 Dina Vorkapic1
Dina Vorkapic1 Fabian Mitterer1
Fabian Mitterer1 Katharina Pressler1
Katharina Pressler1 Deborah R. Leitner1Jan Haug Anonsen2Laura Liesinger3,4
Deborah R. Leitner1Jan Haug Anonsen2Laura Liesinger3,4 Lisa-Maria Mauerhofer1
Lisa-Maria Mauerhofer1 Torben Kuehnast1Manuela Toeglhofer1
Torben Kuehnast1Manuela Toeglhofer1 Adina Schulze1
Adina Schulze1 Franz G. Zingl1
Franz G. Zingl1 Mario F. Feldman5
Mario F. Feldman5 Joachim Reidl1,6
Joachim Reidl1,6 Ruth Birner-Gruenberger3,4,7
Ruth Birner-Gruenberger3,4,7 Michael Koomey2
Michael Koomey2 Stefan Schild1,6*
Stefan Schild1,6*- 1Institute of Molecular Biosciences, University of Graz, Graz, Austria
- 2Centre for Ecological and Evolutionary Synthesis, Department of Biosciences, University of Oslo, Oslo, Norway
- 3Institute of Pathology, Medical University of Graz, Graz, Austria
- 4Omics Center Graz, BioTechMed-Graz, Graz, Austria
- 5Department of Molecular Microbiology, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
- 6BioTechMed-Graz, Graz, Austria
- 7Institute of Chemical Technologies and Analytics, Vienna University of Technology, Vienna, Austria
Protein secretion plays a crucial role for bacterial pathogens, exemplified by facultative human-pathogen Vibrio cholerae, which secretes various proteinaceous effectors at different stages of its lifecycle. Accordingly, the identification of factors impacting on protein secretion is important to understand the bacterial pathophysiology. PglLVc, a predicted oligosaccharyltransferase of V. cholerae, has been recently shown to exhibit O-glycosylation activity with relaxed glycan specificity in an engineered Escherichia coli system. By engineering V. cholerae strains to express a defined, undecaprenyl diphosphate-linked glycoform precursor, we confirmed functional O-linked protein glycosylation activity of PglLVc in V. cholerae. We demonstrate that PglLVc is required for the glycosylation of multiple V. cholerae proteins, including periplasmic chaperones such as DegP, that are required for efficient type II-dependent secretion. Moreover, defined deletion mutants and complementation strains provided first insights into the physiological role of O-linked protein glycosylation in V. cholerae. RbmD, a protein with structural similarities to PglLVc and other established oligosaccharyltransferases (OTases), was also included in this phenotypical characterization. Remarkably, presence or absence of PglLVc and RbmD impacts the secretion of proteins via the type II secretion system (T2SS). This is highlighted by altered cholera toxin (CT) secretion, chitin utilization and biofilm formation observed in ΔpglLVc and ΔrbmD single or double mutants. This work thus establishes a unique connection between broad spectrum O-linked protein glycosylation and the efficacy of type II-dependent protein secretion critical to the pathogen’s lifecycle.
Introduction
The Gram-negative pathogen Vibrio cholerae, the causative agent of the severe secretory diarrheal disease cholera, transits between two dissimilar habitats along its life cycle. V. cholerae efficiently colonizes the human gastrointestinal tract upon oral ingestion of contaminated food or water, but also persists as a natural inhabitant in aquatic ecosystems during inter-epidemic periods (Schild et al., 2008; Nelson et al., 2009). V. cholerae employs a number of strategies to quickly adapt to the differential conditions faced along its life cycle, which ensures survival even under unfavorable conditions.
A hallmark of environmental survival and transmission of V. cholerae is the ability to form biofilms on chitinous surfaces in the aquatic reservoirs. Ubiquitous chitin particles in the nutrient-limited aquatic reservoir not only act as an attachment platform for subsequent biofilm formation, but also serve as a carbon and nitrogen source. Accordingly, V. cholerae expresses a complex chitin utilization program including several secreted chitinases (Meibom et al., 2004). Once ingested by humans, V. cholerae passes the acidic barrier in the stomach and reaches the small intestine, the primary site of colonization. During this passage, V. cholerae activates a complex regulatory cascade to induce the expression of virulence factors and achieve full colonization fitness (Childers and Klose, 2007). The pathology of cholera is mainly due to the activity of the secreted cholera toxin (CT), which induces a massive efflux of chloride ions and water into the intestinal lumen resulting in a secretory diarrhea (Childers and Klose, 2007).
Several proteins crucial for survival fitness along the different stages of the life cycle are secreted via the type II secretion system (T2SS). These include CT, the biofilm matrix proteins (RbmA, RbmC, and Bap1), and chitinases along with complementary proteins involved in chitin metabolism (Sikora et al., 2011; Johnson et al., 2014). Adaptation to different conditions has so far been mainly investigated through transcriptional profiling, although there is growing evidence that V. cholerae also takes advantage of post-translational regulatory strategies. These are exemplified by proteolysis control of transcription factors TcpP and ToxR affecting virulence factor expression, RpoS responsible for the mucosal escape response and FliA, which inversely controls flagella gene transcription and virulence gene expression (Matson and DiRita, 2005; Nielsen et al., 2006; Almagro-Moreno et al., 2015; Pressler et al., 2016; Rogers et al., 2016; Wurm et al., 2017).
O-linked protein glycosylation appears to be widely distributed in bacteria and represents a post-translational modification whereby oligosaccharyltransferases (OTases) transfer a pre-assembled glycan onto target proteins (Iwashkiw et al., 2013). Based on current models for Gram-negative bacteria, the glycan is synthesized on an undecaprenyl pyrophosphate (Und-PP) lipid carrier in the cytoplasm, flipped across the inner membrane and utilized by O-OTases to glycosylate serine or threonine residues of target proteins in the periplasm (Nothaft and Szymanski, 2010). Probably the best studied O-glycosylation system of Gram-negative bacteria is the protein glycosylation (pgl) in Neisseria gonorrhoeae. Briefly, PglB, PglC, and PglD are required for synthesis of Und-PP linked di-N-acetyl bacillosamine (diNAcBac), the proximal sugar in the oligosaccharide (Aas et al., 2007; Hartley et al., 2011). The fully synthesized glycan is flipped to the periplasmic space by the flippase PglF and subsequently transferred by the OTase PglL/O on diverse target proteins via en block transfer (Aas et al., 2007; Vik et al., 2009).
Intrigued by a potential, intrinsic V. cholerae post-translational modification system (Gebhart et al., 2012), we constructed and characterized a pglLVc null mutant. Using a reverse glycoengineering strategy, we found that PglLVc is required for the glycosylation of multiple V. cholerae proteins. In addition, the pglLVc null mutant showed altered biofilm features and a trend toward enhanced type II substrate secretion. Remarkably, these phenotypes were significantly augmented in a background simultaneously lacking RbmD, a protein of unknown function with limited structural similarities to PglLVc and other established oligosaccharyltransferases (Supplementary Figure S1). Thus, this work reveals genetic interactions between pglLVc and rbmD, and establishes clear connections between broad spectrum O-linked protein glycosylation and secretion processes important in diverse pathogen life styles.
Materials and Methods
Strain Construction and Growth Conditions
Bacterial strains and plasmids or oligonucleotides used in this study are listed in Tables 1, 2, respectively. The clinical isolate V. cholerae O1 El Tor C6709 was used as WT strain in all experiments. Escherichia coli strains DH5αλpir and SM10λpir were used for genetic manipulations. Unless stated otherwise, all strains were grown with aeration in lysogeny broth (LB, 1% tryptone; 1% NaCl; 0.5% yeast extract) on 37°C, on LB agar plates at 37°C, or for biofilm formation under static conditions at 24°C. To assess CT expression and secretion, V. cholerae strains were grown under virulence gene inducing conditions at 37°C for 4 h anaerobically using AKI broth, followed by 4 h aerobic growth with shaking at 180 rpm (Iwanaga and Yamamoto, 1985; Iwanaga et al., 1986). Minimal media M9 was prepared according to standard recipe (Miller, 1972), and is indicated as M9-X, whereby X represents the sole carbon source used. Antibiotics and other supplements were used in the following final concentrations: streptomycin (Sm, 100 μg/ml), ampicillin (Ap, 50 μg/ml in combination with other antibiotics, otherwise 100 μg/ml), kanamycin (Km, 50 μg/ml), chloramphenicol (Cm, 20 μg/ml for E. coli strains or 2 μg/ml for V. cholerae strains), sucrose (10%), glucose (Glc, 0.2%), chitin (0,2%, Sigma-Aldrich), N-acetylglucosamine (GlcNAc, 0.2%).
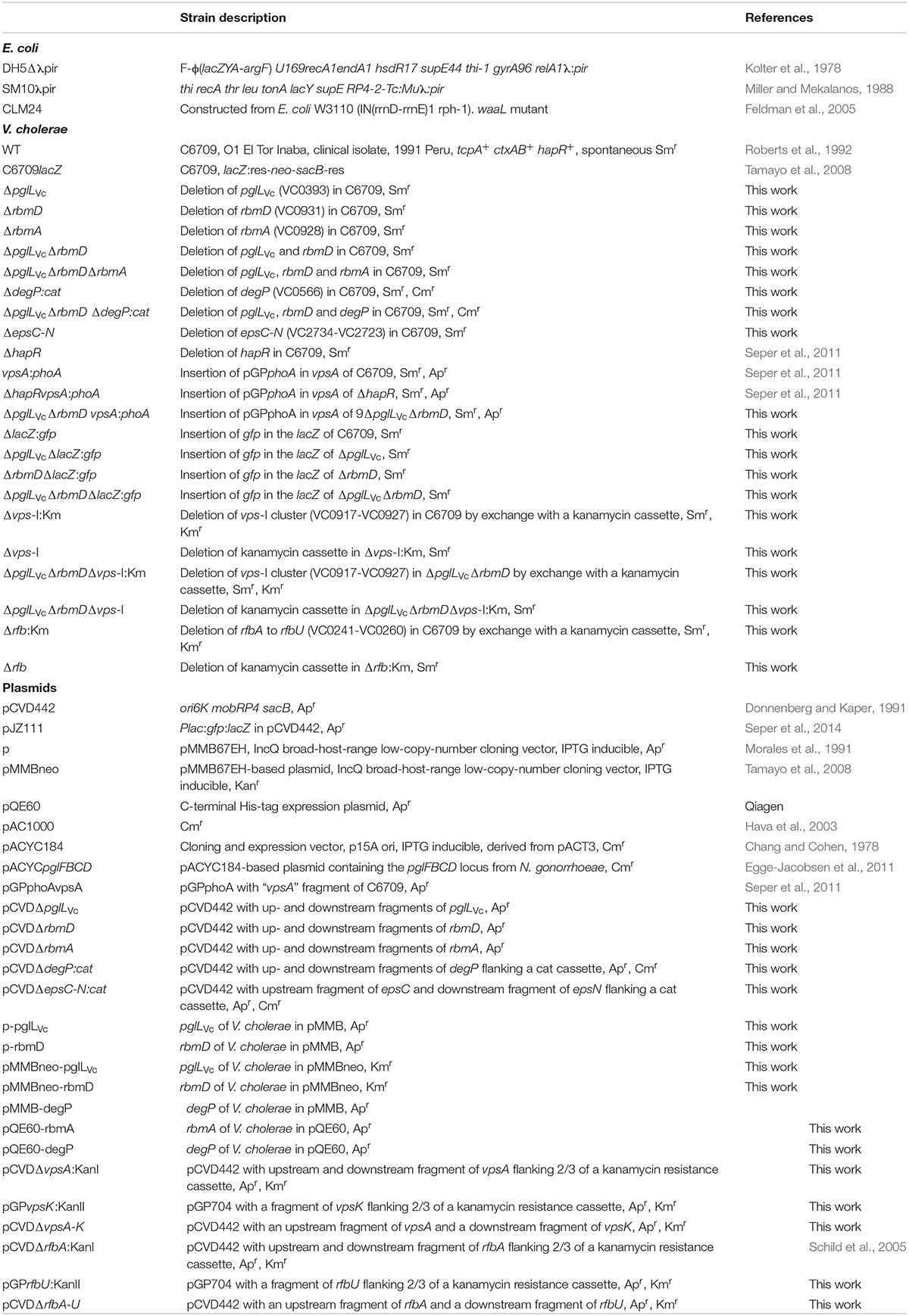
Table 1. Strains and plasmids used in this study.

Table 2. Oligonucleotides used in this study.
DNA Manipulations, Construction of Suicide Plasmids, Reporter Fusions, Mutant Strains, and Expression Plasmids
Qiaquick® Gel extraction and Qiaquick® PCR Purification kits (Qiagen) were used for purifying PCR products and digested plasmid DNA. PCR reactions for subcloning were carried out using the Q5® High-Fidelity DNA Polymerase (NEB), whereas Taq DNA Polymerase (NEB) was used for all other PCRs. Plasmid DNA was isolated using plasmid DNA purification kits (Qiagen).
Constructions of in frame deletion mutants were carried out as described by Donnenberg and Kaper (1991). 800 bp long DNA fragments flanking the gene of interest were amplified using the oligonucleotide pairs x_y_1 and x_y_2 or x_y_3 and x_y_4, where x represents the gene and y the restriction site/enzyme used (Table 2). For the construction of pCVDΔdegP:cat the cat-fragment was additionally amplified using pAC1000 as template and the oligonucleotide pairs cat_EcoRI_fw and cat_EcoRI_rev. After digestion with the appropriate restriction enzyme, indicated by the name of oligonucleotide, PCR fragments were ligated into pCVD442 restricted with similar restriction enzymes (NEB). Obtained corresponding knockout plasmids are listed in Table 1. Generated plasmids were first transformed into E.coli Sm10λpir and mobilized into V.cholerae via conjugation. Exconjugants were purified by Smr and Apr selection, followed by sucrose selection, as previously described (Donnenberg and Kaper, 1991). Correct chromosomal deletions were confirmed by PCR. GFP expressing strains were generated by insertion of gfp in the lacZ locus using the suicide plasmid pJZ111 (Seper et al., 2014).
For the construction of Δvps-I deletion mutant a method by Schild et al. (2005) was used, which enabled us to remove a relatively large chromosomal gene cluster (VC0917-VC0927). Briefly, the first step was the construction of a vpsA mutant carrying an insertion of the kanR’ gene (originating from plasmid pACYC177), consisting of about two-thirds of its gene length (KanI), and including the promoter and Shine-Dalgarno sequences. 800 bp long upstream and downstream sequences of vpsA were amplified by PCR using oligonucleotide pairs vpsA_SacI_1 and vpsA_BamHI_2 or vpsA_EcoRI_3 and vpsA_XbaI_4 (Table 2). The 800-bp PCR fragment of KanI was generated using the oligonucleotide pairs kanI_BamHI and kanI_EcoRI (Schild et al., 2005). The three PCR fragments were digested with appropriate restriction enzymes (NEB) and ligated together into a SacI/XbaI-digested pCVD442, resulting in the plasmid pCVDΔvpsA:KanI. The plasmid pCVDΔvpsA:KanI was mobilized into V. cholerae via conjugation, obtaining the mutant C6709ΔvpsA:KanI after sucrose selection. In the second step, the suicide plasmid pGP704 harboring downstream sequence of vpsK and the last two-thirds (C′ terminal) of the ′kanR gene (KanII) was constructed. The PCR fragments were obtained using the oligonucleotide pairs vpsK_NcoI and vpsK_XbaI for 800 bp downstream of vpsK or KanII_SacI and KanII_NcoI for KanII (Schild et al., 2005) (Table 2), digested with Xba/NcoI or NcoI/SacI, and ligated into a SacI/XbaI-digested pGP704, resulting in pGPvpsK:KanII. After conjugation of pGPvpsK:KanII in C6709ΔvpsA:KanI, we obtained Apr colonies. Colony purification of some of these cells in absence of Ap and subsequent selection for Kmr resulted in Kmr/Aps colonies. Deletion of kanamycin cassette was achieved by a pCVD442 suicide vector mutagenesis using pCVDΔvpsA-K, which constructed by amplifying PCR fragments using the oligonucleotide pairs vpsA_SacI_1 and vpsA_NcoI as well as vpsK_NcoI and vpsK_XbaI, digested with Xba/NcoI or NcoI/SacI, and ligated into a SacI/XbaI-digested pGP704, resulting in pGPvpsK:KanII. The correct chromosomal deletion of genes vpsA-K was confirmed by PCR.
The Δrfb deletion mutant was generated as described above (see Δvps-I deletion mutant construction for details) and comprises a deletion spanning from rfbA to rfbU (VC0241-VC0260). This required the pCVDΔrfbA:KanI (Schild et al., 2005) and pGPrfbU:KanII (Table 2). The latter was constructed by amplifying PCR fragments using the oligonucleotide pairs rfbU_BamHI and rfbU_XbaI for 800 bp downstream of rfbU or KanII_SacI and KanII_BamHI for KanII (Schild et al., 2005) (Table 2), digested with Xba/BamHI or BamHI/SacI, and ligated into a SacI/XbaI-digested pGP704, resulting in pGPrfbU:KanII. Deletion of kanamycin cassette was achieved by a pCVD442 suicide vector mutagenesis using pCVDΔrfbA-U, which was constructed by amplifying PCR fragments using the oligonucleotide pairs rfbA_SacI_1 and rfbA_BamHI as well as rfbU_BamHI and rfbU_XbaI, subsequent digestion with SacI/BamHI/or BamHI/XbaI, and ligation into a SacI/XbaI-digested pGP704, resulting in pGPrfbU:KanII. The correct chromosomal deletion of genes rfbA-U was confirmed by PCR.
Construction of ΔepsC-N mutant was done as previously described by Sikora et al. (2007) using the appropriate oligonucleotide pairs listed in the Table 2.
All expression plasmids were constructed in a similar manner. PCR fragments of the respective genes containing their own ribosomal binding sites were generated using the oligonucleotide pairs x_y_z_fwd and x_y_z_rev, where x represents the gene, y the restriction site/enzyme used and z the respective expression plasmid (Table 2). PCR fragments digested with the respective restriction enzymes were ligated into the similarly digested IPTG-inducible expression vectors pMMB67EH, pMMBneo or pQE60. Expression constructs were transformed into DH5αλpir, and Apr colonies were characterized by colony PCR.
Immunoprecipitation
Immunoprecipitation was performed using Dynabeads® Protein G Immunoprecipitation Kit (Invitrogen) according to manufacturer’s manual. 4 μl of npg1 antibody was incubated with 196 μl of binding and washing buffer and subsequently bound to the Dynabeads. Periplasmic protein fractions of respective strains were precipitated using ammonium sulfate and 200 μg of protein in 500 μl sample buffer was used as antigen. Proteins in the immunoprecipitations were incubated in 5× Laemmli buffer for 10 min at 100°C, separated with SDS-PAGE and analyzed by mass spectrometry.
SDS-PAGE and Immunoblot Analysis
To separate proteins the standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) procedure was used with 12 or 15% gels and the Prestained Protein Marker Broad Range (New England Biolabs) as a molecular mass standard. Proteins were visualized by conventional colloidal Coomassie staining according to Kang et al. (2002) or transferred to a nitrocellulose membrane (Amersham) for immunoblot analysis using mouse anti-His antisera (Qiagen, 1:2,000 diluted in 3% BSA) for detection of His-tagged proteins, anti-CT antisera (Sigma-Aldrich, 1:5,000 diluted in 10% skim milk), npg1 (1:10,000 diluted in 10% skim milk) or rabbit anti-BSA antisera (Thermo Fisher Scientific, 1:2,000 diluted in 3.7% soy milk) as primary antibodies. Peroxidase-conjugated goat anti-mouse (Dianova GmbH, diluted 1:7,500 in 10% skim milk) or peroxidase conjugated goat anti-rabbit (Dianova GmbH, 1:7,500 in 10% skim milk or 1:7,500 diluted in 3.7% soy milk in case of BSA detection) were used as secondary antibodies. Loading of equal amounts proteins was achieved by Bradford quantification and verified by Kang staining of SDS-PAGE gels in parallel to the immunoblot analysis. Chemiluminescence detection was performed by incubating the membrane in an ECL solution (Bio-Rad Laboratories) with subsequent exposure in a ChemiDoc XRS system (Bio-Rad Laboratories) in combination with the Quantity One software (Bio-Rad Laboratories).
His-Purification of Proteins Using Immobilized Metal Ion Affinity Chromatography
Proteins with C-terminal His-tag were purified using a Ni SepharoseTM Fast Flow (GE Healthcare) according to the manufacturer’s manual for batch purification. Briefly, appropriate amounts of V. cholerae cultures grown over night (ON) were inoculated in fresh LB to an OD600 of 0.1, grown to and OD600 of 0.5 following induction with IPTG (1 mM) for 6 h at 37°C. Cells were pelleted for 30 min at 17,000 × g and pellets were stored at −20°C. On the following day, pellets were thawed in 5 ml lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.5, 1% Triton-X) with the addition of protease inhibitor cocktail tablet (cOmpleteTM, Roche). After sonification to lyse the cells, the soluble fraction was collected by centrifugation (30 min, 17,000 × g) and filtered through a syringe filter (0.45 μm). The resin was washed once with ddH2O and twice with binding and washing buffer pH 7.4 (81 mM Na2HPO4, 19 mM NaH2PO4, 0.5 M NaCl, 30 mM imidazole). Finally, 1 ml 50% Ni Sepharose slurry was added to 5 ml sample and incubated 1 h at 4°C with rotation. To wash the unbound protein, Ni Sepharose/sample was incubated three times 2 min with 1 mL binding and washing buffer on a rocker. Each time, Ni Sepharose/sample was collected by centrifugation 5 min at 500 × g. His-tagged protein was eluted stepwise with 500 μL elution buffer containing 100 mM, 200, 300or 500 mM imidazole, respectively. Each elution step was performed for 30 min at 4°C with rotation. All fractions were collected in the separate tubes and subjected to SDS-PAGE and/or immunoblot analyses.
Protein Analysis by Mass Spectrometry
Protein bands excised from the SDS gel were reduced, alkylated and digested with Promega modified trypsin according to the method of Shevchenko et al. (1996). Peptide extracts were dissolved in 0.1% formic acid (FA), 5% acetonitrile (ACN) and separated by nano-HPLC (Dionex Ultimate 3000) equipped with an enrichment column (C18, 5 μm, 100 Å, 5 × 0.3 mm) and an Acclaim PepMap RSLC nanocolumn (C18, 2 μm, 100 Å, 500 × 0.075 mm) (all Thermo Fisher Scientific, Vienna, Austria). Samples were concentrated on the enrichment column for 6 min at a flow rate of 5 μl/min with 0.1% formic acid as isocratic solvent. Separation was carried out on the nanocolumn at a flow rate of 250 nl/min at 60°C using the following gradient, where solvent A is 0.1% formic acid in water and solvent B is acetonitrile containing 0.1% formic acid: 0–6 min: 4% B; 6–94 min: 4–25% B; 94–99 min: 25–95% B, 99–109 min: 95% B; 109.1–124 min: 4% B; The sample was ionized in the nanospray source equipped with stainless steel emitters (ES528, Thermo Fisher Scientific, Vienna, Austria) and analyzed in an Orbitrap velos pro mass spectrometer (Thermo Fisher Scientific, Waltham, MA, United States) operated in positive ion mode, applying alternating full scan MS (m/z 400–2,000) in the ion cyclotron and MS/MS by CID of the 20 most intense peaks with dynamic exclusion enabled. The LC-MS/MS data were analyzed by searching a database containing all V. cholerae sequences of the Uniprot database (downloaded on December 5th, 2016, 92555 sequences) and all common contaminants with Proteome Discoverer 1.4 (Thermo Fisher Scientific) and Mascot 2.4.1 (MatrixScience, London, United Kingdom). Carbamidomethylation on cysteine was entered as fixed and oxidation on methionine and 2,4-diacetamido-2,4,6-trideoxyhexose (DATDH) with a mass of 228.111 Da on serine as variable modification. Detailed search criteria were used as follows: semitrypsin; max. missed cleavage sites: 2; search mode: MS/MS ion search with decoy database search included; precursor mass tolerance ±10 ppm; product mass tolerance ±0.7 Da; acceptance parameters: 1% false discovery rate (FDR); only rank 1 peptides; minimum Mascot ion score 20; minimum 2 peptides per protein.
PTM Analysis by Mass Spectrometry
Kang stained protein bands of affinity purified DegP was washed, de-stained and digested with both trypsin and GluC as previously described (Anonsen et al., 2012). Dried samples were redissolved in 0.1% FA prior to LC-MS/MS analysis. The LC-MS/MS analysis was performed as for protein analysis by MS with the following modification: Solvent B was changed to 90% ACN/9.9% H2O containing 0.1% FA. The column temperature was set at 40°C. The separation gradient was modified to, 0–3 min: 3% B; 3–53 min: 3–55% B; 55–59 min: 80% B, 60.3–63.3 min: 3% B. The sample was ionized in the nanospray source and analyzed in an Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, United States). Mass spectra were acquired in the positive ion mode applying alternating full scan MS (m/z 200–2,000) at a resolution of 70000 (at m/z 400) and MS/MS by HCD with NCE@28 of the 12 most intense peaks at a resolution of 17500 (at m/z 400) with dynamic exclusion for 30 s. Generated mass spectra were analyzed with the DegP protein sequence. Glycopeptides mass spectra were extracted based on the presence of the three diNAcBac reporter ions at m/z 229.118, at m/z 211.107 and at m/z 169.096 using Thermo Xcalibur (version 3.0.63) and manually assigned to DegP peptide sequences.
Alkaline Phosphatase Assays
To determine the enzymatic activities for the transcriptional phoA fusions, alkaline phosphatase assays were performed as described previously (Manoil, 1991). Bacterial cultures were grown to mid-log phase (∼OD600 = 0.5) in LB media at 24°C. The activities were expressed in Miller units, given by (A405 × 1,000)/(A600 × ml × min).
Static Biofilm Assay With Crystal Violet Staining
Biofilm assays under static conditions were essentially performed as previously described (Seper et al., 2011). Briefly, the respective strains were grown ON) in LB-Sm or LB-Ap/Glc liquid media (for plasmid containing strains). On the following day, a fresh culture LB-Sm or LB-Ap/IPTG (in case of strains harboring pMMB67EH or its derivatives), was adjusted to an OD600 = 0.01 and inoculated in a 96 well microtiter plate (U bottom, Sterilin). Biofilm was grown in a 24°C climate chamber for 24, 48 or 72 h. Wells were subsequently rinsed using a microplate washer (anthos Mikrosysteme GmbH, Fluido2). Biofilm was stained with 0.1% crystal violet, solubilized in 96% ethanol and the OD595 was measured (microplate reader: BMG Labtech SPECTROstarNano) to quantify the amount of biofilm.
Flow Cell Biofilm Experiments
For visualization and quantification of dynamically formed biofilm, a three channel flow cell system using 2% LB-Sm broth (24 h, RT) was used as described previously (Seper et al., 2011, 2014; Pressler et al., 2016). The respective GFP expressing V. cholerae strains were used for biofilm formation to allow acquisition of fluorescent images with confocal laser scanning microscopy. A coverslip 24 × 50 mm (Menzel-Glaeser) was used as substratum for biofilm growth. The respective ON cultures were adjusted to OD600 = 0.1 using 2% LB Approximately 300 μl of the dilutions were inoculated per channel. After static incubation for 2 h at RT, initial attachment of cells to the abiotic surface was analyzed in the attachment assay. For analysis of mature biofilm, the static incubation for 2 h at RT was followed by a constant flow of 2% LB at the rate of 3 ml h–1 for 22 h. Images of attached bacteria or biofilms were recorded by confocal laser scanning microscopy.
Confocal Laser Scanning Microscopy
Images of biofilms were acquired using a Leica SP5 confocal microscope (Leica Microsystems, Mannheim, Germany) with spectral detection and a Leica HCX PL APO CS 63x water objective (NA 1.2). Optical sectioning was performed in 0.13 μm steps. GFP was excited at 488 nm, fluorescence emission was detected between 500 and 560 nm, and images were recorded without differential interference contrast (DIC) optics. For visualization and processing of image data the Leica LAS AF Lite and ImageJ 1.46 software was used. Quantification and morphological analysis of image stacks was performed using the computer program COMSTAT1 (Heydorn et al., 2000; Vorregaard, 2008).
Cell Fractionation
A cell fractionation was performed essentially as described by Neu and Heppel (1965). Briefly, V. cholerae cultures grown ON were inoculated in fresh LB to an OD600 of 0.1, grown until OD600 of 0.5 and subsequently induced with 1 mM IPTG for 4 h. Supernatant (SUP) of the culture was collected by centrifugation 10 min, 5,000 × g and filter-sterilized using a 0.22 μm syringe filter. In parallel, cell equivalents reflecting 1 l of a culture with OD600 of 1 were pelleted by centrifugation (10 min, 5,000 × g) and washed in buffer 20 mM Tris–HCl pH 8. After another round of centrifugation, cells were resuspended in 20% sucrose and 1 mM Na-EDTA and incubated for 10 min. Cells were then centrifuged for 10 min, 5,000 × g at 4°C and resuspended in ice cold 0.5 mM Mg2SO4 by slow pipetting. The combination of cold temperature and hypotonic solution caused the burst of the bacterial outer membrane and release of the periplasmic fraction (PF) in the supernatant. After incubation for ∼5 min, samples were centrifuged for 5 min, 10,000 × g at 4°C to remove cell debris and intact cells. The supernatant was recovered as the periplasmic fraction. Periplasmic fractions were directly used for CT quantification by ELISA, while in all other cases proteins in the collected SUP and PF were either precipitated using trichloroacetic acid (TCA)/acetone for immunoblot analyses or ammonium sulfate in case of immunoprecipitation. In case of ELISA and immunoblot analyses, periplasmic fractions were controlled for cytoplasmic contamination and cell lysis via dot blot analyses detecting RpoA, the α-subunit of the RNA-Polymerase (for details see section “Dot Blot Analysis”). A representative dot blot is shown in Supplementary Figure S4A. In general, similar intensities for periplasmic fractions used for ELISA were observed. Moreover, intensities in all periplasmic fractions were magnitudes lower compared to whole cell extracts (WCE). This indicates equally low levels of cytoplasmic contamination for all periplasmic fractions used for ELISA.
Dot Blot Analysis
Dot blot analyses was essentially performed as described recently using a 96-well Whatman Minifold I Dot-Blot System (GE Healthcare) (Roier et al., 2016). Briefly, equal protein amounts (according to Bradford) of periplasmic factions or dilutions (3-fold steps) thereof were spotted onto a nitrocellulose membrane. Dilutions (3-fold steps) of a whole cell extract derived from the WT served as positive control (for details see section “Generation of Whole Cell Extracts”). Then, each membrane was dried, incubated in TBS (20 mM Tris/HCl, pH 7.5, 150 mM NaCl) for 2 min, and blocked in 10% skim milk in TBS for 2 h. Afterward, the membrane was incubated with a monoclonal antibody to the α-subunit of the RNA polymerase (RpoA, NeoClone Biotechnology, diluted 1:2,000 in 10% skim milk) overnight. The next day, membranes were washed three times in TBS for 10 min, incubated with the secondary antibody (horseradish peroxidase-conjugated goat anti-mouse IgG antibody from Dianova, diluted 1:7,500 in 10% skim milk in TBS) for 2 h, washed once in TBS-T (20 mM Tris/HCl, pH 7.5, 250 mM NaCl, 0.05% Tween-20), and twice in TBS for 10 min each. Finally, chemiluminescence detection was performed as described above (for details see section “SDS-PAGE and Immunoblot Analysis”).
Precipitation of Proteins Using TCA/Acetone
Protein precipitation using TCA and acetone was essentially performed as described by Link and LaBaer (2011), with following modifications. Samples were spiked with BSA (0.1 μg/ml), before a solution of 100% TCA was added to the protein samples (SUP or the PF) to the final TCA concentration of 20%. Samples were mixed and stored ON at −20°C. On the next day, samples were thawed and centrifuged 30,000 × g for 1 h. Supernatant was carefully decanted and 20 ml of acetone was added to the pellet. Samples were again centrifuged 30,000 × g for 1 h. A second washing step with 20 ml of acetone was performed with the final centrifugation step of 50,000 × g. After decanting the supernatant, pellets were air-dried for 30 min and subsequently suspended in 200 μl of PBS buffer. In attempt to completely dissolve the pellet, samples were placed in a sonification bath for 30 min. Undissolved particles were removed by short centrifugation. Concentration of proteins was determined with Bradford assay (BioRad). Similar efficiency of protein precipitation using TCA was controlled by immunoblot analyses detecting BSA (for details see section “SDS-PAGE and Immunoblot Analysis”).
Precipitation of Proteins Using Ammonium Sulfate
Periplasmic protein content was also subjected to precipitation using ammonium sulfate, which yields a relatively soluble protein extract essential for immunoprecipitation. Following cell fractionation, ammonium sulfate was added to the periplasmic fraction to final concentration of 50% (Wood, 1976) and stirred for 1 h at RT. Sample was then subjected to centrifugation 10,000 × g for 30 min at RT. After decanting supernatant, pellet was suspended in 500 μl PBS buffer. Sample was dialyzed ON against PBS in a dialysis tube with pore size 3.5 (Roth).
Growth Assays
Growth kinetics were performed as previously described in M9-chitin or M9-GlcNAc with following modifications (Schild et al., 2007). Briefly, ΔpglLVcΔrbmD or WT were grown in a M9-Glc pre-culture ON and inoculated in M9-chitin or M9-GlcNAc to a starting OD600 = 0.025. To determine the exact CFUs appropriate dilutions were plated on LB plates at 0, 24, 48, and 72 h. Before plating each culture was rigorously mixed by vortexing. The cultures were incubated at 37°C with aeration and samples were collected after 24, 48, and 72 h. Results are given as CFU/ml normalized to a starting concentration of 107 CFU/ml at t = 0 h.
Generation of Whole Cell Extracts
Whole cell extracts were generated by collecting the pellet of 20 ml culture by centrifugation 5 min at 5,000 × g. Pellet was suspended in 1 ml of PBS buffer and transferred to a cryotube. Glass beads (0.5 mm in diameter, Roth) were added to the sample and lysed using the PowerLyserTM Bench Top Bead-Based Homogenizer (MO BIO Laboratories, Inc.) 3 times 1 min, with 1 min incubation steps on ice after each round of lysis. Samples were centrifuged 2 min at 10,000 × g to remove cell debris and unbroken cells. Supernatant was separated from glass beads and used as WCE.
CT ELISA
Cholera toxin production in culture supernatants or periplasmic fractions was determined by the ganglioside GM1 ELISA (Pressler et al., 2016). V. cholerae strains were grown ON in LB medium supplemented with 5% glucose, which is beneficial for growth of the ΔepsC-N (Sikora et al., 2007). On the following day, strains were shifted to AKI broth with 5% glucose, which represents virulence gene inducing conditions. Strains were grown at 37°C for 4 h anaerobically, followed by 4 h aerobic growth with shaking at 180 rpm (Iwanaga et al., 1986). For complementation strains, which have been grown and induced with IPTG on 37°C, neither LB medium nor AKI broth contained additional glucose to avoid interference with Ptac promotor expression. Subsequently, cells were removed from CT containing supernatants by centrifugation and supernatants were stored at –20°C. Periplasmic fractions were obtained from the cell pellet as described above (see section “Cell Fractionation”) and adjusted to protein equivalents (Bradford). ELISA plates (BRANDplates®, 96-well, immunoGradeTM) were coated with 10 μg/ml GM1 ganglioside (Calbiochem) in 60 mM Na2CO3 ON at 37°C and washed four times with PBS pH 7.4 and 0.5% Tween (PBS-T). Free binding sites were blocked with 4 mg/ml BSA in PBS (BSA-PBS) for 1 h at RT. After washing as described above, CT containing supernatants were diluted in PBS and added to the plate. Additionally, purified CT in PBS (Sigma-Aldrich) was inoculated in separate wells to generate a standard curve. ELISA plates were incubated with supernatants and purified CT for 1 h at RT and were washed again as described above. After incubation with the primary antibody (Sigma-Aldrich, anti-CT antibody), diluted 1:10,000 in BSA-PBS for 1 h at RT, ELISA plates were washed four times with PBS-T. After incubation with the secondary antibody (Dianova GmbH, peroxidase conjugated goat anti-rabbit), diluted 1:2,000 in BSA-PBS for 1 h at RT and subsequent washing, the ELISA plates were incubated with TMB Substrate Reagent Set (BioLegend, Vienna) for detection of CT. The reaction was stopped by the addition of 1 M H3PO4 and ELISA plates were measured at OD450 by using SPECTROstarNano microplate reader (BMG Labtech).
Statistical Analysis
Data were analyzed using the Mann–Whitney U test or a Kruskal–Wallis test followed by post hoc Dunn’s multiple comparisons. Differences were considered significant at P-values of <0.05. For all statistical analyses, GraphPad Prism version 6 was used.
Results
Reverse Glycoengineering Reveals That PglLVc Glycosylates Multiple V. cholerae Proteins
The activity of PglLVc has only been shown in a heterologous E. coli system via simultaneous expression of PglLVc along with a defined glycan and protein acceptors for O-linked protein glycosylation from foreign bacteria (Gebhart et al., 2012). Although this demonstrated functionality of PglLVc with relaxed glycan specificity, O-linked protein glycosylation has not been documented in V. cholerae. Hence, neither the transferred glycan structure nor glycosylated proteins of V. cholerae are known. Along this study we focused on the latter and exploited the relaxed glycan specificity of PglLVc along with a well-characterized glycan biosynthesis locus from N. gonorrhoeae to identify a first set of glycosylated proteins in V. cholerae. Therefore, we introduced a plasmid harboring the core pglFBCD glycosylation locus from N. gonorrhoeae into V. cholerae to express the lipid-linked monosaccharide precursor Und-PP-diNAcBac. Proteins glycosylated with diNAcBac monosaccharide could then be readily detected using the specific monoclonal antibody npg1 (Aas et al., 2007; Faridmoayer et al., 2007; Egge-Jacobsen et al., 2011; Anonsen et al., 2015). Immunoblotting of WCE revealed a number of distinct npg1-reactive proteins whose detection was dependent on the presence of both PglLVc and the N. gonorrhoeae pgl plasmid (Figure 1A). Moreover, these findings were independent on the presence or absence of RbmD. This indicates that PglLVc, in contrast to RbmD, can utilize the diNAcBac glycan to modify a defined subset of V. cholerae proteins. Hence, we focused on the identification of proteins glycosylated via PglLVc.
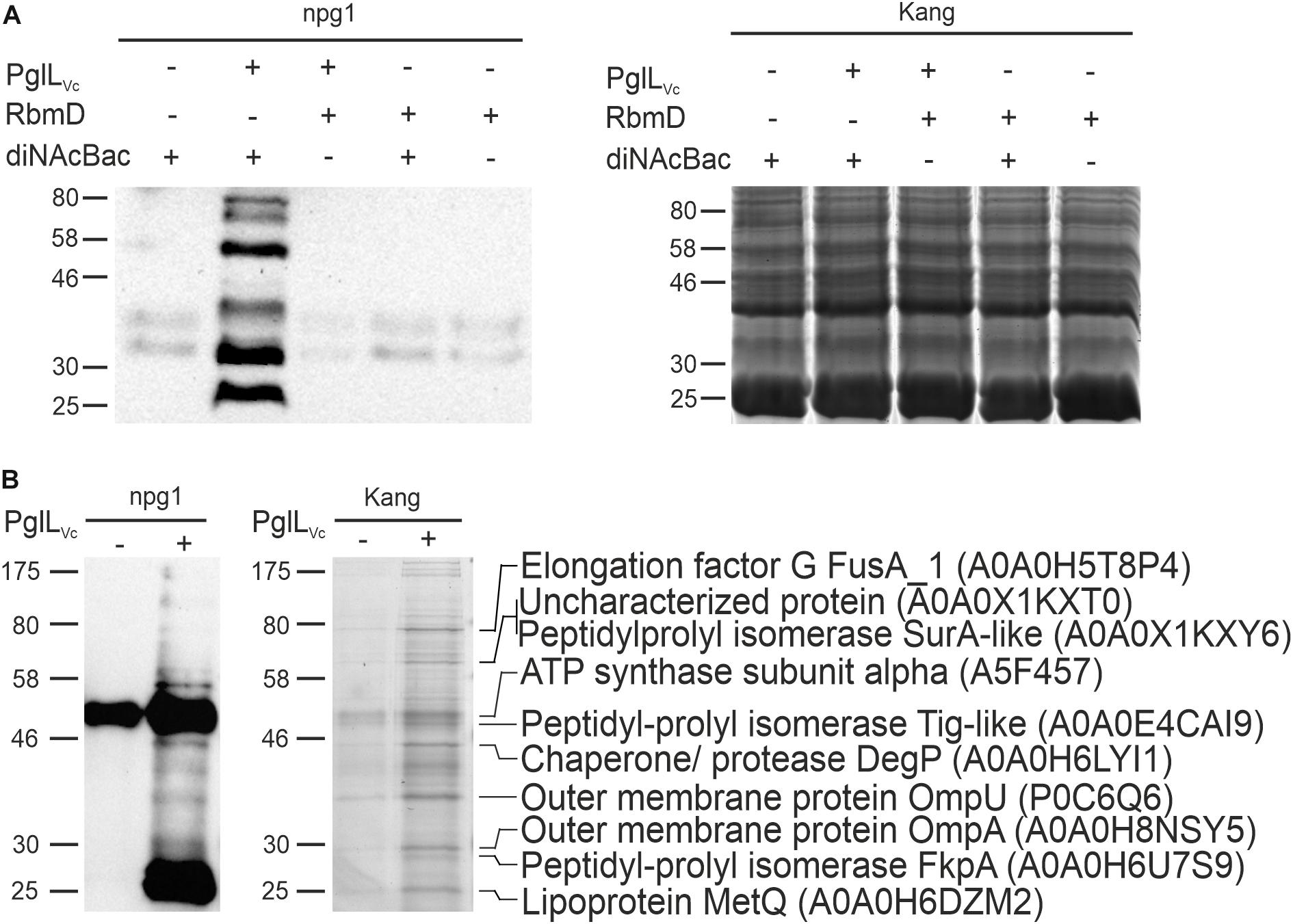
Figure 1. Expression of the diNAcBac glycan in V. cholerae results in multiple glycosylated proteins in a PglLVc-dependent manner. (A) Shown is a representative immunoblot using the anti-diNAcBac antibody (npg1) to detect diNAcBac-glycosylated proteins and the corresponding Kang-stained SDS-gel of whole cell extracts derived from ΔpglLVcΔrbmD with or without in trans expression of respective O-OTases PglLVc or RbmD (+ = pMMBneo-pglLVc or pMMBneo-rbmD; – = pMMBneo) as well as the diNAcBac glycan (+ = pACYCpglFBCD; – = pACYC184). Only the presence of PglLVc and diNAcBAc yielded in the detection of multiple bands. (B) Shown is an immunoblot using the npg1-antibody to detect diNAcBac-glycosylated proteins and the corresponding Kang-stained SDS-gel of immunopreciptated samples of ΔpglLVcΔrbmD expressing the diNAcBac glycan (pACYCpglFBCD) in the presence (+ = pMMBneo-pglLVc) or absence (- = pMMBneo) of PglLVc. Immunoprecipitation was performed using npg1 immobilized onto Dynabeads coupled with protein G in combination with membrane and periplasmic enriched protein samples from the respective strains. Selected protein bands, which appear to be enriched upon presence of PglLVc on the SDS-gel and resulted in a decent signal on the immunoblot, were excised and subjected to MS. Identified proteins are indicated on the right with their respective position on the gel, protein identities and accession numbers. (A,B) Lines to the left indicate the molecular masses of the protein standards in kDa.
To identify V. cholerae proteins bearing the diNAcBac monosaccharide, immunoaffinity purification was employed as previously described (Anonsen et al., 2012) using samples enriched for membrane and periplasmic proteins from various backgrounds as prey (Figure 1B). Immunoblotting using the npg1 monoclonal antibody of immunoprecipitated samples revealed a number of proteins specifically detected in the presence of both PglLVc and the N. gonorrhoeae pgl plasmid. Besides several bands with low intensity the immunoblot shows two intense bands at approximately 50 and 25 kDa. The 50 kDa band is likely a cross-reacting band as it also detected in absence of PglLVc. In contrast, the 25 kDa band only appears in presence of PglLVc, but its exact nature remains to be elucidated.
Robust signals obtained by immunoblot indicating immunoaffinity purified proteins or complexes thereof, were aligned to protein bands on a conventionally stained gel that were subsequently excised and identified by mass spectrometric (MS) techniques. The analyses revealed several proteins, such as abundant outer membrane proteins OmpA and OmpU as well as periplasmic chaperones, i.e., the peptidyl-prolyl cis-trans isomerases (SurA-like and FkpA) and the chaperone/protease DegP (Figure 1B).
DegP Glycosylation Can Be Reconstituted in an E. coli Background
In order to further validate the glycosylation status of the proteins identified above, the ability of DegP as a representative candidate to be glycosylated in a heterologous system was examined. Plasmids individually expressing PglLVc, a C-terminal His-tagged DegP and Und-PP-diNAcBac were introduced into the glycosylation-deficient E. coli strain CLM24. Following a His-tag purification, samples were subjected to immunoblot analyses using penta-His or npg1 antibodies as well as conventional gel staining (Figure 2A). Co-expression of PglLVc and Und-PP-diNAcBac was both necessary and sufficient to establish reactivity of DegP with npg1, indicative of diNAcBac modification (Figure 2A). In addition, a cross-reactive band (∼20 kDa) was detected by the npg1 antibody upon expression of the diNAcBac. It could be speculated that this band reflects the lipid-linked monosaccharide precursor Und-PP-diNAcBac (Und-PP-diNAcBac), but the exact origin remains to be elucidated.
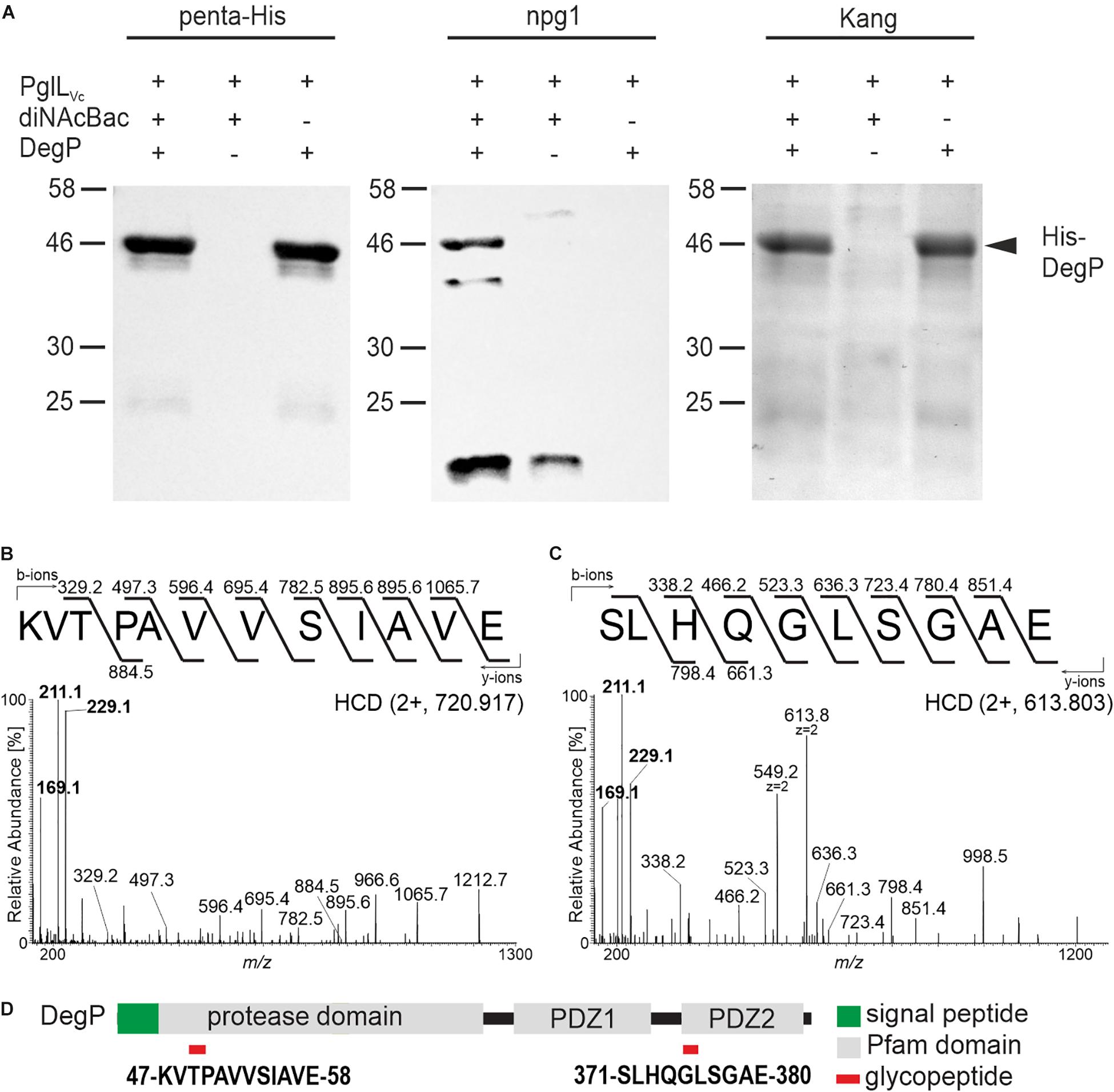
Figure 2. PglLVc glycosylates DegP with the diNAcBac glycan. (A) Ni-sepharose purified samples of E. coli CLM24 with varying expression of PglLVc (pMMBneo-pglLVc), the diNAcBac glycan (+ = pACYCpglFBCD; – = pACYC184) and/or His-tagged DegP (+ = pQE60-degP; – = pQE60), respectively, were subjected for immunoblot analyses using the penta-His antibody or the npg1 antibody. A corresponding Kang-stained SDS-gel served as control. The arrow highlights the location of the His-DegP band (approx. 46 kDa). Only in the presence of PglLVc, diNAcBac, and His-DegP a band of similar size can be detected with the npg1 antibody, suggesting a diNAcBac-modification of His-DegP. Lines to the left indicate the molecular masses of the protein standards in kDa. (B,C) Identification of GluC-derived glycopeptides from DegP. HCD MS/MS spectra of the [M+2H+]2+ precursor ions at m/z 720.917 and 613.803 of the 47KVTPAVVSIAVE58 and 371SLHQGLSGAE380 peptides respectively, modified with one glycan. Spectra show the reporter ions of diNAcBac at m/z 229.1, at m/z 211.1 and at m/z 169.1 in the low mass area. (D) Domain organization and location of DegP glycopeptides. Glycopeptides are numbered according to the DegP sequence (generated by in-house sequencing). Red rectangles denote the location of the glycopeptide.
Gel slices bearing His-tagged DegP were excised and processed for MS analyses to detect glycan attachment sites. Utilizing LC-MS/MS employing higher-energy collision dissociation in conjunction with in-gel digestion with trypsin and GluC (HCD) (Anonsen et al., 2012) peptides covering 98% of DegP including two glycopeptides were identified (Supplementary Figure S2A). Specifically, the MS/MS spectrum of the doubly charged precursor ion at m/z 720.917 (charge adjusted mass 1440.827 [M+H+]) corresponds to the peptide 47KVTPAVVSIAVE58 (theoretical monoisotopic mass 1212.720 [M+H+]) modified with a single diNAcBac moiety (228.111 Da) (Figure 2B). Similarly, the MS/MS spectrum of the doubly charged precursor ion at m/z 613.803 (charge adjusted mass 1226.599 [M+H+]) corresponds to the peptide 371SLHQGLSGAE380 (theoretical monoisotopic mass 998.490 [M+H+]) with a single diNAcBac modification (Figure 2C). In addition to extensive y- and b-ions series that were detected in both MS/MS spectra enabling the accurate amino acid sequence determination for both peptides, the diNAcBac specific reporter ions at m/z 229.118, at m/z 211.107 and at m/z 169.096 (Anonsen et al., 2012) was detected in the low mass area. The MS results therefore clearly establish both the 47KVTPAVVSIAVE58 and the 371SLHQGLSGAE380 peptides as modified by a diNAcBac moiety at a single site (Figures 2B,C and Supplementary Figures S2B,C).
Interestingly, both DegP glycopeptides are located within defined domain structures. The 47KVTPAVVSIAVE58 glycopeptide is localized within the protease domain whereas the 371SLHQGLSGAE380 glycopeptide is positioned in the carbohydrate binding loop of the PDZ2 domain (Figure 2D), which is suggested to be involved in substrate binding (Krojer et al., 2002). Thus, if identical sites of glycan attachment occur in V. cholerae, DegP activities might be altered.
PglLVc and RbmD Status Affect Secretion of Cholera Toxin
Based on the confirmed O-glycosylation of DegP, the identification of other periplasmic chaperones to be targets for O-glycosylation in V. cholerae (Figure 1B) and a recent report implying that DegP affects type II-dependent secretion in V. cholerae (Altindis et al., 2014), we sought to examine if PglLVc influences T2SS-associated phenotypes. Given the structural similarities of RbmD to PglLVc and other established oligosaccharyltransferases (Supplementary Figure S1) we also examined the potential effects of RbmD, although the O-OTase activity of RbmD remains to be confirmed. Thus, respective single and double mutants (i.e., ΔpglLVc, ΔrbmD, and ΔpglLVcΔrbmD) as well as complementation strains for both gene products were included in the following phenotypical analyses.
V. cholerae secretes several important T2SS effectors along its lifecycle (Sikora, 2013). The most prominent example is probably CT, which is predominantly responsible for the secretory diarrhea during human colonization. To investigate whether secretion of CT is affected in the context of the PglLVc, and RmbD status, secretion levels of CT were compared for WT, ΔdegP, ΔpglLVc, ΔrbmD, ΔpglLVcΔrbmD, and ΔpglLVcΔrbmDΔdegP. As a control, CT secretion was measured in a ΔepsC-N background that lacks essential parts of the T2SS machinery and is therefore severely impaired for secretion. Of note, CT expression remained equal in all strains tested as indicated by comparable signal intensities in the WCE of respective strains (Supplementary Figure S3A). Quantification of secreted CT revealed significant higher levels in the supernatants of ΔpglLVc, ΔrbmD, and ΔpglLVcΔrbmD backgrounds compared to WT (Figure 3A).
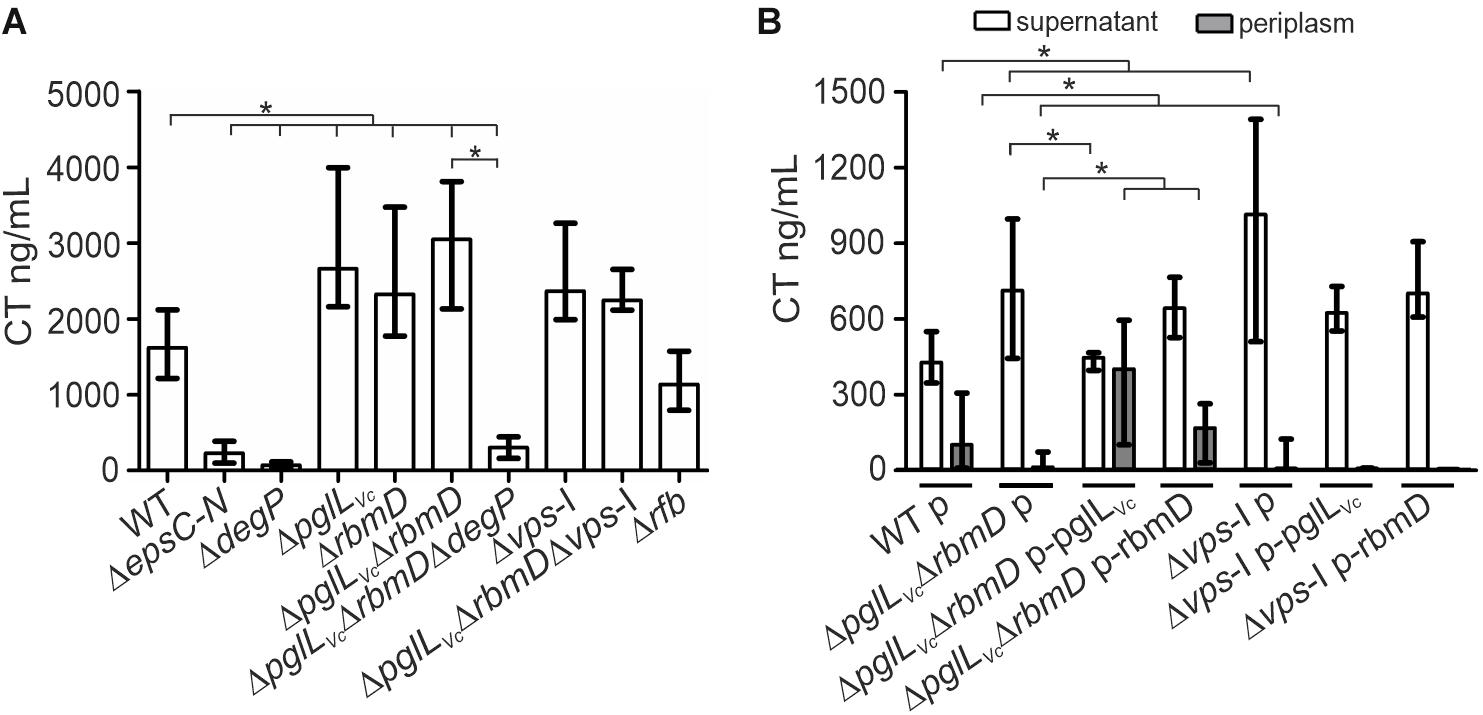
Figure 3. O-OTases impact CT secretion. (A) The amount of secreted CT was determined in the culture supernatants by ELISA for V. cholerae WT, ΔepsC-N, ΔdegP, ΔpglLVc, ΔrbmD, ΔpglLVcΔrbmD, ΔpglLVcΔrbmDΔdegP, Δvps-I, ΔpglLVcΔrbmDΔvps-I, and Δrfb. Strains were grown under virulence gene factor expressing conditions (AKI conditions) to induce CT expression. (B) The amounts of secreted (open bars) and periplasmic (gray bars) CT for strains carrying empty vector or complementation plasmids were determined in the culture supernatants and periplasmic fractions by ELISA. Periplasmic fractions were controlled for cytoplasmic contamination and cell lysis via dot blot analyses detecting RpoA, the α-subunit of the RNA-Polymerase (for details see section “Dot Blot Analysis”). A representative dot blot is shown in Supplementary Figure S4A, indicating equally low levels of cytoplasmic contamination for all periplasmic fractions. (A,B) The data is given as median with interquartile range (n ≥ 8). Asterisks highlight significant differences between respective data sets (∗P < 0.05 Kruskal–Wallis test followed by post hoc Dunn’s multiple comparisons).
Restoration of PglLVc expression significantly lowered the CT secretion compared to the ΔpglLVcΔrbmD with an empty vector control, while with restoration of RbmD expression, only a mild reduction was observed (Figure 3B). In addition, the CT levels in the periplasm were quantified for theses strains showing an inverse pattern compared to secreted CT. Periplasmic CT levels were significantly lower for ΔpglLVcΔrbmD with an empty vector compared to WT with empty vector. Moreover, PglLVc or RbmD expression significantly increased periplasmic CT levels compared to ΔpglLVcΔrbmD with an empty vector. Again, equal CT expression was observed in these strains with plasmids indicated by comparable signal intensities in the respective WCE (Supplementary Figure S3B). In comparison to the WT a marked decrease of CT secretion for the T2SS-mutant ΔepsC-N and ΔdegP was observed (Figure 3A). Moreover, deletion of degP in the ΔpglLVcΔrbmD background (ΔpglLVcΔrbmDΔdegP) also reduced CT secretion compared to ΔpglLVcΔrbmD. Thus, degP is epistatic to pglLVc and rbmD. In summary, CT is more efficiently secreted in the absence of PglLVc and RbmD, while presence of PglLVc and RbmD is associated with reduced translocation into the supernatant resulting in accumulation of CT in the periplasm.
Next, we tried to pinpoint the potential origin of an endogenous protein glycosylation by assuming that a defect in endogenous glycan availability might phenocopy pglLVc- and rbmD-mutants. In principle, V. cholerae encodes two major glycan biosynthesis pathways, represented by the vps- (Vibrio exopolysaccharide, VPS) and rfb- (O-antigen glycan) gene clusters (Manning et al., 1995). To assess the potential influence of these two pathways, CT secretion was quantified for strains lacking either the rfb or vps genes. Similar to ΔpglLVc, ΔrbmD, and ΔpglLVcΔrbmD, mutants lacking the entire vps-I gene cluster (Δvps-I and ΔpglLVcΔrbmDΔvps-I) showed increased CT levels in the supernatant compared to WT, whereas deletion of rfb genes (Δrfb) had no effect (Figure 3A). Deletion of pglLVc and rbmD in addition to the vps-I gene cluster did not further enhance the secreted CT levels. Moreover, Δvps-I and ΔpglLVcΔrbmD showed a similar secretion efficacy of CT with almost no detectable CT amounts in the periplasm and high levels in the supernatant (Figure 3B). Notably, expression of PglLVc and RbmD in Δvps-I (Δvps-I p-pglLVc and Δvps-I p-rbmD) did not restore detectable CT levels in the periplasm (Figure 3B). These phenotypes provide a first hint that the vps cluster could be involved in providing an endogenous glycan.
PglLVc and RbmD Affect Growth on Chitin
In the aquatic lifestyle, V. cholerae secrets several proteins of the chitin utilization program via the T2SS (Sikora et al., 2011). As PglLVc and RbmD may generally affect secretion of T2SS-dependent substrates including chitinases, mutants thereof might exhibit altered growth on chitin. Therefore, we compared growth dynamics of ΔpglLVc, ΔrbmD, and ΔpglLVcΔrbmD to the WT in minimal media supplemented with chitin or its corresponding monomer GlcNAc as sole carbon sources, respectively (Figures 4A,B). All strains showed similar growth on GlcNAc demonstrating equal uptake and metabolic usage of the monosaccharide (Figure 4B). In contrast, the ΔpglLVc, ΔrbmD and especially the ΔpglLVcΔrbmD mutant showed enhanced growth on chitin that gradually increased over the course of 72 h growth period compared to the WT (Figure 4A). Similar to ΔpglLVcΔrbmD mutant, the Δvps-I mutant also showed enhanced growth on chitin, but similar growth on GlcNAc, if compared to the WT (Figures 4A,B). In trans expression of both PglLVc and RbmD in the double mutant did not affect growth on GlcNAc, but significantly reduced growth on chitin and allowed (partial) restoration of the WT phenotypes at 48 and 72 h (Figures 4C,D). In conclusion, single and double mutants of pglLVc and rbmD have a fitness advantage compared to WT while growing on chitin, consistent with the notion of a more efficient secretion of chitinases as T2SS substrates.
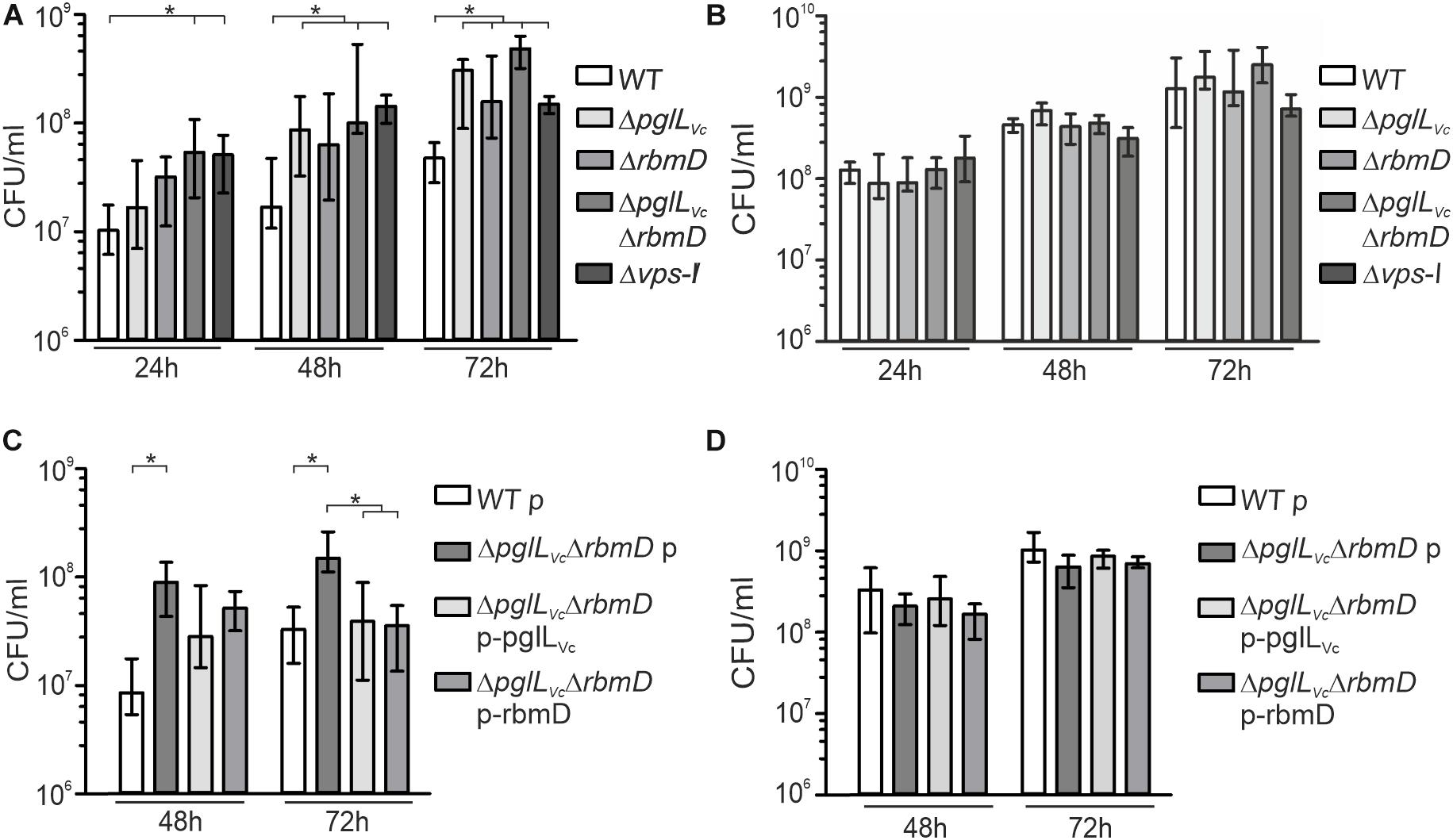
Figure 4. Deletion of O-OTases affects chitin utilization. (A,B) Shown is growth (CFU/ml) at indicated time points of WT, ΔpglLVc, ΔrbmD, ΔpglLVcΔrbmD, and Δvps-I in minimal media M9 supplemented with chitin (A) or supplemented with GlcNAc (B) as a sole carbon source. (C,D) Shown is growth (CFU/ml) at indicated time points of WT with empty vector (WT p), ΔpglLVcΔrbmD with empty vector (ΔpglLVcΔrbmD p), ΔpglLVcΔrbmD p-pglLVc, and ΔpglLVcΔrbmD p-rbmD in minimal media M9 supplemented with chitin (C) or supplemented with GlcNAc (D) as a sole carbon source. (A–D) The data is given as median with interquartile range (n ≥ 8). Asterisks highlight significant differences between respective data sets (∗P < 0.05 Kruskal–Wallis test followed by post hoc Dunn’s multiple comparisons).
PglLVc and RbmD Affect Biofilm Formation
Both, pglLVc and rbmD, have been previously identified in a screen for genes up-regulated during dynamic biofilm formation (Seper et al., 2014). In that report, they were excluded from subsequent characterization as they did not meet the stringent criteria for bona fide in biofilm induced and were at time only annotated with unknown function. Of note, rbmD is part of the rbm-gene cluster (rugosity and biofilm structure modulators) that harbors the biofilm matrix protein genes rbmA and rbmC (Fong et al., 2006; Fong and Yildiz, 2007). Based on this, we analyzed the pglLVc and rbmD mutants for alterations in biofilm formation. First, we focused on static biofilm formation after 24 and 48 h, representing two time points frequently used for biofilm analyses in V. cholerae (Seper et al., 2011). In general, altered biofilm formation can already be observed at 24 h (Supplementary Figure S5A), but phenotypes are more pronounced after 48 h (Figure 5A). Here, the ΔpglLVc mutant showed a slight, but significant increase in static biofilm formation, while the ΔrbmD mutant and especially the ΔpglLVcΔrbmD double mutant exhibited pronounced increases in biofilm formation compared to the WT (Figure 5A). In trans expression of both PglLVc and RbmD in the double mutant significantly reduced biofilm formation and allowed partial restoration of the WT phenotypes at both time points (Figure 5B and Supplementary Figure S5B).
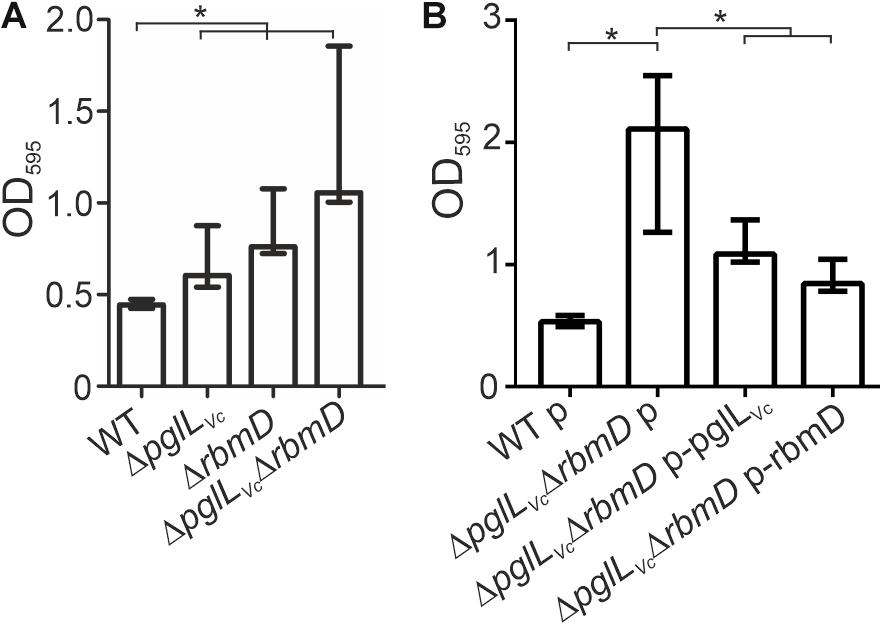
Figure 5. O-OTases impact static biofilm formation. The biofilm formation capacity of the strains indicated on the x-axis was assayed under static conditions by crystal violet staining and subsequent determination of the OD595 (n ≥ 24). (A) Biofilms of WT, ΔpglLVc, ΔrbmD and ΔpglLVcΔrbmD were quantified after 48 h. (B) In trans expression of PglLVc or RbmD partially restores biofilm to WT levels at 48 h. (A,B) The data is given as median with interquartile range (n ≥ 24). Asterisks highlight significant differences between the indicated data sets (∗P < 0.05 Kruskal–Wallis test followed by post hoc Dunn’s multiple comparisons).
Three-dimensional biofilm morphology was microscopically analyzed in a flow cell system using GFP-expressing backgrounds (Seper et al., 2014). The ΔpglLVc mutant and the ΔpglLVc,ΔrbmD double mutant exhibited a significant increased attachment capability, while the surface coverage of the ΔrbmD mutant was unaltered compared to the WT (Figures 6A,B). In mature biofilms, distinct differences could be observed upon deletion of pglLVc and rbmD (Figure 6C). Most pronounced phenotypes in comparison to the WT were the increased biomass in case of ΔpglLVc biofilms and the enhanced maximum thickness and higher roughness coefficient for ΔrbmD biofilms (Figure 6D). Biofilm formation of the double mutant ΔpglLVcΔrbmD combines characteristics of both single mutants with an increase in attachment, biomass and maximum thickness (Figure 6D). V. cholerae mutants exhibiting increased biofilm formation can be frequently correlated with enhanced expression of vps genes encoding proteins for the V. cholerae exopolysaccharide (VPS) matrix synthesis and secretion (Yildiz and Schoolnik, 1999). To assess whether the enhanced biofilm formation was related to enhanced vps expression here, chromosomal transcriptional fusions of a promoterless phoA reporter gene to vpsA representing one of the first genes in the vps-I locus, were constructed in the WT and mutant backgrounds, as well as in the ΔhapR mutant, which served as a positive control for de-repressed vpsA expression (Seper et al., 2011). Thus, PhoA activities reflect the transcription levels of vpsA in the respective strains. O-OTase mutants and WT exhibited comparable levels of PhoA activity indicating similar transcription levels of vpsA in these strains (Supplementary Figure S5C). Thus, the alterations in biofilms and attachment cannot be simply explained by elevated transcription of VPS genes.
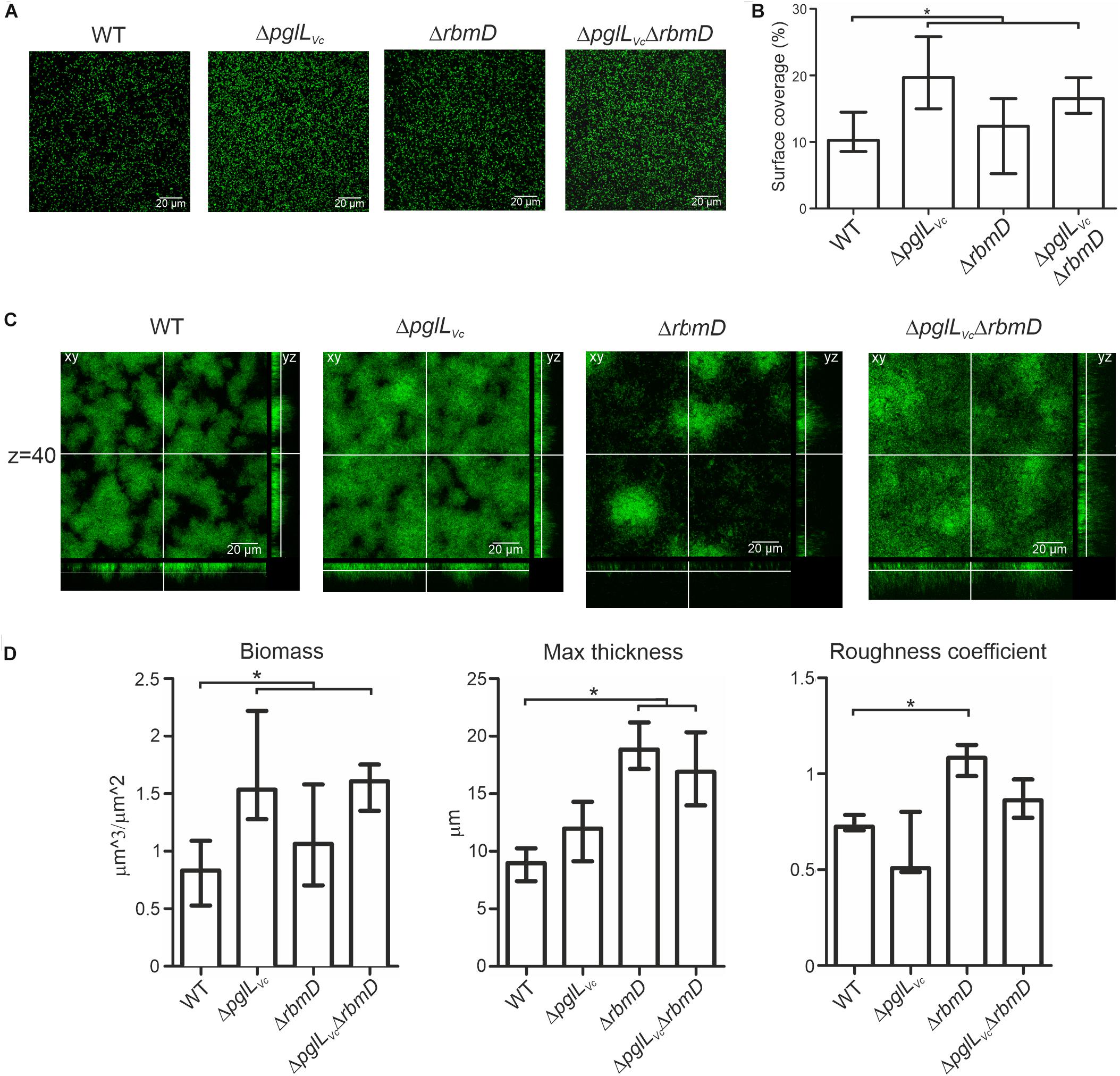
Figure 6. O-OTases alter attachment on abiotic surfaces and 3-dimensional biofilm architecture. (A) Shown are representative confocal microscopy images of surface coverage after 2 h incubation time reflecting attachment properties. (B) The median surface coverage of GFP-expressing WT and deletion mutants after 2 h incubation in flow cell chambers was determined by the COMSTAT software (n ≥ 6). (C) Shown are representative confocal laser scanning microscopy images of GFP-expressing WT and deletion mutant biofilms as horizontal (xy) and vertical (xz and xy) projections. Biofilms were allowed to mature for 24 h in flow cell chambers with constant 2% LB medium flow. The xy panels represent selected single optical sections through the three-dimensional data sets at the indicated z position (z = 40, steps = 0.13 μm). (D) Biofilm parameters such as biomass, the maximum thickness and the roughness coefficient of WT and mutants were analyzed using COMSTAT software (n ≥ 13). (A,C) Images were obtained with Leica SP5 confocal microscope. (B,D) Data is presented as median with interquartile range. Asterisks highlight significant differences between respective data sets (∗P < 0.05 Kruskal–Wallis test followed by post hoc Dunn’s multiple comparisons).
RbmD Restricts RbmA Secretion in a VPS Dependent Manner
As vps expression was unaltered in the ΔpglLVcΔrbmD mutant, we hypothesized that the alterations in biofilm amounts and architecture could be due to differential secretion of the T2SS substrates RbmA, RbmC or Bap1, which represent three major adhesive protein components of the V. cholerae biofilm matrix (Absalon et al., 2011; Berk et al., 2012). We therefore deleted rbmA, rbmC and bap1 independently in the WT, ΔpglLVc, ΔrbmD, and ΔpglLVcΔrbmD backgrounds and tested biofilm formation. Individual mutants ΔrbmA, ΔrbmC, and Δbap1 exhibit similar biofilm levels compared to the WT in the static biofilm assay (Figure 7A), which is concordant with a previous report as single deletion of each of the three matrix proteins does not impact biofilm formation (Fong et al., 2006). The absence of PglLVc and RbmD still resulted in increased biofilm formation in the ΔrbmC and Δbap1 backgrounds (Figure 7A). Similarly, ΔpglLVc and ΔrbmAΔpglLVc showed equally enhanced biofilm levels compared to the WT. Deletion of rbmA in the ΔrbmD or ΔpglLVcΔrbmD background negated the RbmD-dependent biofilm phenotypes and resulted in significant lower biofilm amounts compared to the parental ΔrbmD or ΔpglLVcΔrbmD strains (Figure 7A). This phenotypic analysis thus revealed synthetic genetic interactions between rbmA and rbmD.
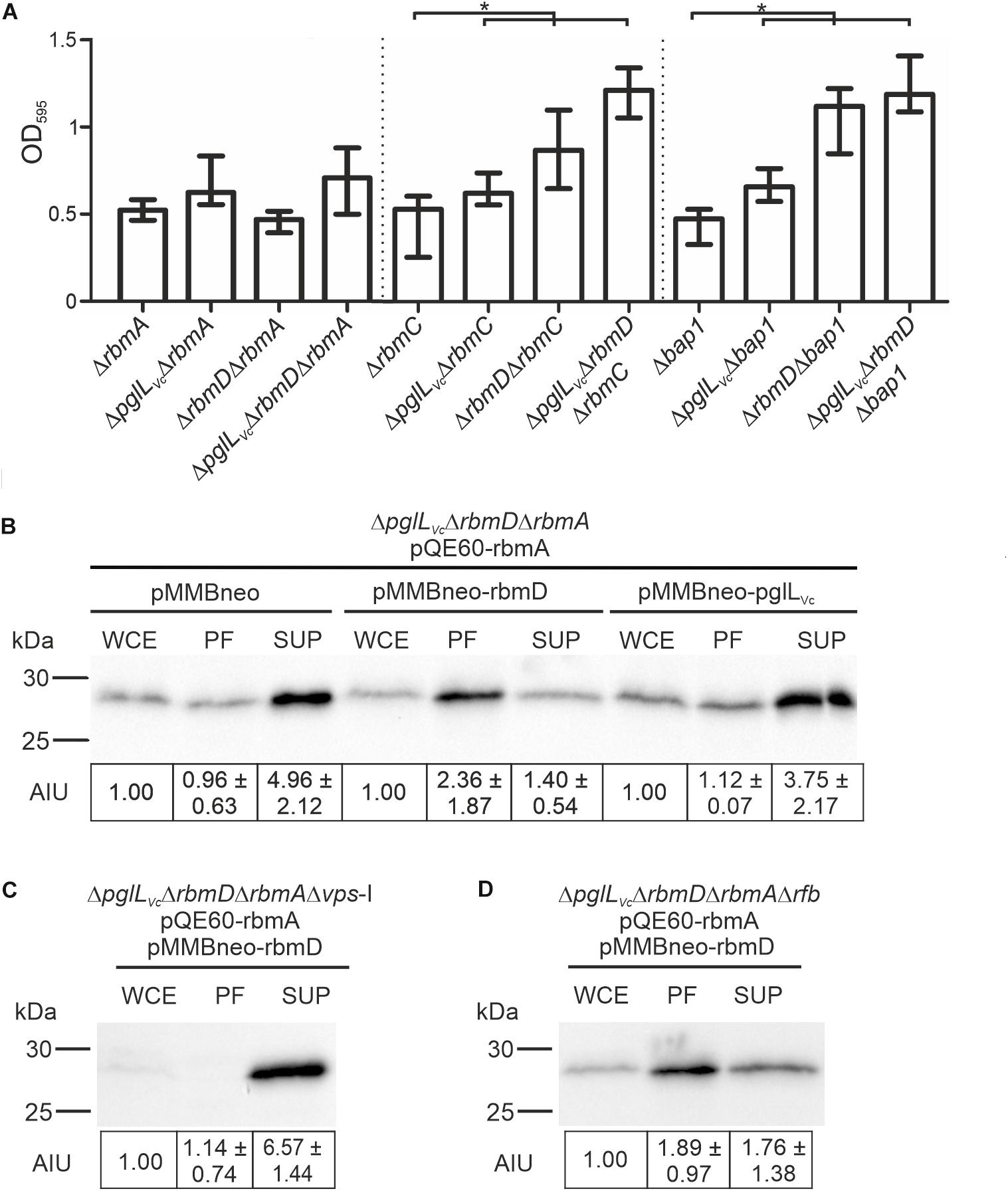
Figure 7. O-OTases impact RbmA secretion. (A) Deletion of rbmA, but not rbmC or bap1, negates the increased biofilm formation in O-OTase mutants. The biofilm formation capacity of the strains indicated on the x-axis were quantified after 48 h under static conditions by crystal violet staining and subsequent determination of the OD595 (n ≥ 24). (B–D) Shown are representative immunoblots detecting His-RbmA in whole cell extracts (WCE), periplasmic fractions (PF), and supernatants (SUP) of strain ΔpglLVcΔrbmDΔrbmA (B), ΔpglLVcΔrbmDΔrbmAΔvps-I (C), or ΔpglLVcΔrbmDΔrbmAΔrfb (D) expressing His-tagged RbmA in trans (pQE60-rbmA) in the presence or absence of RbmD (pMMBneo-rbmD or pMMBneo), respectively. Equal amounts of proteins for WCE, PF or SUP were loaded according to Bradford to allow direct comparison of the fractions. Semiquantitative densitometric evaluation of detected His-RbmA was performed with the Quantity One software (Bio-Rad Laboratories) and is indicated below the immunoblots as arbitrary intensity units [mean AIU with standard deviation (n ≥ 3)] normalized to WCE of the respective strain, which was always set to 1. Notably, TCA precipitation of PF and SUP was necessary to ensure stable detection. Similar efficiency of protein precipitation for PF and SUP using TCA was controlled by spiking the samples with BSA prior to the precipitation and subsequent immunoblot analyses detecting BSA in the precipitated samples. A representative immunoblot is shown in Supplementary Figure S6, indicating equal precipitation efficiency in all samples. Moreover, PF obtained after TCA precipitation were controlled for differential cytoplasmic contamination and cell lysis via dot blot analyses detecting RpoA, the α-subunit of the RNA-Polymerase (for details see section “Dot Blot Analysis”). A representative dot blot is shown in Supplementary Figure S4B, indicating equally low levels of cytoplasmic contamination for all periplasmic fractions.
To further investigate the relationships between RbmD and RbmA, we examined RbmA trafficking using strains expressing a His-tagged derivative allowing immunoblot analysis. In the absence of PglLVc and RbmD, RbmA was relatively efficiently secreted into the supernatant with a reciprocal minority localized in the periplasmic fraction. Remarkably, expression of solely RbmD, but not PglLVc, effectively reversed RbmA localization resulting in its relative accumulation in the periplasmic fraction and reduced translocation into the supernatant (Figure 7B). RbmD thus diminishes the secretion efficacy of RbmA, providing an example for differential secretion of a T2SS substrate modulated by the status of RbmD.
Having revealed a RbmD-dependent periplasmic localization of RbmA, we tried to pinpoint the origin of the glycan potentially transferred by RbmD. Based on the differential secretion pattern of CT in presence or absence of the VPS, we focused on a strain lacking the vps-I gene cluster. In the Δvps-I background the RbmD-dependent periplasmic accumulation of His-tagged RbmA was negated and a robust translocation of His-tagged RbmA was observed (Figure 7C). Concordant to the CT data, RbmD-dependent periplasmic accumulation of His-tagged RbmA was still observed in a Δrfb background (Figure 7D). This suggests that the vps cluster can provide a glycan precursor transferred by RbmD.
Discussion
Here, we establish a clear association between O-linked protein glycosylation mediated by the oligosaccharyltransferase PglLVc and the efficacy of type II protein secretion-dependent processes in the human pathogen V. cholerae. These findings define a first connection between protein glycosylation and processes critical to V. cholerae fitness in and outside of its host, such as differential secretion of T2SS-dependent substrates including CT, chitinases and the biofilm matrix protein RbmA. Moreover, we show that mutants lacking RbmD, whose functions remain unknown but shares structural features with PglLVc, exhibit phenotypic perturbations analogous to those lacking PglLVc.
These findings raise the obvious question as to how O-linked protein glycosylation status mediated by PglLVc might impact on the translocation of type II protein secretion substrates across the outer membrane. Perhaps the simplest explanation would be that one or more of the glycoproteins targeted by PglLVc influences type II protein secretion proficiency and that the activity or functionality of that target protein is influenced by its glycosylation status. Here, obvious candidates for such glycoproteins would be DegP as well as other periplasmic chaperones such as SurA and FkpA acting alone or in concert. Nonetheless, we hypothesize that glycosylation of these and/or other proteins might diminish their intrinsic functionality or abundance. In this model, lack of glycosylation would thus enhance their activity or levels to foster the terminal steps of the T2SS. Confirmation of this model requires a more thorough characterization of the V. cholerae glycoproteome and the effects of O-linked protein glycosylation on glycoprotein abundance and functionality. Notably, pglLVc is located upstream of the msh-operon encoding a surface pilus termed the mannose-sensitive hemagglutinin (MSHA). Although not identified along our study, it is tempting to speculate that O-glycosylation also targets components of MSHA or affect their secretion as O-glycosylation of pili components has been demonstrated for other bacteria (Aas et al., 2007; Egge-Jacobsen et al., 2011). Notably, MSHA is important for the first steps in biofilm formation by facilitating adherence to abiotic and chitinous surfaces as well as for the pharyngeal colonization of nematodes (Watnick et al., 1999; Chiavelli et al., 2001; Meibom et al., 2004; List et al., 2018). Thus, it cannot be excluded that the biofilm phenotypes of pglLVc and rbmD mutants observed herein are at least partially associated to differential MSHA functionality.
An intriguing aspect of this work relates to role of RbmD whose mutation phenocopies that of the pglLVc null mutant. To our knowledge, this is one of the first reports of these phenotypes associated with the failure to express this protein in V. cholerae WT. Notably, V. cholerae can undergo phenotypic variation generating smooth and rugose variants. The later shows elevated VPS-production, increased biofilm formation and enhanced resistance to several environmental stresses. While the work herein is based on a smooth V. cholerae isolate, Fong and Yildiz (2007) analyzed biofilm formation of a rbmD-mutant in a rugose variant. In the rugose background deletion of rbmD resulted in less compact biofilms and larger cell aggregates compared to the parental rugose variant. However, direct comparison of the results from our study and the work by Fong et al. is difficult, due to the differential phenotypic background and different V. cholerae isolates used. Based on the results presented herein, we surmise that RbmD impacts on the same pathway as that modulated by PglLVc in a non-redundant fashion. Besides similarity PglLVc, RbmD is also related to WaaL-type O-antigen ligases and members of the SEDS (shape, elongation, division and sporulation) protein family members that serve as bacterial cell wall polymerases (Meeske et al., 2016). The potential for RbmD to have a role other than in protein glycosylation is intriguing especially given its inability to use the UndPP-diNACBac donor that can be used by virtually all O-OTases tested to date.
Another obvious point of interest relates to the nature of the endogenous glycan used by PglLVc and potentially RbmD. Computational analyses of the Sequence Similarity DataBase revealed no obvious similarities of the entire gonococcal pgl gene cluster in V. cholerae. However, similarity searches for orthologs to the individual gonococcal pgl genes revealed several hits in two gene clusters (VC0240–VC0263 and VC917–VC927) encoding enzymes for the O-antigen biosynthesis and the Vibrio exopolysacharide (VPS), respectively. Deletion of the vps-I gene cluster (but not that for the O-antigen biosynthesis) phenocopied pglLVc and rbmD mutants in the enhanced secretion of both CT and RbmA, providing first evidence that the diverse sugars present in the VPS [i.e., α-GulNAcAGly, α-Glcp, β-Glcp, α-Galp, and α-D-GlcNAc (Yildiz et al., 2014)] or combinations thereof may serve as at least one source for the transferred O-glycan in V. cholerae. Notably, the VPS oligosaccharide is notoriously recalcitrant to biochemical characterization as it is bound to a yet unidentified component, which gives it high viscosity and which would theoretically complicate its structural characterization by MS as a post-translational protein modification.
Further insights into the mechanisms operating here will undoubtedly require elucidation of the cognate glycoform utilized by PglLVc and potentially by RbmD. Along this line we tried to identify the glycan present in V. cholerae. Briefly, His-tagged DegP was isolated in absence and presence of PglLVc and/or RbmD and subjected to MS analysis, but no defined modification could be identified. It should be emphasized that detection and identification of a glycan with unknown composition is not straightforward and requires substantial expertise and instrumentation. A series of unfavorable limitations decreased the likelihood of glycan identification: (i) Overexpression of periplasmic His-tagged DegP reduced fitness of V. cholerae. Thus, the yield of purified protein was relatively low. (ii) The chemical composition of the natural glycan is not only unknown, but might exhibit heterogeneity. Thus, the glycosylated population may not represent a single peak in the MS. (iii) O-glycosylation might render protein stability. (iv) Based on our results using the gonoccocal glycan only a minor population of substrate is glycosylated, e.g., ∼1% in case of DegP in presence of the diNAcBac glycan and PglLVc. It should be noted, that such low modification rates could massively affect overall DegP chaperone activity in the periplasm as DegP monomers assemble from trimers and hexamers to multimeric polyhedral cages with up to 24 subunits (Krojer et al., 2002; Kim and Sauer, 2014). However, such modification rates further reduce the chances for identification of the natural glycan. Based on these limitations the identification of the cognate glycan seems to be a challenging task for the future.
In summary, we herein reveal multiple gene interactions that ultimately impact on the efficacy of the V. cholerae T2SS at the level of translocation of substrates from the periplasm to the extracellular milieu. T2SS modulation affects several stages of the pathogens’ lifecycle as highlighted by the diverse phenotypes reported herein. It is tempting to speculate that O-glycosylation provides a post-translational mechanism allowing bacterial cells to accumulate effector proteins, like RbmA, chitinases or CT in the periplasm to be readily available for release when the cells settle down on the new substratum for biofilm formation or at the colonization site in the gastrointestinal tract. Thus, O-glycosylation via PglLVc and possibly RbmD could represent a fine-tuned feedback mechanism controlling release of T2SS effectors by modulation of secretion efficacy.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
DV, FM, KP, JA, MF, JR, MK, and SS designed the study. DV, FM, KP, DL, JA, LL, L-MM, TK, MT, AS, FZ, RB-G, JR, MK, and SS performed the experiments and analysis. DV, FM, KP, JA, FZ, MF, JR, MK, and SS contributed to the discussion and data evaluation. DV, FM, JR, MK, and SS wrote the manuscript.
Funding
This work was supported by the Austrian Science Fund (FWF) grants: W901 (DK Molecular Enzymology) to DV, FM, KP, FZ, and SS as well as P27654 to SS and KLI425 to RB-G. MK and JA were supported by the University of Oslo and its associated Center for Integrative Microbial Evolution. AS was supported by the Doctoral Academy Graz.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to M. Aebi and colleagues for the opportunity to test alternative bacterial glycosylation systems.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02780/full#supplementary-material
Footnotes
References
Aas, F. E., Vik, A., Vedde, J., Koomey, M., and Egge-Jacobsen, W. (2007). Neisseria gonorrhoeae O-linked pilin glycosylation: functional analyses define both the biosynthetic pathway and glycan structure. Mol. Microbiol. 65, 607–624. doi: 10.1111/j.1365-2958.2007.05806.x
Absalon, C., Van Dellen, K., and Watnick, P. I. (2011). A communal bacterial adhesin anchors biofilm and bystander cells to surfaces. PLoS Pathog. 7:e1002210. doi: 10.1371/journal.ppat.1002210
Almagro-Moreno, S., Kim, T. K., Skorupski, K., and Taylor, R. K. (2015). Proteolysis of virulence regulator ToxR is associated with entry of Vibrio cholerae into a dormant state. PLoS Genet. 11:e1005145. doi: 10.1371/journal.pgen.1005145
Altindis, E., Fu, Y., and Mekalanos, J. J. (2014). Proteomic analysis of Vibrio cholerae outer membrane vesicles. Proc. Natl. Acad. Sci. U.S.A. 111, E1548–E1556. doi: 10.1073/pnas.1403683111
Anonsen, J. H., Vik, A., Borud, B., Viburiene, R., Aas, F. E., Kidd, S. W., et al. (2015). Characterization of a unique tetrasaccharide and distinct glycoproteome in the o-linked protein glycosylation system of Neisseria elongata subsp. glycolytica. J. Bacteriol. 198, 256–267. doi: 10.1128/JB.00620-615
Anonsen, J. H., Vik, A., Egge-Jacobsen, W., and Koomey, M. (2012). An extended spectrum of target proteins and modification sites in the general O-linked protein glycosylation system in Neisseria gonorrhoeae. J. Proteome Res. 11, 5781–5793. doi: 10.1021/pr300584x
Berk, V., Fong, J. C., Dempsey, G. T., Develioglu, O. N., Zhuang, X., Liphardt, J., et al. (2012). Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science 337, 236–239. doi: 10.1126/science.1222981
Chang, A. C., and Cohen, S. N. (1978). Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134, 1141–1156.
Chiavelli, D. A., Marsh, J. W., and Taylor, R. K. (2001). The mannose-sensitive hemagglutinin of Vibrio cholerae promotes adherence to zooplankton. Appl. Environ. Microbiol. 67, 3220–3225. doi: 10.1128/aem.67.7.3220-3225.2001
Childers, B. M., and Klose, K. E. (2007). Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol. 2, 335–344. doi: 10.2217/17460913.2.3.335
Donnenberg, M. S., and Kaper, J. B. (1991). Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59, 4310–4317.
Egge-Jacobsen, W., Salomonsson, E. N., Aas, F. E., Forslund, A. L., Winther-Larsen, H. C., Maier, J., et al. (2011). O-linked glycosylation of the PilA pilin protein of Francisella tularensis: identification of the endogenous protein-targeting oligosaccharyltransferase and characterization of the native oligosaccharide. J. Bacteriol. 193, 5487–5497. doi: 10.1128/JB.00383-311
Faridmoayer, A., Fentabil, M. A., Mills, D. C., Klassen, J. S., and Feldman, M. F. (2007). Functional characterization of bacterial oligosaccharyltransferases involved in O-linked protein glycosylation. J. Bacteriol. 189, 8088–8098. doi: 10.1128/JB.01318-1317
Feldman, M. F., Wacker, M., Hernandez, M., Hitchen, P. G., Marolda, C. L., Kowarik, M., et al. (2005). Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021. doi: 10.1073/pnas.0500044102
Fong, J. C., Karplus, K., Schoolnik, G. K., and Yildiz, F. H. (2006). Identification and characterization of RbmA, a novel protein required for the development of rugose colony morphology and biofilm structure in Vibrio cholerae. J. Bacteriol. 188, 1049–1059. doi: 10.1128/jb.188.3.1049-1059.2006
Fong, J. C., and Yildiz, F. H. (2007). The rbmBCDEF gene cluster modulates development of rugose colony morphology and biofilm formation in Vibrio cholerae. J. Bacteriol. 189, 2319–2330. doi: 10.1128/jb.01569-06
Gebhart, C., Ielmini, M. V., Reiz, B., Price, N. L., Aas, F. E., Koomey, M., et al. (2012). Characterization of exogenous bacterial oligosaccharyltransferases in Escherichia coli reveals the potential for O-linked protein glycosylation in Vibrio cholerae and Burkholderia thailandensis. Glycobiology 22, 962–974. doi: 10.1093/glycob/cws059
Hartley, M. D., Morrison, M. J., Aas, F. E., Borud, B., Koomey, M., and Imperiali, B. (2011). Biochemical characterization of the O-linked glycosylation pathway in Neisseria gonorrhoeae responsible for biosynthesis of protein glycans containing N,N’-diacetylbacillosamine. Biochemistry 50, 4936–4948. doi: 10.1021/bi2003372
Hava, D. L., Hemsley, C. J., and Camilli, A. (2003). Transcriptional regulation in the Streptococcus pneumoniae rlrA pathogenicity islet by RlrA. J. Bacteriol. 185, 413–421. doi: 10.1128/jb.185.2.413-421.2003
Heydorn, A., Nielsen, A. T., Hentzer, M., Sternberg, C., Givskov, M., Ersboll, B. K., et al. (2000). Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146(Pt 10), 2395–2407. doi: 10.1099/00221287-146-10-2395
Iwanaga, M., and Yamamoto, K. (1985). New medium for the production of cholera toxin by Vibrio cholerae O1 biotype El Tor. J. Clin. Microbiol. 22, 405–408.
Iwanaga, M., Yamamoto, K., Higa, N., Ichinose, Y., Nakasone, N., and Tanabe, M. (1986). Culture Conditions for Stimulating Cholera Toxin Production by Vibrio cholerae O1 El Tor. Microbiol. Immunol. 30, 1075–1083. doi: 10.1111/j.1348-0421.1986.tb03037.x
Iwashkiw, J. A., Vozza, N. F., Kinsella, R. L., and Feldman, M. F. (2013). Pour some sugar on it: the expanding world of bacterial protein O-linked glycosylation. Mol. Microbiol. 89, 14–28. doi: 10.1111/mmi.12265
Johnson, T. L., Fong, J. C., Rule, C., Rogers, A., Yildiz, F. H., and Sandkvist, M. (2014). The Type II secretion system delivers matrix proteins for biofilm formation by Vibrio cholerae. J. Bacteriol. 196, 4245–4252. doi: 10.1128/JB.01944-1914
Kang, D., Gho, Y. S., Suh, M., and Kang, C. (2002). Highly sensitive and fast protein detection with coomassie brilliant blue in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Bull. Kor. Chem. Soc. 23, 1511–1512. doi: 10.5012/bkcs.2002.23.11.1511
Kim, S., and Sauer, R. T. (2014). Distinct regulatory mechanisms balance DegP proteolysis to maintain cellular fitness during heat stress. Genes Dev. 28, 902–911. doi: 10.1101/gad.238394.114
Kolter, R., Inuzuka, M., and Helinski, D. R. (1978). Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell 15, 1199–1208. doi: 10.1016/0092-8674(78)90046-6
Krojer, T., Garrido-Franco, M., Huber, R., Ehrmann, M., and Clausen, T. (2002). Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature 416, 455–459. doi: 10.1038/416455a
Link, A. J., and LaBaer, J. (2011). Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb. Protoc. 2011, 993–994. doi: 10.1101/pdb.prot5651
List, C., Grutsch, A., Radler, C., Cakar, F., Zingl, F. G., Schild-Prufert, K., et al. (2018). Genes activated by Vibrio cholerae upon exposure to Caenorhabditis elegans reveal the mannose-sensitive hemagglutinin to be essential for colonization. mSphere 3:e00238-18. doi: 10.1128/mSphereDirect.00238-218
Manning, P. A., Stroeher, U. H., Karageorgos, L. E., and Morona, R. (1995). Putative O-antigen transport genes within the rfb region of Vibrio cholerae O1 are homologous to those for capsule transport. Gene 158, 1–7. doi: 10.1016/0378-1119(95)00124-o
Manoil, C. (1991). Analysis of membrane protein topology using alkaline phosphatase and beta-galactosidase gene fusions. Methods Cell Biol. 34, 61–75. doi: 10.1016/s0091-679x(08)61676-3
Matson, J. S., and DiRita, V. J. (2005). Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 102, 16403–16408. doi: 10.1073/pnas.0505818102
Meeske, A. J., Riley, E. P., Robins, W. P., Uehara, T., Mekalanos, J. J., Kahne, D., et al. (2016). SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638. doi: 10.1038/nature19331
Meibom, K. L., Li, X. B., Nielsen, A. T., Wu, C. Y., Roseman, S., and Schoolnik, G. K. (2004). The Vibrio cholerae chitin utilization program. Proc. Natl. Acad. Sci. U.S.A. 101, 2524–2529. doi: 10.1073/pnas.0308707101
Miller, J. H. (1972). Experiments in Molecular Genetics. New York, NY: Cold Spring Harbor Laboratory.
Miller, V. L., and Mekalanos, J. J. (1988). A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170, 2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988
Morales, V. M., Backman, A., and Bagdasarian, M. (1991). A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97, 39–47. doi: 10.1016/0378-1119(91)90007-x
Nelson, E. J., Harris, J. B., Morris, J. G. Jr., Calderwood, S. B., and Camilli, A. (2009). Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat. Rev. Microbiol. 7, 693–702. doi: 10.1038/nrmicro2204
Neu, H. C., and Heppel, L. A. (1965). The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J. Biol. Chem. 240, 3685–3692.
Nielsen, A. T., Dolganov, N. A., Otto, G., Miller, M. C., Wu, C. Y., and Schoolnik, G. K. (2006). RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog. 2:e109. doi: 10.1371/journal.ppat.0020109
Nothaft, H., and Szymanski, C. M. (2010). Protein glycosylation in bacteria: sweeter than ever. Nat. Rev. Microbiol. 8, 765–778. doi: 10.1038/nrmicro2383
Pressler, K., Vorkapic, D., Lichtenegger, S., Malli, G., Barilich, B. P., Cakar, F., et al. (2016). AAA+ proteases and their role in distinct stages along the Vibrio cholerae lifecycle. Int. J. Med. Microbiol. 306, 452–462. doi: 10.1016/j.ijmm.2016.05.013
Roberts, A., Pearson, G. D., and Mekalanos, J. J. (1992). “Cholera vaccines strains derived from a 1991 peruvian isolate of Vibrio cholerae and other El Tor strains,” in Proceedings of the 28th Joint Conference, U.S.-Japan Cooperative Medical Science Program on Cholera and Related Diarrheal Diseases, Tokyo.
Rogers, A., Townsley, L., Gallego-Hernandez, A. L., Beyhan, S., Kwuan, L., and Yildiz, F. H. (2016). The LonA protease regulates biofilm formation, motility, virulence, and the type VI secretion system in Vibrio cholerae. J. Bacteriol. 198, 973–985. doi: 10.1128/JB.00741-715
Roier, S., Zingl, F. G., Cakar, F., Durakovic, S., Kohl, P., Eichmann, T. O., et al. (2016). A novel mechanism for the biogenesis of outer membrane vesicles in Gram-negative bacteria. Nat. Commun. 7:10515. doi: 10.1038/ncomms10515
Schild, S., Bishop, A. L., and Camilli, A. (2008). Ins and outs of Vibrio cholerae. Microbe 3, 131–136. doi: 10.1016/j.mib.2017.01.002
Schild, S., Lamprecht, A. K., Fourestier, C., Lauriano, C. M., Klose, K. E., and Reidl, J. (2005). Characterizing lipopolysaccharide and core lipid A mutant O1 and O139 Vibrio cholerae strains for adherence properties on mucus-producing cell line HT29-Rev MTX and virulence in mice. Int. J. Med. Microbiol. 295, 243–251. doi: 10.1016/j.ijmm.2005.05.002
Schild, S., Tamayo, R., Nelson, E. J., Qadri, F., Calderwood, S. B., and Camilli, A. (2007). Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe 2, 264–277. doi: 10.1016/j.chom.2007.09.004
Seper, A., Fengler, V. H., Roier, S., Wolinski, H., Kohlwein, S. D., Bishop, A. L., et al. (2011). Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol. Microbiol. 82, 1015–1037. doi: 10.1111/j.1365-2958.2011.07867.x
Seper, A., Pressler, K., Kariisa, A., Haid, A. G., Roier, S., Leitner, D. R., et al. (2014). Identification of genes induced in Vibrio cholerae in a dynamic biofilm system. Int. J. Med. Microbiol. 304, 749–763. doi: 10.1016/j.ijmm.2014.05.011
Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858. doi: 10.1021/ac950914h
Sikora, A. E. (2013). Proteins secreted via the type II secretion system: smart strategies of Vibrio cholerae to maintain fitness in different ecological niches. PLoS Pathog. 9:e1003126. doi: 10.1371/journal.ppat.1003126
Sikora, A. E., Lybarger, S. R., and Sandkvist, M. (2007). Compromised outer membrane integrity in Vibrio cholerae Type II secretion mutants. J. Bacteriol. 189, 8484–8495. doi: 10.1128/JB.00583-587
Sikora, A. E., Zielke, R. A., Lawrence, D. A., Andrews, P. C., and Sandkvist, M. (2011). Proteomic analysis of the Vibrio cholerae type II secretome reveals new proteins, including three related serine proteases. J. Biol. Chem. 286, 16555–16566. doi: 10.1074/jbc.M110.211078
Tamayo, R., Schild, S., Pratt, J. T., and Camilli, A. (2008). Role of cyclic Di-GMP during el tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic Di-GMP phosphodiesterase CdpA. Infect. Immun. 76, 1617–1627. doi: 10.1128/IAI.01337-07
Vik, A., Aas, F. E., Anonsen, J. H., Bilsborough, S., Schneider, A., Egge-Jacobsen, W., et al. (2009). Broad spectrum O-linked protein glycosylation in the human pathogen Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U.S.A. 106, 4447–4452. doi: 10.1073/pnas.0809504106
Vorregaard, M. (2008). Comstat2 - a Modern 3D Image Analysis Environment for Biofilms. Lyngby: Technical University of Denmark.
Watnick, P. I., Fullner, K. J., and Kolter, R. (1999). A role for the mannose-sensitive hemagglutinin in biofilm formation by Vibrio cholerae El Tor. J. Bacteriol. 181, 3606–3609.
Wood, W. I. (1976). Tables for the preparation of ammonium sulfate solutions. Anal. Biochem. 73, 250–257. doi: 10.1016/0003-2697(76)90165-2
Wurm, P., Tutz, S., Mutsam, B., Vorkapic, D., Heyne, B., Grabner, C., et al. (2017). Stringent factor and proteolysis control of sigma factor RpoS expression in Vibrio cholerae. Int. J. Med. Microbiol. 307, 154–165. doi: 10.1016/j.ijmm.2017.01.006
Yildiz, F., Fong, J., Sadovskaya, I., Grard, T., and Vinogradov, E. (2014). Structural characterization of the extracellular polysaccharide from Vibrio cholerae O1 El-Tor. PLoS One 9:e86751. doi: 10.1371/journal.pone.0086751
Keywords: post-translational modification, O-OTase, Vibrio cholerae, biofilm, virulence, chaperone
Citation: Vorkapic D, Mitterer F, Pressler K, Leitner DR, Anonsen JH, Liesinger L, Mauerhofer L-M, Kuehnast T, Toeglhofer M, Schulze A, Zingl FG, Feldman MF, Reidl J, Birner-Gruenberger R, Koomey M and Schild S (2019) A Broad Spectrum Protein Glycosylation System Influences Type II Protein Secretion and Associated Phenotypes in Vibrio cholerae. Front. Microbiol. 10:2780. doi: 10.3389/fmicb.2019.02780
Received: 06 August 2019; Accepted: 14 November 2019;
Published: 03 December 2019.
Edited by:
Konstantin V. Korotkov, University of Kentucky, United StatesReviewed by:
David Salvador Zamorano Sanchez, National Autonomous University of Mexico, MexicoMichel Gilbert, National Research Council Canada, Canada
Copyright © 2019 Vorkapic, Mitterer, Pressler, Leitner, Anonsen, Liesinger, Mauerhofer, Kuehnast, Toeglhofer, Schulze, Zingl, Feldman, Reidl, Birner-Gruenberger, Koomey and Schild. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Schild, c3RlZmFuLnNjaGlsZEB1bmktZ3Jhei5hdA==