
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 February 2020
Sec. Infectious Agents and Disease
Volume 11 - 2020 | https://doi.org/10.3389/fmicb.2020.00117
This article is part of the Research Topic Genetics Architecture and Underlying Molecular Mechanisms in Host-Pathogen Interactions View all 19 articles
 Cheng-Lu Hsieh1
Cheng-Lu Hsieh1 Shu-Ying Hsieh2
Shu-Ying Hsieh2 Hsuan-Min Huang3Shiou-Ling Lu4Hiroko Omori5
Hsuan-Min Huang3Shiou-Ling Lu4Hiroko Omori5 Po-Xing Zheng6Yen-Ning Ho7
Po-Xing Zheng6Yen-Ning Ho7 Yi-Lin Cheng7Yee-Shin Lin1,6,8
Yi-Lin Cheng7Yee-Shin Lin1,6,8 Chuan Chiang-Ni9,10
Chuan Chiang-Ni9,10 Pei-Jane Tsai1,3
Pei-Jane Tsai1,3 Shu-Ying Wang1,8
Shu-Ying Wang1,8 Ching-Chuan Liu1,6,11
Ching-Chuan Liu1,6,11 Takeshi Noda4
Takeshi Noda4 Jiunn-Jong Wu1,7*
Jiunn-Jong Wu1,7*Group A streptococcus (GAS) is a versatile pathogen that causes a wide spectrum of diseases in humans. Invading host cells is a known strategy for GAS to avoid antibiotic killing and immune recognition. However, the underlying mechanisms of GAS resistance to intracellular killing need to be explored. Endothelial HMEC-1 cells were infected with GAS, methicillin-resistant Staphylococcus aureus (MRSA) and Salmonella Typhimurium under nicotinamide (NAM)-supplemented conditions. The intracellular NAD+ level and cell viability were respectively measured by NAD+ quantification kit and protease-based cytotoxicity assay. Moreover, the intracellular bacteria were analyzed by colony-forming assay, transmission electron microscopy, and confocal microscopy. We found that supplementation with exogenous nicotinamide during infection significantly inhibited the growth of intracellular GAS in endothelial cells. Moreover, the NAD+ content and NAD+/NADH ratio of GAS-infected endothelial cells were dramatically increased, whereas the cell cytotoxicity was decreased by exogenous nicotinamide treatment. After knockdown of the autophagy-related ATG9A, the intracellular bacterial load was increased in nicotinamide-treated endothelial cells. The results of Western blot and transmission electron microscopy also revealed that cells treated with nicotinamide can increase autophagy-associated LC3 conversion and double-membrane formation during GAS infection. Confocal microscopy images further showed that more GAS-containing vacuoles were colocalized with lysosome under nicotinamide-supplemented conditions than without nicotinamide treatment. In contrast to GAS, supplementation with exogenous nicotinamide did not effectively inhibit the growth of MRSA or S. Typhimurium in endothelial cells. These results indicate that intracellular NAD+ homeostasis is crucial for controlling intracellular GAS infection in endothelial cells. In addition, nicotinamide may be a potential new therapeutic agent to overcome persistent infections of GAS.
Group A streptococcus (GAS) is recognized as one of the major human pathogens that not only provokes mild skin and throat infections such as impetigo or pharyngitis, but also occasionally causes serious infections such as streptococcal toxic shock syndrome and necrotizing fasciitis (Cunningham, 2008; Walker et al., 2014). Although antibiotics are effective for treating GAS infections, accumulating evidence has shown that invading host cells could be one of the strategies used by GAS to avoid killing by host immune responses and antibiotics, which may contribute to persistent infection seen in 5–30% of individuals (Osterlund et al., 1997; Marouni et al., 2004; Kaplan et al., 2006).
Nicotinamide adenine dinucleotide (NAD+) and its reduced form (NADH) have been known as important cofactors that are coupled to the redox reactions for energy production, or used as enzymatic cofactors involving in hundreds of biochemical functions within living cells (Canto et al., 2015). Nicotinamide (NAM) is a vitamin B3 derivative that has been used as a metabolic precursor of NAD+ to raise intracellular NAD+ level for improvement of metabolic diseases involving energy production (Fricker et al., 2018). Autophagy, the cellular degradation of ubiquitinated proteins in lysosomes under stress conditions, is one of the most tightly regulated homeostatic processes involving intracellular NAD+ (Klionsky et al., 2016; Kocaturk and Gozuacik, 2018). Moreover, autophagy also plays a crucial role in innate immune defenses against invading pathogens including Listeria monocytogenes, Salmonella Typhimurium, and GAS (Castrejón-Jiménez et al., 2015; Kimmey and Stallings, 2016; Bah and Vergne, 2017). In order to survive in host cells, GAS expresses various virulence factors to impair autophagic clearance, including streptococcal cysteine protease SpeB, streptolysin O (SLO), and NAD-glycohydrolase (NADase) (Sakurai et al., 2010; Barnett et al., 2013; Mestre and Colombo, 2013; O’Seaghdha and Wessels, 2013; O’Neill et al., 2016; Sharma et al., 2016). NADase is a potent hydrolase involved in the consumption of NAD+ that leads to intracellular energy collapse and programmed necrosis of infected cells (Chandrasekaran and Caparon, 2015, 2016; Pajuelo et al., 2018). In addition, several studies have indicated that NADase is involved with the structural and functional stabilization of SLO, which contributes to enhance GAS pathogenesis and global dissemination of serotype M1 and M89 GAS, indicating that NADase plays an important role during GAS infection (Michos et al., 2006; Turner et al., 2015; Zhu et al., 2015; Velarde et al., 2017; Barnett et al., 2018). However, the mechanisms of NAD+ homeostasis controlling GAS survival in the host are complicated and need to be explored.
Previously, we have found that defective acidification of autophagosomes allows GAS growth in endothelial cells (Lu et al., 2015). NADase is responsible for the depletion of intracellular NAD+ and inhibition of autophagosomal acidification, which results in the multiplication of GAS in endothelial cells (Hsieh et al., 2018). In this study, we demonstrate that supplementation with exogenous NAM significantly restores the intracellular NAD+ content and NAD+/NADH ratio, which enhances the acidification of GAS-containing autophagosomes and clearance of intracellular GAS within endothelial cells.
Human microvascular endothelial cell line-1 (HMEC-1) cells were cultured in endothelial growth medium M200 with low serum growth factors (Gibco Life Technologies, Grand Island, NY, United States) and 10% fetal bovine serum (FBS) at 37°C in a humidified incubator with 5% CO2. When the cell confluence reached 80%, cells were detached with trypsin-EDTA (Gibco Life Technologies) and seeded at the density of 0.75 × 106 cells/dish in 10-cm dishes for maintenance or 3 × 105 cells/well in 6-well plates for the intracellular bacteria survival assay and confocal microscopy.
Group A streptococcus strains SF370 (M1 serotype) and NZ131 (M49 serotype) were purchased from the American Type Culture Collection (Manassas, VA, United States). GAS strain A20 (M1 serotype) was isolated from the blood of a patient with necrotizing fasciitis (Zheng et al., 2013). Methicillin-resistant Staphylococcus aureus (MRSA) and Salmonella Typhimurium were isolated from patients with bacteremia. All strains were susceptible to gentamicin and cultured on tryptic soy agar containing 5% defibrinated sheep blood or tryptic soy broth (Becton Dickinson, Sparks, MD, United States) supplemented with 0.5% yeast extract (TSBY).
The cell infection was described in the previous study with modifications (Hsieh et al., 2018). In brief, the overnight bacterial cultures were transferred and grown to the exponential growth phase, and then resuspended in endothelial growth medium M200. HMEC-1 cells were seeded in 6-well plates at a density of 3 × 105 cells/well and infected with bacteria at different multiplicities of infection (M.O.I.) to ensure equivalent intracellular bacterial load at 1 h of infection. Plates were then centrifuged at 500 g for 5 min to synchronize the infection and incubated in humidified 5% CO2 at 37°C for 30 min. After infection, the infected cells were treated with gentamicin (125 μg/mL) to kill extracellular bacteria. Subsequently, cells were maintained in M200 medium with or without β-NAD, NADH, or NAM for additional 2 and 4 h. At indicated times post infection, cells were lysed by deionized water and plated on TSBY agar plates to evaluate the intracellular GAS growth within endothelial cells.
The NADase activity in GAS culture supernatants was measured by the qualitative fluorescence assay according to the method described above by Bricker et al. (2002). Briefly, the bacterial supernatants from different culture treatments were harvested by centrifugation at 2,330 g for 10 min. The supernatants were reacted with 1 mM of β-NAD (Sigma-Aldrich, St. Louis, MO, United States) at 37°C in 5% CO2 for 1 h. After incubation, sodium hydroxide (PanReac AppliChem, Barcelona, Spain) was added to develop reactions at room temperature in the dark for 1 h. The fluorescence intensity of β-NAD was determined by Tecan M200 Pro Infinite plate reader (Tecan, Crailsheim, Germany) at excitation wavelength 360 nm. The NADase activity of each sample was expressed as a relative percentage compared with uninoculated culture medium.
The change of intracellular NAD+ content was determined by the NAD+/NADH quantification kit (Sigma-Aldrich), according to the manufacturer’s instructions. Briefly, a total of 2 × 105 cells were harvested by trypsinization and suspended in NAD+/NADH extraction buffer to lyse the cells by freezing and thawing three times. The 10-kDa cut-off spin column (GE Healthcare, Buckinghamshire, United Kingdom) was used to remove NADH degrading enzymes by centrifugation at 21,000 g, 4°C for 60 min. The intracellular NAD+ of each sample was decomposed in 60°C for 30 min and then transferred to microtiter plates to determine the concentration of intracellular NADH. In addition, total intracellular NADH of each sample was reacted with NAD cycling enzyme to convert NAD+ to NADH. Subsequently, all samples were loaded into microtiter plates and mixed with NADH developer and incubated at room temperature for 1 h. After reaction, the absorbance of each sample was measured by ELISA reader (Tecan, infinite M200) at the wavelength of 450 nm. The NAD+/NADH ratio was calculated to evaluate the metabolic status of cells following a formula: [(NADHtotal−NADH)/NADH].
To determine cell cytotoxicity of GAS-infected cells under NAM treatment, the activity of dead-cell protease was measured by CytoTox-Glo cytotoxicity assay kit according to the manufacturer’s instructions (Promega, Madison, WI, United States). Briefly, cell were infected with GAS as described above. The cell culture supernatants were harvested at indicated times post infection, and co-incubated with luminogenic substrate at room temperature for 15 min. The luminescent intensity was measured by Tecan M200 Pro Infinite plate reader (Tecan). The cell cytotoxicity of each sample was expressed as a relative percentage compared with non-infected cells.
The expression of ATG9A in endothelial HMEC-1 cells was downregulated through lentiviral expression of a short hairpin RNA (clone 81, TRCN0000244081 containing target sequence 5′-AGTCACCTTGGCACCATATTG-3′; clone 82, TRCN0000244082 containing target sequence 5′-GTGGACTATGACATCCTATTT -3′; clone 83, TRCN0000244083 containing target sequence 5′-TGTAGGAGCAGGATGGAAATA-3′). The shRNA lentiviral clones were obtained from the National RNAi Core Facility at the Academia Sinica in Taiwan. Endothelial HMEC-1 cells were seeded in 6-well plates at a density of 2 × 105 cells/well and infected with lentivirus at a M.O.I of 3 in M200 medium containing 8 μg/mL of polybrene at 37°C in 5% CO2 for 24 h. After transduction, cells were treated with 5 μg/mL of puromycin (Sigma-Aldrich) for three generations to select the stable clone. The efficiency of ATG9A knockdown was evaluated by Western blot analysis.
Endothelial HMEC-1 cells were infected with GAS at M.O.I. of 1 for 30 min, and treated with gentamicin to kill extracellular bacteria. After treatment, cells were maintained in NAM-supplemented medium for additional 2 h. Cells were detached by trypsinization and gently resuspended in phenol red-free medium containing 5% FBS and 40% Dextran T2000. Cells were kept on ice and cryo-fixed in a high-pressure freezer (Leica EM HPM100) according to manufacturer’s instructions. After fixation, samples were dehydrated by freeze substitution and embedded in plastic (Epon812, TAAB Laboratories Equipment, Aldermaston, United Kingdom). The embedded samples were cut into 70 nm ultrathin sections, and stained with saturated uranyl acetate and Reynolds lead citrate solution. TEM images were acquired with a JEOL JEM-1011 transmission electron microscope (JEOL, JEM-1011, Tokyo, Japan).
Cells were infected with GAS as described above. At indicated times post infection, cells were lysed with RIPA lysis buffer containing protease inhibitor (Promega). Cell lysates were quantified using Bradford protein assay (Bio-Rad, Hercules, CA, United States) and boiled in SDS sample buffer for 10 min. All samples were then separated by 12% SDS-polyacrylamide gel and transferred onto the polyvinylidene difluoride (PVDF) membranes (Millipore, Boston, MA, United States). After blocking with 5% skim milk, the membranes were incubated with primary antibody against ATG9A (Abcam Technology, Cambridge, MA, United States), LC3 (MBL, Nagoya, Japan), or β-actin (novusbio, Littleton, CO) in blocking buffer at 4°C for overnight. After incubation, horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA, United States) were used to visualize the Western blot. All images were acquired with the ImageQuant LAS-4000 imaging system (GE Healthcare Life Sciences, Pittsburgh, PA, United States).
The confocal microscopy was performed as previously described (Hsieh et al., 2018). All images were acquired and analyzed with the confocal microscope LSM 880 (Carl Zeiss, Jena, Germany). The percentage of colocalization between GAS, autophagosomal LC3 (MBL), lysosomal marker LAMP1 (Cell Signaling Technology, Danvers, MA, United States), and acidotropic probe Lysotracker (Invitrogen Molecular Probes, Eugene, OR, United States) was quantified from five independent fields of each experiment.
Data are representative of three independent experiments and expressed as the mean ± standard error of the mean (SEM). GraphPad Prism 5.0 was used to plot all graphs and evaluate statistical significance by 1-way or 2-way ANOVA with post hoc Tukey’s or Bonferroni multiple comparison test, respectively. Results were considered as statistically significant when the p-value was less than 0.05, and marked by an asterisk (∗) in figures.
NAD-glycohydrolase is a potent glycohydrolase that cleaves β-NAD to produce nicotinamide and ADP-ribose, leading to disrupted energy homeostasis and promoting bacterial survival in host cells (Bastiat-Sempe et al., 2014; Hsieh et al., 2018; Pajuelo et al., 2018). However, the underlying mechanisms associating NAD+ metabolism with intracellular bacterial survival are complicated and poorly understood. To clarify whether NAD+ depletion or accumulated metabolic products contributed to GAS survival in endothelial cells, exogenous β-NAD, NADH and nicotinamide (NAM) were supplemented in culture medium, and the GAS survival in endothelial HMEC-1 cells was evaluated by a colony-forming assay. The results revealed no significant difference in the bacterial count of intracellular GAS within endothelial HMEC-1 cells under different concentrations of β-NAD+ and NADH after 5 h of infection (Figures 1A,B). In contrast, supplementation with 2.5 to 40 mM of NAM was able to inhibit the intracellular GAS growth within endothelial HMEC-1 cells in a time and dose-dependent manner (Figure 1C).
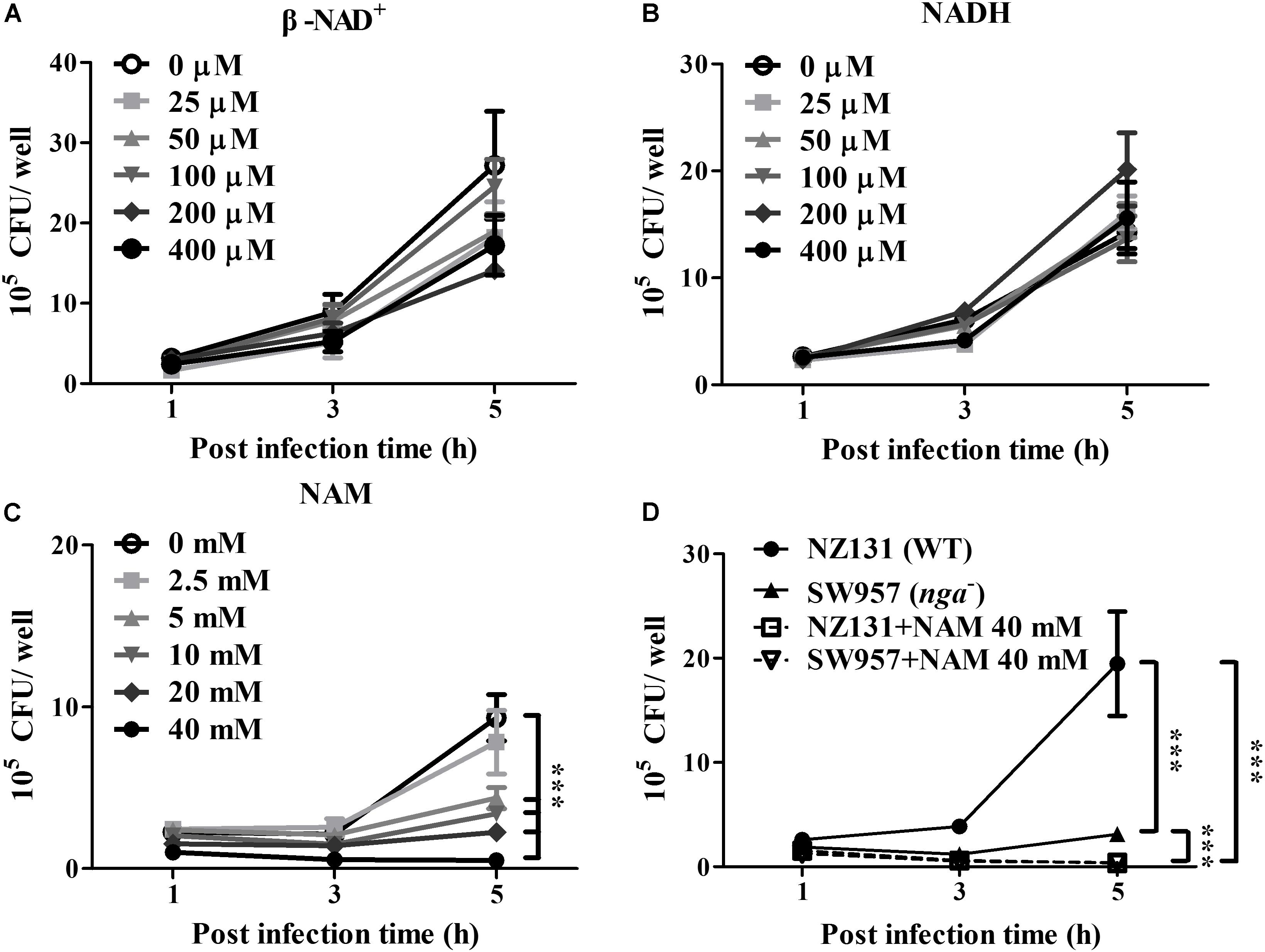
Figure 1. Nicotinamide inhibits intracellular GAS growth in endothelial cells. The intracellular growth of GAS was analyzed in endothelial HMEC-1 cells under exogenous NAD+ substrate supplementation. HMEC-1 cells were infected with wild-type NZ131 at M.O.I. of 1 and gentamicin was used to kill extracellular bacteria. Cells subsequently were maintained in the fresh M200 medium with/without different concentrations of (A) β-NAD, (B) NADH, or (C) nicotinamide (NAM) for an additional 2 and 4 h. The intracellular bacterial load was counted by CFU-based assay. (D) The intracellular growth of GAS was analyzed in the NAM-treated endothelial HMEC-1 cells. HMEC-1 cells were infected with wild-type NZ131 or NADase-knockout strain SW957, treated with gentamicin, and maintained in 40 mM of NAM. The intracellular bacterial load was counted by CFU-based assay. The data represent the means ± SEM of at least three independent experiments. ***p < 0.001 (2-way ANOVA).
Nicotinamide has been extensively used to treat various clinical disorders and skin infections (Rolfe, 2014). To investigate whether NAM directly inhibits bacterial survival, the TSBY broth was supplemented with NAM to measure the growth of GAS in vitro. The results showed that the bacterial growth was not affected in the presence of 2.5 to 40 mM of NAM, when compared with the control medium (Supplementary Figure S1A). Previous reports showed that the enzymatic activity of NADase is required for GAS survival in host cells (O’Seaghdha and Wessels, 2013; Sharma et al., 2016). To explore whether NAM could mediate intracellular GAS growth through repressing NADase activity, the GAS was cultured to mid-logarithmic phase in TSBY broth supplemented with different concentrations of NAM. The results showed that deletion of nga (SW957) caused loss of the NADase activity, compared to wild-type NZ131. Moreover, NADase activity of wild-type NZ131 in GAS culture supernatant was decreased to 45 and 60.8% in the presence of 40 mM NAM at 4 and 6 h of incubation, respectively (Supplementary Figure S1B), when compared with untreated group. In order to clarify whether NAM reduced NADase activity to mediate intracellular GAS survival, the NADase-encoding gene (nga) was deleted by homologous recombination (Hsieh et al., 2018). The endothelial HMEC-1 cells were then infected with nga mutant under NAM treatment. The results showed that the intracellular growth of the NADase mutant was significant decreased when compared with the wild-type NZ131 in non-NAM-treated HMEC-1 cells. Moreover, supplementation with exogenous NAM further reduced both wild-type NZ131 and NADase mutant growth in HMEC-1 cells during infection (Figure 1D). These results imply that NADase activity contributes to intracellular GAS survival, but NAM may trigger other mechanisms to inhibit intracellular GAS growth in endothelial cells.
Nicotinamide can be used as a precursor for the biosynthesis of NAD+ through the salvage pathway in mammalian cells (Canto et al., 2015). We were interested to understand whether NAM could increase the intracellular NAD+ content in endothelial cells. NAM was supplemented in culture medium, and then intracellular NAD+ and NAD+/NADH ratio were evaluated at indicated time points post infection. As expected, the results revealed that NZ131-infected cells have lower NAD+ content and NAD+/NADH ratios than non-infected cells at 1 and 5 h of infection. In contrast, supplementation with exogenous NAM could significantly increase intracellular NAD+ content and the NAD+/NADH ratio in endothelial cells at 1 and 5 h of infection, compared with untreated cells (Figures 2A,B). In order to illustrate the correlation between exogenous NAM supplementation and intracellular NAD+ content in endothelial cells during GAS infection, the intracellular NAD+ content and NAD+/NADH ratio of HMEC-1 cells were analyzed by treatment with different concentrations of NAM (2.5 to 40 mM). The results showed that supplementation with exogenous NAM can gradually elevate intracellular NAD+ content and the NAD+/NADH ratio in NZ131-infected HMEC-1 cells (Figures 2C,D), suggesting that both intracellular NAD+ level and NAD+/NADH ratio are correlated with NAM treatment in endothelial cells during GAS infection. In addition to NAM, the intracellular NAD+ content of NZ131-infected cells was also measured after exogenous β-NAD and NADH supplementation. The results showed that the intracellular NAD+ content was not increased after β-NAD and NADH treatment, when compared with untreated and NAM-treated cells at 5 h of infection (Supplementary Figure S2).

Figure 2. Nicotinamide increases intracellular NAD+ content and NAD+/NADH ratio of endothelial cells. (A,C) The intracellular NAD+ content of endothelial HMEC-1 cells was measured after nicotinamide (NAM) treatment. The cell lysates were extracted from GAS-infected HMEC-1 cells with/without NAM treatment and analyzed by a NAD+/NADH quantification kit. (B,D) The NAD+/NADH ratio of GAS-infected HMEC-1 cells was calculated following a formula: [(NADHtotal – NADH)/NADH]. (E) The cell viability of GAS-infected cells after NAM treatment were measured by CytoTox-Glo cytotoxicity assay kit. The cell culture supernatants were harvested after GAS infection with/without NAM treatment at indicated time point, and then co-incubated with luminogenic substrate. The cell cytotoxicity was expressed as relative percentage compared to 1 h of cell alone. The data represent the means ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 (1- or 2-way ANOVA).
Nicotinamide adenine dinucleotide (NAD+) is considered as a critical molecule involved in diverse physiological functions including energy production and cell viability (Filomeni et al., 2015). To illustrate whether increased NAD+ contributed to cell survival, the protease-based cytotoxicity assay was used to measure the cell viability of GAS-infected cells under NAM treatment. The results showed that the GAS infection induced higher cytotoxicity than non-infected cells, but NAM treatment can dramatically decrease cytotoxicity in both non-infected and GAS-infected cells at 1 and 5 h of infection (Figure 2E).
Several studies have indicated that NAD+ homeostasis plays a prominent role in the regulation of autophagy responses (Preyat and Leo, 2016; Zhang et al., 2016). Accordingly, the autophagy-related ATG9A was knocked down by lentiviral short hairpin RNA (shRNA) to explore whether the increasing NAD+ was associated with autophagy-mediated intracellular GAS clearance in endothelial cells. The results of Western blot analysis showed that the clone 82 had the highest knockdown efficiency of ATG9A, compared to wild-type and negative control shLuc-infeced HMEC-1 cells (Figure 3A). In addition, the autophagy-related LC3-II form was decreased in ATG9A-knockdown cells when compared to wild-type cells (Supplementary Figure S3A). The clone 82 was subsequently chosen to infect with GAS for analyzing bacterial intracellular growth under NAM treatment. The results showed that the intracellular bacterial load was significantly increased in ATG9A-knockdown HMEC-1 cells under 40 mM of NAM supplementation, when compared to wild-type and shLuc-infected cells (Figure 3B). In order to clarify whether autophagy could be activated by NAM treatment, the LC3 conversion was analyzed in endothelial HMEC-1 cells treated with NAM during GAS infection. The results of the Western blotting showed that a trend of increasing level of LC3-II form was observed in NAM-treated cells at 5 h of infection, however, it has no significance when compared to non-treated cells (lane 3 versus lane 4, p-value = 0.1, Figures 3C,D). In contrast, the LC3-II form was not induced in ATG9A-knockdown HMEC-1 cells after NAM treatment (Supplementary Figure S3B). Next, transmission electron microscopy (TEM) was further used to visualize the autophagosome membrane structure in GAS-infected HMEC-1 cells. The images revealed that intracellular GAS was surrounded by the single membrane structure, but double membrane-engulfed GAS was markedly observed in NAM-treated HMEC-1 cells (Figure 3E). Theses evidences suggest that intracellular GAS can be sequestered by autophagy in endothelial cells after NAM treatment.
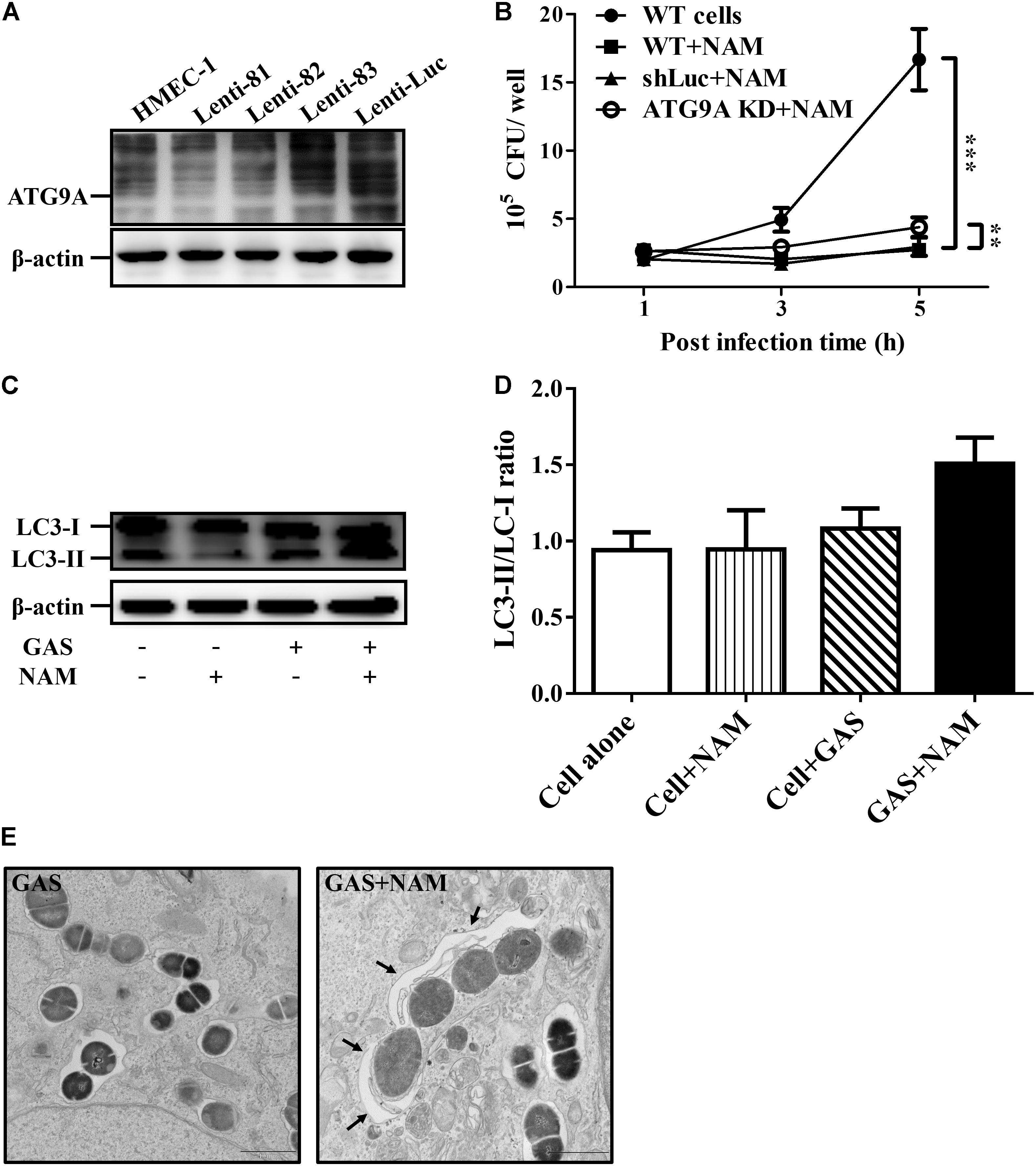
Figure 3. Nicotinamide activates autophagy to inhibit intracellular GAS growth in endothelial cells. (A) The expression of ATG9A in endothelial HMEC-1 cells. HMEC-1 cells were transfected with three lentivirus-based shRNAs (shATG9A 81, 82, and 83) to silence the ATG9A expression or Luciferase shRNA (shLuc) for a negative control. The expression of ATG9A was examined by Western blot analysis. (B) The intracellular growth of GAS was evaluated in ATG9A-knockdown (KD) HMEC-1 cells under nicotinamide (NAM) treatment. The wild-type and lentivirus-infected HMEC-1 cells (shLuc and clone 82) were infected with GAS at M.O.I. of 1, and gentamicin was used to kill extracellular bacteria. The intracellular bacterial load was counted by CFU-based assays. The data represent the means ± SEM of at least three independent experiments. **p < 0.01; ***p < 0.001 (2-way ANOVA). (C) The LC3 conversion of GAS-infected HMEC-1 cells under NAM treatment were analyzed by Western blotting. (D) The relative band intensities of LC3-I and LC3-II were measured by densitometry analysis using ImageJ software and expressed as the ratio of LC3-II/LC3-I. (E) Cells were infected with GAS and treated with/without NAM at 1 h of infection. The membrane structure of GAS-containing vacuoles were observed by conventional transmission electron microscopy (TEM). Black arrow indicate the double membrane compartment. Scale bar = 1 μm.
Previous studies have shown that GAS can survive in host cells through avoidance of lysosomal degradation with bacteria-containing vacuoles (O’Seaghdha and Wessels, 2013; Sharma et al., 2016; Hsieh et al., 2018). Therefore, whether NAM could enhance lysosomal recruitment with GAS-containing vacuoles to inhibit GAS growth in endothelial cells was further analyzed. The intracellular localization of bacteria, LC3 decorated vesicles, and lysosomes in endothelial HMEC-1 cells were observed by confocal microscopy after NAM treatment. Confocal images showed that the intracellular bacteria were colocalized with LC3-positive vacuoles, but the colocalization of intracellular bacteria with LC3 puncta was reduced in HMEC-1 cells without NAM treatment after 5 h of infection. In contrast, more intracellular bacteria were colocalized with LC3-positive vacuoles in NAM-treated HMEC-1 cells compared to without NAM treatment after 5 h of infection (Figure 4A). Furthermore, the membrane glycoprotein LAMP-1 was used as a marker for visualizing the colocalization of lysosomes with intracellular GAS in endothelial HMEC-1 cells. The results showed that only 25 and 17% of intracellular GAS was associated with LAMP-1 in untreated cells at 3 and 5 h of infection, respectively. Under the NAM treatment, 61 and 74% of intracellular GAS was colocalized with LAMP-1 at 3 and 5 h of infection, respectively (Figures 4A,B).
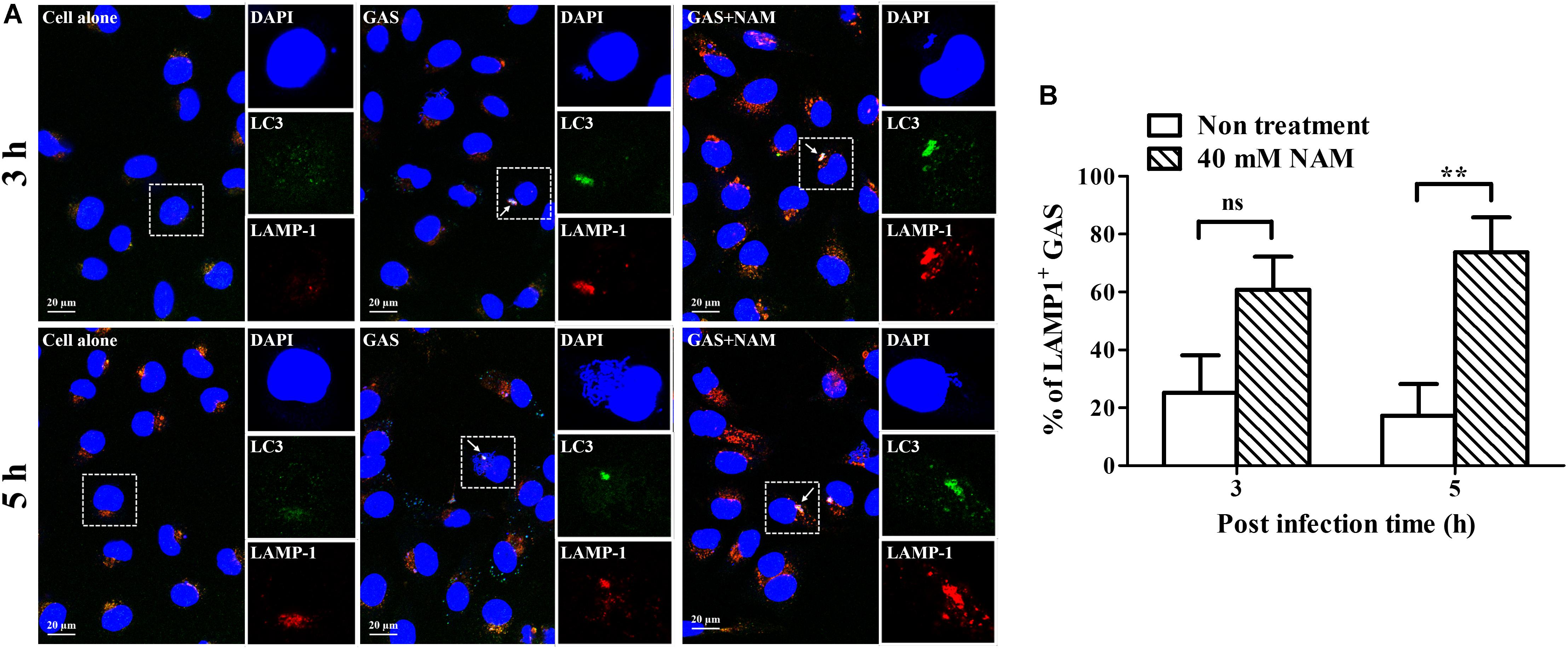
Figure 4. Nicotinamide enhances GAS-containing vacuole trafficking to lysosomes in endothelial cells. (A) The colocalization of intracellular GAS with autophagosomes and lysosomes was analyzed in nicotinamide (NAM)-treated endothelial HMEC-1 cells. Confocal microscopy was used to observe the colocalization of GAS (DAPI, blue) with LC3 (Alexa 488, green) and LAMP-1 (Alexa 594, red) in HMEC-1 cells under NAM treatment at 3 and 5 h of infection. Representative images are shown from three independent experiments. Scale bar = 20 μm. (B) The intracellular bacteria associated with LAMP-1 at 3 and 5 h of infection were quantified from confocal microscopy images. At least 100 intracellular GAS were quantified for each time point in at least three independent experiments. The data represent the means ± SEM of at least three independent experiments. ns, not significant; **p < 0.01 (2-way ANOVA).
Sufficient acidification is required for lysosomal enzyme activity to eliminate intracellular bacteria in endothelial cells and macrophages (Bastiat-Sempe et al., 2014; Valderrama and Nizet, 2018). In order to clarify whether the GAS-containing vacuoles were successfully acidified after NAM treatment, the fluorescent indicator Lysotracker was used to observe the acidification within NAM-treated endothelial HMEC-1 cells during GAS infection. The results revealed that about 40% of intracellular bacteria were colocalized with Lysotracker under NAM-treated and untreated conditions at 3 h of infection. However, the colocalization of intracellular GAS with Lysotracker was higher in NAM-treated endothelial HMEC-1 cells compared to cells without NAM treatments at 5 h of infection (51 vs. 13%, respectively) (Figures 5A,B). The activity of lysosomal H+-ATPase was inhibited by bafilomycin A1 to evaluate the roles of acidification for intracellular survival of GAS in NAM-treated endothelial HMEC-1 cells. The results showed that the intracellular bacterial load was significantly increased following treatment with bafilomycin A1 at 5 h of infection compared to untreated cells under NAM-supplemented conditions (Figure 5C). These results indicate that treatment with exogenous NAM markedly enhances lysosome-mediated acidification to inhibit intracellular GAS growth in endothelial cells.
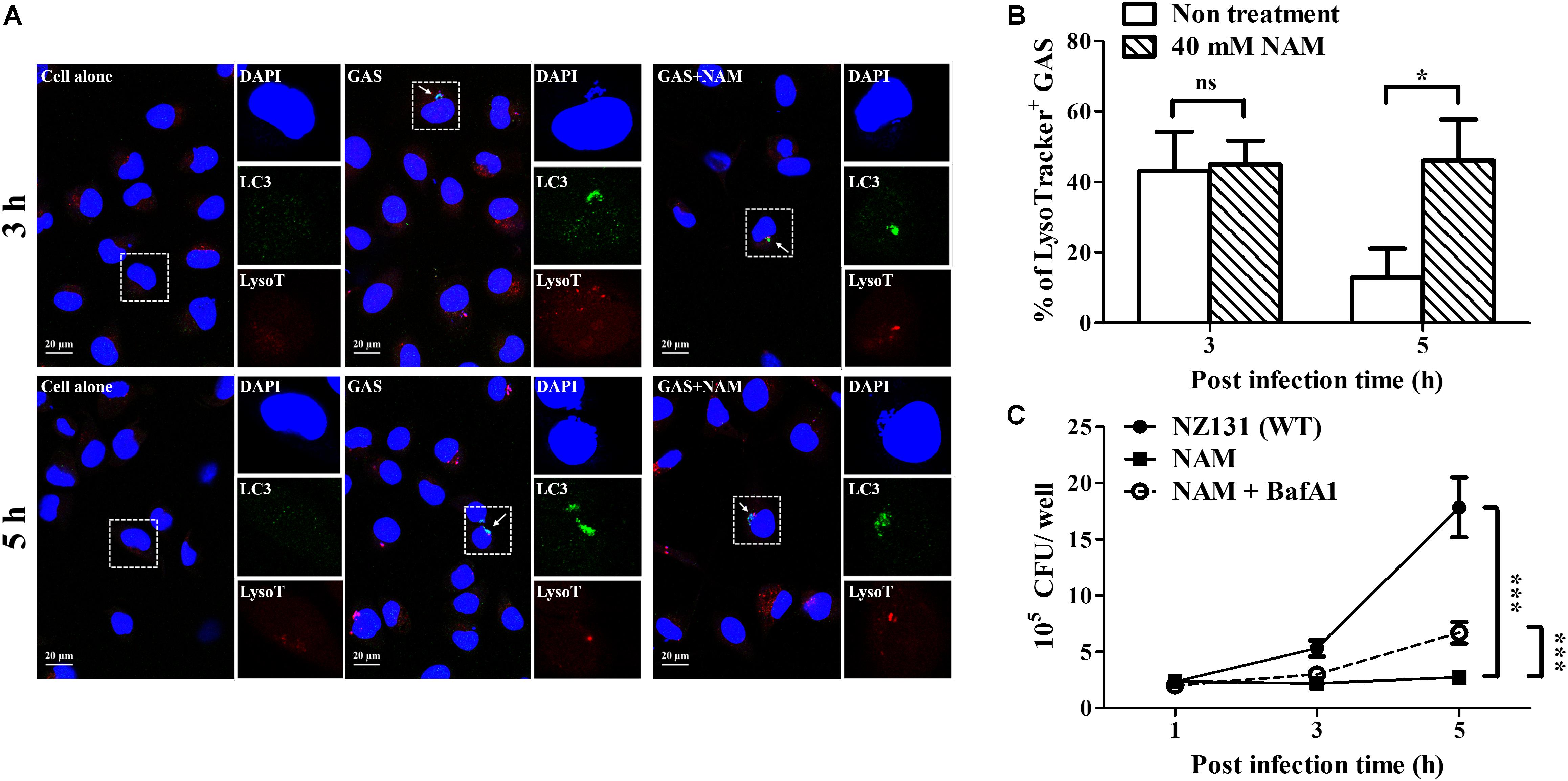
Figure 5. Nicotinamide increases acidification of GAS-containing vacuole in endothelial cells. (A) The colocalization of intracellular GAS with acidified autophagosomes was analyzed in nicotinamide (NAM)-treated HMEC-1 cells. The intracellular localization of GAS (DAPI, blue), LC3 (Alexa 488, green), and acidification (acidotropic indicator LysoTracker Red DND-99, red) in endothelial cells was observed by confocal microscopy at 3 and 5 h of infection. Representative images are shown from three independent experiments. Scale bar = 20 μm. (B) The percentage of intracellular bacteria associated with LysoTracker was quantified at 3 and 5 h of infection. At least 100 intracellular GAS were quantified for each time point in at least three independent experiments. (C) The intracellular growth of GAS was analyzed in endothelial HMEC-1 cells under bafilomycin A1 (BafA1) and NAM cotreatment. HMEC-1 cells with/without pretreatment with 200 nM BafA1, were then infected with GAS at M.O.I. of 1 in presence or absence of 40 mM NAM. The intracellular bacterial load was counted by CFU-based assays. The data represent the means ± SEM of at least three independent experiments. ns, not significant; *p < 0.05; ***p < 0.001 (2-way ANOVA).
Nicotinamide and its derivatives have been extensively used to treat bacterial and viral infections (Murray, 2003; Singhal and Cheng, 2019). Based on these findings, we sought to examine whether NAM could inhibit other pathogens’ survival in endothelial cells. The serotype M1 and M49 GAS, MRSA, and S. Typhimurium were utilized to infect endothelial HMEC-1 cells and the intracellular bacterial growth was measured by a colony-forming assay. The results showed that the intracellular growth of both serotype M1 (strain A20) and M49 (strain NZ131) was significantly reduced in NAM-treated endothelial HMEC-1 cells at 5 h of infection, compared to non-treated cells (Figure 6A). However, supplementation with exogenous NAM did not significantly inhibit the growth of MRSA and S. Typhimurium in endothelial HMEC-1 cells (Figure 6B), implying that these pathogens may use different mechanisms to escape NAM-mediated clearance in endothelial cells.
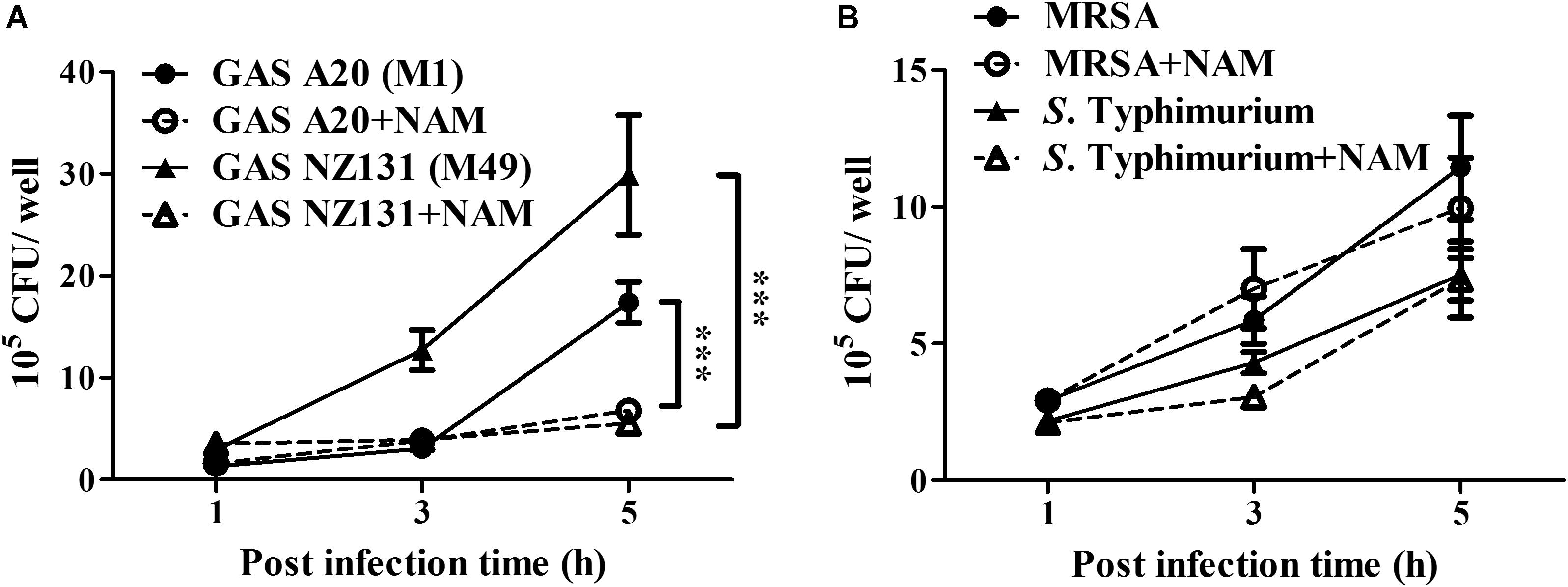
Figure 6. Nicotinamide specifically inhibits intracellular multiplication of GAS in endothelial cells. The intracellular growth of GAS, MRSA, and S. Typhimurium was analyzed in nicotinamide (NAM)-treated endothelial HMEC-1 cells. HMEC-1 cells were infected with (A) serotype M1 A20 at M.O.I. of 10, M49 NZ131 at M.O.I of 1 or (B) MRSA and S. Typhimurium at M.O.I of 1. The extracellular bacteria were killed by gentamicin and intracellular bacterial load was counted by CFU-based assays. The data represent the means ± SEM of at least three independent experiments. ***p < 0.001 (2-way ANOVA).
Nicotinamide adenine dinucleotide (NAD+) is one of the most important molecules involved in energy generation and signal transduction to control cellular metabolism in live cells (Canto et al., 2015). However, several studies found that many pathogens express multiple toxins to influence intracellular NAD+ homeostasis of infected cells, which results in energy collapse, impaired host defense, immunopathology, and increased pathogen survival (Bastiat-Sempe et al., 2014; Sun et al., 2015; Bhat et al., 2016; Mesquita et al., 2016; Pajuelo et al., 2018). In this study, we found that supplementation with exogenous NAM could increase intracellular NAD+ levels and the NAD+/NADH ratio that contributed to enhance intracellular GAS clearance by autophagy in endothelial cells (Figure 7).
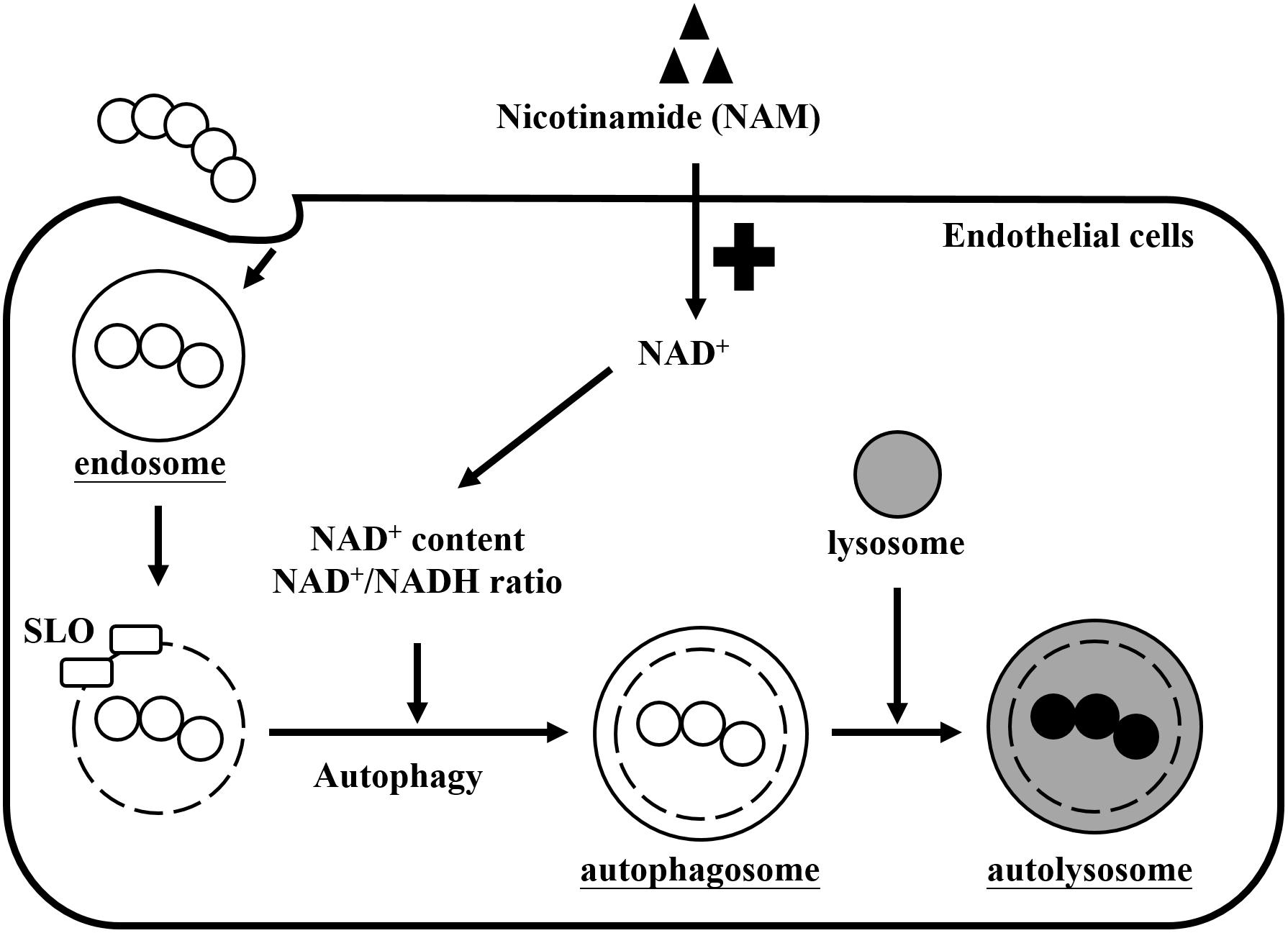
Figure 7. Effects of nicotinamide on GAS survival in endothelial cells. GAS is internalized into cells through endocytosis to form the endosomes. However, SLO is expressed to damage the endosomal membrane and further induces autophagy responses. GAS depletes intracellular NAD+ promoting their intracellular survival, but cells supplemented with exogenous nicotinamide (NAM) can restore the intracellular NAD+ content and NAD+/NADH ratio, resulting in GAS clearance by autophagy in endothelial cells.
In order to clarify the correlation between the intracellular NAD+ content and bacterial survival of infected cells, the exogenous NAD+-associated substrates were supplemented in GAS-infected endothelial cells and bacterial survival was analyzed by a colony-formation assay. Our results demonstrated that supplementation with exogenous NAM not only dramatically increased intracellular NAD+ level and NAD+/NADH ratio, but also inhibited GAS growth in endothelial cells in a dose- and time-dependent manner (Figures 1, 2). In contrast to NAM, the intracellular NAD+ content and bacterial load were not affected by the exogenous β-NAD and NADH treatments (Supplementary Figure S2), which might be due to the membrane impermeability (Canto et al., 2015). In addition to NAM, intracellular NAD+ is also generated from tryptophan through a de novo biosynthetic pathway in the cells (Moffett and Namboodiri, 2003). Recent studies have shown that tryptophan catabolism plays a crucial role against bacterial and viral infections, including those caused by Clostridium difficile, Mycobacterium tuberculosis, and hepatitis B and C viruses (Larrea et al., 2007; El-Zaatari et al., 2014; Schmidt and Schultze, 2014). However, whether tryptophan is involved in NAD+ fluctuation influencing GAS survival in endothelial cells needs to be evaluated. These results indicate that the importance of intracellular NAD+ homeostasis on pathogens’ survival in the host.
Our results have shown that the given exogenous NAM can increase the conversion of LC3-I to LC3-II and induce autophagic double membrane formation to inhibit GAS survival in endothelial cells (Figures 3–5). The LC3-II level and ratio of LC3-II/LC3-I have been considered as a reliable marker for reflecting the activation of autophagy (Kadowaki and Karim, 2009). Recently, several studies have shown that supplementation with NAM can induce the conversion of LC3-I to LC3-II in chronic hypoxic myocardial cells, which support our results that the LC3-II was increased after NAM treatment in GAS-infected endothelial cells (Kang and Hwang, 2009; Hwang and Song, 2017; Li et al., 2019). These evidences indicate that NAM can increase autophagic activation under different stress conditions. However, the LC3-II-decorated single-membrane phagosomes or vacuoles (LAP) recently has been found in microbial survival within phagocytic and non-phagocytotic cells (Schille et al., 2018; Upadhyay and Philips, 2019). Therefore, the signaling pathways and membrane structure can be utilized to define the difference between the conventional autophagosome and LC3-associated phagosome (LAPosome) (Martinez et al., 2015; Schille et al., 2018). Autophagy is a tightly regulated process influenced by many NAD+-consuming enzymes including ADP-ribose polymerase-1 (PARP-1) and sirtuins (Zhang et al., 2016). Several studies have demonstrated that the NAD+-dependent sirtuins are involved in bacterial survival by regulating autophagy via activation of the AMPK/mTOR signaling pathway in intestinal epithelial cells and macrophages (Ganesan et al., 2017; Al Azzaz et al., 2018; Li et al., 2019; Yang et al., 2019). Moreover, exogenous NAM treatment can enhance Sirtuin-1 activity to control autophagy-mediated mitochondrial quality in human fibroblasts (Kang and Hwang, 2009; Jang et al., 2012; Shen et al., 2017). On the other hand, reactive oxygen species (ROS) are considered as another critical mediators of NAD+ in autophagy responses (Filomeni et al., 2015; Preyat and Leo, 2016). We recently found that GAS infection can induce ROS production to stimulate single membrane LAPosome formation for bacterial survival and activate the inhibition of mTOR for GAS escaping from autophagic clearance in endothelial cells. In contrast, suppression of ROS level significantly enhance GAS clearance through converting defective LAPosome to conventional double membrane autophagosome in endothelial cells (Lu et al., 2017; Cheng et al., 2019). Moreover, autophagy could be upregulated by supplementation with exogenous NAD+, which can suppress PARP-1-mediated necrotic death of retinal pigment epithelium cells under oxidative stress (Zhu et al., 2016). These evidences suggest that NAM may modulate sirtuins activity or ROS production to regulate signaling transduction of conventional autophagy for intracellular GAS clearance in endothelial cells, but the underlying mechanisms require further investigation.
Nicotinamide has been considered as a safe and readily available therapeutic agent to improve the metabolic diseases, neurodegeneration, and skin infection through raising intracellular NAD+ level (Knip et al., 2000; Fricker et al., 2018). In this study, we showed that supplementation with exogenous NAM greatly reduced the intracellular growth of both serotype M1 and M49 GAS in endothelial cells, but did not dramatically inhibit the growth of MRSA and S. Typhimurium (Figure 6). Recent studies have shown that the M. tuberculosis depletes intracellular NAD+ content to enhance their intracellular survival, but treatment with NAM can restore intracellular NAD+ level and reduce the bacterial load of M. tuberculosis in macrophages (Sun et al., 2015; Pajuelo et al., 2018). Interestingly, NADase depleting intracellular NAD+ is required for the intracellular survival of GAS and M. tuberculosis in host cells, but the NADase activity of MRSA and S. Typhimurium cannot be determined in the culture supernatants (data not shown). These results imply that GAS and M. tuberculosis may share similar NAD+-associated mechanisms for intracellular survival in host cells.
In summary, we conclude that intracellular GAS survival within endothelial cells is linked to NAD+ homeostasis, indicating that NAM and its analogs may provide novel therapeutic strategies against GAS infection.
All datasets generated for this study are included in the article/Supplementary Material.
Y-SL, CC-N, P-JT, S-YW, C-CL, TN, and J-JW conceived and designed the study. C-LH, S-YH, H-MH, Y-NH, P-XZ, and J-JW analyzed the intracellular bacterial survival. C-LH, S-YH, Y-LC, and J-JW analyzed the cell response. C-LH, S-YH, S-LL, HO, TN, and J-JW did the images acquisition. C-LH, CC-N, P-XZ, and J-JW wrote the manuscript.
This work was supported by grants MOST105-2320-B006-009, MOST106-2320-B010-039, MOST 107-2320-B010-021, and MOST 108-2320-B010-004 from Ministry of Science and Technology, Taiwan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are very grateful to Prof. Robert M. Jonas for their helpful comments on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00117/full#supplementary-material
Al Azzaz, J., Rieu, A., Aires, V., Delmas, D., Chluba, J., Winckler, P., et al. (2018). Resveratrol-induced xenophagy promotes intracellular bacteria clearance in intestinal epithelial cells and macrophages. Front. Immunol. 9:3149. doi: 10.3389/fimmu.2018.03149
Bah, A., and Vergne, I. (2017). Macrophage autophagy and bacterial infections. Front. Immunol. 8:1483. doi: 10.3389/fimmu.2017.01483
Barnett, T. C., Bowen, A. C., and Carapetis, J. R. (2018). The fall and rise of group A Streptococcus diseases. Epidemiol. Infect. 15, 1–6. doi: 10.1017/S0950268818002285
Barnett, T. C., Liebl, D., Seymour, L. M., Gillen, C. M., Lim, J. Y., Larock, C. N., et al. (2013). The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 14, 675–682. doi: 10.1016/j.chom.2013.11.003
Bastiat-Sempe, B., Love, J. F., Lomayesva, N., and Wessels, M. R. (2014). Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio 5:e1690-14. doi: 10.1128/mBio.01690-14
Bhat, S. A., Iqbal, I. K., and Kumar, A. (2016). Imaging the NADH:NAD+ homeostasis for understanding the metabolic response of Mycobacterium to physiologically relevant stresses. Front. Cell. Infect. Microbiol. 6:145. doi: 10.3389/fcimb.2016.00145
Bricker, A. L., Cywes, C., Ashbaugh, C. D., and Wessels, M. R. (2002). NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol. Microbiol. 44, 257–269. doi: 10.1046/j.1365-2958.2002.02876.x
Canto, C., Menzies, K. J., and Auwerx, J. (2015). NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53. doi: 10.1016/j.cmet.2015.05.023
Castrejón-Jiménez, N. S., Leyva-Paredes, K., Hernandez-González, J. C., Luna-Herrera, J., and García-Pérez, B. E. (2015). The role of autophagy in bacterial infections. Biosci. Trends 9, 149–159. doi: 10.5582/bst.2015.01035
Chandrasekaran, S., and Caparon, M. G. (2015). The Streptococcus pyogenes NAD+ glycohydrolase modulates epithelial cell PARylation and HMGB1 release. Cell. Microbiol. 17, 1376–1390. doi: 10.1111/cmi.12442
Chandrasekaran, S., and Caparon, M. G. (2016). The NADase-negative variant of the Streptococcus pyogenes toxin NAD+ glycohydrolase induces JNK1-mediated programmed cellular necrosis. mBio 7:e2215. doi: 10.1128/mBio.02215-15
Cheng, Y. L., Kuo, C. F., Lu, S. L., Hiroko, O., Wu, Y. N., Hsieh, C. L., et al. (2019). Group A Streptococcus induces LAPosomes via SLO/β1 integrin/NOX2/ROS pathway in endothelial cells that are ineffective in bacterial killing and suppress xenophagy. mBio 10:e2148-19. doi: 10.1128/mBio.02148-19
Cunningham, M. W. (2008). Pathogenesis of group A streptococcal infections and their sequelae. Adv. Exp. Med. Biol. 609, 29–42. doi: 10.1007/978-0-387-73960-1_3
El-Zaatari, M., Chang, Y. M., Zhang, M., Franz, M., Shreiner, A., McDermott, A. J., et al. (2014). Tryptophan catabolism restricts IFN-γ-expressing neutrophils and Clostridium difficile immunopathology. J. Immunol. 193, 807–816. doi: 10.4049/jimmunol.1302913
Filomeni, G., De Zio, D., and Cecconi, F. (2015). Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388. doi: 10.1038/cdd.2014.150
Fricker, R. A., Green, E. L., Jenkins, S. I., and Griffin, S. M. (2018). The influence of nicotinamide on health and disease in the central nervous system. Int. J. Tryptophan Res. 11:1178646918776658. doi: 10.1177/1178646918776658
Ganesan, R., Hos, N. J., Gutierrez, S., Fischer, J., Stepek, J. M., Daglidu, E., et al. (2017). Salmonella typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS Pathog. 13:e1006227. doi: 10.1371/journal.ppat.1006227
Hsieh, C. L., Huang, H. M., Hsieh, S. Y., Zheng, P. X., Lin, Y. S., Chiang-Ni, C., et al. (2018). NAD-glycohydrolase depletes intracellular NAD+ and inhibits acidification of autophagosomes to enhance multiplication of group A Streptococcus in endothelial cells. Front. Microbiol. 9:1733. doi: 10.3389/fmicb.2018.01733
Hwang, E. S., and Song, S. B. (2017). Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci. 74, 3347–3362. doi: 10.1007/s00018-017-2527-8
Jang, S. Y., Kang, H. T., and Hwang, E. S. (2012). Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 287, 19304–19314. doi: 10.1074/jbc.M112.363747
Kadowaki, M., and Karim, M. R. (2009). Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 452, 199–213. doi: 10.1016/S0076-6879(08)03613-6
Kang, H. T., and Hwang, E. S. (2009). Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell. 8, 426–438. doi: 10.1111/j.1474-9726.2009.00487.x
Kaplan, E. L., Chhatwal, G. S., and Rohde, M. (2006). Reduced ability of penicillin to eradicate ingested group A streptococci from epithelial cells: clinical and pathogenetic implications. Clin. Infect. Dis. 43, 1398–1406. doi: 10.1086/508773
Kimmey, J. M., and Stallings, C. L. (2016). Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol. Med. 22, 1060–1076. doi: 10.1016/j.molmed.2016.10.008
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Knip, M., Douek, I. F., Moore, W. P., Gillmor, H. A., McLean, A. E., Bingley, P. J., et al. (2000). Safety of high-dose nicotinamide: a review. Diabetologia 43, 1337–1345. doi: 10.1007/s001250051536
Kocaturk, N. M., and Gozuacik, D. (2018). Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 6:128. doi: 10.3389/fcell.2018.00128
Larrea, E., Riezu-Boj, J. I., Gil-Guerrero, L., Casares, N., Aldabe, R., Sarobe, P., et al. (2007). Upregulation of indoleamine 2,3-dioxygenase in hepatitis C virus infection. J. Virol. 81, 3662–3666. doi: 10.1128/JVI.02248-06
Li, W., Zhu, L., Ruan, Z. B., Wang, M. X., Ren, Y., and Lu, W. (2019). Nicotinamide protects chronic hypoxic myocardial cells through regulating mTOR pathway and inducing autophagy. Eur. Rev. Med. Pharmacol. Sci. 23, 5503–5511. doi: 10.26355/eurrev_201906_18220
Lu, S. L., Kawabata, T., Cheng, Y. L., Omori, H., Hamasaki, M., Kusaba, T., et al. (2017). Endothelial cells are intrinsically defective in xenophagy of Streptococcus pyogenes. PLoS Pathog. 13:e1006444. doi: 10.1371/journal.ppat.1006444
Lu, S. L., Kuo, C. F., Chen, H. W., Yang, Y. S., Liu, C. C., Anderson, R., et al. (2015). Insufficient acidification of autophagosomes facilitates group A Streptococcus survival and growth in endothelial cells. mBio 6:e1435-15. doi: 10.1128/mBio.01435-15
Marouni, M. J., Barzilai, A., Keller, N., Rubinstein, E., and Sela, S. (2004). Intracellular survival of persistent group A streptococci in cultured epithelial cells. Int. J. Med. Microbiol. 294, 27–33. doi: 10.1016/j.ijmm.2004.01.001
Martinez, J., Malireddi, R. K., Lu, Q., Cunha, L. D., Pelletier, S., Gingras, S., et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906. doi: 10.1038/ncb3192
Mesquita, I., Varela, P., Belinha, A., Gaifem, J., Laforge, M., Vergnes, B., et al. (2016). Exploring NAD+ metabolism in host-pathogen interactions. Cell. Mol. Life Sci. 73, 1225–1236. doi: 10.1007/s00018-015-2119-4
Mestre, M. B., and Colombo, M. I. (2013). Autophagy and toxins: a matter of life or death. Curr. Mol. Med. 13, 241–251. doi: 10.2174/1566524011313020002
Michos, A., Gryllos, I., Hakansson, A., Srivastava, A., Kokkotou, E., and Wessels, M. R. (2006). Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J. Biol. Chem. 281, 8216–8223. doi: 10.1074/jbc.M511674200
Moffett, J. R., and Namboodiri, M. A. (2003). Tryptophan and the immune response. Immunol. Cell Biol. 81, 247–265. doi: 10.1046/j.1440-1711.2003.t01-1-01177.x
Murray, M. F. (2003). Nicotinamide: an oral antimicrobial agent with activity against both Mycobacterium tuberculosis and human immunodeficiency virus. Clin. Infect. Dis. 36, 453–460. doi: 10.1086/367544
O’Neill, A. M., Thurston, T. L. M., and Holden, D. W. (2016). Cytosolic replication of group A Streptococcus in human macrophages. mBio 7:e00020-16. doi: 10.1128/mBio.00020-16
O’Seaghdha, M., and Wessels, M. R. (2013). Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from xenophagic killing. PLoS Pathog. 9:e1003394. doi: 10.1371/journal.ppat.1003394
Osterlund, A., Popa, R., Nikkila, T., Scheynius, A., and Engstrand, L. (1997). Intracellular reservoir of Streptococcus pyogenes in vivo: a possible explanation for recurrent pharyngotonsillitis. Laryngoscope 107, 640–647. doi: 10.1097/00005537-199705000-00016
Pajuelo, D., Gonzalez-Juarbe, N., Tak, U., Sun, J., Orihuela, C. J., and Niederweis, M. (2018). NAD+ depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep. 24, 429–440. doi: 10.1016/j.celrep.2018.06.042
Preyat, N., and Leo, O. (2016). Complex role of nicotinamide adenine dinucleotide in the regulation of programmed cell death pathways. Biochem. Pharmacol. 101, 13–26. doi: 10.1016/j.bcp.2015.08.110
Rolfe, H. M. (2014). A review of nicotinamide: treatment of skin diseases and potential side effects. J. Cosmet. Dermatol. 13, 324–328. doi: 10.1111/jocd.12119
Sakurai, A., Maruyama, F., Funao, J., Nozawa, T., Aikawa, C., Okahashi, N., et al. (2010). Specific behavior of intracellular Streptococcus pyogenes that has undergone autophagic degradation is associated with bacterial streptolysin O and host small G proteins Rab5 and Rab7. J. Biol. Chem. 285, 22666–22675. doi: 10.1074/jbc.M109.100131
Schille, S., Crauwels, P., Bohn, R., Bagola, K., Walther, P., and van Zandbergen, G. (2018). LC3-associated phagocytosis in microbial pathogenesis. Int. J. Med. Microbiol. 308, 228–236. doi: 10.1016/j.ijmm.2017.10.014
Schmidt, S. V., and Schultze, J. L. (2014). New insights into IDO biology in bacterial and viral infections. Front. Immunol. 5:384. doi: 10.3389/fimmu.2014.00384
Sharma, O., O’Seaghdha, M., Velarde, J. J., and Wessels, M. R. (2016). NAD+-glycohydrolase promotes intracellular survival of group A Streptococcus. PLoS Pathog. 12:e1005468. doi: 10.1371/journal.ppat.1005468
Shen, C., Dou, X., Ma, Y., Ma, W., Li, S., and Song, Z. (2017). Nicotinamide protects hepatocytes against palmitate-induced lipotoxicity via SIRT1-dependent autophagy induction. Nutr. Res. 40, 40–47. doi: 10.1016/j.nutres.2017.03.005
Singhal, A., and Cheng, C. Y. (2019). Host NAD+ metabolism and infections: therapeutic implications. Int. Immunol. 31, 59–67. doi: 10.1093/intimm/dxy068
Sun, J., Siroy, A., Lokareddy, R. K., Speer, A., Doornbos, K. S., Cingolani, G., et al. (2015). The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 22, 672–678. doi: 10.1038/nsmb.3064
Turner, C. E., Abbott, J., Lamagni, T., Holden, M. T., David, S., Jones, M. D., et al. (2015). Emergence of a new highly successful acapsular group A Streptococcus clade of genotype emm89 in the United Kingdom. mBio 6:e00622. doi: 10.1128/mBio.00622-15
Upadhyay, S., and Philips, J. A. (2019). LC3-associated phagocytosis: host defense and microbial response. Curr. Opin. Immunol. 60, 81–90. doi: 10.1016/j.coi.2019.04.012
Valderrama, J. A., and Nizet, V. (2018). Group A Streptococcus encounters with host macrophages. Future Microbiol. 13, 119–134. doi: 10.2217/fmb-2017-0142
Velarde, J. J., O’Seaghdha, M., Baddal, B., Bastiat-Sempe, B., and Wessels, M. R. (2017). Binding of NAD+-glycohydrolase to streptolysin O stabilizes both toxins and promotes virulence of group A Streptococcus. mBio 8:e1382-17. doi: 10.1128/mBio.01382-17
Walker, M. J., Barnett, T. C., McArthur, J. D., Cole, J. N., Gillen, C. M., Henningham, A., et al. (2014). Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin. Microbiol. Rev. 27, 264–301. doi: 10.1128/CMR.00101-13
Yang, H., Hu, J., Chen, Y. J., and Ge, B. (2019). Role of Sirt1 in innate immune mechanisms against Mycobacterium tuberculosis via the inhibition of TAK1 activation. Arch. Biochem. Biophys. 667, 49–58. doi: 10.1016/j.abb.2019.04.006
Zhang, D. X., Zhang, J. P., Hu, J. Y., and Huang, Y. S. (2016). The potential regulatory roles of NAD+ and its metabolism in autophagy. Metabolism 65, 454–462. doi: 10.1016/j.metabol.2015.11.010
Zheng, P. X., Chung, K. T., Chiang-Ni, C., Wang, S. Y., Tsai, P. J., Chuang, W. J., et al. (2013). Complete genome sequence of emm1 Streptococcus pyogenes A20, a strain with an intact two-component system, CovRS, isolated from a patient with necrotizing fasciitis. Genome Announc. 1:e149-12. doi: 10.1128/genomeA.00149-12
Zhu, L., Olsen, R. J., Nasser, W., Beres, S. B., Vuopio, J., Kristinsson, K. G., et al. (2015). A molecular trigger for intercontinental epidemics of group A Streptococcus. J. Clin. Invest. 125, 3545–3559. doi: 10.1172/JCI82478
Keywords: Group A streptococcus (GAS), nicotinamide (NAM), NAD+ homeostasis, intracellular survival, endothelial cells (ECs)
Citation: Hsieh C-L, Hsieh S-Y, Huang H-M, Lu S-L, Omori H, Zheng P-X, Ho Y-N, Cheng Y-L, Lin Y-S, Chiang-Ni C, Tsai P-J, Wang S-Y, Liu C-C, Noda T and Wu J-J (2020) Nicotinamide Increases Intracellular NAD+ Content to Enhance Autophagy-Mediated Group A Streptococcal Clearance in Endothelial Cells. Front. Microbiol. 11:117. doi: 10.3389/fmicb.2020.00117
Received: 10 October 2019; Accepted: 20 January 2020;
Published: 11 February 2020.
Edited by:
Diego Robledo, The University of Edinburgh, United KingdomReviewed by:
Blanca Estela Garcia-Perez, National Polytechnic Institute (Mexico), MexicoCopyright © 2020 Hsieh, Hsieh, Huang, Lu, Omori, Zheng, Ho, Cheng, Lin, Chiang-Ni, Tsai, Wang, Liu, Noda and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiunn-Jong Wu, amp3dTEwMTlAeW0uZWR1LnR3
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.