Corrigendum: sraX: A Novel Comprehensive Resistome Analysis Tool
 Leonardo G. Panunzi
Leonardo G. Panunzi- 1Institut Pasteur, Biodiversity and Epidemiology of Bacterial Pathogens, Paris, France
- 2Institut Français de Bioinformatique, CNRS UMS 3601, Evry, France
The accurate identification of the assortment of antibiotic resistance genes within a collection of genomes enables the discernment of intricate antimicrobial resistance (AMR) patterns while depicting the diversity of resistome profiles of the analyzed samples. The availability of large amount of sequence data, owing to the advancement of novel sequencing technologies, have conceded exciting possibilities for developing suitable AMR exploration tools. However, the level of complexity of bioinformatic analyses has raised as well, since the achievement of desired results involves executing several challenging steps. Here, sraX is proposed as a fully automated analytical pipeline for performing a precise resistome analysis. Our nominated tool is capable of scrutinizing hundreds of bacterial genomes in-parallel for detecting and annotating putative resistant determinants. Particularly, sraX presents unique features: genomic context analysis, validation of known mutations conferring resistance, illustration of drug classes and type of mutated loci proportions and integration of results into a single hyperlinked navigable HTML-formatted file. Furthermore, sraX also exhibits relevant operational features since the complete analysis is accomplished by executing a single-command step. The capacity and efficacy of sraX was demonstrated by re-analyzing 197 strains belonging to Enterococcus spp., from which we confirmed 99.15% of all detection events that were reported in the original study. sraX can be downloaded from https://github.com/lgpdevtools/srax.
1. Introduction
Antimicrobial resistance (AMR) constitutes a serious menace to global public health, since its rise is being detected in samples from a wide variety of environmental sources (Munk et al., 2018). In addition, a growing number of imputable deaths per year is evidenced and is calculated to surpass the 10 million by 2050 (O'Neill, 2016). Under these circumstances, the accompanying development of novel sequencing technologies—along with continually decreasing costs—have raised the amount of available sequence data, and consequently, have led to devise viable AMR exploration tools. Particularly, whole-genome sequencing (WGS) and whole-metagenome sequencing (WMS) approaches have demonstrated enormous capabilities of epidemiological surveillance, outbreak detection, and infection control of bacterial pathogens (Didelot et al., 2012). In relation to the type of demanded sequence data, two main methodological approaches have materialized: those capable of processing raw reads—read-based methods—and those requiring contig-assembled genome sequences—assembly-based methods. Ultimately, both procedures exhaustively examine for AMR determinants by aligning the input sequence data to curated antibiotic resistance genes (ARG) from custom or public dedicated reference AMR databases (DB). Essentially, read-based methods are faster and less computationally demanding. However, false positives originated from spurious mapping might arise. Moreover, since the genomic context is generally missed, the arrangement of adjacent genes can not be evinced and it constitutes a major drawback (Boolchandani et al., 2019). Quite contrary, assembly-based methods are computationally expensive and time consuming because of the de novo assembly step. Nonetheless, when sequencing at adequate genome coverage, known or novel ARGs bearing low sequence similarity with AMR DBs are normally detected, and remarkably, genomic context, and regulatory sequence elements are captured (Boolchandani et al., 2019).
In consequence, applying any of previously indicated techniques, several bioinformatic tools tailored to annotate ARGs have been produced (see Table 1). For instance, considering read-based methods, SRST2 (Inouye et al., 2014) and KmerResistance (Clausen et al., 2016) are not confined to any specific microbial species or AMR type, but both approaches completely neglect resistance conferred by single–nucleotide polymorphisms (SNPs). By contrast, Mykrobe predictor (Bradley et al., 2015) is suitable for detecting sequence variants but, regrettably, is restricted to Staphylococcus aureus and Mycobacterium tuberculosis and to 12 types of antibiotics. On the contrary, ARIBA (Hunt et al., 2017) can identify not merely ARGs corresponding to AMR from any type but SNPs linked to resistance as well. However, some visualization files (like gene/SNP presence) must be obtained via the Phandango server (Hadfield et al., 2017). Additionally, before mapping the reads, ARIBA performs a clustering procedure for finding a single sequence representative from each ARG locus on the employed reference AMR DBs. Albeit reducing ambiguous alignments, this strategy unaccount for substantial sequence variation existing within gene families (Munk et al., 2017). To circumvent this lack of accuracy, a novel method called GROOT (Rowe and Winn, 2018) has been proposed and it relies on building variation graphs of previously clustered ARGs from AMR DBs, before mapping the reads to them. Despite succeeding at characterizing variation through sequence graphs, GROOT is mainly limited to properly perform the annotation of ARGs on metagenome samples, without any graphical output or further analysis. Other read-based tools include: SEAR (Rowe et al., 2015)—already archived, SSTAR (de Man and Limbago, 2016), PATRIC (Antonopoulos et al., 2017)—not standalone mode, and DeepARG (Arango-Argoty et al., 2018). Regarding assembly-based tools, certain of them are capable of elucidating SNPs developing AMR but, to our knowledge, none of them is suited for providing a genomic context analysis of identified ARGs. In addition, the output information and its visualization is generally limited (see Table 1). Current implementations entirely relying on already assembled sequences include: ResFinder (Zankari et al., 2012), ARG-ANNOT (Gupta et al., 2014), RAST (Davis et al., 2016), RGI (Jia et al., 2016), PointFinder (Zankari et al., 2017), ARGs-OAP (Yin et al., 2018), and NCBI-AMRFinder (Feldgarden et al., 2019).
TABLE 1
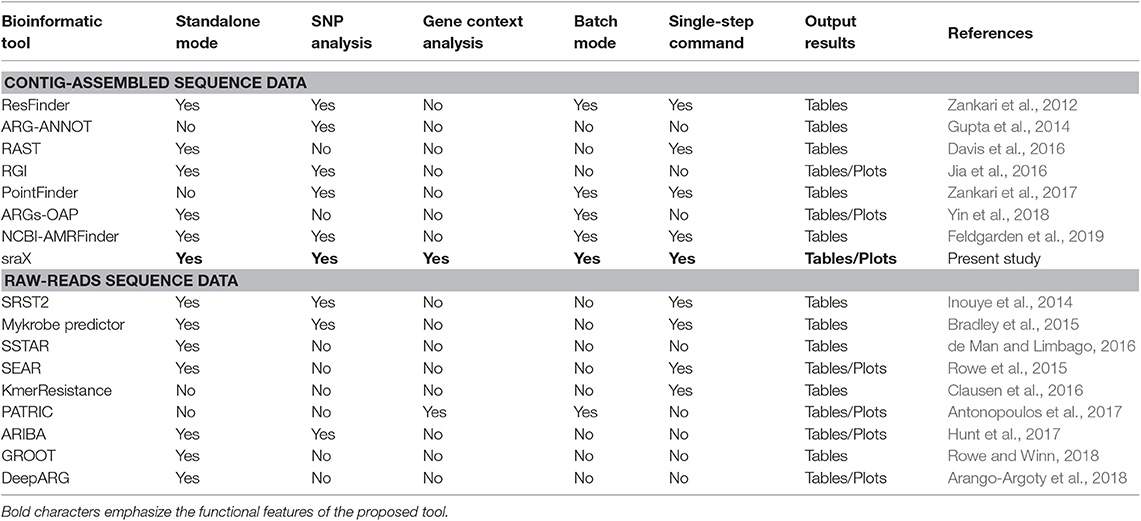
Table 1. Features of different resistome analysis pipelines, in comparison to sraX.
In this context, and attempting to address the mentioned limitations, a suit of federated modular functions was developed and integrated into a user-friendly tool named sraX. It has been devised as a fully automated pipeline for performing a systematic resistome profiling analysis through a series of operational steps, which are conveniently concatenated for achieving a greater computational efficiency (see Figure 1 for a schematic diagram). sraX follows this analytical workflow by executing a montage of custom Perl and R scripts that opportunely call external open source software and make use of reference AMR DBs. The main capabilities of sraX and a detailed comparison with other pipelines is shown in Table 1. Apart from the usual strategy of retrieving AMR data from public—or privately owned—repositories, compiling a local DB and detecting resistance determinants in analyzed samples, sraX has unique and noteworthy built-in features, like: gene context exploration, SNP analysis, complete graphical output—including drug classes and type of mutated loci—and integration of results into a fully navigable HTML report file. In addition, sraX is proposed as a single-command tool—envisaging that inexperienced users without any technical or bioinformatic knowledge would run it—and it has been devised for running on desktop computer systems, under limited RAM and processing resources. Wherefore, sraX operates as a standalone tool and a simple and straightforward deployment is achieved from source code—available at https://github.com/lgpdevtools/srax. In addition, easier instances have been produced in the form of a bioconda package—https://anaconda.org/lgpdevtools/srax—and a docker image—https://hub.docker.com/r/lgpdevtools/srax. The complete procedures for properly installing and using sraX are described in the User Manual, Supplementary Information.
FIGURE 1
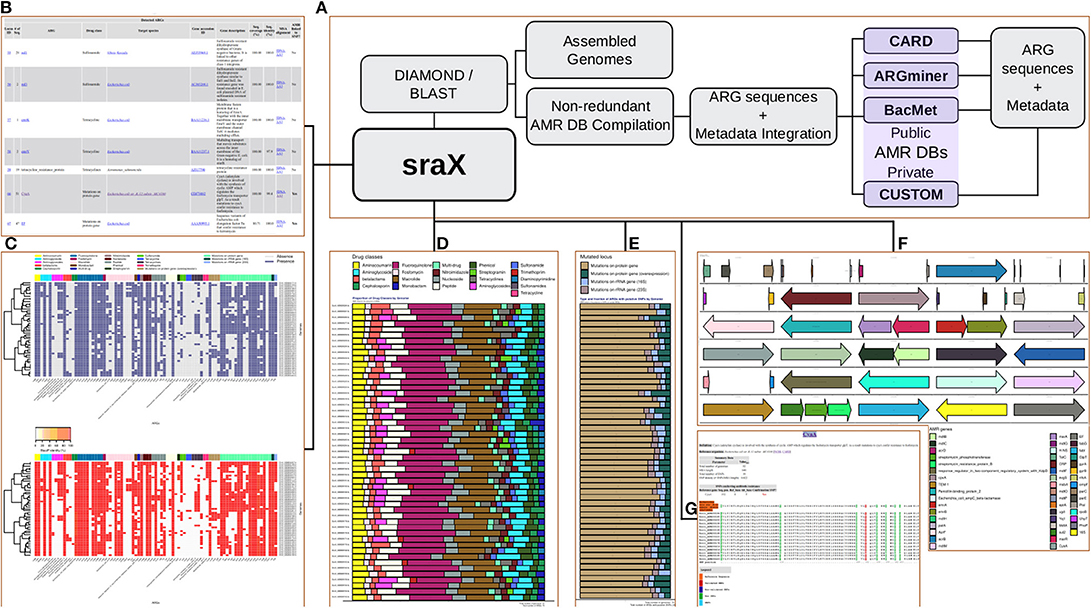
Figure 1. (A) sraX workflow. (B) ARG repertoire. (C) Heat-maps of gene presence and sequence identity. (D) Proportion of drug classes. (E) Type of mutated loci. (F) Spatial distribution of detected ARGs per genome (gene context analysis). (G) SNP validation and detection of putative new variants.
2. Materials and Methods
2.1. Third-Party Data and Software Requirements
2.1.1. Reference Databases
sraX depend upon a locally compiled AMR DB, which is obtained by gathering sequence data and extensive metadata from external reference AMR DBs. When launching the default resistome analysis, CARD v3.0.7 (Jia et al., 2016) constitutes the primary data source of genetic determinants. The reasoning behind our choice is that CARD not only provides regularly updated and curated sequence data, but additional complementary information that is specifically organized into ontology entries. Ultimately, sraX benefits from this level of organization for the straightforward access and retrieval of collected AMR data. Nevertheless, in order to conduct a more extensive and thorough ARG homology search, the ARGminer v1.1.1 (Argoty et al., 2019) and BacMet v2.0 (Pal et al., 2014) DBs—or even custom-provided ARGs—are allowed to be eventually incorporated into the sraX analysis. A noteworthy feature of ARGminer (Argoty et al., 2019) is that it aggregates AMR data from several dedicated repositories, including: ResFinder (Zankari et al., 2012), ARG-ANNOT (Gupta et al., 2014), CARD (Jia et al., 2016), MEGARes (Lakin et al., 2016), ARDB (Liu and Pop, 2008), SARG (Yang et al., 2016), NDARO (https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/), and DeepARG (Arango-Argoty et al., 2018). In consequence, a larger collection of resistance determinants is acquired by combining curated AMR data from CARD, ARGminer and BacMet that ensures a massive search through a wider space.
2.1.2. Software Dependencies
Perl v5.26.x and the following complementary Perl libraries are required for having operative sraX modules: LWP::Simple, Data::Dumper, JSON, File::Slurp, FindBin, and Cwd. For aligning the ARGs to the analyzed genomes, sraX makes use of DIAMOND dblastx v0.9.29 (Buchfink et al., 2015) and NCBI blastx/blastn v2.10.0 (Altschul et al., 1990). In addition, prior to validating known polymorphic positions conferring AMR, multiple-sequence alignment (MSA) files are created using MUSCLE v3.8.31 (Edgar, 2004a,b), MAFFT v7.450 (Katoh et al., 2002), or CLUSTAL Ω v1.2.4 (Sievers et al., 2011). The figures generated during sraX analysis are achieved using R v.3.6.1 and the additional following packages: ggplot2 (Wickham, 2016), dplyr (Wickham et al., 2019), and gridExtra (Auguie, 2017). Importantly, these same software versions are currently employed for producing the docker image file.
2.2. Systematic Resistome Analysis
sraX accomplishes a series of challenging tasks for completing the resistome profiling analysis (see Figure 1).
The main assignments are fully described as follows:
2.2.1. Creation of the Required Arrangement of Directories
Output results and temporary files are allocated within a defined configuration of specific folders and sub-folders, which is systematically produced at the beginning of sraX analysis (see Figure S1).
2.2.2. Acquisition of Data Sources for AMR DB Compilation
The core collection of reference ARG sequences and their associated metadata is automatically retrieved only from CARD (Jia et al., 2016) when sraX is executed under default parameters. Nevertheless, a more exhaustive search can be performed by selecting the proper option, for including the ARGminer (Argoty et al., 2019) and BacMet (Pal et al., 2014) DBs into the analysis. Moreover, alternative user-provided and curated ARG (nucleotide or protein) sequences in FASTA format can be added (see the User Manual, Supplementary Information). In order to create the corresponding hyperlinks in the final report file, the header should include the following arrayed metadata: gene name, NCBI Accession ID, gene description, type of evinced AMR (protein homolog, protein variant, protein over-expression or rRNA gene variant), drug class, and species name. The added metadata is certainly not mandatory for sraX to incorporate the user-provided ARG sequences, though it allows the pertinent interrelation of acquired results. Ultimately, after appending the user-provided ARGs some redundancy might arise, thus identical sequences are excluded before compiling the final AMR DB.
2.2.3. Detection of ARGs
A directory containing the assembled genome files in FASTA format is the only requirement for accomplishing the sraX analysis. Prokaryote and eukaryote WGS data at any assembly level (e.g., complete genome, chromosome, scaffolds, or contigs) can be utilized. The homologous genomic regions are detected by aligning the genome assemblies to previously compiled AMR DB. DIAMOND dblastx (Buchfink et al., 2015) is applied by default for making the procedure faster. However, for a higher detection accuracy, NCBI blastx (Altschul et al., 1990) can be chosen as well. Importantly, NCBI blastn (Altschul et al., 1990) is regularly used for aligning rRNA gene sequences and subsequently identifying the variants conferring resistance.
2.2.4. Analytical Processing
Genome files are examined in-parallel for optimizing speed processing—up to 100 files are simultaneously queried, while a series of concerted actions are performed simultaneously for accomplishing the systematic resistome analysis. These actions constitute the core of the AMR scanning procedure and includes:
1. Depletion of redundant BLAST hits and compilation of the final ARG inventory. Concurrent matching ARGs—constituting variants of large gene families—are eliminated according to their sequence identity and coverage. Only those hits—or gene variants—revealing the highest values of both parameters are kept.
2. Detection of putative paralog copies of identified ARGs. The selected gene variants—from the previously compiled list of ARGs—which are detected in non-overlapping genome regions, are presumed paralog copies and cataloged on a separate file for further analyses.
3. Production of heatmaps ellucidating the gene presence and its sequence identity with respect to the reference. The devoted modularized function employs binary data (0 = absence, 1 = presence) and sequence similarity (0–100%) values for generating the corresponding heatmaps.
4. Calculation of drug classes and type of mutated locus proportions, for subsequent production of interpretive stacked barplots.
5. Extensive gene context exploration and production of resulting elucidative arrowplots, on every single genome (see Figure S3).
6. Clustering of homologous genomic sequences and their consecutive alignment by running MUSCLE (Edgar, 2004a,b), MAFFT (Katoh et al., 2002), or Clustal Ω (Sievers et al., 2011) on each ARG.
7. Detection of sequence variants on previously obtained MSA files, for the subsequent validation of known SNPs conferring AMR.
8. Creation of the readily navigable HTML-formatted final report, by integrating the corresponding text and plot files.
2.3. Genome Collection: Phenotyping and Genotyping Assays
The genome sequence files from 100 Enterococcus faecium and 97 Enterococcus faecalis strains that were previously obtained (Tyson et al., 2018a,b), and whose AMR profiles were further characterized employing a panel of 9 antibiotic drugs (Tyson et al., 2018b), were downloaded from the NCBI repository (Accession: PRJNA292665 and PRJNA292669) in July 2019, and alternatively, have been made available from a dedicated Zenodo repository (Panunzi, 2019). The main biological and genomic features, including their genome, assembly and BioSample Accession IDs, sequence coverage, N50 values as well as isolation sources and antibiotic drug susceptibility characterization can be found in Table S1.
The authors (Tyson et al., 2018b) appraised the antimicrobial susceptibilities of the selected strains by comparing the minimum inhibitory concentration (MIC) values, which were determined using an automated broth microdilution method (Trek Diagnostics, Independence, OH) with the US National Antimicrobial Resistance Monitoring System (NARMS) CMV3AGPF plates, to the breakpoints defined in the 2015 Clinical and Laboratory Standards Institute (CLSI) guidelines (CLSI, 2015). However, since the analyzed dataset is mainly composed of bacterial isolates derived from animals, the zone diameter interpretations should be made using veterinary standards. Regrettably, this relevant information was not provided by Tyson et al. (2018b). Therefore, in our present study we specify that these reported susceptibilities were employed only for validation purposes. Afterwards, the contrast between the measured and the MIC breakpoint values allowed the determination of the “resistant” and “susceptible” phenotypes. With reference to the ARG determination at genomic level, Tyson et al. (2018b) employed the ResFinder (Zankari et al., 2012) and the NCBI Pathogen Detection (https://www.ncbi.nlm.nih.gov/pathogens/) DBs. Thereafter, Tyson et al. (2018b) added two further categories—“resistant*” and “susceptible*”—when differences between genotype (presence or absence of corresponding ARG) and phenotype (resistant or susceptible) occurred. The “resistant*” label indicated a resistant phenotype but the absence of a verified ARG, while the “susceptible*” label indicated the opposite: a susceptible phenotype but the presence of a verified ARG. Nevertheless, in our study we have replaced this latter category for “silenced resistance.”
2.4. Genome Annotation, Pan-Genome Estimation, and Phylogeny Assessment
The automated annotation of genomes from both datasets (Accession: PRJNA292665 and PRJNA292669) was performed using PROKKA v1.11 (Seemann, 2014), while the core-genome and pan-genome were estimated using CD-HIT (Fu et al., 2012) by clustering predicted genes when ortholog loci shared ≥ 95% of amino acid sequence identity and ≥ 95% of alignment coverage. A total of 424 genes existing in all strains were estimated to compose the core set. Afterwards, those 424 genes were aligned using MUSCLE (Edgar, 2004a,b) and concatenated into a final MSA file comprising 327,376 bps of DNA sequence. A phylogenetic tree was built from this core-genome MSA after filtering recombination regions using GUBBINS (Croucher et al., 2014) under default parameters. Visualization of the tree and AMR phenotypes was conducted using the Interactive Tree Of Life (iTOL) v4 online tool (Letunic and Bork, 2019).
3. Results
3.1. Phylogeny and Distribution of AMR Phenotypes
A total of 100 E. faecium and 97 E. faecalis isolates constituted our validation set. This collection of genomes had been sequenced and phenotyped–phenotyping is described in Methods, while genome samples accession codes and phenotyping data are included in Table S1—in previous studies (Tyson et al., 2018a,b). However, the authors did not include a phylogeny of the analyzed samples.
In our present study, the annotation of the 197 genome assemblies produced a sum of 531,915 putative protein-coding sequences, that were subsequently grouped into 13,702 clusters of orthologous genes (COGs). The pan-genome calculation evinced that 424 of these COGs were present in all the samples and constituted the core-genome. The successive alignment and catenation of core-genome COGs procured a MSA composed of 327,376 nucleotides that was later employed in the phylogeny reconstruction (illustrated on the uppermost of Figure 2).
FIGURE 2

Figure 2. AMR activities of the collection of 197 Enterococcus spp. strains. A phylogenetic tree is shown on top of the heatmap. Red and light-green colors indicate resistance and susceptibility to corresponding antibiotics. Dark-red color indicates an exhibited resistance phenotype in the absence of an in silico detected ARG, while a dark-green color indicates an exhibited susceptible phenotype in the presence of an in silico detected ARG.
The phylogeny showed a large evolutionary distance between both species—faecium and faecalis—that produced, as awaited, a complete break inside the tree. Briefly, in terms of AMR distribution, the original study (Tyson et al., 2018b) found that 46 isolates were pan-susceptible, with 90 isolates resistant to drugs in at least three antimicrobial classes (Table S1). We further compared the number of resistant isolates between both species according to the antimicrobial classes, and found important differences in: aminoglycosides (E. faecalis: 50, E. faecium: 38), β-lactams (E. faecalis: 6, E. faecium: 23), fluoroquinolones (E. faecalis: 1, E. faecium: 0), lipopeptides (E. faecalis: 2, E. faecium: 12), macrolides (E. faecalis: 55, E. faecium: 34), nitrofuran (E. faecalis: 1, E. faecium: 41), phenicol (E. faecalis: 20, E. faecium: 2), and streptogramin (E. faecalis: 0, E. faecium: 30). On the contrary, tetracyclines (E. faecalis: 69, E. faecium: 64), glycopeptides (all susceptibles) and oxazolidinones (all susceptibles) indicated somewhat similar AMR profiles.
3.2. Efficiency of sraX in Resolving AMR Determinants: Concordance With Phenotypes
The automated in silico resistome analysis using sraX was performed for establishing the one-to-one correspondence between the phenotyping and genotyping data (see Figure 2).
As previously mentioned, all isolates were vancomycin and linezolid susceptibles, and consistent with this phenotype, neither related ARG nor 23S rRNA mutations were discovered. Regarding ciprofloxacin, only one E. faecalis isolate evinced the resistant phenotype. The sraX's SNP analysis detected the gyrA S84F mutation, in agreement with previous studies accounting for its association with the resistant phenotype (El Amin et al., 1999) (see the corresponding MSA file inside the “SNPs_conferring_AMR” folder, Supplementary Information).
Considering chloramphenicol resistance, phenotyping data revealed a remarkable species restraint that made E. faecalis almost exclusive. The chloramphenicol acetyltransferase genes cat, cat(pC221), and cat(pC194) were responsible for the exhibited resistance, although 2 isolates had the genes but were phenotyped as susceptibles (see Table 2).
TABLE 2
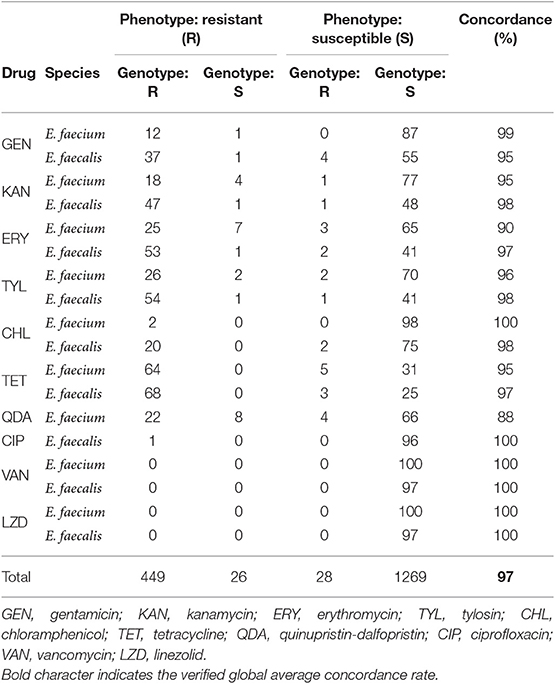
Table 2. Genotype–phenotype concordance of sraX analysis for selected E. faecalis and E. faecium strains.
In respect to erythromycin and tylosin resistance, it was revealed that both phenotypes consistently co-occurred: 80 tylosin-resistant isolates out of 86 erythromycin-resistant isolates were the same samples. Among these isolates, 81 harbored the erm(B) gene, while 3 of them the erm(A) gene. In addition, the msr(C) gene was also present in macrolide-resistant isolates.
Regarding streptomycin resistance, many strains were phenotyped as susceptible but harbored the aadE and str genes. In the original study the authors mentioned to have employed the same resistance cutoff for both species, a decision that probably was the source of the experimental error (Tyson et al., 2018b). For this reason, we had not further considered the streptomycin phenotyping data in our present study.
Concerning kanamycin and gentamicin resistance, the aac(6')-aph(2”), aph(2”)-Id, aph(3')-III, and aadD genes were responsible for the resulting phenotype in 96.75% of resistant isolates. Remarkably, the co-occurrence of the aac(6')-aph(2”), aph(3')-III, and erm(B) genes was revealed, indicating their putative transmission on a mobile element.
In view of tetracycline resistance, the tet(L), tet(M), tet(S), and tet(O) genes were detected in 96% of 133 resistant strains. On the contrary, eight susceptible isolates held a copy of tet(L) or tet(M) genes. In respect to streptogramin resistance, E. faecalis is considered constitutionally resistant, while the vat(D) and/or vat(E) genes were found in E. faecium in 22 out of 30 resistant strains and in 4 out of 70 susceptible isolates, what produced a 88% of concordance.
Considering penicillin resistance, previous studies have shown that mutations on the pbp4 (Duez et al., 2001) and pbp5 (Zorzi et al., 1996; Rice et al., 2004) genes accounted for the observed resistant phenotypes in E. faecalis and E. faecium, respectively. In accordance with these results, our SNP analysis (see the corresponding MSA file, Supplementary Information) verified the occurrence of the T500I and E630V mutations in 17 out of 21 resistant E. faecium strains. Additionally, the co-occurrence of the serine or aspartate codon insertion at position 467 was detected in 4 resistant isolates harboring the E630V mutation. The event of concurrent mutations was detected likewise in the M486A and P668S mutations, which were found in 3 and 6 resistant isolates harboring the E630V mutation, respectively. However, the T500I and P668S mutations were also found in several susceptible isolates and 2 resistant E. faecium strains neither evidenced any recognized mutations on the pbp5 gene nor revealed alternative genetic determinants, like β-lactamases enzymes. In view of recognized pbp4 mutations, our SNP analysis has validated the D573E amino-acid change that has been recently reported (Conceição et al., 2014; Infante et al., 2016) in 11 E. faecalis strains (see the corresponding MSA file, Supplementary Information). Furthermore, we detected an alternative mutation (I50T) in the same sequence position where a validated mutation (I50Y) was formerly reported (Ono et al., 2005). Nevertheless, none of the E. faecalis strains were phenotyped as penicillin-resistant in the original study (Tyson et al., 2018b). For this reason, and because of the apparent inconsistencies in the pbp5 phenotypes, we did not include the penicillin in the concordance estimation study.
Regarding daptomycin resistance, some mutations occurring on the lia(F), lia(S), lia(R), YybT, gsh(F), gdp(D), and cls genes were previously reported (Arias et al., 2011). However, a similar situation was found: our SNP analysis was capable of confirming the E192G mutation on the lia(S) gene in 5 E. faecium strains (see the corresponding MSA file, Supplementary Information), but none of these samples were phenotyped as resistant in the original study (Tyson et al., 2018b). Still, the putative existence of not yet recognized alternative mutations or unknown mechanisms is a possibility, since several other strains were certainly phenotyped as resistant.
Finally, tigecycline and nitrofurantoin susceptibilities were assayed as well. With regard to tigecycline resistance, only 8 resistant strains were detected and all of them possessed the tet(L) and tet(M) genes. A previous study linked the upregulation of these tetracycline resistance genes with the only known resistance mechanism (Fiedler et al., 2015). However, regarding nitrofurantoin resistance, the discovery of genomic AMR determinants is impracticable since any clear mechanism has been established so far.
In addition, the in silico analysis allowed to identify further ARGs which confer resistance to trimethoprim, lincomycin, spectinomycin, biocides and metals, and some other antimicrobials. However, none of them are currently employed for treating Enterococcus spp. infections. Probably, these ARGs are involved in conferring resistance to susceptible organisms via their horizontal transference. Notably, trimethoprim resistance genes dfr(D), dfr(E), and dfr(G) were only detected in E. faecalis strains, as well as the efr(A) and efr(B) genes conferring resistance to multiple drugs and the macrolide resistance gene mph(D). Additionally, the bacitracin resistance gene bac(A) and the streptogramin resistance gene lsa(A), along with the efp(A), eme(A) and major facilitator superfamily transporter genes conferring resistance to multiple drugs, were only found in E. faecalis strains likewise. Conversely, the macrolide resistance genes mph(B) and msr(C) were only found in E. faecium strains, as well as the aac(6')-li and spc genes conferring resistance to aminoglycosides. In addition, the ade(C) and efm(A) genes conferring resistance to multiple drugs were also detected only in E. faecium strains. In contrast, the lsa(E) and lnu(B) genes conferring resistance to streptogramin and lincosamide, respectively, were observed in both E. faecalis and E. faecium strains. Lastly, the bcr(A), bcr(B), and bcr(C) biocide genes, which are involved in benzalkonium chloride—a quaternary ammonium compound—resistance (Dutta et al., 2013), were detected in both E. faecalis and E. faecium strains as well as the tcr(A), tcr(B), tcr(Y), and tcr(Z) copper resistance genes. Additionally, the cht(R), cht(S), cop(B), and cop(Y) copper resistance genes were only detected in E. faecium strains, while the mer(A) mercury resistance gene was only found in E. faecalis strains, respectively. For a thorough and comparative analysis of in silico detected ARGs (see Figure 2, Figure S2, and Table S1).
3.3. Confirmation Rate of Detected AMR Determinants
Our previous results have proven a close correspondence between the in silico AMR genotype predictions and the experimentally assessed phenotypes. These findings were in accordance with the original study (Tyson et al., 2018b). Following the fidelity assessment of sraX, we next calculated the proportion of matching ARGs in corresponding genomes (see Table 3) and contrasted with the original values (Tyson et al., 2018b). A total of 25 ARGs conferring resistance to nine antimicrobial classes were distinguished across the samples. Among identified ARGs, 18 of them had a direct connection with phenotyping assays, while the remaining were related to drugs not usually employed for treating Enterococcus spp. infections. Comparing to original findings (Tyson et al., 2018b), we globally verified 425 out of 436 detection events that were reported. Our results implied almost an exact correspondence with the original study (validation rate: 99.15%), suggesting a considerable accuracy of our proposed tool.
TABLE 3
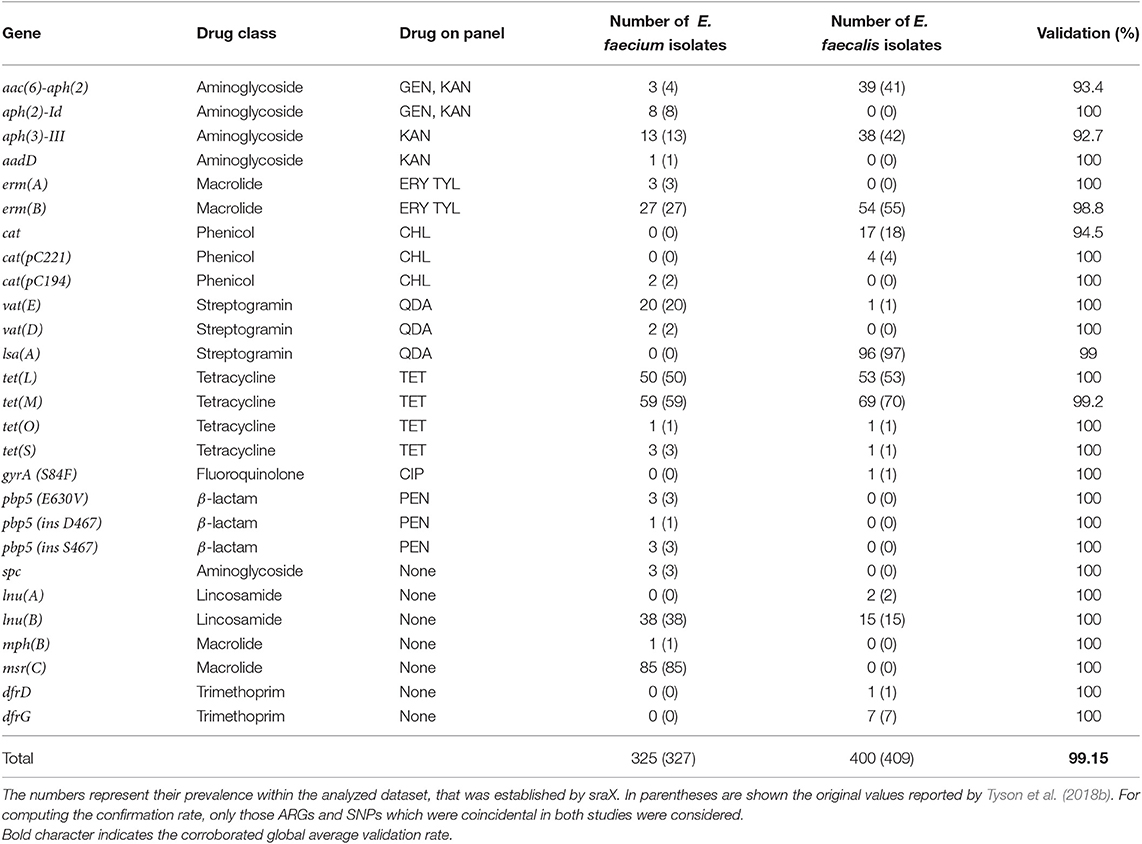
Table 3. List of detected ARGs in isolates.
4. Discussion
Several studies have proven the significant advantages of WGS approaches over traditional methods for the molecular disease characterization and further epidemiological surveillance of bacterial pathogens. Nevertheless, major technical difficulties are encountered not only with implementing WGS in clinical and reference laboratories, but when analysing the obtained data as well. The amount and complexity of data impose the requirement of specific bioinformatics skills. However, these tough challenges are largely overcome in the laboratories by employing automated and user-friendly bioinformatics pipelines. In addition, the application of standardized analytical methods allows the reproducibility of results that might be subsequently shared among laboratories.
Aiming at fulfilling these technical needs, in the current study we present sraX as a novel bioinformatic tool for detecting, characterizing, plotting and producing a final comprehensive report about the presence of AMR determinants on assembled genomes. It constitutes a comprehensive, automated, and efficient standalone tool for performing diverse complex analyses and producing graphical results embedded in a HTML-based summary. The simplicity of its computational operation allow users to easily download, install, configure and finally complete several difficult assignments, by orchestrating diverse algorithms through a primary single-step command. It can properly be deployed from source code, by running an assistance bash script, or even simpler, as a docker image. Regarding its functionalities, despite apparent similarities with previously developed tools, sraX offers several unique advantages over them as it supplies non-redundant ARG annotations, SNP analysis through multiple-sequence alignment (MSA) files and graphic representation of discovered ARGs illustrating their existence and corresponding sequence identity percentage on each genome, their subsequent genomic context analysis and the global proportions of drug classes and type of mutated locus.
Concerning the performance of our tool, we have evaluated the walltime and memory required to achieve the resistome analysis. Both metrics were obtained after processing with sraX increasing amount of data, that was acquired by randomly sampling intervals of 100 up to 1,000 genome sequence files, in a five replicates bootstrapped operation (see Figure 3). These sequence files were selected from a set composed of 5,000 Klebsiella pneumoniae distinct genomes. We have chosen this organism since its genome size (≈5.5 Mbp) is typical from a Gram-negative bacteria. Our results indicated a linear scale of running time (≈12.5 min) and memory consumption (≈250 MB) with the processing of every 100-genome interval (Figures 3A,B, respectively). Defaults parameters were employed to ensure consistency during the testing. Each test was independently performed on a desktop computer IntelⓇ XeonⓇ W-2104 processor (4-core, 3.20 GHz) with 32 GB RAM. The fact of employing a simple desktop computer and completing the resistome analysis quite rapidly, envisages a greater performance when running sraX under more powerful computational resources.
FIGURE 3
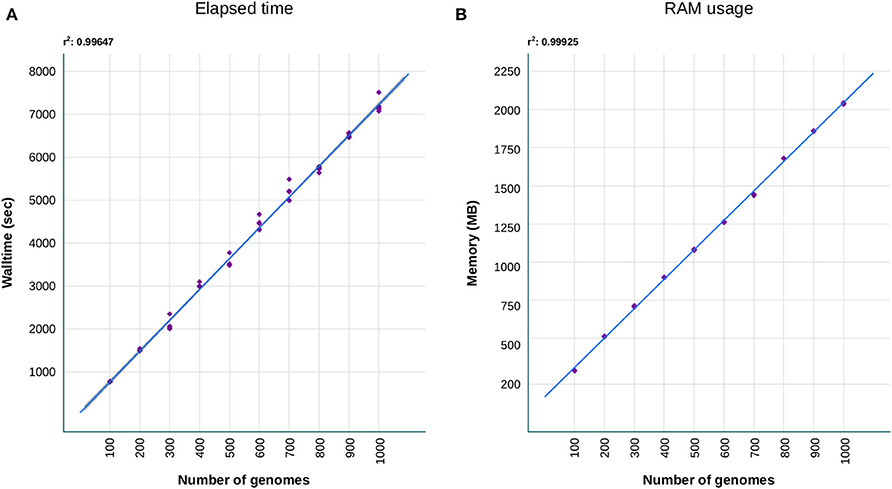
Figure 3. Benchmark comparisons of (A) walltime and (B) RAM consuming that were required by sraX to complete the resistome analysis. A collection of 5,000 Klebsiella pneumoniae genomes (average size: 5.5 Mbp) were randomly sampled five times at increasing 100-genome intervals and run on a desktop computer (4 cores, 32GB RAM). The processing of additional genomes evidenced a linear scaling of sraX performance, based on running time and RAM usage.
Afterwards, in order to assess the functionality and verify the accuracy of our proposed tool, we selected a genome dataset belonging to Enterococcus spp. for which AMR has been previously analyzed and genome sequences were available (Tyson et al., 2018b). When examining the genotype-phenotype correspondence for each assayed antimicrobial drug, sraX achieved an overall 97% of success and completely matched original results (Tyson et al., 2018b). Regarding the overall accuracies, our implementation was able to confirm a global 99.15% of all detection events—involving AMR determinants and genomes—reported in the original study (Tyson et al., 2018b). In addition, sraX confirmed the existence of distinct AMR profiles according to the involved species. For example, phenotyping and genotyping data evinced that phenicol resistance was almost exclusively determined in E. faecalis strains, while streptogramin resistance was only detected in E. faecium strains. However, in both species, tetracycline resistance genes were extremely frequent. Regarding not assayed drugs, the in silico AMR profiling analysis showed that trimethoprim resistance genes dfrD and dfrE were detected only in E. faecium, while macrolide and streptogramin resistance genes mph(B) and msr(C) were only found in E. faecalis. Globally, except for a few discrepancies, the results were entirely compatible with published findings (Tyson et al., 2018b). On that account, sraX has demonstrated a proven capability for discovering AMR determinants and has supported the utility of WGS applications in clinical microbiology. In addition, sraX was able to validate known SNPs conferring resistance.
Considering the drawbacks, sraX is wholly dependent on assembled genomic contigs that must be provided. Since the de novo assembly is a time consuming and computationally expensive procedure which, in addition, its accuracy is overwhelmingly influenced by the nature of the genome, we have finally decided to omit this step and give priority to AMR detection facets. In general, the tools that exploit read mapping-based methods or generate de novo or reference-based assemblies are targeted for metagenomic samples. The main problem with these samples is their high microbial diversity and unbalanced abundance that clearly limit the AMR profiling analysis.
5. Conclusions
sraX facilitates the resistome analysis of all-levels assembled genomes through a series of automated procedures. In the end, the obtained results are easily visualized as fully navigable HTML-formatted files containing summarized data and embedded plots. Challenging analysis, such as the SNP validation or gene context examination, can be performed in a one-step systematic and user-friendly manner. Additional tough assignments on ARG multiple-hits depletion and complex graphical representations are effectively completed as well. Lastly, sraX is capable of analyzing a variety of curated data-sets of properly formatted ARG sequences.
Data Availability Statement
The datasets generated for this study can be found in a purposeful Zenodo repository, available at: http://doi.org/10.5281/zenodo.3571224.
Author Contributions
LP conceived and designed the tool, created the software, performed all the necessary testing, and wrote the manuscript.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is indebted to the Institut Français de Bioinformatique and the CONECT-AML (COllaborative NEtwork on research for Children and Teenagers with Acute Myeloid Leukemia) consortium for their support. In addition, the author acknowledges Mr. Mario R. Panunzi and MS. Brenda N. Henriquez for their invaluable help and inspiration, and specially dedicates this paper to them.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00052/full#supplementary-material
Abbreviations
AMR, antimicrobial resistance; ARG, antibiotic resistance gene; DB, data base.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Antonopoulos, D. A., Assaf, R., Aziz, R. K., Brettin, T., Bun, C., Conrad, N., et al. (2017). Patric as a unique resource for studying antimicrobial resistance. Brief. Bioinform. 20, 1094–1102. doi: 10.1093/bib/bbx083
Arango-Argoty, G., Garner, E., Pruden, A., Heath, L. S., Vikesland, P., and Zhang, L. (2018). Deeparg: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Metagenomics 6:23. doi: 10.1186/s40168-018-0401-z
Arango-Argoty, G. A., Guron, G. K. P., Garner, E., Riquelme, M., Heath, L. S., Pruden, A., et al. (2019). Argminer: a web platform for crowdsourcing-based curation of antibiotic resistance genes. bioRxiv. doi: 10.1101/274282
Arias, C. A., Panesso, D., McGrath, D. M., Qin, X., Mojica, M. F., Miller, C., et al. (2011). Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med. 365, 892–900. doi: 10.1056/NEJMoa1011138
Auguie, B., and Antonov, A. (2017). gridExtra: Miscellaneous Functions for Grid Graphics. R Package Version 2.3. Available online at: https://CRAN.R-project.org/package=gridExtra
Boolchandani, M., D'Souza, A. W., and Dantas, G. (2019). Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 20, 356–370. doi: 10.1038/s41576-019-0108-4
Bradley, P., Gordon, N. C., Walker, T. M., Dunn, L., Heys, S., Huang, B., et al. (2015). Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 6:10063. doi: 10.1038/ncomms10063
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using diamond. Nat. Methods 12:59. doi: 10.1038/nmeth.3176
Clausen, P. T. L. C., Lund, O., Zankari, E., and Aarestrup, F. M. (2016). Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother. 71, 2484–2488. doi: 10.1093/jac/dkw184
CLSI (2015). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard–Tenth Edition Wayne, PA: Clinical and Laboratory Standards Institute.
Conceição, N., da Silva, L. E. P., da Costa Darini, A. L., Pitondo-Silva, A., and de Oliveira, A. G. (2014). Penicillin-resistant, ampicillin-susceptible Enterococcus faecalis of hospital origin: pbp4 gene polymorphism and genetic diversity. Infect. Genet. Evol. 28, 289–295. doi: 10.1016/j.meegid.2014.10.018
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2014). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Davis, J. J., Boisvert, S., Brettin, T., Kenyon, R. W., Mao, C., Olson, R., et al. (2016). Antimicrobial resistance prediction in patric and rast. Sci. Rep. 6:27930. doi: 10.1038/srep27930
de Man, T. J. B., and Limbago, B. M. (2016). Sstar, a stand-alone easy-to-use antimicrobial resistance gene predictor. mSphere 1:e00050-15. doi: 10.1128/mSphere.00050-15
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E., and Crook, D. W. (2012). Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13, 601–612. doi: 10.1038/nrg3226
Duez, C., Zorzi, W., Sapunaric, F., Amoroso, A., Thamm, I., and Coyette, J. (2001). The penicillin resistance of Enterococcus faecalis JH2-2R results from an overproduction of the low-affinity penicillin-binding protein pbp4 and does not involve a PSR-like gene. Microbiology 147, 2561–2569. doi: 10.1099/00221287-147-9-2561
Dutta, V., Elhanafi, D., and Kathariou, S. (2013). Conservation and distribution of the benzalkonium chloride resistance cassette bcrabc in listeria monocytogenes. Appl. Environ. Microbiol. 79, 6067–6074. doi: 10.1128/AEM.01751-13
Edgar, R. C. (2004a). Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Edgar, R. C. (2004b). Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
El Amin, N., Jalal, S., and Wretlind, B. (1999). Alterations in GYRA and PARC associated with fluoroquinolone resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 43, 947–949. doi: 10.1128/AAC.43.4.947
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019). Validating the AMRfinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63:e00483–19. doi: 10.1128/AAC.00483-19
Fiedler, S., Bender, J., Klare, I., Halbedel, S., Grohmann, E., Szewzyk, U., et al. (2015). Tigecycline resistance in clinical isolates of enterococcus faecium is mediated by an upregulation of plasmid-encoded tetracycline determinants tet (L) and tet (M). J. Antimicrob. Chemother. 71, 871–881. doi: 10.1093/jac/dkv420
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). Arg-annot, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2017). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Hunt, M., Mather, A. E., Sánchez-Busó, L., Page, A. J., Parkhill, J., Keane, J. A., et al. (2017). ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genomics 3:e000131. doi: 10.1099/mgen.0.000131
Infante, V. H. P., Conceição, N., de Oliveira, A. G., and da Costa Darini, A. L. (2016). Evaluation of polymorphisms in pbp4 gene and genetic diversity in penicillin-resistant, ampicillin-susceptible Enterococcus faecalis from hospitals in different states in Brazil. FEMS Microbiol. Lett. 363:fnw044. doi: 10.1093/femsle/fnw044
Inouye, M., Dashnow, H., Raven, L.-A., Schultz, M. B., Pope, B. J., Tomita, T., et al. (2014). Srst2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2016). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Katoh, K., Misawa, K., Kuma, K.-I., and Miyata, T. (2002). Mafft: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Lakin, S. M., Dean, C., Noyes, N. R., Dettenwanger, A., Ross, A. S., Doster, E., et al. (2016). Megares: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 45, D574–D580. doi: 10.1093/nar/gkw1009
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Liu, B., and Pop, M. (2008). ARDB—antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Munk, P., Andersen, V. D., de Knegt, L., Jensen, M. S., Knudsen, B. E., Lukjancenko, O., et al. (2017). A sampling and metagenomic sequencing-based methodology for monitoring antimicrobial resistance in swine herds. J. Antimicrob. Chemother. 72, 385–392. doi: 10.1093/jac/dkw415
Munk, P., Knudsen, B. E., Lukjancenko, O., Duarte, A. S. R., Van Gompel, L., Luiken, R. E. C., et al. (2018). Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine european countries. Nat. Microbiol. 3, 898–908. doi: 10.1038/s41564-018-0192-9
O'Neill (2016). Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance. London: Wellcome Trust and the UK Department of Health.
Ono, S., Muratani, T., and Matsumoto, T. (2005). Mechanisms of resistance to imipenem and ampicillin in Enterococcus faecalis. Antimicrob. Agents Chemother. 49, 2954–2958. doi: 10.1128/AAC.49.7.2954-2958.2005
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E., and Larsson, D. G. J. (2014). BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 42, D737–D743. doi: 10.1093/nar/gkt1252
Panunzi, L. G. (2019). AMR Datasets From Diverse Spp (Version v0.1.0) [Data set]. Zenodo. doi: 10.5281/zenodo.3571224
Rice, L. B., Bellais, S., Carias, L. L., Hutton-Thomas, R., Bonomo, R. A., Caspers, P., et al. (2004). Impact of specific pbp5 mutations on expression of β-lactam resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 48, 3028–3032. doi: 10.1128/AAC.48.8.3028-3032.2004
Rowe, W., Baker, K. S., Verner-Jeffreys, D., Baker-Austin, C., Ryan, J. J., Maskell, D., et al. (2015). Search engine for antimicrobial resistance: a cloud compatible pipeline and web interface for rapidly detecting antimicrobial resistance genes directly from sequence data. PLoS ONE 10:e0133492. doi: 10.1371/journal.pone.0133492
Rowe, W. P. M., and Winn, M. D. (2018). Indexed variation graphs for efficient and accurate resistome profiling. Bioinformatics 34, 3601–3608. doi: 10.1093/bioinformatics/bty387
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Tyson, G. H., Nyirabahizi, E., Crarey, E., Kabera, C., Lam, C., Rice-Trujillo, C., et al. (2018a). Prevalence and antimicrobial resistance of enterococci isolated from retail meats in the united states, 2002 to 2014. Appl. Environ. Microbiol. 84:e01902-17. doi: 10.1128/AEM.01902-17
Tyson, G. H., Sabo, J. L., Rice-Trujillo, C., Hernandez, J., and McDermott, P. F. (2018b). Whole-genome sequencing based characterization of antimicrobial resistance in enterococcus. Pathog. Dis. 76:fty018. doi: 10.1093/femspd/fty018
Wickham, H., François, R., Henry, L., and Müller, K. (2019). dplyr: A Grammar of Data Manipulation. R Package Version 0.8.3. Available online at: https://CRAN.R-project.org/package=dplyr
Yang, Y., Jiang, X., Chai, B., Ma, L., Li, B., Zhang, A., et al. (2016). ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured arg-database. Bioinformatics 32, 2346–2351. doi: 10.1093/bioinformatics/btw136
Yin, X., Jiang, X., Chai, B., Li, L., Yang, Y., Cole, J. R., et al. (2018). ARGs-OAP v2.0 with an expanded SARG database and hidden Markov models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34, 2263–2270. doi: 10.1093/bioinformatics/bty053
Zankari, E., Allesøe, R., Joensen, K. G., Cavaco, L. M., Lund, O., and Aarestrup, F. M. (2017). Pointfinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72, 2764–2768. doi: 10.1093/jac/dkx217
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: antimicrobial resistance, antibiotic resistance gene, resistome profiling, stand-alone software, sequence analysis, gene context visualization
Citation: Panunzi LG (2020) sraX: A Novel Comprehensive Resistome Analysis Tool. Front. Microbiol. 11:52. doi: 10.3389/fmicb.2020.00052
Received: 18 September 2019; Accepted: 13 January 2020;
Published: 05 February 2020.
Edited by:
Jose L. Martinez, Spanish National Research Council (CSIC), SpainReviewed by:
Teresa M. Coque, Ramón y Cajal Institute for Health Research, SpainSonia Agrawal, University of Maryland, Baltimore, United States
Xiaoting Hua, Zhejiang University, China
Felipe Lira, Centro Nacional de Biotecnología (CNB), Spain
Copyright © 2020 Panunzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonardo G. Panunzi, bGdwYW51bnppJiN4MDAwNDA7Z21haWwuY29t