 Kristin Grøsvik1Almaz Nigatu Tesfahun1Izaskun Muruzábal-Lecumberri1
Kristin Grøsvik1Almaz Nigatu Tesfahun1Izaskun Muruzábal-Lecumberri1 Gyri Teien Haugland2
Gyri Teien Haugland2 Ingar Leiros3
Ingar Leiros3 Peter Ruoff1Jan Terje Kvaløy4Ingeborg Knævelsrud1Hilde Ånensen1Marina Alexeeva1Kousuke Sato5Akira Matsuda6Ingrun Alseth7Arne Klungland7,8
Peter Ruoff1Jan Terje Kvaløy4Ingeborg Knævelsrud1Hilde Ånensen1Marina Alexeeva1Kousuke Sato5Akira Matsuda6Ingrun Alseth7Arne Klungland7,8 Svein Bjelland1*
Svein Bjelland1*- 1Department of Chemistry, Bioscience and Environmental Technology, Centre for Organelle Research, Faculty of Science and Technology, University of Stavanger, Stavanger, Norway
- 2Department of Biological Sciences, University of Bergen, Bergen, Norway
- 3The Norwegian Structural Biology Centre, Department of Chemistry, UiT – The Arctic University of Norway, Tromsø, Norway
- 4Department of Mathematics and Physics, Faculty of Science and Technology, University of Stavanger, Stavanger, Norway
- 5Faculty of Pharmaceutical Sciences, Health Sciences University of Hokkaido, Tobetsu, Japan
- 6Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo, Japan
- 7Department of Microbiology, Oslo University Hospital, Oslo, Norway
- 8Department of Molecular Medicine, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
The cellular methyl donor S-adenosylmethionine (SAM) and other endo/exogenous agents methylate DNA bases non-enzymatically into products interfering with replication and transcription. An important product is 3-methyladenine (m3A), which in Escherichia coli is removed by m3A-DNA glycosylase I (Tag) and II (AlkA). The tag gene is constitutively expressed, while alkA is induced by sub-lethal concentrations of methylating agents. We previously found that AlkA exhibits activity for the reactive oxygen-induced thymine (T) lesion 5-formyluracil (fU) in vitro. Here, we provide evidence for AlkA involvement in the repair of oxidized bases by showing that the adenine (A) ⋅ T → guanine (G) ⋅ cytosine (C) mutation rate increased 10-fold in E. coli wild-type and alkA– cells exposed to 0.1 mM 5-formyl-2′-deoxyuridine (fdU) compared to a wild-type specific reduction of the mutation rate at 0.2 mM fdU, which correlated with alkA gene induction. G⋅C → A⋅T alleviation occurred without alkA induction (at 0.1 mM fdU), correlating with a much higher AlkA efficiency for fU opposite to G than for that to A. The common keto form of fU is the AlkA substrate. Mispairing with G by ionized fU is favored by its exclusion from the AlkA active site.
Introduction
Reactive oxygen species (ROS) are formed as by-products in the electron transport chain of bacterial plasma membranes, mitochondria, chloroplasts, and some other eukaryotic organelles. Thus, oxidative damage to DNA and its precursors is substantial in all aerobic cells. ROS are also formed by photosensitization reactions involving both ultraviolet and visible light. The most reactive ROS is the hydroxyl radical (⋅OH), which is formed from hydrogen peroxide (H2O2) and Fe2+ in the Fenton reaction, and by ionizing radiation. Singlet oxygen is another important reactive, but non-radical, oxygen form [for a review see Halliwell and Gutteridge (1989)].
Several different nucleic acid base modifications are formed by ROS (Cadet et al., 1997). Many of these, including ring-saturated and ring-fragmented thymine (T) residues, are mainly cytotoxic lesions that contribute slightly to mutation induction (Bjelland and Seeberg, 2003). In contrast, guanine (G) oxidized at the 8-position (8-oxo-7,8-dihydroguanine; oxoG) is a principal mutagenic ROS-induced lesion (Lindahl, 1993; Freudenthal et al., 2015). The pyrimidine oxidation products 5-hydroxycytosine, 5-hydroxyuracil, and uracil glycol also cause mispairing and contribute to mutation induction (Feig et al., 1994; Purmal et al., 1994; Kreutzer and Essigmann, 1998). In Escherichia coli, oxidized bases are primarily removed from DNA by three DNA glycosylases: formamidopyrimidine-DNA glycosylase (Fpg), endonuclease III (Nth), and endonuclease VIII (Nei) (Zharkov et al., 2003; Boiteux et al., 2017; Lee and Wallace, 2017). How cells repair and tolerate DNA lesions has considerable impact on human diseases, including cancer and neurodegeneration (Berquist and Wilson, 2012; Dizdaroglu, 2015).
Almost 30 years ago, 5-formyluracil (fU) was identified as an abundant oxidized form of thymine in DNA, generated in yields similar to oxoG (Kasai et al., 1990; Douki et al., 1996). Later, a potential fU mutagenic or cytotoxic effect was suggested from the observation that fU is removed efficiently from DNA by 3-methyladenine (m3A)-DNA glycosylase II (AlkA) in E. coli (Bjelland et al., 1994) and by DNA glycosylase present in mammalian and human cell-free extracts (Bjelland et al., 1995; Zhang et al., 1995). The KM and Vmax values for AlkA release of fU are similar to m3A (Bjelland et al., 1994). AlkA is the E. coli homolog to the extensively characterized mammalian methylpurine-DNA glycosylase (MPG) (Labahn et al., 1996; Yamagata et al., 1996; Hollis et al., 2000; O’Brien and Ellenberger, 2004; Sedgwick, 2004), which exhibits no activity for fU (Bjelland et al., 1994). DNA glycosylases initiate the base excision repair (BER) pathway, which is predominantly completed by reinsertion of a single nucleotide (nt) via 5′-acting apurinic/apyrimidinic (AP) endonuclease (Xth and Nfo proteins in E. coli) (Doetsch and Cunningham, 1990), DNA deoxyribose-5′-phosphate lyase, DNA polymerase, and DNA ligase activity (Lindahl and Wood, 1999; Friedberg et al., 2006; Zharkov, 2008). Several in vitro and in vivo studies have demonstrated that fU generated in DNA or incorporated into DNA from the nucleotide precursor pool induces several base substitutions, except the G ⋅ cytosine (C) → C⋅G transversion (Yoshida et al., 1997; Zhang et al., 1997; Fujikawa et al., 1998; Terato et al., 1999; Ånensen et al., 2001). Oxidized bases or nucleosides may be taken up from the environment, anabolized, and incorporated into DNA. Importantly, this metabolic pathway for base damage generation has been mostly neglected.
Methyl transfer reactions are involved in numerous cellular processes. S-adenosylmethionine (SAM) is the most common methyl donor used by methyltransferases. However, in addition to its function as a methyltransferase co-substrate, SAM and other endogenous and exogenous agents non-enzymatically methylate normal DNA bases into products that interfere with replication and transcription (Sedgwick, 2004; Shrivastav et al., 2010; Soll et al., 2017). An important product is m3A, which is removed by m3A-DNA glycosylase I (Tag) (Sakumi et al., 1986; Bjelland and Seeberg, 1987) and II (AlkA) in E. coli (Nakabeppu et al., 1984). While the tag gene is constitutively expressed, alkA is induced by sub-lethal methylating agent concentrations (Evensen and Seeberg, 1982; Karran et al., 1982). Because of this, the discovery that AlkA efficiently removes fU from DNA in vitro was unexpected (Bjelland et al., 1994). Although this was confirmed by other studies (Masaoka et al., 1999; Terato et al., 1999), no supporting in vivo evidence exists for AlkA involvement in counteracting the consequences of ROS-induced DNA damage.
Here, we present the first in vivo evidence that AlkA is involved in the processing of fU lesions in DNA. When 5-formyl-2′-deoxyuridine (fdU) is added to exponentially growing E. coli cells to incorporate fU into DNA, repair-proficient wild-type and repair-deficient alkA gene-lacking cells respond differently, indicating that AlkA is a key factor in determining the base substitution induced by fdU. In addition, we demonstrate that fdU increases alkA gene expression.
Materials and Methods
Materials
5-Formyl-2′-deoxyuridine was prepared as described (Ono et al., 1994). Rifampicin was obtained from Sigma (Cat. No. R-3501).
Bacterial Cells
Three E. coli K-12 derivatives were investigated: AB1157 (wild-type) (Boyce and Howard-Flanders, 1964); MS23 [alkA–; AB1157 transduced by P1(his+ alkA1–)] (Yamamoto et al., 1978); and BW528 (xth– nfo–) (Gu et al., 1993). Frozen glycerol cultures were stored at −80°C.
Mutagenesis
Mutagenesis was performed as previously described (Cupples and Miller, 1989), with all growth carried out at 37°C. Cells were transferred from freeze culture to 5 mL LB (lysogeny-broth) medium, grown overnight, and spread on LB agar plates for further growth and short-term storage at 4°C. All cells in a mutagenesis culture were derived from a single colony picked from the LB agar plate and grown overnight in 2 mL A medium [A buffer (see below) containing 1 mM MgSO4, 0.2% (w/v) glucose, 0.04 mg/mL L-amino acids (Thr, Arg, Pro, Leu, His), and vitamin B1 (5 μg/mL)] (contamination controls without cells added). The number of cells after overnight culture was determined from the OD600. Overnight cultures (10 μL) were diluted in A buffer [K2HPO4 (10.5 g/L), KH2PO4 (4.5 g/L), (NH4)2SO4 (1 g/L), and C6H5Na3O7 × 2H2O (0.5 g/L)] to (usually) 100,000–200,000 cells/mL. Then, 9000–45,000 cells were added to 2 mL A medium. The cultures grew for 2 h to allow for adaptation of cells and increase cell number. fdU addition (controls without fdU determined the spontaneous mutagenesis rate) defined the start of the mutagenesis experiment/culture (2 mL). Cells cultured with fdU were grown for 20–50 h. The rifR mutants and viable cells were selected on minimal agar plates containing rifampicin (150 μg/mL).
Mutation Rate
The experimental procedures to determine the mutation rate μ follow recommendations described elsewhere (Rosche and Foster, 2000) for the p0 method (Luria and Delbrück, 1943), which is frequently used to estimate low mutation rates, such as deoxynucleoside analog (like fdU)-induced mutations. Thus, mutagenesis cultures grown to a similar final cell concentration (Nt) of 1.5–1.8 × 109 per 2 mL were selected from each experimental group (i.e. a certain E. coli strain/mutant growing in the presence of a defined fdU concentration). Then, μ was calculated according to the p0 method, where p0 (the mutant-free culture proportion) was between 0.1 and 0.3 for all groups. From each culture, we usually plated 100 μL on each rifampicin-containing plate (six replicates per culture), which resulted in a z (fraction of a culture plated) value of 0.3. Only cultures with z-values identical or very close to 0.3 were included (see the section “Mutagenesis Experiments” in the Supplementary Material). The estimator mobs (observed number of mutations per culture) was calculated from the observed p0 value and ranged from 1.1 to 2.3, resulting in mact (actual number of mutations per culture) values from 2.1 to 4.5, following calculation by equation [18] in the cited article (Rosche and Foster, 2000; see Supplementary Table S2A).
DNA Amplification and Sequencing
The rifR colonies were grown at 37°C in 2 mL LB medium containing 150 μL/mL rifampicin (only one mutant was characterized from each mutagenesis culture). Enriched chromosomal DNA was prepared for polymerase chain reaction (PCR) by heating 5 μL bacterial culture in 100 μL sterile water at 100°C for 5 min, followed by cooling on ice. Cultures were centrifuged and the supernatant collected. The rpoB rifR region was amplified by PCR using the forward primer 5′-GCCAAGCCGATTTCC-3′ (F-1021) and the reverse primer 5′-GTATTCGTTAGTCTG-3′ (R-1022) (0.2 pmol/μL each) using a KOD Hot Start DNA Polymerase kit (VWR) or GoTaq HotStart Polymerase (Promega) in a 50 μL reaction volume. The PCR products (10–80 ng DNA) were purified using illustra MicroSpin S-400 HR columns (GE Healthcare) or NucleoSpin Gel and PCR Clean-up (Macherey-Nagel). DNA sequencing (using F-1021 as primer) was performed by the Genome Analysis Centre, Norwich, United Kingdom and GATC Biotech, Cologne, Germany (using an Applied Biosystems 3730xl DNA Analyzer).
alkA Wild-Type Gene Cloning and Expression and AlkA Protein Purification
The alkA gene was amplified from DNA isolated from E. coli K-12 (AB1157) by PCR using the following primers: forward, 5′-GCCGTCATATGTATACCCTGAACTGGCAGCC-3′, Tm 67.26°C; reverse, 5′-CCGCTCGAGTCATCATGCTTCGTCT GGTTGCCA-3′, Tm 71.46°C (Tm was determined by the Thermo Scientific Tm calculator web tool. The first stop codon is outlined in bold). The PCR product was inserted into the pET21a vector (NEB), using the restriction enzymes NdeI and XhoI (recognition sites underlined in the forward and reverse primer sequences, respectively) and T4 DNA ligase (all three enzymes from NEB). Plasmid was transformed into One shot chemical competent TOP10 E. coli cells (Invitrogen). Expressed plasmid was purified from three positive colonies identified by PCR screening using Dynazyme and Finnzymes (ThermoFisher Scientific). Correct insert sequences were confirmed by sequencing both DNA strands at the DNA Sequencing Facility at the High Technology Centre in Bergen, Norway. After testing the E. coli cells BL-21-CodonPlus (DE3)-RIL (Stratagene), BL21(DE3) (Invitrogen), and EB106DE3 for alkA gene overexpression and AlkA production, CodonPlus and BL21 cells were found to be superior. alkA was overexpressed in BL21 cells, since they produced the highest amount of soluble AlkA. The bacteria were grown in 1-l ZYP-5052 medium according to Studier’s auto-induction method (Studier, 2005) and incubated at 37°C for 24 h. After harvesting by centrifugation (4 g wet weight, stored at −20°C), the cells were resuspended in 0.1 M Tris–HCl pH 8.5, 1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM β-mercaptoethanol, and 20% (v/v) glycerol (1 g cells per 10 mL), disrupted by sonication and the cell debris was removed by centrifugation. Soluble proteins were precipitated with ammonium sulfate (40–70% fraction). AlkA protein was purified using a previously described procedure (Bjelland et al., 1994), with slight modifications. The ammonium sulfate precipitate was dissolved in 5 mL 0.1 M Tris–HCl pH 8.5, 1 mM EDTA, 10 mM β-mercaptoethanol, and 20% (v/v) glycerol and filtered with a 26/60 Superdex 200 prep grade column (Cat. No. 17-1071-01, GE Healthcare) in 330 mL of the same buffer. AlkA eluted after 248 mL, and six 3-mL fractions were collected and pooled. The protein was concentrated by centrifugation using a 20 mL Vivaspin concentrator with a 3000 MWCO PES membrane (Sartorius Stedim Biotech). The elution buffer was exchanged to 50 mM Mes pH 6.0, 1 mM EDTA, 10 mM β-mercaptoethanol, and 20% (v/v) glycerol with PD-10 desalting columns (GE Healthcare). Cation exchange chromatography was carried out on a 5 mL Bio-Scale Mini Macro-Prep High S column (Bio-Rad) equilibrated with the same buffer. Proteins were eluted with a 0–1.0 M NaCl gradient. AlkA started to elute at 0.24 M NaCl, and 1-mL fractions were collected (total volume: 21 mL). The protein was concentrated using the same concentrator and membrane as above, and the buffer exchanged to 0.2 M Tris–HCl pH 7.27, 1 mM EDTA, 1 mM dithiothreitol (DTT), and 20% (v/v) glycerol with PD Spin Trap G25 columns (GE Healthcare, Cat. No. 28-9180-04). Protein fractions from each step during the purification procedure were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) (Supplementary Figure S3). Protein concentration was measured using Bio-Rad Protein Assay (BioRad, Cat. No. 500-0006).
Incision of 5-Formyluracil-Containing DNA
Polydeoxynucleotides containing fdU monophosphate (fdUMP) at a specific site were synthesized as described (Ono et al., 1994), and fluorescently labeled with Cy3 at the 5′-end (Sigma and Eurofins). The labeled strand [Cy3] 5′-GGCGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAG TfUGACCTGCAGGCATGCAAGCTTGAGT-3′ (64 nt) was annealed to equimolar amounts of a complementary strand with A or G opposite fU (called fU⋅A and fU⋅G substrate, respectively) by heating at 90°C for 3 min followed by cooling at 37°C for 15 min and to room temperature for 30 min. Increasing AlkA amounts were incubated with 11 nM fU⋅A or fU⋅G substrates in 70 mM MOPS [3-(N-morpholino)propanesulfonic acid] pH 7.0, 1 mM EDTA, 1 mM DTT, and 5% (v/v) glycerol (reaction buffer) at 37°C for 1 h in a final volume of 50 μL. Reactions were terminated by the addition of 20 mM EDTA, 0.5% (w/v) SDS, and 190 μg/mL proteinase K. DNA was precipitated with ethanol and resuspended in 10 μL water. The abasic sites formed by AlkA were incised by 0.1 M NaOH treatment at 90°C for 10 min. Samples were mixed with 10 μL loading solution containing 80% (v/v) formamide, 1 mM EDTA, and 0.05% (w/v) Blue Dextran, and incubated at 95°C for 5 min to denature DNA. After cooling on ice, 10 samples were subjected to denaturing PAGE [20% (w/v) polyacrylamide gels with 7 M urea]. Experimental data were fitted to Michaelis–Menten kinetics by non-linear regression using KaleidaGraph (Synergy Software).
pH Dependence of the Excision of fU and m3A From DNA by AlkA
The m3A-DNA glycosylase activity was determined as a function of pH as previously described (Bjelland and Seeberg, 1987). This also applies to the determination of fU-DNA glycosylase activity using substrate and enzyme conditions as described elsewhere (Bjelland et al., 1994). To prepare [3H]methylated DNA, calf thymus DNA (Sigma, code D3664) was treated with [3H]N-methyl-N′-nitrosourea (MNU; 18.4 Ci/mmol, GE Healthcare), as previously described (Alseth et al., 2005). E. coli [methyl-3H]thymine-labeled DNA, which accumulates [3H]fU residues upon storage (Bjelland et al., 1995), was provided by New England Nuclear (Cat. No. NET-561). Essentially, [3H]MNU-treated [2000 disintegrations per min (dpm)] and aged [methyl-3H]thymine-labeled DNA (10,000 dpm; 4000 dpm [3H]fU) were incubated with AlkA (0.084–0.168 and 1.969 pmol, respectively) (Bjelland et al., 1994, 1995) in a modified universal buffer (Johnson and Lindsey, 1939; Bjelland and Seeberg, 1987) at 37°C for 10 min, followed by DNA precipitation with ethanol. Supernatant radioactivity was measured in a Tri-CarbTM liquid scintillation analyzer (Model 1900 TR) from Packard.
fU and m3A Docking in the AlkA Active Site
AlkA served as the receptor in automated docking simulations using AutoDock software (Morris et al., 1998), based on the AlkA crystal structure in complex with 10 nt double-stranded DNA containing the modified abasic nt 1-azaribose (Hollis et al., 2000). A single-stranded trinucleotide with the 1-azaribose part substituted by fdU was modified into the docking ligand to reduce the molecular complexity and computing power. Nevertheless, super-positioning the docked trinucleotide back onto the DNA fragment in the crystal structure of the DNA–protein complex (Hollis et al., 2000) resulted in similar conformations, indicating that the DNA–protein interactions were adequately mimicked in the automatic docking trials. Since m3A is the preferred AlkA substrate (Lindahl, 1993), m3A-containing trinucleotide was included as a positive control. To allow for some degree of ligand reorientation during docking, four fU and three m3A freely rotational bonds were selected.
alkA and ada Gene Expression Analysis
RT-qPCR was used to determine the relative alkA and ada expression in wild-type E. coli (AB1157) 1, 3, 6, and 24 h after adding 0.1 or 0.2 mM fdU to the growth culture. A bacterial growth culture was prepared by adding 500 μL cells from an overnight culture (OD600 ∼1.5) to 10 mL LB medium. After 2 h incubation to OD600 ∼0.5, 3 mL growth culture was treated with 0.1 or 0.2 mM fdU (control samples without treatment). Then, the samples were incubated for 1, 3, 6, and 24 h. All incubations were performed at 37°C with shaking. Before RNA isolation, 1.5 mL culture (1 × 108 cells) from each sample was mixed with 500 μL RNAprotect Cell Reagent (Qiagen) for RNA stabilization. Total RNA was extracted using a TRIzol Max Bacterial RNA Isolation Kit (Ambion Life Technologies). Genomic DNA contamination was removed by DNase I treatment using a RNA purification kit (Zymo Research), followed by resuspension of RNA pellets in 50 μL DNase/RNase-free water. Next, synthesis of double-stranded cDNA and integrated removal of genomic DNA was conducted using a QuantiTect reverse transcription reagent kit (Qiagen). To normalize the target gene expression level for each sample, 1 μg total RNA was used for cDNA synthesis in a 20 μL final reaction volume. cDNA was synthesized at 42°C for 15 min and 95°C for 3 min. Diluted cDNA (fivefold) was used as a template for amplifications using 10 μM target primer mix (Primerdesign) and SYBR green master mix (Qiagen) in a 20 μL reaction mixture. Test samples without reverse transcriptase and without template were used to test for genomic DNA and other technical contaminations. PCR assays were performed with a LightCycler 96 instrument (Roche, Germany) with the following conditions: 95°C for 2 min, followed by 45 cycles of 95°C for 15 s, 60°C for 60 s, and 65°C for 30 s. All data shown resulted from three independent replicates. Real-time PCR runs were measured in duplicate. Relative target gene expression was normalized to reference gene (tag) expression.
Statistical Analysis
Continuous data are reported as averages and standard errors. Categorical data are reported as counts and percentages. Chi-square or Fisher’s exact tests, as appropriate, were used for testing differences in base substitution distributions between different mutant types and different fdU levels. P-values ≤ 0.05 were considered statistically significant. The statistical analyses were done in R version 3.4.3 (R Core Team, 2017). Confidence intervals for mutation rates were calculated as described (Rosche and Foster, 2000).
Results
AlkA Influences fdU Mutagenicity in E. coli
When fdU is converted to fdUMP in vivo, the latter inhibits thymidylate synthase causing cellular toxicity (Park et al., 1980; Ahmad et al., 1998; Masaoka et al., 1999; Klungland et al., 2001). However, mutagenesis experiments using an initial cell number (N0) of 9000 (as determined by OD600) indicated no significant growth inhibition by 0.1 or 0.2 mM fdU (Supplementary Figure S1 and Supplementary Table S1A) showing moderate toxicity in both the wild-type and the repair-deficient cells. Further, these results agree with the notion that fU in DNA is a mutagenic lesion (Bjelland and Seeberg, 2003).
Our goal was to investigate whether there is any connection between fdU mutagenesis and BER, focusing on AlkA as the most active DNA glycosylase that removes fU from DNA (Bjelland et al., 1994; Bjelland and Seeberg, 2003). We systematically determined the mutation rate (μ) following the recommendations presented elsewhere (Rosche and Foster, 2000). Accordingly, wild-type (alkA+) was compared to the alkA– and xth– nfo– strains. The latter is deficient in exonuclease III (Xth) and endonuclease IV (Nfo), and thus should have increased unrepaired AP sites following fU excision. The spontaneous mutation rates in the alkA– and xth– nfo– cells were identical, and only marginally higher than in wild-type cells (Table 1A), indicating efficient back-up repair systems for AlkA, Xth, and Nfo under normal physiological conditions. Supplementing the culture medium with 0.1 mM fdU roughly doubled the mutation rate in the wild-type and alkA– cells, demonstrating a moderate mutagenicity both in the presence and absence of AlkA. Interestingly, 0.1 mM fdU caused a smaller increase (1.6-fold) in mutation rate in the xth– nfo– cells than in the wild-type and alkA– cells. This largely rules out the possibility that fdU mutagenesis occurs through trans-lesion bypass of AP sites caused by increased excision of fU from DNA at this concentration, rather than via the fdU lesion itself. Increasing fdU to 0.2 mM unexpectedly caused a noticeable decrease in the wild-type mutation rate, while the alkA– cells remained unaffected. This indicates that AlkA counteracts fdU mutation induction. In xth– nfo– cells, the mutation rate remained the same when the concentration increased from 0.1 to 0.2 mM fdU (Table 1A).
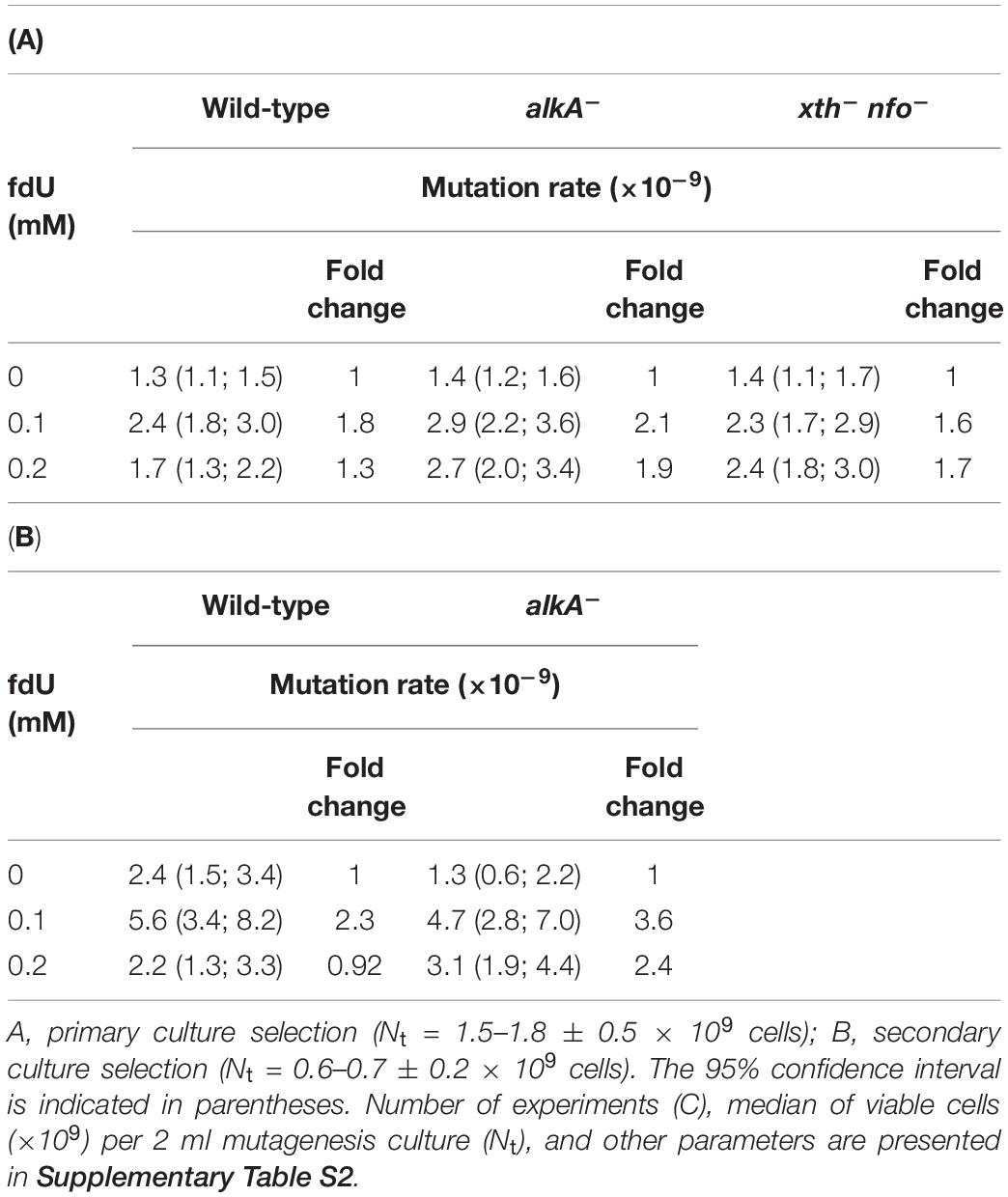
Table 1. Mutation rates for rifR in exponentially growing wild-type, alkA–, and xth– nfo– E. coli in the presence of 0–0.2 mM fdU.
Another selection of independent cultures with 0.6 ± 0.2 (median = 0.6) × 109 cells per 2 mL (Supplementary Tables S1B, S2B, and the section “Mutagenesis Experiments, Secondary Selection” in the Supplementary Material), although resulting in less accurate mutation rate values due to the lower number of cultures (Table 1B), largely confirmed the above results (Table 1A). Importantly, the increased mutation rate at 0.1 mM fdU was larger for both wild type and alkA– cells. When 0.2 mM fdU was added, the rate decreased to the spontaneous level in wild-type cells, but not alkA– cells (Table 1B).
In conclusion, adding 0.1 mM fdU to the growth medium doubles the mutation rate in both wild-type and alkA– cells, while 0.2 mM fdU causes a lower mutation rate in wild-type cells, but not alkA– cells, suggesting that AlkA alleviates fdU-mediated mutagenesis.
fdU Alters the Distribution of Base Substitutions Independent of AlkA
We next investigated whether AlkA deficiency influences the mutation spectrum at different fdU concentrations. Consequently, we determined the mutation types induced by fdU in E. coli by sequencing the rifampicin resistant (rifR) region of at least 60 mutants exposed to the same mutagen concentration (Table 2A and Figure 1). The results verified that most rifR mutations in all investigated cell types are base substitutions, with very few insertions and deletions (indels) (Table 2A and Supplementary Figure S2A; see the sections “Base Substitutions” and “Indels” in the Supplementary Material for a more detailed description of the mutations). Therefore, neither indels nor the uncharacterized fraction outside the sequenced rifR region are included in the statistical analysis.
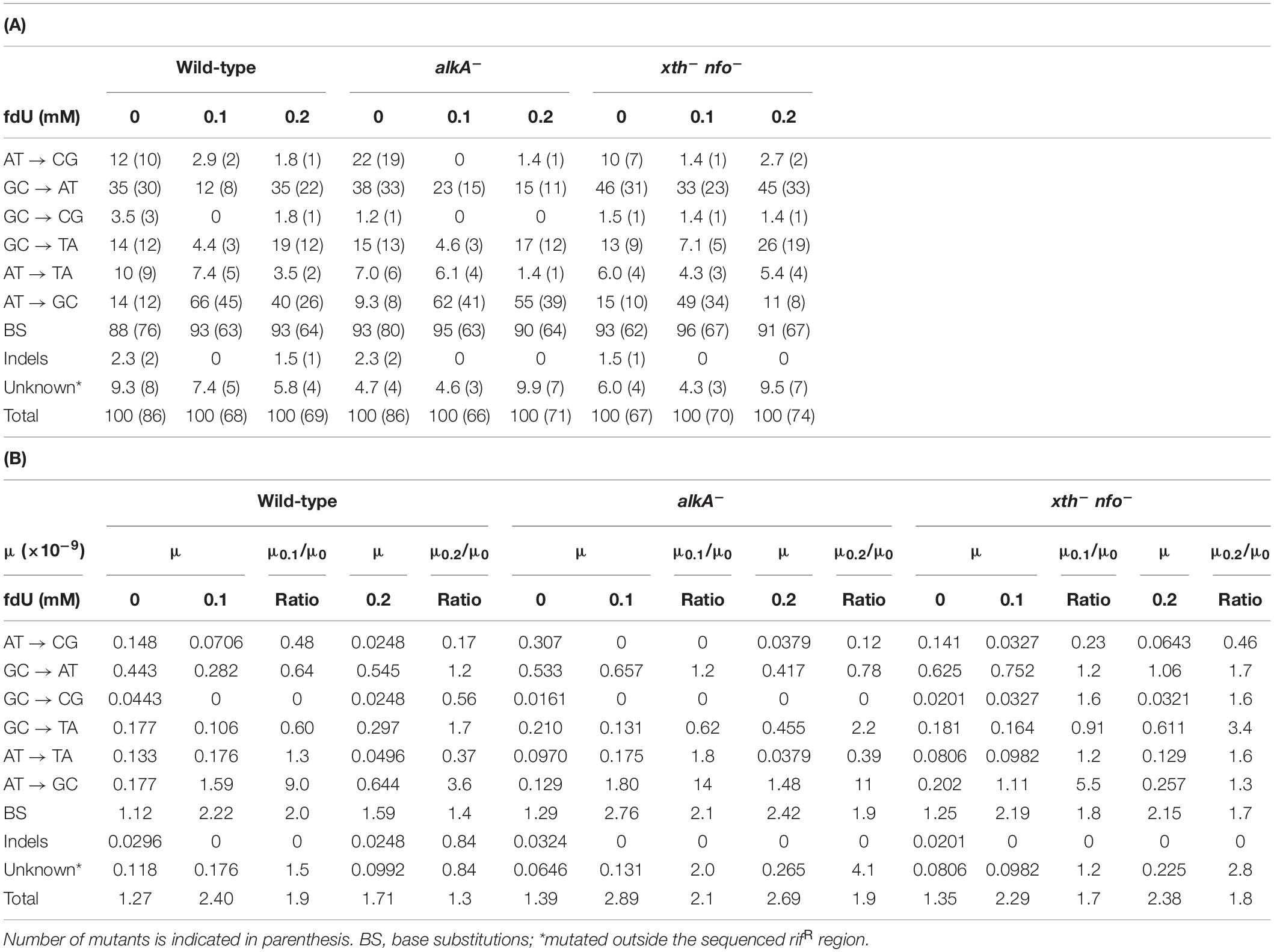
Table 2. Base substitutions [A, in percent; B, their mutation rates (μ)] among E. coli rifR mutants induced by 0.1 and 0.2 mM fdU.
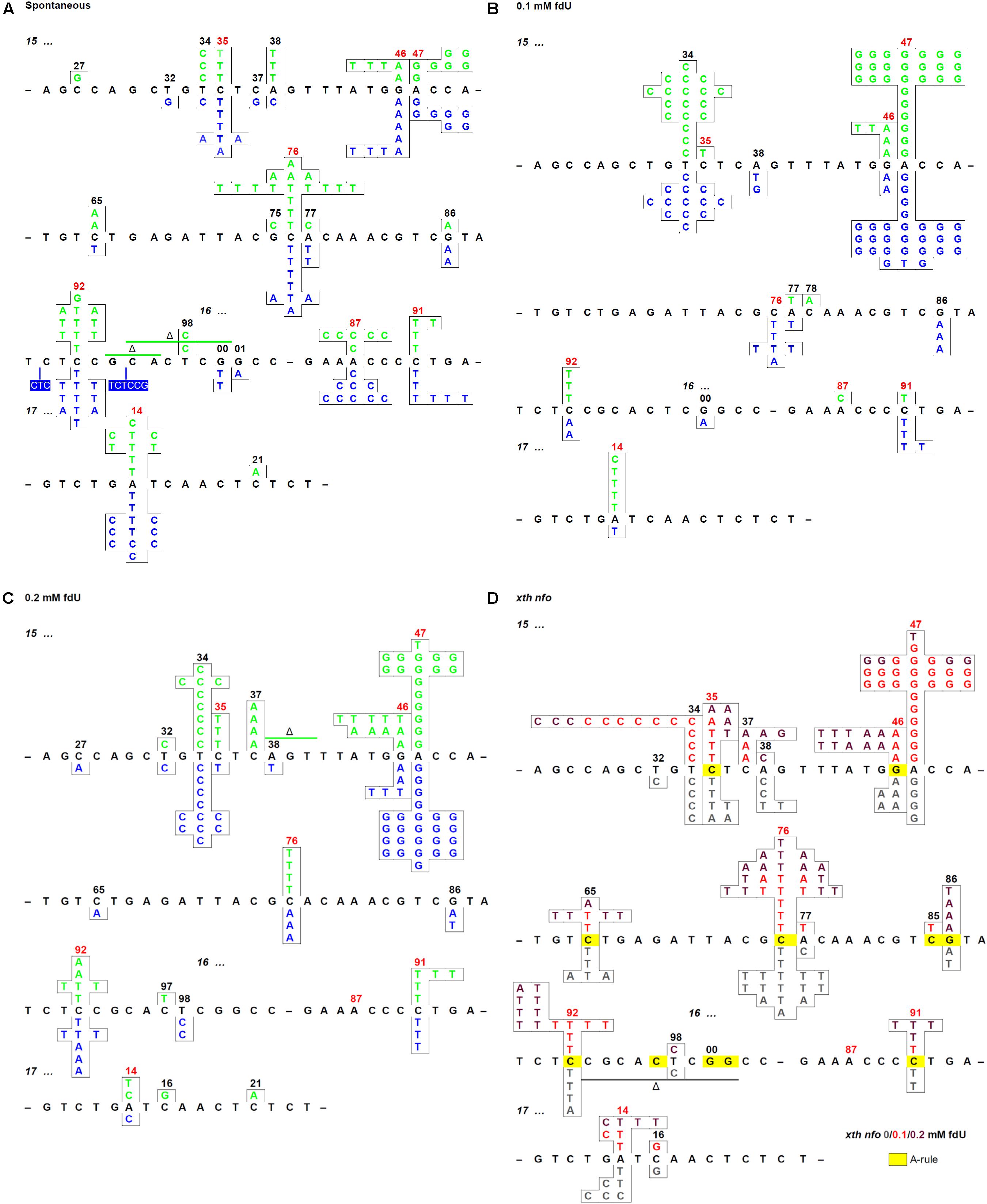
Figure 1. Mutation spectra of the E. coli repair deficient and proficient cells with and without fdU in the growth culture. (A) Spontaneous mutations in the rpoB gene rifR region of wild-type (alkA+; green) and alkA– (blue) cells. (B) Mutations in the rifR region of wild-type (green) and alkA– (blue) cells grown with 0.1 mM fdU, (C) 0.2 mM fdU. (D) xth– nfo– cells grown with 0 (gray), 0.1 (red), and 0.2 mM (dark red) fdU. Spots where the A-rule should apply are marked in yellow. In omitted regions (indicated by a short black line), no mutation was detected. Spontaneous mutation hot spots are indicated by red numbers. See Table 2A for the number of mutations presented.
The results showed no significant difference in distribution of spontaneous base substitutions between wild-type and alkA– cells (p = 0.40), or between wild-type and the xth– nfo– cells (p = 0.76). Thus, a similar baseline was used to score the effect of fdU in wild-type and mutant cell lines. As expected, the base substitutions in wild-type cells changed significantly after exposure to 0.1 mM (p = 0.000001) and 0.2 mM fdU (p = 0.0015; Table 2A), confirming fdU mutagenicity (Fujikawa et al., 1998). Similar results were obtained for the alkA– cells at 0.1 mM (p = 0.000001) and 0.2 mM fdU (p = 0.000001), and for the xth– nfo– cells at 0.1 mM fdU (p = 0.0005), when compared to the spontaneous distribution. Unexpectedly, 0.2 mM fdU in the xth– nfo– cells caused no significant change in the base substitution distribution (p = 0.27; Table 2A).
Although there was a strong effect on the base substitutions distribution induced by fdU, no significant difference in distribution was observed between the wild-type and alkA– cells when treated with 0.1 mM (p = 0.36) or 0.2 mM (p = 0.14) fdU, which implies that the AlkA effect on mutagenesis is quantitative rather than qualitative.
The A⋅T → G⋅C Transition Is Primarily Induced by fdU and Is Alleviated by AlkA
Most mutations in the wild-type and alkA– cells following fdU exposure were adenine (A)⋅ T to G⋅C transitions. While representing around 10–15% of the spontaneous mutations in the wild-type and alkA– cells, this transition increased to 66% (p = 0.000001) and 62% (p = 0.000001) at 0.1 mM, and to 40% (p = 0.0012) and 55% (p = 0.000001) at 0.2 mM, respectively (Table 2A). This result corroborates our previous report showing that A⋅T → G⋅C is the most frequent base substitution induced by fdU in wild-type E. coli (Ånensen et al., 2001). Although this difference in substitution abundance between the wild-type and alkA– cells was not significant in controls (p = 0.34) and at 0.1 mM fdU (p = 0.57), 0.2 mM fdU caused significantly different substitution levels (p = 0.034). The fraction of A⋅T → G⋅C in wild-type cells decreased to almost the half from 0.1 to 0.2 mM fdU, while being quite similar at the two fdU concentrations in alkA– cells (Table 2A).
In the unexposed xth– nfo– cells, the A⋅T → G⋅C fraction was equal to wild-type cells (p = 1). The fraction increased to 49% (p = 0.00003) with 0.1 mM fdU, which was significantly lower than in wild-type cells (p = 0.020; Table 2A). This supports the notion that A⋅T → G⋅C mutations cannot arise via AP site formation, according to the A-rule, because of fU excision (Figure 2). This conclusion was supported by 0.2 mM fdU treatment, where the A⋅T → G⋅C fraction in the xth– nfo– cells (11%) was similar to that without fdU (p = 0.61), but significantly lower than in wild-type cells (p = 0.0003; Table 2A).
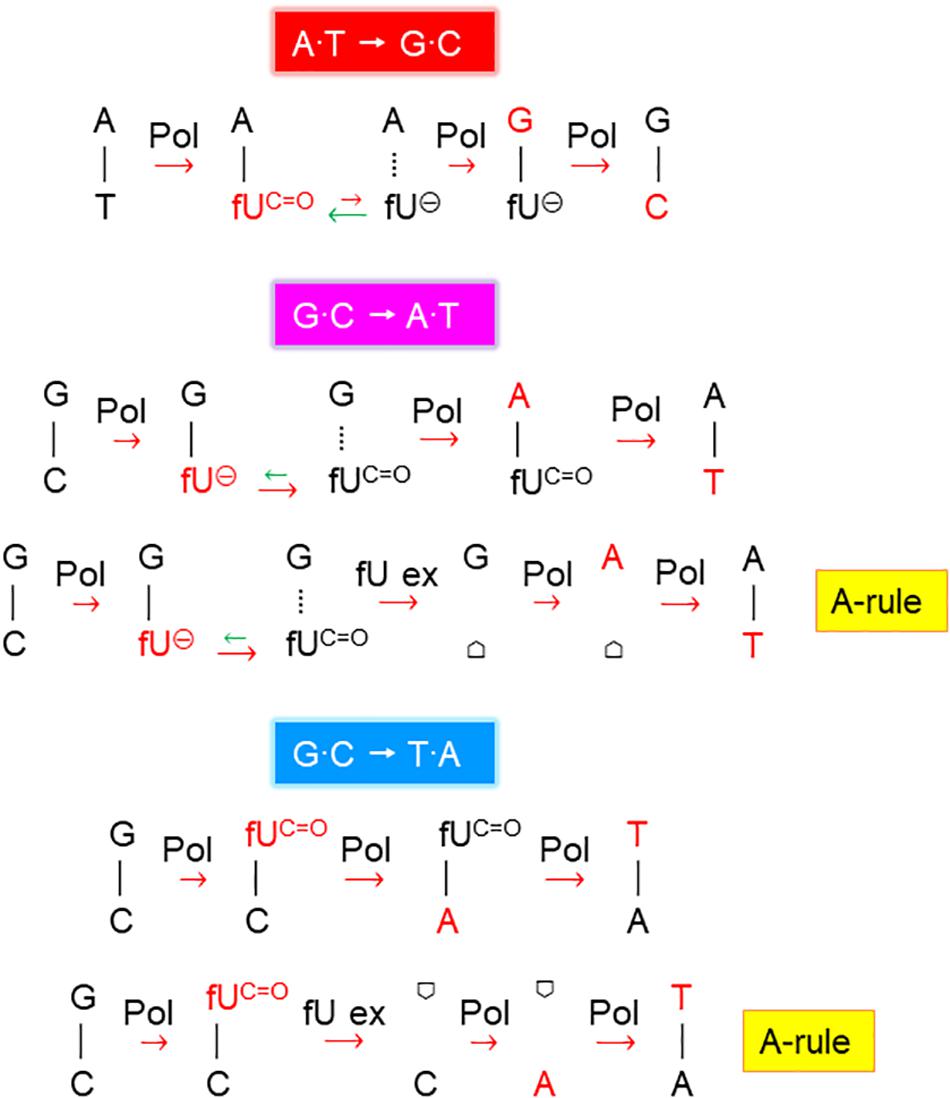
Figure 2. Proposed fdU mutagenesis mechanisms. Schemes suggesting the major molecular events inducing A⋅T → G⋅C, G⋅C → A⋅T, and G⋅C → T⋅A mutations by fdU. Pol, DNA polymerase; fUC=O, keto form of fU, which pairs with A (and less efficiently with C); fU⊖, ionized form of fU, which pairs with G;  , AP site/deoxyribose; template and newly inserted bases are presented in black and red, respectively; the major processes or events alleviating (green arrows) and contributing (red arrows) to mutagenesis are indicated (short, weak effect; long, stronger effect).
, AP site/deoxyribose; template and newly inserted bases are presented in black and red, respectively; the major processes or events alleviating (green arrows) and contributing (red arrows) to mutagenesis are indicated (short, weak effect; long, stronger effect).
Allocating the mutation rate (Table 1A) to the percent occurrence of each type of mutation (Table 2A) determines the quantitative contribution by each mutation and substitution type to mutagenesis (Table 2B and Supplementary Figure S2). This shows that the twofold increase in the mutation rate in the wild-type and alkA– cells caused by 0.1 mM fdU (Table 1A) was mostly due to induction of the A⋅T → G⋅C transition (Table 2B). This substitution rate increased 9- and 14-fold higher than the spontaneous level in wild-type and alkA– cells, respectively (Supplementary Figures S2B,C). However, while this rate was modestly lower at 0.2 than at 0.1 mM fdU in the alkA– cells (Supplementary Figure S2C), it was less than half of the rate at 0.1 at 0.2 mM fdU in wild-type cells (Supplementary Figure S2B). Since the difference in A⋅T → G⋅C abundance in wild-type and alkA– cells (Table 2A) was statistically significant, we conclude that AlkA alleviates A⋅T → G⋅C induction at 0.2 mM fdU (Figure 3A), resulting in a lower total mutation rate (Table 1A).
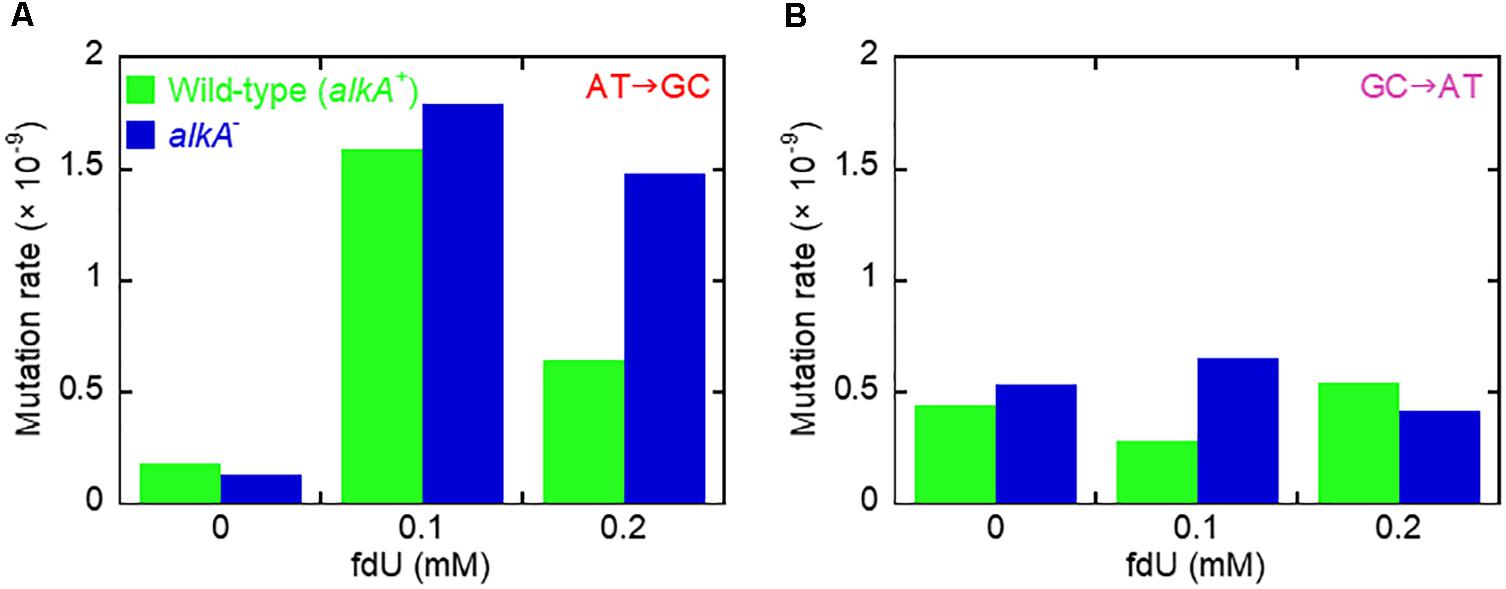
Figure 3. Mutation induction by fdU involves mutagen concentration-dependent alleviation by AlkA fU-DNA glycosylase. (A) The major base substitution induced by fdU, A⋅T → G⋅C, is alleviated by AlkA at 0.2 mM fdU. (B) AlkA seems to alleviate G⋅C → A⋅T transitions at 0.1 mM fdU.
In xth– nfo– cells, the A⋅T → G⋅C transition contributed to half the total base substitution rate at 0.1 mM fdU, while the A⋅T → G⋅C transition rate decreased to the spontaneous level at 0.2 mM fdU (Table 2B). The higher fdU load caused a bias toward the A-rule mutations, G⋅C → A⋅T and G⋅C → T⋅A (Figure 2), since the base substitution rate was the same at the two fdU concentrations (Table 2B and Supplementary Figure S2D). The higher number of unrepaired AP sites expected at 0.2 mM fdU is likely unable to be repaired efficiently in the absence of the major AP endonucleases, thus leaving behind more unrepaired AP sites to be bypassed by trans-lesion synthesis (Figure 2).
In conclusion, the A⋅T → G⋅C transition is the principal mutation induced by 0.1 mM fdU in E. coli, due to its large increase in abundance compared to other base substitutions (Table 2A). A significantly reduced A⋅T → G⋅C transition rate in the xth– nfo– cells (Table 2B and Supplementary Figure S2D) suggests that the accumulation of unrepaired AP sites favors the A-rule mutations (Figure 2). The increased formation and A⋅T → G⋅C rate in alkA– compared to wild-type cells at 0.2 mM fdU (Figure 3A), which theoretically should be connected to poor fU repair (Bjelland et al., 1994; Masaoka et al., 1999), suggests that AlkA initiates fU repair in DNA in vivo to alleviate fdU-induced mutations.
G⋅C → A⋅T and G⋅C → T⋅A Were the Second and Third Most Common Mutations Induced by fdU
The G⋅C → A⋅T transition was the most abundant spontaneous base substitution, occurring at a similar fraction in wild-type and alkA– cells (p = 0.87; Table 2A). Although the higher fraction in the xth– nfo– cells accords with the A-rule (Figure 2), the difference was not significant (p = 0.23).
The G⋅C → A⋅T fraction at 0.1 mM fdU in wild-type was about half the fraction in alkA– cells (p = 0.17; Table 2A), resulting in a 2.3-fold higher mutation rate in repair-deficient cells (Table 2B, Supplementary Figures S2B,C, and Figure 3B). However, a larger sample size is required to verify the possible G⋅C → A⋅T alleviation by AlkA. In contrast, the G⋅C → A⋅T fraction was twice as high in wild-type than in alkA– cells at 0.2 mM fdU. However, this significant difference (p = 0.042) resulted in an only slightly higher transition rate (Figure 3B).
In xth– nfo– cells, the G⋅C → A⋅T fraction at 0.1 mM fdU was significantly higher (p = 0.0042) than in wild-type cells. However, this difference decreased with 0.2 mM fdU (p = 0.11; Table 2A). The resulting two to threefold increase in the G⋅C → A⋅T rate with fdU in xth– nfo– cells compared to wild-type cells (Supplementary Figures S2B,D, and Table 2B) could be explained if mutations formed by fU are alleviated by AlkA-initiated BER in wild-type cells, while mutations are formed via AP sites in xth– nfo– cells lacking AP endonucleases (Figure 2). However, no firm conclusion can be made whether G⋅C → A⋅T is alleviated by AlkA (Figure 3B).
We previously showed that the G⋅C → A⋅T transition is the second most common base substitution induced by fdU in wild-type E. coli (Ånensen et al., 2001). Our present results support this finding.
The G⋅C → T⋅A transversion amounted to 13–15% of the spontaneous base substitutions in all three genotypes (Table 2A). Addition of 0.1 mM fdU significantly decreased the mutated fraction to about 4.5% in wild-type and alkA– cells (p = 0.053 and 0.034, respectively). In contrast, addition of 0.2 mM fdU slightly increased the G⋅C → T⋅A fraction in wild-type (p = 0.66) and alkA– cells (p = 0.82). Our results show no difference between the G⋅C → T⋅A transition in wild-type and alkA– cells with 0, 0.1, or 0.2 mM fdU (p = 1) suggesting that AlkA minimally influences mutagenesis.
In xth– nfo– cells, 0.1 mM fdU decreased the G⋅C → T⋅A fraction by about half (p = 0.26), while 0.2 mM fdU doubled the fraction (p = 0.086; Table 2A) indicating a slight mutation induction. Compared to the wild-type fractions at 0.1 and 0.2 mM fdU, these values are not significant (p = 0.72 and 0.22, respectively). However, at 0.2 mM fdU, the resulting G⋅C → T⋅A rate in xth– nfo– cells doubled compared to wild-type cells (Table 2B and Supplementary Figures S2B,D), according to the A-rule (Figure 2).
The G⋅C → T⋅A transversion rate decreased similarly (times 0.6–0.9) with 0.1 mM fdU in wild-type, alkA–, and xth– nfo– cells, but increased 1. 7-, 2. 2-, and 3.4-fold, respectively, with 0.2 mM fdU (Table 2B and Supplementary Figures S2B–D). This confirms our previous observation that the G⋅C→T⋅A transversion is the third most prevalent base substitution in wild-type E. coli (Ånensen et al., 2001), and suggests that unrepaired AP site accumulation also slightly affects transversion generation.
A⋅T → C⋅G, G⋅C → C⋅G, and A⋅T → T⋅A Were Not Significantly Induced by fdU
The A⋅T → C⋅G transversion was the most distinct spontaneous mutation observed, as it roughly occurred one order of magnitude more often without mutagen compared to the few mutants observed in fdU-exposed cells, while the G⋅C → C⋅G transversion was the most rare mutation no matter whether fdU was present or not. The A⋅T → T⋅A transversion was the second least common spontaneous base substitution recorded, and in the presence of fdU its fraction decreased rather than increased (Table 2A). These results largely accord with our previous study showing no induction of G⋅C → C⋅G and weak induction of A⋅T → C⋅G and A⋅T → T⋅A by fdU in wild-type E. coli (Ånensen et al., 2001).
Exposure to fdU Causes Increased alkA Expression
Since fdU-induced mutagenesis is alleviated by AlkA, we asked whether alkA gene expression is also induced by fdU. Thus, relative alkA gene expression was determined by RT-qPCR in wild-type E. coli with and without fdU treatment up to 24 h. alkA transcript remained at the same level after treatment with 0.1 mM fdU, even after 24 h exposure. This contrasted with a clear time-dependent induction of alkA with the highest fdU concentration (Figure 4A). Exposure to 0.2 mM fdU for 6 h increased alkA expression 2.2-fold, and 3.7-fold after 24 h.
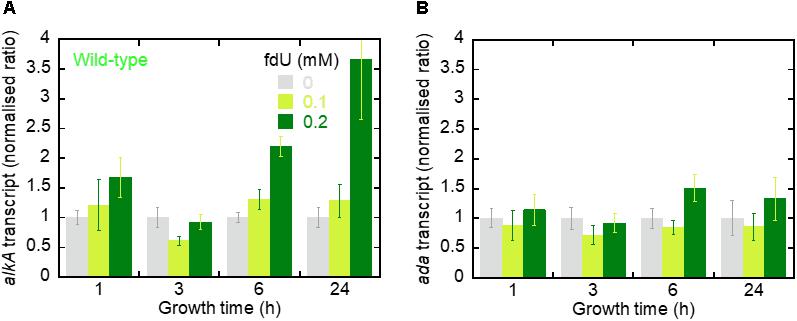
Figure 4. The alkA transcript is induced by fdU exposure. (A) The alkA transcript expression levels (fold changes) in E. coli wild-type as a function of growth time with 0, 0.1, and 0.2 mM fdU. Target gene expression (alkA) is normalized to the endogenous tag gene. The columns represent the mean value ± SD for three independent experiments. (B) ada transcript expression levels (fold changes). Target gene expression (ada) is normalized to the endogenous tag gene. The columns represent the mean value ± SD for three independent experiments.
We then analyzed ada gene expression, which controls the alkA gene (Lindahl et al., 1988; Landini and Volkert, 2000; Mielecki et al., 2015). We observed no difference in ada expression across most fdU concentrations and time durations (Figure 4B). The only exception was after 6 h, where 0.2 mM fdU treatment increased ada expression 1.5-fold. Thus, treating E. coli wild-type cells with 0.2 mM fdU induces alkA gene transcription.
AlkA-Mediated fU Excision From DNA Is Dependent on the Complementary Base
AlkA protein was purified (Supplementary Table S3) to apparent physical homogeneity (Supplementary Figure S3) as described in the section “Materials and Methods.” To analyze the efficiency of fU excision, a defined, 64 nt double-stranded DNA oligonucleotide with a central fU opposite A or G (Figure 5A) was used as an AlkA substrate. Significant NaOH-mediated damaged strand cleavage was observed in the presence of enzyme. However, the amount of enzyme needed to create alkaline labile AP sites in DNA was highly dependent on the opposite base. Kinetic analysis showed that the fU⋅G substrate was cleaved 17 times more efficiently than the fU⋅A substrate (Figures 5B,C), according to the kcat/KD value (Table 3). More efficient fU excision opposite G compared to A was also reported for human SMUG1 (Knævelsrud et al., 2009), and agrees with the widely accepted flipping-out mechanism for DNA glycosylase action. In this mechanism, the enzyme has to disrupt fU base pairing and base-stacking interactions in double-stranded DNA to be accommodated in the active site pocket (McCullough et al., 1999; Wibley et al., 2003).
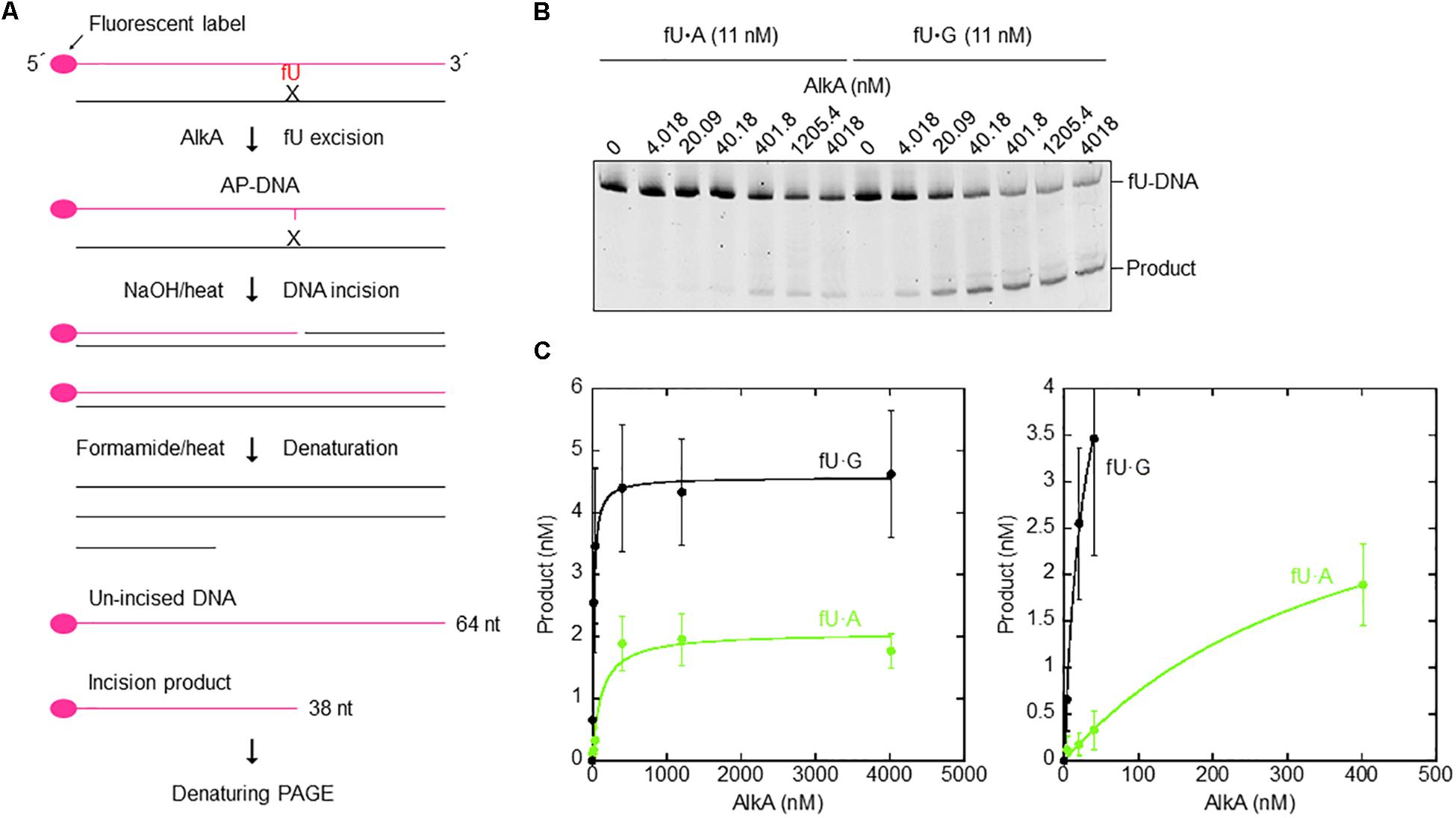
Figure 5. Single-turnover kinetics for AlkA-mediated fU excision opposite A and G in DNA. (A) DNA substrates and assays (see the section “Materials and Methods”). (B) AlkA was incubated with 11 nM fU⋅A (Left) or fU⋅G (Right) substrate at 37°C for 60 min in 50 μL reaction buffer. Electrophoresis was performed on a 20% (w/v) polyacrylamide gel containing 7 M urea at 500 V for ∼1.5 h. (C) Product (fU) formation as a function of enzyme concentration (Left); a magnified image of product formation when approaching enzyme saturation (Right). Each value represents the average of seven independent measurements.
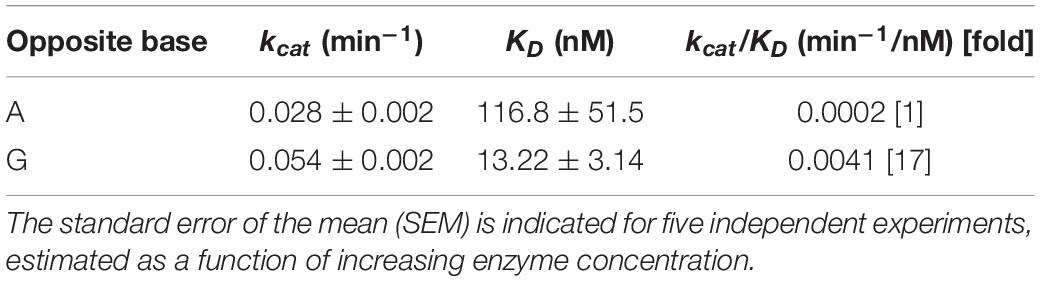
Table 3. Kinetic parameters for the opposite-base dependent fU excision by AlkA.
The Keto, but Not the Ionic Form, of fU Is Efficiently Accommodated in the AlkA Active Site Pocket, and Is Indicated by the fU Release pH Dependency Profile
To indicate how AlkA accommodates fU in the substrate-binding pocket (Figure 6A, upper panel), AlkA served as receptor in automated docking simulations. Following sampling of 250 possible docking runs, fU showed a docking energy distribution, with several runs clustering into the lowest energy bin. For the primary AlkA substrate, m3A, this result was even more pronounced. The fewer successful docking runs obtained with fU may reflect a reduced binding affinity compared to m3A. However, successful m3A docking was dependent on its positive charge (extensively described in Supplementary Material, section “Docking m3A and fU in the Binding Pocket of E. coli AlkA” and Supplementary Figures S4–S10). In the specific conformation with the lowest docking energy, the formyl group in the 5-position of the uracil ring forms a hydrogen bond to the Arg22 side chain in the bottom of the binding pocket (Figure 6A, upper panel), which appears to stabilize binding of the neutral fU keto form (fUC=O). Unlike m3A (Supplementary Figure S4), π–π stacking with the Trp272 side chain does not appear to be important for fU binding. Importantly, if fU adopts the ionic form (fU⊖), where the negative charge is delocalized to the uracil ring (Figure 6A, lower panel), the overall sum of repulsive forces to the catalytic residues Asp238 and Trp272 could outweigh the potentially attractive forces to Arg22 (Figure 6A, upper panel), thus excluding fU⊖ from the active site pocket.
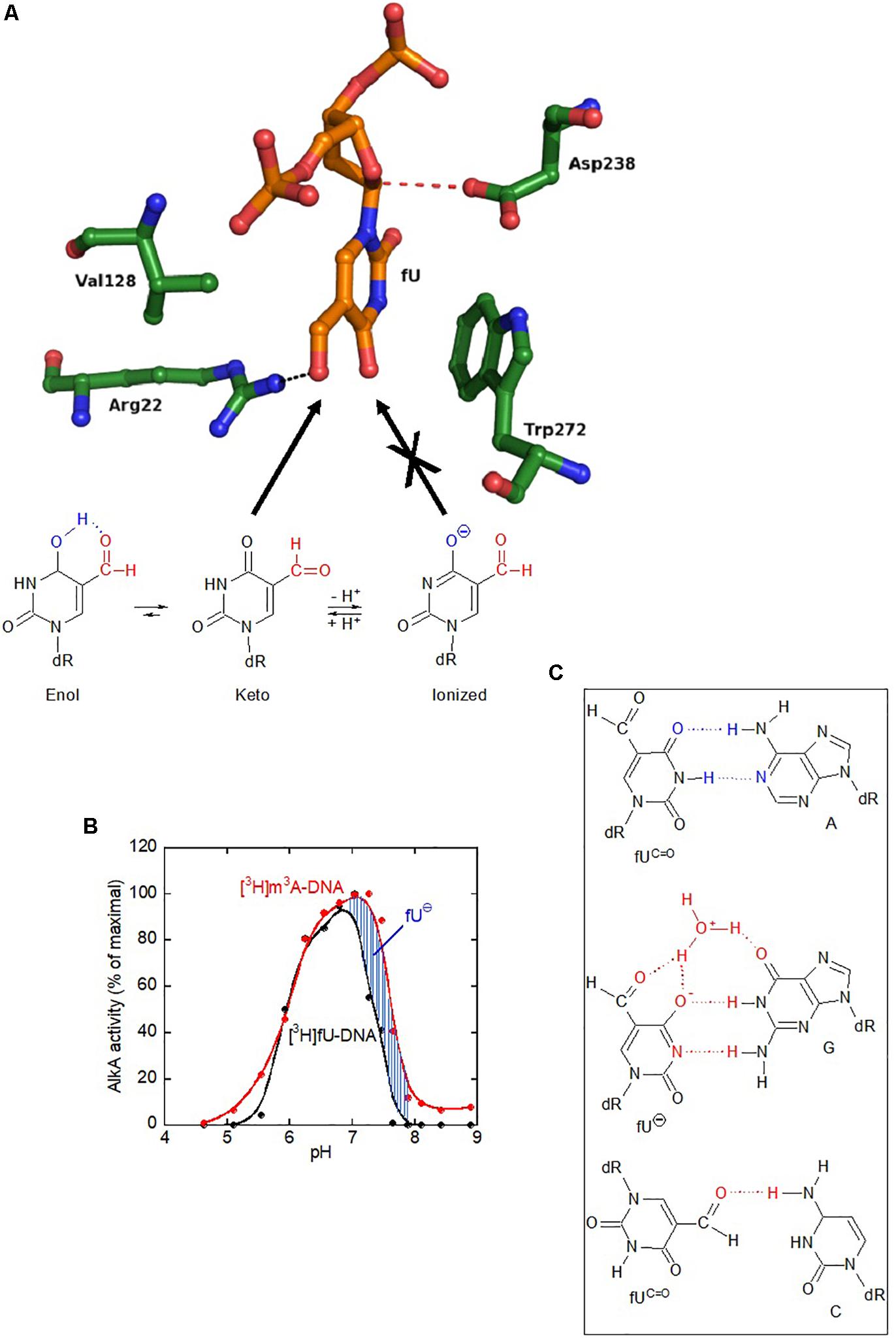
Figure 6. The AlkA active site does not accommodate the ionized form of fU, which erroneously pairs with guanine. (A) Docking of the keto, but not the ionic form of fU, into the AlkA substrate-binding pocket. Unlike primary substrate m3A docking, no stacking occurs between fU and Trp272. Instead, the fU formyl group forms a hydrogen bond to Arg22. Regarding m3A, Asp238 is positioned to attack the N1–C1 bond. The abundance of the ionized (⊖) form of fU increases with pH (lower panel). While the AlkA active site pocket accommodates the fU keto form, it excludes the ionized form (not shown). (B) Exclusion of fU⊖ from the AlkA active site was inferred from enzymatic fU release. AlkA fU- and m3A-DNA glycosylase activity as a function of pH shows a much stronger decline in the activity for fU than for m3A at high pH, which is explained by the higher abundance of non-excisable fU⊖. AlkA (2 pmol and 0.08/0.16 pmol) was incubated with aged [methyl-3H]thymine-labeled (11 independent experiments) and [3H]MNU-treated DNA (4 independent experiments), respectively. (C) Postulated base pairing properties of fU residues in DNA. Since fU is a thymine analog that efficiently pairs with adenine, the fU⋅A pair is most likely to occur in vivo. The fU⋅G mispair formed by the ionized form (fU⊖) is stabilized by a water bridge. Both these base pairs have been demonstrated by X-ray analyses of DNA oligomers with fU inserted at a specific site (Tsunoda et al., 2001, 2002). However, the fU⋅C mispair is tentative (Kamiya et al., 2002). Cognate base pairing with adenine is due to the keto form (fUC=O). Cognate hydrogen bonding pattern is shown in blue; non-cognate hydrogen bonding/base pairing pattern in red.
To provide chemical evidence for in silico fU⊖ exclusion from the active site pocket, AlkA activity for radioactively labeled fU in DNA was measured as a function of pH. Since fU⊖ abundance increases with pH, its rejection from active site should cause decreased enzyme activity for fU, compared to m3A, which exists in one chemical form. Interestingly, a large decrease in DNA glycosylase activity occurred at pH 7.25 for fU, rather than pH 7.6 for m3A (Figure 6B), which supports the molecular docking simulation results showing fU⊖ exclusion from the active site pocket.
Discussion
In spite of several studies establishing AlkA as the major glycosylase to initiate fU BER in DNA (Bjelland et al., 1994; Masaoka et al., 1999; Terato et al., 1999), no evidence has emerged indicating such a role in vivo. In fact, it appeared strange to allocate this function to a protein whose expression is induced by sub-lethal methylating agent concentrations, and is dependent on enhanced ada expression (Evensen and Seeberg, 1982; Karran et al., 1982; Sedgwick, 2004; Shrivastav et al., 2010). To gain insight into this issue, we investigated whether BER influences fU-induced mutagenesis. We added fdU to E. coli growth cultures to specifically introduce fU into the DNA of repair proficient (wild-type; alkA+ xth+ nfo+) and deficient (alkA– and xth– nfo–) cells, and systematically determined the mutation rate and spectrum. To bind the E. coli plasma membrane transporter NupC, a C3′-hydroxyl group is required, while binding to NupG is dependent on a hydroxyl group at both the C3′ and C5′-position (Patching et al., 2005). Thus, fdU is most likely taken up from the culture medium by both these proteins (Figure 7A). We chose to select for rifampicin-resistant cells formed as a result of a targeted mutation in the rpoB rifR region, which was then sequenced using a single mutant clone collected from each experiment. The spontaneous mutation spectrum obtained from wild-type cells (Figure 1A, green) shows the distribution of the six possible base substitutions (Table 2A). The distribution was very similar to recent results from a comprehensive whole-genome sequencing analysis of several E. coli wild-type strains (Foster et al., 2015). These data confirm that the rifR system is adequate for such investigations.
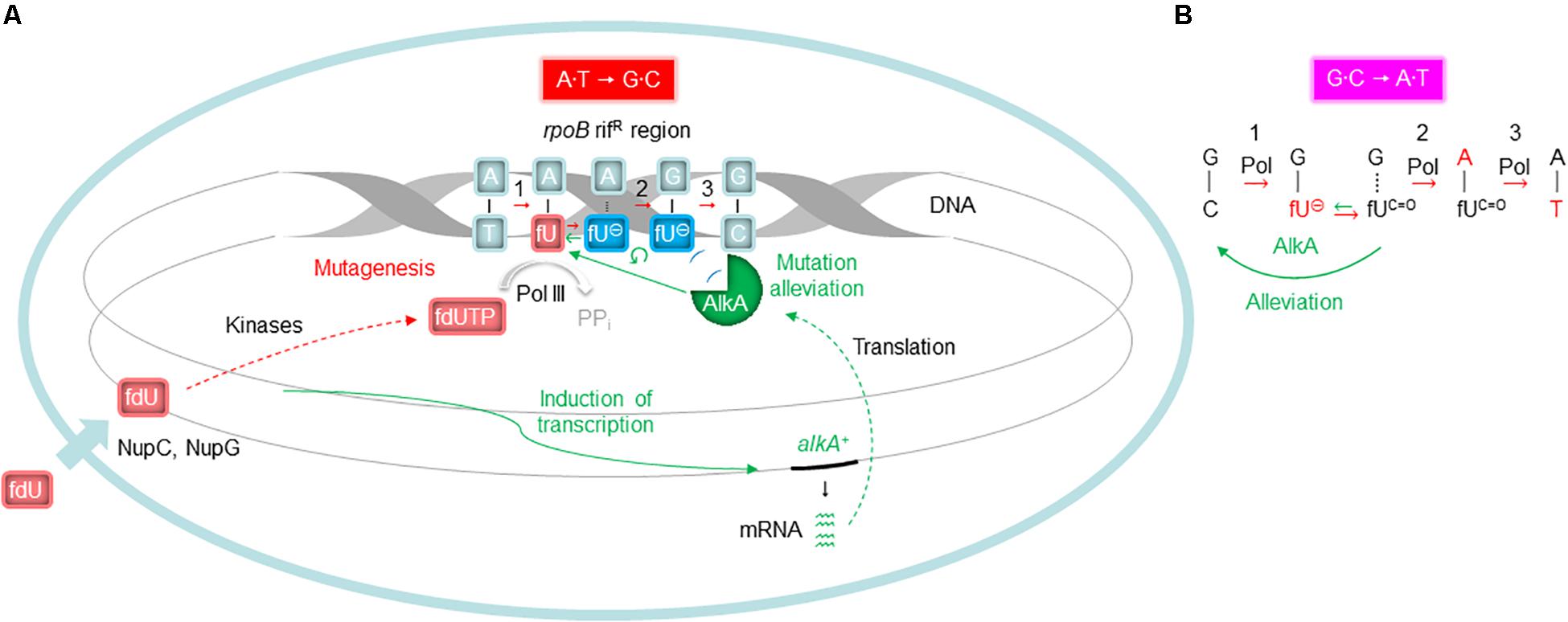
Figure 7. Proposed molecular events influencing fdU mutagenesis alleviation by AlkA. (A) Alleviation of A⋅T → G⋅C (Figure 3A) needs alkA induction (Figure 4A), due to low kcat/KD for fU⋅A (Table 3). Since the common fU keto form (fUC=O) primarily behaves like thymine, the corresponding fdUMP is most efficiently incorporated into DNA opposite adenine (replicative event 1). If this newly inserted fU is removed by AlkA, the mutation-initiating event is reversed or alleviated. Alternatively, when some fUC=O paired with A transforms to the ionic state (fU⊖), AlkA recognition is excluded (Figure 6A) in favor of polymerase binding, leading to guanine insertion opposite fU⊖, causing mutagenesis (replicative events 2 and 3). (B) The possible alleviation of G⋅C → A⋅T (Figure 3B) takes place without alkA induction (Figure 4A) because of high kcat/KD for fU⋅G (Table 3). Although the most frequent replicative event (event 1 in A) is fdUTP pairing with adenine, fdUTP will pair with template guanine if it adopts the ionic state (replicative event 1). When the newly inserted fU⊖ reverts to fUC=O, it will be an efficient target for AlkA reversing or alleviating the mutation-initiating event. If replication takes place before AlkA reaches fU, an adenine will usually be inserted, causing mutagenesis (replicative events 2 and 3). Events leading to base substitutions are in red; events promoting alleviation of mutagenesis are in green. Processes demonstrated in this study are shown by unbroken arrows and otherwise by broken arrows; ↺, exclusion from AlkA active site. Plasma membrane with nucleoside transporter (short thick arrow at the bottom left) is shown in light blue. PPi, pyrophosphate. See Figure 2 for definitions for the other symbols.
Our results confirm that the A⋅T → G⋅C transition is the principal mutation arising from fU base lesions in DNA (Table 2A and Figures 1B–D) due to occasional mispairing with guanine (Figure 6C, middle panel). The connection with the fU base is indicated by lower formation in the xth– nfo– mutant (Table 2B and Supplementary Figure S2D). The predominance of A⋅T → G⋅C at low fdU (0.1 mM) is challenged by G⋅C → A⋅T at higher (0.2 mM) fdU concentration (Table 2A) in the wild-type, but not the alkA– cells (Supplementary Figures S2B,C, respectively, and Table 2B). The G⋅C → A⋅T transition is a typical A-rule mutation that is also formed via AP sites (Figure 2), which is underscored by its consistently higher rates in the xth– nfo– cells (Table 2B and Supplementary Figure S2D). However, trans-lesion synthesis past AP sites formed following enzymatic fU removal does not appear to be the single major mechanism generating G⋅C → A⋅T after fdU incorporation into DNA, because AlkA deficiency causes a similar increase in the (0.1 mM) fdU mutation rate as AP endonuclease deficiency (Supplementary Figures S2C,D, respectively). Indeed, fewer AP sites should form from fU in alkA– cells compared to wild-type cells. As previously shown (Ånensen et al., 2001), the present results confirm that the G⋅C → A⋅T transition is an important fU-induced mutation formed by occasional non-cognate fdUMP incorporation opposite guanine in DNA (Figure 2). The G⋅C → T⋅A transversion is also an A-rule mutation (Figure 2), as confirmed by its higher rate in xth– nfo– cells compared to wild-type cells at 0.2 mM fdU (Table 2B and Supplementary Figures S2B,D). The increased mutation rate in alkA– cells indicates that mechanisms other than trans-lesion synthesis past AP sites are involved in G⋅C → A⋅T transition generation. Although our data lacks statistical significance due to the insufficient number of experiments required for this particular mutant, the results corroborate previous studies showing that G⋅C → T⋅A transversions arise in E. coli (Fujikawa et al., 1998; Ånensen et al., 2001) because of mispairing between fU and cytosine (Zhang et al., 1997; Figure 6C, lower panel). In our study, this was likely due to occasional non-cognate fdUMP incorporation opposite cytosine in DNA (Figure 2). Consequently, mutations change from mostly A⋅T → G⋅C at low fdU (0.1 mM) to additional G⋅C → A⋅T and G⋅C → T⋅A at higher fdU (0.2 mM) concentrations. The other possible base substitutions, A⋅T→C⋅G, A⋅T → T⋅A, and G⋅C → C⋅G, were not significantly induced by fdU in this investigation (Table 2A), which confirms our previous results showing that the two former are less efficiently formed, while the latter is not formed by fdU (Ånensen et al., 2001).
We made some mechanistic conclusions regarding the fdU-induced base substitutions based on the abundance in repair proficient and deficient strains. However, the major topic of the investigation was to investigate whether AlkA-initiated BER counteracts fdU-mediated mutagenesis. The results show that AlkA significantly influences mutagenesis, and that this in vivo effect is dependent on the mutagen concentration in the culture medium. At the lowest fdU concentration (0.1 mM), alkA– cells experienced a similar mutation rate as wild-type cells (Table 1A). Although we observed an alleviating AlkA effect on G⋅C → A⋅T induction that was not statistically significant (Figure 3B), such lack of phenotype for fU repair can be explained by the presence of several back-up systems, like Fpg, Nth, and Nei glycosylases (Zhang et al., 2000). However, this study employed oligonucleotides with fU inserted at a specific site, which is more sensitive to glycosylase fU recognition due to lack of competition from other lesions than the multiple-lesion containing [3H]thymine-DNA we used in our first study comparing Fpg, Nth, and AlkA, which showed no activity for fU (Bjelland et al., 1994). Because Nei exhibits similar fU activity as Nth (Zhang et al., 2000), and only AlkA exhibited clearly detectable fU activity using [3H]thymine-DNA, we conclude that AlkA is superior to Fpg, Nth, and Nei in removing fU from DNA. In contrast, we observed significantly different A⋅T → G⋅C transition levels between wild-type and alkA– cells at 0.2 mM fdU (Table 2A) resulting in a more than twice as high mutation rate (Figure 3A). These results provide evidence that AlkA alleviates fdU-induced mutagenesis (Figure 7A). It was especially interesting that the alkA gene is indeed induced at 0.2 mM fdU (Figure 4A), explaining the higher impact of AlkA on mutagenesis at this concentration. It should be noted that the A⋅T → G⋅C rate increase in alkA– compared to wild-type at 0.2 mM fdU is in the same order of magnitude as the more than fivefold increase in total methyl methanesulfonate (MMS)-induced mutagenesis in the former compared with that in the latter cell type, although this was investigated under different conditions using another mutagenicity tester system (Grzesiuk et al., 2001).
The alkA gene is induced by sub-lethal concentrations of alkylating agents (Evensen and Seeberg, 1982; Karran et al., 1982; Teo et al., 1986; Sakumi and Sekiguchi, 1989; Saget and Walker, 1994; Landini and Volkert, 2000; Kleibl, 2002; Mielecki et al., 2015). While SN1 and SN2 methylating agents like N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and MMS (Sedgwick, 2004) cause a 5–20-fold increase in alkA expression or AlkA activity (Evensen and Seeberg, 1982; Karran et al., 1982; Baek et al., 2009; Booth et al., 2013), we detected a nearly fourfold increase by fdU when normalized to the constitutively expressed tag gene (Figure 4A), which encodes m3A-DNA glycosylase I (Sakumi et al., 1986; Bjelland and Seeberg, 1987). Whether this increase is dependent on regulation by the Ada protein is questionable, since fdU only caused a minimal increase in ada expression (Figure 4B). The hypothesis that alkA is only induced by alkylation exposure was also challenged by a previous study, showing that RNA polymerase associated with the stationary phase σs instead of the normal σ70 transcription factor transcribes alkA independent of ada (Landini and Busby, 1999). It has been calculated that an un-adapted E. coli cell contains ∼50 AlkA molecules (Bjelland et al., 1994). If we consider that the 3.7-fold increase in transcripts (Figure 4A) causes a similar increase in protein concentration, fdU increased AlkA expression to ∼200 molecules per cell in our experiments, which equals the concentration of Tag in E. coli (Bjelland and Seeberg, 1987). Tag and AlkA have similar activity for m3A, which is the primary substrate for both glycosylases. Thus, both the cellular concentration of AlkA caused by fdU and the significant effects on fdU-mediated mutagenesis caused by AlkA inactivation suggest a significant contribution by this enzyme in modulating some ROS-mediated effects.
Although we have demonstrated a connection between the fU-DNA glycosylase activity of AlkA, alkA gene induction, and fdU-induced mutagenesis, in vivo evidence for the participation of other glycosylases like Fpg, Nth, Nei, and Mug in E. coli fU repair is elusive (Zhang et al., 2000; Liu et al., 2003). Since the latter enzymes seem to exhibit significantly lower in vitro activity than AlkA (see above), it is probably challenging to detect phenotypic differences in fdU-mediated mutagenesis between wild-type and cells with only one of these glycosylase genes inactivated. Instead, investigation of doubly and/or triply inactivated cells like fpg–nth– and fpg–nth–nei– might be more fruitful. Because mutagenesis is expected to mainly take place during the exponential growth phase, we judge Mug as less important due to its function during the late stationary phase of cells (Mokkapati et al., 2001). Interestingly, the KsgA enzyme exhibits DNA glycosylase activity for C opposite fU and potentially might promote G⋅C → T⋅A transversions (Figure 2). In addition, mismatch repair is probably involved in mutation prevention because the fU⋅G mismatch is recognized by MutS protein (Terato et al., 1999). The mammalian nucleotide excision repair component XPC-HR23B recognizes fU in DNA (Kino et al., 2004), which calls for examination of the analogous UvrABC system of E. coli (Van Houten et al., 2005).
In addition to showing alkA gene induction by fdU and demonstrating that AlkA alleviates mutagenesis caused by this oxidized nucleoside, our findings also provide mechanistic understanding of how fdU shifts the E. coli mutation spectrum to mainly A⋅T → G⋅C transitions (Table 2A). A major breakthrough came with the description of stable reversed wobble type pairing between fU⊖ and guanine (Figure 6C). In silico modeling showed this pairing is accommodated by DNA polymerase (Tsunoda et al., 2001, 2010), providing the molecular basis for both A⋅T → G⋅C and G⋅C → A⋅T mutations. Here, we substantiate this finding with biochemical evidence (Figure 6B) that fU⊖ is also excluded from the AlkA active site pocket (Labahn et al., 1996; Yamagata et al., 1996), further leading to polymerase 2′-deoxyguanosine monophosphate (dGMP) mis-insertion, which primarily causes A⋅T → G⋅C transitions due to the ability of the major fUC=O species to first pair with A. Actually, we believe that the fU⊖ inaccessibility to the AlkA active site pocket is essential for allowing polymerase access and enough time for dGMP mis-incorporation (Figure 7A). It is interesting to note that to alleviate this mutation at 0.2 mM fdU, alkA gene expression is required (Figure 4A), which might be related with the low kcat/KD for fU⋅A (Table 3). This can be observed by the slight AlkA alleviation offered by 0.1 mM fdU without alkA induction (Figure 3A). Similarly, the possible alleviation of the G⋅C → A⋅T transition (Figure 7B) at 0.1 mM fdU (Figure 3B), which occurs without alkA induction (Figure 4A), may be explained by the nearly 20 times higher kcat/KD for fU⋅G compared to fU⋅A (Table 3). As indicated by the predominance of the A⋅T → G⋅C transition, most fdUMP is inserted opposite adenine in DNA during replication. When inserted fU is converted to the ionized form, DNA polymerase processes the lesion, inserting a G opposite fU, instead of AlkA (Figure 7A). By contrast, how may the efficient excision of fU opposite G (Table 3) occur, when the fU⊖ ⋅G pair should be stable (Figure 6C, middle panel)? This can be explained by the favored conversion of fU⊖ to the keto form, which disrupts the hydrogen bonding to G. This results in efficient “flipping out” and fU excision by AlkA, possibly alleviating the G⋅C → A⋅T transition.
Conclusion
This is the first report demonstrating that an oxidized deoxynucleoside (fdU) at a certain concentration in the culture medium induces expression of the alkA gene in E. coli, resulting in alleviation of mutagenesis caused by that agent. Specifically, the occurrence of the A⋅T → G⋅C transition, which is the primary mutation caused by fdU, is reduced to half by alkA gene induction. Thus, for the first time we show in vivo evidence for the participation of the AlkA DNA glycosylase in the repair of base lesions damaged by oxidation. Our study also elaborates a novel mutation mechanism in E. coli, which may be used to explain other instances of fU-mediated mutagenesis. Further studies are required to clarify the molecular mechanisms involved in the fdU-mediated alkA gene induction and search for links to concomitant changes in cell physiology.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
KG and IM-L designed and carried out the mutagenesis experiments, analyzed the data, and wrote parts of the manuscript. IK, HÅ, and IA designed and carried out the mutagenesis experiments and analyzed the data. AT designed and carried out the RT-PCR experiments, analyzed the data, and wrote parts of the manuscript. MA carried out the RT-PCR experiments and analyzed the data. GH produced and characterized the AlkA enzyme and wrote parts of the manuscript. IL performed the in silico analyses and wrote parts of the manuscript. KG, IK, and PR performed the kinetic analysis. JK performed the statistical analyses and wrote parts of the manuscript. KS and AM produced and characterized fdU and the fdU-containing DNA. IA and AK suggested and contributed to experiments, and revised the manuscript. SB conceived the project, designed the experiments, analyzed the data, supervised and managed the study, and wrote the manuscript. All authors took part in the analysis of data and approved the final version of the manuscript.
Funding
This study was supported by the University of Stavanger.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to E. I. Baardsen, H. Bratthammer, K. L. Bringsjord, T. T. Dao, B. Domuz, L. H. Endresen, S. O. Engevik, B. A. Erland, G. Falkeid, T. Gulbrandsen, M. L. Hagseth, B. Hop, M. D. Ivesdal, S. Malmstrøm, A. Oftedal, K. M. Olsen, J. Ringvoll, B. Røiland, A. M. Sagen, E. Skårland, K. Sortland, Y. Ueno, A. Underhaug, A.-L. Vatne, A. M. Østhus, and M. Østvoll for technical assistance. We would like to thank Editage (www.editage.com) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00263/full#supplementary-material
References
Ahmad, S. I., Kirk, S. H., and Eisenstark, A. (1998). Thymine metabolism and thymineless death in prokaryotes and eukaryotes. Annu. Rev. Microbiol. 52, 591–625. doi: 10.1146/annurev.micro.52.1.591
Alseth, I., Osman, F., Korvald, H., Tsaneva, I., Whitby, M. C., Seeberg, E., et al. (2005). Biochemical characterization and DNA repair pathway interactions of Mag1-mediated base excision repair in Schizosaccharomyces pombe. Nucleic Acids Res. 33, 1123–1131. doi: 10.1093/nar/gki259
Ånensen, H., Provan, F., Lian, A. T., Reinertsen, S.-H. H. S., Ueno, Y., Matsuda, A., et al. (2001). Mutations induced by 5-formyl-2′-deoxyuridine in Escherichia coli include base substitutions that can arise from mispairs of 5-formyluracil with guanine, cytosine and thymine. Mutat. Res. 476, 99–107. doi: 10.1016/s0027-5107(01)00086-0
Baek, J. H., Han, M. J., Lee, S. Y., and Yoo, J. S. (2009). Transcriptome and proteome analyses of adaptive responses to methyl methanesulfonate in Escherichia coli K-12 and ada mutant strains. BMC Microbiol. 9:186. doi: 10.1186/1471-2180-9-186
Berquist, B. R., and Wilson, D. M. III (2012). Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 327, 61–72. doi: 10.1016/j.canlet.2012.02.001
Bjelland, S., Birkeland, N.-K., Benneche, T., Volden, G., and Seeberg, E. (1994). DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J. Biol. Chem. 269, 30489–30495.
Bjelland, S., Eide, L., Time, R. W., Stote, R., Eftedal, I., Volden, G., et al. (1995). Oxidation of thymine to 5-formyluracil in DNA: mechanisms of formation, structural implications, and base excision by human cell free extracts. Biochemistry 34, 14758–14764. doi: 10.1021/bi00045a017
Bjelland, S., and Seeberg, E. (1987). Purification and characterization of 3-methyladenine DNA glycosylase I from Escherichia coli. Nucleic Acids Res. 15, 2787–2801. doi: 10.1093/nar/15.7.2787
Bjelland, S., and Seeberg, E. (2003). Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 531, 37–80. doi: 10.1016/j.mrfmmm.2003.07.002
Boiteux, S., Coste, F., and Castaing, B. (2017). Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free Radic. Biol. Med. 107, 179–201. doi: 10.1016/j.freeradbiomed.2016.11.042
Booth, J. A., Thomassen, G. O., Rowe, A. D., Weel-Sneve, R., Lagesen, K., Kristiansen, K. I., et al. (2013). Tiling array study of MNNG treated Escherichia coli reveals a widespread transcriptional response. Sci. Rep. 3:3053. doi: 10.1038/srep03053
Boyce, R. P., and Howard-Flanders, P. (1964). Release of ultraviolet light-induced thymine dimers from DNA in E. coli K-12. Proc. Natl. Acad. Sci. U.S.A. 51, 293–300. doi: 10.1073/pnas.51.2.293
Cadet, J., Berger, M., Douki, T., and Ravanat, J.-L. (1997). Oxidative damage to DNA: formation, measurement, and biological significance. Rev. Physiol. Biochem. Pharmacol. 131, 1–87. doi: 10.1007/3-540-61992-5_5
Cupples, C. G., and Miller, J. H. (1989). A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl. Acad. Sci. U.S.A. 86, 5345–5349. doi: 10.1073/pnas.86.14.5345
Dizdaroglu, M. (2015). Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Rev. Mutat. Res. 763, 212–245. doi: 10.1016/j.mrrev.2014.11.002
Doetsch, P. W., and Cunningham, R. P. (1990). The enzymology of apurinic/apyrimidinic endonucleases. Mutat. Res. 236, 173–201. doi: 10.1016/0921-8777(90)90004-o
Douki, T., Delatour, T., Paganon, F., and Cadet, J. (1996). Measurement of oxidative damage at pyrimidine bases in γ-irradiated DNA. Chem. Res. Toxicol. 9, 1145–1151. doi: 10.1021/tx960095b
Evensen, G., and Seeberg, E. (1982). Adaptation to alkylation resistance involves the induction of a DNA glycosylase. Nature 296, 773–775. doi: 10.1038/296773a0
Feig, D. I., Sowers, L. C., and Loeb, L. A. (1994). Reverse chemical mutagenesis: identification of the mutagenic lesions resulting from reactive oxygen species-mediated damage to DNA. Proc. Natl. Acad. Sci. U.S.A. 91, 6609–6613. doi: 10.1073/pnas.91.14.6609
Foster, P. L., Lee, H., Popodi, E., Townes, J. P., and Tang, H. (2015). Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 112, E5990–E5999. doi: 10.1073/pnas.1512136112
Freudenthal, B. D., Beard, W. A., Perera, L., Shock, D. D., Kim, T., Schlick, T., et al. (2015). Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature 517, 635–639. doi: 10.1038/nature13886
Friedberg, E. C., Walker, G. C., Siede, W., Wood, R. D., Schultz, R. A., and Ellenberger, T. (2006). DNA Repair and Mutagenesis. Washington, DC: ASM Press.
Fujikawa, K., Kamiya, H., and Kasai, H. (1998). The mutations induced by oxidatively damaged nucleotides, 5-formyl-dUTP and 5-hydroxy-dCTP, in Escherichia coli. Nucleic Acids Res. 26, 4582–4587. doi: 10.1093/nar/26.20.4582
Grzesiuk, E., Gozdek, A., and Tudek, B. (2001). Contribution of E. coli AlkA, TagA glycosylases and UvrABC-excinuclease in MMS mutagenesis. Mutat. Res. 480–481, 77–84. doi: 10.1016/s0027-5107(01)00171-3
Gu, L., Huang, S.-M., and Sander, M. (1993). Drosophila Rrp1 complements E. coli xth nfo mutants: protection against both oxidative and alkylation-induced DNA damage. Nucleic Acids Res. 21, 4788–4795. doi: 10.1093/nar/21.20.4788
Halliwell, B., and Gutteridge, J. M. C. (1989). Free Radicals in Biology and Medicine. Oxford: Clarendon Press.
Hollis, T., Ichikawa, Y., and Ellenberger, T. (2000). DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 19, 758–766. doi: 10.1093/emboj/19.4.758
Kamiya, H., Murata-Kamiya, N., Karino, N., Ueno, Y., Matsuda, A., and Kasai, H. (2002). Induction of T → G and T → A transversions by 5-formyluracil in mammalian cells. Mutat. Res. 513, 213–222. doi: 10.1016/s1383-5718(01)00312-6
Karran, P., Hjelmgren, T., and Lindahl, T. (1982). Induction of a DNA glycosylase for N-methylated purines is part of the adaptive response to alkylating agents. Nature 296, 770–773. doi: 10.1038/296770a0
Kasai, H., Iida, A., Yamaizumi, Z., Nishimura, S., and Tanooka, H. (1990). 5-Formyldeoxyuridine: a new type of DNA damage induced by ionizing radiation and its mutagenicity to Salmonella strain TA102. Mutat. Res. 243, 249–253. doi: 10.1016/0165-7992(90)90139-b
Kino, K., Shimizu, Y., Sugasawa, K., Sugiyama, H., and Hanaoka, F. (2004). Nucleotide excision repair of 5-formyluracil in vitro is enhanced by the presence of mismatched bases. Biochemistry 43, 2682–2687. doi: 10.1021/bi0361416
Kleibl, K. (2002). Molecular mechanisms of adaptive response to alkylating agents in Escherichia coli and some remarks on O6-methylguanine DNA-methyltransferase in other organisms. Mutat. Res. 512, 67–84. doi: 10.1016/s1383-5742(02)00025-x
Klungland, A., Paulsen, R., Rolseth, V., Yamada, Y., Ueno, Y., Wiik, P., et al. (2001). 5-Formyluracil and its nucleoside derivatives confer toxicity and mutagenicity to mammalian cells by interfering with normal RNA and DNA metabolism. Toxicol. Lett. 119, 71–78. doi: 10.1016/s0378-4274(00)00308-8
Knævelsrud, I., Slupphaug, G., Leiros, I., Matsuda, A., Ruoff, P., and Bjelland, S. (2009). Opposite-base dependent excision of 5-formyluracil from DNA by hSMUG1. Int. J. Radiat. Biol. 85, 413–420. doi: 10.1080/09553000902818915
Kreutzer, D. A., and Essigmann, J. M. (1998). Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc. Natl. Acad. Sci. U.S.A. 95, 3578–3582. doi: 10.1073/pnas.95.7.3578
Labahn, J., Schärer, O. D., Long, A., Ezaz-Nikpay, K., Verdine, G. L., and Ellenberger, T. E. (1996). Structural basis for the excision repair of alkylation-damaged DNA. Cell 86, 321–329. doi: 10.1016/s0092-8674(00)80103-8
Landini, P., and Busby, S. J. (1999). Expression of the Escherichia coli Ada regulon in stationary phase: evidence for rpoS-dependent negative regulation of alkA transcription. J. Bacteriol. 181, 6836–6839. doi: 10.1128/jb.181.21.6836-6839.1999
Landini, P., and Volkert, M. R. (2000). Regulatory responses of the adaptive response to alkylation damage: a simple regulon with complex regulatory features. J. Bacteriol. 182, 6543–6549. doi: 10.1128/jb.182.23.6543-6549.2000
Lee, A. J., and Wallace, S. S. (2017). Hide and seek: how do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radic. Biol. Med. 107, 170–178. doi: 10.1016/j.freeradbiomed.2016.11.024
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature 362, 709–715. doi: 10.1038/362709a0
Lindahl, T., Sedgwick, B., Sekiguchi, M., and Nakabeppu, Y. (1988). Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem. 57, 133–157. doi: 10.1146/annurev.bi.57.070188.001025
Lindahl, T., and Wood, R. D. (1999). Quality control by DNA repair. Science 286, 1897–1905. doi: 10.1126/science.286.5446.1897
Liu, P., Burdzy, A., and Sowers, L. C. (2003). Repair of the mutagenic DNA oxidation product, 5-formyluracil. DNA Repair 2, 199–210. doi: 10.1016/s1568-7864(02)00198-2
Luria, S. E., and Delbrück, M. (1943). Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 491–511.
Masaoka, A., Terato, H., Kobayashi, M., Honsho, A., Ohyama, Y., and Ide, H. (1999). Enzymatic repair of 5-formyluracil. I. Excision of 5-formyluracil site-specifically incorporated into oligonucleotide substrates by AlkA protein (Escherichia coli 3-methyladenine DNA glycosylase II). J. Biol. Chem. 274, 25136–25143. doi: 10.1074/jbc.274.35.25136
McCullough, A. K., Dodson, M. L., and Lloyd, R. S. (1999). Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem. 68, 255–285. doi: 10.1146/annurev.biochem.68.1.255
Mielecki, D., Wrzesiński, M., and Grzesiuk, E. (2015). Inducible repair of alkylated DNA in microorganisms. Mutat. Res. Rev. Mutat. Res. 763, 294–305. doi: 10.1016/j.mrrev.2014.12.001
Mokkapati, S. K., Fernandez, De Henestrosa, A. R., and Bhagwat, A. S. (2001). Escherichia coli DNA glycosylase Mug: a growth-regulated enzyme required for mutation avoidance in stationary-phase cells. Mol. Microbiol. 41, 1101–1111. doi: 10.1046/j.1365-2958.2001.02559.x
Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., et al. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662. doi: 10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b
Nakabeppu, Y., Kondo, H., and Sekiguchi, M. (1984). Cloning and characterization of the alkA gene of Escherichia coli that encodes 3-methyladenine DNA glycosylase II. J. Biol. Chem. 259, 13723–13729.
Ono, A., Okamoto, T., Inada, M., Nara, H., and Matsuda, A. (1994). Nucleosides and nucleotides. 131. Synthesis and properties of oligonucleotides containing 5-formyl-2′-deoxyuridine. Chem. Pharm. Bull. 42, 2231–2237. doi: 10.1248/cpb.42.2231
O’Brien, P. J., and Ellenberger, T. (2004). The Escherichia coli 3-methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J. Biol. Chem. 279, 26876–26884. doi: 10.1074/jbc.m403860200
Park, J. S., Chang, C. T.-C., Schmidt, C. L., Golander, Y., De Clercq, E., Descamps, J., et al. (1980). Oxime and dithiolane derivatives of 5-formyl-2′-deoxyuridine and their 5′-phosphates: antiviral effects and thymidylate synthetase inhibition. J. Med. Chem. 23, 661–665. doi: 10.1021/jm00180a016
Patching, S. G., Baldwin, S. A., Baldwin, A. D., Young, J. D., Gallagher, M. P., Henderson, P. J., et al. (2005). The nucleoside transport proteins, NupC and NupG, from Escherichia coli: specific structural motifs necessary for the binding of ligands. Org. Biomol. Chem. 3, 462–470. doi: 10.1039/b414739a
Purmal, A. A., Kow, Y. W., and Wallace, S. S. (1994). Major oxidative products of cytosine, 5-hydroxycytosine and 5-hydroxyuracil, exhibit sequence context-dependent mispairing in vitro. Nucleic Acids Res. 22, 72–78. doi: 10.1093/nar/22.1.72
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rosche, W. A., and Foster, P. L. (2000). Determining mutation rates in bacterial populations. Methods 20, 4–17. doi: 10.1006/meth.1999.0901
Saget, B. M., and Walker, G. C. (1994). The Ada protein acts as both a positive and a negative modulator of Escherichia coli’s response to methylating agents. Proc. Natl. Acad. Sci. U.S.A. 91, 9730–9734. doi: 10.1073/pnas.91.21.9730
Sakumi, K., Nakabeppu, Y., Yamamoto, Y., Kawabata, S., Iwanaga, S., and Sekiguchi, M. (1986). Purification and structure of 3-methyladenine-DNA glycosylase I of Escherichia coli. J. Biol. Chem. 261, 15761–15766.
Sakumi, K., and Sekiguchi, M. (1989). Regulation of expression of the ada gene controlling the adaptive response. Interactions with the ada promoter of the Ada protein and RNA polymerase. J. Mol. Biol. 205, 373–385. doi: 10.1016/0022-2836(89)90348-3
Sedgwick, B. (2004). Repairing DNA-methylation damage. Nat. Rev. Mol. Cell. Biol. 5, 148–157. doi: 10.1038/nrm1312
Shrivastav, N., Li, D., and Essigmann, J. M. (2010). Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis 31, 59–70. doi: 10.1093/carcin/bgp262
Soll, J. M., Sobol, R. W., and Mosammaparast, N. (2017). Regulation of DNA alkylation damage repair: lessons and therapeutic opportunities. Trends Biochem. Sci. 42, 206–218. doi: 10.1016/j.tibs.2016.10.001
Studier, F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. doi: 10.1016/j.pep.2005.01.016
Teo, I., Sedgwick, B., Kilpatrick, M. W., Mccarthy, T. V., and Lindahl, T. (1986). The intracellular signal for induction of resistance to alkylating agents in E. coli. Cell 45, 315–324. doi: 10.1016/0092-8674(86)90396-x
Terato, H., Masaoka, A., Kobayashi, M., Fukushima, S., Ohyama, Y., Yoshida, M., et al. (1999). Enzymatic repair of 5-formyluracil. II. Mismatch formation between 5-formyluracil and guanine during DNA replication and its recognition by two proteins involved in base excision repair (AlkA) and mismatch repair (MutS). J. Biol. Chem. 274, 25144–25150. doi: 10.1074/jbc.274.35.25144
Tsunoda, M., Kondo, J., Karino, N., Ueno, Y., Matsuda, A., and Takénaka, A. (2002). Water mediated Dickerson-drew-type crystal of DNA dodecamer containing 2′-deoxy-5-formyluridine. Biophys. Chem. 95, 227–233. doi: 10.1016/s0301-4622(01)00259-9
Tsunoda, M., Sakaue, T., Naito, S., Sunami, T., Abe, N., Ueno, Y., et al. (2010). Insights into the structures of DNA damaged by hydroxyl radical: crystal structures of DNA duplexes containing 5-formyluracil. J. Nucleic Acids 2010:107289. doi: 10.4061/2010/107289
Tsunoda, M., Sakaue, T., Naito, S., Sunami, T., Karino, N., Ueno, Y., et al. (2001). X-ray analyses of DNA dodecamers containing 2′-deoxy-5-formyluridine. Nucleic Acids Res. Suppl. 61, 279–280. doi: 10.1093/nass/1.1.279
Van Houten, B., Croteau, D. L., Dellavecchia, M. J., Wang, H., and Kisker, C. (2005). ‘Close-fitting sleeves’: DNA damage recognition by the UvrABC nuclease system. Mutat. Res. 577, 92–117. doi: 10.1016/j.mrfmmm.2005.03.013
Wibley, J. E. A., Waters, T. R., Haushalter, K., Verdine, G. L., and Pearl, L. H. (2003). Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol. Cell 11, 1647–1659. doi: 10.1016/s1097-2765(03)00235-1
Yamagata, Y., Kato, M., Odawara, K., Tokuno, Y., Nakashima, Y., Matsushima, N., et al. (1996). Three-dimensional structure of a DNA repair enzyme, 3-methyladenine DNA glycosylase II, from Escherichia coli. Cell 86, 311–319. doi: 10.1016/s0092-8674(00)80102-6
Yamamoto, Y., Katsuki, M., Sekiguchi, M., and Otsuji, N. (1978). Escherichia coli gene that controls sensitivity to alkylating agents. J. Bacteriol. 135, 144–152. doi: 10.1128/jb.135.1.144-152.1978
Yoshida, M., Makino, K., Morita, H., Terato, H., Ohyama, Y., and Ide, H. (1997). Substrate and mispairing properties of 5-formyl-2′-deoxyuridine 5′-triphosphate assessed by in vitro DNA polymerase reactions. Nucleic Acids Res. 25, 1570–1577. doi: 10.1093/nar/25.8.1570
Zhang, Q.-M., Fujimoto, J., and Yonei, S. (1995). Enzymatic release of 5-formyluracil by mammalian liver extracts from DNA irradiated with ionizing radiation. Int. J. Radiat. Biol. 68, 603–607. doi: 10.1080/09553009514551601
Zhang, Q. M., Miyabe, I., Matsumoto, Y., Kino, K., Sugiyama, H., and Yonei, S. (2000). Identification of repair enzymes for 5-formyluracil in DNA. Nth, Nei, and MutM proteins of Escherichia coli. J. Biol. Chem. 275, 35471–35477. doi: 10.1074/jbc.m006125200
Zhang, Q.-M., Sugiyama, H., Miyabe, I., Matsuda, S., Saito, I., and Yonei, S. (1997). Replication of DNA templates containing 5-formyluracil, a major oxidative lesion of thymine in DNA. Nucleic Acids Res. 25, 3969–3973. doi: 10.1093/nar/25.20.3969
Zharkov, D. O. (2008). Base excision DNA repair. Cell. Mol. Life Sci. 65, 1544–1565. doi: 10.1007/s00018-008-7543-2
Keywords: 5-formyluracil, oxidized base repair, base substitution, AlkA, DNA glycosylase
Citation: Grøsvik K, Tesfahun AN, Muruzábal-Lecumberri I, Haugland GT, Leiros I, Ruoff P, Kvaløy JT, Knævelsrud I, Ånensen H, Alexeeva M, Sato K, Matsuda A, Alseth I, Klungland A and Bjelland S (2020) The Escherichia coli alkA Gene Is Activated to Alleviate Mutagenesis by an Oxidized Deoxynucleoside. Front. Microbiol. 11:263. doi: 10.3389/fmicb.2020.00263
Received: 09 December 2019; Accepted: 04 February 2020;
Published: 25 February 2020.
Edited by:
Marie-Joelle Virolle, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Modesto Redrejo-Rodríguez, Spanish National Research Council, SpainRobert G. Fowler, San José State University, United States
Copyright © 2020 Grøsvik, Tesfahun, Muruzábal-Lecumberri, Haugland, Leiros, Ruoff, Kvaløy, Knævelsrud, Ånensen, Alexeeva, Sato, Matsuda, Alseth, Klungland and Bjelland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svein Bjelland, svein.bjelland@uis.no