 Voula Alexandraki
Voula Alexandraki Maria Kazou
Maria Kazou Jochen Blom
Jochen Blom Bruno Pot
Bruno Pot Konstantinos Papadimitriou
Konstantinos Papadimitriou Effie Tsakalidou
Effie Tsakalidou- 1Laboratory of Dairy Research, Department of Food Science and Human Nutrition, Agricultural University of Athens, Athens, Greece
- 2Bioinformatics and Systems Biology, Justus Liebig University Giessen, Giessen, Germany
- 3Research Group of Industrial Microbiology and Food Biotechnology (IMDO), Department of Bioengineering Sciences (DBIT), Vrije Universiteit Brussel, Brussels, Belgium
Streptococcus thermophilus is a major starter for the dairy industry with great economic importance. In this study we analyzed 23 fully sequenced genomes of S. thermophilus to highlight novel aspects of the evolution, biology and technological properties of this species. Pan/core genome analysis revealed that the species has an important number of conserved genes and that the pan genome is probably going to be closed soon. According to whole genome phylogeny and average nucleotide identity (ANI) analysis, most S. thermophilus strains were grouped in two major clusters (i.e., clusters A and B). More specifically, cluster A includes strains with chromosomes above 1.83 Mbp, while cluster B includes chromosomes below this threshold. This observation suggests that strains belonging to the two clusters may be differentiated by gene gain or gene loss events. Furthermore, certain strains of cluster A could be further subdivided in subgroups, i.e., subgroup I (ASCC 1275, DGCC 7710, KLDS SM, MN-BM-A02, and ND07), II (MN-BM-A01 and MN-ZLW-002), III (LMD-9 and SMQ-301), and IV (APC151 and ND03). In cluster B certain strains formed one distinct subgroup, i.e., subgroup I (CNRZ1066, CS8, EPS, and S9). Clusters and subgroups observed for S. thermophilus indicate the existence of lineages within the species, an observation which was further supported to a variable degree by the distribution and/or the architecture of several genomic traits. These would include exopolysaccharide (EPS) gene clusters, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs)-CRISPR associated (Cas) systems, as well as restriction-modification (R-M) systems and genomic islands (GIs). Of note, the histidine biosynthetic cluster was found present in all cluster A strains (plus strain NCTC12958T) but was absent from all strains in cluster B. Other loci related to lactose/galactose catabolism and urea metabolism, aminopeptidases, the majority of amino acid and peptide transporters, as well as amino acid biosynthetic pathways were found to be conserved in all strains suggesting their central role for the species. Our study highlights the necessity of sequencing and analyzing more S. thermophilus complete genomes to further elucidate important aspects of strain diversity within this starter culture that may be related to its application in the dairy industry.
Introduction
Lactic acid bacteria (LAB) include several species, which are extensively used as starters in dairy fermentations (Kongo, 2013). Among them, Streptococcus thermophilus constitutes a major starter for the dairy industry. It is primarily used in the production of yogurt, alongside with Lactobacillus delbrueckii subsp. bulgaricus, but also in the production of several cheese varieties, such as Feta and Mozzarella (Purwandari et al., 2007; Rantsiou et al., 2008; Anbukkarasi et al., 2013). S. thermophilus is the only species which was granted the generally recognized as safe (GRAS) status according to the Food and Drug Administration [FDA], 2007 and the qualified presumption of safety (QPS) status according to the European Food Safety Authority [EFSA], 2007 within the Streptococcus genus, which consists mainly of commensals and pathogenic species. As it is attested by the large number of pseudogenes identified in the genomes of the S. thermophilus strains sequenced so far, the species has undergone significant genome decay probably due to its adaptation to the dairy environment, which is particularly rich in nutrients (Bolotin et al., 2004; Hols et al., 2005; Goh et al., 2011). The regressive evolution of the species has led to genome reduction and simplification of its metabolism (Mayo et al., 2008). The latter is reflected in the deterioration of genes involved, among others, in sugar utilization. S. thermophilus has also lost typical streptococcal pathogenic features presumably through strain selection during domestication toward a starter culture (Bolotin et al., 2004; Hols et al., 2005; Goh et al., 2011; Papadimitriou et al., 2015b). Furthermore, the protocooperation with L. bulgaricus during the production of yogurt has further shaped the metabolic properties of S. thermophilus toward this symbiotic relationship (Mayo et al., 2008).
Typical technological features of S. thermophilus, such as milk acidification, lactose and galactose utilization, proteolytic activity and exopolysaccharide (EPS) production, contribute in shaping the organoleptic characteristics of the final products (Cui et al., 2016). In addition, the stress responses of the species define its performance under the unfavorable conditions prevailing during food production (Zotta et al., 2008; Cui et al., 2016). S. thermophilus also carries Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs)-CRISPR associated (Cas) (CRISPR-Cas) and restriction-modification (R-M) systems, which may contribute to competitiveness in microbial food ecosystems and resistance against bacteriophages and other parasitic DNA (Horvath and Barrangou, 2010; Dupuis et al., 2013). Moreover, genes in genomic islands (GIs), which have been acquired most probably through horizontal gene transfer (HGT) events, may ascribe a number of adaptive traits to S. thermophilus and could be related to technological characteristics, such as EPS production, bacteriocin biosynthesis, and protocooperation (Liu et al., 2009; Eng et al., 2011).
Another topic that has attracted some attention concerning S. thermophilus was the biodiversity of strains within the species. Original studies used typing techniques like random amplification of polymorphic DNA-PCR (RAPD-PCR) or pulsed-field gel electrophoresis (PFGE), while more recent ones used multilocus sequence typing (MLST) (Moschetti et al., 1998; Giraffa et al., 2001; Mora et al., 2002; Ercolini et al., 2005; Delorme et al., 2010, 2017; Yu et al., 2015). Application of MLST in S. thermophilus had to be optimized to increase discriminating power, given the fact that the species may exhibit limited genetic variability (Delorme et al., 2017). In this study the authors reported 116 sequence types and the existence of groups of strains based on phylogenetic analysis of concatenated sequences of housekeeping genes. Additional analysis revealed clustering of strains based on core genome and CRISPR spacer analysis of 25 sequenced strains (both complete and partial). The authors reported that the clustering based on MLST and whole genome analysis was in agreement but differed from that of CRISPR analysis. With the MLST scheme developed and the wide sample of S. thermophilus strains (n = 178), it was feasible to detect relationship between strains and geographic location.
Furthermore, due to the economic importance of S. thermophilus as a starter, a number of groundbreaking studies have been conducted in an attempt to elucidate the genetic basis behind the physiological and the metabolic properties of the species, which define its technological and probiotic potential. Comparative genomics of S. thermophilus was carried out early on and provided significant information about its adaptation to the milk environment and technological traits (Bolotin et al., 2004; Hols et al., 2005; Goh et al., 2011). However, these studies relied only on a limited number of genome sequences. The current accumulation of completely sequenced S. thermophilus genomes can increase the predictive power of comparative analysis and enhance the interpretation of the acquired data about the genome architecture, functionality and evolution. Furthermore, the advancement of bioinformatics tools and the demand of the dairy industry for novel starter strains render an updated analysis of the species essential. In the present study, the results of an in depth analysis of 23 complete S. thermophilus genomes are presented, focusing on main technological features of the species.
Materials and Methods
Strains
The 23 S. thermophilus genomes designated as “complete” up to RefSeq release 88, were selected for analysis in this study (Table 1). The majority of S. thermophilus strains have been isolated from yogurt (strains LMG 18311, CNRZ1066, LMD-9, MN-ZLW-002, MN-BM-A01, KLDS SM, KLDS 3.1003, and ACA-DC 2) and milk (strains JIM 8232, SMQ-301, ND03, ND07, B59671, EPS, GABA, and NCTC12958T). Furthermore, three isolates, namely strains S9, MN-BM-A02, and CS8, derived from traditional Chinese dairy products. More specifically, MN-BM-A02 was isolated from Fan, a traditional Chinese cheese-like product, while CS8 from Rubing, a Chinese fresh goat milk cheese. Finally, strains APC151 and ST3 were isolated from fish intestine and commercial dietary supplements, respectively.

Table 1. Streptococcus thermophilus strains with complete genomes analyzed in this study.
Comparative and Evolutionary Genomics
ProgressiveMauve was used for the whole genome alignment of the 23 S. thermophilus strains analyzed in this study (Darling et al., 2010). GenSkew online application was employed in the evaluation of the chromosomal inversions in strains EPS, MN-BM-A01, and MN-ZLW-0021. The pan/core genome analysis was performed with the bacterial pan genome analysis (BPGA) pipeline v.1.3 using USEARCH v.9.2.64 for clustering gene families (Edgar, 2010) with a 60% sequence identity cut-off and 20 random permutations of genomes to avoid any bias in the sequential addition of new genomes. The protein coding sequences assigned in the core, accessory and unique gene families were further analyzed for clusters of orthologous groups (COG) categories within the BPGA pipeline (Chaudhari et al., 2016). Alternatively, protein coding sequences of S. thermophilus strains were also analyzed for COG categories with the eggnog-mapper based on eggNOG v.4.5 orthology database, as highlighted in the text (Huerta-Cepas et al., 2016, 2017). The EDGAR tool was also employed to assist analysis of orthologs whenever necessary, as well as for core genome phylogenetic analysis among S. thermophilus strains (Blom et al., 2016). For the latter, the alignments of the core gene sets were executed with MUSCLE and concatenated to one complete core alignment, which was used to generate the phylogenetic tree by the neighbor-joining method as implemented in the PHYLIP package. The consensus tree topology was verified by 100 bootstrap iterations. The EDGAR software was also exploited for the investigation of the relatedness among S. thermophilus strains through the construction of average nucleotide identity (ANI) heat map. The ANI values were computed as described by Goris et al. (2007) and as implemented in the JSpecies package (Richter and Rossello-Mora, 2009). The resulting phylogenetic distance values were arranged in an ANI matrix, clustered according to their distance patterns and visualized as a color-coded heatmap, with dark and light orange for high and low similarity regions, respectively. Box Plot Generator was employed for the visualization of genome size differences between the two clusters of the S. thermophilus strains2. Statistical differences in genome size were accessed with the Mann–Whitney U Test for p < 0.05. The quality of the genome assemblies was evaluated with the microbial genomes atlas (MiGA) webserver (Rodriguez-R et al., 2018). The COG frequency and the accessory genes presence/absence heatmaps were generated with the RStudio using the heatmap.2 function included in the Gplots package3. Kyoto encyclopedia of genes and genomes (KEGG) orthology and links Annotation (KOALA) was employed for K number assignment to S. thermophilus protein coding sequences (Kanehisa et al., 2016b), while KEGG Mapper tools were exploited for further processing of KO annotations (Kanehisa et al., 2016a). The PHASTER web server was used for the identification of putative prophages (Arndt et al., 2016). The comparison of the EPS gene clusters was performed with the Easyfig tool (Sullivan et al., 2011). The transporters were determined using the TransportDB database (Elbourne et al., 2017). The CRISPRs were identified with CRISPRFinder web tool (Grissa et al., 2007), while comparison of the predicted spacers was performed with CD-HIT Suite (Huang et al., 2010). The REBASE database was used for verifying the R-M systems (Roberts et al., 2015). Finally, the GIs were obtained through the IslandViewer 4 web-based resource (Bertelli et al., 2017). For our analysis, GIs characterized as integrated by the IslandViewer tool were analyzed.
Results and Discussion
General Genomic Features
The general genome features of the 23 S. thermophilus strains used in this study are presented in Table 2. The chromosome length of the strains ranges between 1.73 and 2.10 Mbp, with an average of 1.85 Mbp, while the % GC content is around 39.0. The number of genes varied between 1,847 and 2,237 including protein coding sequences that varied between 1,555 and 1,854. The percentage of pseudogenes ranged between 9.64 and 13.97%. These variations in genome size, gene and pseudogene content indicate important differences in both gene gain and gene loss events during the evolution of the different strains. It has been previously reported that S. thermophilus owns some of the smallest genomes within streptococci while Streptococcus salivarius some of the largest (Delorme et al., 2015). Based on the complete genome sequences within the salivarius group we found that the percentage of pseudogenes of S. salivarius (12 complete genomes) may reach up to 4% while the percentage of pseudogenes of Streptococcus vestibularis NCTC12167, the only strain with a complete genome, was around 8%. These findings suggest a variable degree of evolution through genome decay within the group. Beyond the salivarius group, high percentages of pseudogenes have also been reported for Streptococcus macedonicus and Streptococcus infantarius that are also associated with the dairy environment (Jans et al., 2013a; Papadimitriou et al., 2014). A high number of pseudogenes has also been reported for certain strains of Streptococcus pneumoniae (see for example the studies by Junges et al., 2019; Scott et al., 2019). Interestingly, extensive genome decay seems to be compatible with adaptation in milk (Bolotin et al., 2004; Hols et al., 2005; Jans et al., 2013a; Papadimitriou et al., 2014) or a pathogenic lifestyle (Lerat and Ochman, 2005). Obviously more research is needed to appreciate the strains/species within streptococci that have evolved through reductive processes and to test whether this evolution path can be correlated with the niches they occupy.
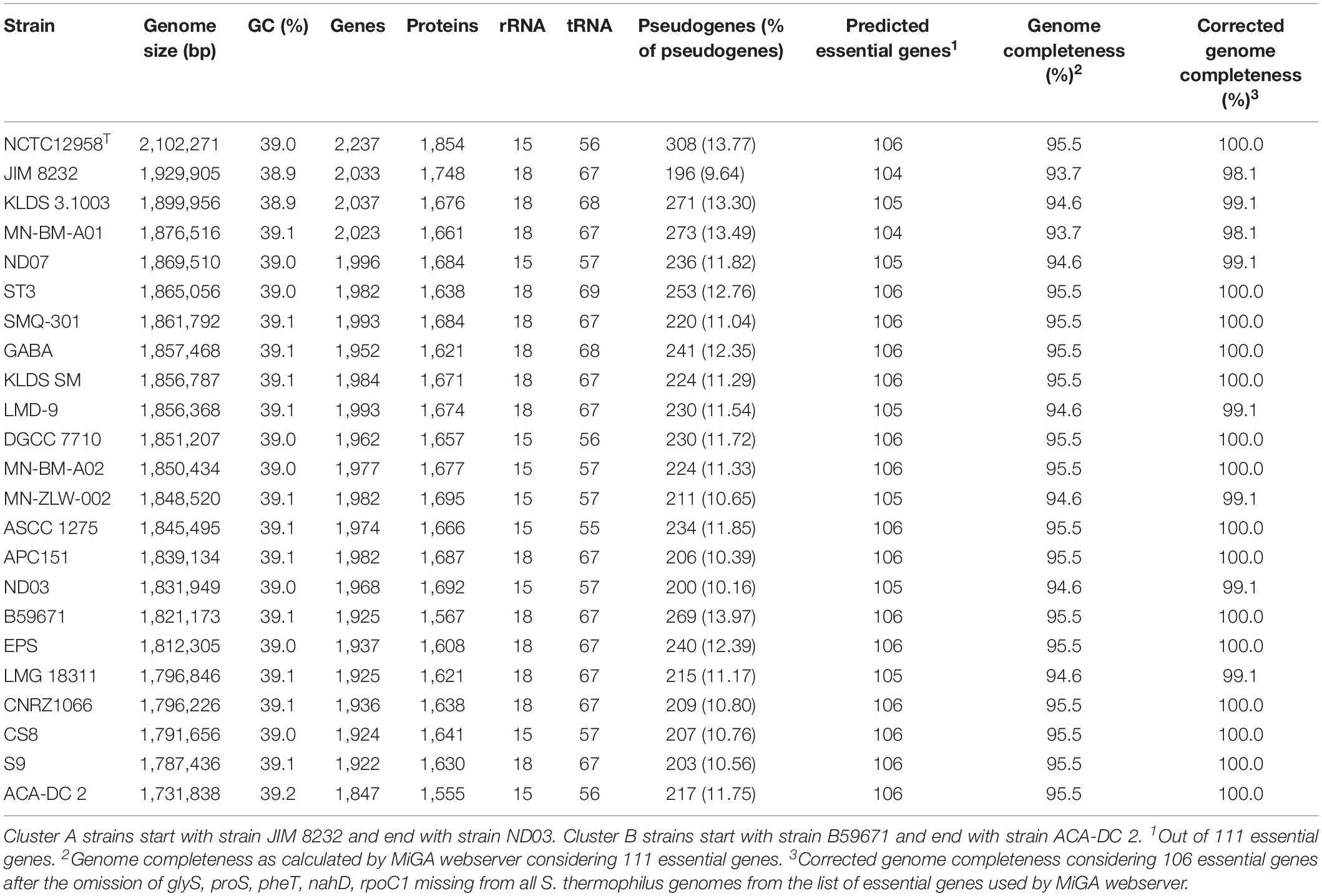
Table 2. General genome features of S. thermophilus strains with complete genomes analyzed in this study.
Fourteen out of 23 strains carry 18 rRNA genes and the rest carry 15. Interestingly, strains with 18 rRNA genes also own a higher number of tRNA genes (ranging from 67 to 69) compared to strains with 15 rRNAs which own fewer tRNA genes (ranging from 55 to 57). A general comment that can be made about this difference is that strains with a higher number of rRNA and tRNA genes could potentially exhibit a higher growth/metabolic rate (Wassenaar and Lukjancenko, 2014).
Comparison of the chromosomal architecture of the 23 S. thermophilus strains was performed through full-length sequence alignments (Supplementary Figure S1). All strains were synchronized from the dnaA so as to simplify the alignment. Analysis revealed a high degree of conservation among different strains. However, strain-specific differences could also be detected. More specifically, low similarity regions, represented as white regions inside the local collinear blocks (LCBs), were found in all strains. Furthermore, many unique regions, represented as blank spaces between the LCBs, were also identified in all strains. In strain EPS a large inversion (1.47 Mbp) was present, while in strains MN-BM-A01 and MN-ZLW-002, a ∼300 kbp inverted region was identified between coordinates 768,310–1,068,868 and 740,416–1,040,999 bp, respectively. These inversions could be either genuine or could be ascribed to assembly artifacts. If the first is true, our observations may correspond to an inversion around the origin of replication for strain EPS, or to an inversion around the terminus of replication for strains MN-BM-A01 and MN-ZLW-002. Such inversions have been described before for bacterial genomes as part of their evolution (Eisen et al., 2000; Darling et al., 2008; Repar and Warnecke, 2017).
Pan/Core Genome Analysis and Phylogenomics
The pan genome of the 23 S. thermophilus strains contains a total number of 2,516 genes, including 1,082 and 997 genes in the core and accessory genomes, respectively (Figure 1A). The number of genes in the accessory genome of each strain varied between 432 and 568 and a total of 437 unique genes (singletons) were identified in 14 strains (Supplementary Table S1 and Figure 1A). According to BPGA analysis, the b value of 0.14 in the power-law regression model is indicative of an open pan genome for S. thermophilus that is probably going to be closed soon (Figure 1B). This may also be supported by the fact that within the total of unique genes identified in S. thermophilus strains, 71% belong to three strains, namely KLDS 3.1003 (n = 41), JIM 8232 (n = 67), and NCTC12958T (n = 204), while strains APC151, ASCC 1275, CNRZ1066, CS8, MN-BM-A01, MN-BM-A02, ND03, ND07, and S9 have no unique genes (Supplementary Tables S1, S2). BPGA analysis also revealed the number of exclusively absent genes per strain (Supplementary Table S1). Core, accessory and unique genes were further classified into COG categories, as implemented within the BPGA pipeline (Supplementary Figure S2). The analysis revealed that approximately 90% of the core, 60% of the accessory and 40% of the unique genes were assigned to various COG categories, with the rest having no prediction. We then excluded the poorly characterized categories R and S from further analysis. The majority of core genes encode proteins involved primarily in housekeeping and metabolic processes. The three most abundant COG categories were J (translation, ribosomal structure, and biogenesis, 12.7%), E (amino acid transport and metabolism, 11.8%), and L (replication, recombination, and repair, 7.3%). In the case of the accessory and unique genes the categories with the highest percentages included categories E, L, and K (transcription) and L, K, and V (defense mechanisms), respectively. In general, accessory and unique genes encoded among others transposases, Cas proteins, R-M systems, glycosyltranferases, polysaccharide biosynthesis proteins, amino acid biosynthesis proteins, proteolytic enzymes, stress related proteins, as well as transporters which may contribute to strain-specific technological traits (please see below).
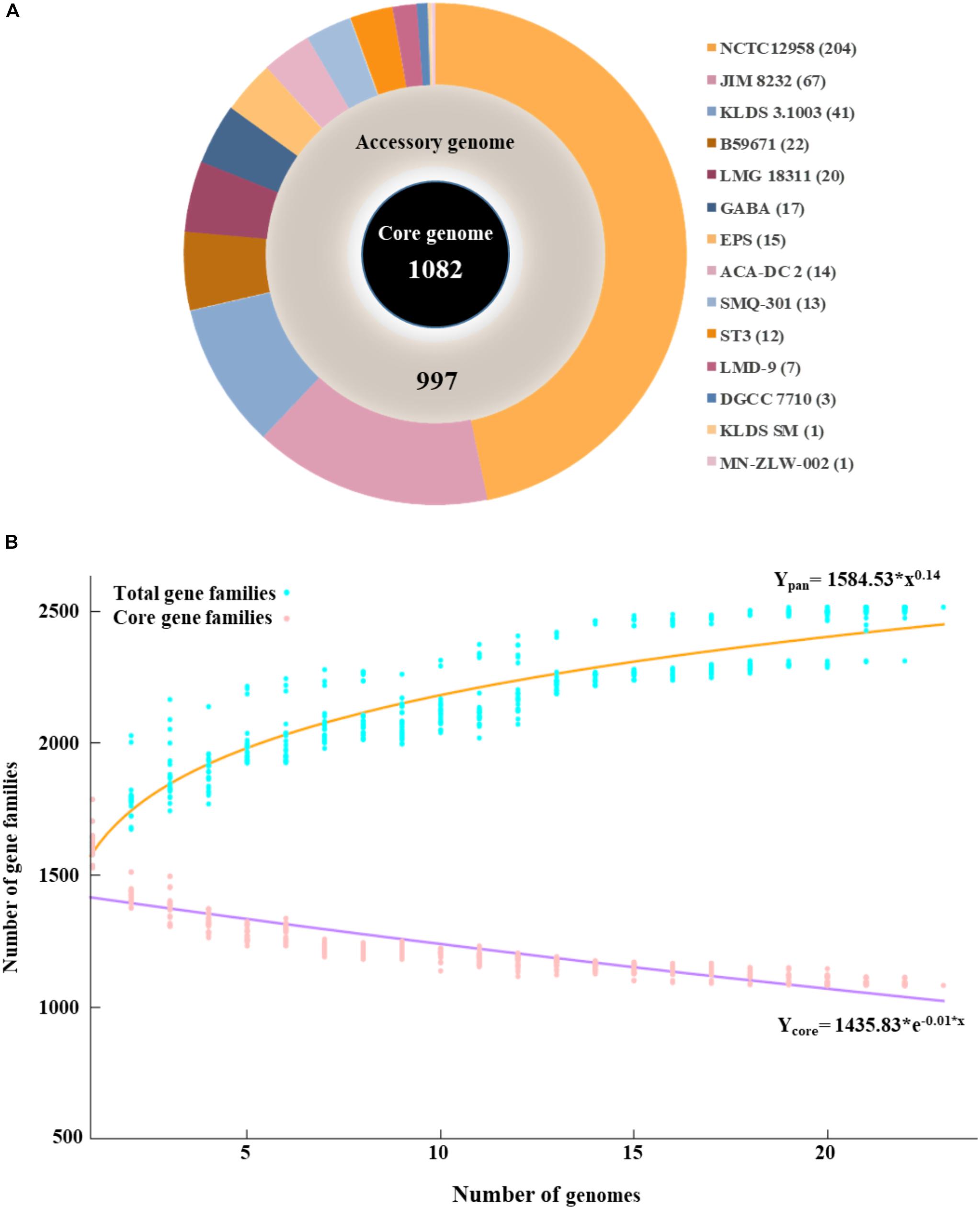
Figure 1. Graphical presentation of core genome (inner black circle), accessory genome (middle gray circle) and unique genes (outer multicolored circle) of the 23 S. thermophilus strains. The number of unique genes for each strain is presented in the parentheses (A). Pan and core genome plots of S. thermophilus strains analyzed (B).
The phylogenetic relationship among the S. thermophilus strains was determined based on the core genome of the strains and revealed two main clusters containing 15 (APC151, ASCC 1275, DGCC 7710, GABA, JIM 8232, KLDS 3.1003, KLDS SM, LMD-9, MN-BM-A01, MN-BM-A02, MN-ZLW-002, ND03, ND07, SMQ-301, and ST3; cluster A) and seven (ACA-DC 2, B59671, CNRZ1066, CS8, EPS, LMG 18311, and S9; cluster B) strains, respectively, while strain NCTC12958T was placed separately (Figure 2). Moreover, the ANI phylogenetic tree had practically an identical topology to that of the phylogenetic tree (Figure 3). There was only one exception with strain KLDS 3.1003 being placed in cluster B. A more detailed inspection of the potential differences between strains in the two clusters revealed that cluster A strains had larger genomes beyond 1.83 Mbp, while those in cluster B had smaller genomes (Table 2 and Supplementary Figure S3). This difference was found to be statistically significant (p < 0.05) suggesting that strains in the two clusters may have been separated by distinct gene gain and/or gene loss events. Within these two main clusters, subgroups of S. thermophilus strains could also be identified during both phylogenetic and ANI analysis. These subgroups include strains ASCC 1275, DGCC 7710, KLDS SM, MN-BM-A02, and ND07 (subgroup AI), MN-BM-A01 and MN-ZLW-002 (subgroup AII), LMD-9 and SMQ-301 (subgroup AIII), APC151 and ND03 (subgroup AIV), and finally CNRZ1066, CS8, EPS, and S9 (subgroup BI) (Figures 2, 3). As already mentioned, core genome phylogeny was also previously performed in a dataset of 25 S. thermophilus strains employing genomes sequenced to a variable degree of completeness (Delorme et al., 2017). In this study 1,311 core proteins were reported. Of note, an earliest study was performed based on three S. thermophilus genome sequences reporting 1,487 core genes (Lefébure and Stanhope, 2007). Our core genome was estimated to consist of 1,082 core proteins. This may suggest a more stringent selection of core proteins during our analysis. Despite the fact that several different strains were analyzed in our study and the study by Delorme et al. (2017), phylogenetic clustering of strains exhibited similarities supporting more or less the distinction we propose between cluster A and B strains and the subgroups observed within them. Differences in the topology of the two phylogenetic trees can be attributed to the different dataset of genomes analyzed as well as the different methods employed to construct the trees. The fact that we concentrated our analysis solely on strains with complete genome sequences presents an important advantage, since we were able to support clustering of strains based on the comparative genomic analysis of additional genomic traits as follows. Completeness of genome sequence is of utmost importance when the presence/absence of specific loci or their exact organization are the main factors for strain diversification.
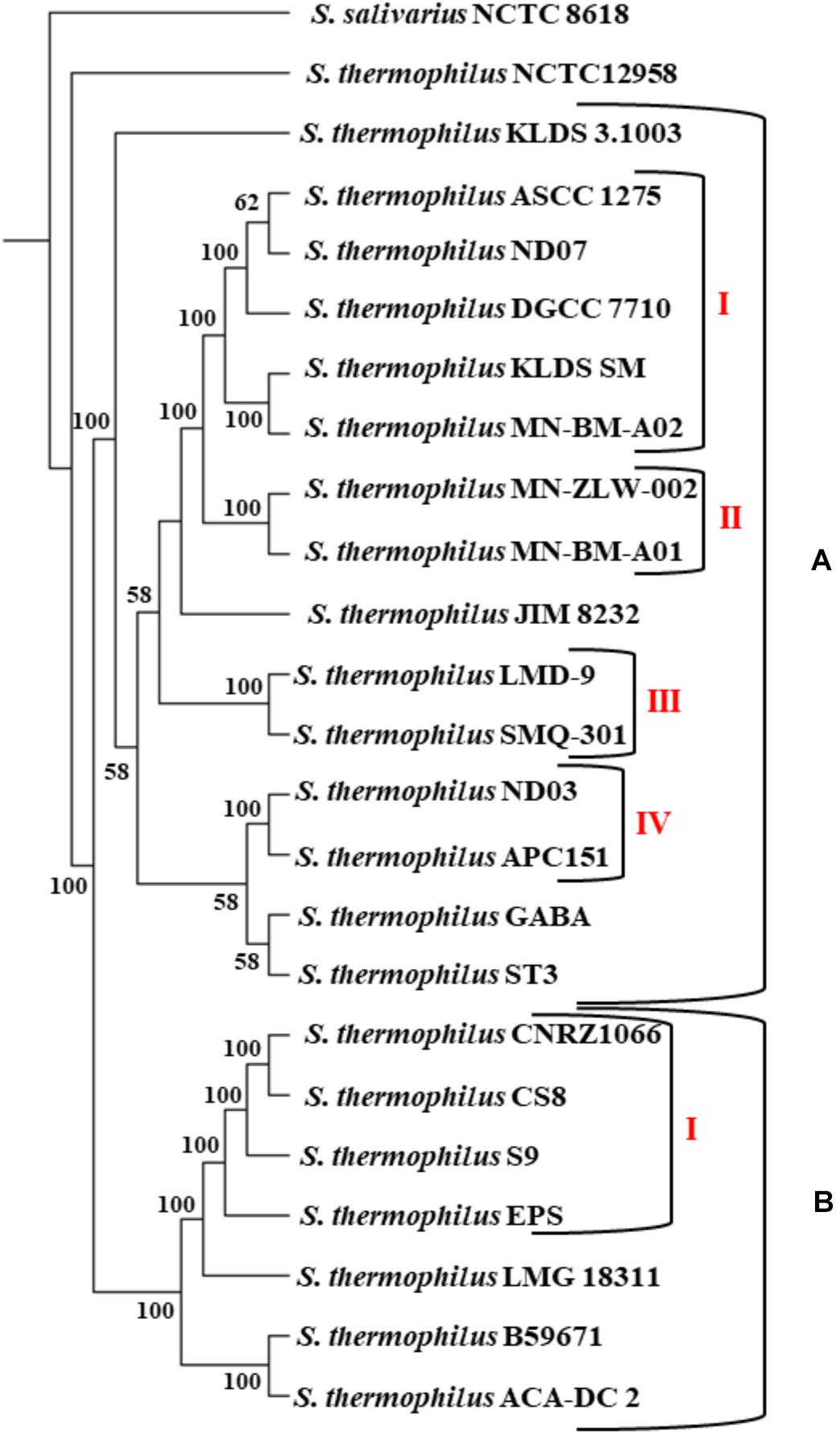
Figure 2. Core genome phylogenetic tree of the 23 S. thermophilus strains. Strains were grouped in two clusters (A,B). Subgroups within the clusters are also highlighted (AI, AII, AIII, AIV, and BI). S. salivarius NCTC 8618 was used as an outgroup. For branches with less than 50% bootstrap support the bootstrap values are not shown.
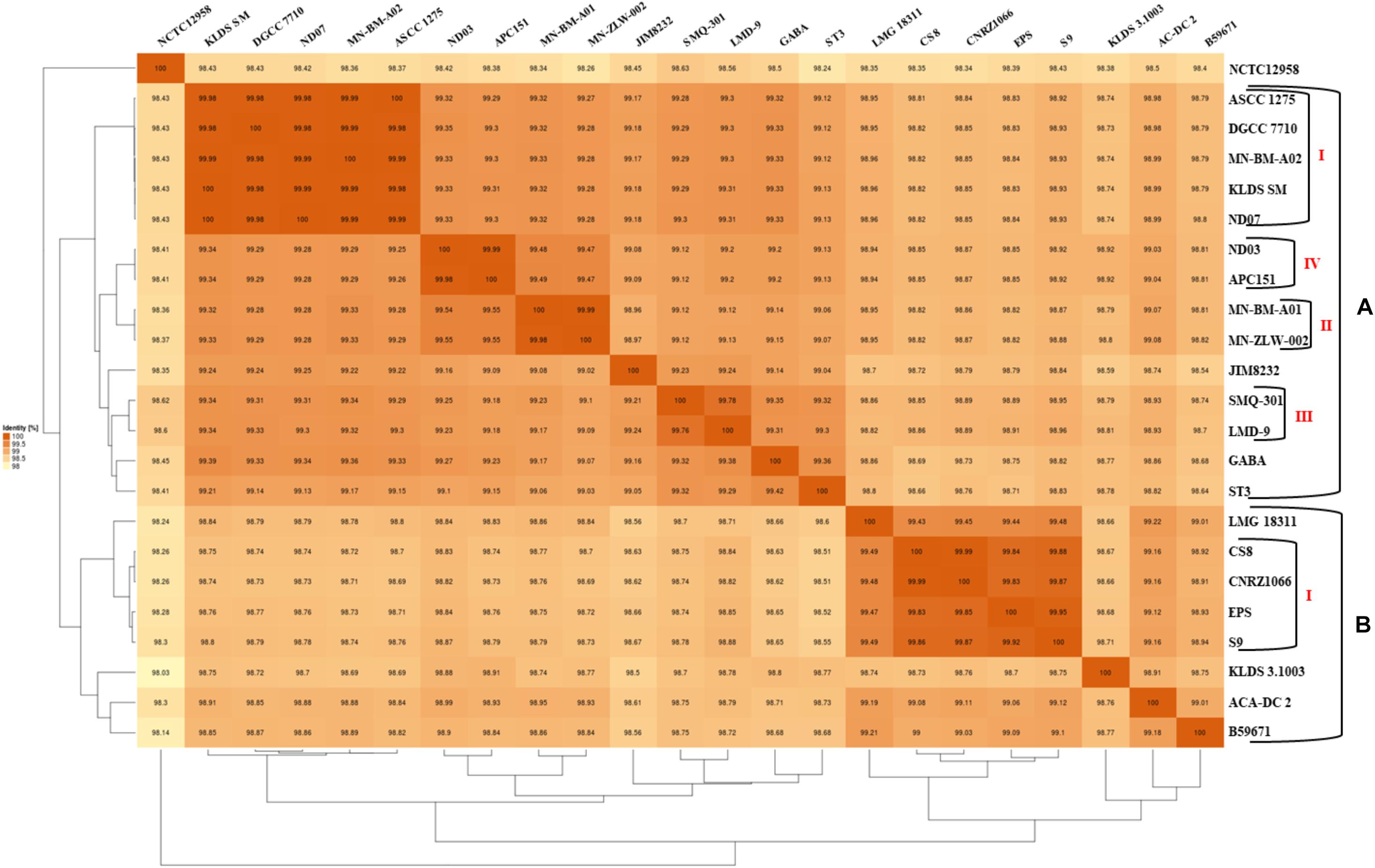
Figure 3. Average nucleotide identity (ANI) phylogenetic tree and heat map visualization of the 23 S. thermophilus strains.
The subgroups mentioned above appeared at high ANI values (>99.9%) which may suggest relatively subtle genomic differences. Such differences may indicate that strains of the same subgroup may be very similar but may deviate from the strict definition of clones. However, clonal relationships may be masked among strains due to aberrations in genome assembly that may come into play at such high ANI values (Burall et al., 2016). To avoid this pitfall, we investigated the quality of the assemblies of all S. thermophilus genomes analyzed in this study using the MiGA webserver (Table 2). Our analysis indicated that from the list of the 111 essential genes used to access genome completeness by MiGA, five (i.e., glyS, proS, pheT, nhaD, and rpoC1) were systematically missing from all S. thermophilus genomes. This observation suggested that they do not belong to the gene pool of the species, which is also supported by data presented previously for essential genes in Firmicutes (Albertsen et al., 2013). We thus corrected the completeness score of the genomes by calculating a total of 106 essential genes. Fifteen genomes received 100% genome completeness. Five genomes missed only secE, two missed secE plus an additional gene (rpiX or uvrb) and one missed only ychF receiving scores above 98.1%. The presence/absence frequency of secE may indicate that it is an accessory gene for S. thermophilus. In all cases the completeness scores of S. thermophilus genomes suggest perfect or nearly perfect assemblies. This is also corroborated by the quality scores for the genome assemblies that were all found “excellent” by MiGA webserver.
Hierarchical clustering of the COG frequency heat map generated for all S. thermophilus strains also supported the existence of the clusters and subgroups mentioned above, with minor alterations (Figure 4). Strains GABA and B59671 were placed in opposite clusters, while strains of the BI subgroup were associated more loosely (i.e., not forming a distinct subgroup). The most abundant category in all strains was E, followed by J and L. The prevalence of the E category may support adaptation of S. thermophilus to milk and the necessity of the organism to use amino acids from the environment.
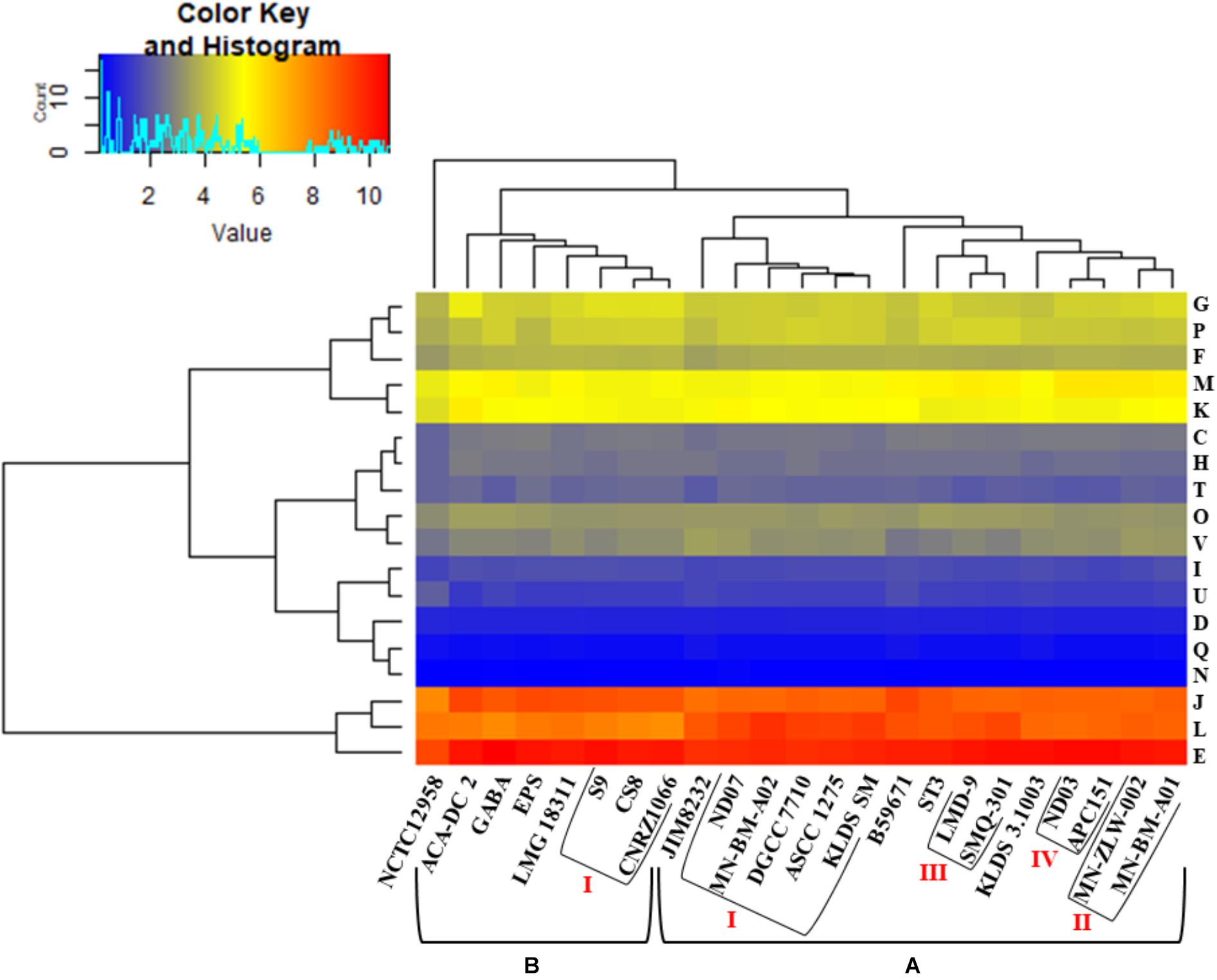
Figure 4. Clusters of orthologous groups (COG) frequency heat map based on a two-dimensional hierarchical clustering. The horizontal axis corresponds to the percentage frequency of proteins involved in the respective COG functional categories: Information storage and processing: translation, ribosomal structure, and biogenesis (J), transcription (K), replication, recombination, and repair (L); cellular processes and signaling: cell cycle control, cell division, chromosome partitioning (D), cell wall/membrane/envelope biogenesis (M), cell motility (N), post-translational modification, protein turnover, chaperones (O), signal transduction mechanisms (T), intracellular trafficking, secretion, and vesicular transport (U), defense mechanisms (V); metabolism: energy production and conversion (C), amino acid transport and metabolism (E), nucleotide transport and metabolism (F), carbohydrate transport and metabolism (G), coenzyme transport and metabolism (H), lipid transport and metabolism (I), inorganic ion transport and metabolism (P), secondary metabolites biosynthesis, transport and catabolism (Q). The vertical axis shows the 23 S. thermophilus strains. Strains were grouped in two clusters (A,B). Subgroups within the clusters are also highlighted (AI, AII, AIII, AIV, and BI). Categories R and S, concerning poorly characterized proteins, were not included in the analysis.
The presence/absence heat map of the accessory genes of S. thermophilus strains supported once again the existence of clusters A and B (Figure 5A). The analysis allowed the identification of genes, which may contribute to the grouping of the strains. As shown in the horizontal axis of the heat map, genes within clusters 4 and 6 are characteristic of clusters B and A, respectively. Moreover, genes of clusters 1, 2, 3, 5, and 4 seem to be present in specific subgroups, namely AII, AIV, AIII, AI, and BI, respectively. Further analysis of the accessory proteins, specifically of those involved in metabolic processes, revealed that cluster A strains (including NCTC12958T) carry the entire set of genes responsible for the biosynthesis of histidine that are basically absent from cluster B (Figure 5B). Based on these findings it is plausible to state that strains of S. thermophilus exhibit lineage-type relationships.
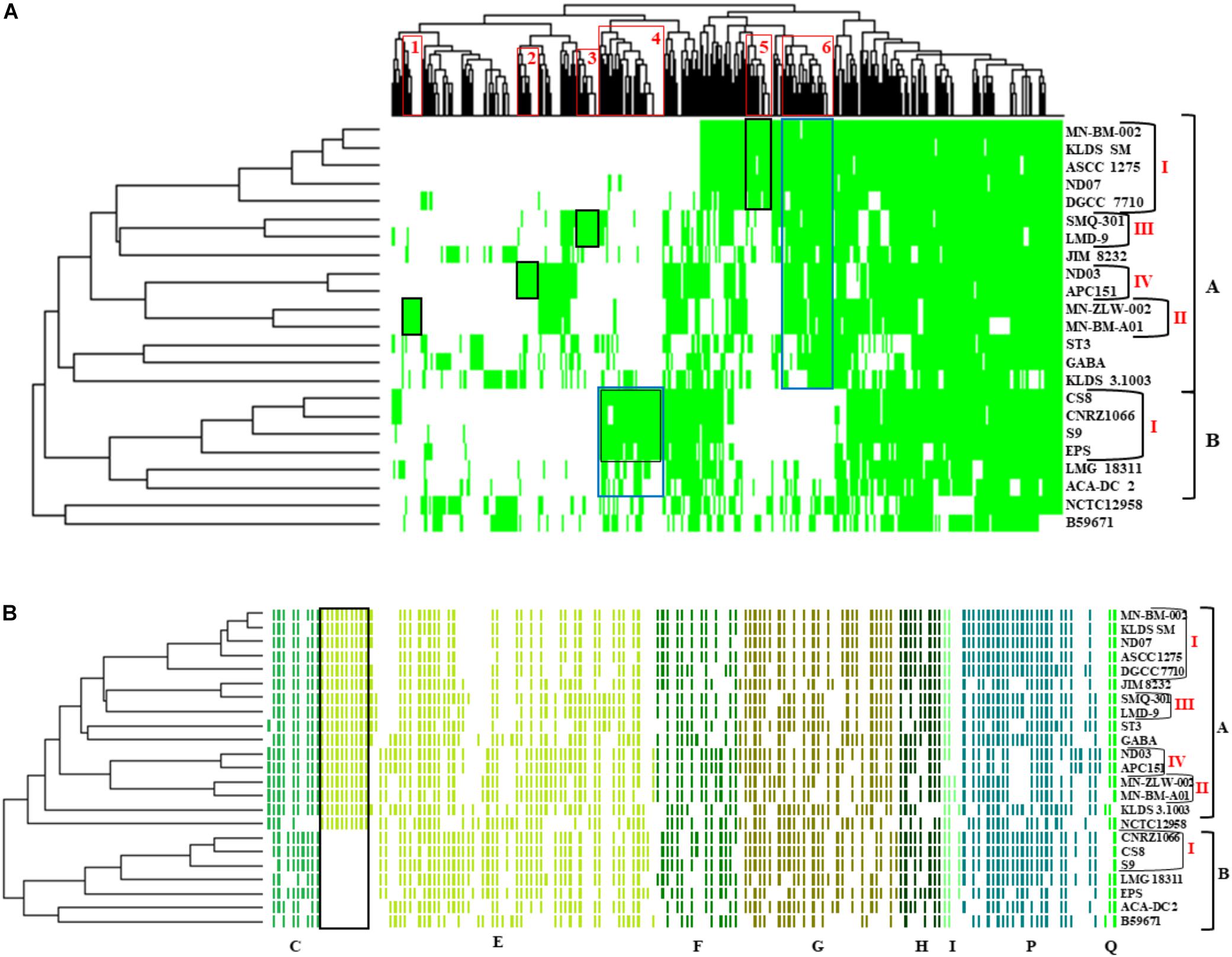
Figure 5. Presence/absence heat map and two-dimensional hierarchical clustering of the 23 S. thermophilus strains accessory genome (A). Colored areas represent the presence of genes in the respective S. thermophilus strains, while white areas indicate the absence of genes. Accessory genes clustered according to their presence in specific subgroups or clusters of strains (gene clusters 1 to 6) are highlighted with red frames on the x axis. The presence of a specific gene cluster in cluster A or B as well as specific subgroups of strains is highlighted with blue or black frames, respectively. Presence/absence heat map and hierarchical clustering of S. thermophilus strains based on accessory genes with clusters of orthologous groups (COG) assignment involved in metabolism (categories C, E, F, G, H, I, P, and Q) (B). Colored areas represent the presence of genes in the respective S. thermophilus strains, while white areas indicate the absence of genes. Genes implicated in the biosynthesis of histidine are highlighted with a black frame. Of note, panel (B) is a composite figure of an excel generated table manually colored, while clustering was exported from RStudio. This was necessary to achieve clustering of genes grouped based on COG categories.
Lactose and Galactose Metabolism
Streptococcus thermophilus ferments preferentially lactose over glucose (Geertsma et al., 2005). Lactose is the main carbohydrate of milk and therefore constitutes the primary carbon and energy source for S. thermophilus, due to the adaptation of the microorganism to this particular niche (Bolotin et al., 2004; Hols et al., 2005; Goh et al., 2011). The genes implicated in the fermentation of lactose and galactose are organized in two adjacent operons (galRKTEM-lacSZ) (Vaughan et al., 2001). We found the complete locus in all S. thermophilus strains analyzed, with the exception of three strains in which lacS (strains B59671 and KLDS 3.1003) or galR (strain NCTC12958T) are putative pseudogenes (Supplementary Table S3). The importance of these inactivations needs to be experimentally investigated, but the high degree of conservation of the gal-lac gene clusters among the different S. thermophilus strains, both at sequence and organization levels, reveals its importance in the catabolism of lactose in milk. Apart from galE coding for the enzyme UDP-glucose 4-epimerase that is located in the Leloir gene cluster, a second or even a third distal galE gene was identified in certain strains (Supplementary Table S3). It has been demonstrated that the activity of this enzyme is positively correlated with the biosynthesis of precursors for EPS production in EPS producing Gal– S. thermophilus strains (Degeest and De Vuyst, 2000). Furthermore, the galactose moiety generated by the hydrolysis of lactose is translocated outside the cell via the dedicated antiporter LacS, which is implicated in the uptake of lactose in exchange to galactose (Vaughan et al., 2003). The majority of S. thermophilus strains are unable to metabolize both free and intracellularly produced galactose, probably either due to insufficient activities of galK and galM genes or due to mutations in the galR-galK promoter region, which may interfere with the expression levels of the respective enzymes (De Vin et al., 2005; Vaillancourt et al., 2008; Anbukkarasi et al., 2014; Sørensen et al., 2016). Recently, Xiong et al. (2019b) demonstrated that the Gal+ phenotype of S. thermophilus depends upon the expression of the gal operon, which can be widely affected by a single point mutation at the -9 box in the galK promoter. Since the accumulation of galactose in the medium by S. thermophilus may be important from a technological or nutritional perspective (Giaretta et al., 2018), we examined the presence of the mutation at the -9 box in the galK promoter in the strains analyzed. Accordingly, only B59671, CS8, EPS, and NCTC12958T seem to be able to catabolize galactose, as they own the relevant G to A mutation in the position -9 of the -10 box related Gal+ phenotype (data not shown). However, experimental verification is required to validate this prediction.
Biosynthesis of EPS
One of the key technological properties of S. thermophilus is the production of EPS, which has been related to desirable textural properties and reduced syneresis in fermented dairy products (Lluis-Arroyo et al., 2014; Han et al., 2016). In a recent study, the EPS clusters of several strains were compared suggesting variations in the gene content of these loci (Cui et al., 2017). Our analysis revealed the presence of EPS gene clusters in all S. thermophilus strains examined. The size of the clusters ranged between 18,661 and 35,973 bp and the % GC content (34.3–36.4%) was found to be lower than the % GC content calculated for the complete genomes of all strains (Supplementary Table S4). All clusters are flanked by a purine-nucleoside phosphorylase (deoD) and a transporter protein as their boundaries (Figure 6). The alignment of the EPS loci showed that they are highly conserved at the 5′ and the 3′ ends and their differences are located mainly in the middle of the clusters. At the 5′ end, genes epsA, epsB, epsC, and epsD were found in all EPS gene clusters and their role has been associated with the regulation of eps genes and chain elongation of the EPS molecules (Cui et al., 2017). The adjacent epsE gene coding a galactosyl-1-phosphate transferase was found in five out of 23 EPS gene clusters (strains CNRZ1066, CS8, EPS, S9, and SMQ-301). In the rest EPS clusters, epsE seems to encode a glycosyl-1-phosphate transferase. These enzymes initiate the assembly of the EPS repeating components through the transfer of phosphorylated sugars to the undecaprenyl-phosphate lipid carrier on the cytoplasmic side of the bacterial membrane (Broadbent et al., 2003; Wu et al., 2014). The sugar is transferred to the outer side of the membrane and this translocation process is probably facilitated by a flippase protein (Manat et al., 2014). All cluster A strains, including strain NCTC12958T, carried one flippase coding gene with the exception of strain ST3 which carried two. In contrast, all strains from cluster B seem to lack the respective gene with the exception of strain B59671.
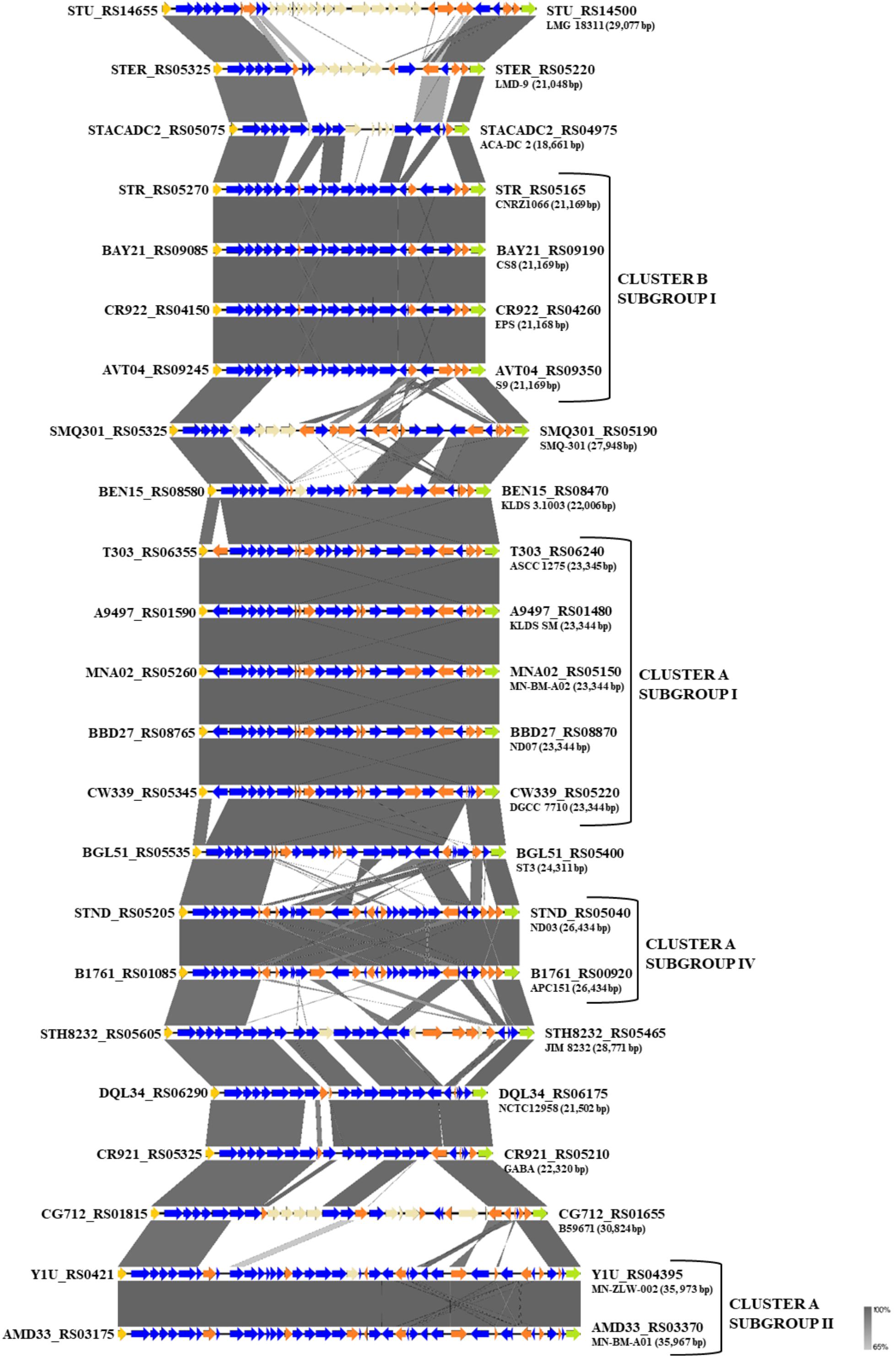
Figure 6. Multiple sequence alignment of the exopolysaccharide (EPS) gene clusters of the 23 S. thermophilus strains after BLASTN analysis. Gray shading represents the % identity among the nucleotide sequences according to the color gradient presented at the lower right corner of the figure. Protein coding genes are highlighted in dark blue, putative pseudogenes in orange, the deoD in yellow, the transporter gene in green and the unique genes for each S. thermophilus strain in beize. Clusters and subgroups of strains are highlighted.
The genes downstream epsE encode proteins with various functions related to EPS biosynthesis. Among them, glycosyltransferases are involved in the consecutive transport of nucleotide sugar moieties to the lipid carrier. Both the number and the type of the respective genes in the EPS clusters are variable and may influence the composition of the produced EPS (Cui et al., 2017). The current analysis revealed the presence of transferases commonly encountered in S. thermophilus EPS clusters, such as glucosyltransferases, galactosyltransferases, and rhamnosyltransferases. A UDP-galactopyranose mutase involved in the synthesis of UDP-galactofuranose was identified in half of the EPS clusters. Interestingly, only strains JIM 8232, GABA, and NCTC12958T were found to carry a gene encoding a putative galactofuranosyl-transferase. Finally, genes implicated in the polymerization and translocation of the EPS repeating units have been also identified in all EPS clusters, as reported previously for S. thermophilus strains (Goh et al., 2011; Wu et al., 2014; Cui et al., 2017; Evivie et al., 2017).
Based on synteny, the EPS gene clusters can be categorized practically in distinct groups, supporting AI, AII, AIV, and BI subgroups. EPS clusters of strains KLDS 3.1003 and ST3 were highly similar to subgroup AI. Similarities in EPS clusters were also observed beyond lineages, as in the case of strains GABA and NCTC12958T. Certain EPS gene clusters, namely those of strains ACA-DC 2, LMD-9, LMG 18311, and B59671, presented higher structural variability due to the presence of many unique genes, which are coding mostly hypothetical proteins and glycosyltransferases. These observations are in accordance with previous findings for strains LMD-9 and LMG 18311 (Goh et al., 2011). Of note, three recent studies have been performed to highlight molecular mechanisms of EPS production in strains ASCC 1275 and KLDS SM (Li et al., 2018; Padmanabhan et al., 2018; Wu and Shah, 2018), while a fourth study suggests a protective role of purified EPS isolated from strain MN-BM-A01 against colitis in mice (Chen et al., 2019). The lineage like-patterns we observed among EPS gene clusters could potentially be useful for extrapolating findings from one strain to another. In all cases understanding of the EPS biosynthesis in S. thermophilus may allow a better selection of strains or even their engineering for improved dairy and probiotic products (Xiong et al., 2019a).
Proteolytic System
The proteolytic system of LAB has been extensively investigated. A number of studies have revealed the diversity of its components, i.e., cell-wall bound proteinases, peptide and amino acid transporters and peptidases, among various LAB species (Savijoki et al., 2006; Liu et al., 2010). In the present study, the proteolytic system of S. thermophilus strains was examined on the basis of the scheme published by Liu et al. (2010) and the recent work of Tian et al. (2018). The results acquired from the TransportDB database were also employed.
Due to the limited availability of free amino acids and peptides in milk, the degradation of caseins is essential for growth. In S. thermophilus, the cell-wall associated proteinase PrtS is implicated in the initiation of the proteolytic cascade (Hols et al., 2005; Goh et al., 2011; Tian et al., 2018). prtS is present in almost half of the strains examined. The analysis showed that the respective gene is present (intact or truncated) solely in cluster A strains with the exception of strains APC151, KLDS 3.1003, and ND03 (Supplementary Table S5A). As it has been previously reported, PrtS presents 95% identity to the PrtS protein of Streptococcus suis and the distribution of prtS in S. thermophilus strains is infrequent in historical collections compared to industrial ones, indicating acquisition by lateral transfer in the species population (Delorme et al., 2010). PrtS has been related to the rapid growth of S. thermophilus in milk as a mono-culture and therefore in the rapid acidification of milk, which is a desirable technological trait. However, the sole presence of prtS is not sufficient for the rapid milk acidification by S. thermophilus. Milk acidification seems to be a complex phenotypic trait, which involves the overexpression of several genes (Galia et al., 2016). Furthermore, it was demonstrated that S. thermophilus strains, irrespective of the prtS+/– status, may present cell-associated extracellular peptidase activities. These activities, albeit weaker than that of PrtS, could probably provide amino acids essential for S. thermophilus growth (Hafeez et al., 2015). The extracellular presence of PepX aminopeptidase in S. thermophilus was recently suggested (Hafeez et al., 2019). Nevertheless, it has been supported that only prtS– S. thermophilus strains can perform protocooperation with L. bulgaricus (Settachaimongkon et al., 2014).
Several peptide and amino acid transporters of various families have been predicted in all S. thermophilus strains (Supplementary Table S5B). The majority of these transporters belong to the ATP-binding cassette (ABC) superfamily and include one oligopeptide Opp ABC transporter, one branched-chain amino acid ABC transporter, one glutamine ABC transporter, four amino acid ABC transporters, one spermidine/putrescine ABC transporter and one methionine ABC transporter. In a number of instances, the gene clusters of these transporters may contain putative pseudogenes and thus may be not functional. It has been previously reported that strain LMD-9 carries a second Opp ABC transporter, which is homologous to that of Bifidobacterium species (Goh et al., 2011). This transporter is also present in strains SMQ-301 and ST3. Strains B59671, GABA, and NCTC12958T have one extra amino acid ABC transporter, which displays high identity (90%) with the respective one of S. salivarius (data not shown). Furthermore, all strains carry four amino acid permeases of the amino acid-polyamine-organocation (APC) family. Additionally, strains ACA-DC 2, APC151, B59671, GABA, KLDS 3.1003, and ND03 carry a glutamate/GABA antiporter (gadC) (Supplementary Table S5B). The latter gene along with glutamate decarboxylase gene (gadB) are responsible for gamma-aminobutyric acid (GABA) production. It was recently demonstrated that strain APC151 is a high-yield GABA producer (Linares et al., 2016). In strain KLDS 3.1003 a unique histidine/histamine antiporter has been also identified (hdcP) (Supplementary Table S5B). The respective gene is located adjacently to a unique histidine decarboxylase gene (hdcA) and along with hdcB form the hdc cluster, probably acquired by HGT (please see below) which has been previously described in two other strains of S. thermophilus (Calles-Enríquez et al., 2010). From a physiological point of view, this gene cluster is probably implicated in cell protection under acidic conditions (De Angelis and Gobetti, 2011). The use of histamine-producing S. thermophilus strains should be avoided in dairy manufacture, since it has been demonstrated that hdcA+ S. thermophilus used as starter in cheese production was associated with the accumulation of histamine in the final product (Gardini et al., 2012). One di-tripeptide transporter is present in all strains. A branched-chain amino acid permease and an amino efflux protein are also present in all strains, but for B59671 and ST3, respectively. The transport of the branched-chain amino acids leucine, isoleucine, and valine, as well as alanine, serine/threonine and glutamate/aspartate is probably facilitated by four symporters, three of them being present in all strains and only one in six strains (Supplementary Table S5B). In addition, a number of incomplete ABC transporters has been also predicted in all the strains analyzed (data not shown).
Besides PrtS, 12 highly conserved cytoplasmic peptidases have been identified in all strains, namely pepA, pepC, pepF, pepM, pepN, pepO, pepP, pepQ, pepS, pepT, pepV, and pepX (Supplementary Table S5A). Moreover, a number of peptidases, which have been identified in several LAB species, are missing from all S. thermophilus strains (Liu et al., 2010). More specifically, pyrrolidone-carboxylate peptidase (pcp) and proline peptidases pepI, pepR, and pepL are absent. Cysteine aminopeptidase (pepE/pepG) presents 40% identity with aminopeptidase C in all S. thermophilus strains, while a putative dipeptidase pepD is present but truncated in 14 S. thermophilus strains. It should be mentioned that the universal distribution of the majority of genes encoding proteins of the proteolytic system of S. thermophilus supports the essential role of the system.
Amino Acids Biosynthesis
The in silico analysis of amino acid biosynthetic pathways has been addressed in S. thermophilus (Hols et al., 2005). Experimental data for the species have been acquired for the biosynthesis of proline, branched-chain amino acids, glutamine and aspartate (Limauro et al., 1996; Garault et al., 2000; Monnet et al., 2005; Arioli et al., 2007). Furthermore, Pastink et al. (2009) studied the amino acid metabolism and amino acid dependency of strain LMG 18311 through amino acid omission experiments, concluding that the minimal amino acid auxotrophy for the strain involves histidine and one of the sulfur-containing amino acids (methionine or cysteine). In some S. thermophilus strains amino acid requirements for growth involve at least four amino acids (Glu, Cys, His, and Met; Letort and Juillard, 2001). It seems that amino acid auxotrophy may be a strain dependent trait.
Most amino acid biosynthetic pathways are highly conserved in the 23 S. thermophilus strains (Supplementary Figure S4 and Supplementary Table S6). Analysis of S. thermophilus protein coding sequences, based on KEGG orthology assignments and Hols et al. (2005), revealed that the majority of the amino acid biosynthetic pathways are present in all strains examined. Complete biosynthetic pathways in all S. thermophilus strains were predicted for threonine, cysteine, glycine, proline, glutamine, asparagine, phenylalanine, alanine, aspartate, and glutamate. Current annotations of all S. thermophilus strains in Refseq with prokaryotic genome annotation pipeline (PGAP) do not seem to support biosynthesis of lysine due to the absence of dapE, dapH, and dapF (Supplementary Table S6). An incomplete Dap-pathway was also reported for strains LMG 18311 (Hols et al., 2005) and LMD-9 (Goh et al., 2011). However, experimental evidence suggests biosynthesis of lysine in strains LMG 18311 (Pastink et al., 2009) and MN-ZLW-002 (Qiao et al., 2018) presumably through a complete Dap-pathway. We found that this discrepancy may be an artifact of annotation with the PGAP tool. Older S. thermophilus GenBank files, annotated with tools other than PGAP included a locus with three genes, the second of which is identified as a (truncated) dapE (data not shown). In contrast, in the same locus, PGAP predicts a single gene corresponding to a putative M20 peptidase pseudogene (e.g., locus_tag Y1U_RS01580 in strain MN-ZLW-002). We also tested other annotation tools, like rapid annotation using subsystem technology (RAST; Aziz et al., 2008) and FGenesB (Solovyev and Salamov, 2011) that also supported a three-gene architecture in the same locus, suggesting that further investigation is required to resolve this matter.
The most striking difference in the biosynthesis of amino acids among S. thermophilus strains examined concerns histidine. Hols et al. (2005) reported absence of this gene cluster in strains CNR1066 and LMG 18311 but its presence in strain LMD-9. As mentioned above, the respective pathway is complete in strains of cluster A and strain NCTC12958T, while strains of cluster B carry only one related gene, namely hisK (Supplementary Table S6 and Figure 5B). Furthermore, several amino acid biosynthetic pathways seem to be incomplete in a number of strains. Analysis revealed that in strain B59671 several genes involved in amino acid biosynthesis are putative pseudogenes or absent compared to the other strains. In this strain glutamate, serine, methionine and tyrosine biosynthetic pathways may be non-functional. Concerning the rest of the strains analyzed, incomplete biosynthetic pathways have been identified for methionine in NCTC12958T and ST3, arginine in MN-BM-A01, branched-chain amino acids in JIM 8232 and tryptophan in EPS (Supplementary Table S6).
In some cases, differences among genes involved in specific biosynthetic steps during amino acid biosynthesis have been also identified. In tryptophan biosynthesis, two adjacent genes, namely aroG1 and aroG2 (Hols et al., 2005), encoding 70% identical proteins, have been identified in all strains except for strains ST3, CNRZ1066, and CS8. The first strain carries only aroG1, while the last two only aroG2. These genes are involved in the first step of chorismate synthesis, an intermediate product during tryptophan biosynthesis. Concerning the biosynthesis of branched-chain amino acids, in all S. thermophilus genomes two ilvD genes have been identified; one belongs to the ilvDBNC operon, while the second is located remotely from the ilvDBNC locus and its functionality is yet to be studied (Hols et al., 2005). The ilvD within the operon is a putative pseudogene in most strains and it seems to be functional only in KLDS 3.1003, LMD-9, NCTC12958T, and SMQ-301. These observations need further experimental investigation.
Urea Metabolism
Streptococcus thermophilus is perhaps the sole species among the dairy LAB with the ability to hydrolyze urea, a phenotypic trait, which affects adversely the milk acidification rate (Pernoud et al., 2004; Iyer et al., 2010). The urease gene cluster is highly conserved in all S. thermophilus strains analyzed and comprises 11 genes in the form of an operon of 8.2 kbp size (Supplementary Table S7). It includes the acid-activated ureI gene, the structural genes ureABC, the accessory genes ureEFGD and the genes encoding the cobalt/nickel uptake system ureMQO (or cbiMQO) (Mora et al., 2004; Iyer et al., 2010). The ureI gene is located upstream the structural genes and is coding a pH-dependent urea channel, which is probably activated for compensating the increase of the extracellular acidity. The ureABC genes are coding the three structural subunits of the enzyme, with ureC coding the large subunit and the remaining two genes coding the two smaller subunits (Ninova-Nikolova and Urshev, 2013). The auxiliary genes ureEFGD encode metallochaperones involved in nickel metallocenter biosynthesis and the delivery of nickel ions to the active site of the urease. More specifically, the urease apoenzyme forms a complex with the UreD, UreF, and UreG proteins, which is activated by the addition of nickel, bicarbonate and the metallochaperone UreE (Sujoy and Aparna, 2013). The ureMQO system is probably responsible for the translocation of nickel ions into the bacterial cell as indicated by functional analysis of the homologous genes in S. salivarius (Chen and Burne, 2003).
The physiological role of S. thermophilus urease has not been thoroughly evaluated. Although it is considered a response mechanism to acid stress, it has been demonstrated that urease is produced at low levels also at neutral pH (Mora et al., 2005). The ureolytic activity of S. thermophilus is probably related not only to the biosynthesis of essential amino acids, e.g., glutamine, but to the overall nitrogen metabolism of the species, with the expression of the ure operon depending on aspartate, glutamate, glutamine, and NH3 concentrations (Monnet et al., 2005; Arioli et al., 2007). However, the rather uncommon urease-negative phenotype has been also reported for S. thermophilus strains, indicating that urease activity may not hold a vital role in milk fermentation (Mora et al., 2002). Recently, spontaneous urease-deficient mutants of S. thermophilus were isolated from S. thermophilus populations deriving from industrial yogurt starters. The stability of the mutated phenotype was confirmed, providing promising results regarding the potential use of urease-deficient strains as starters in dairy fermentations (Ninova-Nikolova and Urshev, 2013). However, in a recent study employing urease deficient mutants it was suggested that urease activity is important for yogurt acidification and that its absence inhibits fermentation acceleration during protocooperation with L. bulgaricus (Yamauchi et al., 2019).
CRISPR-Cas Systems
The CRISPR-Cas systems are defense mechanisms widely distributed in prokaryotes, providing acquired immunity against foreign genetic elements like viruses and plasmids (Horvath and Barrangou, 2010). This immunity mechanism has been extensively studied in S. thermophilus, providing information concerning the environmental adaptability and the anti-phage activity of this microorganism (Sapranauskas et al., 2011; Louis et al., 2017; Hao et al., 2018). In addition, in certain studies spacers within CRISPR arrays in S. thermophilus were employed for assessing diversity among strains of the species (Horvath et al., 2008; Delorme et al., 2017). As mentioned above, Delorme et al. (2017) reported that MLST and whole genome based phylogeny differed from those inferred by CRISPR analysis. Here we revisit clustering of S. thermophilus strains based on CRISPR analysis in the context of complete genome sequences that allowed us further validation of the diversity scheme we propose in this study.
As reported previously (Horvath and Barrangou, 2010), up to four distinct CRISPR-Cas loci, i.e., CRISPR1, CRISPR2, CRISPR3, and CRISPR4 were identified in our S. thermophilus strains (Supplementary Tables S8, S9A and Figure 7). CRISPR1 and CRISPR3 both belong to Class 2/subtype II-A CRISPR-Cas systems, while CRISPR2 and CRISPR4 belong to Class 1/subtype III-A and Class 1/subtype I-E CRISPR-Cas systems, respectively (Horvath et al., 2008; Makarova et al., 2015; Hao et al., 2018). Furthermore, one putative orphan CRISPR array structure was predicted by CRISPRFinder in strains JIM 8232 and LMG 18311, characterized by the absence of adjacent Cas proteins. The direct repeats (DRs) of this array in JIM 8232 were identical to the DRs of CRISPR3 in other strains, suggesting that it must have owned the relevant Cas proteins originally and subsequently lost them. In contrast, the DRs of LMG 18311 in the orphan array did not match any other DRs.

Figure 7. Spacer sequences alignment of the various clustered regularly interspaced short palindromic repeats-CRISPR associated (CRISPR-Cas) system types found in the 22 S. thermophilus strains. In the alignments only the spacer sequences have been used. In each type of CRISPR-Cas system each spacer is represented by the combination of a character and a font color. The spacers represented in black font with the letter U correspond to unique spacers. Spacers represented by the same combination of a character and a font color correspond to identical spacers. Spacers of CRISPR1 (A), CRISPR2 (B), CRISPR4 (C), and CRISPR3 (D).
CRISPR1 was found in 22 out of the 23 S. thermophilus strains analyzed here, with ACA-DC 2 carrying no CRISPR array despite retaining CRISPR-related genes (Alexandraki et al., 2017). CRISPR1 array size ranged between 760 and 2,805 bp. This size variability is associated with the number of spacers (11–42) in the arrays of the different strains. This is the largest CRISPR array within the S. thermophilus strains analyzed with the exception of strain ST3 (Figure 7) and it has been reported to be ubiquitous in S. thermophilus strains (Horvath et al., 2008). In strains B59671 and KLDS 3.1003 the gene coding the Cas9 protein is a putative pseudogene, indicating that the respective CRISPR-Cas systems might have been inactivated. Strains ASCC 1275, APC151, DGCC 7710, GABA, KLDS 3.1003, KLDS SM, LMD-9, MN-BM-A02, MN-ZLW-002, ND03, ND07, NCTC12958T, SMQ-301, and ST3 also carry CRISPR3. This CRISPR contains 8 to 26 spacers and in most cases is shorter than CRISPR1 (Figure 7). A higher activity for CRISPR1 in comparison to CRISPR3 has been experimentally validated (Horvath et al., 2008). In the case of CRISPR3, cas9 is a putative pseudogene in strain MN-BM-A01, indicating that the specific system may have been also inactivated. It should be emphasized that CRISPR1 is detected in both cluster A and B strains, while CRISPR3 is present only in cluster A (apart from strain JIM 8232) and it is totally absent from cluster B strains. Based on analysis of the LMD-9 genome sequence, Horvath et al. (2008) proposed that the entire CRISPR3-Cas system may have been deleted or inserted in S. thermophilus strains through a recombination event between a repeat present in the terminal repeat of CRISPR3 and a repeat close to serB which flanks the system from one side.
CRISPR2 was found in strains ASCC 1275, DGCC 7710, GABA, KLDS 3.1003, KLDS SM, JIM 8232, LMD-9, LMG 18311, MN-BM-A02, ND07, SMQ-301, and ST3. Thus, CRISPR2 was present only in cluster A strains, apart from strain LMG 18311 which belongs to cluster B. Among the Cas proteins of CRISPR2, cas1 is a putative pseudogene in strains KLDS 3.1003 and LMG 18311, while cas10 is a putative pseudogene in strains LMD-9 and SMQ-301. Furthermore, the respective CRISPR-Cas systems of strains GABA and ST3 carry only three CRISPR-associated genes (cas1, cas2, and csm6) which indicates that they are incomplete. All these CRISPR2 systems carried a CRISPR array. However, additional “possible” CRISPR2 systems were predicted by CRISPRFinder owning an incomplete set of Cas proteins followed by a single spacer within two DRs (Supplementary Table S9A). Our findings suggest inactivation and/or degeneration of CRISPR2 in several strains. Horvath et al. (2008) reported that CRISPR2 may indeed be inactivated in certain strains, however Tamulaitis et al. (2014) were able to demonstrate its activity in at least another strain. CRISPR4 was identified in strains ASCC 1275, B59671, DGCC 7710, KLDS SM, MN-BM-A02, and ND07. Genes cse1 and cas2 in strains ASCC 1275 and B59671, respectively, are putative pseudogenes. Interestingly, the CRISPR4 was basically found in subgroup AI strains. Further subgrouping could be supported not only through the presence/absence of CRISPR-Cas systems, but also through the distribution of different spacers, as discussed below.
A total of 997 spacers were found in the confirmed CRISPR-Cas systems of the 22 S. thermophilus strains with 93% being assigned in CRISPR1 and CRISPR3. Analysis of the respective sequences revealed that 258 are unique among 11 strains, namely NCTC12958T, JIM 8232, GABA, KLDS 3.1003, ST3, B59671, LMG 18311, LMD-9, SMQ-301, S9, and EPS, while 253 appeared more than once in the CRISPR arrays. As shown previously, CRISPR arrays may be employed for accessing strain diversity within S. thermophilus (Horvath et al., 2008; Delorme et al., 2017). Indeed, looking into the architecture of the CRISPR arrays we could identify once more patterns that are not shared by all S. thermophilus strains, but they are specific to the grouping of strains we have already described. For example, CRISPR1 supports subgroups AI, AII, AIII, and AIV. Subgroup BI is partially supported, since only strains CNRZ1066 and CS8 share the same CRISPR array. CRISPR3 supports subgroups AI, AII, AIII, and AIV. CRISPR4 has a unique pattern of spacers for subgroup AI. As mentioned above CRISPR2 is present only in cluster A strains, apart from strain LMG 18311 which belongs to cluster B, but the spacer pattern in the arrays could not distinguish any subgroup (Figure 7). Most spacers were unique for each subgroup and were present in a specific order in the array. This observation suggests that this part of the array was present in the common ancestor of these subgroups of strains. However, in certain instances, a specific spacer could be found common between two seemingly unrelated arrays belonging to different subgroups of strains. Most probably such spacers were acquired by the common ancestor of each subgroup due to exposure to the same exogenous DNA that resulted in the acquisition of the same part of sequence into the specific CRISPR array. Evidently, these spacers were identified only in arrays of the same class and subtype CRISPR-Cas systems. Similar analysis of spacers to infer evolutionary relationships among S. thermophilus strains have been reported previously (Horvath et al., 2008). However, when looking solely to the architecture of the CRISPR array it is very difficult to distinguish between clones or complexes of very similar strains that are not actual clones.
BLASTN analysis of the spacers showed that 317 sequences matched several different S. thermophilus bacteriophages (Supplementary Table S9B). Almost half of the spacers analyzed could be related to phages 7201, Sfi19, Sfi21, DT1, and Sfi11. This finding may indicate a high frequency of exposure of S. thermophilus to the specific phages. Finally, six spacers were highly identical to Lactococcus phages, while 12 spacers were highly identical to plasmids of Enterococcus faecium, S. suis, Streptococcus pyogenes, S. pneumoniae, Lactobacillus salivarius and Lactococcus lactis. These findings indicate that S. thermophilus has been found in the same environment with these bacteria. Furthermore, it could be hypothesized that at least some potential HGT events of plasmid donation toward S. thermophilus were aborted through the activity of CRISPR-Cas systems. Overall our findings are in agreement with previous results (Bolotin et al., 2005; Horvath et al., 2008).
R-M Systems and Prophages
Another immunity mechanism employed by the prokaryotes against foreign DNA are the R-M systems. All S. thermophilus strains analyzed carry several R-M systems, classified into four types (Roberts et al., 2005, 2015; Supplementary Table S10 and Supplementary Figure S5). The majority of strains carry one complete type I R-M system with strains EPS, NCTC12958T, GABA, and KLDS 3.1003 carrying two. No type I R-M system was predicted for strain B59671, while in strains MN-ZLW-002, MN-BM-A01, and ST3 the predicted type I R-M system was incomplete due to the absence or inactivation of one or more of the necessary genes. This was the case for additional predicted type I systems in several strains. Certain S. thermophilus strains carry at least one type II system with strains LMD-9, MN-BM-A01, ND03, and APC151 owning three such systems. Unlike type I R-M systems, most type II systems seem to be complete and potentially active. One type III system is present in strains ACA-DC 2, CNRZ1066, CS8, EPS, S9, LMG 18311, NCTC12958T, and GABA. Finally, a type IV system has been predicted in almost half of the strains analyzed, which contains only a restriction enzyme that recognizes and cuts modified DNA.
A more detailed investigation revealed that type II and type III R-M systems are absent or inactivated from S. thermophilus strains of subgroup AI. For this reason, we wanted to examine whether the presence/absence pattern of R-M systems in S. thermophilus is lineage specific. As demonstrated in Supplementary Figure S5 in several instances R-M systems are distributed on the chromosome in a manner that is characteristic for a potential lineage. This is particularly obvious for cluster A and more specifically for subgroups I, II, III, and IV. The R-M systems of strains in cluster B presented some similarities within the same subgroup, but they were more variable.
Despite the presence of the aforementioned defense mechanisms, complete prophages were also predicted for strains APC151, ND03, and NCTC12958T, while in the rest of the examined genomes only remnants of prophages have been identified (data not shown). In strains APC151 and ND03 the same prophage was predicted located within the EPS cluster of each strain. In strain NCTC12958T the intact prophage was previously described as phage 20617 by Arioli et al. (2018). Interestingly, the authors of this study demonstrated that the lysogenic strain NCTC12958T (DSM 20617T) exhibited higher adhesion to solid surfaces and heat resistance compared to the phage-cured derivative strain, suggesting some competitive advantage due to the stable association of the phage and the host.
Genomic Islands
Genomic islands acquired through HGT can provide adaptive and technological traits to the host microorganism (Juhas et al., 2009). In silico prediction of HGT in S. thermophilus has been previously reported (Hols et al., 2005; Liu et al., 2009; Eng et al., 2011). In this study, the GIs predicted by IslandViewer 4 in S. thermophilus ranged from 5 to 23 per strain, with sizes between 3.5 and 58 kbp and variable GC content from 26.1 to 45.2% (Supplementary Table S11). A total of 253 GIs were predicted, 31 of which were unique in 11 strains. The rest of the GIs have been identified in at least two S. thermophilus strains, either complete or partial. Of note, the genome array of ribosomal proteins was predicted as part of a GI in a number of strains. This is a false positive result, since it has been reported that the nucleotide composition of these arrays differentiates significantly from the rest protein coding genes (Hols et al., 2005; Fernandez-Gomez et al., 2012). Thus, these GIs (14 in total) were excluded from further analysis (Supplementary Tables S11, S12A). Several GIs were found to be present in both clusters A and B strains, while others were present either in cluster A or B strains. The first type of GIs was most probably acquired earlier than the second, i.e., before clusters A and B were separated. In accordance with what has been reported above for other genomic features, certain subgroups of strains display a unique distribution pattern of specific GIs that can support subgroups AI–AIV and BI (Supplementary Figure S6).
BLASTN analysis of the predicted GIs could not always reveal a potential donor. Nevertheless, a number of GIs could be traced back to specific microorganisms (coverage >70%, identity >90%; Supplementary Table S12B). The majority of species acting as potential donors belongs to the Streptococcus genus but also to other LAB like L. lactis, Lactobacillus casei, and Leuconostoc gelidum. In these last three cases GIs present high identity to plasmids carried by these organisms. In detail, specific GIs in subgroups AI, AII, and one GI in strain NCTC12958T present high identity to plasmids pLd7/p229C of L. lactis subsp. lactis (Kelleher et al., 2017; Van Mastrigt et al., 2018), pBD-II/pLC2W of L. casei (Ai et al., 2011; Chen et al., 2011; Song et al., 2018) and plasmid 1 of L. gelidum subsp. gasicomitatum (Andreevskaya et al., 2016), respectively. It is interesting to highlight that strains of S. thermophilus seem to have also interacted with members of the Streptococcus bovis/Streptococcus equinus complex (SBSEC), namely S. macedonicus, S. infantarius subsp. infantarius, Streptococcus gallolyticus, and S. equinus. Members of the complex are established members of the gastrointestinal tract (GIT) of ruminants, while certain species like S. macedonicus and S. infantarius are increasingly associated with fermented foods, especially of dairy origin (Jans et al., 2013a, b; Papadimitriou et al., 2014, 2015a).
A detailed investigation of the annotated features of S. thermophilus GIs revealed that they could be involved in EPS biosynthesis in accordance with previous findings reported for strains CNRZ1066, LMD-9, and LMG 18311 (Liu et al., 2009). CRISPR-Cas and complete R-M systems have been also identified in GIs. This would include CRIPR3 and CRISPR4 and type I and III R-M systems. In addition, the 38.5 kbp GI 9 contains most part of the intact prophage in strain NCTC12958T (Supplementary Table S12A). Our analysis supports the presence of bacteriocin coding genes in the GIs of a number of strains. However, Hols et al. (2005) suggested that the activity of these antimicrobial peptides may not be always guaranteed due to the absence of genes coding for transport or immunity proteins or other differences. For example, the locus of a class II bacteriocin-like peptide (blp) was experimentally studied in strains CNRZ1066, LMG 18311, and LMD-9 and it was concluded that it is only functional in the last strain (Hols et al., 2005). In strain B59671, GI 5 carries genes of the blp gene cluster involved in the production of the bacteriocin thermophilin 110 (Renye et al., 2017). Finally, in GI 6 of strain GABA we found a locus containing several genes coding for leader peptides (including mutacin IV, BlpU, and bovicin 255), but transport or immunity proteins seem to be inactive or absent (Supplementary Table S12A). Moreover, several genes involved in amino acid transport have been found in the predicted GIs of S. thermophilus strains. Some of these include a glutamate:GABA antiporter in strains APC151, GABA, and ND03, a dicarboxylate/amino acid:cation symporter in strains APC151, KLDS 3.1003, MN-BM-A01, MN-ZLW-002, ND03, and ST3 and a complete amino acid ABC transporter in strains CS8, EPS, KLDS 3.1003, and S9. The hdc cluster of strain KLDS 3.1003 was also identified in a GI and BLASTN analysis revealed possible HGT from a satellite phage. Furthermore, GI 7 of strain JIM 8232 corresponds to the biosynthetic gene cluster of histidine. As already mentioned, this region is also present in all cluster A S. thermophilus strains (plus strain NCTC12958T) but for unknown reasons it was assigned as a GI only in JIM 8232. BLASTN analysis revealed that this region presents high identity to the SBSEC member S. equinus (92%) supporting its potential acquisition by HGT in S. thermophilus chromosome. In addition, genes involved in fatty acid biosynthesis were identified in GIs of strains APC151, GABA, MN-BM-A01, MN-ZLW-002, and ND03, while stress response genes, e.g., coding for cold-shock proteins were also identified in a number of strains, including ASCC 1275, CNRZ1066, KLDS 3.1003, LMG 18311, ND03, and ST3. Finally, the gene cluster cbs-cblB-cysE involved in the metabolism of sulfur-containing amino acids has been previously suggested to have been transmitted by HGT from L. bulgaricus or Lactobacillus helveticus to S. thermophilus (Liu et al., 2009). Current analysis revealed that the respective cluster was predicted as part of a bigger GI in 17 S. thermophilus strains. More specifically, this GI along with the three genes were identified in strains APC151, GABA, KLDS 3.1003, LMD-9, LMG 18311, MN-BM-A01, MN-ZLW-002, ND03, and SMQ-301, while in strains ACA-DC 2, ASCC 1275, CNRZ1066, CS8, MN-BM-A02, ND07, S9, and ST3 the cysE is a putative pseudogene (Supplementary Table S12A).
It should be mentioned that Selle et al. (2015) identified four expendable GIs in the genome of strain LMD-9 with variable distribution in other sequenced strains. IslandViewer 4 did not predict GIs 1 and 2 reported in that study, while it detected GIs overlapping or included in GIs 3 and 4. These differences can be explained by the in silico methods employed to detect GIs. Selle et al. (2015) employed a strategy combining the location of potentially essential open reading frames (ORFs) and highly similar insertion sequences (ISs) which is distinct from the strategies employed by the tools included in IslandViewer 4.
S. thermophilus Genes Implicated in Protocooperation With L. bulgaricus
The bacterial pair of S. thermophilus and L. bulgaricus is routinely employed in yogurt production. The mutually beneficial interaction between these bacteria in the yogurt ecosystem, known as protocooperation, is based on the exchange of metabolites and results in improved metabolic performance related to accelerated acidification, enhanced EPS production and abundance of aroma volatiles. Initially, S. thermophilus boosts the growth of L. bulgaricus by lowering the pH and providing formic, pyruvic and folic acid as well as carbon dioxide. Subsequently, L. bulgaricus stimulates S. thermophilus growth by producing peptides and free amino acids (Settachaimongkon et al., 2014). Transcriptome analysis of a mixed S. thermophilus and L. bulgaricus culture also supports that metabolites like formic and folic acid produced by S. thermophilus are utilized by L. bulgaricus as precursors in purine biosynthesis (Sieuwerts et al., 2010). S. thermophilus carries genes encoding pyruvate formate lyase (PFL) and pyruvate formate-lyase activating (PFLA) enzyme, while L. bulgaricus lacks these genes (Nishimura et al., 2013). Our analysis revealed the presence of both pfl and pflA in all S. thermophilus strains examined (Supplementary Table S13).
In addition, a number of studies have been performed concerning the role of PrtS produced by S. thermophilus during manufacture of dairy products, especially yogurt. For example, PrtS production may positively affect S. thermophilus growth in a pure culture, but it may be neutral in a mixture with L. bulgaricus strains producing the protease PrtB (Courtin et al., 2002). In a more recent study, it was demonstrated that only non-proteolytic S. thermophilus strains performed protocooperation with L. bulgaricus (Settachaimongkon et al., 2014). As already mentioned, the majority of cluster A strains carries prtS, while it is absent from all cluster B strains, indicating that the latter may be more appropriate for protocooperation. However, specific S. thermophilus strains carrying the prtS have been shown to exhibit weak or no PrtS activity (Galia et al., 2009; Cui et al., 2016). In our dataset in strains MN-BM-A01 and SMQ-301 prtS was found to be truncated, an observation that may support to a degree the findings by Galia et al. (2009). Furthermore, it was recently reported that prtS+ strains may also present some technological advantages (Tian et al., 2018). We thus believe that more research is needed to establish the actual role of prtS regarding protocooperation.
The response of S. thermophilus to H2O2 produced by L. bulgaricus has also been studied. It appears that there is an inverse correlation between iron intake by S. thermophilus and H2O2 production by L. bulgaricus, and that S. thermophilus in the presence of H2O2 is regulating iron metabolism in order to diminish the production of harmful reactive oxygen species (ROS) (Herve-Jimenez et al., 2009; Sieuwerts et al., 2010). However, the results of two different studies are rather diverge. In one study, the expression patterns of S. thermophilus genes related to iron transport in the presence of L. bulgaricus were found to be upregulated (Sieuwerts et al., 2010), while in another study downregulated (Herve-Jimenez et al., 2009). Only dpr (peroxide resistance protein) and fur (ferric transport regulator protein) were found upregulated in both studies. In silico analysis of the 23 S. thermophilus strains revealed that dpr and fur belong to the core genome, while the iron ABC transporter is absent from strains JIM 8232, MN-ZLW-002, ND03, APC151, MN-BM-A01, and ST3 (Supplementary Table S13).
A novel protocooperation relationship between S. thermophilus and L. bulgaricus in yogurt fermentation concerns the bi-functional glutathione (GSH) synthetase gene of S. thermophilus, which produces GSH (Wang et al., 2016). The respective gene was found to be conserved in all 23 S. thermophilus strains analyzed (Supplementary Table S13). In a recent study, it was demonstrated that GSH produced by S. thermophilus provided protection to both S. thermophilus and L. bulgaricus cells toward acid stress. Additionally, the secreted GSH could enhance the growth of L. bulgaricus (Wang et al., 2016). Finally, genes related to EPS production were found to be upregulated in both microorganisms in a mixed culture when compared to monocultures, and thus they may play an important role in the texture of the final product (Sieuwerts et al., 2010). Given the heterogeneity observed in the EPS gene cluster of S. thermophilus strains, no mechanistic insight could be inferred.
Conclusion
Streptococcus thermophilus is a starter of great economic significance for the dairy industry contributing to the production of world-wide consumed dairy products like yogurt and cheeses. A number of studies have been published in an attempt to explore and interpret various features of the species biology related to its technological potential. This became more feasible during the last two decades with the sequencing of genomes of S. thermophilus strains. In this study we analyzed 23 fully sequenced genomes of S. thermophilus in order to examine features of the species related to technological and evolutionary traits. Even from the beginning of our study, it became evident that strains of S. thermophilus present some variability considering the properties of the genomes (e.g., size, gene content, % of pseudogenes, rRNA and tRNA content). Core genome and ANI phylogenetic analysis revealed a specific pattern of clustering of strains (Figure 8). A main observation was that most strains could be separated in two major clusters. Cluster A was characterized by larger genomes, the presence of prtS in the majority of strains, the inclusion of a histidine biosynthesis gene cluster, as well as the presence of certain CRISPR-Cas system types and specific GIs. Strains in cluster B diversified from those in cluster A in all these aspects. These observations indicated the existence of at least two major lineages in S. thermophilus that appear at ANI values >98%. Further investigation suggested the presence of subgroups within the two clusters, i.e., subgroups AI–AIV and BI. The existence of these subgroups was also supported to a variable degree during COG analysis as well as the presence/absence pattern of specific loci and/or their organization, i.e., EPS clusters, CRISPR arrays, R-M systems and GIs. Clustering of S. thermophilus strains based on the spacers of CRISPR arrays has been performed before (Horvath et al., 2008; Delorme et al., 2017). Given the fact that CRISPR arrays can provide a retrospective view of the history of each strain based on the parasitic DNA it was exposed to, spacer sequences of the CRISPR1 which is present practically in all strains support the existence of evolutionary distinct lineages in S. thermophilus. Biodiversity within strains of S. thermophilus has been previously suggested using CRISPR array and/or MLST clustering (Horvath et al., 2008; Delorme et al., 2010, 2017; Yu et al., 2015). In our opinion, clustering of strains according to CRISPR array architecture or even MLST has important advantages (e.g., the ability to screen many strains), but these approaches may derive more easily to the characterization of potential clonal strains due to the use of limited genomic information. In contrast, whole genome phylogeny based on core genes should be more robust, while analysis of complete genome sequences may provide even more information concerning the discrimination of strains based on loci beyond core genome, like accessory genes or even unique genes. The subgroups we describe appeared at ANI values well above 99%, an observation that could indicate that they derive from clonal strains. A closer investigation of the data presented in this study suggests in some cases differences among strains of the same subgroup. For example, this becomes obvious when considering the exact sizes of the chromosome of the strains, the exact gene content (including accessory genes but also genes that are exclusively absent from a specific strain). In some instances, differences were observed in the EPS clusters, the distribution of R-M systems and GIs of strains within the same subgroup. Even though the differences among strains of the same subgroup may be rather subtle thus justifying the high ANI values at which their relatedness appears, they diversify strains beyond the strict definition of clones. Our analysis concerning the genome assemblies of the strains suggested a quality level that may not interfere with the grouping scheme we describe. Nonetheless, apart from the differences identified among the strains, our analysis also validated common features or features beyond the clustering pattern mentioned above (Figure 8). These would include characteristic traits for the adaptation of S. thermophilus to milk, like the conserved gal-lac and urease operons, the extended arsenal of peptidases and amino acid/peptide transporters in parallel to genes related to protocooperation. The high percentage of pseudogenes has been related to the reductive evolution of S. thermophilus during adaptation to rich in nutrients dairy niches (Bolotin et al., 2004; Hols et al., 2005; Goh et al., 2011). This trait was also apparent in all strains analyzed here. Interestingly, features related to milk adaptation seem to be also present in APC151. The strain does not diversify from the dairy strains, even though it was the only strain in our dataset that was isolated from a non-dairy environment, i.e., the fish intestine. This was also suggested previously (Linares et al., 2017). This relatively odd observation highlights the need to study strains found in environments different than milk and dairy products to fully apprehend the evolution of the species. Finally, the pan genome of the species is not closed yet, suggesting that sequencing of additional strains will be important. Certain new complete genomes have appeared in the databases since the initiation of our analysis (Proust et al., 2018; Renye et al., 2019), but more are required to further expand and validate any lineage-like patterns that may exist and could be related to the technological/probiotic repertoire of S. thermophilus.
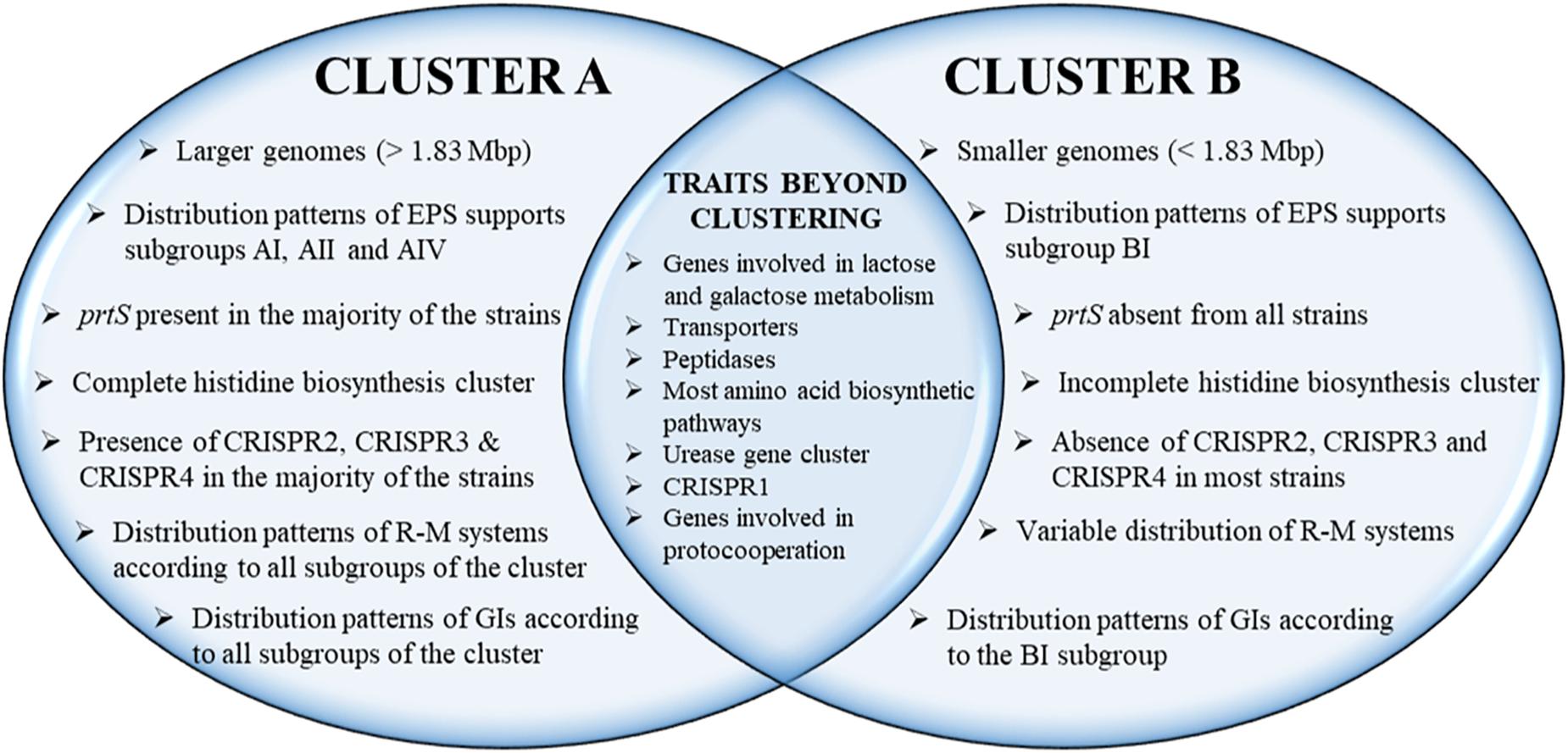
Figure 8. Schematic representation of structural and functional genomic traits that support the distinction of S. thermophilus strains in clusters A and B. The genomic features included in the common area of the Venn diagram are either present to all strains or they exhibit a presence/absence pattern beyond clusters A and B.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
VA and MK performed genome analysis and participated in the writing of the manuscript. JB and BP performed genome analysis. KP conceived the project, performed genome analysis, and participated in the writing of the manuscript. ET conceived the project and participated in the writing of the manuscript. All authors read and approved the final manuscript.
Funding
The present work was co-financed by the European Social Fund and the National Resources EPEAEK and YPEPTH through the Thales project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02916/full#supplementary-material
Abbreviations
ABC, ATP-binding cassette; ANI, average nucleotide identity; APC, amino acid-polyamine-organocation; blp, bacteriocin-like peptide; BPGA, bacterial pan genome analysis; COG, clusters of orthologous groups; CRISPR-Cas, clustered regularly interspaced short palindromic repeats-CRISPR associated; DRs, direct repeats; EFSA, European Food Safety Authority; EPS, exopolysaccharide; FDA, Food and Drug Administration; GABA, gamma-aminobutyric acid; GIs, genomic islands; GIT, gastrointestinal tract; GRAS, generally recognized as safe; GSH, glutathione; HGT, horizontal gene transfer; ISs, insertion sequences; KEGG, Kyoto Encyclopedia of Genes and Genomes; KOALA, KEGG orthology and links annotation; LAB, lactic acid bacteria; LCBs, local collinear blocks; MiGA, microbial genomes atlas; MLST, multilocus sequence typing; ORFs, open reading frames; PFGE, pulsed-field gel electrophoresis; PFL, pyruvate formate lyase; PFLA, pyruvate formate-lyase activating; PGAP, prokaryotic genome annotation pipeline; QPS, qualified presumption of safety; RAPD, random amplification of polymorphic DNA; RAST, rapid annotation using subsystem technology; R-M, restriction-modification; ROS, reactive oxygen species; SBSEC, Streptococcus bovis/Streptococcus equinus complex.
Footnotes
References
Ai, L., Chen, C., Zhou, F., Wang, L., Zhang, H., Chen, W., et al. (2011). Complete genome sequence of the probiotic strain Lactobacillus casei BD-II. J. Bacteriol. 193, 3160–3161. doi: 10.1128/JB.00421-11
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., and Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538. doi: 10.1038/nbt.2579
Alexandraki, V., Kazou, M., Blom, J., Pot, B., Tsakalidou, E., and Papadimitriou, K. (2017). The complete genome sequence of the yogurt isolate Streptococcus thermophilus ACA-DC 2. Stand. Genomic Sci. 12:18. doi: 10.1186/s40793-017-0227-5
Anbukkarasi, K., Nanda, D. K., Umamaheswari, T., Hemalatha, T., Singh, P., and Singh, R. (2014). Assessment of expression of Leloir pathway genes in wild-type galactose-fermenting Streptococcus thermophilus by real-time PCR. Eur. Food Res. Technol. 239, 895–903. doi: 10.1007/s00217-014-2286-9
Anbukkarasi, K., Umamaheswari, T., Hemalatha, T., Nanda, D. K., Singh, P., Rashmi, H. M., et al. (2013). Production of low browning Mozzarella cheese: screening and characterization of wild galactose fermenting Streptococcus thermophilus strains. Int. J. Adv. Res. 1, 83–96.
Andreevskaya, M., Hultman, J., Johansson, P., Laine, P., Paulin, L., Auvinen, P., et al. (2016). Complete genome sequence of Leuconostoc gelidum subsp. gasicomitatum KG16-1, isolated from vacuum-packaged vegetable sausages. Stand. Genomic Sci. 11:40. doi: 10.1186/s40793-016-0164-8
Arioli, S., Eraclio, G., Della Scala, G., Neri, E., Colombo, S., Scaloni, A., et al. (2018). Role of temperate bacteriophage ϕ20617 on Streptococcus thermophilus DSM 20617T autolysis and biology. Front. Microbiol. 9:2719. doi: 10.3389/fmicb.2018.02719
Arioli, S., Monnet, C., Guglielmetti, S., Parini, C., De Noni, I., Hogenboom, J., et al. (2007). Aspartate biosynthesis is essential for the growth of Streptococcus thermophilus in milk, and aspartate availability modulates the level of urease activity. Appl. Environ. Microbiol. 73, 5789–5796. doi: 10.1128/AEM.00533-07
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bai, Y., Sun, E., Shi, Y., Jiang, Y., Chen, Y., Liu, S., et al. (2016). Complete genome sequence of Streptococcus thermophilus MN-BM-A01, a strain with high exopolysaccharides production. J. Biotechnol. 224, 45–46. doi: 10.1016/j.jbiotec.2016.03.003
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Blom, J., Kreis, J., Spanig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bolotin, A., Quinquis, B., Renault, P., Sorokin, A., Ehrlich, S. D., Kulakauskas, S., et al. (2004). Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat. Biotechnol. 22, 1554–1558. doi: 10.1038/nbt1034
Bolotin, A., Quinquis, B., Sorokine, A., and Dusko Ehrlich, S. (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. doi: 10.1099/mic.0.28048-0
Broadbent, J. R., Mcmahon, D. J., Welker, D. L., Oberg, C. J., and Moineau, S. (2003). Biochemistry, genetics, and applications of exopolysaccharide production in Streptococcus thermophilus: a review. J. Dairy Sci. 86, 407–423. doi: 10.3168/jds.S0022-0302(03)73619-4
Burall, L. S., Grim, C. J., Mammel, M. K., and Datta, A. R. (2016). Whole genome sequence analysis using JSpecies tool establishes clonal relationships between Listeria monocytogenes strains from epidemiologically unrelated listeriosis outbreaks. PLoS One 11:e0150797. doi: 10.1371/journal.pone.0150797
Calles-Enríquez, M., Eriksen, B. H., Andersen, P. S., Rattray, F. P., Johansen, A. H., Fernández, M., et al. (2010). Sequencing and transcriptional analysis of the Streptococcus thermophilus histamine biosynthesis gene cluster: factors that affect differential hdcA expression. Appl. Environ. Microbiol. 76, 6231–6238. doi: 10.1128/AEM.00827-10
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chen, C., Ai, L., Zhou, F., Wang, L., Zhang, H., Chen, W., et al. (2011). Complete genome sequence of the probiotic bacterium Lactobacillus casei LC2W. J. Bacteriol. 193, 3419–3420. doi: 10.1128/JB.05017-11
Chen, Y., Zhang, M., and Ren, F. (2019). A role of exopolysaccharide produced by Streptococcus thermophilus in the intestinal inflammation and mucosal barrier in Caco-2 monolayer and dextran sulphate sodium-induced experimental murine colitis. Molecules 24:513. doi: 10.3390/molecules24030513
Chen, Y.-Y. M., and Burne, R. A. (2003). Identification and characterization of the nickel uptake system for urease biogenesis in Streptococcus salivarius 57.I. J. Bacteriol. 185, 6773–6779. doi: 10.1128/jb.185.23.6773-6779.2003
Courtin, P., Monnet, V., and Rul, F. (2002). Cell-wall proteinases PrtS and PrtB have a different role in Streptococcus thermophilus/Lactobacillus bulgaricus mixed cultures in milk. Microbiology 148, 3413–3421. doi: 10.1099/00221287-148-11-3413
Cui, Y., Jiang, X., Hao, M., Qu, X., and Hu, T. (2017). New advances in exopolysaccharides production of Streptococcus thermophilus. Arch. Microbiol. 199, 799–809. doi: 10.1007/s00203-017-1366-1
Cui, Y., Xu, T., Qu, X., Hu, T., Jiang, X., and Zhao, C. (2016). New insights into various production characteristics of Streptococcus thermophilus strains. Int. J. Mol. Sci. 17:E1701. doi: 10.3390/ijms17101701
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Darling, A. E., Miklós, I., and Ragan, M. A. (2008). Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 4:e1000128. doi: 10.1371/journal.pgen.1000128
De Angelis, M., and Gobetti, M. (2011). “Stress responses of lactobacilli,” in Stress Responses of Lactic Acid Bacteria, eds E. Tsakalidou and K. Papadimitriou, (New York, NY: Springer Science+Business Media), 219–249. doi: 10.1007/978-0-387-92771-8_11
De Vin, F., Radstrom, P., Herman, L., and De Vuyst, L. (2005). Molecular and biochemical analysis of the galactose phenotype of dairy Streptococcus thermophilus strains reveals four different fermentation profiles. Appl. Environ. Microbiol. 71, 3659–3667. doi: 10.1128/AEM.71.7.3659-3667.2005
Degeest, B., and De Vuyst, L. (2000). Correlation of activities of the enzymes alpha-phosphoglucomutase, UDP-galactose 4-epimerase, and UDP-glucose pyrophosphorylase with exopolysaccharide biosynthesis by Streptococcus thermophilus LY03. Appl. Environ. Microbiol. 66, 3519–3527. doi: 10.1128/aem.66.8.3519-3527.2000
Delorme, C., Abraham, A. L., Renault, P., and Guédon, E. (2015). Genomics of Streptococcus salivarius, a major human commensal. Infect. Genet. Evol. 33, 381–392. doi: 10.1016/j.meegid.2014.10.001
Delorme, C., Bartholini, C., Bolotine, A., Ehrlich, S. D., and Renault, P. (2010). Emergence of a cell wall protease in the Streptococcus thermophilus population. Appl. Environ. Microbiol. 76, 451–460. doi: 10.1128/AEM.01018-09
Delorme, C., Bartholini, C., Luraschi, M., Pons, N., Loux, V., Almeida, M., et al. (2011). Complete genome sequence of the pigmented Streptococcus thermophilus strain JIM8232. J. Bacteriol. 193, 5581–5582. doi: 10.1128/JB.05404-11
Delorme, C., Legravet, N., Jamet, E., Hoarau, C., Alexandre, B., El-Sharoud, W. M., et al. (2017). Study of Streptococcus thermophilus population on a world-wide and historical collection by a new MLST scheme. Int. J. Food Microbiol. 242, 70–81. doi: 10.1016/j.ijfoodmicro.2016.11.016
Dupuis, M.-È., Villion, M., Magadán, A. H., and Moineau, S. (2013). CRISPR-Cas and restriction–modification systems are compatible and increase phage resistance. Nat. Commun. 4:2087. doi: 10.1038/ncomms3087
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Eisen, J. A., Heidelberg, J. F., White, O., and Salzberg, S. L. (2000). Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 1:RESEARCH0011. doi: 10.1186/gb-2000-1-6-research0011
Elbourne, L. D., Tetu, S. G., Hassan, K. A., and Paulsen, I. T. (2017). TransportDB 2.0: a database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 45, D320–D324. doi: 10.1093/nar/gkw1068
Eng, C., Thibessard, A., Danielsen, M., Rasmussen, T. B., Mari, J. F., and Leblond, P. (2011). In silico prediction of horizontal gene transfer in Streptococcus thermophilus. Arch. Microbiol. 193, 287–297. doi: 10.1007/s00203-010-0671-8
Ercolini, D., Fusco, V., Blaiotta, G., and Coppola, S. (2005). Sequence heterogeneity in the lacSZ operon of Streptococcus thermophilus and its use in PCR systems for strain differentiation. Res. Microbiol. 156, 161–172. doi: 10.1016/j.resmic.2004.09.005
European Food Safety Authority [EFSA], (2007). Opinion of the scientific committee on a request from EFSA on the introduction of a Qualified Presumption of Safety (QPS) approach for assessment of selected microorganisms referred to EFSA. EFSA J. 587, 1–16. doi: 10.2903/j.efsa.2007.587
Evivie, S. E., Li, B., Ding, X., Meng, Y., Yu, S., Du, J., et al. (2017). Complete genome sequence of Streptococcus thermophilus KLDS 3.1003, a strain with high antimicrobial potential against foodborne and vaginal pathogens. Front. Microbiol. 8:1238. doi: 10.3389/fmicb.2017.01238
Fernandez-Gomez, B., Fernandez-Guerra, A., Casamayor, E. O., Gonzalez, J. M., Pedros-Alio, C., and Acinas, S. G. (2012). Patterns and architecture of genomic islands in marine bacteria. BMC Genomics 13:347. doi: 10.1186/1471-2164-13-347
Food and Drug Administration [FDA] (2007). 21 CFR Part 131: Microorganisms & Microbial-Derived Ingredients Used in Food (Partial List). Silver Spring, MA: FDA.
Galia, W., Jameh, N., Perrin, C., Genay, M., and Dary-Mourot, A. (2016). Acquisition of PrtS in Streptococcus thermophilus is not enough in certain strains to achieve rapid milk acidification. Dairy Sci. Technol. 96, 623–636. doi: 10.1007/s13594-016-0292-3
Galia, W., Perrin, C., Genay, M., and Dary, A. (2009). Variability and molecular typing of Streptococcus thermophilus strains displaying different proteolytic and acidifying properties. Int. Dairy J. 19, 89–95. doi: 10.1016/j.idairyj.2008.08.004
Garault, P., Letort, C., Juillard, V., and Monnet, V. (2000). Branched-chain amino acid biosynthesis is essential for optimal growth of Streptococcus thermophilus in milk. Appl. Environ. Microbiol. 66, 5128–5133. doi: 10.1128/aem.66.12.5128-5133.2000
Gardini, F., Rossi, F., Rizzotti, L., Torriani, S., Grazia, L., Chiavari, C., et al. (2012). Role of Streptococcus thermophilus PRI60 in histamine accumulation in cheese. Int. Dairy J. 27, 71–76. doi: 10.1016/j.idairyj.2012.07.005
Geertsma, E. R., Duurkens, R. H., and Poolman, B. (2005). The activity of the lactose transporter from Streptococcus thermophilus is increased by phosphorylated IIA and the action of β-galactosidase. Biochemistry 44, 15889–15897. doi: 10.1021/bi051638w
Giaretta, S., Treu, L., Vendramin, V., Da Silva Duarte, V., Tarrah, A., Campanaro, S., et al. (2018). Comparative transcriptomic analysis of Streptococcus thermophilus TH1436 and TH1477 showing different capability in the use of galactose. Front. Microbiol. 9:1765. doi: 10.3389/fmicb.2018.01765
Giraffa, G., Paris, A., Valcavi, L., Gatti, M., and Neviani, E. (2001). Genotypic and phenotypic heterogeneity of Streptococcus thermophilus strains isolated from dairy products. J. Appl. Microbiol. 91, 937–943. doi: 10.1046/j.1365-2672.2001.01464.x
Goh, Y. J., Goin, C., O’flaherty, S., Altermann, E., and Hutkins, R. (2011). Specialized adaptation of a lactic acid bacterium to the milk environment: the comparative genomics of Streptococcus thermophilus LMD-9. Microb. Cell Fact. 10(Suppl. 1):S22. doi: 10.1186/1475-2859-10-S1-S22
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hafeez, Z., Cakir-Kiefer, C., Girardet, J.-M., Lecomte, X., Paris, C., Galia, W., et al. (2015). New insights into the proteolytic system of Streptococcus thermophilus: use of isracidin to characterize cell-associated extracellular peptidase activities. J. Agric. Food Chem. 63, 7522–7531. doi: 10.1021/acs.jafc.5b01647
Hafeez, Z., Cakir-Kiefer, C., Lecomte, X., Miclo, L., and Dary-Mourot, A. (2019). The X-prolyl dipeptidyl-peptidase PepX of Streptococcus thermophilus initially described as intracellular is also responsible for peptidase extracellular activity. J. Dairy Sci. 102, 113–123. doi: 10.3168/jds.2018-14823
Han, X., Yang, Z., Jing, X., Yu, P., Zhang, Y., Yi, H., et al. (2016). Improvement of the texture of yogurt by use of exopolysaccharide producing lactic acid bacteria. Biomed. Res. Int. 2016:7945675. doi: 10.1155/2016/7945675
Hao, M., Cui, Y., and Qu, X. (2018). Analysis of CRISPR-Cas system in Streptococcus thermophilus and its application. Front. Microbiol. 9:257. doi: 10.3389/fmicb.2018.00257
Hatmaker, E. A., Riley, L. A., O’dell, K. B., Papanek, B., Graveley, B. R., Garrett, S. C., et al. (2018). Complete genome sequence of industrial dairy strain Streptococcus thermophilus DGCC 7710. Genome Announc. 6:e01587-17. doi: 10.1128/genomeA.01587-17
Herve-Jimenez, L., Guillouard, I., Guedon, E., Boudebbouze, S., Hols, P., Monnet, V., et al. (2009). Postgenomic analysis of Streptococcus thermophilus cocultivated in milk with Lactobacillus delbrueckii subsp. bulgaricus: involvement of nitrogen, purine, and iron metabolism. Appl. Environ. Microbiol. 75, 2062–2073. doi: 10.1128/AEM.01984-08
Hols, P., Hancy, F., Fontaine, L., Grossiord, B., Prozzi, D., Leblond-Bourget, N., et al. (2005). New insights in the molecular biology and physiology of Streptococcus thermophilus revealed by comparative genomics. FEMS Microbiol. Rev. 29, 435–463. doi: 10.1016/j.fmrre.2005.04.008
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Horvath, P., Romero, D. A., Coute-Monvoisin, A. C., Richards, M., Deveau, H., Moineau, S., et al. (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190, 1401–1412. doi: 10.1128/JB.01415-07
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Iyer, R., Tomar, S. K., Uma Maheswari, T., and Singh, R. (2010). Streptococcus thermophilus strains: multifunctional lactic acid bacteria. Int. Dairy J. 20, 133–141. doi: 10.1016/j.idairyj.2009.10.005
Jans, C., Follador, R., Hochstrasser, M., Lacroix, C., Meile, L., and Stevens, M. J. A. (2013a). Comparative genome analysis of Streptococcus infantarius subsp. infantarius CJ18, an African fermented camel milk isolate with adaptations to dairy environment. BMC Genomics 14:200. doi: 10.1186/1471-2164-14-200
Jans, C., Kaindi, D. W. M., Böck, D., Njage, P. M. K., Kouamé-Sina, S. M., Bonfoh, B., et al. (2013b). Prevalence and comparison of Streptococcus infantarius subsp. infantarius and Streptococcus gallolyticus subsp. macedonicus in raw and fermented dairy products from East and West Africa. Int. J. Food Microbiol. 167, 186–195. doi: 10.1016/j.ijfoodmicro.2013.09.008
Juhas, M., Van Der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Junges, R., Maienschein-Cline, M., Morrison, D. A., and Petersen, F. C. (2019). Complete genome sequence of Streptococcus pneumoniae serotype 19F strain EF3030. Microbiol. Resour. Announc. 8:e00198-19. doi: 10.1128/MRA.00198-19
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016a). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kanehisa, M., Sato, Y., and Morishima, K. (2016b). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kang, X., Ling, N., Sun, G., Zhou, Q., Zhang, L., and Sheng, Q. (2012). Complete genome sequence of Streptococcus thermophilus strain MN-ZLW-002. J. Bacteriol. 194, 4428–4429. doi: 10.1128/JB.00740-12
Kelleher, P., Bottacini, F., Mahony, J., Kilcawley, K. N., and Van Sinderen, D. (2017). Comparative and functional genomics of the Lactococcus lactis Taxon; insights into evolution and niche adaptation. BMC Genomics 18:267. doi: 10.1186/s12864-017-3650-5
Kongo, J. M. (2013). “Lactic acid bacteria as starter-cultures for cheese processing: past, present and future developments,” in Lactic Acid Bacteria - R & D for Food, Health and Livestock Purposes, ed. J. M. Kongo, (London: IntechOpen), doi: 10.5772/55937
Labrie, S. J., Tremblay, D. M., Plante, P. L., Wasserscheid, J., Dewar, K., Corbeil, J., et al. (2015). Complete genome sequence of Streptococcus thermophilus SMQ-301, a model strain for phage-host interactions. Genome Announc. 3:e00480-15. doi: 10.1128/genomeA.00480-15
Lefébure, T., and Stanhope, M. J. (2007). Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8:R71. doi: 10.1186/gb-2007-8-5-r71
Lerat, L., and Ochman, H. (2005). Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 33, 3125–3132. doi: 10.1093/nar/gki631
Letort, C., and Juillard, V. (2001). Development of a minimal chemically-defined medium for the exponential growth of Streptococcus thermophilus. J. Appl. Microbiol. 91, 1023–1029. doi: 10.1046/j.1365-2672.2001.01469.x
Li, B., Ding, X., Evivie, S. E., Jin, D., Meng, Y., Huo, G., et al. (2018). Short communication: genomic and phenotypic analyses of exopolysaccharides produced by Streptococcus thermophilus KLDS SM. J. Dairy Sci. 101, 106–112. doi: 10.3168/jds.2017-13534
Limauro, D., Falciatore, A., Basso, A. L., Forlani, G., and De Felice, M. (1996). Proline biosynthesis in Streptococcus thermophilus: characterization of the proBA operon and its products. Microbiology 142, 3275–3282. doi: 10.1099/13500872-142-11-3275
Linares, D. M., Arboleya, S., Ross, R. P., and Stanton, C. (2017). Complete genome sequence of the gamma-aminobutyric acid-producing strain Streptococcus thermophilus APC151. Genome Announc. 5:e00205-17. doi: 10.1128/genomeA.00205-17
Linares, D. M., O’callaghan, T. F., O’connor, P. M., Ross, R. P., and Stanton, C. (2016). Streptococcus thermophilus APC151 strain is suitable for the manufacture of naturally GABA-enriched bioactive yogurt. Front. Microbiol. 7:1876. doi: 10.3389/fmicb.2016.01876
Liu, M., Bayjanov, J. R., Renckens, B., Nauta, A., and Siezen, R. J. (2010). The proteolytic system of lactic acid bacteria revisited: a genomic comparison. BMC Genomics 11:36. doi: 10.1186/1471-2164-11-36
Liu, M., Siezen, R. J., and Nauta, A. (2009). In silico prediction of horizontal gene transfer events in Lactobacillus bulgaricus and Streptococcus thermophilus reveals protocooperation in yogurt manufacturing. Appl. Environ. Microbiol. 75, 4120–4129. doi: 10.1128/AEM.02898-08
Lluis-Arroyo, D., Flores-Nájera, A., Cruz-Guerrero, A., Gallardo-Escamilla, F., Lobato-Calleros, C., Jiménez-Guzmán, J., et al. (2014). Effect of an exopolysaccharide-producing strain of Streptococcus thermophilus on the yield and texture of Mexican Manchego-type cheese. Int. J. Food Prop. 17, 1680–1693. doi: 10.1080/10942912.2011.599091
Louis, E. P., Wei, Y., and Terns, M. P. (2017). Investigating the molecular mechanism of CRISPR-Cas adaptation of Streptococcus thermophilus. J. Immunol. 198(Suppl. 1):67.11.
Makarova, K., Slesarev, A., Wolf, Y., Sorokin, A., Mirkin, B., Koonin, E., et al. (2006). Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 15611–15616. doi: 10.1073/pnas.0607117103
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Manat, G., Roure, S., Auger, R., Bouhss, A., Barreteau, H., Mengin-Lecreulx, D., et al. (2014). Deciphering the metabolism of undecaprenyl-phosphate: the bacterial cell-wall unit carrier at the membrane frontier. Microb. Drug Resist. 20, 199–214. doi: 10.1089/mdr.2014.0035
Mayo, B., Van Sinderen, D., and Ventura, M. (2008). Genome analysis of food grade lactic Acid-producing bacteria: from basics to applications. Curr. Genomics 9, 169–183. doi: 10.2174/138920208784340731
Monnet, C., Mora, D., and Corrieu, G. (2005). Glutamine synthesis is essential for growth of Streptococcus thermophilus in milk and is linked to urea catabolism. Appl. Environ. Microbiol. 71, 3376–3378. doi: 10.1128/AEM.71.6.3376-3378.2005
Mora, D., Fortina, M. G., Parini, C., Ricci, G., Gatti, M., Giraffa, G., et al. (2002). Genetic diversity and technological properties of Streptococcus thermophilus strains isolated from dairy products. J. Appl. Microbiol. 93, 278–287. doi: 10.1046/j.1365-2672.2002.01696.x
Mora, D., Maguin, E., Masiero, M., Parini, C., Ricci, G., Manachini, P. L., et al. (2004). Characterization of urease genes cluster of Streptococcus thermophilus. J. Appl. Microbiol. 96, 209–219. doi: 10.1046/j.1365-2672.2003.02148.x
Mora, D., Monnet, C., Parini, C., Guglielmetti, S., Mariani, A., Pintus, P., et al. (2005). Urease biogenesis in Streptococcus thermophilus. Res. Microbiol. 156, 897–903. doi: 10.1016/j.resmic.2005.04.005
Moschetti, G., Blaiotta, G., Aponte, M., Catzeddu, P., Villani, F., Deiana, P., et al. (1998). Random amplified polymorphic DNA and amplified ribosomal DNA spacer polymorphism: powerful methods to differentiate Streptococcus thermophilus strains. J. Appl. Microbiol. 85, 25–36. doi: 10.1046/j.1365-2672.1998.00461.x
Ninova-Nikolova, N., and Urshev, Z. (2013). Fast acidifying urease-deficient Streptococcus thermophilus isolate shows spontaneous deletion of its complete urease operon. Bulg. J. Agric. Sci. 19, 112–116.
Nishimura, J., Kawai, Y., Aritomo, R., Ito, Y., Makino, S., Ikegami, S., et al. (2013). Effect of formic acid on exopolysaccharide production in skim milk fermentation by Lactobacillus delbrueckii subsp. bulgaricus OLL1073R-1. Biosci. Microbiota Food Health 32, 23–32. doi: 10.12938/bmfh.32.23
Padmanabhan, A., Tong, Y., Wu, Q., Zhang, J., and Shah, N. P. (2018). Transcriptomic insights into the growth phase- and sugar-associated changes in the exopolysaccharide production of a high EPS-producing Streptococcus thermophilus ASCC 1275. Front. Microbiol. 9:1919. doi: 10.3389/fmicb.2018.01919
Papadimitriou, K., Anastasiou, R., Maistrou, E., Plakas, T., Papandreou, N. C., Hamodrakas, S. J., et al. (2015a). Acquisition through horizontal gene transfer of plasmid pSMA198 by Streptococcus macedonicus ACA-DC 198 points towards the dairy origin of the species. PLoS One 10:e0116337. doi: 10.1371/journal.pone.0116337
Papadimitriou, K., Pot, B., and Tsakalidou, E. (2015b). How microbes adapt to a diversity of food niches. Curr. Opin. Food Sci. 2, 29–35. doi: 10.1016/j.cofs.2015.01.001
Papadimitriou, K., Anastasiou, R., Mavrogonatou, E., Blom, J., Papandreou, N. C., Hamodrakas, S. J., et al. (2014). Comparative genomics of the dairy isolate Streptococcus macedonicus ACA-DC 198 against related members of the Streptococcus bovis/Streptococcus equinus complex. BMC Genomics 15:272. doi: 10.1186/1471-2164-15-272
Pastink, M. I., Teusink, B., Hols, P., Visser, S., De Vos, W. M., and Hugenholtz, J. (2009). Genome-scale model of Streptococcus thermophilus LMG18311 for metabolic comparison of lactic acid bacteria. Appl. Environ. Microbiol. 75, 3627–3633. doi: 10.1128/AEM.00138-09
Pernoud, S., Fremaux, C., Sepulchre, A., Corrieu, G., and Monnet, C. (2004). Effect of the metabolism of urea on the acidifying activity of Streptococcus thermophilus. J. Dairy Sci. 87, 550–555. doi: 10.3168/jds.S0022-0302(04)73196-3
Proust, L., Loux, V., Martin, V., Magnabosco, C., Pedersen, M., Monnet, V., et al. (2018). Complete genome sequence of the industrial fast-acidifying strain Streptococcus thermophilus N4L. Microbiol. Resour. Announc. 7:e01029-18. doi: 10.1128/MRA.01029-18
Purwandari, U., Shah, N. P., and Vasiljevic, T. (2007). Effects of exopolysaccharide-producing strains of Streptococcus thermophilus on technological and rheological properties of set-type yoghurt. Int. Dairy J. 17, 1344–1352. doi: 10.1016/j.idairyj.2007.01.018
Qiao, Y., Liu, G., Leng, C., Zhang, Y., Lv, X., Chen, H., et al. (2018). Metabolic profiles of cysteine, methionine, glutamate, glutamine, arginine, aspartate, asparagine, alanine and glutathione in Streptococcus thermophilus during pH-controlled batch fermentations. Sci. Rep. 8:12441. doi: 10.1038/s41598-018-30272-5
Rantsiou, K., Urso, R., Dolci, P., Comi, G., and Cocolin, L. (2008). Microflora of Feta cheese from four Greek manufacturers. Int. J. Food Microbiol. 126, 36–42. doi: 10.1016/j.ijfoodmicro.2008.04.031
Renye, J. A. Jr., Needleman, D. S., Somkuti, G. A., and Steinberg, D. H. (2017). Complete genome sequence of Streptococcus thermophilus strain B59671, which naturally produces the broad-spectrum bacteriocin thermophilin 110. Genome Announc. 5:e01213-17. doi: 10.1128/genomeA.01213-17
Renye, J. A. Jr., Needleman, D. S., and Steinberg, D. H. (2019). Complete genome sequences of bacteriocin-producing Streptococcus thermophilus strains ST106 and ST109. Microbiol. Resour. Announc. 8:e01336-18. doi: 10.1128/MRA.01336-18
Repar, J., and Warnecke, T. (2017). Non-random inversion landscapes in prokaryotic genomes are shaped by heterogeneous selection pressures. Mol. Biol. Evol. 34, 1902–1911. doi: 10.1093/molbev/msx127
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2005). REBASE–restriction enzymes and DNA methyltransferases. Nucleic Acids Res. 33, D230–D232. doi: 10.1093/nar/gki029
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. doi: 10.1093/nar/gku1046
Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Sapranauskas, R., Gasiunas, G., Fremaux, C., Barrangou, R., Horvath, P., and Siksnys, V. (2011). The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 39, 9275–9282. doi: 10.1093/nar/gkr606
Savijoki, K., Ingmer, H., and Varmanen, P. (2006). Proteolytic systems of lactic acid bacteria. Appl. Microbiol. Biotechnol. 71, 394–406. doi: 10.1007/s00253-006-0427-1
Scott, E. J. II, Luke-Marshall, N. R., Campagnari, A. A., and Dyer, D. W. (2019). Draft genome sequence of pediatric otitis media isolate Streptococcus pneumoniae strain EF3030, which forms in vitro biofilms that closely mimic in vivo biofilms. Microbiol. Resour. Announc. 8:e01114-18. doi: 10.1128/MRA.01114-18
Selle, K., Klaenhammer, T. R., and Barrangou, R. (2015). CRISPR-based screening of genomic island excision events in bacteria. Proc. Natl. Acad. Sci. U.S.A. 112, 8076–8081. doi: 10.1073/pnas.1508525112
Settachaimongkon, S., Nout, M. J., Antunes Fernandes, E. C., Hettinga, K. A., Vervoort, J. M., Van Hooijdonk, T. C., et al. (2014). Influence of different proteolytic strains of Streptococcus thermophilus in co-culture with Lactobacillus delbrueckii subsp. bulgaricus on the metabolite profile of set-yoghurt. Int. J. Food Microbiol. 177, 29–36. doi: 10.1016/j.ijfoodmicro.2014.02.008
Shi, Y., Chen, Y., Li, Z., Yang, L., Chen, W., and Mu, Z. (2015). Complete genome sequence of Streptococcus thermophilus MN-BM-A02, a rare strain with a high acid-producing rate and low post-acidification ability. Genome Announc. 3:e00979-15. doi: 10.1128/genomeA.00979-15
Sieuwerts, S., Molenaar, D., Van Hijum, S. A., Beerthuyzen, M., Stevens, M. J., Janssen, P. W., et al. (2010). Mixed-culture transcriptome analysis reveals the molecular basis of mixed-culture growth in Streptococcus thermophilus and Lactobacillus bulgaricus. Appl. Environ. Microbiol. 76, 7775–7784. doi: 10.1128/AEM.01122-10
Solovyev, V., and Salamov, A. (2011). “Automatic annotation of microbial genomes and metagenomic sequences,” in Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies, ed. R. W. Li, (New York, NY: Nova Science Publishers), 61–78.
Song, X., Huang, H., Xiong, Z., Xia, Y., Wang, G., Yin, B., et al. (2018). Characterization of a cryptic plasmid isolated from Lactobacillus casei CP002616 and construction of shuttle vectors based on its Replicon. J. Dairy Sci. 101, 2875–2886. doi: 10.3168/jds.2017-13771
Sørensen, K. I., Curic-Bawden, M., Junge, M. P., Janzen, T., and Johansen, E. (2016). Enhancing the sweetness of yoghurt through metabolic remodeling of carbohydrate metabolism in Streptococcus thermophilus and Lactobacillus delbrueckii subsp. bulgaricus. Appl. Environ. Microbiol. 82, 3683–3692. doi: 10.1128/AEM.00462-16
Sujoy, B., and Aparna, A. (2013). Enzymology, immobilization and applications of urease enzyme. Int. Res. J. Biol. Sci. 2, 51–56.
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Sun, Z., Chen, X., Wang, J., Zhao, W., Shao, Y., Wu, L., et al. (2011). Complete genome sequence of Streptococcus thermophilus strain ND03. J. Bacteriol. 193, 793–794. doi: 10.1128/JB.01374-10
Tamulaitis, G., Kazlauskienë, M., Manakova, E., Venclovas, Č, Nwokeoji, A., Dickman, J., et al. (2014). Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol. Cell 56, 506–517. doi: 10.1016/j.molcel.2014.09.027
Tian, H., Li, B., Evivie, S. E., Sarker, S. K., Chowdhury, S., Lu, J., et al. (2018). Technological and genomic analysis of roles of the cell-envelope protease PrtS in yoghurt starter development. Int. J. Mol. Sci. 19:E1068. doi: 10.3390/ijms19041068
Vaillancourt, K., Bedard, N., Bart, C., Tessier, M., Robitaille, G., Turgeon, N., et al. (2008). Role of galK and galM in galactose metabolism by Streptococcus thermophilus. Appl. Environ. Microbiol. 74, 1264–1267. doi: 10.1128/AEM.01585-07
Van Mastrigt, O., Di Stefano, E., Hartono, S., Abee, T., and Smid, E. J. (2018). Large plasmidome of dairy Lactococcus lactis subsp. lactis biovar diacetylactis FM03P encodes technological functions and appears highly unstable. BMC Genomics 19:620. doi: 10.1186/s12864-018-5005-2
Vaughan, E. E., Kleerebezem, M., and de Vos, W. M. (2003). “Genetics of the metabolism of lactose and other sugars,” in Genetics of Lactic Acid Bacteria, eds B. J. B. Wood and P. J. Warner, (New York, NY: Kluwer Academic), 95–119. doi: 10.1007/978-1-4615-7090-5_4
Vaughan, E. E., Van Den Bogaard, P. T., Catzeddu, P., Kuipers, O. P., and De Vos, W. M. (2001). Activation of silent gal genes in the lac-gal regulon of Streptococcus thermophilus. J. Bacteriol. 183, 1184–1194. doi: 10.1128/JB.183.4.1184-1194.2001
Wang, T., Xu, Z., Lu, S., Xin, M., and Kong, J. (2016). Effects of glutathione on acid stress resistance and symbiosis between Streptococcus thermophilus and Lactobacillus delbrueckii subsp. bulgaricus. Int. Dairy J. 61, 22–28. doi: 10.1016/j.idairyj.2016.03.012
Wassenaar, T. M., and Lukjancenko, O. (2014). “Comparative genomics of Lactobacillus and other LAB,” in Lactic Acid Bacteria: Biodiversity and Taxonomy, eds W. H. Holzapfel and B. J. B. Wood, (Hoboken, NJ: John Wiley & Sons), 55–69. doi: 10.1002/9781118655252.ch5
Wu, Q., and Shah, N. P. (2018). Comparative mRNA-Seq analysis reveals the improved EPS production machinery in Streptococcus thermophilus ASCC 1275 during optimized milk fermentation. Front. Microbiol. 9:445. doi: 10.3389/fmicb.2018.00445
Wu, Q., Tun, H. M., Leung, F. C., and Shah, N. P. (2014). Genomic insights into high exopolysaccharide-producing dairy starter bacterium Streptococcus thermophilus ASCC 1275. Sci. Rep. 4:4974. doi: 10.1038/srep04974
Xiong, Z.-Q., Kong, L.-H., Lai, P. F. H., Xia, Y.-J., Liu, J.-C., Li, Q.-Y., et al. (2019a). Genomic and phenotypic analyses of exopolysaccharide biosynthesis in Streptococcus thermophilus S-3. J. Dairy Sci. 102, 4925–4934. doi: 10.3168/jds.2018-15572
Xiong, Z.-Q., Kong, L.-H., Meng, H.-L., Cui, J.-M., Xia, Y.-J., Wang, S.-J., et al. (2019b). Comparison of gal-lac operons in wild-type galactose-positive and -negative Streptococcus thermophilus by genomics and transcription analysis. J. Ind. Microbiol. Biotechnol. 46, 751–758. doi: 10.1007/s10295-019-02145-x
Yamauchi, R., Maguin, E., Horiuchi, H., Hosokawa, M., and Sasaki, Y. (2019). The critical role of urease in yogurt fermentation with various combinations of Streptococcus thermophilus and Lactobacillus delbrueckii ssp. bulgaricus. J. Dairy Sci. 102, 1033–1043. doi: 10.3168/jds.2018-15192
Yu, J., Sun, Z., Liu, W., Xi, X., Song, Y., Xu, H., et al. (2015). Multilocus sequence typing of Streptococcus thermophilus from naturally fermented dairy foods in China and Mongolia. BMC Microbiol. 15:236. doi: 10.1186/s12866-015-0551-0
Keywords: lineage, horizontal gene transfer, genomic islands, milk, yogurt, cheese, pan genome, CRISPR
Citation: Alexandraki V, Kazou M, Blom J, Pot B, Papadimitriou K and Tsakalidou E (2019) Comparative Genomics of Streptococcus thermophilus Support Important Traits Concerning the Evolution, Biology and Technological Properties of the Species. Front. Microbiol. 10:2916. doi: 10.3389/fmicb.2019.02916
Received: 09 August 2019; Accepted: 03 December 2019;
Published: 20 December 2019.
Edited by:
Nikos Kyrpides, Lawrence Berkeley National Laboratory, United StatesReviewed by:
Stefano Campanaro, University of Padua, ItalyAnastasia Chasapi, Centre for Research & Technology Hellas, Greece
Copyright © 2019 Alexandraki, Kazou, Blom, Pot, Papadimitriou and Tsakalidou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantinos Papadimitriou, a3BhcGFkaW1pdHJpb3VAYXVhLmdy
†Present address: Konstantinos Papadimitriou, Department of Food Science and Technology, Faculty of Agriculture and Foods, University of Peloponnese, Kalamata, Greece