 Sébastien O. Leclercq
Sébastien O. Leclercq Axel Cloeckaert
Axel Cloeckaert Michel S. Zygmunt
Michel S. Zygmunt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 30 January 2020
Sec. Infectious Agents and Disease
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.03083
This article is part of the Research TopicPathogenomics of the genus Brucella and beyondView all 31 articles
Deciphering the evolutionary history of pathogenic bacteria and their near neighbors may help to understand the genetic or ecological bases which led to their pathogenic behavior. The Brucellaceae family comprises zoonotic pathogenic species belonging to the genus Brucella as well as the environmental genus Ochrobactrum for which some species are considered as opportunistic pathogens. Here, we used a phylogenomic approach including a set of 145 Brucellaceae genomes representative of the family diversity and more than 40 genomes of the order Rhizobiales to infer the taxonomic relationships between the family’s species. Our results clarified some unresolved phylogenetic ambiguities, conducting to the exclusion of Mycoplana spp. out of the family Brucellaceae and the positioning of all Brucella spp. as a single genomic species within the current Ochrobactrum species diversity. Additional analyses also revealed that Ochrobactrum spp. separate into two clades, one comprising mostly environmental species while the other one includes the species considered as pathogens (Brucella spp.) or opportunistic pathogens (mainly O. anthropi, O. intermedium, and O. pseudintermedium). Finally, we show that O. intermedium is undergoing a beginning of genome reduction suggestive of an ongoing ecological niche specialization, and that some lineages of O. intermedium and O. anthropi may shift toward an adaption to the human host.
The Brucellaceae is a family of Gram negative bacteria, member of the order Rhizobiales within the class Alphaproteobacteria. The family was named after the genus Brucella, a facultative intracellular pathogen responsible for the zoonotic disease brucellosis, which causes major economical burden in livestock and human health concerns worldwide (Ficht, 2010; Garin-Bastuji et al., 2014). First described Brucella species were specifically associated with livestock animals, such as sheep and goats for B. melitensis, cattle for B. abortus, and pigs for B. suis, in which they can cause abortions and other reproductive diseases. These species are highly transmissible to humans through direct contacts with infected animals, aerosols, or consumption of raw-milk dairy products, and can produce chronic, debilitating infections. Other pathogenic Brucella species were later described, such as B. canis causing infection in dogs and which is also pathogenic in humans, and B. ovis causing epididymitis in rams (Buddle, 1956; Carmichael and Bruner, 1968). Other species have been isolated from wildlife and consist in their order of description of (i) B. neotomae isolated from rodents (Stoenner and Lackman, 1957), (ii) B. ceti and B. pinnipedialis isolated from marine mammals (Foster et al., 2007), (iii) B. microti initially isolated from the common vole but later found in a wider variety of animals such as red foxes, wild boar and even frogs (Jay et al., 2018; Scholz et al., 2008b), (iv) B. papionis isolated from baboons (Whatmore et al., 2014), and (v) B. vulpis isolated from red foxes (Scholz et al., 2016). More genetically distant Brucella strains comprising several potential new species, have been also reported (Tiller et al., 2010; Eisenberg et al., 2012, 2016). To date only one species of this group of strains has validly been published as B. inopinata, and consists of a single human isolate for which the animal or environmental reservoir has not yet been identified (De et al., 2008; Scholz et al., 2010). The pathogenic status for humans of the newly described wildlife or environmental species is currently not well understood although some human clinical isolates with a similar genetic background have been reported (Sohn et al., 2003; McDonald et al., 2006; Scholz et al., 2010; Tiller et al., 2010; Cloeckaert et al., 2011). One of the striking specificity of the Brucella genus is its extreme homogeneity at the genetic level, with more than 90% DNA-DNA hybridization between species (Verger et al., 1985). Despite this lack of diversity, the phylogenetic relationship between species was extensively investigated using various genetic tools and more recently with the availability of whole-genome sequences, and are now well resolved (Foster et al., 2009; Wattam et al., 2014, Al Dahouk et al., 2017). The current classification includes 12 Brucella species commonly classified as ‘classical’ or ‘atypical’, depending on their phenotypical characteristics and their phylogenetic relationships (Scholz et al., 2018).
The other well-studied genus in the family Brucellaceae is Ochrobactrum. There are currently 18 Ochrobactrum species described, with the recent re-classification of O. lupini as an O. anthropi synonym (Gazolla Volpiano et al., 2019), which are mostly considered as environmental bacteria. For example, O. tritici, O. oryzae, and O. cystisi were first isolated from plant rhizosphere (Lebuhn et al., 2000; Tripathi et al., 2006; Zurdo-Pineiro et al., 2007), O. anthropi, O. grignonense, O. intermedium can be isolated from bulk soil (Lebuhn et al., 2000; Scholz et al., 2008a), while the type strain of O. thiophenivorans was isolated from wastewater (Kämpfer et al., 2007). Nonetheless, some few species, such as O. anthropi, O. intermedium, O. pseudintermedium, O. haematophilum, were initially isolated from human clinical cases (Holmes et al., 1988; Velasco et al., 1998; Kämpfer et al., 2007, 2008; Teyssier et al., 2007) and among them the two first seem to be the main species involved in opportunistic human infections (Teyssier et al., 2005). The other members of the family Brucellaceae family are the more distant Pseudochrobactrum, Paenochrobactrum, Falsochrobactrum, Mycoplana and Daeguia genera, composed of four, three, two, two, and one species, respectively (Yoon et al., 2008; Kämpfer et al., 2014; Kämpfer and Glaeser, 2015; Urakami and Segers, 2015; Sun et al., 2019). Although Paenochrobactrum, Pseudochrobactrum, and Falsochrobactrum genera seem to be true Brucellaceae, the inclusion of the two latter genera in the family is still a matter of debate (Kämpfer and Glaeser, 2015). The phylogenetic relationships between Brucellaceae species have been much less thoroughly investigated than those belonging strictly to the genus Brucella, and most studies relied on the comparison of the small subunit 16S rRNA or the recA gene. The most comprehensive analysis of Brucellaceae species to date, that included more than one hundred strains, showed that the use of these single markers results in different tree topologies depending on the locus and the phylogenetic reconstruction algorithm, and with low to very low branching support throughout the topology (Scholz et al., 2008a). Such lack of resolution was confirmed by a number of other studies which each reported a unique tree topology using the same markers (Teyssier et al., 2007; Huber et al., 2009; Kämpfer et al., 2013, 2015; Chai et al., 2015; Gazolla Volpiano et al., 2019; Krzyżanowska et al., 2019). Nonetheless, a striking common observation of these various studies is that the genus Brucella was consistently embedded within the Ochrobactrum genus diversity, although no definitive conclusion about the taxonomic status of Brucella spp. could be drawn because of the underlying lack of phylogenetic resolution. Limited multilocus sequence typing (MLST) or Internal Transcribed Spacer (ITS) region-based analyses were also performed but were hampered by a small set of genes, a small set of species, or poorly supported branching, and still resulted in conflicting results (Zurdo-Pineiro et al., 2007; Aujoulat et al., 2014; Burygin et al., 2019; Gazolla Volpiano et al., 2019; Krzyżanowska et al., 2019). More recently, whole genome phylogenetic studies started to emerge but generally focused on a limited number of isolates of few species and could not provide a comprehensive view of the phylogenetic relationships at the family level or even at the Ochrobactrum genus level (Gazolla Volpiano et al., 2019; Krzyżanowska et al., 2019). Finally, the question of the Brucellaceae positioning among the Rhizobiales order have not been thoroughly investigated to date. Depending on the studies, the Brucellaceae family is a sister group of the Bartonellaceae, the Rhizobiaceae, or the Phyllobacteriaceae (Gupta and Mok, 2007; Scholz et al., 2008a; Kämpfer et al., 2015). In the present study, the taxonomic organization of the Brucellaceae genera and their species among the Rhizobiales order was determined at the genomic scale using a representative sample of genome sequences available in GenBank, and the inter-relationship between members of the family Brucellaceae was investigated using a comprehensive phylogenomic analysis including most GenBank genomes and newly sequenced isolates.
The 101 non-Brucella Brucellaceae genomes available in the GenBank assembly summary file as of 07/14/2019 were downloaded from the NCBI FTP repository1 in fasta format. Since almost 800 Brucella genomes were available at the time of this study and that the phylogenomic organization of the genus was already thoroughly investigated before (Wattam et al., 2014; Al Dahouk et al., 2017), only 12 genomes spanning the diversity of the classical and non-classical Brucella species were used in our analyses (Supplementary Table S1). A set of 43 diverse non-Brucellaceae Rhizobiales genomes listed as ‘reference’ or ‘representative’ genomes in the NCBI database were also downloaded, as well as the genome of two Caulobacteriales (Caulobacter vibrioides and Parvularcula bermudensis) to serve as outgroups. GenBank assembly identifiers of all downloaded genomes are given in Supplementary Table S1. Additionally, the genomes of 32 Ochrobactrum spp. isolates were sequenced and added to the analysis. For the 32 isolates, bacterial DNA was extracted with DNeasy Blood & Tissue kit from QIAGEN. Genome sequencing was performed by Genoscreen (Lille, France) on an Illumina MiSeq apparatus (paired end 250 bp), with an average of 60x coverage per genome, except for the Ochrobactrum sp. strain Kaboul genome which was sequenced on a Pacific Bioscience SMRT device. Resulting reads were quality trimmed with Trimmomatic v.0.33 (Bolger et al., 2014) and assembled using the Spades v.3.11.1 assembly software (Bankevich et al., 2012). Information and accession/assembly number of these newly sequenced genomes are given in Supplementary Table S2.
All genomic nucleotide sequences were first annotated with PROKKA 1.12 (Seemann, 2014) using the same parameters, i.e., default parameters with no search of ribosomal and transfer RNA, to ensure consistent ORF detection among all sequences. At this stage, the genome sequence of O. anthropi SUBG007 was excluded from further analysis because of an excessive number of very short predicted proteins, suggesting a poor assembly quality. The remaining annotated proteomes were then used to create two datasets. Dataset 1 included all non-Brucellaceae Rhizobiales proteomes, the two Caulobacteriales proteomes, and 25 Brucellaceae proteomes representative of the family diversity. Dataset 2 included all downloaded and locally sequenced Brucellaceae proteomes as well as four other Rhizobiales proteomes to serve as outgroups. Proteomes of Bartonella species were not selected as outgroups despite being the closest Brucellaceae’s family because their highly reduced genomes would lower the number of proteins shared with the Brucellaceae and reduce the power of the analysis. Protein orthology relationships were inferred for both datasets independently using the OrthoMCL pipeline (Li et al., 2003) based on all-against-all BlastP hits with an e-value of 10–20. Among the 20,319 and 16,717 orthology groups returned, respectively 145 and 195 were retained because they included only orthologs (detected only once in each proteome) and were present in 100% and at least 95% of all proteomes, including all outgroups. We used 95% ortholog prevalence threshold for the Brucellaceae analysis because this dataset included a number of genomes originating from metagenomic data (Supplementary Table S1) for which some conserved genomic regions were missing. Proteins of each orthogroup were aligned independently using MAFFT v.7.310 (Katoh and Standley, 2013) with the L-INS-I method and the resulting alignments were concatenated in a single large alignment for each dataset, of 78512 and 79,793 residues respectively, in which missing proteins in some orthogroups were replaced by tracks of gaps. To select the model of protein evolution that best fitted our two datasets, we performed a PROTTEST 3.4.2 preanalysis (Darriba et al., 2011). The program returned the LG model (Le and Gascuel, 2008) with a gamma model of rate heterogeneity, a proportion of invariable sites, and an empirical residue frequency as the best model for both datasets. RAxML v8.2.11 (Stamatakis, 2006) was used to compute a maximum likelihood tree for each concatenated alignment, using the rapid bootstrap algorithm with the protein model returned by the PROTTEST analysis (GAMMAILGF) and 100 bootstraps. Additional phylogenomic reconstructions were computed to test the positioning of Falsochrobactrum spp. genomes within the resulting trees. Each reconstruction was performed on the same selection of 25 Brucellaceae genomes used in Dataset 1 (and the genome of Mesorhizobium loti as an outgroup), for which F. ovis genome, F. shanghaiense genome, or none were discarded, respectively. Trees were calculated for these three combinations using each of the 145 and 195 orthogroups alignments obtained from Dataset 1 (Rhizobiales analysis) and Dataset 2 (Brucellaceae analysis), with RaxML and the same parameters as in the two first calculations. All trees were displayed and annotated using the iTOL v.4 web interface (Letunic and Bork, 2019).
The 16S rRNA sequence was extracted from each Brucellaceae genome used in Dataset 1 and were completed with 15 16S rRNA sequences from type strains of all Brucellaceae species with no genome available, downloaded from NCBI. The sequence of some other Rhizobiales were also added for comparison purposes. Pooled fasta were aligned using MUSCLE (Edgar, 2004) with default parameters using Ugene v.1.30 (Okonechnikov et al., 2012). The alignment was manually inspected and unexpectedly similar sequence stretches found in phylogenetically distant strains were considered as hallmarks of past recombination events.
All Brucellaceae annotated genomes from Dataset 2 were subjected to a pairwise gANI calculation using ANIcalculator v.1 (Varghese et al., 2015) with rRNA genes excluded. As recommended in Varghese et al. (2015), all genome pairs with a gANI value > 96.5 were considered belonging to the same species.
Core and pan proteomes were computed independently on Brucellaceae phylogenetic clades including at least five isolates and corresponding to species according to the gANI criterion. Five genomes of O. intermedium, 10 genomes of O. anthropi and 4 genomes of O. pseudogrignonense clades were excluded from the analysis because they were reconstructed from metagenomic sequencing and lacked large core-genome regions, which resulted in inconsistent results. Additionally, no contig of strain BH3 (O. pituitosum clade) matched the second chromosome of Ochrobactrum, suggesting incomplete sequencing. This genome was therefore also excluded from the analysis. Calculations were performed using a in-house procedure (detailed below) and were then confirmed using the Roary software (Page et al., 2015) with a 95% blastp minimum identity, and genes defined as core when present in 100% of isolates. The in-house procedure started with a within-species all-vs.-all blastp comparison with an e-value of 10–20, at least 80% coverage of both sequences, and a post-treatment filtering to keep only hits with ≥95% amino-acid identity. All filtered result files were parsed to group proteins into homology clusters, by recursively combining hits matching to the same proteins. For each homology group, the number of core and pan protein-coding genes were defined as the minimum and maximum number of homologues in a single genome, respectively. Core and pan numbers were summed over all homology groups to obtain the total core and pan genomes. Both calculations (in-house and Roary) returned core and pan genomes of each genomic species. Because each species has a different number of sequenced genomes, accumulation curves for the core and accessory (pan minus core) genomes were calculated as follows: for each x between 1 and N, with N the total number of genomes in the group, 50 random combinations of x genomes among N were selected, and their core and accessory genomes were estimated.
Forty-three complete and draft genomes representative of the Rhizobiales diversity and 25 genomes representative of the Brucellaceae diversity from the NCBI ftp repository (Supplementary Table S1) were selected, and a phylogenomic tree from a set of 145 proteins shared by all genomes (core proteins) was constructed. As seen in Figure 1, the high number of compared proteins resulted in an almost completely resolved tree, with all but one bootstrap values being ≥98%. The tree structure generally reflects the currently accepted Rhizobiales families grouping, with few exceptions. Consistent with 16S rRNA and recA analyses, Pseudochrobactrum and Falsochrobactrum spp. form a monophyletic group with other Ochrobactrum and Brucella spp. On the contrary, the Mycoplana dimorpha genome investigated here branches with maximum bootstrap support distant to the Brucellaceae family, and actually within the family Rhizobiaceae (Figure 1). Mycoplana stricto sensu have been variously included into the Mycobacteriaceae, the Pseudomonadaceae, or even left unclassified before ending up into the Brucellaceae (Urakami and Segers, 2015). However, this classification was not based on phylogenetic studies, and most current phylogenies actually branches the genus outside the Brucellaceae (Yanagi and Yamasato, 1993; Scholz et al., 2008a; Kämpfer and Glaeser, 2015). This results provide the first robust taxonomic position for Mycoplana, which indicates that the genus should be transferred from the Brucellaceae to the Rhizobiaceae. Our fully resolved tree also indicates that the sister group of the Brucellaceae family is the Bartonellaceae family with 100% confidence. This result is consistent with another phylogenomic study that spanned the whole α-Proteobacteria diversity and therefore included only a few Brucella species as representative of the Brucellaceae (Gupta and Mok, 2007). Finally, the extensive Rhizobiales genome diversity used here provide a clear delineation of the Brucellaceae family and confirms that no other Rhizobiales species/genus are part of the family.
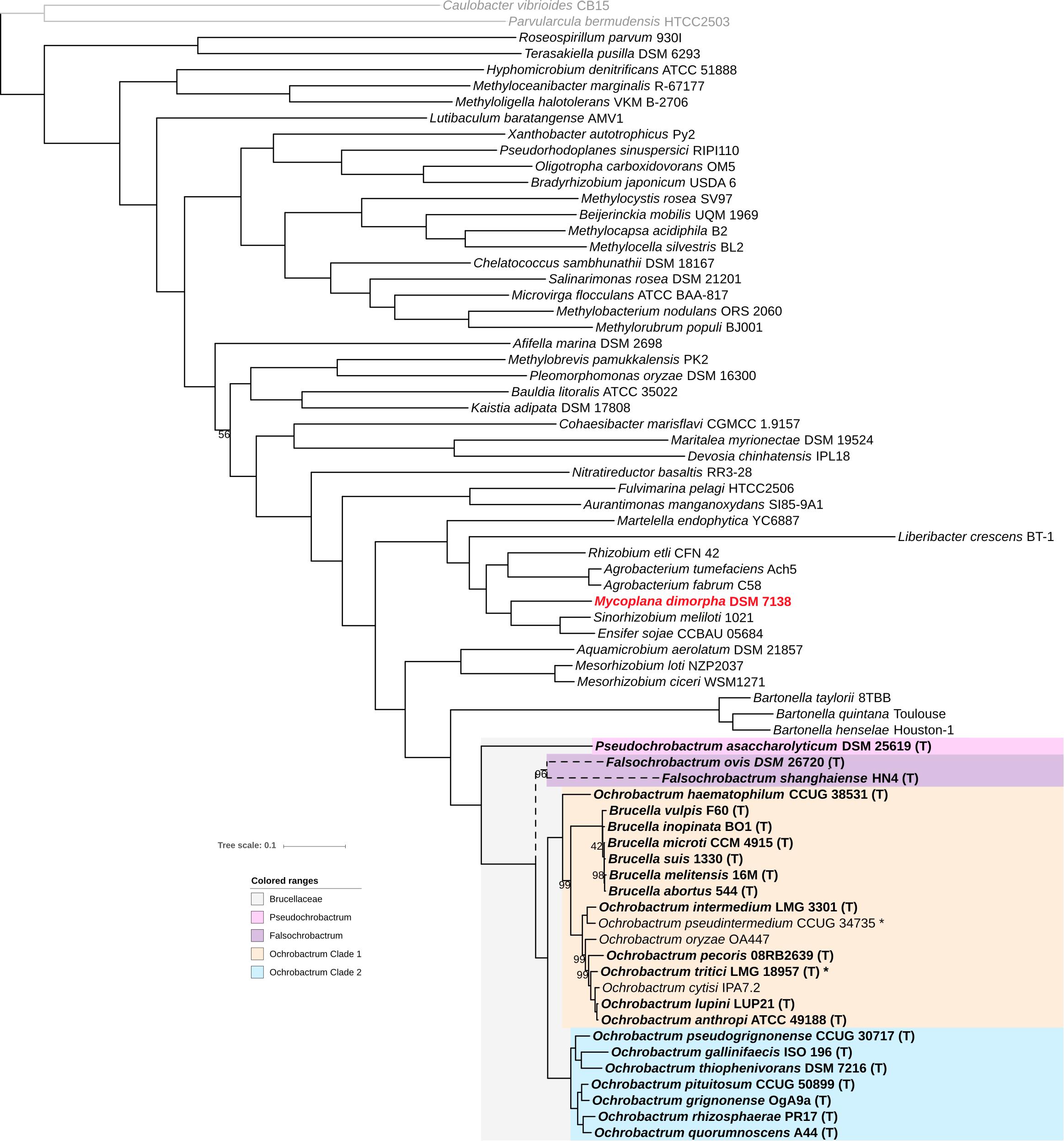
Figure 1. Position of the Brucellaceae genomes in the Rhizobiales phylogeny. The ML phylogenetic tree was computed using RaxML from an alignment of 145 core proteins, and rooted using two Caulobacteriales genomes (depicted in gray). Branching support was estimated using 100 bootstrap replicates, with only values lower than 100% displayed. The Brucellaceae clade is indicated with gray background. Brucellaceae type strains are highlighted with bold labeling. A star indicates a genome sequenced in the current study. Pseudochrobactrum, Falsochrobactrum, and Ochrobactrum clade 1 and clade 2 are indicated with pink, purple, orange, and blue backgrounds, respectively. The misclassified Mycoplana dimorpha genome is labeled in red. Branching of Falsochrobactrum spp. is depicted in dashed lines to reflect conflicting topology with the phylogeny displayed in Figure 2.
Our next objective was to better understand the intra-family phylogenetic relationships between Brucellaceae species. In the topology observed in Figure 1, Pseudochrobactrum asaccharolyticum branches at the root of the Brucellaceae, and Falsochrobactrum spp. branch as a sister clade of all Ochrobactrum and Brucella species. All these other species are then separated into two main clades. The first clade includes Ochrobactrum species haematophilum, pseudintermedium, intermedium, oryzae, pecoris, tritici, cytisi, and anthropi-lupini. The triad anthropi-lupini/cytisi/tritici was generally well resolved in most previous phylogenies, but the position of O. oryzae, O. pecoris, and O. haematophilum was much less consistent. 16S rRNA-based analyses usually branch O. oryzae with O. pseudintermedium and O. gallinifaecis, away from the anthropi/tritici clade, although recA analyses provided results in agreement with our phylogenomic analysis (Scholz et al., 2008a; Huber et al., 2009; Gazolla Volpiano et al., 2019; Krzyżanowska et al., 2019). Similarly, O. pecoris usually branches within or as a sister taxon of Ochrobactrum Clade 2 (see below) in 16S rRNA-based phylogenies, with sometimes a close connection to O. rhizospherae and O. pituitosum (Kämpfer et al., 2011, 2013, 2015; Krzyżanowska et al., 2019), while the MLST phylogeny more accurately branched it with the other members of Ochrobactrum Clade 1 (Aujoulat et al., 2014). For O. haematophilum, no single-locus or even multi-locus analysis could have resolved its correct position with high support (Scholz et al., 2008a; Aujoulat et al., 2014; Gazolla Volpiano et al., 2019), suggesting that its deep branching at the root of Clade 1 precludes from retrieving enough phylogenetic signal when only few genes are used. It should also be noted that all Brucella species form a monophyletic cluster inside Clade 1 with maximum support. The inclusion of Brucella among Ochrobactrum species was recurrently observed within previous studies, but the exact positioning have never been conclusively resolved (Zurdo-Pineiro et al., 2007; Huber et al., 2009; Kämpfer et al., 2015; Burygin et al., 2019). Specifically, the species O. intermedium was named after its supposed closest relationship to Brucella spp. using serological cross-reactivity, polymyxin resistance, and 16S rRNA-based phylogeny (Velasco et al., 1998), a clustering also observed in most 16S rRNA-based analyses (Kämpfer et al., 2007, 2008; Teyssier et al., 2007; Zurdo-Pineiro et al., 2007; Scholz et al., 2008a; Huber et al., 2009). Here we show that Brucella spp. branch as a sister group of a clade including the Ochrobactrum species pseudintermedium, intermedium, oryzae, perocis, cytisi, tritici, and anthropi-lupini. It indicates that O. intermedium is actually more genetically related to most Ochrobactrum spp. of Clade 1 than to Brucella spp.
Clade 2 includes Ochrobactrum species gallinifaecis, thiophenivorans, pseudorignonense, quorumnoscens, rhizospherae, grignonense, and pituitosum. These species consistently group together in most 16S rRNA-based phylogenetic analyses, but usually as an internal Ochrobactrum clade and with O. gallinifaecis excluded (Chai et al., 2015; Gazolla Volpiano et al., 2019; Krzyżanowska et al., 2019). On the contrary, recA and multilocus phylogenies usually separated correctly this group from the other Ochrobactrum spp. (Aujoulat et al., 2014; Kämpfer et al., 2015; Gazolla Volpiano et al., 2019), suggesting that some recombination events in the 16S rRNA region have blurred the phylogenetic signal for this clade.
The numerous discrepancies revealed in the previous section between our phylogenomic analysis and 16S rRNA-based phylogenies prompted us to investigate in more detail the 16S rRNA sequence of the Brucellaceae. We constructed an alignment using the 16S rRNA sequence extracted from all Brucellaceae genomes and several outgroups from Figure 1, completed with 16S rRNA sequences for the Brucellaceae type strains not available as genome sequences in the NCBI database. The resulting alignment show numerous likely recombined regions involving diverse Ochrobactrum and non-Ochrobactrum species (see Table 1 and Supplementary Figure S1). For instance, the O. gallinifaecis ISO 196 16S rRNA sequence is highly recombined, with three independent inferred recombined regions, all three of them shared with non-Ochrobactrum species or possibly with Aquamicrobium aerolatum from the family Phyllobacteriaceae (Table 1). These observed recombination events are likely the reason for the incorrect positioning of O. gallinifaecis outside Ochrobactrum clade 2 in previous 16S rRNA-based phylogenies. Interestingly, one of them is shared with O. pseudintermedium and two with O. oryzae, two species usually grouped along with O. gallinifaecis in 16S rRNA phylogenies. The grouping of O. pecoris with Ochrobactrum clade 2 in 16S rRNA phylogenies is also probably caused by the recombination event inferred at positions 75–91 of the alignment, which also involves O. rhizospherae, O. pituitosum, and O. daejeonense (Table 1). Another recombined region is inferred at pos. 927–979 in the alignment and involves the whole clade including O. tritici, O. cytisi, and O. anthropi-lupini with Ochrobactrum Clade 2 (Table 1). Since this region is one of the highly variable region of the 16S rRNA sequence (see Supplementary Figure S1), the resulting inter-species similarity likely weighted on the positioning of Ochrobactrum Clade 2 within Ochrobactrum Clade 1 in former studies. Likewise, it forces the O. anthropi-tritici clade to branch out of Ochrobactrum Clade 1, leaving O. intermedium as an apparent sister clade of the Brucella spp. in these studies. Finally, a number of other discrepancies were noticed in the 16S rRNA alignment (green boxes in Supplementary Figure S1), which contributes to the general unreliability of phylogenies based on the 16S rRNA sequence. In contrast, no obvious recombined region involving specifically Mycoplana and any member of the Brucellaceae was observed, consistent with its highly supported positioning among the family Rhizobiaceae.
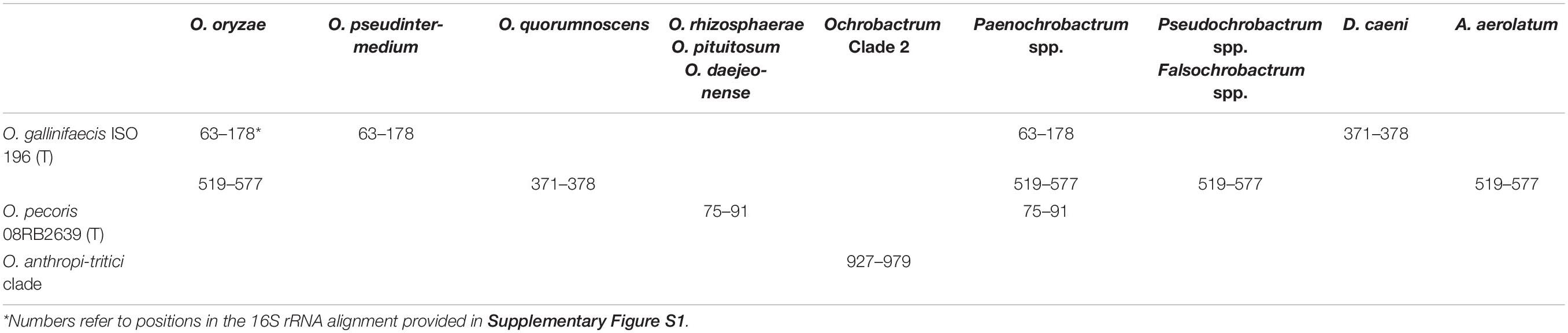
Table 1. Inferred recombination regions in 16S rRNA sequence of Brucellaceae isolates.
To go further in the taxonomic analysis of the Brucellaceae, a new core-proteins tree was constructed, including all non-Brucella Brucellaceae genomes available in GenBank at the time of the study (15th of July, 2019), 12 Brucella genomes representative of the diversity of the genus, and 32 novel Ochrobactrum genomes sequenced in the present study (see Supplementary Tables S1, S2). The topology of this new tree is highly congruent with the topology observed for the Brucelleaceae type strains in the Rhizobiales tree, except for the position of Falsochrobactrum spp. (Figure 2 and Supplementary Figure S2). Indeed, although the two Falsochrobactrum species cluster together in both topologies, they branch with maximum support as a distinct genus in the Rhizobiales tree (Figure 1) but within the Ochrobactrum diversity in the Brucellaceae tree (Figure 2). To test whether this discrepancy was caused by the set of included genomes or by the set of core proteins used, additional phylogenetic trees were constructed using the same representative Brucellaceae type strains with/without Falsochrobactrum genomes, and with each core protein dataset. When the two Falsochrobactrum genomes are included in the analysis, both calculations provide the same topology than the original trees (see Supplementary Figures S3A,B). When each single Falsochrobactrum genome is included alone, the branching is similar whatever the protein dataset: outside the Ochrobactrum clade for F. ovis, and inside the Ochrobactrum clade for F. shanghaiense (see Supplementary Figures S3C–F). This inconsistent positioning suggests that extensive recombination events occurred in F. shanghaiense or F. ovis, or both, and questions the classification of Falsochrobactrum spp. as a new genus, at least from a genomic point of view. Unfortunately, the deep branching of these genomes within the Brucellaceae and the lack of sequenced close relatives hamper any further investigation using existing algorithm of recombination detection. The Falsochrobactrum spp. phylogenetic position in the family Brucellaceae thus cannot be accurately determined in the present study, and its resolution will require additional sequencing effort.
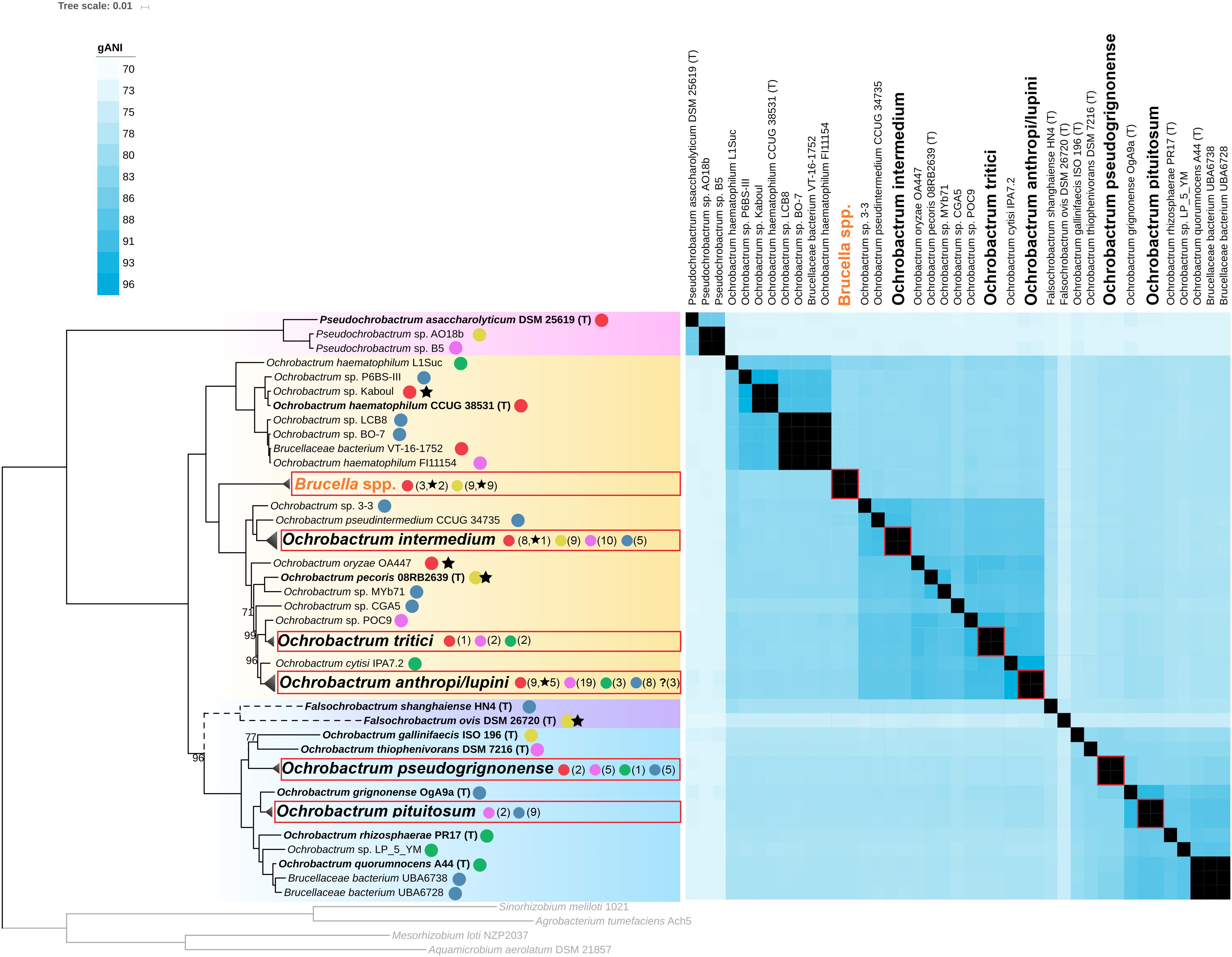
Figure 2. ML Phylogenetic tree of the Brucellaceae clade. The tree was computed using RaxML from an alignment of 195 core proteins, and rooted using four members of the Rhizobiaceae and Phyllobacteriaceae families (depicted in gray). Branching support was estimated using 100 bootstrap replicates, with only values lower than 100% displayed. Genomes of type strains are in bold font. The Pseudochrobactrum clade is indicated with pink background, the Falsochrobactrum in purple background, and the Ochrobactrum Clades 1 and 2 are indicated with orange and blue backgrounds, respectively. Monophyletic clades corresponding to genomic species and including 5 genomes or more are collapsed and boxed in red. Branching of Falsochrobactrum spp. is depicted in dashed lines to reflect conflicting topology with the phylogeny displayed in Figure 1. Origin of strain isolation is indicated with a colored dot: red, human; yellow, animal; pink, anthropized environment; green, plant rhizosphere; blue, other (environmental). Numbers in parenthesis indicate the number of genomes of each origin for collapsed clades. Stars indicate human or animal clinical isolates. The heatmap represent pairwise genome gANI values from 70 (light blue) to 96.5 (deep blue). Values larger than 96.5 (the genomic species threshold) are displayed in black. Collapsed gANI values are boxed in red.
The high resolution of our Brucellaceae core protein phylogeny emphasized the presence of clusters that generally overlap with the species names given to isolates (see Supplementary Figure S2). To delineate even more precisely species boundaries at the genomic level, an all-versus-all gANI comparison was conducted on the Brucellaceae genomes. A gANI score is a measure of genomic relatedness which can be viewed as an in silico DNA-DNA hybridization (Goris et al., 2007), and benchmarking on more than 13,000 genomes representative of most of the accepted bacterial species indicated that a genome pair with a gANI value > 96.5 can be considered belonging to the same species (Varghese et al., 2015). In the following sections, cluster of genomes with a gANI higher than this threshold are named genomic species, to avoid any ambiguity with taxonomically accepted species. In our phylogenomic framework, most clusters with genomes harboring the same species name match a single genomic species, supporting the robustness of the gANI indicator to detect species boundaries. The O. haematophilum cluster is an exception, with two genomes (O. haematophilum L1Suc and O. haematophilum FI11154) distantly related to the type strain genome, with 86.37 and 90.21 gANI values, respectively (Figure 2). The strain FI11154 was identified as O. haematophilum solely on the closest relationship of its 16S rRNA sequence to the O. haematophilum type strain (Diaz et al., 2018) while no information about the characterization process is available for strain L1Suc. It is thus likely that these two strains do not belong to the species O. haematophilum and should return to an unidentified Ochrobactrum sp. status until more thorough characterization. The gANI analysis also indicated that O. cytisi IPA7.2 can be clearly separated from O. anthropi genomes (gANI in the range 95.47–95.99) despite a very close phylogenetic relationship (Figure 2). Although the sequenced strain is not the type strain of the species, a multilocus analysis showed previously that IPA7.2 is phylogenetically closer to the O. cytisi type strain than to any O. anthropi strain (Burygin et al., 2019). These results support the status of O. cytisi as a separate species, a status which have been recently questioned based on 16S rRNA, MLSA and DDH analyses (Gazolla Volpiano et al., 2019). On the contrary, gANI calculation indicates that all Brucella genomes included in this study, which span the whole diversity of currently known Brucella species, actually belong to the same genomic species. This result is fully consistent with previous DNA-DNA hybridization and 16S rRNA analyses and support the concept that all currently known members of the genus Brucella constitute in fact a single species, at least from a genetic point of view (Verger et al., 1985). The genomic species delineation finally confirmed that O. lupini isolates fall in the diversity of the O. anthropi species (Gazolla Volpiano et al., 2019) and revealed that a number of genome sequences available in GenBank were incorrectly identified. Indeed, phylogenomic and gANI analyses indicate that O. anthropi FRAF13 belongs to the tritici clade, O. pituitosum AA2 to the pseudogrignonense clade, and both O. rhizospherae SJY1 and O. thiophenivorans MYb6 to the pituitosum clade (see Supplementary Figure S2).
After considering these species boundaries at the genomic level, five Ochrobactrum species have more than four sequenced genomes, with a diversity suitable for core and pan genome estimations. Among them, O. anthropi and O. intermedium were the most heavily sequenced, with 43 and 33 genomes available, respectively. O. pituitosum, O. pseudogrignonense, and O. tritici followed, with 11, 8, and 5 genomes available, respectively. All these species have ∼4300–5000 protein-coding genes, with no significant difference between species (Figure 3A, ANOVA test, p = 0.061). By comparison, the genomes of the 12 Brucella isolates, known to have experienced genomic reduction (Wattam et al., 2014), harbor ∼3200 protein-coding genes on average. When the core genome of each species is estimated, differences emerge. While the species anthropi, pituitosum, pseudogrignonense, and tritici species show a similar decreasing curve with an increasing number of observed genomes, O. intermedium exhibits a faster and stronger decrease (Figure 3A). When 28 genomes are included in the comparison (the maximum possible for O. intermedium after discarding incompletely sequenced genomes), the core genome of O. intermedium is almost 1000 genes smaller than those of O. anthropi, a result supported by two independent calculation methods (see Section Materials and Methods, Supplementary Figure S4A). Such difference is not compensated by a larger accessory genome (all non-core protein-coding genes) for which all Ochrobactrum species except O. tritici show a similar increase in the accessory genome with increasing number of genomes observed (Figure 3B and see Supplementary Figure S4B). Overall, these results suggest that O. intermedium could be subject of pervasive gene disruption, with no or very few gene inactivation yet fixed in the population. Genome-wide gene disruption is considered as one of the first steps of genome reduction, usually caused by the reduction of effective population size following ecological niche specialization, such as host-adaptation (Toft and Andersson, 2010). Consistent with our observation, an ongoing process of genome reduction in O. intermedium have already been proposed, based on the recurrent deletion of large genomic regions in different strains (Aujoulat et al., 2014). The authors of this study compared the population structure and isolation site of 65 O. intermedium isolates and concluded that the species may undergo a niche specialization toward an environment related to medical and industrial technologies. Another study suggested that O. intermedium may be specializing to the animal gastric niche, based on co-isolation and genetic features shared with the stomach-adapted human pathogen Helicobacter pylori (Kulkarni et al., 2014). The large number of human-related O. intermedium isolates (Scholz et al., 2008a; Aujoulat et al., 2014), sometimes from human clinical cases (Velasco et al., 1998; Moller et al., 1999), as well as their intrinsic resistance to polymyxin E (Velasco et al., 1998), a trait shared with Brucella spp., also suggest that O. intermedium may shift to an animal-associated and potential pathogenic lifestyle.
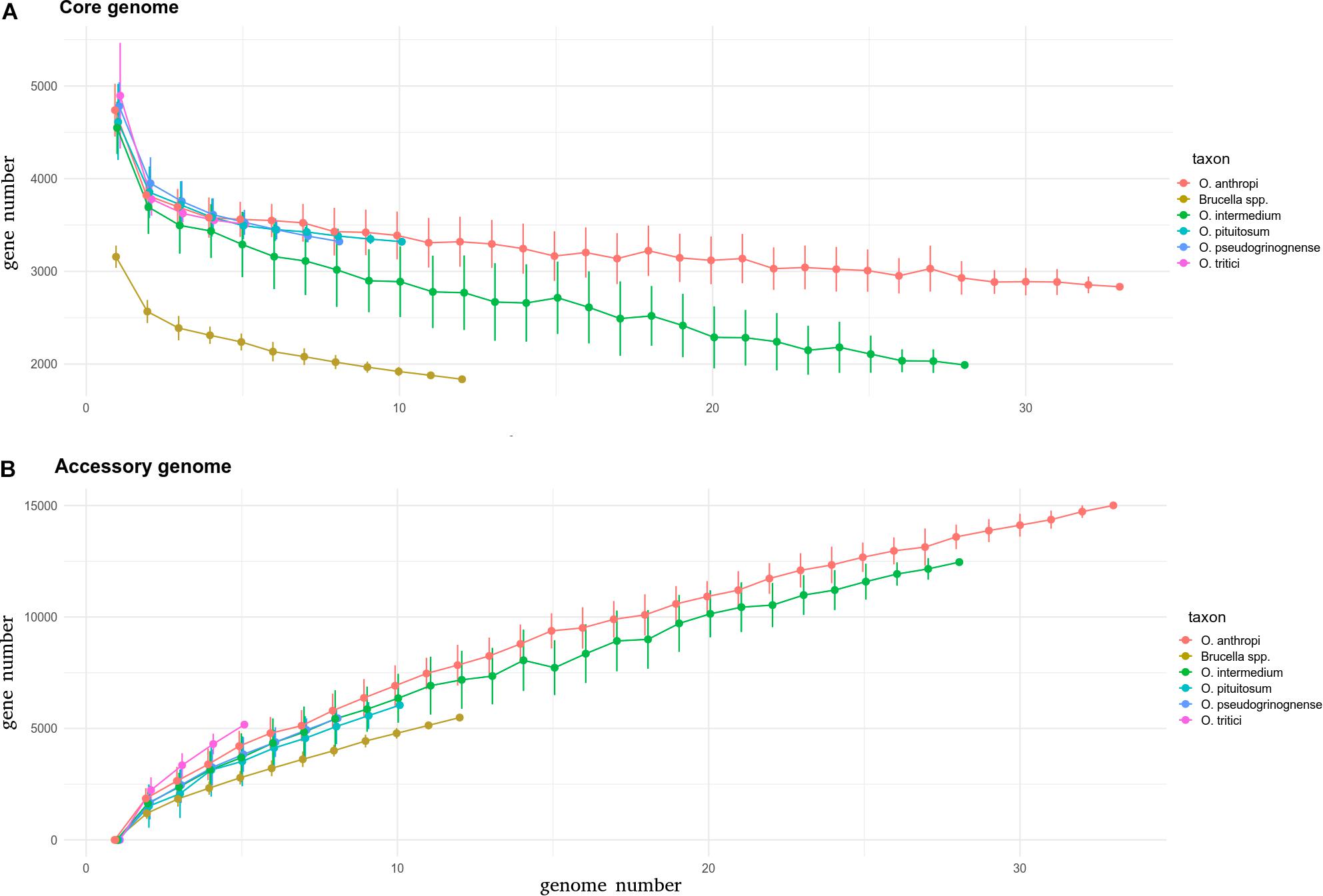
Figure 3. Core (A) and accessory (B) genomes of Brucella and various Ochrobactrum species calculated using an in-house pipeline. For each species, the graph shows the number of genes in core and accessory genomes estimated for an increasing number of considered genomes, up to the number of genomes available for the taxon. The core genome is defined as the set of genes detected in all genomes under consideration. The accessory genome is defined as the total of distinct genes detected in the considered genomes (pan genome) minus the core genome. Since not all taxa have the same number of sequenced genomes, accumulation curves were produced as follows: the number of considered genomes increases from one to the total number of genomes in the taxon, and at each step, the considered genomes are selected randomly in the pool of genomes available for the taxon. This random selection was repeated up to 50 times at each step and median and standard error were estimated for core and accessory gene numbers (see section Materials and Methods).
The intriguing observation made for O. intermedium genomes suggests that isolation sources for the sequenced strains may inform us on its potential niche specialization. We therefore compiled this information for all the Brucellaceae and classified them into five main sources: human clinical, human-related environments, vertebrate animals (including strains causing diseases), plant rhizosphere, and natural environment (including invertebrate animals such as worms). The distribution of these main sources among the Brucellaceae tree revealed a clear separation between Ochrobactrum Clade 1 and Clade 2, with almost all human, human-related, and animal isolates being localized in Clade 1 while Clade 2 comprises mainly environmental and plant-associated isolates (Figure 2). Strikingly, Clade 1 is actually those in which the Brucella spp. belong to, suggesting that this taxonomic lineage may harbor genetic determinants facilitating a preferential association with animal hosts, including humans. At the species level, O. intermedium shows the lowest proportion of environmental isolates and many isolates sampled in poultry (all isolates identified as xx/2009 and xx/2015 in Supplementary Figure S2), in line with a niche specialization toward a human-associated environment. Moreover, an internal O. intermedium highly supported clade is exclusively composed of human isolates which includes two strains isolated from gastric disorder in association with H. pylori (Kulkarni et al., 2013, 2014) and the type strain of clinical origin (see Supplementary Figure S2). Whether this clade represents an ongoing pathogenic lineage should be carefully investigated with a more comprehensive epidemiological analysis. Similarly, more than 50% of the O. anthropi human isolates cluster in a well-supported clade that includes only human-related isolates (see Supplementary Figure S2). This cluster matches remarkably well the clonal complex CC4 observed by Romano et al. (2009) with 3 common strains, a complex exclusively composed of human clinical isolates in their study. Our results thus fully support the proposal of these authors that this lineage may represent a human-specialized opportunistic pathogen lineage among the environmental O. anthropi species.
In summary, this study used a phylogenomic approach to investigate the taxonomic relationship of the family Brucellaceae. Our results confirmed the taxonomic position of the family Brucellaceae within the order of Rhizobiales, with the family Bartonellaceae containing the closest relatives. The family Brucellaceae includes, using this approach, the following current genera: Pseudochrobactrum, Falsochrobactrum, Ochrobactrum, and Brucella. The genus Mycoplana, described previously as belonging to the Brucellaceae, is actually positioned within the Rhizobiaceae and thus outside the Brucellaceae. Within the family Brucellaceae, while our approach confirmed the genus status of Ochrobactrum and Pseudochrobactrum, that of Falsochrobactrum was not totally resolved and that of Brucella appeared questionable. The various Ochrobactrum species clustered into two main clades. The first one includes Brucella spp. and the main opportunistic pathogenic Ochrobactrum species, such as O. anthropi, O. intermedium, O. pseudintermedium, and O. haematophilum. The second clade comprises the other Ochrobactrum species that seem currently not involved in opportunistic infections and are only exceptionally associated with humans. The robustness of the phylogenetic inferences we obtained here using two set of marker proteins and two sets of input genomes, compared to what was previously reported with single locus markers, emphasizes the help that genomic information can bring to characterize the taxonomic status of potential new species or genera. Nevertheless, according to our gANI analysis, the use of the 16S rRNA marker appears to be quite reliable to assign isolates to known species and should still be used for this purpose. Finally, the present work provides new elements on the question of the Brucella species delineation, and its status as a genus. Species definition has been problematic for numerous bacterial taxa because of a lack of a strong and universally accepted theoretical basis (Gevers et al., 2005). Our finding that all Brucella species can be considered as a single species according to gANI scores is actually also supported by the approach of the Genome Taxonomy Database (GTDB2) (Parks et al., 2018). This approach is an alternative to the official prokaryotic taxonomic nomenclature in which taxonomic ranks are defined according to a strict monophyly among groups and thresholds based on corrected genomic distances. Interestingly, under the GTDB framework, Brucella remains a genus, consisting of a single species as initially proposed by Verger et al. (1985). However, Ochrobactrum would be separated into three genera corresponding to the O. haematophilum clade, the O. anthropi/tritici/intermedium clade, and the Ochrobactrum clade 2 of the present study. The resulting GTDB classification remains, however, to be validated.
The datasets generated for this study can be found in the ncbi Biosample, SAMN12821270, SAMN12821271, SAMN12821272, SAMN12821273, SAMN12821274, SAMN12821275, SAMN1282 1276, SAMN12821277, SAMN12821278, SAMN12821279, SAMN12821280, SAMN12821281, SAMN12821282, SAMN 12821283, SAMN12821284, SAMN12821285, SAMN12821286, SAMN12821287, SAMN12821288, SAMN12821289, SAMN 12821290, SAMN12821291, SAMN12821292, SAMN12821293, SAMN12821294, SAMN12821295, SAMN12821296, SAMN 12821297, SAMN12821298, SAMN12821299, SAMN12821300, and SAMN12821301.
AC and MZ conceived and designed the study. SL performed in silico data analyses. SL, AC, and MZ analyzed the data and drafted the manuscript.
This work was funded by Agence Nationale de la Recherche Grant ANR-14-ASMA-0002-02.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Holger Scholz and Michel Thibault for supplying Ochrobactrum strains. We would also like to thank Nelly Bernardet and Isabelle Foubert for expert technical assistance and Mathieu Gonnet for preliminary genome analyses.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.03083/full#supplementary-material
Al Dahouk, S., Köhler, S., Occhialini, A., de Bagues, M. P. J., Hammerl, J. A., Eisenberg, T., et al. (2017). Brucella spp. of amphibians comprise genomically diverse motile strains competent for replication in macrophages and survival in mammalian hosts. Sci. Rep. 7:44420. doi: 10.1038/srep44420
Aujoulat, F., Romano-Bertrand, S., Masnou, A., Marchandin, H., and Jumas-Bilak, E. (2014). Niches, population structure and genome reduction in Ochrobactrum intermedium: clues to technology-driven emergence of pathogens. PLoS One 9:e83376. doi: 10.1371/journal.pone.0083376
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buddle, M. B. (1956). Studies on Brucella ovis (n.sp.), a cause of genital disease of sheep in New Zealand and Australia. J. Hyg. 54, 351–364. doi: 10.1017/s0022172400044612
Burygin, G. L., Kargapolova, K. Y., Kryuchkova, Y. V., Avdeeva, E. S., Gogoleva, N. E., Ponomaryova, T. S., et al. (2019). Ochrobactrum cytisi IPA7.2 promotes growth of potato microplants and is resistant to abiotic stress. World J. Microbiol. Biotechnol. 35:55. doi: 10.1007/s11274-019-2633-x
Carmichael, L. E., and Bruner, D. W. (1968). Characteristics of a newly-recognized species of Brucella responsible for infectious canine abortions. Cornell Vet. 484, 579–592.
Chai, L., Jiang, X., Zhang, F., Zheng, B., Shu, F., Wang, Z., et al. (2015). Isolation and characterization of a crude oil degrading bacteria from formation water: comparative genomic analysis of environmental Ochrobactrum intermedium isolate versus clinical strains. J. Zhejiang Univ. Sci. B 16, 865–874. doi: 10.1631/jzus.b1500029
Cloeckaert, A., Bernardet, N., Koylass, M. S., Whatmore, A. M., and Zygmunt, M. S. (2011). Novel IS711 chromosomal location useful for identification of marine mammal Brucella genotype ST27, which is associated with zoonotic infection. J. Clin. Microbiol. 49, 3954–3959. doi: 10.1128/JCM.05238-11
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
De, B. K., Stauffer, L., Koylass, M. S., Sharp, S. E., Gee, J. E., Helsel, L. O., et al. (2008). Novel Brucella strain (BO1) associated with a prosthetic breast implant infection. J. Clin. Microbiol. 46, 43–49. doi: 10.1128/JCM.01494-07
Diaz, M., Wegmann, U., Akinyemi, N., Oguntoyinbo, F. A., Sayavedra, L., Mayer, M. J., et al. (2018). Complete genome sequence of Ochrobactrum haematophilum FI11154, isolated from kunu-zaki, a nigerian millet-based fermented food. Genome Announc. 6:e00428-18. doi: 10.1128/genomeA.00428-18
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eisenberg, T., Hamann, H.-P., Kaim, U., Schlez, K., Seeger, H., Schauerte, N., et al. (2012). Isolation of potentially novel Brucella spp. from frogs. Appl. Environ. Microbiol. 78, 3753–3755. doi: 10.1128/aem.07509-11
Eisenberg, T., Riße, K., Schauerte, N., Geiger, C., Blom, J., and Scholz, H. C. (2016). Isolation of a novel “atypical” Brucella strain from a bluespotted ribbontail ray (Taeniura lymma). Antonie Van Leeuwenhoek 110, 221–234. doi: 10.1007/s10482-016-0792-4
Ficht, T. A. (2010). Brucella taxonomy and evolution. Future Microbiol. 5, 859–866. doi: 10.2217/fmb.10.52
Foster, G., Osterman, B. S., Godfroid, J., Jacques, I., and Cloeckaert, A. (2007). Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int. J. Syst. Evol. Microbiol. 57, 2688–2693. doi: 10.1099/ijs.0.65269-0
Foster, J. T., Beckstrom-Sternberg, S. M., Pearson, T., Beckstrom-Sternberg, J. S., Chain, P. S. G., Roberto, F. F., et al. (2009). Whole-genome-based phylogeny and divergence of the genus Brucella. J. Bacteriol. 191, 2864–2870. doi: 10.1128/jb.01581-08
Garin-Bastuji, B., Hars, J., Drapeau, A., Cherfa, M. A., Game, Y., Le Horgne, J. M., et al. (2014). Reemergence of Brucella melitensis in wildlife. France. Emerg. Infect. Dis. 20, 1570–1571. doi: 10.3201/eid2009.131517
Gazolla Volpiano, C., Hayashi Sant’Anna, F., Ambrosini, A., Brito Lisboa, B., Kayser Vargas, L., and Passaglia, L. M. P. (2019). Reclassification of Ochrobactrum lupini as a later heterotypic synonym of Ochrobactrum anthropi based on whole-genome sequence analysis. Int. J. Syst. Evol. Microbiol. 69, 2312–2314. doi: 10.1099/ijsem.0.003465
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., et al. (2005). Opinion: re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Gupta, R. S., and Mok, A. (2007). Phylogenomics and signature proteins for the alpha Proteobacteria and its main groups. BMC Microbiol. 7:106. doi: 10.1186/1471-2180-7-106
Holmes, B., Popoff, M., Kiredjian, M., and Kersters, K. (1988). Ochrobactrum anthropi gen. nov., sp. nov. from human clinical specimens and previously known as group vd. Int. J. Syst. Evol. Microbiol. 38, 406–416. doi: 10.1099/00207713-38-4-406
Huber, B., Scholz, H. C., Kämpfer, P., Falsen, E., Langer, S., and Busse, H.-J. (2009). Ochrobactrum pituitosum sp. nov., isolated from an industrial environment. Int. J. Syst. Evol. Microbiol. 60, 321–326. doi: 10.1099/ijs.0.011668-0
Jay, M., Girault, G., Perrot, L., Taunay, B., Vuilmet, T., Rossignol, F., et al. (2018). Phenotypic and molecular characterization of Brucella microti-like bacteria from a domestic marsh frog (Pelophylax ridibundus). Front. Vet. Sci. 5:283. doi: 10.3389/fvets.2018.00283
Kämpfer, P., Glaeser, S., Busse, H.-J., Eisenberg, T., and Scholz, H. (2013). Falsochrobactrum ovis gen. nov., sp. nov., isolated from a sheep. Int. J. Syst. Evol. Microbiol. 63, 3841–3847. doi: 10.1099/ijs.0.049627-0
Kämpfer, P., and Glaeser, S. P. (2015). “Brucellaceae,” in Bergey’s Manual of Systematics of Archaea and Bacteria. eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, and P. DeVos, (Hoboken, NJ: John Wiley & Sons, Inc.) doi: 10.1002/9781118960608.fbm00166.pub2
Kämpfer, P., Glaeser, S. P., and Holmes, B. (2015). “Ochrobactrum,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, and P. DeVos, (Hoboken, NJ: John Wiley & Sons, Inc.) doi: 10.1002/9781118960608.gbm00809.pub2
Kämpfer, P., Poppel, M. T., Wilharm, G., Glaeser, S. P., and Busse, H.-J. (2014). Paenochrobactrum pullorum sp. nov. isolated from a chicken. Int. J. Syst. Evol. Microbiol. 64, 1724–1728. doi: 10.1099/ijs.0.061101-0
Kämpfer, P., Scholz, H. C., Huber, B., Falsen, E., and Busse, H.-J. (2007). Ochrobactrum haematophilum sp. nov. and Ochrobactrum pseudogrignonense sp. nov., isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 58:1271. doi: 10.1099/ijs.0.65899-0
Kämpfer, P., Sessitsch, A., Schloter, M., Huber, B., Busse, H.-J., and Scholz, H. C. (2008). Ochrobactrum rhizosphaerae sp. nov. and Ochrobactrum thiophenivorans sp. nov., isolated from the environment. Int. J. Syst. Evol. Microbiol. 58, 1426–1431. doi: 10.1099/ijs.0.65407-0
Kämpfer, P., Huber, B., Busse, H.-J., Scholz, H. C., Tomaso, H., Hotzel, H., et al. (2011). Ochrobactrum pecoris sp. nov., isolated from farm animals. Int. J. Syst. Evol. Microbiol. 61, 2278–2283. doi: 10.1099/ijs.0.027631-0
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Krzyżanowska, D. M., Maciag, T., Ossowicki, A., Rajewska, M., Kaczynski, Z., Czerwicka, M., et al. (2019). Ochrobactrum quorumnocens sp. nov., a quorum quenching bacterium from the potato rhizosphere, and comparative genome analysis with related type strains. PLoS One 14:e0210874. doi: 10.1371/journal.pone.0210874
Kulkarni, G., Dhotre, D., Dharne, M., Shetty, S., Chowdhury, S., Misra, V., et al. (2013). Draft genome of Ochrobactrum intermedium strain M86 isolated from non-ulcer dyspeptic individual from India. Gut Pathogens 5:7. doi: 10.1186/1757-4749-5-7
Kulkarni, G. J., Shetty, S., Dharne, M. S., and Shouche, Y. S. (2014). Genome sequencing analysis reveals virulence-related gene content of Ochrobactrum intermedium strain 229E, a urease-positive strain isolated from the human gastric niche. FEMS Microbiol. Lett. 359, 12–15. doi: 10.1111/1574-6968.12549
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Lebuhn, M., Achouak, W., Schloter, M., Berge, O., Meier, H., Barakat, M., et al. (2000). Taxonomic characterization of Ochrobactrum sp. isolates from soil samples and wheat roots, and description of Ochrobactrum tritici sp. nov. and Ochrobactrum grignonense sp. nov. Int. J. Syst. Evol. Microbiol. 50, 2207–2223. doi: 10.1099/00207713-50-6-2207
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, L., Stoeckert, C. J. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
McDonald, W. L., Jamaludin, R., Mackereth, G., Hansen, M., Humphrey, S., Short, P., et al. (2006). Characterization of a Brucella sp. strain as a marine-mammal type despite isolation from a patient with spinal osteomyelitis in New Zealand. J. Clin. Microbiol. 44, 4363–4370. doi: 10.1128/jcm.00680-06
Moller, L. V., Arends, J. P., Harmsen, H. J., Talens, A., Terpstra, P., and Slooff, M. J. (1999). Ochrobactrum intermedium infection after liver transplantation. J. Clin. Microbiol. 37, 241–244.
Okonechnikov, K., Golosova, O., and Fursov, M. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Romano, S., Aujoulat, F., Jumas-Bilak, E., Masnou, A., Jeannot, J.-L., Falsen, E., et al. (2009). Multilocus sequence typing supports the hypothesis that Ochrobactrum anthropi displays a human-associated subpopulation. BMC Microbiol. 9:267. doi: 10.1186/1471-2180-9-267
Scholz, H. C., Banai, M., Cloeckaert, A., Kämpfer, P., and Whatmore, A. M. S. (2018). Brucella in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, and P. DeVos, (Hoboken, NJ: John Wiley & Sons, Inc.) doi: 10.1002/9781118960608.gbm00807.pub2
Scholz, H. C., Dahouk, S. A., Tomaso, H., Neubauer, H., Witte, A., Schloter, M., et al. (2008a). Genetic diversity and phylogenetic relationships of bacteria belonging to the Ochrobactrum-Brucella group by recA and 16S rRNA gene-based comparative sequence analysis. Syst Appl. Microbiol. 31, 1–16. doi: 10.1016/j.syapm.2007.10.004
Scholz, H. C., Hofer, E., Hammerl, J. A., Zygmunt, M. S., Cloeckaert, A., Koylass, M., et al. (2016). Brucella vulpis sp. nov., a novel Brucella species isolated from mandibular lymph nodes of red foxes (Vulpes vulpes) in Austria. Int. J. Syst. Evol. Microbiol. 66, 2090–2098. doi: 10.1099/ijsem.0.000998
Scholz, H. C., Hubalek, Z., Sedlacek, I., Vergnaud, G., Tomaso, H., Dahouk, S. A., et al. (2008b). Brucella microti sp. nov., isolated from the common vole Microtus arvalis. Int. J. Syst. Evol. Microbiol. 58, 375–382. doi: 10.1099/ijs.0.65356-0
Scholz, H. C., Nockler, K., Gollner, C., Bahn, P., Vergnaud, G., Tomaso, H., et al. (2010). Brucella inopinata sp. nov., isolated from a breast implant infection. Int. J. Syst. Evol. Microbiol. 60, 801–808. doi: 10.1099/ijs.0.011148-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sohn, A. H., Probert, W. S., Glaser, C. A., Gupta, N., Bollen, A. W., Wong, J. D., et al. (2003). Human neurobrucellosis with intracerebral granuloma caused by a marine mammal Brucella spp. Emerg Infect. Dis. 9, 485–488. doi: 10.3201/eid0904.020576
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stoenner, H. G., and Lackman, D. B. (1957). A new species of Brucella isolated from the desert wood rat Neotoma lepida. Am. J. Vet. Res. 18, 947–951.
Sun, L., Yao, L., Gao, X., Huang, K., Bai, N., Lyu, W., et al. (2019). Falsochrobactrum shanghaiense sp. nov., isolated from paddy soil and emended description of the genus Falsochrobactrum. Int. J. Syst. Evol. Microbiol. 69, 778–782. doi: 10.1099/ijsem.0.003236
Teyssier, C., Marchandin, H., Jean-Pierre, H., Diego, I., Darbas, H., Jeannot, J. L., et al. (2005). Molecular and phenotypic features for identification of the opportunistic pathogens Ochrobactrum spp. J. Med. Microbiol. 54, 945–953. doi: 10.1099/jmm.0.46116-0
Teyssier, C., Marchandin, H., Jean-Pierre, H., Masnou, A., Dusart, G., and Jumas-Bilak, E. (2007). Ochrobactrum pseudintermedium sp. nov., a novel member of the family Brucellaceae, isolated from human clinical samples. Int. J. Syst. Evol. Microbiol. 57, 1007–1013. doi: 10.1099/ijs.0.64416-0
Tiller, R. V., Gee, J. E., Lonsway, D. R., Gribble, S., Bell, S. C., Jennison, A. V., et al. (2010). Identification of an unusual Brucella strain (BO2) from a lung biopsy in a 52 year-old patient with chronic destructive pneumonia. BMC Microbiol. 10:23. doi: 10.1186/1471-2180-10-23
Toft, C., and Andersson, S. G. E. (2010). Evolutionary microbial genomics: insights into bacterial host adaptation. Nat. Rev. Genet. 11, 465–475. doi: 10.1038/nrg2798
Tripathi, A. K., Verma, S. C., Chowdhury, S. P., Lebuhn, M., Gattinger, A., and Schloter, M. (2006). Ochrobactrum oryzae sp. nov., an endophytic bacterial species isolated from deep-water rice in India. Int. J. Syst. Evol. Microbiol. 56, 1677–1680. doi: 10.1099/ijs.0.63934-0
Urakami, T., and Segers, P. (2015). “Mycoplana,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. Rainey, P. Kämpfer, M. Trujillo, J. Chun, and P. DeVos, (Hoboken, NJ: John Wiley & Sons, Inc.) doi: 10.1002/9781118960608.gbm00808
Varghese, N. J., Mukherjee, S., Ivanova, N., Konstantinidis, K. T., Mavrommatis, K., Kyrpides, N. C., et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771. doi: 10.1093/nar/gkv657
Velasco, J., Romero, C., Lopez-Goni, I., Leiva, J., Diaz, R., and Moriyon, I. (1998). Evaluation of the relatedness of Brucella spp. and Ochrobactrum anthropi and description of Ochrobactrum intermedium sp. nov., a new species with a closer relationship to Brucella spp. Int. J. Syst. Bacteriol. 48, 759–768. doi: 10.1099/00207713-48-3-759
Verger, J. M., Grimont, F., Grimont, P. A. D., and Grayon, M. (1985). Brucella, a monospecific genus as shown by deoxyribonucleic acid hybridization. Int. J. Syst. Evol. Microbiol. 35, 292–295.
Wattam, A. R., Foster, J. T., Mane, S. P., Beckstrom-Sternberg, S. M., Beckstrom-Sternberg, J. M., Dickerman, A. W., et al. (2014). Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J. Bacteriol. 196, 920–930. doi: 10.1128/jb.01091-13
Whatmore, A. M., Davison, N., Cloeckaert, A., Dahouk, S. A., Zygmunt, M. S., Brew, S. D., et al. (2014). Brucella papionis sp. nov., isolated from baboons (Papio spp.). Int. J. Syst. Evol. Microbiol. 64, 4120–4128. doi: 10.1099/ijs.0.065482-0
Yanagi, M., and Yamasato, K. (1993). Phylogenetic analysis of the family Rhizobiaceae and related bacteria by sequencing of 16S rRNA gene using PCR and DNA sequencer. FEMS Microbiol. Lett. 107, 115–120. doi: 10.1111/j.1574-6968.1993.tb06014.x
Yoon, J. H., Kang, S.-J., Park, S., and Oh, T.-K. (2008). Daeguia caeni gen. nov., sp. nov., isolated from sludge of a textile dye works. Int. J. Syst. Evol. Microbiol. 58, 168–172. doi: 10.1099/ijs.0.65483-0
Keywords: Brucella, Ochrobactrum, Rhizobiales, Mycoplana, phylogenomic reconstruction
Citation: Leclercq SO, Cloeckaert A and Zygmunt MS (2020) Taxonomic Organization of the Family Brucellaceae Based on a Phylogenomic Approach. Front. Microbiol. 10:3083. doi: 10.3389/fmicb.2019.03083
Received: 02 October 2019; Accepted: 20 December 2019;
Published: 30 January 2020.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Jens Andre Hammerl, Federal Institute for Risk Assessment (BfR), GermanyCopyright © 2020 Leclercq, Cloeckaert and Zygmunt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel S. Zygmunt, bWljaGVsLnp5Z211bnRAaW5yYWUuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.