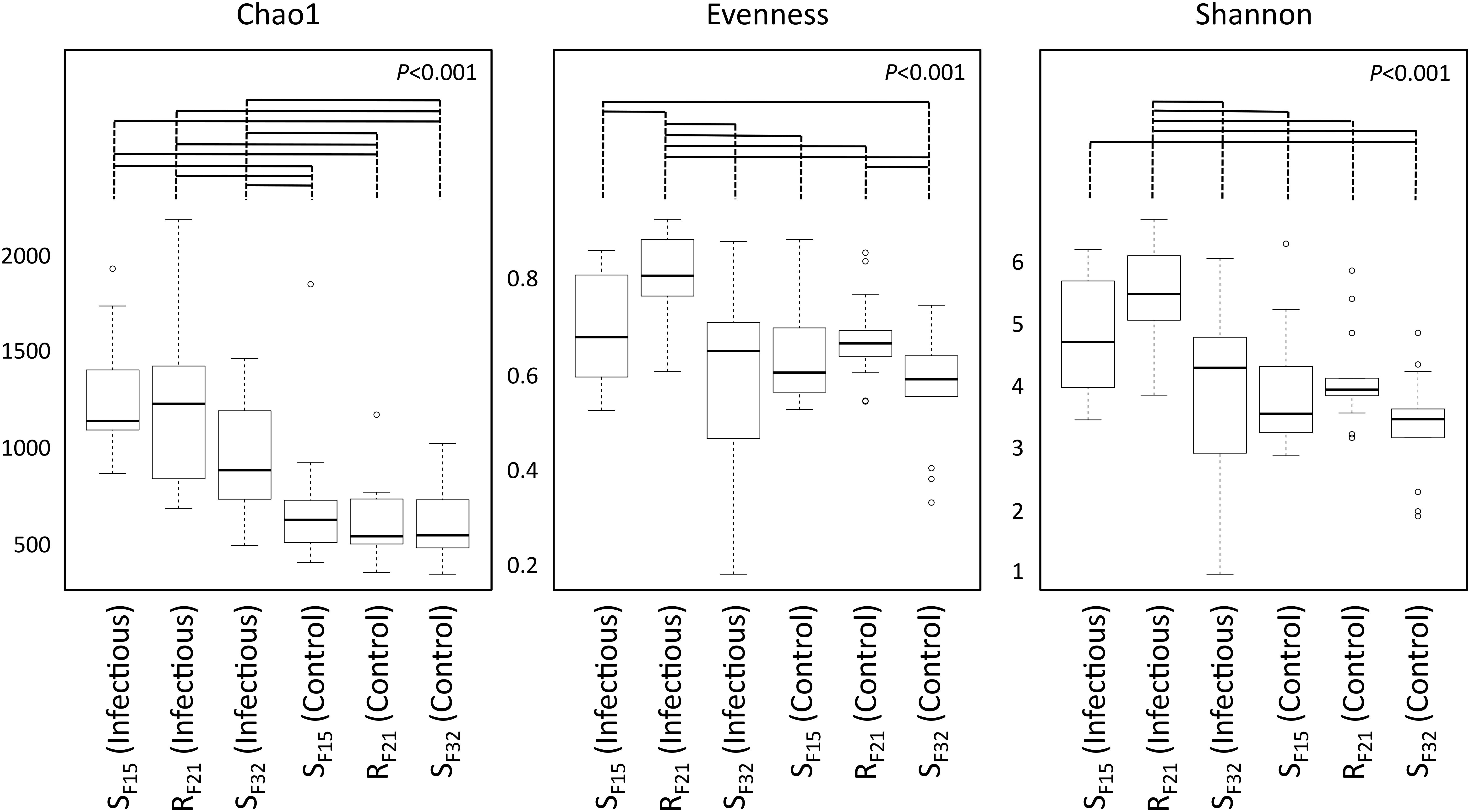
Figure 4. Comparison of alpha diversity indices between oyster families in controlled (hatchery) and infectious (field) conditions. P-values correspond to global tests. Horizontal lines above boxplots indicate significant pairwise comparisons between group levels (post hoc analyses) (n = 80).
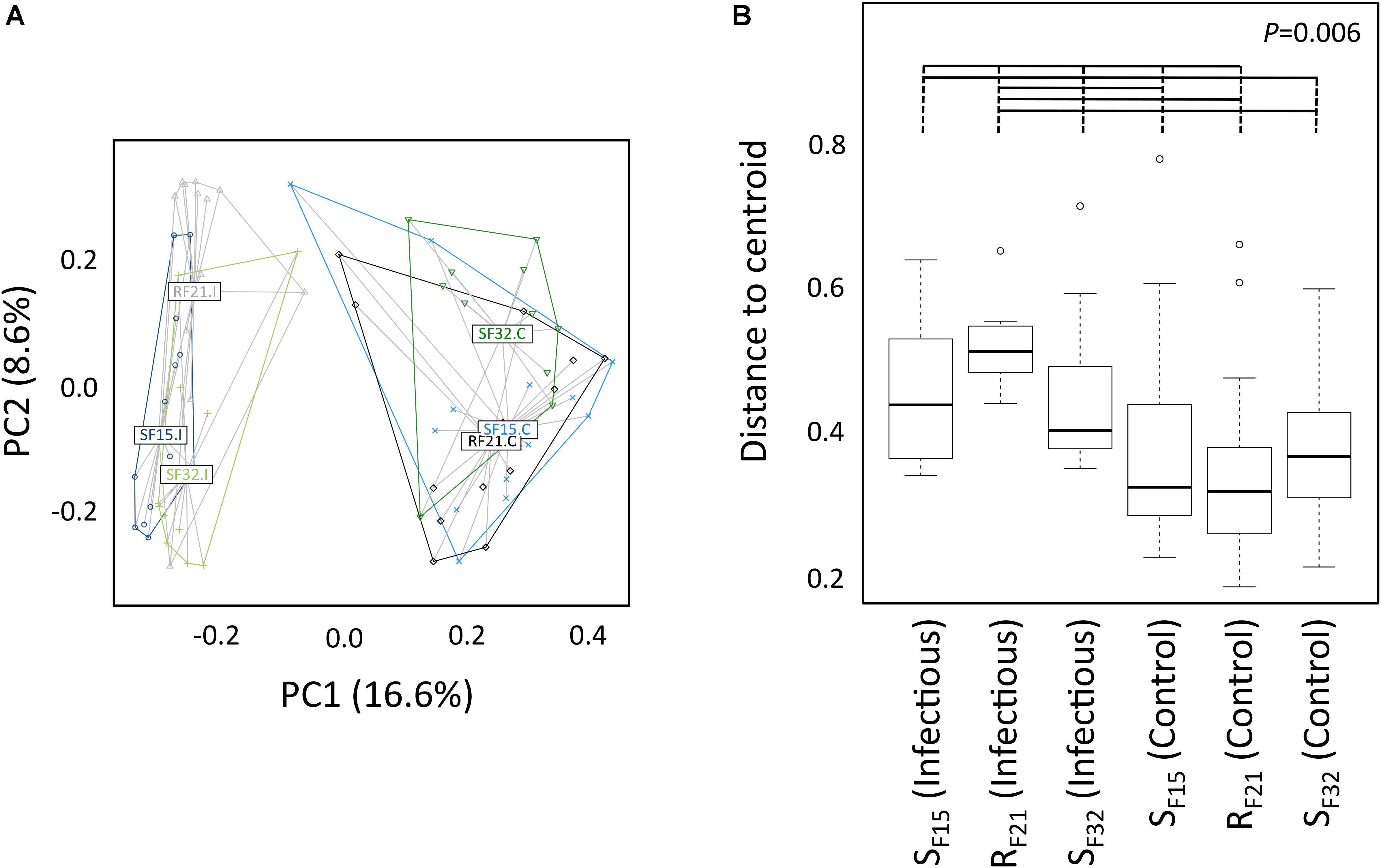
Figure 5. Microbiota compositions between oyster families in controlled (hatchery) and infectious (field) conditions. (A) Principal coordinate analysis (Bray–Curtis dissimilarities between samples). Each dot corresponds to one individual. Colors correspond to oyster families in controlled (C) and infectious (I) conditions. Labels are displayed at the barycenter of dots. (B) Multivariate homogeneity of group dispersions. P-value corresponds to the global test. Horizontal lines above boxplots indicate significant pairwise comparisons between group levels (post hoc analyses) (n = 80).
Altogether, these results suggested that microbiota assemblages highly changed between hatchery and the infectious environment for the three families. Although we did not expect to find higher microbiota dispersion for RF21, our analyses revealed that the resistant family had higher evenness than the susceptible families before the onset of disease development.
Because resistant and susceptible oyster families had different microbiota characteristics (evenness and dispersion) at the early steps of infection, we compared abundances of bacterial taxa within hatchery to identify putative pathogens, opportunists or mutualists that were consistently presents in microbiota before transplantation. Abundances were compared at two taxonomic ranks, bacterial genera (Supplementary Table S4) and families (Supplementary Table S5). Two genera (Colwellia and Photobacterium) (Supplementary Figure S2) and four families (Anaplasmataceae, Colwelliaceae, Mycoplasmataceae, and Vibrionaceae) (Figure 6) showed high abundances (>4% in at least one sample) and were significantly different between resistant and susceptible oyster families.
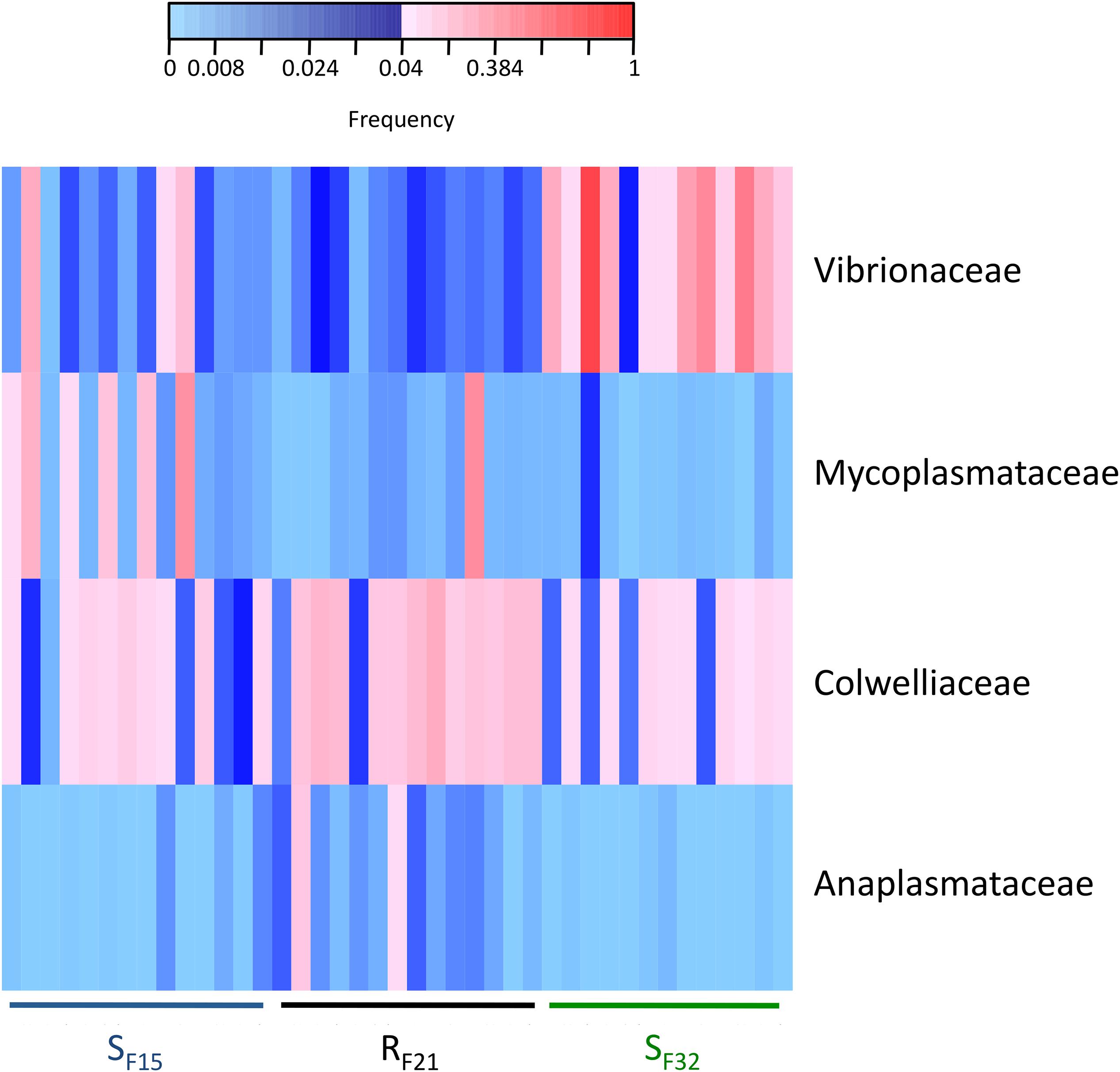
Figure 6. Heatmap of bacterial families that were significantly different between resistant and susceptible oyster families in the controlled condition (hatchery). Only bacterial families with a frequency above 4% in at least one sample are shown. Frequencies above and below 4% are displayed in red and blue, respectively (n = 41).
We also compared bacterial taxa before disease development between resistant and susceptible oyster families that were placed in the infectious environment. Except for Mycoplasmataceae, significant bacterial taxa were not similar to hatchery-identified candidates (Figure 7 and Supplementary Figure S3). Four genera (Phormidium, Pseudomonas, Thalassospira, and Umboniibacter) (Supplementary Figure S3) and seven families (Cyanobacteria (Subsection I, family I), Cyanobacteria (Subsection III, family I), Mycoplasmataceae, Pseudomonadaceae, Rhodobacteraceae, Rhodospirillaceae, and Spirochaetaceae) (Figure 7) had high abundances (>4% in at least one sample) and were significantly different between resistant and susceptible oyster families.
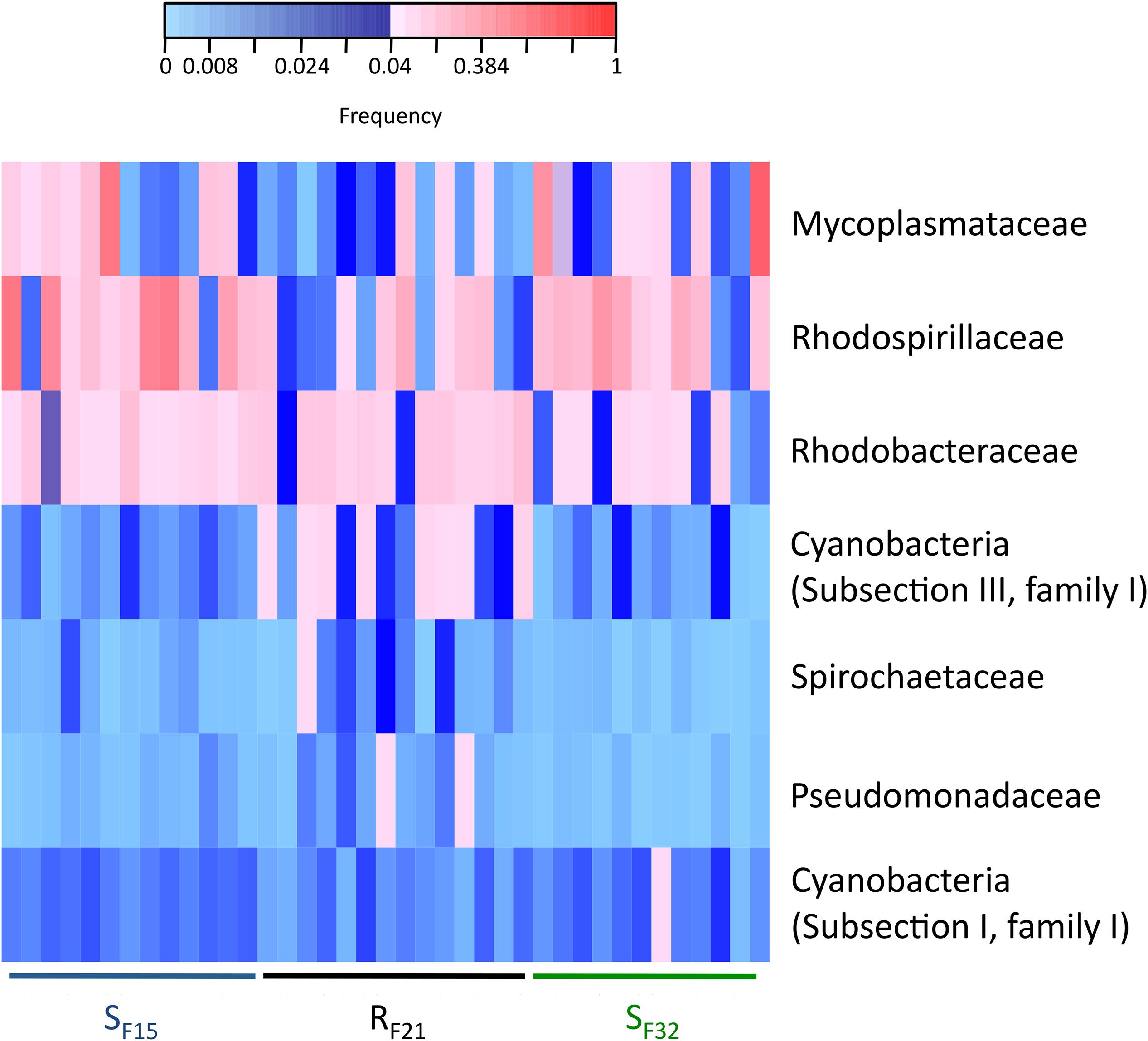
Figure 7. Heatmap of bacterial families that were significantly different between resistant and susceptible oyster families in the infectious condition (field). Only bacterial families with a frequency above 4% in at least one sample are shown. Frequencies above and below 4% are displayed in red and blue, respectively (n = 39).
Oysters were transplanted for 5 days in the field during an infectious period. According to previous observations (Dégremont, 2011; Petton et al., 2015), this time period was considered sufficient for oyster contamination. We did not observed dead oysters when they were sampled, but all individuals of susceptible families died 8 days after they were placed back to laboratory tanks. This observation showed that oysters were contaminated during transplantation, and this exposure was sufficient for the further development of the complete pathogenesis process.
High abundances of OsHV-1 and total bacteria were previously observed for another susceptible family (SF11) during disease development (de Lorgeril et al., 2018). In our study, most oysters (except three) did not exhibit high abundances of OsHV-1 or bacteria after 5 days of transplantation. After exclusion of these three oysters, we thus analyzed oyster microbiota before the onset of disease development (i.e., at the early steps of infection).
According to the previous holistic study (de Lorgeril et al., 2018), we expected to observe many modifications of microbiota for susceptible oyster families during transplantation, but not or few for the resistant one. In particular, we expected increase of alpha diversity indices (Chao1 and Shannon), and of microbiota dispersion for the susceptible families. Furthermore, we hypothesized that the resistant family might have stable microbiota, because this characteristic was linked to host homeostasis in many studies (Faith et al., 2013; Rungrassamee et al., 2016; Zaneveld et al., 2017).
Unexpectedly, we found that alpha diversity indices and dispersion of microbiota highly changed for the three oyster families (susceptible as well as resistant) when they were transferred from hatchery to the infectious environment. As a consequence, stable microbiota was not a characteristic of the resistant family, suggesting either a neutral effect or that flexibility of microbiota might also be advantageous in a changing environment, such as proposed for Nematostella vectensis (Mortzfeld et al., 2016). Microbiota evenness was the only index that discriminated resistant and susceptible families. It highlighted that OTU abundances within resistant oysters were more equally distributed than within susceptible oysters. This index may possibly have important effects on oyster homeostasis, because it was already found to be positively linked to ecosystem productivity (Wilsey and Potvin, 2000; Zhang et al., 2012), functional stability (Balvanera et al., 2005; Wittebolle et al., 2009), and invasion resistance (Wilsey and Polley, 2002; De Roy et al., 2013).
Several resistant (e.g., RF21) and susceptible (e.g., SF15 and SF32) families were produced previously to describe POMS in the Atlantic Ocean (de Lorgeril et al., 2018). In this previous study, a holistic approach combining metabarcoding, transcriptomic, qPCR and histology successfully described disease dynamic using a resistant (RF21) and a susceptible (RF11) family. In particular, after viral burst, several bacterial genera significantly contributed to the observed bacteraemia such as Arcobacter, Cryomorphaceae, Fusibacter, Marinobacterium, Marinomonas, Psychrilyobacter, Psychrobium, Psychromonas, Salinirepens, and Vibrio. In our study, we thus analyzed if these genera were present in oyster microbiota. We particularly evaluated if their abundances were high within the three diseased oysters, but also significantly different between resistant and susceptible families (in hatchery and before the onset of disease development in the field). Most of these genera were identified within the oyster that displayed high abundances of OsHV-1 and bacteraemia (SF15.I.R14). Psychromonas occurred within the three diseased oysters, and might possibly be involved early in disease development, even if its pathogenic role was not demonstrated so far.
Furthermore, three bacterial families (Mycoplasmataceae, Rhodospirillaceae, and Vibrionaceae) were linked to susceptible oysters in hatchery and/or before disease development in the field, suggesting putative roles of opportunists or pathogens. Among Vibrionaceae, Photobacterium genus was mostly associated with SF32 in hatchery, and as well with low disease resistance oysters in a previous study (King et al., 2019). Among Rhodospirillaceae, Thalassospira genus was already identified in C. gigas oysters (Fernandez-Piquer et al., 2012). Even though Rhodospirillaceae were found in diseased tissues of Platygyra carnosus corals (Ng et al., 2015), there is no evidence that they may act as pathogens for oysters until now. Lastly, Mycoplasmataceae family was abundant in C. virginica oysters from Hackberry Bay (King et al., 2012), and decreased in oysters during experimental infections (de Lorgeril et al., 2018). However, it was intriguing to find in this study that Mycoplasmataceae abundances were significantly lower for resistant than susceptible oyster families in both hatchery and field conditions, at the early steps of infection. As a consequence, this family might have negative effects on oyster survival, but future studies should experimentally test its role within oyster microbiota.
Some observations suggested that bacteria might protect oysters from pathogens. For example, Aeromonas media A199, Phaeobacter gallaeciensis, and Pseudoalteromonas sp. D41 improved survival of C. gigas against Vibrio strains when they were tested as probiotics (Gibson et al., 1998; Kesarcodi-Watson et al., 2012). In particular, the Pseudoalteromonas genus is known to produce a wide variety of biologically active secondary metabolites (Kalinovskaya et al., 2004; Bowman, 2007). Finding beneficial bacteria could be interesting and pave the way for prophylactic measures in oyster farming.
Here we identified three bacterial families significantly associated with resistant oysters: Colwelliaceae, Cyanobacteria (Subsection III, family I), and Rhodobacteraceae. Among Colwelliaceae, the Colwellia genus was already found within C. gigas microbiota (Madigan et al., 2014). In this study, this family and genus were significantly different between resistant and susceptible oysters in hatchery. However, some susceptible oysters also contained high abundances of these bacteria, and they were not significantly different between resistant and susceptible oyster families during transplantation. Altogether, these observations thus suggested a limited role of Colwelliaceae in POMS. Moreover, although Rhodobacteraceae was associated to juvenile oyster disease (Boettcher, 2005), this bacterial family was commonly associated with oysters (Asmani et al., 2016; Takenaka et al., 2017; Arahal et al., 2018). Strikingly, Cyanobacteria (Subsection III, family I) had low abundances in all susceptible oyster microbiota before disease development, and were abundant in most resistant oysters. Although cyanobacteria had a negative effect on other marine invertebrates such as scleractinian corals (Meyer et al., 2016), they were already observed at high relative abundances within oyster microbiota (Chauhan et al., 2014). Particularly, they were abundant in the digestive gland, connective tissue, mantle, and gonad of oysters (Avila-Poveda et al., 2014). Because cyanobacteria persisted in oyster tissues without signs of alterations, a possible endosymbiotic relationship was even proposed (Avila-Poveda et al., 2014). Notably, cyanobacteria were also negatively correlated to culturable Vibrio abundance in free-living microbial communities (Turner et al., 2009). Hence, cyanobacteria might play a role of barrier against pathogens from the genus Vibrio. In particular, photosynthetic activity of cyanobacteria could inhibited pathogen growth through the production of reactive oxygen species, such as observed for Plasmodium infection in Anopheles gambiae (Cirimotich et al., 2011).
To conclude, we placed oysters from resistant and susceptible families in field condition for 5 days during a period in favor of POMS. The analyses of oyster microbiota at the early steps of infection revealed differences between resistant and susceptible oyster families. These differences suggested that both the structure (evenness) and the composition (putative opportunists, pathogens, mutualists) might predict oyster mortalities. Future studies should test the role of putative pathogens (Mycoplasmataceae and Rhodospirillaceae), but also of putative symbionts, such as Cyanobacteria (Subsection III, family I) and Rhodobacteraceae within C. gigas microbiota. In particular, these studies should evaluate the possibility of using Cyanobacteria (Subsection III, family I) as probiotics for oyster farming.
The datasets generated during the current study are available in the Sequence Read Archive repository under BioProject ID PRJNA419907.
CC, JL, J-ME, YG, LD, GM, and ET were involved in the study concept and design. BP was involved in the generation and maintaining of all animals used in this study. JL, BP, J-ME, YG, GM, and ET were involved in the collection of samples. CC, JL, AL, and ET were involved in data acquisition and analysis. CC and ET drafted the manuscript. All authors contributed to critical revisions and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CC benefited from post-doctoral fellowships from CNRS and IFREMER. This work was supported by the French National Research Agency ANR, project ANR-14-CE19-0023 DECIPHER (coordinator GM) and the DHOF program of the UMR5244/IHPE (http://ihpe.univ-perp.fr/en/ihpe-transversal-holobiont/). This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement no. 678589 (VIVALDI project). This study is set within the framework of the “Laboratoires d’Excellence (LABEX)” TULIP (ANR-10-LABX-41) and CeMEB (ANR-10-LABX-04-01).
We thank the staff of the Ifremer stations of Argenton (LPI, PFOM) and Sète (LER), and the Comité Régional de Conchyliculture de Méditerranée (CRCM) for technical support in the collection of the oyster genitors and reproduction. We are grateful to IHPE members for stimulating discussions. We also thank the genotoul bioinformatics platform Toulouse Midi-Pyrenees and Sigenae group for providing help and computing resources thanks to Galaxy instance http://sigenae-workbench.toulouse.inra.fr. This manuscript has been released as a Pre-Print (Clerissi et al., 2018) at https://www.biorxiv.org/content/10.1101/378125v2.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00311/full#supplementary-material
Arahal, D. R., Lucena, T., Rodrigo-Torres, L., and Pujalte, M. J. (2018). Ruegeria denitrificans sp. nov., a marine bacterium in the family Rhodobacteraceae with the potential ability for cyanophycin synthesis. Int. J. Syst. Evol. Microbiol. 68, 2515–2522. doi: 10.1099/ijsem.0.002867
Asmani, K., Petton, B., Le Grand, J., Mounier, J., Robert, R., and Nicolas, J.-L. (2016). Establishment of microbiota in larval culture of Pacific oyster, Crassostrea gigas. Aquaculture 464, 434–444. doi: 10.1016/j.aquaculture.2016.07.020
Avila-Poveda, O. H., Torres-Ariño, A., Girón-Cruz, D. A., and Cuevas-Aguirre, A. (2014). Evidence for accumulation of Synechococcus elongatus (Cyanobacteria: Cyanophyceae) in the tissues of the oyster Crassostrea gigas (Mollusca: Bivalvia). Tissue Cell 46, 379–387. doi: 10.1016/j.tice.2014.07.001
Azéma, P., Lamy, J.-B., Boudry, P., Renault, T., Travers, M.-A., and Dégremont, L. (2017). Genetic parameters of resistance to Vibrio aestuarianus, and OsHV-1 infections in the Pacific oyster, Crassostrea gigas, at three different life stages. Genet. Sel. Evol. 49:23. doi: 10.1186/s12711-017-0297-2
Balvanera, P., Kremen, C., and Martínez-Ramos, M. (2005). Applying community structure analysis to ecosystem function: examples from pollination and carbon storage. Ecol. Appl. 15, 360–375. doi: 10.1890/03-5192
Barbosa Solomieu, V., Renault, T., and Travers, M.-A. (2015). Mass mortality in bivalves and the intricate case of the Pacific oyster, Crassostrea gigas. J. Invertebr. Pathol. 131, 2–10. doi: 10.1016/j.jip.2015.07.011
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Boettcher, K. J. (2005). Roseovarius crassostreae sp. nov., a member of the Roseobacter clade and the apparent cause of juvenile oyster disease (JOD) in cultured Eastern oysters. Int. J. Syst. Evol. Microbiol. 55(Pt 4), 1531–1537. doi: 10.1099/ijs.0.63620-0
Bowman, J. P. (2007). Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 5, 220–241. doi: 10.3390/md504220
Chauhan, A., Wafula, D., Lewis, D. E., and Pathak, A. (2014). Metagenomic assessment of the Eastern oyster-associated microbiota. Genome Announc. 2:e01083-14. doi: 10.1128/genomeA.01083-14
Cirimotich, C. M., Dong, Y., Clayton, A. M., Sandiford, S. L., Souza-Neto, J. A., Mulenga, M., et al. (2011). Natural microbe-mediated refractoriness to plasmodium infection in Anopheles gambiae. Science 332, 855–858. doi: 10.1126/science.1201618
Clerissi, C., de Lorgeril, J., Petton, B., Lucasson, A., Escoubas, J.-M., Gueguen, Y., et al. (2018). Microbiota composition and evenness predict survival rate of oysters confronted to Pacific Oyster Mortality Syndrome. bioRxiv [Preprint]. doi: 10.1101/378125
Davison, A. J., Trus, B. L., Cheng, N., Steven, A. C., Watson, M. S., Cunningham, C., et al. (2005). A novel class of herpesvirus with bivalve hosts. J. Gen. Virol. 86(Pt 1), 41–53. doi: 10.1099/vir.0.80382-0
de Lorgeril, J., Lucasson, A., Petton, B., Toulza, E., Montagnani, C., Clerissi, C., et al. (2018). Immune-suppression by OsHV-1 viral infection causes fatal bacteraemia in Pacific oysters. Nat. Commun. 9:4215. doi: 10.1038/s41467-018-06659-3
De Roy, K., Marzorati, M., Negroni, A., Thas, O., Balloi, A., Fava, F., et al. (2013). Environmental conditions and community evenness determine the outcome of biological invasion. Nat. Commun. 4:1383. doi: 10.1038/ncomms2392
Dégremont, L. (2011). Evidence of herpesvirus (OsHV-1) resistance in juvenile Crassostrea gigas selected for high resistance to the summer mortality phenomenon. Aquaculture 317, 94–98. doi: 10.1016/j.aquaculture.2011.04.029
Dégremont, L., Bédier, E., Soletchnik, P., Ropert, M., Huvet, A., Moal, J., et al. (2005). Relative importance of family, site, and field placement timing on survival, growth, and yield of hatchery-produced Pacific oyster spat (Crassostrea gigas). Aquaculture 249, 213–229. doi: 10.1016/j.aquaculture.2005.03.046
Dégremont, L., Nourry, M., and Maurouard, E. (2015). Mass selection for survival and resistance to OsHV-1 infection in Crassostrea gigas spat in field conditions: response to selection after four generations. Aquaculture 446, 111–121. doi: 10.1016/j.aquaculture.2015.04.029
Desriac, F., Le Chevalier, P., Brillet, B., Leguerinel, I., Thuillier, B., Paillard, C., et al. (2014). Exploring the hologenome concept in marine bivalvia: haemolymph microbiota as a pertinent source of probiotics for aquaculture. FEMS Microbiol. Lett. 350, 107–116. doi: 10.1111/1574-6968.12308
Escudié, F., Auer, L., Bernard, M., Mariadassou, M., Cauquil, L., Vidal, K., et al. (2017). FROGS: find, rapidly, OTUs with Galaxy Solution. Bioinformatics 34, 1287–1294. doi: 10.1093/bioinformatics/btx791
Faith, J. J., Guruge, J. L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A. L., et al. (2013). The long-term stability of the human gut microbiota. Science 341:1237439. doi: 10.1126/science.1237439
Fernandez-Piquer, J., Bowman, J. P., Ross, T., and Tamplin, M. L. (2012). Molecular analysis of the bacterial communities in the live Pacific oyster (Crassostrea gigas) and the influence of postharvest temperature on its structure. J. Appl. Microbiol. 112, 1134–1143. doi: 10.1111/j.1365-2672.2012.05287.x
Gibson, L. F., Woodworth, J., and George, A. M. (1998). Probiotic activity of Aeromonas media on the Pacific oyster, Crassostrea gigas, when challenged with Vibrio tubiashii. Aquaculture 169, 111–120. doi: 10.1016/S0044-8486(98)00369-X
Kalinovskaya, N. I., Ivanova, E. P., Alexeeva, Y. V., Gorshkova, N. M., Kuznetsova, T. A., Dmitrenok, A. S., et al. (2004). Low-molecular-weight, biologically active compounds from marine Pseudoalteromonas species. Curr. Microbiol. 48, 441–446. doi: 10.1007/s00284-003-4240-0
Kesarcodi-Watson, A., Miner, P., Nicolas, J.-L., and Robert, R. (2012). Protective effect of four potential probiotics against pathogen-challenge of the larvae of three bivalves: Pacific oyster (Crassostrea gigas), flat oyster (Ostrea edulis) and scallop (Pecten maximus). Aquaculture 344–349, 29–34. doi: 10.1016/j.aquaculture.2012.02.029
King, G. M., Judd, C., Kuske, C. R., and Smith, C. (2012). Analysis of stomach and gut microbiomes of the Eastern oyster (Crassostrea virginica) from coastal Louisiana, USA. PLoS One 7:e51475. doi: 10.1371/journal.pone.0051475
King, W. L., Siboni, N., Williams, N. L. R., Kahlke, T., Nguyen, K. V., Jenkins, C., et al. (2019). Variability in the composition of Pacific oyster microbiomes across oyster families exhibiting different levels of susceptibility to OsHV-1 μvar disease. Front. Microbiol. 10:473. doi: 10.3389/fmicb.2019.00473
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Lokmer, A., and Wegner, K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9, 670–682. doi: 10.1038/ismej.2014.160
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Madigan, T. L., Bott, N. J., Torok, V. A., Percy, N. J., Carragher, J. F., de Barros Lopes, M. A., et al. (2014). A microbial spoilage profile of half shell Pacific oysters (Crassostrea gigas) and Sydney rock oysters (Saccostrea glomerata). Food Microbiol. 38, 219–227. doi: 10.1016/j.fm.2013.09.005
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2015). Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ 3:e1420. doi: 10.7717/peerj.1420
Mansergh, S., and Zehr, J. P. (2014). Vibrio diversity and dynamics in the Monterey Bay upwelling region. Front. Microbiol. 5:48. doi: 10.3389/fmicb.2014.00048
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meyer, J. L., Gunasekera, S. P., Scott, R. M., Paul, V. J., and Teplitski, M. (2016). Microbiome shifts and the inhibition of quorum sensing by Black Band Disease cyanobacteria. ISME J. 10, 1204–1216. doi: 10.1038/ismej.2015.184
Mortzfeld, B. M., Urbanski, S., Reitzel, A. M., Künzel, S., Technau, U., and Fraune, S. (2016). Response of bacterial colonization in Nematostella vectensis to development, environment and biogeography. Environ. Microbiol. 18, 1764–1781. doi: 10.1111/1462-2920.12926
Ng, J. C. Y., Chan, Y., Tun, H. M., Leung, F. C. C., Shin, P. K. S., and Chiu, J. M. Y. (2015). Pyrosequencing of the bacteria associated with Platygyra carnosus corals with skeletal growth anomalies reveals differences in bacterial community composition in apparently healthy and diseased tissues. Front. Microbiol. 6:1142. doi: 10.3389/fmicb.2015.01142
Oliver, K. M., Russell, J. A., Moran, N. A., and Hunter, M. S. (2003). Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc. Natl. Acad. Sci. U.S.A. 100, 1803–1807. doi: 10.1073/pnas.0335320100
Pernet, F., Barret, J., Le Gall, P., Corporeau, C., Dégremont, L., Lagarde, F., et al. (2012). Mass mortalities of Pacific oysters Crassostrea gigas reflect infectious diseases and vary with farming practices in the Mediterranean Thau lagoon, France. Aquac. Environ. Interact. 2, 215–237. doi: 10.3354/aei00041
Pernet, F., Lupo, C., Bacher, C., and Whittington, R. J. (2016). Infectious diseases in oyster aquaculture require a new integrated approach. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20150213. doi: 10.1098/rstb.2015.0213
Pernet, F., Tamayo, D., Fuhrmann, M., and Petton, B. (2019). Deciphering the effect of food availability, growth and host condition on disease susceptibility in a marine invertebrate. J. Exp. Biol. 222(Pt 17):jeb210534. doi: 10.1242/jeb.210534
Petton, B., Bruto, M., James, A., Labreuche, Y., Alunno-Bruscia, M., and Le Roux, F. (2015). Crassostrea gigas mortality in France: the usual suspect, a herpes virus, may not be the killer in this polymicrobial opportunistic disease. Front. Microbiol. 6:686. doi: 10.3389/fmicb.2015.00686
Petton, B., Pernet, F., Robert, R., and Boudry, P. (2013). Temperature influence on pathogen transmission and subsequent mortalities in juvenile Pacific oysters Crassostrea gigas. Aquac. Environ. Interact. 3, 257–273. doi: 10.3354/aei00070
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. doi: 10.1093/nar/29.9.e45
R Development Core Team (2008). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rico-Villa, B., Pouvreau, S., and Robert, R. (2009). Influence of food density and temperature on ingestion, growth and settlement of Pacific oyster larvae, Crassostrea gigas. Aquaculture 287, 395–401. doi: 10.1016/j.aquaculture.2008.10.054
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rubio, T., Oyanedel, D., Labreuche, Y., Toulza, E., Luo, X., Bruto, M., et al. (2019). Species-specific mechanisms of cytotoxicity toward immune cells determine the successful outcome of Vibrio infections. Proc. Natl. Acad. Sci. U.S.A. 116, 14238–14247. doi: 10.1073/pnas.1905747116
Rungrassamee, W., Klanchui, A., Maibunkaew, S., and Karoonuthaisiri, N. (2016). Bacterial dynamics in intestines of the black tiger shrimp and the Pacific white shrimp during Vibrio harveyi exposure. J. Invertebr. Pathol. 133, 12–19. doi: 10.1016/j.jip.2015.11.004
Samain, J. F., Dégremont, L., Soletchnik, P., Haure, J., Bédier, E., Ropert, M., et al. (2007). Genetically based resistance to summer mortality in the Pacific oyster (Crassostrea gigas) and its relationship with physiological, immunological characteristics and infection processes. Aquaculture 268, 227–243. doi: 10.1016/j.aquaculture.2007.04.044
Takenaka, S., Yoshikawa, T., Kadowaki, S., Okunishi, S., and Maeda, H. (2017). Microflora in the soft tissue of the Pacific oyster Crassostrea gigas exposed to the harmful microalga Heterosigma akashiwo. Biocontrol Sci. 22, 79–87. doi: 10.4265/bio.22.79
Turner, J. W., Good, B., Cole, D., and Lipp, E. K. (2009). Plankton composition and environmental factors contribute to Vibrio seasonality. ISME J. 3, 1082–1092. doi: 10.1038/ismej.2009.50
Wang, A., Ran, C., Wang, Y., Zhang, Z., Ding, Q., Yang, Y., et al. (2019). Use of probiotics in aquaculture of China–a review of the past decade. Fish Shellfish Immunol. 86, 734–755. doi: 10.1016/j.fsi.2018.12.026
Wendling, C. C., Fabritzek, A. G., and Wegner, K. M. (2017). Population-specific genotype x genotype x environment interactions in bacterial disease of early life stages of Pacific oyster larvae. Evol. Appl. 10, 338–347. doi: 10.1111/eva.12452
Wilsey, B. J., and Polley, H. W. (2002). Reductions in grassland species evenness increase dicot seedling invasion and spittle bug infestation. Ecol. Lett. 5, 676–684. doi: 10.1046/j.1461-0248.2002.00372.x
Wilsey, B. J., and Potvin, C. (2000). Biodiversity and ecosystem functioning: importance of species evenness in an old field. Ecology 81, 887–892. doi: 10.2307/177163
Wittebolle, L., Marzorati, M., Clement, L., Balloi, A., Daffonchio, D., Heylen, K., et al. (2009). Initial community evenness favours functionality under selective stress. Nature 458, 623–626. doi: 10.1038/nature07840
Woodhams, D. C., Vredenburg, V. T., Simon, M.-A., Billheimer, D., Shakhtour, B., Shyr, Y., et al. (2007). Symbiotic bacteria contribute to innate immune defenses of the threatened mountain yellow-legged frog, Rana muscosa. Biol. Conserv. 138, 390–398. doi: 10.1016/j.biocon.2007.05.004
Zaneveld, J. R., McMinds, R., and Vega Thurber, R. (2017). Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2:17121. doi: 10.1038/nmicrobiol.2017.121
Keywords: holobiont, microbiome, metabarcoding, fitness, bacteria
Citation: Clerissi C, de Lorgeril J, Petton B, Lucasson A, Escoubas J-M, Gueguen Y, Dégremont L, Mitta G and Toulza E (2020) Microbiota Composition and Evenness Predict Survival Rate of Oysters Confronted to Pacific Oyster Mortality Syndrome. Front. Microbiol. 11:311. doi: 10.3389/fmicb.2020.00311
Received: 21 November 2019; Accepted: 12 February 2020;
Published: 27 February 2020.
Edited by:
Alfonso Benítez-Páez, Institute of Agrochemistry and Food Technology (IATA), SpainReviewed by:
Aide Lasa, University of Santiago de Compostela, SpainCopyright © 2020 Clerissi, de Lorgeril, Petton, Lucasson, Escoubas, Gueguen, Dégremont, Mitta and Toulza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camille Clerissi, Y2FtaWxsZS5jbGVyaXNzaUBlcGhlLnBzbC5ldQ==; Eve Toulza, ZXZlLnRvdWx6YUB1bml2LXBlcnAuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.