 Chao Yuan1,2,3,4,5†Zhiqiu Yin1,2,3,4,5†Junyue Wang1,2,3,4,5†Chengqian Qian1,2,3,4Yi Wei1,2,3,4Si Zhang1,2,3,4Lingyan Jiang1,2,3,4*
Chao Yuan1,2,3,4,5†Zhiqiu Yin1,2,3,4,5†Junyue Wang1,2,3,4,5†Chengqian Qian1,2,3,4Yi Wei1,2,3,4Si Zhang1,2,3,4Lingyan Jiang1,2,3,4* Bin Liu1,2,3,4*
Bin Liu1,2,3,4*- 1Key Laboratory of Molecular Microbiology and Technology, Ministry of Education, TEDA College, Nankai University, Tianjin, China
- 2TEDA Institute of Biological Sciences and Biotechnology, Nankai University, Tianjin, China
- 3Tianjin Research Center for Functional Genomics and Biochips, TEDA College, Nankai University, Tianjin, China
- 4Tianjin Key Laboratory of Microbial Functional Genomics, TEDA College, Nankai University, Tianjin, China
- 5College of Life Sciences, Nankai University, Tianjin, China
Citrobacter species are opportunistic bacterial pathogens that have been implicated in both nosocomial and community-acquired infections. Among the genus Citrobacter, Citrobacter koseri is often isolated from clinical material, and has been known to cause meningitis and brain abscess in neonates and immunocompromised individuals. The virulence determinants of Citrobacter, however, remain largely unknown. Based on traditional methods, the genus Citrobacter has been divided into 11 species, but this has been problematic. Here, we determined an improved, detailed, and more accurate phylogeny of the genus Citrobacter based on whole genome sequence (WGS) data from 129 Citrobacter genomes, 31 of which were sequenced in this study. A maximum likelihood (ML) phylogeny constructed with core genome single-nucleotide polymorphisms (SNPs) classified all Citrobacter isolates into 11 distinct groups, with all C. koseri strains clustering into a single group. For comprehensive and systematic comparative genomic analyses, we investigated the distribution of virulence factors, resistance genes, and macromolecular secretion systems among the Citrobacter genus. Moreover, combined with group-specific genes analysis, we identified a key gene cluster for iron transport, which is present in the C. koseri group, but absent in other the groups, suggesting that the high-pathogenicity island (HPI) cluster may be important for the pathogenicity of C. koseri. Animal experiments showed that loss of the HPI cluster significantly decreased C. koseri virulence in mice and rat. Further, we provide evidence to explain why Citrobacter freundii is less susceptible than C. koseri to several antibiotics in silico. Overall, our data reveal novel virulence clusters specific to the predominantly pathogenic C. koseri strains, which form the basis for elucidating the virulence mechanisms underlying these important pathogens.
Introduction
The Citrobacter genus belongs to the Enterobacteriaceae family, which is a distinct group of aerobic, Gram-negative, non-spore-forming, rod-shaped bacteria that typically utilize citrate as their primary carbon source (Janda et al., 1994; Sakazaki, 1997). Citrobacter species are commonly found in water, soil, and food, and occasionally colonize the gastrointestinal tract of animals and humans (Arens and Verbist, 1997). Although Citrobacter strains colonizing the human gastrointestinal tract are generally considered to have low virulence (Pepperell et al., 2002), they may cause a wide range of diseases in the urinary tract, respiratory tract, bone, peritoneum, endocardium, meninges, intestines, bloodstream, and central nervous system, particularly in infants, young children, and immunocompromised adults (Altmann et al., 1976; Fincher et al., 1990).
The majority of infections are associated with Citrobacter koseri (Hodges et al., 1978; Lipsky et al., 1980; Lavigne et al., 2007; Mohanty et al., 2007). Citrobacter infections usually supervene upon debilitated, hospitalized patients, with multiple comorbidities (Gross et al., 1973; Williams et al., 1984; Kline and Kaplan, 1987; Mohanty et al., 2007; Samonis et al., 2009). Urinary tract infections caused by Citrobacter account for approximately half of all infections, although there are no reports of statistically significant associations between infection sites and Citrobacter species, except for C. koseri, which exhibits a remarkable degree of tropism for the brain (Gross et al., 1973; Ribeiro et al., 1976; Curless, 1980; Williams et al., 1984; Kline and Kaplan, 1987; Doran, 1999).
Based on traditional methods, the Citrobacter genus has been divided into 11 species (Brenner et al., 1993, 1999). Phylogenetic relationships based on 16S ribosomal RNA (rRNA) sequences and multilocus sequence analysis (MLSA) based on partial sequences of rpoB, pyrG, fusA, and leuS have been used to discriminate Citrobacter species (Warren et al., 2000; Clermont et al., 2015). However, gene sequence variation provides limited resolution to discriminate between closely related members of the Enterobacteriaceae family (Naum et al., 2008). Whole genome sequence (WGS) is now being employed for routine surveillance and for the detection of possible outbreaks in many countries due to lower cost, simpler protocols, and reduced time investments (Kaas et al., 2012; Dallman et al., 2015), which provides the opportunity to resolve bacterial strains at the single-nucleotide resolution needed for identifying cases linked to a common infection source (Ashton et al., 2016) and for clustering isolates into higher taxonomic groups.
Our past research has established experimental and in silico serotyping systems for Citrobacter based on specific genes in O-antigen biosynthesis gene clusters (Qian et al., 2018). In this work, we aimed to generate a fine-scaled, more accurate phylogeny and population structure based on whole genome data of Citrobacter species. Moreover, we performed an affiliation of Citrobacter species and comparative genomic analysis to gain a better understanding of virulence and resistance gene distribution in Citrobacter. Furthermore, we identified key genes contributing to C. koseri virulence. We also found explored why Citrobacter freundii is considered less susceptible than C. koseri to several antibiotics in silico.
Materials and Methods
Bacterial Strains, Plasmids, and DNA Extraction
Bacterial strains, plasmids, and primers used in this study are listed in Supplementary Table S1. The 31 Citrobacter strains sequenced in this study were obtained from the Polish Collection of Microorganisms (PCM) at the Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences (Wrocław, Poland). All strains were stored at −80°C in Luria-Bertani (LB) broth supplemented with 20% (v/v) glycerol and were cultured at 37°C in LB broth. When necessary, chloramphenicol was used at a final concentrations of 15 μg/ml. A Bacteria Extraction Kit (CWBIO Co., Ltd., China) was used for DNA extractions from each strain according to the manufacturer’s instructions. Mutant strains were generated using a λRed Recombinase System (Kirill and Barry, 2000), and all strains were verified via PCR amplification and sequencing. To generate ΔHPI (high-pathogenicity island) mutant strains, HPI cluster gene fragments were replaced by the chloramphenicol acetyltransferase cassette.
Genome Sequencing and Assembly
The whole genome of strain TBCP-5362 was sequenced using a PacBio RS II (Pacific Biosciences, Menlo Park, CA, United States), with a depth of approximately 100-fold coverage. The reads produced with the PacBio RS II were de novo assembled using MaSuRCA (Tallon et al., 2014; Kuang et al., 2015). The other 30 strains were sequenced using Illumina Paired-End sequencing technology (Illumina, Little Chesterford, Essex, United Kingdom), with a depth of 90–100-fold coverage. A library for Illumina Paired-End sequencing was prepared from 5 μg DNA using a Paired-End DNA Sample Prep Kit (Pe-102-1001, Illumina Inc., Cambridge, United Kingdom). Libraries prepared using Nextera technology and paired end reads of either 100 bp (Illumina HiSeq 2000). DNA was fragmented by nebulization for 6 min at a pressure of 32 psi. For end-repair and phosphorylation, sheared DNA was purified using the QIAquick Nucleotide Removal Kit (Qiagen, Crawley, United Kingdom). The end-repaired DNA was A-tailed and adaptors were ligated according to the manufacturer’s instructions. De novo assembly was performed using Velvet Optimiser v2.2 (Zerbino and Birney, 2008). Genome sequence annotation was conducted using the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline1. In addition, 98 publicly available Citrobacter genomes were obtained from NCBI GenBank.
Identification of Gene Orthologous Groups
OrthoFinder (Emms and Kelly, 2015) was used to determine orthologous families of the pan-genome with default parameter (for BLASTp: outfmt = 6, e-vaule = 0.001; for MCL: I = 1.5). All protein sequences were compared using a BLASTp “all-against-all” search with an E-value cutoff of < 1e−3. The single-copy core gene, pan gene, and core genome families were extracted from the OrthoFinder output file. Nucleotide sequences of single-copy core genes were extracted according to protein ID.
Phylogenetic Analysis
According to the identification of gene orthologous clusters, a total of 1450 single-copy orthologous core genes were found to be shared per genome. To determine the single-nucleotide polymorphisms (SNPs), the nucleotide sequences of single-copy core genes identified by core genome phylogenetic analysis were aligned using MAFFT (Katoh and Standley, 2013). The SNPs were integrated according to the arrangement of the single-copy genes in the C. koseri TBCP-5362 genomes. The phylogeny of SNPs was inferred using the maximum likelihood (ML) algorithm in PhyML (with the GTR model of nucleotide substitution and c-distributed rates among sites). MEGA7 and FigTree v1.4.32 were employed to construct the trees. In consideration of homologous recombination caused by horizontal gene transfer occurring in bacterial populations which can confound phylogenetic analysis, we identified and removed putative recombination regions in the set of SNPs of single-copy core genes using CloneFrameML (Didelot and Wilson, 2015). Neighbor Net (NNet) splits graph based on uncorrected p-distances were constructed and visualized with SplitsTree 4 (Huson and Bryant, 2006).
Whole-Genome Nucleotide Identity
Average nucleotide identity (ANI) and tetramer usage pattern were calculated for the 129 Citrobacter genome datasets using JSpecies 1.2.1 (Richter and Rosselló-Móra, 2009) and Gegenees v3.0 (Agren et al., 2012), using default parameters. The results were visualized using the pheatmap R package.
Core and Pan-Genome Analysis
Core and pan-genome analyses were separately performed using the 129 Citrobacter genomes. The regression analysis for the core gene cluster curve was performed using a weighted least square regression by fitting the power law n = κexp(m ×N) + Θ to means (Bottacini et al., 2010). N is the number of genomes, n is the number of core gene clusters, Θ is a constant value representing the predicted minimum number of core genes, and κ and m are parameters. According to Heap’s law pan-genome model (Tettelin et al., 2008), the total number of gene clusters is shown for increasing number of genomes (N). The curve was a least squares fit of the power law n = :Nγ to averages. An exponent γ > 0 indicates an open pan-genome species.
Species-Specific Core Genome Comparison
To examine the pan-genome in greater detail, we constructed a cluster map of the gene families across all 129 genomes using the heatmap clustering command from the pheatmap R package (Figure 3B). We excluded the core gene families and low frequency gene families that are shared by <10 strains. We termed the results as the “Group 8-specific core genome” (Supplementary Table S5), which represents the set of gene families that are shared across all strains of a species.
Gene Functional Category
We analyzed the functional category of the core gene families and the Group 8-specific core genome using different database (COG/GO/KEGG) and compute the numbers of proteins for each corresponding COG/GO/KEGG term. For different database, we use Cluster of Orthologous Group (COG) assignment (Galperin et al., 2015), InterProScan 5 (Jones et al., 2014), and WEGO 2.0 (Ye et al., 2006), BlastKOALA3, using default parameters, respectively. We determined the main biological function of differential proteins using function enrichment analysis and results visualized by GraphPad Prism 7.0.
Identification of Macromolecular Secretion Systems and Gene Island (GI)
The detection and visualization of macromolecular systems in the Citrobacter genus were performed using the programs MacSyFinder (Abby et al., 2014) and TXSScan (Abby and Rocha, 2017), using default parameters4. Furthermore, the type VI secretion system (T6SS) was predicted as described previously (Boyer et al., 2009) and combined with the results obtained using SecReT6 (Li et al., 2015), using default parameters. Genomic Island (GI) was detected using IslandViewer 4, using default parameters5 (Bertelli et al., 2017).
Calculation of Codon Usage and GC Content
We used CodonW software6 to compute statistical parameters of nucleotide composition of gene clusters and genomes.
Identification of Virulence Genes and Resistance Genes
We made use of the large-scale BLAST score ratio (LS-BSR) tool (Sahl et al., 2014), which was run against the Virulence Factors Database (Liu et al., 2019) and the Comprehensive Antibiotic Resistance Database (Jia et al., 2017). We removed virulence factors of previously studied macromolecular secretion systems and the lipopolysaccharide (LPS). Heatmaps showing the distribution of virulence factors and resistance genes were generated using the pheatmap R package.
Animal Infection
Laboratory animals, BALB/c mice (18 days old) and Sprague Dawley (SD) rats (2 days old), were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All mice were maintained in a specific pathogen-free environment. 18-day-old mice received bacteria in 100 μl phosphate-buffered saline (PBS) via the tail vein (Zhu et al., 2010). Two-day-old rat pups were anesthetized with isoflurane and inoculated with bacteria in 100 μl PBS via intraperitoneal injection. Blood and cerebral spinal fluid (CSF) samples were collected from anesthetized rat pups at the indicated time points. Blood and CSF samples were aseptically collected via intracardiac and cisterna magna punctures, respectively, as previously described (Kim et al., 1992). CSF (5 μl) and blood samples (50 μl) were inoculated into Luria broth and agar plates with antibiotic selection. Rats were then euthanized, and whole brains were removed.
Ethics Statement
The Institutional Animal Care Committee at Nankai University (Tianjin, China) approved all animal research procedures. Every effort was made to minimize animal suffering and to reduce the number of animals used.
Results and Discussion
Genomic Features, Phylogenetic Analysis, and Species Taxonomy
A summary of the features of each of the 129 Citrobacter genomes is shown in Supplementary Table S1. The GC content of the genomes ranged from 51 to 56%. The genome sizes varied from 4.0 to 5.0 Mb, with the number of coding sequences ranging from 4277 to 5904. In order to assess the phylogenetic relationship of Citrobacter species, a ML phylogeny tree was constructed using the concatenated nucleotide sequence of 1450 core genes from 31 newly sequenced and 98 publicly available Citrobacter strains. The core genome tree generated a reliable delineation of phylogenetic relationships across the Citrobacter genus. According to the core genome tree, the 129 strains were divided into 11 lineages (Figure 1A). To further explore the genomic similarities among strains, we supplemented phylogenies with genetic population structure analysis using Bayesian analysis of population structure (BAPS) (Cheng et al., 2013) and the ANI value was calculated to estimate the genetic distance between strains at the genomic level (Richter and Rosselló-Móra, 2009). ANI value was calculated using two software JSpecies 1.2.1 (Figure 2) and Gegenees v3.0 (Supplementary Figure S1). Population structure analysis assigned the Citrobacter genus into 11 groups (Groups 1–11) (Figure 1), which corresponded to our core genome tree. To further group Citrobacter in a phylogenetic context, NNet splits graph was generated (Figure 1B). The splits in the network serve the same purpose as the familiar branches in a phylogenetic tree, that is, they separate the sequences into 11 groups, one either side of the branch or split. We found that Groups 8 and 11 were specific to C. koseri and Citrobacter rodentium, respectively. Citrobacter amalonaticus was distributed in Groups 9 and 10. Though Group 1 only had C. freundii, this species was distributed in all groups except Groups 8 and 11. The distribution of other species was not group-specific. Because of its global presence, intraspecific variation and diverse behavior in strains led to ambiguity in Citrobacter classification. Hence, WGS has been helpful for allowing us to investigate the genomes and classify them accordingly. Based on whole genome data of Citrobacter, we constructed a high-resolution, more accurate phylogeny and population structure, which represented a significant improvement over that based on the conventional classification system.
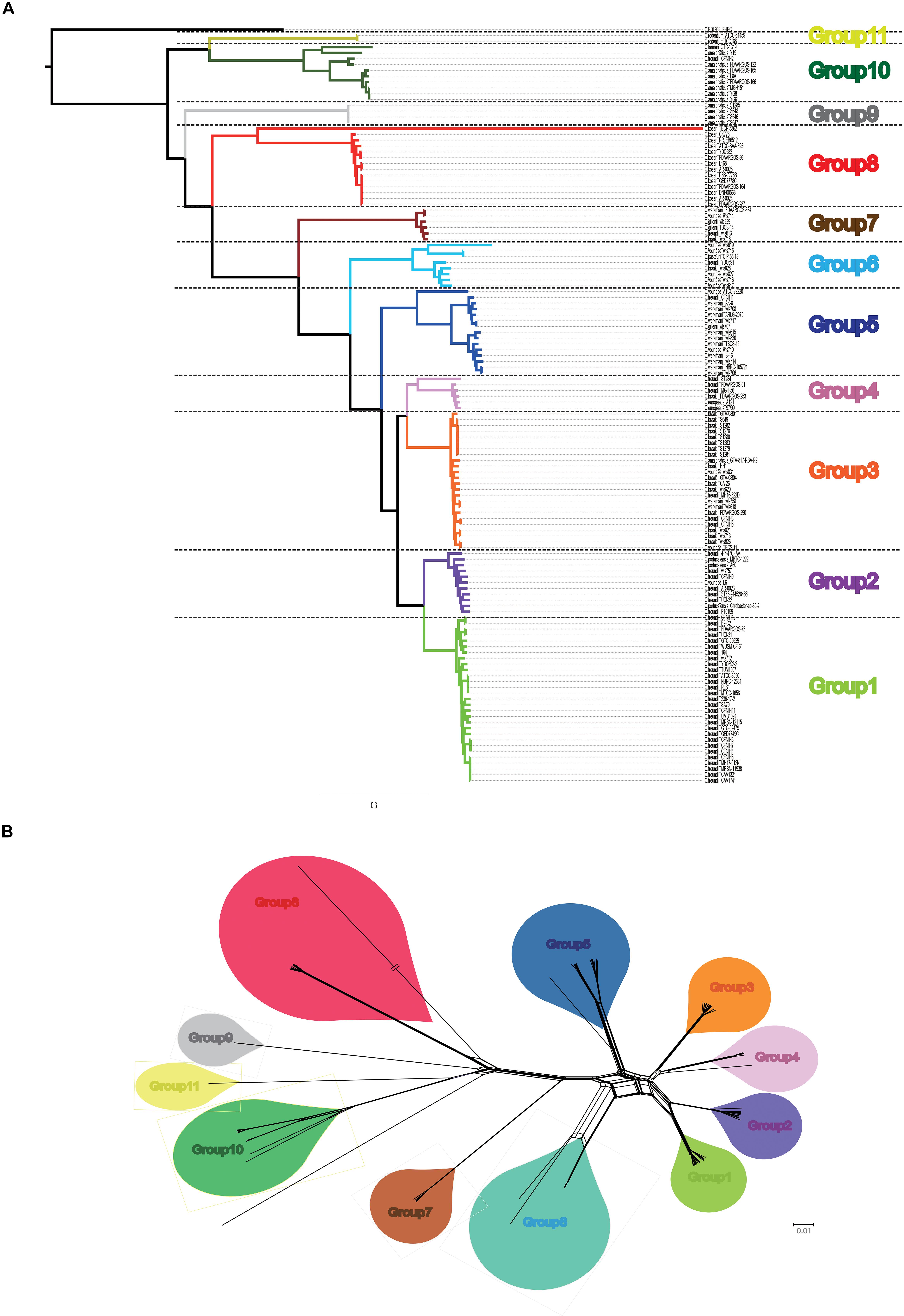
Figure 1. Core genome-based phylogeny. (A) Maximum likelihood tree was constructed using PhyML based on 1450 single-copy core genes shared by 129 Citrobacter strains. Different group are shown in different colors. (B) Neighbor Net (NNet) splits graphs based on uncorrected p-distances inferred from 1450 single-copy core genes shared by 129 Citrobacter strains. NNet splits graphs were constructed and visualized with SplitsTree4. Different group are shown in different colors, which was corresponding to our phylogenetic tree.
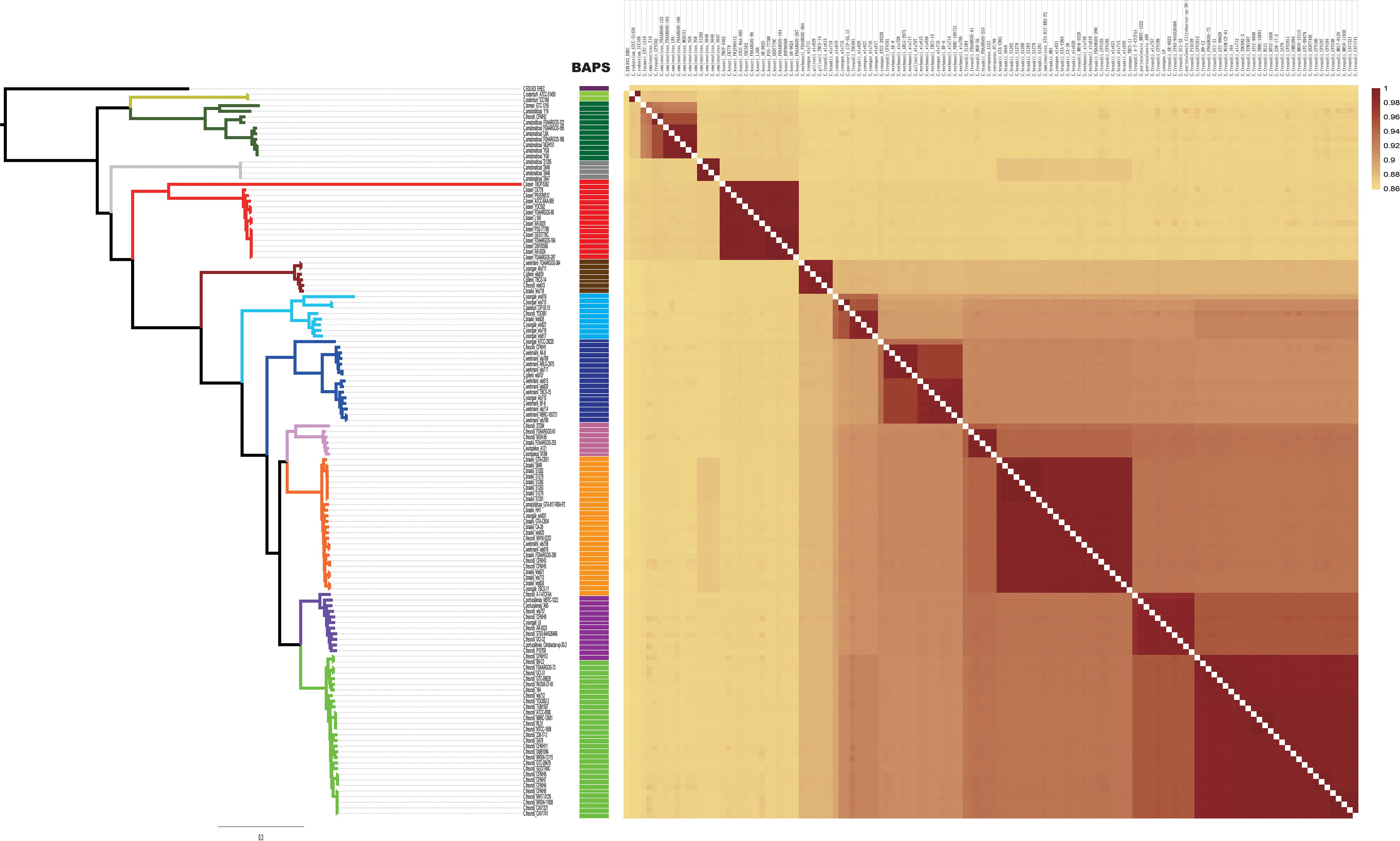
Figure 2. Heatmap chart generated from distances calculated based on the ANI values of 129 Citrobacter strains. The colors in the heatmap represent pairwise average nucleotide identity (ANI) values, with a gradient from yellow (low identity) to dark red (high identity).
Characterizing the Core and Pan-Genomes
To assess genetic diversity, we constructed core and pan-genome plotted of the Citrobacter genus (Figure 3A). From the Citrobacter pan-genome, 20114 gene families were identified across 129 genomes, of which 1450 constituted the core genome. We further used the Cluster of Orthologous Group (COG) assignments to identify the functional categories of the core gene families of all Citrobacter species and those specifically of Group 8 (C. koseri). The core gene families were unevenly distributed across the functional categories (Figure 3C). There were several obvious differences between all groups and Group 8 in the numbers of genes belonging to the same COG category, such as transport and metabolism of carbohydrates (category G); translation, and ribosomal structure and biogenesis (category J); and inorganic ion transport and metabolism (category P). It is noteworthy that most of the core gene differences were related to transport and metabolism. Bacterial signal transduction systems are responsible for sensing environmental cues and adjusting cellular behavior and/or metabolism in response to such cues. Besides, we also analyzed the functional category of the core genes shared by 129 Citrobacter and the Group 8-specific core genes using two other different database (GO/KEGG) and compute the numbers of proteins for each corresponding GO/KEGG term (Figures 3D,E). Results indicate that 8 gene functional categories were enriched in both core gene families and the Group 8-specific core genome based on GO functional annotations. Including “cell,” “cell part,” “membrane,” “binding,” “catalytic,” “localization,” “cellular process,” and “metabolism process” (Figure 3E). For KEGG annotations, two gene functional categories were enriched in both core gene families and the Group 8-specific core genome including “Enzymes” and “Transporters” (Figure 3D). Thus, we hypothesized that genes related to transport and metabolism might provide a competitive advantage to C. koseri infecting humans, which may lead to unique pathogenicity.
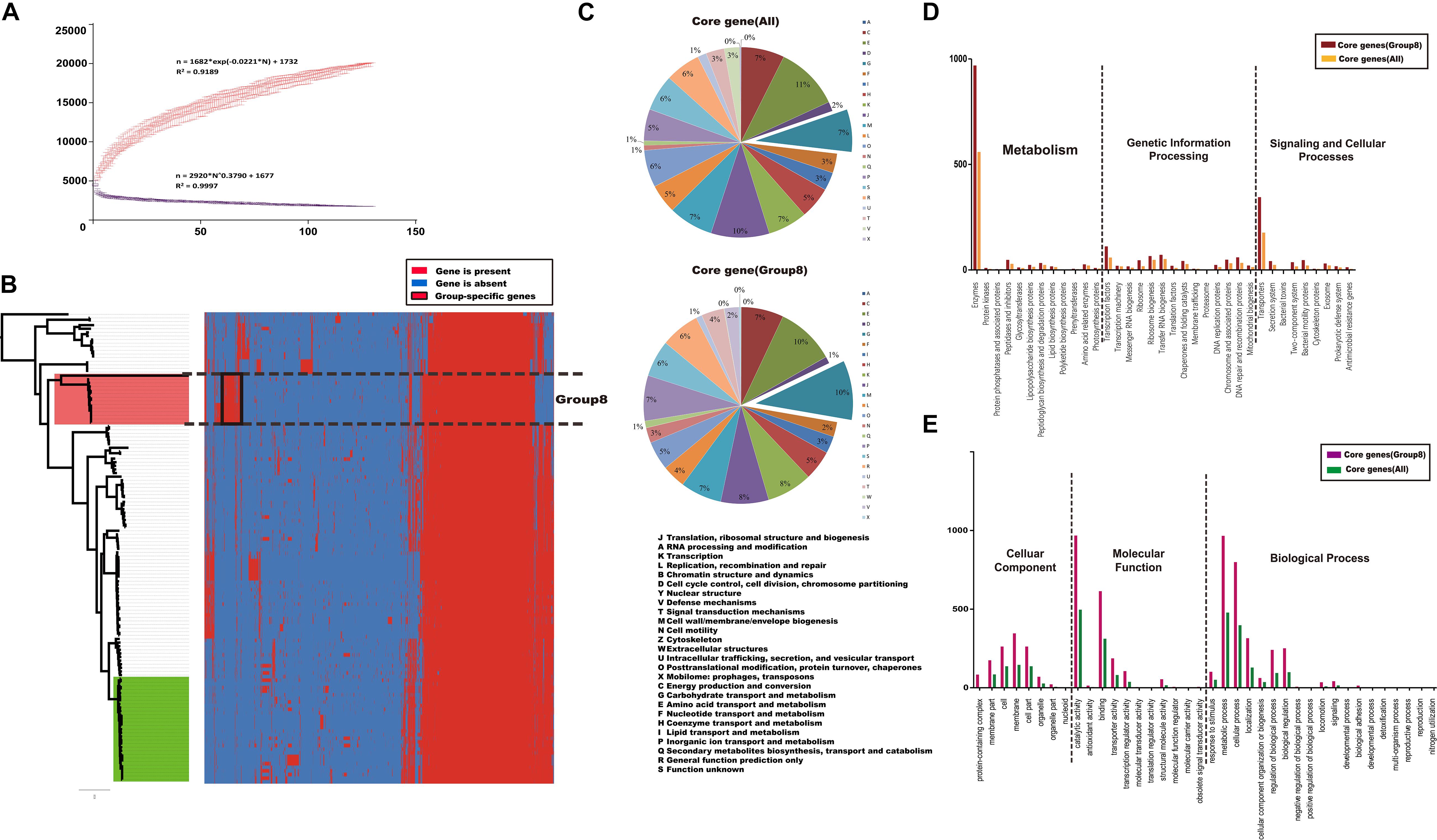
Figure 3. Core and pan-genome analysis of Citrobacter strains. (A) The Citrobacter core and pan-genome plotted were constructed for 129 genome sequences of Citrobacter. (B) Cluster map of the accessory genome of Citrobacter. The gene families that are unique to a species and conserved across most of strains in that species are framed in black. (C–E) Distribution of functional catalogs of core genes of all Citrobacter strains and Group 8 only after COG/GO/KEGG annotation, respectively.
Comparative Genomics Analysis of Virulence Genes in Citrobacter
Previously research made use of infant mouse models to study the pathogenicity of C. koseri (Soriano et al., 1991). Some in vitro studies have shown that C. koseri is able to invade and replicate inside human U937 macrophages and human brain microvascular endothelial cells (Badger et al., 1999; Townsend et al., 2003). It has also been demonstrated that fliP influences the uptake of C. koseri by macrophages, as well as cytokine expression and brain abscess formation in neonatal rats (Townsend et al., 2006). However, there is limited information on the virulence genes of C. koseri. To identify key pathogenicity genes of C. koseri, we investigated the distribution of virulence genes in Citrobacter. All 129 Citrobacter genomes were locally aligned against the Virulence Factors Database (Liu et al., 2019) to detect virulence genes (Supplementary Table S2). We found that the major virulence factors identified in all strains were associated with flagellar apparatus biosynthesis (ompA, csg fimbriae, and the che operon) and iron uptake (chu, fep, and ent). Specifically, Group 8, which consisted of C. koseri only, contained a virulence gene cluster (VFG000358–VFG000368) (Figure 4), which formed a complete HPI found in highly pathogenic strains of the genus Yersinia. This gene cluster encodes a siderophore-mediated iron uptake system that is required for full virulence in Yersinia (Carniel, 1999). Thus, we speculated that this genomic feature may contribute to the pathogenic effects on the central nervous system (CNS) during C. koseri infection, which is rarely observed for other related species. Group 3, which mainly consisted of C. freundii and Citrobacter braakii, contained the genes VFG000423–VFG000430, which encode the Vi capsule polysaccharide of Salmonella enterica subsp. Enterica serovar Typhi str. CT18 (Salmonella Typhi, for short). The Vi capsule enables Salmonella Typhi to avoid host defenses and is important in enhancing infectivity and virulence (Joan and John, 1984; Hone et al., 1988; Arya, 2002). These genes may provide bacteria higher potential for pathogenicity and adaption within humans. Group 11, which consisted of C. rodentium, contained genes that encoded well characterized type III secretion system (T3SS) components (Schauer et al., 1995; Luperchio et al., 2000; Vallance et al., 2003).
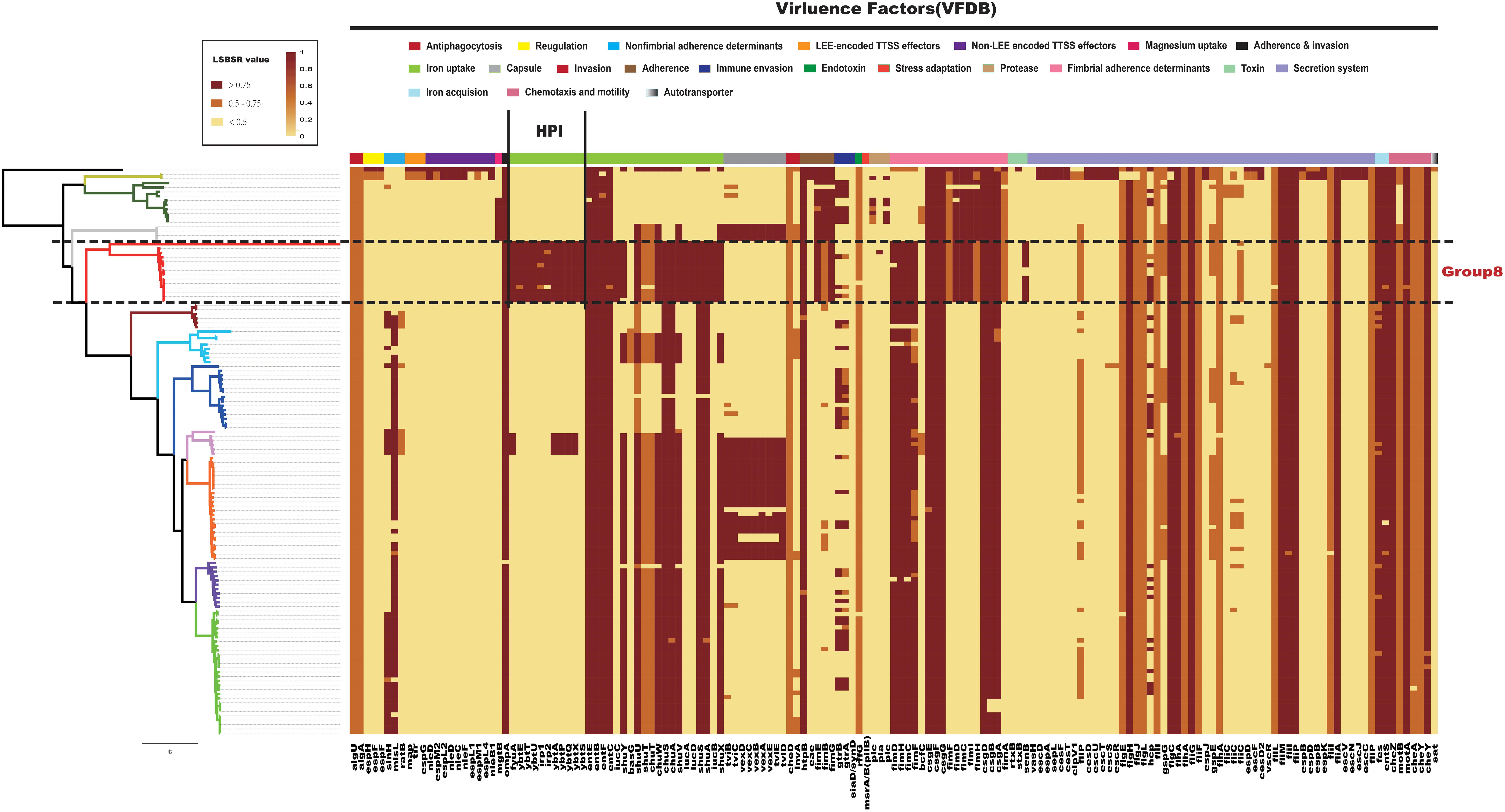
Figure 4. Distribution heatmap of virulence factors across Citrobacter strains. Color coding for virulence factors corresponds to the score ratio recorded for each genome when screened with the reference Virulence Factors Database. For a full outline of sources, see Supplementary Table S2. In the current presentation, we removed the virulence factors of previously studied macromolecular secretion systems and O-antigen/LPS/Capsule.
Genomic islands play an important role in the evolution, adaptation, and diversification of bacterial genomes, carrying genes that encode proteins with diverse functions (Juhas et al., 2009). Distinct properties of GIs allow bacterial organisms to evolve and adapt to different environments, it is possible to understand why they spread rapidly (Juhas et al., 2007). This adaptation process is among the most important factors in generating diversity and facilitating the propagation of genes in bacteria, as the organism receives an already prepared and improved set of genes, increasing its chances of adaptation (Wilson, 2012). We detected GIs in the 129 Citrobacter genomes using IslandViewer 4 (Figure 5). Results indicated that Citrobacter strains harbor diverse and heterogeneous GIs and the distribution of GIs-number of 129 Citrobacter is irregular. The average-GIs-number of Citrobacter was 42, which may provide Citrobacter with diversity functions like ability to adapt to more environment and virulence.
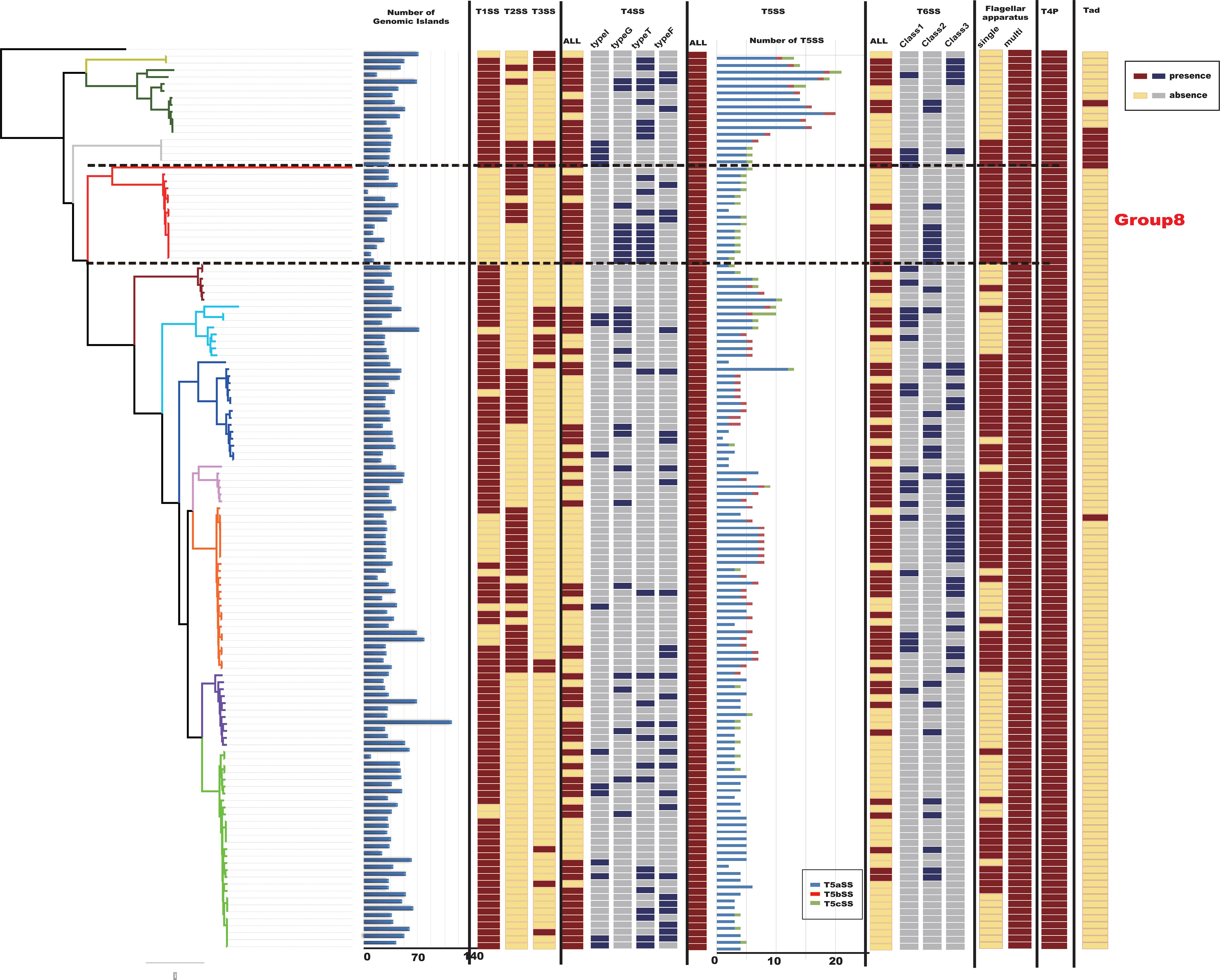
Figure 5. Macromolecular secretion system and genomic island (GI) distribution in Citrobacter strains. Dark red colors and blue boxes represent the presence of a macromolecular system within a genome, while gray colors and yellow boxes indicate the absence of a macromolecular system.
Protein secretion is essential for bacteria to interact with their surrounding environments (Abby and Rocha, 2017). In particular, pathogenic bacteria secrete many virulence factors (Raymond et al., 2013). To further analyze the virulence genes of Citrobacter, we investigated the occurrences of macromolecular secretion systems in the 129 Citrobacter genomes using the MacSyFinder (Figure 5). We found that Flagellar apparatus, Tad pilus, type IV pilus, and type V secretion system (T5SS) are restricted to the Citrobacter genus, while type II secretion system (T2SS), type III secretion system (T3SS), type IV secretion system (T4SS), type V secretion system (T5SS), and type VI secretion system (T6SS) are shared by some strains within the genus. However, the strains in Group 8 did not have type I secretion system (T1SS), type III secretion system (T3SS), one kind of type IV secretion system (T4SS), one kind of type V secretion system (T5SS), and Tad pilus, which might be related to higher success in colonizing the human environment.
In particular, the T6SS is a multiprotein machine, widespread in Gram-negative proteobacteria, and especially common in Gamma-proteobacteria (Coulthurst, 2013; Zoued et al., 2014). Previous work showed that Escherchia coli T6SSs can be categorized into three distinct phylogenetic groups: T6SS-1 to T6SS-3 (Russell et al., 2014). In our study, using the same method in E. coli, we performed a phylogenetic analysis of the Citrobacter genus and other classified strains including Salmonella LT2, NMEC RS218, EAEC 042, APEC TW-XN, Pseudomonas aeruginosa, Vibro cholerae, Edwardsiella tarda, and Francisella tularensis to classify the T6SSs into three classes, as shown in Figure 6A. The phylogenetic tree suggests that T6SS gene cluster in Citrobacter genus might be acquired by horizontal gene transfer (HGT). The functions associated with the three classes are quite different. T6SS-1 proteins are involved in biofilm formation; T6SS-2 proteins are commonly involved in colonization, survival, or invasion (often of human hosts); and T6SS-3 proteins include antibacterial effectors. We found that only T6SS-2 genes were present in Group 8. Nearly half of the strains in Group 8 have T6SS-2 genes, which may be related to the unique pathogenicity of these strains in their host environments. This idea should be further explored in future studies. A model of the three Citrobacter T6SS gene cluster classes is shown in Figure 6B.
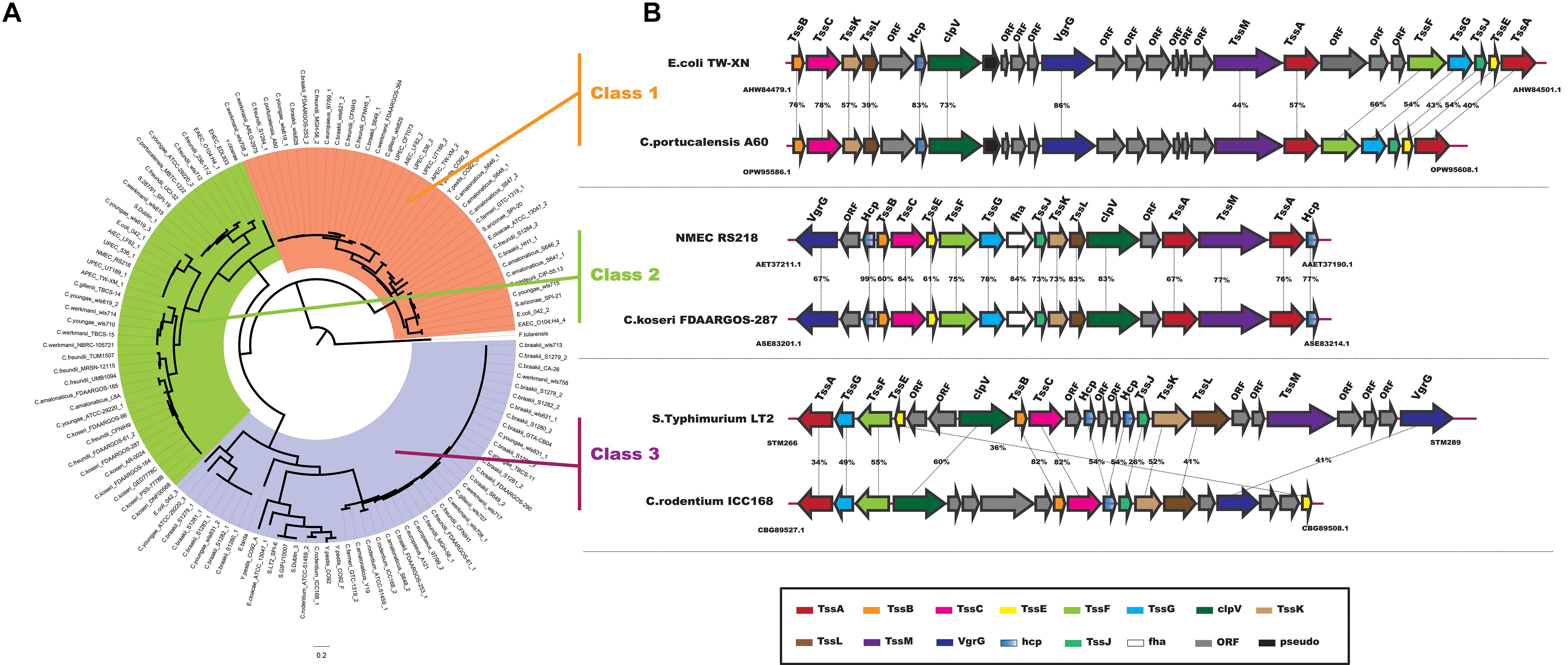
Figure 6. Model of T6SS in Citrobacter strains. (A) Phylogenetic tree of select T6SS gene clusters. The figure was made using the sequences of the TssF core component homologs. (B) Genes encoding the T6SS-1, T6SS-2, and T6SS-3 in the indicated Citrobacter strains are shown schematically. Homologous genes are colored similarly (see box below).
Resistance Genes Distribution of Citrobacter
Previous research has shown that resistance to β-lactams is common among Citrobacter and is mediated through production of β-lactamases. C. freundii is considered inherently resistant to many antimicrobial agents including amoxicillin, amoxicillin-clavulanate, ampicillin, ampicillin/sulbactam, first- and second-generation cephalosporins, and cephamycins (Patel, 2001). It has been suggested that C. freundii is less susceptible than C. koseri to several antibiotics (Samonis et al., 1987). To explore this possibility, all 129 Citrobacter genomes were locally aligned against the ResFinder database (Zankari et al., 2012) to detect resistance genes (Supplementary Table S3). We identified a number of resistance genes within the Citrobacter genus (Figure 7). We found that all species had genes which encoded different types of antibiotic efflux pumps, including resistance-nodulation-cell division (RND) types, major-facilitator superfamily (MFS) types, ATP-binding cassette (ABC) types, which confer resistant to aminoglycoside antibiotics, aminocoumarin antibiotics, fluoroquinolone, lincosamide antibiotics, cephalosporin, cephamycin, and penam (Supplementary Table S4). Genes encoding β-lactamase were distributed in all Citrobacter strains except those in Group 8, which rarely have such genes. Strains in Groups 9 and 10 primarily contained CTX-M β-lactamase genes for resistance to cephalosporin, while strains in Groups 1–7 consistently contained CMY β-lactamase genes. In particular, genes encoding the quinolone resistance protein Qnr were mainly distributed in Groups 1, 2, 4, 5, and 6. This finding is in agreement with a previous study, which suggested that Citrobacter spp. may be the origin of qnrB genes, a hypothesis based on species distribution (>60% in Citrobacter spp.) (Kehrenberg et al., 2008; Jacoby et al., 2011; Saga et al., 2013) and that the qnrB gene is prevalent in C. freundii strains isolated from human clinical specimens (Park et al., 2007). Other work supports Citrobacter as the origin of qnrB as this gene is distributed in strains including C. freundii, C. braakii, Citrobacter youngae, and Citrobacter werkmanii (Ribeiro et al., 2015). Importantly, strains of Group 8 had less antibiotic-resistant genes than other those in the other groups, especially Groups 1–6, which contained C. freundii, C. braakii, C. youngae, and C. werkmanii. Our research showed that C. koseri had less resistance genes than other species, which may explain why C. koseri is considered more susceptible to several antibiotics. Furthermore, we provided a comprehensive comparative genomics analysis on the distribution of resistance genes in Citrobacter, which establishes a foundation for the clinical treatment of Citrobacter in the future.
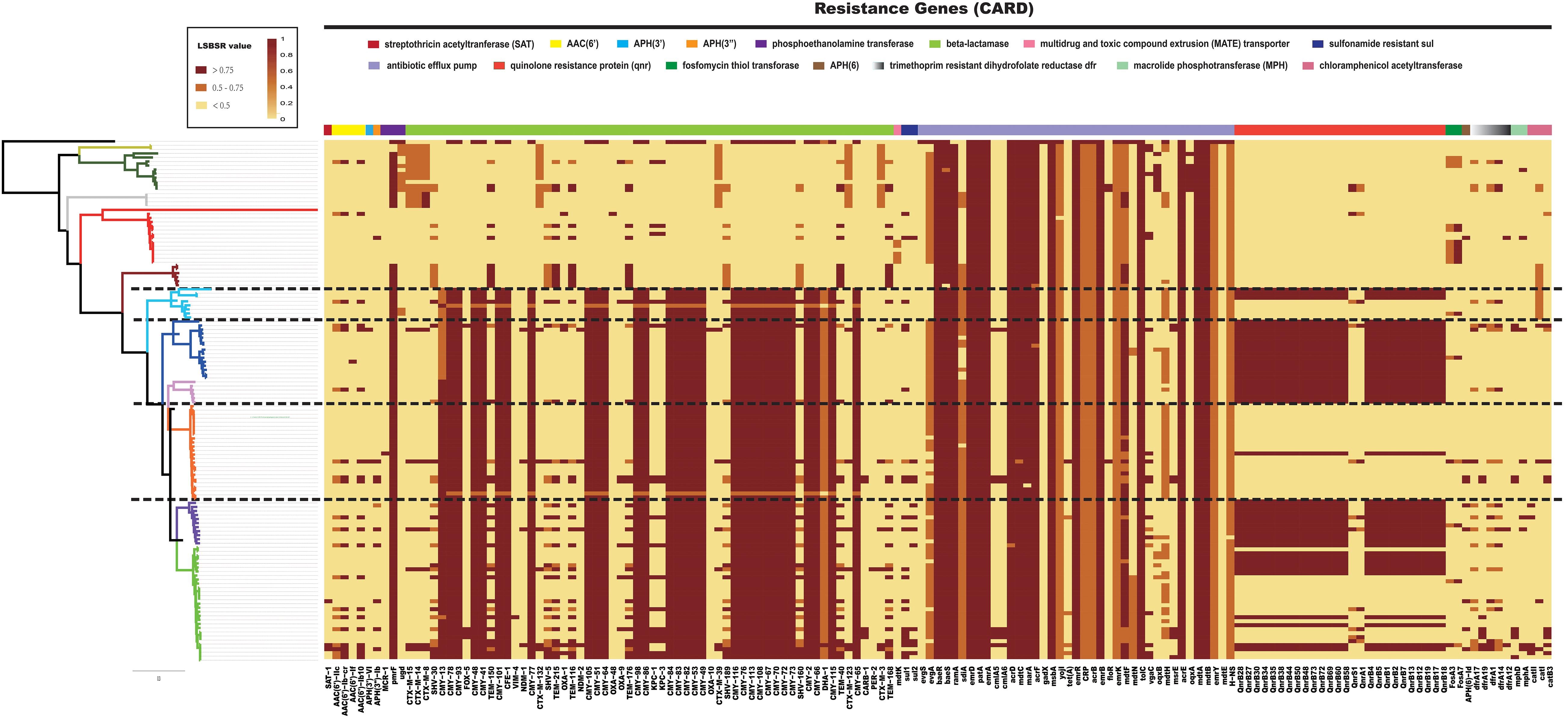
Figure 7. Distribution heatmap of resistance genes across Citrobacter strains. Color coding for resistance genes corresponds to the score ratio recorded for each genome when screened with the reference The Comprehensive Antibiotic Resistance Database. For a full outline of sources, see Supplementary Table S3.
Group-Specific Genes
The group-specific gene analysis revealed underlying profiles of gene families that are conserved among strains within a group, and some of which are unique to a particular group. As such, Group 8-specific genes may provide information not only about virulence of C. koseri, but also about the metabolic features of the host environment. To identity group-specific gene families, we constructed an accessory genome by subtracting the core genome and low frequency genes (existence < 10) from the pan-genome. For Group 8, a total of 285 gene families were identified as the group-specific core genome (Supplementary Table S5). Based on KEGG annotation, the functional categories “Protein families: signaling and cellular process,” “Environment information processing,” and “Amino acid metabolism” were enriched in the Group 8-specific core genome (Figure 8C). These group-specific genes may be related to inherent differences in pathogenicity between C. koseri and strains of the other groups. Through systemic analysis of these genes, four complete pathway modules, including three ABC transporters and one MCP (methyl-accepting chemotaxis protein), were present in the Group 8-specific core genome. Moreover, we found group-specific genes related to HPI clusters, aerobactin biosynthetic clusters, methionine salvage-related clusters, ABC transporters for D-allose, ABC transporters for branched-chain amino acids, and MCP genes (Figure 8D).
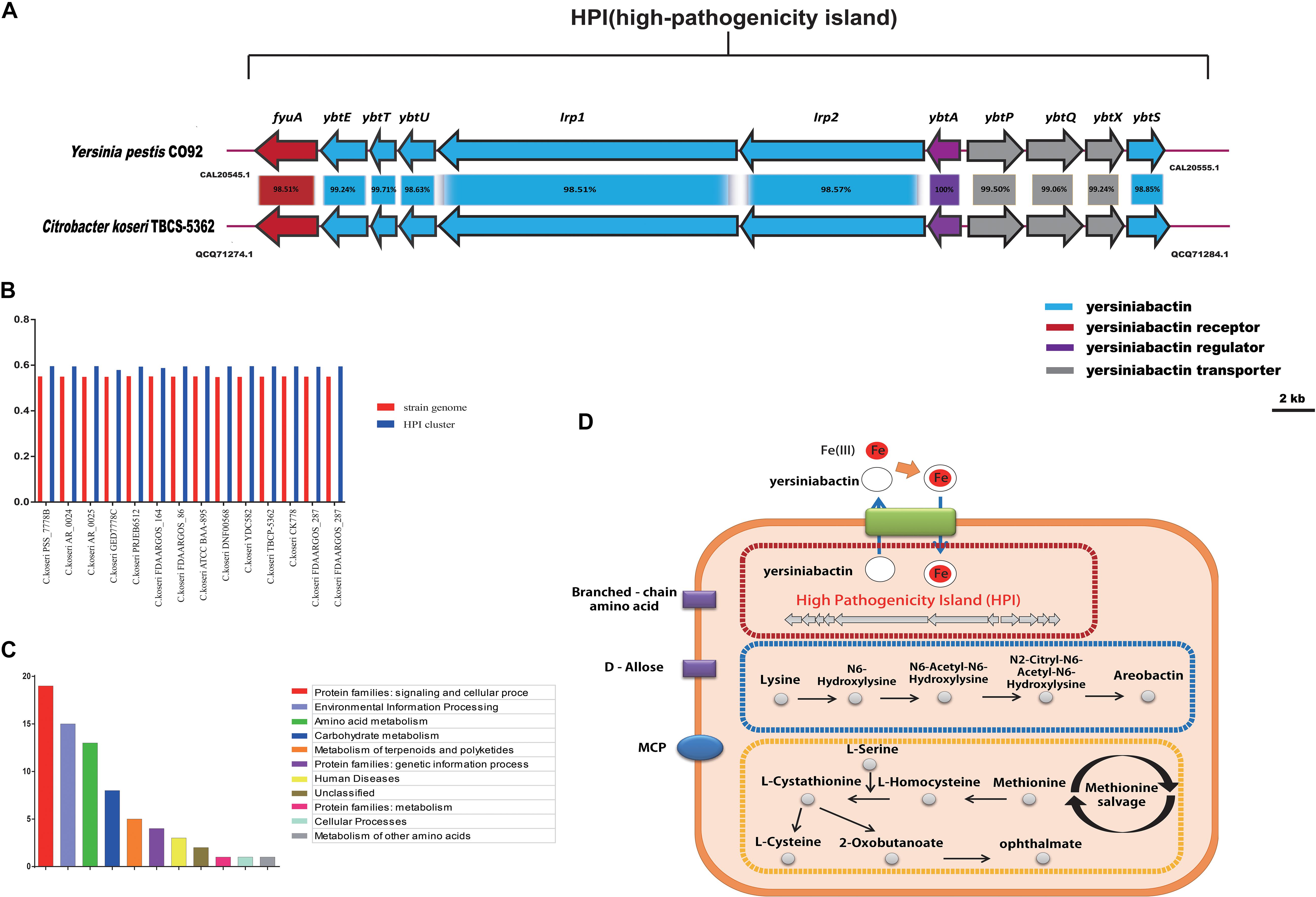
Figure 8. Group 8-specific core genome. (A) Model of HPI in Citrobacter koseri TBCP-5362. The percentage of protein identity for each set of homologous genes is shown. Homologous genes are colored similarly (see box below). (B) GC content of HPI in strains of Group 8. (C) Functional enrichment of Group 8-specific core genome after KEGG annotation. (D) Sketch map of the Group 8-specific core genome after KEGG annotation.
The HPI Clusters Is Essential for C. koseri Pathogenicity
Based on the results of the comparative genomics analysis, we focused on HPI clusters, models of which are shown in Figure 8A. We also examined the GC content of each of the two gene clusters and the three host genomes. Our results showed that the gene clusters displayed apparent deviations in GC content (Figure 8B), which suggests that they may have been acquired by HGT events. Furthermore, we assessed the putative HPI homologs using BLASTp searches in the NCBI non-redundant protein database. We found that the well characterized HPI in Yersinia pestis CO92 was closely related to the HPI cluster in C. koseri TBCP-5362, and that these gene clusters are present in similar genomic locations and encode proteins with more than 95% sequence identity. Previous studies have shown that HPIs are common in highly pathogenic Yersinia strains and that they are required for full virulence (Gehring et al., 1998; Carniel, 1999; Perry et al., 1999). Thus, we speculated that these gene clusters might play a vital role in allowing C. koseri to survive in humans and may explain why C. koseri exhibits a remarkable degree of tropism for the brain.
To test our hypothesis, we generated a mutant strain that lacks all HPI clusters (ΔHPI:chlR) in C. koseri TBCP-5362. As previously described, two animal models were used in our study: 2-day-old SD rats and 18-day-old BALB/c mice (Soriano et al., 1991; Liu et al., 2019). Three groups of each animal model were inoculated with wild-type or ΔHPI mutants of C. koseri TBCP-5362 (∼5 × 105 colony forming units (CFUs) for 2-day-old SD rats and ∼1 × 107 CFUs for 18-day-old BALB/c mice). Blood and cerebral spinal fluid (CSF) were collected analyzed. In 18-day-old mice colonized with ΔHPI mutants, we found that the amount of bacteria in CSF was significantly lower than in mice inoculated with wild type bacteria at 24 h post-infection, while in blood, the bacteria concentrations were very similar (Figures 9D,E). Likewise, in 2-day-old SD rats, although colonization with wild type bacteria resulted in increases in both blood and CSF, the higher level of bacteria in CSF (nearly a 500-fold increase) was much greater than what was observed in blood (a nearly 100-fold increase) (Figures 9A,B). For the ΔHPI mutant, colonization was significantly decreased in both blood and CSF in both animal models after 24 h. Surprisingly, in 18-day-old mice, our results suggest that ΔHPI mutants lost the ability to replicate in brain.
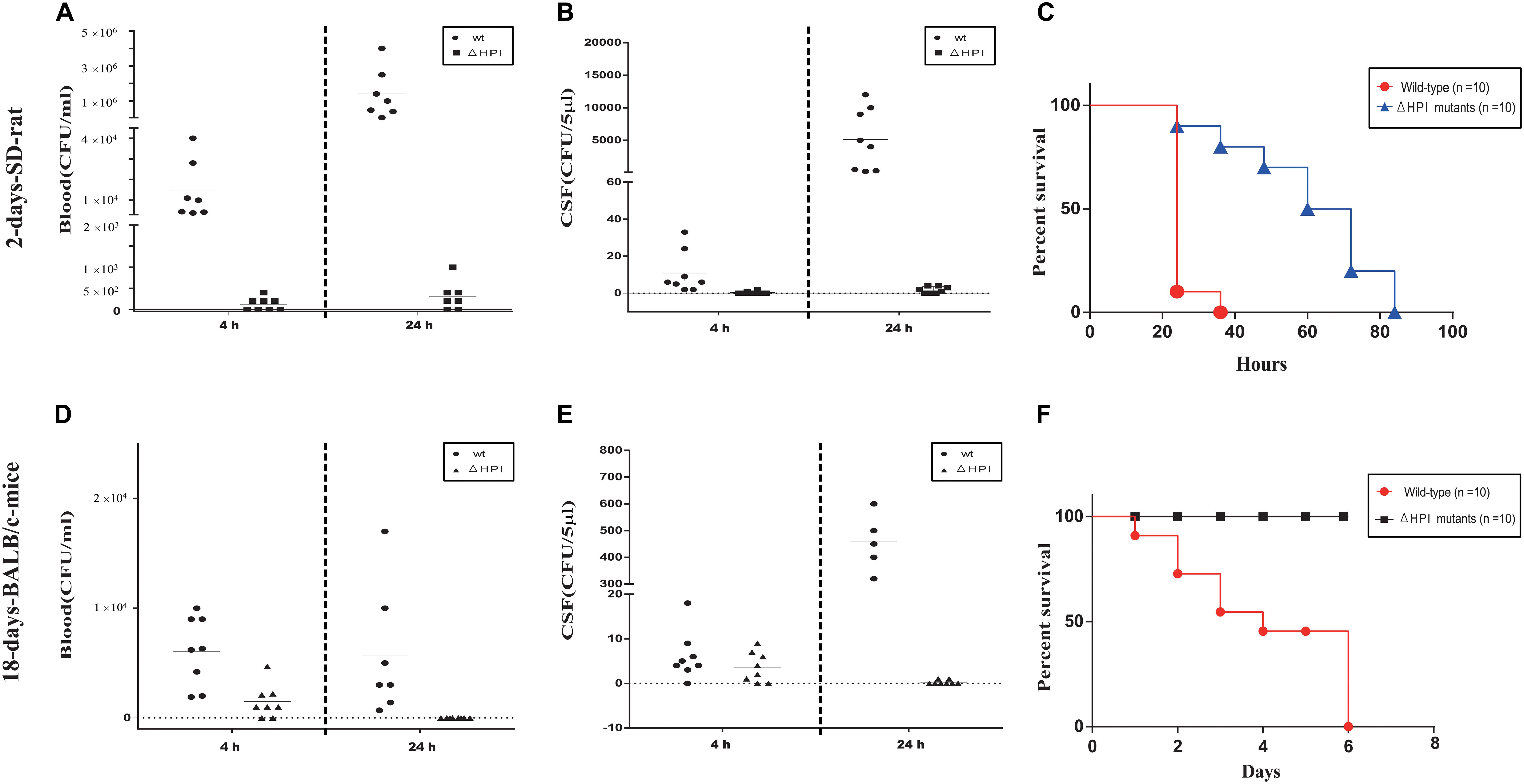
Figure 9. Lack of HPI-cluster decreases C. koseri virulence in host. (A) Bacterial counts recovered from blood of 2-day-old SD rats. (B) Bacterial counts recovered from CSF of 2-day-old SD rats. (D) Bacterial counts recovered from blood of 18-day-old BALB/c mice. (E) Bacterial counts recovered from CSF of 18-day-old BALB/c mice. Data reflect three independent experiments. Bars represent mean CFUs of all mice, with P values determined by the Mann–Whitney U test (∗∗P < 0.01). (C,F) Survival plots of 2-day-old SD rats and 18-day-old BALB/c mice after inoculation with wild type or ΔHPI mutant bacteria (∼1 × 106 CFUs for 2-day-old SD rats and ∼2 × 107 CFUs for 18-day-old BALB/c mice). Data presented are the combination of three independent experiments, ∗∗∗P < 0.001 by log-rank curve comparison test.
In a separate set of experiments, two groups of each animal model were inoculated with wild-type or ΔHPI mutants of C. koseri TBCP-5362 (∼1 × 106 CFUs for 2-day-old SD rats and ∼2 × 107 CFUs for 18-day-old BALB/c mice) and survival was monitored. For 2-day-old SD rats, animals infected with wild type started to die by 24 h and all died within 36 h. In contrast, animals infected with ΔHPI mutants started to die by 24 h and all died within 84 h. Similarly, for 18-day-old BALB/c mice, animals infected with wild type started to die by 24 h and all died within 144 h, while animals infected with ΔHPI mutants did not die within the monitored time. Survival curves identified a significant difference between wild type groups and mutants groups (log-rank test, P < 0.01, Figures 9C,F).
Overall, our results indicated that deletion of the HPI cluster severely attenuated C. koseri virulence in vivo and ΔHPI mutants lost the ability to replicate in brain, which suggests that the HPI cluster is a novel virulence cluster that plays a key role in C. koseri pathogenicity.
Conclusion
The genome sequences of 31 Citrobacter strains generated in this study are important for the broader goal of using WGS data for comparative studies. In this study, phylogenetic and population structure analyses based on a core genome, in combination with whole genome ANI profiling, provide a clear distinction of Citrobacter species into 11 groups.
Our comparative analysis showed differences in macromolecular secretion system profiles among Citrobacter. In particular, for Group 8 (the C. koseri-specific group), we identified several genes vital for pathogenicity and which may contribute to the high degree of tropism for the brain. These include HPI genes, an aerobactin biosynthetic gene cluster, a methionine-salvage related gene cluster, ABC transporters for D-allose, ABC transporters for branched-chain amino acid, and MCP. We also demonstrated through in vivo animal studies that the HPI cluster is essential for C. koseri pathogenicity. On the other hand, our research showed that C. koseri had less resistance genes than other species, which may explain why C. koseri is more susceptible to antibiotics.
Data Availability Statement
The datasets generated for this study can be accessed from the https://card.mcmaster.ca/, http://www.mgc.ac.cn/VFs/main.htm, NCBI, GenBank: GCA_000732965.1, GCA_000835925.1, GCA_000027085.1, GCA_000764735.1, GCA_000981805.1, GCA_002880615.1, GCA_001559075.1, GCA_001558935.1, GCA_000731055.1, GCA_001471655.1, GCA_002151695.1, GCA_001276125.1, GCA_001276105.1, GCA_002918935.1, GCA_002919495.1, GCA_002918555.1, GCA_002918535.1, GCA_005406305.1, GCA_003226155.1, GCA_000939835.1, GCA_000018045.1, GCF_002863965.1, GCA_000783445.1, GCA_003184045.1, GCA_002947035.1, GCA_001546325.1, GCA_001546305.1, GCA_001471775.1, GCA_001552875.1, GCA_002947675.1, GCA_002208985.3, GCA_002386385.1, GCA_005281055.1, GCA_005280825.1, GCA_005280805.1, GCA_005281195.1, GCA_005280945.1, GCA_005281345.1, GCA_005281015.1, GCA_000826205.1, GCF_002863945.1, GCA_005280855.1, GCA_005280875.1, GCA_005280975.1, GCA_005281175.1, GCA_000155975.1, GCA_000648515.1, GCA_002114305.1, GCA_005281255.1, GCA_002185305.2, GCA_005280955.1, GCA_005281125.1, GCA_005281355.1, GCA_005280845.1, GCA_005280815.1, GCA_005281115.1, GCF_002025225.1, GCA_005281025.1, GCA_000759755.1, GCA_005281305.1, GCA_002918505.1, GCA_000783995.1, GCA_000692115.1, GCA_002073755.2, GCA_900080005.1, GCA_900079995.1, GCA_000786265.1, GCA_002918575.1, GCA_002919425.1, GCA_002918495.1, GCA_002918455.1, GCA_002918465.1, GCA_002919455.1, GCA_002919485.1, GCA_000972645.1, GCA_002923765.1, GCA_005281215.1, GCA_000786275.1, GCA_002939255.1, GCA_005281325.1, GCF_003114935.1, GCA_005280915.1, GCA_005281155.1, GCA_002208845.2, GCA_002903215.1, GCA_002919795.1, GCA_005281165.1, GCA_005281265.1, GCA_005281245.1, GCA_005281395.1, GCA_000238735.1, GCA_002843195.2, GCA_002042885.1, GCA_005280935.1, GCA_002903305.1, GCA_003255895.1, GCA_002946635.1, GCA_001317135.1, GCA_000521945.1, GCA_000158355.2, GCA_001281005.1, GCA_002918875.1, GCA_003019835.1, GCA_000783755.1, GCA_000521965.1, GCA_000388155.1, GCA_003195445.1, GCA_003070705.1, GCA_005281045.1, GCF_002864025.1, GCF_003175795.1, GCA_000734905.1, GCA_000759735.1, GCA_000582615.1, GCA_000312465.1, GCA_003015305.1, GCA_000982845.1, GCA_002919825.1, GCA_002871775.1, GCA_000937505.2, GCA_000342325.1, GCA_001546285.1, GCA_002918835.1, GCA_002918855.1, GCA_002934505.1, GCA_002918865.1, GCF_003114915.1, GCA_000937455.2, GCA_001022155.1, and GCA_001022275.1.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care Committee at Nankai University.
Author Contributions
BL and LJ conceived the project. CY and CQ purchased the strains. JW and SZ prepared the sample DNA for sequencing. CY and ZY conducted the comparative genomic analysis. CY, JW, and YW constructed the mutants and preformed the experiments. BL, LJ, and CY prepared the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key Programs for Infectious Diseases of China (2017ZX10303405-001 and 2017ZX10104002-001-006), the National Natural Science Foundation of China (NSFC) Program (31820103002, 31770144, 81772148, and 81871624), and Key Research and Development Project (No. 2018YFA0901000).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02774/full#supplementary-material
FIGURE S1 | Heatmap chart generated from distances calculated based on the ANI values of 129 Citrobacter strains. The colors in the heatmap represent pairwise average nucleotide identity (ANI) values, with a gradient from red (low) to yellow green (high).
TABLE S1 | The list of strains in this research.
TABLE S2 | The result of the Identification of Virulence Genes.
TABLE S3 | The result of the Identification of Resistance Genes.
TABLE S4 | The information of CARD datebase.
TABLE S5 | The information of Group8-specific core genome.
Footnotes
- ^ http://www.ncbi.nlm.nih.gov/genome/annotation_prok
- ^ http://tree.bio.ed.ac.uk/software/figtree
- ^ https://www.kegg.jp/blastkoala/
- ^ http://mobyle.pasteur.fr/cgi-bin/portal.py#forms:txsscan
- ^ http://www.pathogenomics.sfu.ca/islandviewer/
- ^ http://sourceforge.net/projects/codonw
References
Abby, S. S., Neron, B., Menager, H., Touchon, M., and Rocha, E. P. (2014). MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9:e110726. doi: 10.1371/journal.pone.0110726
Abby, S. S., and Rocha, E. P. C. (2017). Identification of protein secretion systems in bacterial genomes using MacSyFinder. Methods Mol. Biol. 1615, 1–21. doi: 10.1007/978-1-4939-7033-9_1
Agren, J., Sundstrom, A., Hafstrom, T., and Segerman, B. (2012). Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS One 7:e39107. doi: 10.1371/journal.pone.0039107
Altmann, G., Sechter, I., Cahan, D., and Gerichter, C. B. (1976). Citrobacter diversus isolated from clinical material. J. Clin. Microbiol. 3, 390–392.
Arens, S., and Verbist, L. (1997). Differentiation and susceptibility of Citrobacter isolates from patients in a university hospital. Clin. Microbiol. Infect. 3, 53–57. doi: 10.1111/j.1469-0691.1997.tb00251.x
Arya, S. C. (2002). Field effectiveness of Vi polysaccharide typhoid vaccine in the People’s Republic of China. J. Infect. Dis. 185, 845; author reply 845–846. doi: 10.1086/339191
Ashton, P. M., Nair, S., Peters, T. M., Bale, J. A., Powell, D. G., Painset, A., et al. (2016). Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ 4:e1752. doi: 10.7717/peerj.1752
Badger, J. L., Stins, M. F., and Kim, K. S. (1999). Citrobacter freundii invades and replicates in human brain microvascular endothelial cells. Infect. Immun. 67, 4208–4015. doi: 10.1007/978-3-540-47648-1_708
Bertelli, C., Laird, M. R., Williams, K. P. Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bottacini, F., Medini, D., Pavesi, A., Turroni, F., Foroni, E., Riley, D., et al. (2010). Comparative genomics of the genus Bifidobacterium. Microbiology 156(Pt 11), 3243–3254. doi: 10.1099/mic.0.039545-0
Boyer, F., Fichant, G., Berthod, J., Vandenbrouck, Y., and Attree, I. (2009). Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics 10:104. doi: 10.1186/1471-2164-10-104
Brenner, D. J., Grimont, P. A. D., Steigerwalt, A. G., Fanning, G. R., Ageron, E., and Riddle, C. F. (1993). Classification of Citrobacteria by DNA hybridization:designation of Citrobacter famzeri sp. nov.,Citrobacter youngae sp. nov., Citrobacter braakii sp. nov.,Citrobacter werkmanii sp. nov., Citrobacter sedlakii sp. nov., and three unnamed Citrobacter genomospecies. Int. J. Syst. Bacteriol. 43, 645–658. doi: 10.1099/00207713-43-4-645
Brenner, D. J., O’Hara, C. M., Grimont, P. A., Janda, J. M., Falsen, E., Aldova, E., et al. (1999). Biochemical identification of Citrobacter species defined by DNA hybridization and description of Citrobacter gillenii sp. nov. (Formerly Citrobacter Genomospecies 10) and Citrobacter murliniae sp. nov. (Formerly Citrobacter Genomospecies 11). J. Clin. Microbiol. 37, 2619–2624.
Carniel, E. (1999). The Yersinia high-pathogenicity island. Microbes Infect. 3, 561–569. doi: 10.1016/S1286-4579(01)01412-5
Cheng, L., Connor, T. R., Siren, J., Aanensen, D. M., and Corander, J. (2013). Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol. Biol. Evol. 30, 1224–1228. doi: 10.1093/molbev/mst028
Clermont, D., Motreff, L., Passet, V., Fernandez, J. C., Bizet, C., and Brisse, S. (2015). Multilocus sequence analysis of the genus Citrobacter and description of Citrobacter pasteurii sp. nov. Int. J. Syst. Evol. Microbiol. 65(Pt 5), 1486–1490. doi: 10.1099/ijs.0.000122
Coulthurst, S. J. (2013). The type VI secretion system - a widespread and versatile cell targeting system. Res. Microbiol. 164, 640–654. doi: 10.1016/j.resmic.2013.03.017
Curless, R. G. (1980). Neonatal intracranial abscess: two cases caused by citrobacter and a literature review. Ann. Neurol. 8, 269–272. doi: 10.1002/ana.410080308
Dallman, T. J., Byrne, L., Ashton, P. M., Cowley, L. A., Perry, N. T., Adak, G., et al. (2015). Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin. Infect. Dis. 61, 305–312. doi: 10.1093/cid/civ318
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Doran, T. I. (1999). The role of Citrobacter in clinical disease of children: review. Clin. Infect. Dis. 28, 384–394. doi: 10.1086/515106
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16:157. doi: 10.1186/s13059-015-0721-2
Fincher, R. M. E., Jackson, M. W., and Fischer, A. Q. (1990). Case report: Citrobacter freundii: a newly reported cause of pyomyositis. Am. J. Med. 299, 331–333. doi: 10.1097/00000441-199005000-00008
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Gehring, A. M., DeMoll, E., Fetherston, J. D., Mori, I., Mayhew, G. F., Blattner, F. R., et al. (1998). Iron acquisition in plague: modular logic in enzymatic biogenesis of yersiniabactin by Yersinia pestis. Chem. Biol. 5, 573–586. doi: 10.1016/s1074-5521(98)90115-6
Gross, R. J., Rowe, B., and Easton, J. A. (1973). Neonatal meningitis caused by Citrobacter koseri. J. Clin. Pathol. 26:552. doi: 10.1136/jcp.26.7.552-c
Hodges, G. R., Degener, C. E., and Barnes, W. G. (1978). Clinical significance of citrobacter isolates. Am. J. Clin. Pathol. 70, 37–40. doi: 10.1093/ajcp/70.1.37
Hone, D. M., Attridge, S. R., Forrest, B., Morona, R., and Daniels, D. (1988). A gale via (Vi antigen-negative) mutant of Salmonella typhi Ty2 retains virulence in humans. Infect. Immun. 56, 1326–1333.
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Jacoby, G. A., Griffin, C. M., and Hooper, D. C. (2011). Citrobacter spp. as a source of qnrB Alleles. Antimicrob. Agents Chemother. 55, 4979–4984. doi: 10.1128/AAC.05187-11
Janda, J. M., Abbott, S. L., Cheung, W. K., and Hanson, D. F. (1994). Biochemical identification of Citrobacteria in the clinical laboratory. J. Clin. Microbiol. 32, 1850–1854.
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Joan, D. R., and John, B. R. (1984). Reexamination of the protective role of the capsular polysaccharide(Vi antigen) of Salmonella typhi. J. Infect. Dis. 150, 436–449. doi: 10.1093/infdis/150.3.436
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Juhas, M., Crook, D. W., Dimopoulou, I. D., Lunter, G., Harding, R. M., Ferguson, D. J., et al. (2007). Novel type IV secretion system involved in propagation of genomic islands. J. Bacteriol. 189, 761–771. doi: 10.1128/JB.01327-06
Juhas, M., van der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Kaas, R. S., Friis, C., Ussery, D. W., and Aarestrup, F. M. (2012). Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics 13:577. doi: 10.1186/1471-2164-13-577
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kehrenberg, C., Friederichs, S., de Jong, A., and Schwarz, S. (2008). Novel variant of the qnrB gene, qnrB12, in Citrobacter werkmanii. Antimicrob. Agents Chemother. 52, 1206–1207. doi: 10.1128/AAC.01042-07
Kim, K. S., Itabashi, H., Gemski, P., Sadoff, J., Warren, R. L., and Cross, A. S. (1992). The KI capsule is the critical determinant in the development of Escherichia coil meningitis in the rat. J. Clin. Invest. 90, 897–905. doi: 10.1172/JCI115965
Kirill, A. D., and Barry, L. W. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Kline, M. W., and Kaplan, S. L. (1987). Citrobacter diversus and neonatal brain abscess. Pediatr. Neurol 3, 178–180. doi: 10.1016/0887-8994(87)90089-0
Kuang, Y., Hu, M., and Wu, Q. (2015). “Multi-level evaluation model for intangible cultural heritage status based on fuzzy set theory,” in Proceedings of the 8th International Symposium on Computational Intelligence and Design (ISCID), Hangzhou.
Lavigne, J. P., Defez, C., Bouziges, N., Mahamat, A., and Sotto, A. (2007). Clinical and molecular epidemiology of multidrug-resistant Citrobacter spp. infections in a French university hospital. Eur. J. Clin. Microbiol. Infect. Dis. 26, 439–441. doi: 10.1007/s10096-007-0315-3
Li, J., Yao, Y., Xu, H. H., Hao, L., Deng, Z., Rajakumar, K., et al. (2015). SecReT6: a web-based resource for type VI secretion systems found in bacteria. Environ. Microbiol. 17, 2196–2202. doi: 10.1111/1462-2920.12794
Lipsky, B. A., Hook, E. W. R., Smith, A. A., and Plorde, J. J. (1980). Citrobacter infections in humans: experience at the seattle veterans administration medical center and a review of the literature. Rev. Infect. Dis. 2, 746–760. doi: 10.1093/clinids/2.5.746
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Luperchio, S. A., Newman, J. V., Dangler, C. A., Schrenzel, M. D., Brenner, D. J., Steigerwalt, A. G., et al. (2000). Citrobacter rodentium, the causative agent of transmissible murine colonic hyperplasia, exhibits clonality: synonymy of C. rodentium and mouse-pathogenic Escherichia coli. J. Clin. Microbiol. 38, 4343–4350.
Mohanty, S., Singhal, R., Sood, S., Dhawan, B., Kapil, A., and Das, B. K. (2007). Citrobacter infections in a tertiary care hospital in Northern India. J. Infect. 54, 58–64. doi: 10.1016/j.jinf.2006.01.015
Naum, M., Brown, E. W., and Mason-Gamer, R. J. (2008). Is 16S rDNA a reliable phylogenetic marker to characterize relationships below the family level in the enterobacteriaceae? J. Mol. Evol. 66, 630–642. doi: 10.1007/s00239-008-9115-3
Park, Y. J., Yu, J. K., Lee, S., Oh, E. J., and Woo, G. J. (2007). Prevalence and diversity of qnr alleles in AmpC-producing Enterobacter cloacae, Enterobacter aerogenes, Citrobacter freundii and Serratia marcescens: a multicentre study from Korea. J. Antimicrob. Chemother. 60, 868–871. doi: 10.1093/jac/dkm266
Patel, J. B. (2001). Performance standards for antimicrobial susceptibility testing. Clin. Microbiol. Newsl. 23:49. doi: 10.1016/s0196-4399(01)88009-0
Pepperell, C., Kus, J. V., Gardam, M. A., Humar, A., and Burrows, L. L. (2002). Low-virulence Citrobacter species encode resistance to multiple antimicrobials. Antimicrob. Agents Chemother. 46, 3555–3560. doi: 10.1128/aac.46.11.3555-3560.2002
Perry, R. D., Balbo, P. B., Jones, H. A., Fetherston, J. D., and DeMoll, E. (1999). Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology 145, 1181–1190. doi: 10.1099/13500872-145-5-1181
Qian, C., Du, Y., Li, H., Wu, P., Wang, L., Wei, Y., et al. (2018). Development of rapid and simple experimental and in silico serotyping systems for Citrobacter. Fut. Microbiol. 13, 1511–1522. doi: 10.2217/fmb-2018-0187
Raymond, B., Young, J. C., Pallett, M., Endres, R. G., Clements, A., and Frankel, G. (2013). Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol. 21, 430–441. doi: 10.1016/j.tim.2013.06.008
Ribeiro, C. D., Davis, P., and Jones, D. M. (1976). Citrobacter koseri meningitis in a special care baby unit. J. Clin. Pathol. 29, 1094–1096. doi: 10.1136/jcp.29.12.1094
Ribeiro, T. G., Novais, A., Branquinho, R., Machado, E., and Peixe, L. (2015). Phylogeny and comparative genomics unveil independent diversification trajectories of qnrB and genetic platforms within particular Citrobacter species. Antimicrob. Agents Chemother. 59, 5951–5958. doi: 10.1128/AAC.00027-15
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Russell, A. B., Wexler, A. G., Harding, B. N., Whitney, J. C., Bohn, A. J., Goo, Y. A., et al. (2014). A type VI secretion-related pathway in bacteroidetes mediates interbacterial antagonism. Cell Host Microbe 16, 227–236. doi: 10.1016/j.chom.2014.07.007
Saga, T., Sabtcheva, S., Mitsutake, K., Ishii, Y., Tateda, K., Yamaguchi, K., et al. (2013). Characterization of qnrB-like genes in Citrobacter species of the American type culture collection. Antimicrob. Agents Chemother. 57, 2863–2866. doi: 10.1128/AAC.02396-12
Sahl, J. W., Caporaso, J. G., Rasko, D. A., and Keim, P. (2014). The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ 2:e332. doi: 10.7717/peerj.332
Sakazaki, R. (1997). Bacterial identification. Clin. Microbiol. Infect. 3, 53–56. doi: 10.1111/j.1469-0691.1997.tb00936.x
Samonis, G., Ho, D. H., Gooch, G. F., Rolston, K. V., and Bodey, G. P. (1987). In vitro susceptibility of Citrobacter species to various antimicrobial agents. Antimicrob. Agents Chemother. 31, 829–830. doi: 10.1128/aac.31.5.829
Samonis, G., Karageorgopoulos, D. E., Kofteridis, D. P., Matthaiou, D. K., Sidiropoulou, V., Maraki, S., et al. (2009). Citrobacter infections in a general hospital: characteristics and outcomes. Eur. J. Clin. Microbiol. Infect. Dis. 28, 61–68. doi: 10.1007/s10096-008-0598-z
Schauer, D. B., Zabel, B. A., Pedraza, I. F., O’Hara, C. M., Steigerwalt, A. G., and Brenner, D. J. (1995). Genetic and biochemical characterization of Citrobacter rodentium sp. nov. J. Clin. Microbiol. 33, 2064–2068.
Soriano, A. L., Russell, R. G., Johnson, D., Lagos, R., Sechter, I., and Morris, J. G. J. (1991). Pathophysiology of Citrobacter diversus neonatal meningitis: comparative studies in an infant mouse model. Infect. Immun. 59, 1352–1358.
Tallon, L. J., Liu, X., Bennuru, S., Chibucos, M. C., Godinez, A., Ott, S., et al. (2014). Single molecule sequencing and genome assembly of a clinical specimen of Loa loa, the causative agent of loiasis. BMC Genomics 15:788. doi: 10.1186/1471-2164-15-788
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Townsend, S. M., Gonzalez-Gomez, I., and Badger, J. L. (2006). fliP influences Citrobacter koseri macrophage uptake, cytokine expression and brain abscess formation in the neonatal rat. J. Med. Microbiol. 55(Pt 12), 1631–1640. doi: 10.1099/jmm.0.46596-0
Townsend, S. M., Pollack, H. A., Gonzalez-Gomez, I., Shimada, H., and Badger, J. L. (2003). Citrobacter koseri brain abscess in the neonatal rat: survival and replication within human and rat macrophages. Infect. Immun. 71, 5871–5880. doi: 10.1128/iai.71.10.5871-5880.2003
Vallance, B. A., Deng, W., Jacobson, K., and Finlay, B. B. (2003). Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect. Immun. 71, 3443–3453. doi: 10.1128/iai.71.6.3443-3453.2003
Warren, R. L., Farmer, J. J., Dewhirst, F. E., Birkhead, K., Zembower, T., Peterson, L. R., et al. (2000). Outbreak of nosocomial infections due to extended-spectrum b-lactamase-producing strains of enteric group 137, a new member of the family Enterobacteriaceae closely related to Citrobacter farmeri and Citrobacter amalonaticus. J. Clin. Microbiol. 38, 3946–3952.
Williams, W. W., Mariano, J., Spurrier, M., Donnell, H. D. Jr., Breckenridge, R. L. Jr., Anderson, R. L., et al. (1984). Nosocomial meningitis due to Citrobacter diversus in neonates: new aspects of the epidemiology. J. Infect. Dis 150, 229–235. doi: 10.1093/infdis/150.2.229
Wilson, D. J. (2012). Insights from genomics into bacterial pathogen populations. PLoS Pathog. 8:e1002874. doi: 10.1371/journal.ppat.1002874
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhu, L., Pearce, D., and Kim, K. S. (2010). Prevention of Escherichia coli K1 penetration of the blood-brain barrier by counteracting the host cell receptor and signaling molecule involved in E. coli invasion of human brain microvascular endothelial cells. Infect. Immun. 78, 3554–3559. doi: 10.1128/IAI.00336-10
Keywords: Citrobacter koseri, whole genome sequence, comparative genomic analysis, pathogenicity, high-pathogenicity island
Citation: Yuan C, Yin Z, Wang J, Qian C, Wei Y, Zhang S, Jiang L and Liu B (2019) Comparative Genomic Analysis of Citrobacter and Key Genes Essential for the Pathogenicity of Citrobacter koseri. Front. Microbiol. 10:2774. doi: 10.3389/fmicb.2019.02774
Received: 07 June 2019; Accepted: 14 November 2019;
Published: 06 December 2019.
Edited by:
Sunil Kumar Lal, Monash University Malaysia, MalaysiaReviewed by:
Adriana Ribeiro Carneiro Folador, Federal University of Pará, BrazilStephen Forsythe, Foodmicrobe.com, United Kingdom
Copyright © 2019 Yuan, Yin, Wang, Qian, Wei, Zhang, Jiang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingyan Jiang, amlhbmdsaW5neWFuQG5hbmthaS5lZHUuY24=; Bin Liu, bGl1YmluMTk4MUBuYW5rYWkuZWR1LmNu
†These authors have contributed equally to this work