 Bo-Shiun Chen
Bo-Shiun Chen Hou-Chen Lee
Hou-Chen Lee Kuo-Ming Lee
Kuo-Ming Lee Yu-Nong Gong
Yu-Nong Gong Shin-Ru Shih
Shin-Ru Shih- 1Research Center for Emerging Viral Infections, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 2Department of Neuroscience and Regenerative Medicine, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 3Department of Laboratory Medicine, Linkou Chang Gung Memorial Hospital, Taoyuan, Taiwan
- 4Department of Medical Biotechnology and Laboratory Science, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 5Research Center for Chinese Herbal Medicine, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
- 6Research Center for Food and Cosmetic Safety, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
- 7Graduate Institute of Health Industry Technology, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
Enterovirus-induced infection of the central nervous system (CNS) results in acute inflammation of the brain (encephalitis) and constitutes a significant global burden to human health. These viruses are thought to be highly cytolytic, therefore normal brain function could be greatly compromised following enteroviral infection of the CNS. A further layer of complexity is added by evidence showing that some enteroviruses may establish a persistent infection within the CNS and eventually lead to pathogenesis of certain neurodegenerative disorders. Interestingly, enterovirus encephalitis is particularly common among young children, suggesting a potential causal link between the development of the neuroimmune system and enteroviral neuroinvasion. Although the CNS involvement in enterovirus infections is a relatively rare complication, it represents a serious underlying cause of mortality. Here we review a selection of enteroviruses that infect the CNS and discuss recent advances in the characterization of these enteroviruses with regard to their routes of CNS infection, tropism, virulence, and immune responses.
Introduction
Enteroviruses belong to the family Picornaviridae, a highly diverse group of small, non-enveloped, icosahedral-shaped viruses with single positive-strand RNA genomes. Based on the sequence diversity, they have been divided into 15 species: enterovirus A to L and rhinovirus A to C. Human enteroviruses containing four enterovirus species (A to D) and three rhinovirus species (A to C) infect millions of people worldwide every year. Although infections are frequently asymptomatic, human enteroviruses can cause a variety of symptoms comprising fever, headache, respiratory illness, sore throat, and, occasionally, vomiting and diarrhea. Importantly, several members of human enteroviruses are neurotropic pathogens with a wide range of clinical disorders ranging from aseptic meningitis to more severe encephalitis. In the United States, enterovirus has been shown to be the most common etiology of meningitis/encephalitis (Hasbun et al., 2017; Balada-Llasat et al., 2019). About 58% of the infected infants and children and 52% of the infected adults diagnosed with meningitis/encephalitis is due to enterovirus. In addition, age is highly associated with the clinical presentation with severe infections including CNS disease, myocarditis and sepsis-like illness occurring most frequently in neonates and infants. The best-known neurotropic enterovirus is the poliovirus (PV), which belongs to the species Enterovirus C and is believed to have almost been eradicated by vaccinations from circulation in human populations. Non-polio enteroviruses are also known to infect the CNS and account for the majority of recent enteroviral infections with neurological disorders. Among the non-polio enteroviruses, the species Enterovirus A like enterovirus 71 (EV-A71), coxsackievirus A6 (CV-A6), and CV-A16 (Goto et al., 2009; Xu et al., 2012; Huang Y. et al., 2015; Holmes et al., 2016; B’Krong et al., 2018; Suresh et al., 2018), the species Enterovirus B like CV-B1 (Sun et al., 2019), CV-B3 (Fan and Liu, 2019), CV-B5 (Mao et al., 2018), CV-A9, Echovirus 6 (E-6), E-7, E-11, and E-13 (Holmes et al., 2016; Chen et al., 2017; B’Krong et al., 2018; Suresh et al., 2018; Chen et al., 2019; Ramalho et al., 2019; Sun et al., 2019), the species Enterovirus C like CV-A24 (Tapparel et al., 2013; B’Krong et al., 2018; Suresh et al., 2018), and the species Enterovirus D like EV-D68 (Tapparel et al., 2013; Messacar et al., 2018) have been shown to involve the CNS and cause diverse neurological complications such as encephalitis, meningitis and acute flaccid paralysis (AFP) (Tapparel et al., 2013) (see Table 1 for details).
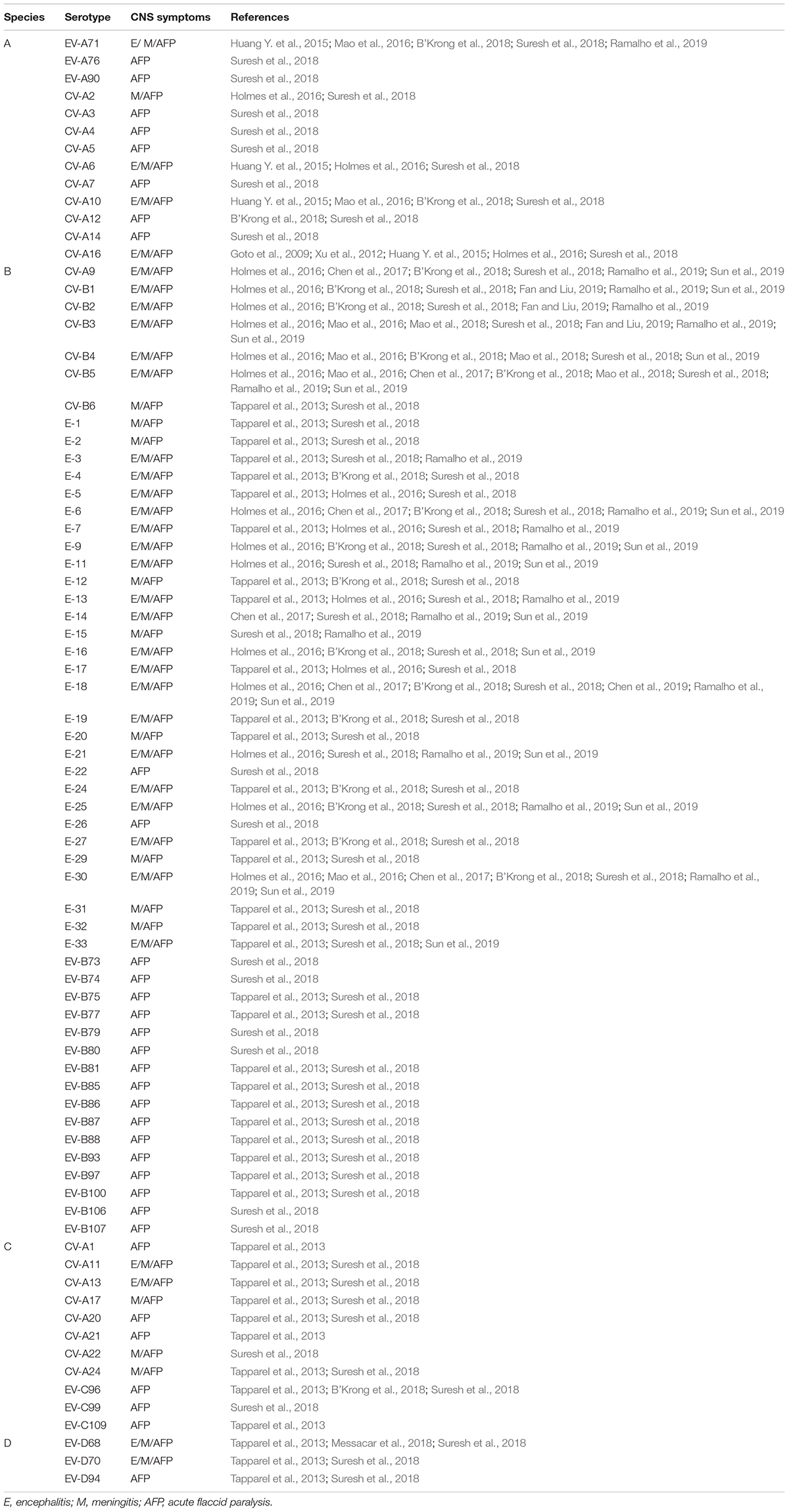
Table 1. Neurological symptoms of non-polio enteroviruses that involve the CNS.
Enterovirus Life Cycle
The enterovirus genome contains approximately 7.5 kb with a single open reading frame flanked by 5′- and 3′-untranslated regions (UTR) and is enclosed in a capsid. While the 5′ end is covalently attached to a viral protein genome-linked (VPg) required for replication, the polyadenylated 3′ terminus is important not only for the negative-stranded RNA synthesis but also for translation and RNA stability (Zoll et al., 2009; Kempf and Barton, 2015). The life cycle of enteroviruses begins with binding to one or more specific receptors on the cell surface. The cell receptors for enteroviruses are quite divergent. For example, the cell surface receptor for PV is cluster of differentiation 155 (CD155), whereas EV-A71 has nine cell surface receptors including human scavenger receptor class B member 2 (hSCARB2), human P-selectin glycoprotein ligand 1 (hPSGL1), annexin II (Anx2), heparan sulfate, sialylated glycan, dendritic cell-specific ICAM3-grabbing non-integrin, vimentin, nucleolin and human tryptophanyl-tRNA synthetase (hWARS) (Baggen et al., 2018; Yeung et al., 2018). Receptor binding allows viruses to enter host cells via receptor-mediated endocytosis. There are several endocytic pathways that can mediate the entry of receptor-bound enterovirus particles at the plasma membrane, including macropinocytosis, clathrin-dependent endocytosis and clathrin-independent uptake such as caveolae-dependent and non-caveolae-dependent endocytosis (Marjomaki et al., 2015). The specific endocytic pathways that virus exploits to enter the cells depend on the viral species and serotype, host cell type and local microenvironment (such as pH and temperature). For instance, EV-A71 enters rhabdomyosarcoma (RD) cells through the clathrin-dependent pathway, whereas the caveolae-mediated pathway is used to enter Jurkat cells. EV-A71 exhibits distinct endocytic pathways in different host cells, partly because of the diversity of the EV-A71 surface receptors (Yamayoshi et al., 2014). Upon entry into host cells, virus uncoating releases its viral RNA genome into the cytoplasm and translation is initiated from an internal ribosome sequence in the 5′ UTR, which is called internal ribosome entry site (IRES). An IRES is a cis-acting RNA element that forms secondary and tertiary structures to allow cap-independent initiation of translation (Shih et al., 2011; Lee et al., 2017). Translation of viral RNA yields a single polyprotein, which is proteolytically processed by viral proteases to produce four capsid proteins (VP4, VP2, VP3, and VP1) required for virion packaging, and seven non-structural proteins (2A-2B-2C and 3A-3B-3C-3Dpol) as well as some stable precursors, which function in virus replication and disrupting the host cellular immune system. Viral RNA replication is catalyzed by the virally encoded RNA-dependent RNA polymerases, 3Dpol, which utilizes Vpg as a protein primer to initiate the replication process. The synthesis of viral RNA takes place on the virus-induced remodeling of intracellular membranes that form replication organelles and leads to double-stranded RNA formation, which in turn will be transcribed into positive single-stranded RNA (Hsu et al., 2010; van der Schaar et al., 2016). Newly synthesized RNA may either serve as a template for translation and replication or be packaged into new infectious virions.
CNS Invasion
Enteroviruses infect humans primarily through the fecal-oral route and replicate in the gastrointestinal tract with the exception of some enteroviruses, e.g., rhinovirus and EV-D68, which can cause respiratory infection and spread via respiratory secretion. After initial infection in the first exposed area, the enteroviruses can gain access to the CNS through multiple pathways, which are not mutually exclusive (Figure 1; Rhoades et al., 2011; Huang and Shih, 2015). First, most neurotropic viruses including enteroviruses spread through the bloodstream to reach the CNS. The spread of virus particles from the blood to the CNS is normally restricted by the blood brain barrier (BBB), which is a highly selective semipermeable barrier between the brain’s blood vessels and the cells in the brain. However, BBB integrity can be compromised by direct infection of the brain microvascular endothelial cells (BMECs) that make up the BBB or by the cytokines that are produced locally in the CNS during viral infections. For example, PV has been shown to invade the CNS via BBB transmission (Yang et al., 1997). Recent evidence has further demonstrated that mouse transferrin receptor 1 is responsible for PV attachment to the cell surface of BMECs, allowing invasion into the CNS via the BBB (Mizutani et al., 2016). Secondly, enteroviruses can invade the CNS through the peripheral circulating immune cells, which carry intracellular viruses (Tabor-Godwin et al., 2010). This is known as the Trojan horse route. Although the brain has been considered a site of immune-privilege, it has an active immune surveillance system that involves the recruitment of non-specific leukocytes such as phagocytes and lymphocytes into the meninges and cerebrospinal fluid (CSF) (Forrester et al., 2018). Indeed, it has been shown that the CSF contains a trafficking population of mononuclear cells, consisting of T cells (∼90%), B cells (∼5%), monocytes (∼5%), and dendritic cells (<1%) (Ransohoff and Engelhardt, 2012). Once these leukocytes are infected, they can act as carriers to bring viruses into the CNS. For instance, CV-B3-infected myeloid cells have been shown to cross the blood-CSF barrier in the choroid plexus (Tabor-Godwin et al., 2010). Upon entry into the CNS, the virus is likely released from myeloid cells and subsequently infect neurons and/or glia in the brain. There is also evidence that EV-A71 can infect leukocytes through binding to hPSGL1, a sialomucin membrane protein primarily expressed on leukocytes (Nishimura et al., 2009). Whether EV-A71-infected leukocytes can bring viruses into the CNS is not clear. Thirdly, certain enteroviruses can enter the CNS through peripheral nerves via retrograde axonal transport and trans-synaptic propagation (Gromeier and Wimmer, 1998; Chen et al., 2007; Ong et al., 2008). Axonal transport is an essential cellular process in neurons required for the movement of synaptic vesicles, lipids, proteins, and organelles including mitochondria, lysosomes, autophagosomes, and endosomes, to and from the cell body. It is well known that some neurotropic viruses can hijack the retrograde axonal transport to invade the CNS. For example, studies have shown that intramuscularly inoculated PV is taken up by endocytosis at the neuromuscular junctions (Ohka et al., 2004). The endocytosed viral particles in the axon terminal are moved in the retrograde direction toward the cell body via dynein-mediated vesicular transport without initiating uncoating (Ohka et al., 2009). The uncoating event takes place upon arrival at the cell body of the motor neuron. EV-A71 and EV-D68 can also enter and infect the CNS by retrograde axonal transport via peripheral spinal motor nerves (Chen et al., 2007; Ong et al., 2008; Hixon et al., 2019). Interestingly, a recent report has demonstrated that EV-A71 can directly infect the brainstem via cranial nerves, suggesting that the virus can use not only the motor components of spinal nerves but also cranial nerves to enter the CNS (Tan et al., 2014).
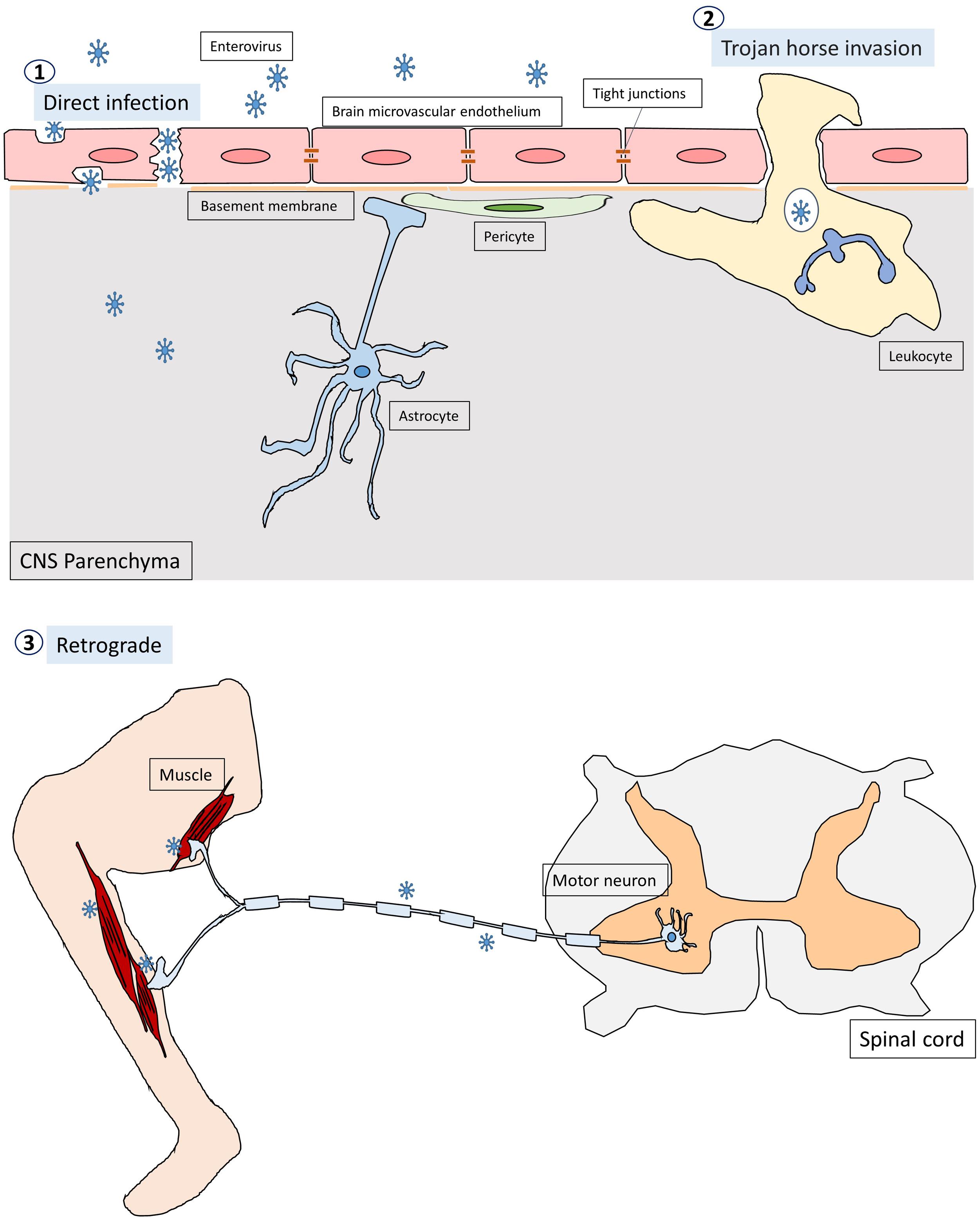
Figure 1. The pathways through which enteroviruses can gain access to the CNS. Enteroviruses can invade the CNS through three pathways. First, they infect BMECs, compromising and crossing the BBB to reach the CNS directly. The second pathway is known as Trojan horse route, in which enterovirus-infected leukocytes act as carriers and transverse the brain barriers bringing virus into the CNS. Thirdly, enteroviruses are able to hijack the retrograde axonal transport to enter the CNS from muscles into the motor neurons in the spinal cord.
Tropism
Each enterovirus has a distinct tropism that is determined by a combination of host and virus factors (Figure 2; Lin and Shih, 2014). Although neurotropic enteroviruses can invade the CNS and are associated with neurological disorders, dissemination of the virus to the CNS seems to occur sporadically and it is not clear how enterovirus targets specific regions and cell types in the brain and spinal cord. Earlier studies on poliovirus have suggested that virus tropism is determined by the cellular receptor for virus entry (Holland, 1961). However, the PV receptor CD155 is found in tissues that are not sites of PV infection (Mendelsohn et al., 1989; Freistadt et al., 1990; Koike et al., 1990), indicating that the cellular receptor is required for susceptibility to PV infection but not the sole determinant for virus tropism. Subsequent studies have suggested that the tissue-specific activity of IRES on viral RNAs also plays an important role in determining virus tropism (Gromeier et al., 1996; Yanagiya et al., 2003). For example, a chimeric PV carrying the IRES of hepatitis C virus replicates well in the liver but not in the brain of a mouse model for poliomyelitis, whereas the control poliovirus replicates well both in the liver and brain (Yanagiya et al., 2003). There is also evidence that innate immune antiviral activities such as the interferon (IFN) response is critical for virus tropism (Wessely et al., 2001; Ida-Hosonuma et al., 2005). In transgenic mice containing human CD155, PV replicates and produces severe lesions in the brain and spinal cord, whereas other tissues did not show severe pathological changes. However, in the CD155 transgenic mice lacking alpha/beta IFN, severe lesions are detected in the liver, spleen, and pancreas in addition to the CNS, suggesting that the alpha/beta IFN system is an important determinant for the differential susceptibility of tissue to PV. Poliovirus invades the CNS, which leads to the development of a paralytic disease in about 1% of virus-infected people (Melnick, 1996). Therefore, it has been suggested that, in 99% of the infected cases, the IFN response limits PV replication in extraneural tissues to prevent the invasion of the CNS (Racaniello, 2006). In the CNS, PV infects and replicates mainly in motor neurons in the anterior horn of the spinal cord, resulting in poliomyelitis (Nagata et al., 2004; Arita et al., 2006). In the most severe cases, PV attacks the neurons of the brainstem, causing bulbar poliomyelitis. In addition to neurons, the CNS contains three major types of non-neuronal cells called glial cells, including astrocytes, oligodendrocytes, and microglia. These glial cells play critical roles in maintaining homeostasis, myelin formation and providing support and protection for neurons. Interestingly, astrocytes and oligodendrocytes are also susceptible to PV infection in primary mouse culture prepared from the cerebral cortex of neonatal CD155 transgenic mice (Couderc et al., 2002). Whether these glial cells are targets for PV infection in the human brain is not known.
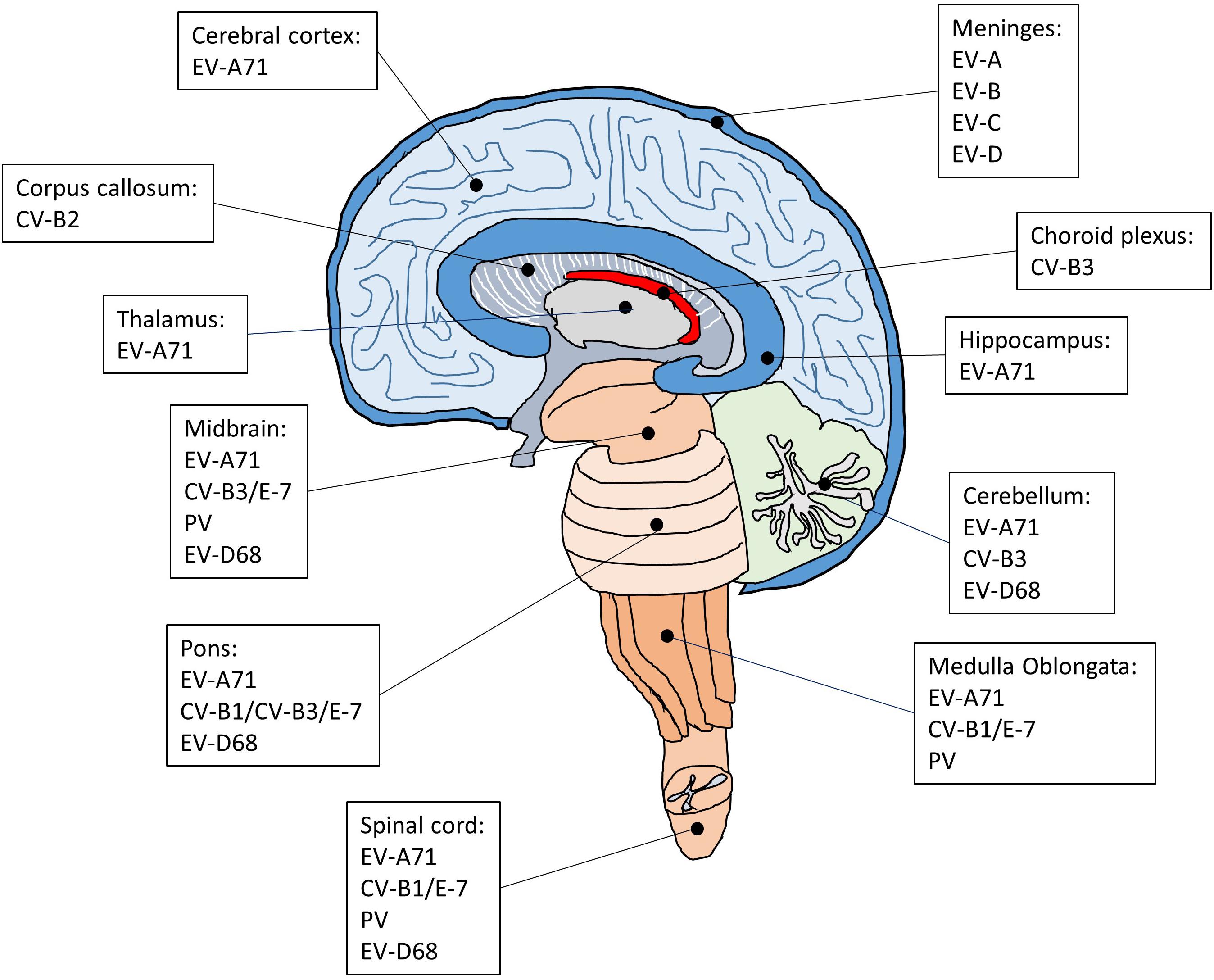
Figure 2. The regions of the CNS affected by enteroviruses that cause encephalitis, meningitis and AFP. The CNS is composed of cerebrum, diencephalon, cerebellum, brainstem, and spinal cord. Following enteroviral infection of the CNS, inflammation can occur in different anatomical regions such as the brain (encephalitis), meninges (meningitis), and spinal cord (myelitis), or simultaneously in multiple regions (meningoencephalitis, encephalomyelitis).
Although EV-A71 also invade the CNS, the infected areas are quite distinct compared with poliovirus. Brainstem encephalitis is the most common neurological presentation of EV-A71 infection. Consistently, viral lesions are mainly observed in the brainstem and predominantly located in the ventral, medial and caudal areas of the medulla oblongata (Kao et al., 2004). A few lesions are also found in the cortex, cerebellum, and spinal cord. In severe cases of EV-A71 infection, the major histopathological changes in the CNS are characterized by inflammatory damage, which selectively leads to neurogenic pulmonary edema and cardiac failure. In agreement with the main CNS lesion locations of EV-A71 infection, the neurons in the medulla oblongata have been implicated in the onset of neurogenic pulmonary edema (Davison et al., 2012). In addition, several autopsy results have suggested that EV-A71 may infect neurons and cause neuronal degeneration, which would activate inflammatory responses in the lesion area and cause encephalitis (Yan et al., 2000; Khong et al., 2012; Yao et al., 2012). Indeed, studies have shown that neurons are susceptible to EV-A71 infection (Huang et al., 2014; Feng et al., 2016). Intriguingly, although neurons can be infected by EV-A71, neural progenitor cells and astrocytes appear to be the main targets for EV-A71 infection in the CNS (Huang et al., 2014; Feng et al., 2016). Both of these cell types are similar in that they are capable of mitosis, which may be critical for virus replication (Yu et al., 2015). Neural progenitor cells are the progenitor cells of the CNS that give rise to many, if not all, of the neuronal and glial cell types and are important for many brain functions including learning and memory and cognition. Thus, the loss of neural progenitor cells due to EV-A71 infection may cause long-term abnormalities of the CNS. This is supported by a long-term follow-up study showing that a large proposition of children after EV-A71 infection with severe CNS involvement and cardiopulmonary failure exhibited delayed neurodevelopment and reduced cognitive function (Chang et al., 2007). Astrocytes also perform many functions in the brain, such as contribution to the formation of the blood-brain barrier, maintenance of extracellular ionic and chemical homeostasis and involvement in the injury response. Since astrocytes are mitotic and localized in a much broader area in the brain than neural progenitor cells, the preferential infection of astrocytes over neurons may create a reservoir for viral proliferation and enable the viral progeny to quickly spread in the CNS and induce massive inflammatory responses. It would be of interest to understand how EV-A71 show a preference for neural progenitor cells and astrocytes.
Similar to EV-A71, coxsackievirus has also been shown to infect neural progenitor cells in addition to neurons (Feuer et al., 2005). Studies have revealed that CV-B3 can infect proliferating neural progenitor cells located in the neonatal subventricular zone and hippocampus. Interestingly, CV-B3 preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells (Tsueng et al., 2011). CV-B3 mediated loss of neural progenitor cells leads to a rapid decline in neurogenesis and may eventually cause developmental defects and CNS dysfunction (Ruller et al., 2012). However, the determinants of CV-B3 tropism in the CNS remain unclear. CV-B3 binds to target cells through two main receptors: decay-accelerating factor (DAF) (Bergelson et al., 1995) and coxsackievirus and adenovirus receptor (CAR) (Bergelson, 2009), which has been found to be highly expressed in the developing brain (Xu and Crowell, 1996). There is evidence that immature neurons express relatively high levels of CAR compare to their fully differentiated counterparts (Ahn et al., 2008), suggesting that the level of virus receptor is one of the critical determinants in preferential virus replication in undifferentiated neural progenitor cells. Besides, CAR exists as multiple isoforms and a specific isoform of CAR that is expressed at high levels in human pancreatic beta cells has been suggested to be prone to coxsackievirus infection (Ifie et al., 2018). Whether this specific isoform of CAR plays a role in coxsackievirus infection in the CNS is not known.
Persistent Infection
Though enteroviruses have been considered as cytolytic viruses and diseases caused by infection with enteroviruses are typically short-lived, several studies have indicated that some enteroviruses may be associated with lifelong disorders including post-polio syndrome (Muir et al., 1995; Julien et al., 1999), schizophrenia (Rantakallio et al., 1997; Suvisaari et al., 2003; Khandaker et al., 2012), amyotrophic lateral sclerosis (Woodall et al., 1994; Berger et al., 2000; Giraud et al., 2001), type 1 diabetes (Richardson and Morgan, 2018), and chronic viral cardiomyopathy (Chapman and Kim, 2008). The cause of these enterovirus-associated lifelong disorders is still not clear, but it has been hypothesized that persistent infection of enterovirus may occur based on the presence of enterovirus RNA and protein in the affected tissues at stages of disease after acute infection (Chapman and Kim, 2008). Consistently, the spread of enteroviruses within tissues is not always accompanied by cell death (Bird et al., 2014). Two major groups of persistent infection have been observed in vitro (Pinkert et al., 2011). One group, called steady-state infection, is characterized by infection of all cells, while the other group, named carrier-state infection, is characterized by a cytolytic infection of a small proportion of cells, which spares the majority of cells in culture from cytolysis. There is evidence that enteroviruses establish carrier-state infection in vitro (Heim et al., 1992; Heim et al., 1995; Pinkert et al., 2011). The carrier-state infection is thought to be induced by the selection of virus mutants that are less cytopathic and may involve the coevolution of both cells and viruses. For example, following CV-B3 infection, expression of the CV-B3 receptor CAR has evolved to be downregulated or eliminated in a subpopulation of cells (Pinkert et al., 2011), which is known to be associated with a decrease of CV-B3 infection and cell lysis (Werk et al., 2005; Fechner et al., 2007). As a result, these evolved cells are protected from virus infection and cell lysis and become dominant in the culture within several passages. In the meantime, the virus also adapts to persist by gaining a CAR-independent entry mechanism (Pinkert et al., 2011). Thus, during the development of persistent infection, viruses and cells have co-evolved such that cellular resistance to viral replication is balanced. In addition to the coevolution model, other viral genomic alterations that cause persistent infection have been reported in the highly conserved 5′ UTR, which is critical for viral replication. For instance, CV-B mutants with deletions in the 5′ end of the viral genome persist in host tissues, and RNA of the variants can be stably detected from heart tissue of mice experimentally inoculated with wild type CV-B3 and from human cases of myocarditis (Kim et al., 2005; Chapman et al., 2008; Kim et al., 2008), suggesting that viral replication is important for persistent infection in vivo. Nonetheless, it has also been suggested that persistence of CV-B1 RNA in skeletal muscle or CV-B3 RNA in the CNS is not facilitated by genetic alterations that give rise to replication-defective forms, but occurs primarily through formation of stable and atypical double-stranded RNA complex (Tam and Messner, 1999; Feuer et al., 2009). Interestingly, besides the reduction of CAR, other cellular factors have also been shown to play an important role in persistent infection of CV-B3. In particular, CV-B3 replication is affected by the cell cycle status, suggesting that the persistence of CV-B3 may dependent on infection of quiescent cells in which viral replication is lowered or suppressed (Feuer et al., 2002; Feuer et al., 2004). Therefore, the differences between these mechanisms are likely due to distinct patterns of virus-host interactions and there may be tissue- or cell-specific mechanisms for establishing a persistent infection.
Early viral infection of the CNS can cause severe physical and intellectual disability and, in some cases, results in unexpected neurological disorders years after acute infection. For example, approximately 30 percent of polio victims experience new symptoms (post-polio syndrome) about 50 years after the primary infection. Some studies have shown that post-polio syndrome is correlated with the presence of viral RNA in the CNS, suggesting that PV can persist and cause long-term damage in the CNS (Muir et al., 1995). In consistent with these findings, it has been shown that poliovirus strains are not fully lytic in neuroblastoma cell lines (Colbere-Garapin et al., 1989). Following PV infection, massive cytopathic effects are observed in cultured cells, but some cells survive infection without further observable cytopathic effects in spite of continuous viral production. During persistent infection, PV mutants are constantly selected, and many of the identified mutations occur at positions known to be involved in the binding of PV receptors, suggesting that the interactions of the virus with its receptor is critical for the establishment of persistent infections (Colbere-Garapin et al., 1998). Although there is not a single mechanism for establishing a persistent infection, it is believed that the virus must evade host’s antiviral immune response. Because enteroviruses have high mutation rates due to the lack of proofreading ability in RNA polymerases, they can generate a variety of mutants not only to affect receptor binding and virus replication but also to evade the immune system. The CNS is relatively inaccessible to immune surveillance compared with other tissues, which makes it particularly vulnerable to persistent infection. Using in vivo imaging, a recent study has demonstrated that EV-A71 infection of AG129 mice, in which alpha/beta and gamma interferon receptors are deficient, shows rapid spread and long-term persistence of the virus in the brain of surviving animals (Caine and Osorio, 2017). Interestingly, high viral loads are maintained in the brain even at 6 weeks following infection, whereas viral loads in other tissues including heart, lung, liver, spleen and intestine are gradually reduced to a lower level. In agreement with these findings, in vitro studies have shown that in contrast to RD cells or neuroblastoma cell lines, motor neuron-like hybrid cell line (NSC-34) infected by EV-A71 does not display cytopathic effect and the viral particles adopt a non-lytic exit pathway through autophagy (Too et al., 2016). Furthermore, there is evidence that EV-A71 RNA is present in stool weeks after initial infection (Han et al., 2010). Taken together, all these findings suggest that similar to PV and CV-B3, EV-A71 can also establish a persistent infection in the CNS, but the long-term impact of EV-A71 infection in the CNS remains elusive.
Neurovirulence
Some enteroviruses can cause diseases within the nervous system. The most thoroughly studied neurovirulent enterovirus is PV. To eradicate global poliomyelitis, much effort has been made to develop polio vaccines including an inactivated PV given by injection and an attenuated PV given by mouth. Although both types of polio vaccines are effective, oral polio vaccines are superior not only in administration but also in providing longer-lasting immunity. The attenuated PV was developed based on the observation that the virus no longer caused disease after many passages in different animals and cell cultures, but replicated sufficiently to induce protective immunity. Genetic analysis of the attenuated PV has shown that a point mutation within the IRES of the vaccine strains is a critical determinant of the attenuation phenotype (Evans et al., 1985; Kawamura et al., 1989; Ren et al., 1991). For instance, the C472U mutation in the IRES of poliovirus type 3 causes a translation defect, which leads to reduced replication in the CNS and attenuation of neurovirulence (La Monica and Racaniello, 1989; Gutierrez et al., 1997; Ohka and Nomoto, 2001). Subsequent studies have shown that the C472U mutation reduces the efficiency of binding of the polypyrimidine-tract binding protein (PTB) to the IRES, which is required for initiation of translation (Guest et al., 2004). Sequence analysis has also identified additional mutations in the capsid region of the vaccine strains. These capsid mutations are likely to disrupt viral particle binding to host cells and reduce the capsid stability, which may contribute to and stabilize the attenuation phenotype. However, PV is notably adaptable and attenuated viruses can increase its virulence through mutation and/or recombination (Jorba et al., 2008; Minor, 2009). Thus, although unusual, immunization with the attenuated vaccine strains may cause vaccine-associated paralytic poliomyelitis, which could be due to the reversion of the mutations in the viral genome that confer the attenuation phenotype and/or acquisition of new mutations with enhanced virulence (Kew et al., 2005; Famulare et al., 2016). Moreover, these vaccine-derived polioviruses (VDPV) have caused outbreaks of poliomyelitis in areas with low routine immunization rates (Burki, 2019). Using approach combining phylogenetic analysis of sequence data from outbreaks of VDPV and an experimental evolution approach in cell culture, a recent study has provided a model describing the evolutionary steps sufficient for the vaccine strain to lose its attenuation and become virulent (Stern et al., 2017). In the first step, the critical mutations for the attenuation phenotype are reverted to increase the viral replication. This is followed by recombination events with co-circulating enterovirus strains, most often a coxsackievirus strain, but in some cases a circulating PV strain to optimize viral replication in the human gut. In the final step, the virus continues to slowly revert to sequences that are conserved across wild-type PV to enhance the fitness of the virus. The information provides a powerful framework for developing safer vaccine strains and for forecasting virulence of viruses.
EV-A71 has emerged as a serious threat to public health across the Asia-Pacific region. EV-A71 causes hand, foot and mouth disease (HFMD) and herpangina, and occasionally severe neurological disorders. Unlike PV in which a point mutation within the IRES can attenuate neurovirulence, the association between EV-A71 neurovirulence and viral genome sequences remains largely unknown. So far, there are only a few reports showing that the neurovirulence can be weakened by mutations in the viral genome of EV-A71. For example, studies have shown that defined genetic manipulation of the EV-A71 genome based on the temperature-sensitive determinants of a poliovirus vaccine strain results in attenuated neurovirulence in monkeys (Arita et al., 2005). The generated EV-A71 mutant strain contains four mutations in the conserved regions of the enterovirus genome including one in the 5′ UTR, two in the 3D polymerase gene and one in the 3′ UTR, and all of them are required for substantial attenuation (Arita et al., 2008). Like PV, the capsid proteins also play an important role in EV-A71 virulence. For instance, the amino acid residue 145 of EV-A71 VP1, which affects receptor usage for cell surface attachment of the viral particles (Nishimura et al., 2013; Tan et al., 2017), has been shown to confer mouse adaptation with a G145E replacement (Arita et al., 2008; Chua et al., 2008) and influence the virulence in mice (Chua et al., 2008; Zaini and McMinn, 2012). Consistent with earlier results, a recent study has further demonstrated that the VP1-145 is a key determinant for EV-A71 neurovirulence in monkeys (Fujii et al., 2018). In addition, changes of nucleotides in the 5′ UTR and an amino acid replacement in the 2A or 3C protein have been identified to play an important role for virulence determination of EV-A71 (Li et al., 2011; Yeh et al., 2011; Li et al., 2017). Whether they attenuate EV-A71 neurovirulence is unknown. Nonetheless, it is expected that all these findings would contribute to not only our understanding of EV-A71 but also the development of live attenuated EV-A71 vaccine in the future.
EV-D68 is another non-polio enterovirus that has an association with a polio-like neurological disorder known as acute flaccid myelitis (AFM) with symptoms such as dysneuria and muscle weakness although the most common clinical symptom of EV-D68 infection is respiratory illness (Holm-Hansen et al., 2016; Messacar et al., 2018). EV-D68 is quite unique among enteroviruses because it carries some characteristic features of respiratory enteroviruses (Rhinovirus A-C) including optimal growth temperature of 33°C allowing better replication in the nasal cavity and inability to survive in the stomach due to acid sensitivity, but is genetically more closely related to enteric enteroviruses based on phylogenetic analysis. EV-D68 has become an emerging pathogen since an outbreak occurred in 2014 in the United States. Studies have shown that six mutations including M291T, V341A, T860N, D297N, S1108G, and R2005K are associated with neurovirulence of the outbreak EV-D68 strains causing AFM in 2014 (Greninger et al., 2015). Interestingly, another study has identified 3 nucleotide variables, C1817T, C3277A, and A4020G, in the 2014 outbreak strains, which differ significantly from previously identified EV-D68 strains (Huang W. et al., 2015). Among these three variables, C3277A causes amino acid substitution T860N in the protease 2A cleavage site between VP1 and 2A, whereas A4020G results in amino acid substitution S1108G in a protease 3C cleavage site between 2B and 2C, suggesting that mutation of these two sites may alter the cleavage efficiency and increase replication and transmission rates. However, using a mouse model of paralytic myelitis caused by EV-D68, a recent study has shown that the 2014 outbreak EV-D68 strains isolated from patients without AFM can also produce paralysis in neonatal mice (Hixon et al., 2017). Thus, further comparative analyses using infectious clones containing different combinations of previously identified mutations will be needed to establish the determinants of EV-D68 neurovirulence.
Immune Responses in the CNS
About 3% of enterovirus infected people will develop encephalitis while most people never show evidence of CNS infection (Koskiniemi et al., 1991). Since encephalitis only occurs in a small percentage of infected people, it is thought that host-pathogen interactions and immune responses in peripheral sites prevent viruses from gaining access to and causing infection within the CNS. Patients with enterovirus encephalitis may present symptoms that range in severity from mild cognitive impairment and memory loss to permanent CNS damage and death. The symptoms are mainly caused by virus-induced inflammatory responses in the brain. Inflammation is the body’s protective immune response against infection, but the brain was thought to be immune-privileged based on two predominant beliefs. First, the BBB prevents the entry of circulating immune cells and antibodies into the brain. Secondly, there is no lymphatic drainage to alert the immune system to the presence of CNS antigens. However, these two beliefs have been questioned by the detection of small numbers of leukocytes in the CSF and the newly identified lymphatics that allow leukocyte egress (Engelhardt et al., 2017). In addition to the BBB, two other brain barrier structures have been described: (1) blood – CSF barrier to the choroid plexus, which is located at the ventricles of the brain, and (2) blood – CSF barrier to the pia arachnoid, which is located at the surface of the brain. It is important to note that CSF spaces in these two brain barriers do not exhibit the same immune privilege as the CNS parenchyma and contain diverse immune cells to monitor the CSF for the presence of immune signals in response to viral pathogens. Therefore, the immune privilege of the brain is not absolute but instead is relative to other organs.
Enterovirus encephalitis is reported to occur more frequently in younger children. Although the exact cause remains unknown, it has been suggested that a compromised or immature immune system may be involved in the observed effect. Studies have shown that expression of CD40-ligand on activated T cells and interleukin 4 (IL-4) production are significantly lower in EV-A71 infected children with meningoencephalitis than those without it (Yang et al., 2001). CD40-ligand plays a pivotal role in co-stimulation and regulation of the immune responses. It binds to the CD40 receptor on antigen-presenting cells including B cells and macrophages to facilitate cell-cell communication and modulate adaptive immunity. Interleukin-4 (IL-4) is also a key regulator in adaptive immunity. It is a cytokine that has many functions including the stimulation of activated B cell and T cell proliferation and the differentiation of naïve helper T cells and B cells into Th2 cells and plasma cells, respectively. Thus, a reduction in CD40-ligand and IL-4 may indicate that adaptive immunity is compromised. In addition, differences in the polymorphism of the cytotoxic T lymphocyte antigen-4 (CTLA-4) have been noted between children with and without meningoencephalitis in the same study (Yang et al., 2001). CTLA-4 is a surface receptor on T cells, which functions to downregulate T cell activity and polymorphism of CTLA-4 has been shown to be associated with certain autoimmune diseases (Marron et al., 1997). Interestingly, a recent study has reported a correlation between human leukocyte antigen (HLA) genotype and enterovirus infectivity in young children (Sioofy-Khojine et al., 2018). Consistently, there is evidence that genetic differences between individuals may affect the immune response to infection (Kim-Hellmuth et al., 2017). Taken together, these results suggest that genetic factors may confer susceptibility to neurological complications following enterovirus infection.
Although genetic susceptibility may provide clues to why enterovirus encephalitis only occurs in some children but not in others, it cannot explain why younger children infected with enteroviruses are at higher risk for developing encephalitis. It has been suggested that the immaturity of the BBB in early development may account, at least in part, for the age-related difference in viral neuroinvasion (Saunders et al., 2014). The development of the BBB is a multistep process that starts with the growth of new vessels into the embryonic neuroectoderm from pre-existing vessels (Blanchette and Daneman, 2015). This is followed by the expression of tight junction proteins and nutrient transporter in BMECs that lines blood vessels in the brain. The BBB becomes mature as nascent vessels come into close contact with pericytes and astrocytes, which provide structural and functional support to the BBB. In addition, the other cell types present at the BBB, including neurons, microglia, and perivascular macrophages also contribute to the properties of the BBB (Banerjee and Bhat, 2007). Interestingly, it has been shown that the BBB is capable of restricting entry of proteins and small molecules during embryogenesis prior to postnatal astrocyte generation and ensheathment of the vessels (Daneman et al., 2010; Saunders et al., 2014), suggesting that the BBB is functionally mature even without astrocytes in early development. Although astrocytes are not required for initial BBB formation, there is strong evidence that astrocytes play an important role in regulating the function of the BBB during postnatal development (Haseloff et al., 2005). For example, astrocytes secrete trophic factors that lead to tighter tight junctions between BMECs (Dehouck et al., 1990; Rubin et al., 1991). In addition to its role in regulating the BBB, astrocytes receive signals from neighboring neurons and responding to them with the release of neuroactive substances to modulate synaptic strength in the CNS (Santello et al., 2019). Moreover, there is also evidence that astrocytes convey signals from neurons to the vasculature, leading to arteriolar dilation and an increase in local blood flow (Anderson and Nedergaard, 2003; Zonta et al., 2003). Thus, astrocytes link neuronal activity to functional properties of the BBB. Given that astrocytes are critical for structural support and the maintenance of the BBB, the developing brain containing immature astrocytes may have higher BBB permeability compared to that in the adult brain and is likely to be more vulnerable to viral neuroinvasion.
The CNS immune system may also be involved in the age-related difference in developing enterovirus encephalitis. Microglia, the primary resident immune cells of the brain, play a key role in regulating signaling pathways during CNS inflammation (Rivest, 2009). Although microglia are phenotypically and developmentally different from peripheral macrophages, they use phagocytic and cytotoxic mechanisms to destroy foreign pathogens and act as antigen-presenting cells to initiate T cell-mediated adaptive immune responses similar to macrophages. In addition, microglia have been shown to induce the recruitment of monocytes into the brain during viral infection (Fekete et al., 2018). However, whether microglia are fully functional during early development is not clear. Microglia in the early postnatal brain exhibit different morphologies compared to the adult brain (Cuadros and Navascues, 1998). They are largely non-ramified and take on an ameboid shape during early development, and gradually differentiate into mature/ramified microglia as the brain mature. Consistently, distinct sets of genes are expressed in microglia during different phases of development (Bennett et al., 2016; Matcovitch-Natan et al., 2016). The ameboid morphology observed in developing microglia is similar to activated phagocytic microglia from the adult brain, suggesting that microglia are in a constitutively “activated” state in the developing brain (Lenz and Nelson, 2018). Interestingly, it has been shown that activated microglia produce inflammatory cytokines such as TNFα and IL-1β (Nishioku et al., 2010; Yang et al., 2015), that increase BBB permeability and downregulate tight-junction proteins between BMECs (Gu et al., 2015; Almutairi et al., 2016). Taken together, these results suggest that developing microglia may increase BBB permeability by releasing inflammatory cytokines. Intriguingly, another study has shown that gene expression profiles are distinct between activated microglia from the adult brain and microglia from a control neonatal brain (Wlodarczyk et al., 2017). Therefore, developing microglia are not the same as adult “activated” microglia even both of them exhibit a remarkable similarity in morphology. Nonetheless, this may provide a logical explanation of why younger children infected with enteroviruses are at higher risk for developing encephalitis.
Conclusion
Enterovirus infection is a major public health concern considering the increase in outbreaks of serious neurological complications. Although there has been considerable progress in studying the complex interplay between enteroviruses and the infected cell in a culture dish, more complicated interaction between virus and host in vivo is largely unknown and how enteroviruses gain access to and spread in the well-protected CNS remains to be explored. For example, a common feature of enteroviral encephalitis is the involvement of brainstem (Wasserstrom et al., 1992; Huang et al., 1999; Shen et al., 1999; Lum et al., 2002; Brecht et al., 2010; Fan and Liu, 2019), but some studies have revealed cerebral white matter lesions without brainstem involvement in neonatal enteroviral encephalitis (Verboon-Maciolek et al., 2006; Hirata et al., 2011; Wu et al., 2014; Correia et al., 2016), suggesting that age and brain maturation may play an important role in the pathogenesis of enteroviral encephalitis. In the current review, we propose that there is a causal link between the development of the neuroimmune system and enteroviral neuroinvasion and have suggested that the age-related difference in developing enterovirus encephalitis may be associated with the development of neuroimmune system such as maturation of astrocytes and/or microglia. Future work will need to include the further characterization of the complex interactions between host and enteroviruses using appropriate animal models and the developmental roles of astrocytes and microglia in regulating BBB permeability. We also need to better understand the regulation of immune responses in the CNS caused by enteroviruses.
Author Contributions
All authors contributed to the writing and discussion of this review article and approved the final version of the manuscript. H-CL made the figure and table.
Funding
This work was financially supported by the Research Center for Emerging Viral Infections from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan, the Ministry of Science and Technology (MOST), Taiwan (MOST 108-3017-F-182-001) and the Chang Gung Memorial Hospital (CORPD1J0061).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, J., Jee, Y., Seo, I., Yoon, S. Y., Kim, D., Kim, Y. K., et al. (2008). Primary neurons become less susceptible to coxsackievirus B5 following maturation: the correlation with the decreased level of CAR expression on cell surface. J. Med. Virol. 80, 434–440. doi: 10.1002/jmv.21100
Almutairi, M. M., Gong, C., Xu, Y. G., Chang, Y., and Shi, H. (2016). Factors controlling permeability of the blood-brain barrier. Cell. Mol. Life Sci. 73, 57–77. doi: 10.1007/s00018-015-2050-8
Anderson, C. M., and Nedergaard, M. (2003). Astrocyte-mediated control of cerebral microcirculation. Trends Neurosci. 26, 340–344; authorrely 344–345.
Arita, M., Ami, Y., Wakita, T., and Shimizu, H. (2008). Cooperative effect of the attenuation determinants derived from poliovirus sabin 1 strain is essential for attenuation of Enterovirus 71 in the NOD/SCID mouse infection model. J. Virol. 82, 1787–1797. doi: 10.1128/jvi.01798-07
Arita, M., Nagata, N., Sata, T., Miyamura, T., and Shimizu, H. (2006). Quantitative analysis of poliomyelitis-like paralysis in mice induced by a poliovirus replicon. J. Gen. Virol. 87, 3317–3327. doi: 10.1099/vir.0.82172-0
Arita, M., Shimizu, H., Nagata, N., Ami, Y., Suzaki, Y., Sata, T., et al. (2005). Temperature-sensitive mutants of Enterovirus 71 show attenuation in cynomolgus monkeys. J. Gen. Virol. 86, 1391–1401. doi: 10.1099/vir.0.80784-0
Baggen, J., Thibaut, H. J., Strating, J., and Van Kuppeveld, F. J. M. (2018). The life cycle of non-polio Enteroviruses and how to target it. Nat. Rev. Microbiol. 16, 368–381. doi: 10.1038/s41579-018-0005-4
Balada-Llasat, J. M., Rosenthal, N., Hasbun, R., Zimmer, L., Bozzette, S., Duff, S., et al. (2019). Cost of managing meningitis and encephalitis among infants and children in the United States. Diagn. Microbiol. Infect. Dis. 93, 349–354. doi: 10.1016/j.diagmicrobio.2018.10.012
Banerjee, S., and Bhat, M. A. (2007). Neuron-glial interactions in blood-brain barrier formation. Annu. Rev. Neurosci. 30, 235–258. doi: 10.1146/annurev.neuro.30.051606.094345
Bennett, M. L., Bennett, F. C., Liddelow, S. A., Ajami, B., Zamanian, J. L., Fernhoff, N. B., et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U.S.A. 113, E1738–E1746. doi: 10.1073/pnas.1525528113
Bergelson, J. M. (2009). Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe 5, 517–521. doi: 10.1016/j.chom.2009.05.009
Bergelson, J. M., Mohanty, J. G., Crowell, R. L., St John, N. F., Lublin, D. M., and Finberg, R. W. (1995). Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 69, 1903–1906. doi: 10.1128/jvi.69.3.1903-1906.1995
Berger, M. M., Kopp, N., Vital, C., Redl, B., Aymard, M., and Lina, B. (2000). Detection and cellular localization of Enterovirus RNA sequences in spinal cord of patients with ALS. Neurology 54, 20–25.
Bird, S. W., Maynard, N. D., Covert, M. W., and Kirkegaard, K. (2014). Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. U.S.A. 111, 13081–13086. doi: 10.1073/pnas.1401437111
B’Krong, N., Minh, N. N. Q., Qui, P. T., Chau, T. T. H., Nghia, H. D. T., Do, L. A. H., et al. (2018). Enterovirus serotypes in patients with central nervous system and respiratory infections in Viet Nam 1997-2010. Virol. J. 15:69. doi: 10.1186/s12985-018-0980-0
Blanchette, M., and Daneman, R. (2015). Formation and maintenance of the BBB. Mech. Dev. 138(Pt 1), 8–16. doi: 10.1016/j.mod.2015.07.007
Brecht, M., Jyoti, R., Mcguire, W., and Chauhan, M. (2010). A case of neonatal coxsackie B virus brainstem encephalitis. J. Paediatr. Child Health 46, 699–701.
Burki, T. (2019). Vaccine derived poliovirus cases exceed wild types. Lancet Infect. Dis. 19:140. doi: 10.1016/s1473-3099(19)30012-x
Caine, E. A., and Osorio, J. E. (2017). In vivo imaging with bioluminescent Enterovirus 71 allows for real-time visualization of tissue tropism and viral spread. J. Virol. 91:e01759-16. doi: 10.1128/JVI.01759-16
Chang, L. Y., Huang, L. M., Gau, S. S., Wu, Y. Y., Hsia, S. H., Fan, T. Y., et al. (2007). Neurodevelopment and cognition in children after Enterovirus 71 infection. N. Engl. J. Med. 356, 1226–1234. doi: 10.1056/nejmoa065954
Chapman, N. M., and Kim, K. S. (2008). Persistent coxsackievirus infection: Enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr. Top. Microbiol. Immunol. 323, 275–292. doi: 10.1007/978-3-540-75546-3_13
Chapman, N. M., Kim, K. S., Drescher, K. M., Oka, K., and Tracy, S. (2008). 5’ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 375, 480–491. doi: 10.1016/j.virol.2008.02.030
Chen, C. S., Yao, Y. C., Lin, S. C., Lee, Y. P., Wang, Y. F., Wang, J. R., et al. (2007). Retrograde axonal transport: a major transmission route of Enterovirus 71 in mice. J. Virol. 81, 8996–9003. doi: 10.1128/jvi.00236-07
Chen, X., Ji, T., Guo, J., Wang, W., Xu, W., and Xie, Z. (2019). Molecular epidemiology of echovirus 18 circulating in mainland China from 2015 to 2016. Virol. Sin. 34, 50–58. doi: 10.1007/s12250-018-0080-8
Chen, X., Li, J., Guo, J., Xu, W., Sun, S., and Xie, Z. (2017). An outbreak of echovirus 18 encephalitis/meningitis in children in Hebei Province, China, 2015. Emerg. Microbes Infect. 6:e54.
Chua, B. H., Phuektes, P., Sanders, S. A., Nicholls, P. K., and Mcminn, P. C. (2008). The molecular basis of mouse adaptation by human Enterovirus 71. J. Gen. Virol. 89, 1622–1632. doi: 10.1099/vir.0.83676-0
Colbere-Garapin, F., Christodoulou, C., Crainic, R., and Pelletier, I. (1989). Persistent poliovirus infection of human neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 86, 7590–7594. doi: 10.1073/pnas.86.19.7590
Colbere-Garapin, F., Duncan, G., Pavio, N., Pelletier, I., and Petit, I. (1998). An approach to understanding the mechanisms of poliovirus persistence in infected cells of neural or non-neural origin. Clin. Diagn. Virol. 9, 107–113. doi: 10.1016/s0928-0197(98)00009-9
Correia, J., Alves, J. E., Ferreira, P., Ferreira, M., Pires, P., and Garrido, C. (2016). Enterovirus 71 meningoencephalitis with extensive white matter damage. Pediatr. Infect. Dis. J. 35, 1277–1278. doi: 10.1097/inf.0000000000001292
Couderc, T., Guivel-Benhassine, F., Calaora, V., Gosselin, A. S., and Blondel, B. (2002). An ex vivo murine model to study poliovirus-induced apoptosis in nerve cells. J. Gen. Virol. 83, 1925–1930. doi: 10.1099/0022-1317-83-8-1925
Cuadros, M. A., and Navascues, J. (1998). The origin and differentiation of microglial cells during development. Prog. Neurobiol. 56, 173–189. doi: 10.1016/s0301-0082(98)00035-5
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Dehouck, M. P., Meresse, S., Delorme, P., Fruchart, J. C., and Cecchelli, R. (1990). An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J. Neurochem. 54, 1798–1801. doi: 10.1111/j.1471-4159.1990.tb01236.x
Engelhardt, B., Vajkoczy, P., and Weller, R. O. (2017). The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131. doi: 10.1038/ni.3666
Evans, D. M., Dunn, G., Minor, P. D., Schild, G. C., Cann, A. J., Stanway, G., et al. (1985). Increased neurovirulence associated with a single nucleotide change in a noncoding region of the sabin type 3 poliovaccine genome. Nature 314, 548–550. doi: 10.1038/314548a0
Famulare, M., Chang, S., Iber, J., Zhao, K., Adeniji, J. A., Bukbuk, D., et al. (2016). Sabin vaccine reversion in the field: a comprehensive analysis of sabin-like poliovirus isolates in Nigeria. J. Virol. 90, 317–331. doi: 10.1128/JVI.01532-15
Fan, Y. K., and Liu, Y. P. (2019). Magnetic resonance imaging features of pediatric coxsackievirus encephalitis. J. Belg. Soc. Radiol. 103:6. doi: 10.5334/jbsr.1685
Fechner, H., Pinkert, S., Wang, X., Sipo, I., Suckau, L., Kurreck, J., et al. (2007). Coxsackievirus B3 and adenovirus infections of cardiac cells are efficiently inhibited by vector-mediated RNA interference targeting their common receptor. Gene Ther. 14, 960–971. doi: 10.1038/sj.gt.3302948
Fekete, R., Cserep, C., Lenart, N., Toth, K., Orsolits, B., Martinecz, B., et al. (2018). Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 136, 461–482. doi: 10.1007/s00401-018-1885-0
Feng, M., Guo, S., Fan, S., Zeng, X., Zhang, Y., Liao, Y., et al. (2016). The preferential infection of astrocytes by Enterovirus 71 plays a key role in the viral neurogenic pathogenesis. Front. Cell. Infect. Microbiol. 6:192. doi: 10.3389/fcimb.2016.00192
Feuer, R., Mena, I., Pagarigan, R., Slifka, M. K., and Whitton, J. L. (2002). Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 76, 4430–4440. doi: 10.1128/jvi.76.9.4430-4440.2002
Feuer, R., Mena, I., Pagarigan, R. R., Hassett, D. E., and Whitton, J. L. (2004). Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med. Microbiol. Immunol. 193, 83–90. doi: 10.1007/s00430-003-0192-z
Feuer, R., Pagarigan, R. R., Harkins, S., Liu, F., Hunziker, I. P., and Whitton, J. L. (2005). Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J. Neurosci. 25, 2434–2444. doi: 10.1523/jneurosci.4517-04.2005
Feuer, R., Ruller, C. M., An, N., Tabor-Godwin, J. M., Rhoades, R. E., Maciejewski, S., et al. (2009). Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J. Virol. 83, 9356–9369. doi: 10.1128/JVI.02382-07
Forrester, J. V., Mcmenamin, P. G., and Dando, S. J. (2018). CNS infection and immune privilege. Nat. Rev. Neurosci. 19, 655–671. doi: 10.1038/s41583-018-0070-8
Freistadt, M. S., Kaplan, G., and Racaniello, V. R. (1990). Heterogeneous expression of poliovirus receptor-related proteins in human cells and tissues. Mol. Cell Biol. 10, 5700–5706. doi: 10.1128/mcb.10.11.5700
Fujii, K., Sudaka, Y., Takashino, A., Kobayashi, K., Kataoka, C., Suzuki, T., et al. (2018). VP1 amino acid residue 145 of Enterovirus 71 is a key residue for its receptor attachment and resistance to neutralizing antibody during cynomolgus monkey infection. J. Virol. 92:e00682-18. doi: 10.1128/JVI.00682-18
Giraud, P., Beaulieux, F., Ono, S., Shimizu, N., Chazot, G., and Lina, B. (2001). Detection of enteroviral sequences from frozen spinal cord samples of Japanese ALS patients. Neurology 56, 1777–1778. doi: 10.1212/wnl.56.12.1777
Goto, K., Sanefuji, M., Kusuhara, K., Nishimura, Y., Shimizu, H., Kira, R., et al. (2009). Rhombencephalitis and coxsackievirus A16. Emerg. Infect. Dis. 15, 1689–1691. doi: 10.3201/eid1510.090594
Greninger, A. L., Naccache, S. N., Messacar, K., Clayton, A., Yu, G., Somasekar, S., et al. (2015). A novel outbreak Enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-14): a retrospective cohort study. Lancet Infect. Dis. 15, 671–682. doi: 10.1016/S1473-3099(15)70093-9
Gromeier, M., Alexander, L., and Wimmer, E. (1996). Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl. Acad. Sci. U.S.A. 93, 2370–2375. doi: 10.1073/pnas.93.6.2370
Gromeier, M., and Wimmer, E. (1998). Mechanism of injury-provoked poliomyelitis. J. Virol. 72, 5056–5060. doi: 10.1128/jvi.72.6.5056-5060.1998
Gu, X., Wei, Z. Z., Espinera, A., Lee, J. H., Ji, X., Wei, L., et al. (2015). Pharmacologically induced hypothermia attenuates traumatic brain injury in neonatal rats. Exp. Neurol. 267, 135–142. doi: 10.1016/j.expneurol.2015.02.029
Guest, S., Pilipenko, E., Sharma, K., Chumakov, K., and Roos, R. P. (2004). Molecular mechanisms of attenuation of the Sabin strain of poliovirus type 3. J. Virol. 78, 11097–11107. doi: 10.1128/jvi.78.20.11097-11107.2004
Gutierrez, A. L., Denova-Ocampo, M., Racaniello, V. R., and Del Angel, R. M. (1997). Attenuating mutations in the poliovirus 5′ untranslated region alter its interaction with polypyrimidine tract-binding protein. J. Virol. 71, 3826–3833. doi: 10.1128/jvi.71.5.3826-3833.1997
Han, J., Ma, X. J., Wan, J. F., Liu, Y. H., Han, Y. L., Chen, C., et al. (2010). Long persistence of EV71 specific nucleotides in respiratory and feces samples of the patients with Hand-Foot-Mouth Disease after recovery. BMC Infect. Dis. 10:178. doi: 10.1186/1471-2334-10-178
Hasbun, R., Rosenthal, N., Balada-Llasat, J. M., Chung, J., Duff, S., Bozzette, S., et al. (2017). Epidemiology of meningitis and encephalitis in the United States, 2011-2014. Clin. Infect. Dis. 65, 359–363. doi: 10.1093/cid/cix319
Haseloff, R. F., Blasig, I. E., Bauer, H. C., and Bauer, H. (2005). In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell. Mol. Neurobiol. 25, 25–39. doi: 10.1007/s10571-004-1375-x
Heim, A., Brehm, C., Stille-Siegener, M., Muller, G., Hake, S., Kandolf, R., et al. (1995). Cultured human myocardial fibroblasts of pediatric origin: natural human interferon-alpha is more effective than recombinant interferon-alpha 2a in carrier-state coxsackievirus B3 replication. J. Mol. Cell. Cardiol. 27, 2199–2208. doi: 10.1016/s0022-2828(95)91515-x
Heim, A., Canu, A., Kirschner, P., Simon, T., Mall, G., Hofschneider, P. H., et al. (1992). Synergistic interaction of interferon-beta and interferon-gamma in coxsackievirus B3-infected carrier cultures of human myocardial fibroblasts. J. Infect. Dis. 166, 958–965. doi: 10.1093/infdis/166.5.985
Hirata, O., Ishikawa, N., Mizoguchi, Y., Nakamura, K., and Kobayashi, M. (2011). A case of neonatal coxsackie B2 meningo-encephalitis in which serial magnetic resonance imaging findings reveal the development of lesions. Neuropediatrics 42, 156–158. doi: 10.1055/s-0031-1285876
Hixon, A. M., Clarke, P., and Tyler, K. L. (2019). Contemporary circulating Enterovirus D68 strains infect and undergo retrograde axonal transport in spinal motor neurons independent of sialic acid. J. Virol. 93:e00578-19. doi: 10.1128/JVI.00578-19
Hixon, A. M., Yu, G., Leser, J. S., Yagi, S., Clarke, P., Chiu, C. Y., et al. (2017). A mouse model of paralytic myelitis caused by Enterovirus D68. PLoS Pathog. 13:e1006199. doi: 10.1371/journal.ppat.1006199
Holland, J. J. (1961). Receptor affinities as major determinants of Enterovirus tissue tropisms in humans. Virology 15, 312–326. doi: 10.1016/0042-6822(61)90363-4
Holmes, C. W., Koo, S. S., Osman, H., Wilson, S., Xerry, J., Gallimore, C. I., et al. (2016). Predominance of Enterovirus B and echovirus 30 as cause of viral meningitis in a UK population. J. Clin. Virol. 81, 90–93. doi: 10.1016/j.jcv.2016.06.007
Holm-Hansen, C. C., Midgley, S. E., and Fischer, T. K. (2016). Global emergence of Enterovirus D68: a systematic review. Lancet Infect. Dis. 16, e64–e75. doi: 10.1016/S1473-3099(15)00543-5
Hsu, N. Y., Ilnytska, O., Belov, G., Santiana, M., Chen, Y. H., Takvorian, P. M., et al. (2010). Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141, 799–811. doi: 10.1016/j.cell.2010.03.050
Huang, C. C., Liu, C. C., Chang, Y. C., Chen, C. Y., Wang, S. T., and Yeh, T. F. (1999). Neurologic complications in children with Enterovirus 71 infection. N. Engl. J. Med. 341, 936–942. doi: 10.1056/nejm199909233411302
Huang, H. I., Lin, J. Y., Chen, H. H., Yeh, S. B., Kuo, R. L., Weng, K. F., et al. (2014). Enterovirus 71 infects brain-derived neural progenitor cells. Virology 468–470, 592–600. doi: 10.1016/j.virol.2014.09.017
Huang, H. I., and Shih, S. R. (2015). Neurotropic Enterovirus infections in the central nervous system. Viruses 7, 6051–6066. doi: 10.3390/v7112920
Huang, W., Wang, G., Zhuge, J., Nolan, S. M., Dimitrova, N., and Fallon, J. T. (2015). Whole-genome sequence analysis reveals the Enterovirus D68 isolates during the United States 2014 outbreak mainly belong to a novel clade. Sci. Rep. 5:15223. doi: 10.1038/srep15223
Huang, Y., Zhou, Y., Lu, H., Yang, H., Feng, Q., Dai, Y., et al. (2015). Characterization of severe hand, foot, and mouth disease in Shenzhen, China, 2009-2013. J. Med. Virol. 87, 1471–1479. doi: 10.1002/jmv.24200
Ida-Hosonuma, M., Iwasaki, T., Yoshikawa, T., Nagata, N., Sato, Y., Sata, T., et al. (2005). The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 79, 4460–4469. doi: 10.1128/jvi.79.7.4460-4469.2005
Ifie, E., Russell, M. A., Dhayal, S., Leete, P., Sebastiani, G., Nigi, L., et al. (2018). Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic beta cells. Diabetologia 61, 2344–2355. doi: 10.1007/s00125-018-4704-1
Jorba, J., Campagnoli, R., De, L., and Kew, O. (2008). Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range. J. Virol. 82, 4429–4440. doi: 10.1128/JVI.02354-07
Julien, J., Leparc-Goffart, I., Lina, B., Fuchs, F., Foray, S., Janatova, I., et al. (1999). Postpolio syndrome: poliovirus persistence is involved in the pathogenesis. J. Neurol. 246, 472–476. doi: 10.1007/s004150050386
Kao, S. J., Yang, F. L., Hsu, Y. H., and Chen, H. I. (2004). Mechanism of fulminant pulmonary edema caused by Enterovirus 71. Clin. Infect. Dis. 38, 1784–1788. doi: 10.1086/421021
Kawamura, N., Kohara, M., Abe, S., Komatsu, T., Tago, K., Arita, M., et al. (1989). Determinants in the 5’ noncoding region of poliovirus Sabin 1 RNA that influence the attenuation phenotype. J. Virol. 63, 1302–1309. doi: 10.1128/jvi.63.3.1302-1309.1989
Kempf, B. J., and Barton, D. J. (2015). Picornavirus RNA polyadenylation by 3D(pol), the viral RNA-dependent RNA polymerase. Virus Res. 206, 3–11. doi: 10.1016/j.virusres.2014.12.030
Kew, O. M., Sutter, R. W., De Gourville, E. M., Dowdle, W. R., and Pallansch, M. A. (2005). Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 59, 587–635. doi: 10.1146/annurev.micro.58.030603.123625
Khandaker, G. M., Zimbron, J., Dalman, C., Lewis, G., and Jones, P. B. (2012). Childhood infection and adult schizophrenia: a meta-analysis of population-based studies. Schizophr. Res. 139, 161–168. doi: 10.1016/j.schres.2012.05.023
Khong, W. X., Yan, B., Yeo, H., Tan, E. L., Lee, J. J., Ng, J. K., et al. (2012). A non-mouse-adapted Enterovirus 71 (EV71) strain exhibits neurotropism, causing neurological manifestations in a novel mouse model of EV71 infection. J. Virol. 86, 2121–2131. doi: 10.1128/JVI.06103-11
Kim, K. S., Chapman, N. M., and Tracy, S. (2008). Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J. Virol. 82, 2033–2037. doi: 10.1128/jvi.01774-07
Kim, K. S., Tracy, S., Tapprich, W., Bailey, J., Lee, C. K., Kim, K., et al. (2005). 5’-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J. Virol. 79, 7024–7041. doi: 10.1128/jvi.79.11.7024-7041.2005
Kim-Hellmuth, S., Bechheim, M., Putz, B., Mohammadi, P., Nedelec, Y., Giangreco, N., et al. (2017). Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun. 8:266. doi: 10.1038/s41467-017-00366-1
Koike, S., Horie, H., Ise, I., Okitsu, A., Yoshida, M., Iizuka, N., et al. (1990). The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 9, 3217–3224. doi: 10.1002/j.1460-2075.1990.tb07520.x
Koskiniemi, M., Rautonen, J., Lehtokoski-Lehtiniemi, E., and Vaheri, A. (1991). Epidemiology of encephalitis in children: a 20-year survey. Ann. Neurol. 29, 492–497. doi: 10.1002/ana.410290508
La Monica, N., and Racaniello, V. R. (1989). Differences in replication of attenuated and neurovirulent polioviruses in human neuroblastoma cell line SH-SY5Y. J. Virol. 63, 2357–2360. doi: 10.1128/jvi.63.5.2357-2360.1989
Lee, K. M., Chen, C. J., and Shih, S. R. (2017). Regulation mechanisms of viral IRES-driven translation. Trends Microbiol. 25, 546–561. doi: 10.1016/j.tim.2017.01.010
Lenz, K. M., and Nelson, L. H. (2018). Microglia and beyond: innate immune cells as regulators of brain development and behavioral function. Front. Immunol. 9:698. doi: 10.3389/fimmu.2018.00698
Li, B., Yue, Y., Zhang, Y., Yuan, Z., Li, P., Song, N., et al. (2017). A novel Enterovirus 71 (EV71) virulence determinant: the 69th residue of 3C protease modulates pathogenicity. Front. Cell. Infect. Microbiol. 7:26. doi: 10.3389/fcimb.2017.00026
Li, R., Zou, Q., Chen, L., Zhang, H., and Wang, Y. (2011). Molecular analysis of virulent determinants of Enterovirus 71. PLoS One 6:e26237. doi: 10.1371/journal.pone.0026237
Lin, J. Y., and Shih, S. R. (2014). Cell and tissue tropism of Enterovirus 71 and other Enteroviruses infections. J. Biomed. Sci. 21:18. doi: 10.1186/1423-0127-21-18
Lum, L. C., Chua, K. B., Mcminn, P. C., Goh, A. Y., Muridan, R., Sarji, S. A., et al. (2002). Echovirus 7 associated encephalomyelitis. J. Clin. Virol. 23, 153–160. doi: 10.1016/s1386-6532(01)00214-1
Mao, Q., Hao, X., Hu, Y., Du, R., Lang, S., Bian, L., et al. (2018). A neonatal mouse model of central nervous system infections caused by Coxsackievirus B5. Emerg. Microbes Infect. 7:185. doi: 10.1038/s41426-018-0186-y
Mao, Q., Wang, Y., Bian, L., Xu, M., and Liang, Z. (2016). EV-A71 vaccine licensure: a first step for multivalent Enterovirus vaccine to control HFMD and other severe diseases. Emerg. Microbes Infect. 5:e75. doi: 10.1038/emi.2016.73
Marjomaki, V., Turkki, P., and Huttunen, M. (2015). Infectious entry pathway of Enterovirus B species. Viruses 7, 6387–6399. doi: 10.3390/v7122945
Marron, M. P., Raffel, L. J., Garchon, H. J., Jacob, C. O., Serrano-Rios, M., Martinez Larrad, M. T., et al. (1997). Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnic groups. Hum. Mol. Genet. 6, 1275–1282. doi: 10.1093/hmg/6.8.1275
Matcovitch-Natan, O., Winter, D. R., Giladi, A., Vargas Aguilar, S., Spinrad, A., Sarrazin, S., et al. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science 353:aad8670. doi: 10.1126/science.aad8670
Melnick, J. L. (1996). Current status of poliovirus infections. Clin. Microbiol. Rev. 9, 293–300. doi: 10.1128/cmr.9.3.293-300.1996
Mendelsohn, C. L., Wimmer, E., and Racaniello, V. R. (1989). Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56, 855–865. doi: 10.1016/0092-8674(89)90690-9
Messacar, K., Asturias, E. J., Hixon, A. M., Van Leer-Buter, C., Niesters, H. G. M., Tyler, K. L., et al. (2018). Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect. Dis. 18, e239–e247. doi: 10.1016/S1473-3099(18)30094-X
Minor, P. (2009). Vaccine-derived poliovirus (VDPV): impact on poliomyelitis eradication. Vaccine 27, 2649–2652. doi: 10.1016/j.vaccine.2009.02.071
Mizutani, T., Ishizaka, A., and Nihei, C. (2016). Transferrin receptor 1 facilitates poliovirus permeation of mouse brain capillary endothelial cells. J. Biol. Chem. 291, 2829–2836. doi: 10.1074/jbc.M115.690941
Muir, P., Nicholson, F., Sharief, M. K., Thompson, E. J., Cairns, N. J., Lantos, P., et al. (1995). Evidence for persistent Enterovirus infection of the central nervous system in patients with previous paralytic poliomyelitis. Ann. N. Y. Acad. Sci. 753, 219–232. doi: 10.1111/j.1749-6632.1995.tb27548.x
Nagata, N., Iwasaki, T., Ami, Y., Sato, Y., Hatano, I., Harashima, A., et al. (2004). A poliomyelitis model through mucosal infection in transgenic mice bearing human poliovirus receptor, TgPVR21. Virology 321, 87–100. doi: 10.1016/j.virol.2003.12.008
Nishimura, Y., Lee, H., Hafenstein, S., Kataoka, C., Wakita, T., Bergelson, J. M., et al. (2013). Enterovirus 71 binding to PSGL-1 on leukocytes: VP1-145 acts as a molecular switch to control receptor interaction. PLoS Pathog. 9:e1003511. doi: 10.1371/journal.ppat.1003511
Nishimura, Y., Shimojima, M., Tano, Y., Miyamura, T., Wakita, T., and Shimizu, H. (2009). Human P-selectin glycoprotein ligand-1 is a functional receptor for Enterovirus 71. Nat. Med. 15, 794–797. doi: 10.1038/nm.1961
Nishioku, T., Matsumoto, J., Dohgu, S., Sumi, N., Miyao, K., Takata, F., et al. (2010). Tumor necrosis factor-alpha mediates the blood-brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. J. Pharmacol. Sci. 112, 251–254. doi: 10.1254/jphs.09292sc
Ohka, S., Matsuda, N., Tohyama, K., Oda, T., Morikawa, M., Kuge, S., et al. (2004). Receptor (CD155)-dependent endocytosis of poliovirus and retrograde axonal transport of the endosome. J. Virol. 78, 7186–7198. doi: 10.1128/jvi.78.13.7186-7198.2004
Ohka, S., and Nomoto, A. (2001). The molecular basis of poliovirus neurovirulence. Dev. Biol. 105, 51–58.
Ohka, S., Sakai, M., Bohnert, S., Igarashi, H., Deinhardt, K., Schiavo, G., et al. (2009). Receptor-dependent and -independent axonal retrograde transport of poliovirus in motor neurons. J. Virol. 83, 4995–5004. doi: 10.1128/JVI.02225-08
Ong, K. C., Badmanathan, M., Devi, S., Leong, K. L., Cardosa, M. J., and Wong, K. T. (2008). Pathologic characterization of a murine model of human Enterovirus 71 encephalomyelitis. J. Neuropathol. Exp. Neurol. 67, 532–542. doi: 10.1097/NEN.0b013e31817713e7
Pinkert, S., Klingel, K., Lindig, V., Dorner, A., Zeichhardt, H., Spiller, O. B., et al. (2011). Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J. Virol. 85, 13409–13419. doi: 10.1128/JVI.00621-11
Racaniello, V. R. (2006). One hundred years of poliovirus pathogenesis. Virology 344, 9–16. doi: 10.1016/j.virol.2005.09.015
Ramalho, E., Sousa, I. Jr., Burlandy, F., Costa, E., Dias, A., Serrano, R., et al. (2019). Identification and phylogenetic characterization of human Enteroviruses isolated from cases of aseptic meningitis in Brazil, 2013-2017. Viruses 11:E690. doi: 10.3390/v11080690
Ransohoff, R. M., and Engelhardt, B. (2012). The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635. doi: 10.1038/nri3265
Rantakallio, P., Jones, P., Moring, J., and Von Wendt, L. (1997). Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int. J. Epidemiol. 26, 837–843. doi: 10.1093/ije/26.4.837
Ren, R. B., Moss, E. G., and Racaniello, V. R. (1991). Identification of two determinants that attenuate vaccine-related type 2 poliovirus. J. Virol. 65, 1377–1382. doi: 10.1128/jvi.65.3.1377-1382.1991
Rhoades, R. E., Tabor-Godwin, J. M., Tsueng, G., and Feuer, R. (2011). Enterovirus infections of the central nervous system. Virology 411, 288–305. doi: 10.1016/j.virol.2010.12.014
Richardson, S. J., and Morgan, N. G. (2018). Enteroviral infections in the pathogenesis of type 1 diabetes: new insights for therapeutic intervention. Curr. Opin. Pharmacol. 43, 11–19. doi: 10.1016/j.coph.2018.07.006
Rivest, S. (2009). Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439. doi: 10.1038/nri2565
Rubin, L. L., Hall, D. E., Porter, S., Barbu, K., Cannon, C., Horner, H. C., et al. (1991). A cell culture model of the blood-brain barrier. J. Cell Biol. 115, 1725–1735.
Ruller, C. M., Tabor-Godwin, J. M., Van Deren, D. A. Jr., Robinson, S. M., Maciejewski, S., Gluhm, S., et al. (2012). Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am. J. Pathol. 180, 1107–1120. doi: 10.1016/j.ajpath.2011.11.016
Santello, M., Toni, N., and Volterra, A. (2019). Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 22, 154–166. doi: 10.1038/s41593-018-0325-8
Saunders, N. R., Dreifuss, J. J., Dziegielewska, K. M., Johansson, P. A., Habgood, M. D., Mollgard, K., et al. (2014). The rights and wrongs of blood-brain barrier permeability studies: a walk through 100 years of history. Front. Neurosci. 8:404. doi: 10.3389/fnins.2014.00404
Shen, W. C., Chiu, H. H., Chow, K. C., and Tsai, C. H. (1999). MR imaging findings of enteroviral encephaloymelitis: an outbreak in Taiwan. AJNR Am. J. Neuroradiol. 20, 1889–1895.
Shih, S. R., Stollar, V., and Li, M. L. (2011). Host factors in Enterovirus 71 replication. J. Virol. 85, 9658–9666. doi: 10.1128/JVI.05063-11
Sioofy-Khojine, A. B., Oikarinen, S., Honkanen, H., Huhtala, H., Lehtonen, J. P., Briese, T., et al. (2018). Molecular epidemiology of Enteroviruses in young children at increased risk of type 1 diabetes. PLoS One 13:e0201959. doi: 10.1371/journal.pone.0201959
Stern, A., Yeh, M. T., Zinger, T., Smith, M., Wright, C., Ling, G., et al. (2017). The evolutionary pathway to virulence of an RNA virus. Cell 169, 35–46.e19. doi: 10.1016/j.cell.2017.03.013
Sun, Y., Miao, Z., Yan, J., Gong, L., Chen, Y., Chen, Y., et al. (2019). Sero-molecular epidemiology of Enterovirus-associated encephalitis in Zhejiang Province, China, from 2014 to 2017. Int. J. Infect. Dis. 79, 58–64. doi: 10.1016/j.ijid.2018.11.002
Suresh, S., Forgie, S., and Robinson, J. (2018). Non-polio Enterovirus detection with acute flaccid paralysis: a systematic review. J. Med. Virol. 90, 3–7. doi: 10.1002/jmv.24933
Suvisaari, J., Mautemps, N., Haukka, J., Hovi, T., and Lonnqvist, J. (2003). Childhood central nervous system viral infections and adult schizophrenia. Am. J. Psychiatry 160, 1183–1185. doi: 10.1176/appi.ajp.160.6.1183
Tabor-Godwin, J. M., Ruller, C. M., Bagalso, N., An, N., Pagarigan, R. R., Harkins, S., et al. (2010). A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J. Neurosci. 30, 8676–8691. doi: 10.1523/JNEUROSCI.1860-10.2010
Tam, P. E., and Messner, R. P. (1999). Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J. Virol. 73, 10113–10121. doi: 10.1128/jvi.73.12.10113-10121.1999
Tan, C. W., Sam, I. C., Lee, V. S., Wong, H. V., and Chan, Y. F. (2017). VP1 residues around the five-fold axis of Enterovirus A71 mediate heparan sulfate interaction. Virology 501, 79–87. doi: 10.1016/j.virol.2016.11.009
Tan, S. H., Ong, K. C., and Wong, K. T. (2014). Enterovirus 71 can directly infect the brainstem via cranial nerves and infection can be ameliorated by passive immunization. J. Neuropathol. Exp. Neurol. 73, 999–1008. doi: 10.1097/NEN.0000000000000122
Tapparel, C., Siegrist, F., Petty, T. J., and Kaiser, L. (2013). Picornavirus and Enterovirus diversity with associated human diseases. Infect. Genet. Evol. 14, 282–293. doi: 10.1016/j.meegid.2012.10.016
Too, I. H., Yeo, H., Sessions, O. M., Yan, B., Libau, E. A., Howe, J. L., et al. (2016). Enterovirus 71 infection of motor neuron-like NSC-34 cells undergoes a non-lytic exit pathway. Sci. Rep. 6:36983. doi: 10.1038/srep36983
Tsueng, G., Tabor-Godwin, J. M., Gopal, A., Ruller, C. M., Deline, S., An, N., et al. (2011). Coxsackievirus preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells. J. Virol. 85, 5718–5732. doi: 10.1128/JVI.02261-10
van der Schaar, H. M., Dorobantu, C. M., Albulescu, L., Strating, J., and Van Kuppeveld, F. J. M. (2016). Fat(al) attraction: picornaviruses usurp lipid transfer at membrane contact sites to create replication organelles. Trends Microbiol. 24, 535–546. doi: 10.1016/j.tim.2016.02.017
Verboon-Maciolek, M. A., Groenendaal, F., Cowan, F., Govaert, P., Van Loon, A. M., and De Vries, L. S. (2006). White matter damage in neonatal Enterovirus meningoencephalitis. Neurology 66, 1267–1269. doi: 10.1212/01.wnl.0000208429.69676.23
Wasserstrom, R., Mamourian, A. C., Mcgary, C. T., and Miller, G. (1992). Bulbar poliomyelitis: MR findings with pathologic correlation. AJNR Am. J. Neuroradiol. 13, 371–373.
Werk, D., Schubert, S., Lindig, V., Grunert, H. P., Zeichhardt, H., Erdmann, V. A., et al. (2005). Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of coxsackievirus B3 and its cognate coxsackievirus-adenovirus receptor. Biol. Chem. 386, 857–863.
Wessely, R., Klingel, K., Knowlton, K. U., and Kandolf, R. (2001). Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation 103, 756–761. doi: 10.1161/01.cir.103.5.756
Wlodarczyk, A., Holtman, I. R., Krueger, M., Yogev, N., Bruttger, J., Khorooshi, R., et al. (2017). A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 36, 3292–3308. doi: 10.15252/embj.201696056
Woodall, C. J., Riding, M. H., Graham, D. I., and Clements, G. B. (1994). Sequences specific for Enterovirus detected in spinal cord from patients with motor neurone disease. BMJ 308, 1541–1543. doi: 10.1136/bmj.308.6943.1541
Wu, T., Fan, X. P., Wang, W. Y., and Yuan, T. M. (2014). Enterovirus infections are associated with white matter damage in neonates. J. Paediatr. Child Health 50, 817–822. doi: 10.1111/jpc.12656
Xu, R., and Crowell, R. L. (1996). Expression and distribution of the receptors for coxsackievirus B3 during fetal development of the Balb/c mouse and of their brain cells in culture. Virus Res. 46, 157–170. doi: 10.1016/s0168-1702(96)01398-6
Xu, W., Liu, C. F., Yan, L., Li, J. J., Wang, L. J., Qi, Y., et al. (2012). Distribution of Enteroviruses in hospitalized children with hand, foot and mouth disease and relationship between pathogens and nervous system complications. Virol. J. 9:8. doi: 10.1186/1743-422X-9-8
Yamayoshi, S., Fujii, K., and Koike, S. (2014). Receptors for Enterovirus 71. Emerg. Microbes Infect. 3:e53. doi: 10.1038/emi.2014.49
Yan, J. J., Wang, J. R., Liu, C. C., Yang, H. B., and Su, I. J. (2000). An outbreak of Enterovirus 71 infection in Taiwan 1998: a comprehensive pathological, virological, and molecular study on a case of fulminant encephalitis. J. Clin. Virol. 17, 13–22. doi: 10.1016/s1386-6532(00)00067-6
Yanagiya, A., Ohka, S., Hashida, N., Okamura, M., Taya, C., Kamoshita, N., et al. (2003). Tissue-specific replicating capacity of a chimeric poliovirus that carries the internal ribosome entry site of hepatitis C virus in a new mouse model transgenic for the human poliovirus receptor. J. Virol. 77, 10479–10487. doi: 10.1128/jvi.77.19.10479-10487.2003
Yang, K. D., Yang, M. Y., Li, C. C., Lin, S. F., Chong, M. C., Wang, C. L., et al. (2001). Altered cellular but not humoral reactions in children with complicated Enterovirus 71 infections in Taiwan. J. Infect. Dis. 183, 850–856. doi: 10.1086/319255
Yang, W. X., Terasaki, T., Shiroki, K., Ohka, S., Aoki, J., Tanabe, S., et al. (1997). Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology 229, 421–428. doi: 10.1006/viro.1997.8450
Yang, Y., Salayandia, V. M., Thompson, J. F., Yang, L. Y., Estrada, E. Y., and Yang, Y. (2015). Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J. Neuroinflammation 12:26. doi: 10.1186/s12974-015-0245-4
Yao, P. P., Qian, L., Xia, Y., Xu, F., Yang, Z. N., Xie, R. H., et al. (2012). Enterovirus 71-induced neurological disorders in young gerbils, Meriones unguiculatus: development and application of a neurological disease model. PLoS One 7:e51996. doi: 10.1371/journal.pone.0051996
Yeh, M. T., Wang, S. W., Yu, C. K., Lin, K. H., Lei, H. Y., Su, I. J., et al. (2011). A single nucleotide in stem loop II of 5’-untranslated region contributes to virulence of Enterovirus 71 in mice. PLoS One 6:e27082. doi: 10.1371/journal.pone.0027082
Yeung, M. L., Jia, L., Yip, C. C. Y., Chan, J. F. W., Teng, J. L. L., Chan, K. H., et al. (2018). Human tryptophanyl-tRNA synthetase is an IFN-gamma-inducible entry factor for Enterovirus. J. Clin. Invest. 128, 5163–5177. doi: 10.1172/JCI99411
Yu, J., Zhang, L., Ren, P., Zhong, T., Li, Z., Wang, Z., et al. (2015). Enterovirus 71 mediates cell cycle arrest in S phase through non-structural protein 3D. Cell Cycle 14, 425–436. doi: 10.4161/15384101.2014.980631
Zaini, Z., and McMinn, P. (2012). A single mutation in capsid protein VP1 (Q145E) of a genogroup C4 strain of human Enterovirus 71 generates a mouse-virulent phenotype. J. Gen. Virol. 93, 1935–1940. doi: 10.1099/vir.0.043893-0
Zoll, J., Heus, H. A., Van Kuppeveld, F. J., and Melchers, W. J. (2009). The structure-function relationship of the Enterovirus 3’-UTR. Virus Res. 139, 209–216. doi: 10.1016/j.virusres.2008.07.014
Keywords: enterovirus, encephalitis, CNS, Picornaviridae, RNA virus
Citation: Chen B-S, Lee H-C, Lee K-M, Gong Y-N and Shih S-R (2020) Enterovirus and Encephalitis. Front. Microbiol. 11:261. doi: 10.3389/fmicb.2020.00261
Received: 06 November 2019; Accepted: 04 February 2020;
Published: 20 February 2020.
Edited by:
Erna Geessien Kroon, Federal University of Minas Gerais, BrazilReviewed by:
Sara Louise Cosby, Agri-Food and Biosciences Institute (AFBI), United KingdomVarpu Seija Marjomäki, University of Jyväskylä, Finland
Copyright © 2020 Chen, Lee, Lee, Gong and Shih. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shin-Ru Shih, c3JzaGloQG1haWwuY2d1LmVkdS50dw==