 Agnieszka Latka
Agnieszka Latka Petr G. Leiman
Petr G. Leiman Zuzanna Drulis-Kawa
Zuzanna Drulis-Kawa Yves Briers
Yves Briers- 1Laboratory of Applied Biotechnology, Department of Biotechnology, Ghent University, Ghent, Belgium
- 2Department of Pathogen Biology and Immunology, Institute of Genetics and Microbiology, University of Wrocław, Wrocław, Poland
- 3Sealy Center for Structural Biology and Molecular Biophysics, Department of Biochemistry and Molecular Biology, The University of Texas Medical Branch, Galveston, TX, United States
Klebsiella pneumoniae carries a thick polysaccharide capsule. This highly variable chemical structure plays an important role in its virulence. Many Klebsiella bacteriophages recognize this capsule with a receptor binding protein (RBP) that contains a depolymerase domain. This domain degrades the capsule to initiate phage infection. RBPs are highly specific and thus largely determine the host spectrum of the phage. A majority of known Klebsiella phages have only one or two RBPs, but phages with up to 11 RBPs with depolymerase activity and a broad host spectrum have been identified. A detailed bioinformatic analysis shows that similar RBP domains repeatedly occur in K. pneumoniae phages with structural RBP domains for attachment of an RBP to the phage tail (anchor domain) or for branching of RBPs (T4gp10-like domain). Structural domains determining the RBP architecture are located at the N-terminus, while the depolymerase is located in the center of protein. Occasionally, the RBP is complemented with an autocleavable chaperone domain at the distal end serving for folding and multimerization. The enzymatic domain is subjected to an intense horizontal transfer to rapidly shift the phage host spectrum without affecting the RBP architecture. These analyses allowed to model a set of conserved RBP architectures, indicating evolutionary linkages.
Introduction
Klebsiella pneumoniae is a Gram-negative bacillus. In spite of being part of the natural human and animal flora, K. pneumoniae is also the widespread cause of both nosocomial and community acquired infections. Since 2013 K. pneumoniae has been marked as a prominent member of the carbapenem-resistant Enterobacteriaceae (CRE), featured by a multidrug-resistant phenotype and labeled as a class of antibiotic-resistant bacteria for which novel ways of therapy are most urgent (Weiner et al., 2016; Calfee, 2017). As natural bacterial predators, bacteriophages have since long been proposed as promising alternatives to antibiotic therapy. The large majority of phages is highly specific with a host spectrum defined at the species/strain level. This high specificity necessitates the selection of a phage sur-mesure for a personalized treatment or the use of a phage cocktail that covers a broader host range. Major determinants of host specificity are the phage receptor binding proteins (RBPs) that mediate the initial contact with the receptor on the host cell envelope (Williams et al., 2008). This initial contact can be based on a direct binding of long tail fibers or shorter tailspikes to the cell surface receptor. Some RBPs possess a depolymerase activity to degrade bacterial exopolysaccharides comprising the capsule (CPS), lipopolysaccharides (LPS) or biofilm matrix (Majkowska-Skrobek et al., 2016, 2018; Olszak et al., 2017). Interaction of RBPs with their cell wall receptors are essential to initiate the infection process (Andres et al., 2010; Broeker et al., 2018).
The primary receptor targeted by RBPs of many Klebsiella specific phages is the thick polysaccharide capsule, which is a hallmark feature of K. pneumoniae. The capsule is a crucial virulence factor as it forms a physical barrier to some antibiotics and host immune mechanisms, enabling bacteria to avoid phagocytosis or complement-mediated killing (Cortés et al., 2002; Lee et al., 2017; Majkowska-Skrobek et al., 2018). Differences in sugar composition, the specific ratio of various sugar components as well as variation in the locus organization are the base to distinguish at least 79 capsular serotypes called K antigens and 134 capsular loci (KL) among Klebsiella species (Pan et al., 2015; Wyres et al., 2016; Wick et al., 2018). This capsular diversity correlates to a correspondingly high variation of Klebsiella phage RBPs that contain a specific polysaccharide-depolymerizing domain (Schmid et al., 2015; Latka et al., 2017). Such domains cleave the O-glycosidic bond of capsular polysaccharides following either a hydrolase or a lyase mechanism. Hydrolases (e.g., sialidases, rhamnosidases, levanases, dextranases, and xylanases) involve a water molecule for cleavage, whereas lyases [e.g., hyaluronate lyases (hyaluronidases), pectin/pectate lyases, alginate lyases, K5 lyases] cleave by β-elimination with introduction of new double bond (Davies and Henrissat, 1995; Sutherland, 1995; Pires et al., 2016). In spite of a high diversity in enzyme specificity and primary amino acid sequence, many known depolymerases contain an elongated, highly interwoven β-helical domain that forms the specific catalytic pocket. In addition, this β-helical domain contributes to a high protein stability in harsh environments (Yan et al., 2014; Majkowska-Skrobek et al., 2016). An overview of (experimentally confirmed) RBPs with depolymerase activity has been recently reported (Latka et al., 2017).
Receptor binding protein with depolymerase activity have a modular structure with the enzymatic domain located in the central part (Figure 1C). The C-terminus of the RBP may comprise a chaperone that assists in a proper folding and trimerization followed by autoproteolytic removal or an additional domain involved in host cell recognition (Weigele et al., 2003; Cornelissen et al., 2011; Schwarzer et al., 2012; Seul et al., 2014; Yan et al., 2014). Autocleavage of the C-terminal chaperone was also reported as a common feature among endosialidases and other tail spikes and tail fibers, necessary to increase the unfolding barrier and to trap the mature trimer in a more kinetically stable conformation (Schwarzer et al., 2007). The N-terminal dome-like domain attaches the RBP to the phage particle by a flexible connector. A modular architecture of RBPs allows for rapid evolution via horizontal gene transfer leading to host range modification. Whereas structural domains responsible for attachment to the tail apparatus are repeatedly present in many phylogenetically related phages, the domains for host cell receptor recognition/degradation are subjected to intense exchanges across phylogenetic borders. In addition, the latter RBP domains undergo further constant modification through vertical transfer and accumulation of mutations (Stummeyer et al., 2006; Barbirz et al., 2008; Leiman and Molineux, 2008; Schwarzer et al., 2012; Latka et al., 2017). The tail fibers of E. coli phage T7 and its relative K1F are type examples of a horizontal transfer of the C-terminal RBP domain. These tail fibers share a conserved N-terminal domain of ∼140 resides that anchors the tail fiber to the phage particle (Figure 1). However, T7 has a C-terminal domain that recognizes and binds lipopolysaccharide, whereas K1F produces an endosialidase specific for recognition and cleavage of E. coli K1 capsular polysaccharide (Steven et al., 1988; Stummeyer et al., 2005).
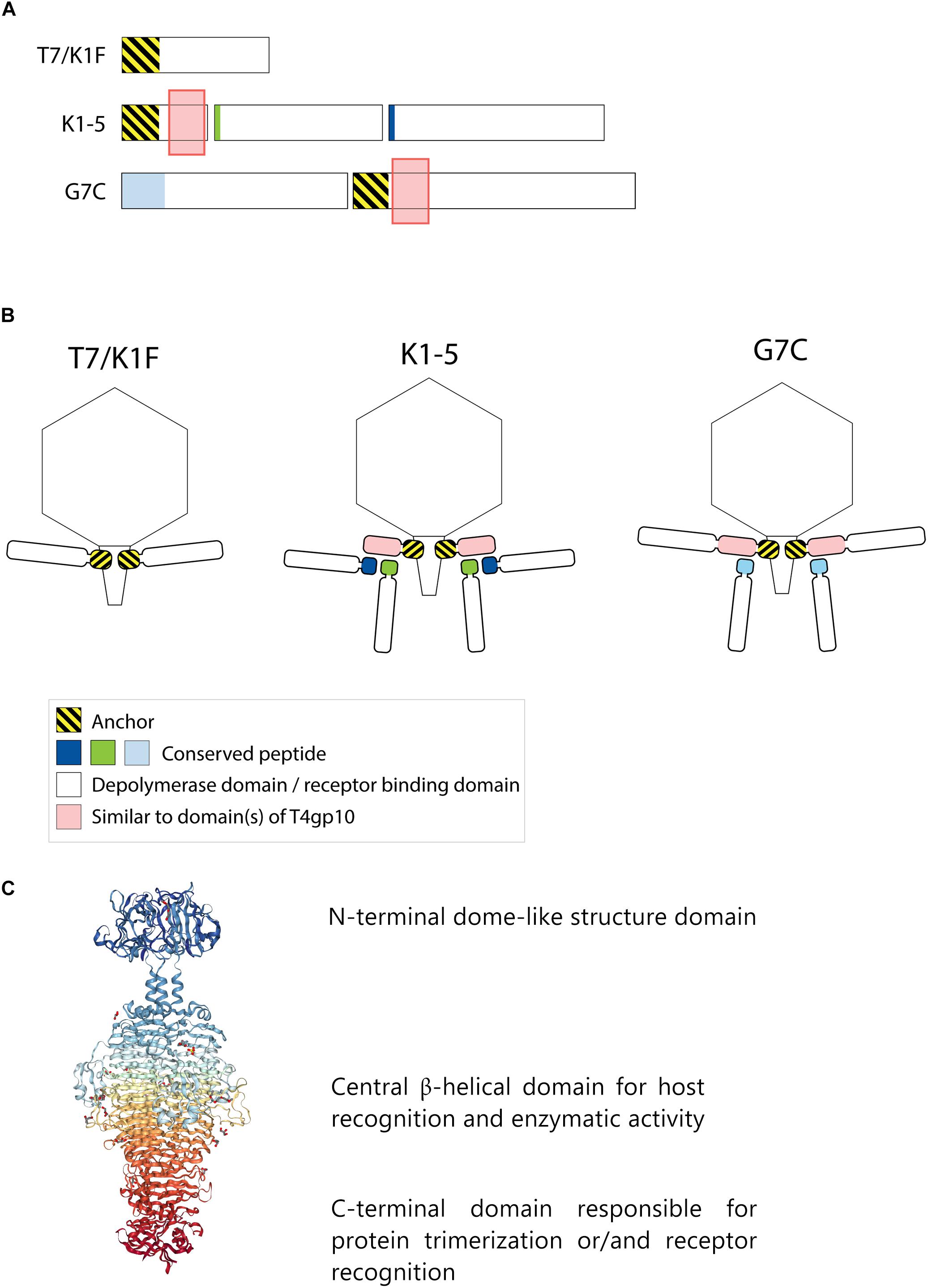
Figure 1. Anchor and anchor-branched receptor binding protein (RBP) complexes confirmed by structural experiments. (A) The modular genetic organization of RBPs in single (T7 and K1F) and double RBP systems (K1-5 and G7C phages). (B) Schematic modeling of four different RBP systems in the virion structure. The T7 tail fiber (gene 17, T7p52) and K1F tail fiber (gene 17, CKV1F_gp36) have only an N-terminal anchor domain; K1-5 uses an adapter protein (gp37 with T4gp10-like domain) interacting with K5 lyase (gp46) and K1 endosialidase (gp47) via a conserved hepta- and undecapeptide, respectively; Phage G7C produces an anchor-branched complex with one anchored RBP (gp66) having a T4gp10-like domain and the second RBP connected via a conserved peptide to theT4gp10-like domain. (C) Modular structure of the model tail spike of Salmonella phage P22 (PDB ID 2XC1), illustrating a typical modular structure of RBPs. A N-terminal dome-like structure domain, a central β-helical domain for host recognition and enzymatic activity and a C-terminal domain responsible for protein trimerization or/and receptor recognition are shown (Berman et al., 2000; Seul et al., 2014; Rose et al., 2018).
Phages with a single RBP such as T7 and K1F are most frequently described in the literature. However, several phages belonging to Podoviridae have also acquired two different RBPs corresponding to a dual receptor-specificity. E.g., K. pneumoniae podoviruses K5-2, K5-4, and KP32 possess two RBPs with a depolymerase domain with different enzymatic specificity (Hsieh et al., 2017; Majkowska-Skrobek et al., 2018). In the last decade, an increasing body of knowledge about the genetic and structural organization of RBPs of such bispecific phages has been acquired, particularly for different T7-like phages such as K1-5 and SP6 (Stummeyer et al., 2006; Leiman et al., 2007; Gebhart et al., 2017; Tu et al., 2017). These phages use a small trimeric adapter protein of approximately 300 amino acids, sharing a high N-terminal sequence identity to T7 and K1F tail fibers (Figure 1). In addition, phage K1-5 encodes a K5 lyase (gp46) and an endosialidase (gp47), which are specific for E. coli K5 and K1 capsule, respectively. CryoEM studies and bioinformatics suggest that K5 lyase binds through a heptapeptide (MAKLTKP) to a specific site in the middle of the K1-5 adapter protein, whereas the second tailspike (endosialidase) binds to a different specific site in its C-terminal part through an undecapeptide (MIQRLGSSLVK) (Leiman et al., 2007). The heptapeptide, undecapeptide, and adapter sequences are conserved among other T7-like phages that infect different bacterial species and that carry two different RBPs on the phage particle (e.g., SP6), demonstrating a conserved mechanism for attachment of two RBPs (Figure 1). Notably, domains recognizing the same host receptor can have highly similar amino acid sequence but can be incorporated into a different RBP architecture. For example, the K1F and K1-5 endosialidase domains specific to K1 capsule show 72% identity with a coverage of 86%, but in phage K1F the endosialidase domain is present in a single RBP with anchor domain, whereas in phage K1-5 the endosialidase is connected to the phage particle via an intermediate adapter protein. A homolog of the endosialidase domain of podovirus K1F is also present in the multivalent E. coli myovirus phi92 (EndoN92; 53% identity with a coverage of 83%), demonstrating exchange of the domain across members of the Podoviridae and Myoviridae families with highly different tail structures (Schwarzer et al., 2012, 2015).
More recently, a different organization of two types of RBPs in a single phage particle has been reported based on structural, genetic and biochemical studies of the RBPs of E. coli N4-like podovirus G7C (Prokhorov et al., 2017). G7C carries two RBPs – a longer G7Cgp66 and a shorter G7Cgp63.1 protein. The specificity of the longer G7cgp66 protein is unknown, but the shorter G7Cgp63.1 RBP was shown to deacetylate the O-antigen of E. coli 4S while leaving the backbone of the sugar intact. G7Cgp63.1 does not interact with the phage particle directly. Instead, it binds to G7Cgp66, which is attached to the phage particle with its N-terminal anchor domain (Figure 1B). The gp63.1 binding region of G7Cgp66 (residues 138–294) is homologous to subdomains D2 and D3 of phage T4 gp10. In phage T4, these subdomains of gp10 serve as an attachment site for two proteins – gp11, which interacts with the long tail fiber RBP or short tail fiber RBP, depending on the state of the phage particle, and gp12, the short tail fiber RBP (Taylor et al., 2016). This protein complex represents a bona fide branched structure involved in the transmission of the signal of reversible host binding, culminating in irreversible binding, sheath contraction and DNA ejection. The T4gp10-like domains are prevalent in RBPs of unrelated phages across Podoviridae and Myoviridae, which may reflect its ancient evolutionary role in the transduction from reversible to irreversible binding during phage adsorption (Prokhorov et al., 2017).
Interestingly, the T4gp10-like region of G7Cgp66 covers both subdomain D2 and D3 of T4gp10 to which T4gp11 and T4gp12 are attached. Though, G7Cgp66 and G7Cgp63.1 form a 1:1 complex, suggesting that G7Cgp63.1 occupies only one of the two RBP binding sites on G7Cgp66. Notably, orthologs of G7Cgp66 in some G7C-like viruses do not contain a putative enzymatic domain but nevertheless retain the N-terminal particle-binding domain and the T4gp10-like domains. As such their attachment apparatus becomes similar to the adapter system of phage K1-5. The N-terminal part of G7Cgp63.1 that interacts with the T4gp10-like domain of G7Cgp66 is also found at the N-terminus of other tail spikes that have a branched structure, such as CBA120 phage tail spike 1 (Chen et al., 2014) and other putative tail spikes of Vil-like phages (Adriaenssens et al., 2012). CBA120 encodes four tail spikes (TSP1-4) from which two (TSP2 and TSP4) are equipped with T4gp10-like domains D2 and D3. These domains provide side or off-axis attachment sites for TSP1 and TSP3. The conserved N-terminal part of TSP4 attaches the whole branched structure composed of four TSPs to the baseplate of the virion (Plattner et al., 2019).
Klebsiella jumbo viruses may also have a multitude of RBPs resulting accordingly in a broader host spectrum. The highest variation of depolymerases has been described for the jumbo ΦK64-1 phage, which is able to infect K. pneumoniae of 10 different capsular serotypes and for which 11 different polysaccharide depolymerases have been identified (Pan et al., 2017). Also, the jumbo vB_KleM-RaK2 phage encodes a multitude of putative depolymerases (Simoliūnas et al., 2013). Electron microscopy images of such jumbo phages typically show an elaborated tail fiber apparatus with a high structural complexity, but for which structural insights are currently lacking.
In this study we present an extensive bioinformatic analysis of the structural and genetic organization of depolymerase-containing RBPs in Klebsiella phages. Next-generation sequencing technologies have recently led to a large number of sequenced phage genomes in public databases including Klebsiella viruses (n = 97). In a large proportion of these phages (59/97; 61%) we could predict an RBP with depolymerase activity. The observed large diversity of depolymerase domains accommodates the high diversity of capsular serotypes among Klebsiella strains. Based on an integrated analysis, we propose diverse RBP architectures in Klebsiella phages.
Materials and Methods
At first, Klebsiella phages were collected from the GenBank database (retrieved at 15.08.2018). A number of 59 phages were finally analyzed (Supplementary Table S1). From these phages proteins annotated as tail fibers or tail spikes were analyzed with BlastP1 (Altschul et al., 1990), Phyre22 (Kelley et al., 2015), SWISS-MODEL3 (Bordoli et al., 2009; Bordoli and Schwede, 2012), HMMER4 (Finn et al., 2011) and HHPred5 (Zimmermann et al., 2018) to identify phages that encode RBPs with putative depolymerase activity (Supplementary Table S2). If neither a tail fiber nor a tail spike gene was found in the genome, we analyzed all genes located in the vicinity of annotated structural genes. BlastP (protein–protein Blast) was performed against the non-redundant protein sequences (nr) database using standard parameters (expect threshold: 10, word size: 6, MATRIX: BLOSUM62, Gap cost: existence 11, extension 1, conditional compositional score matrix adjustment). HMMER was used in the quick search mode against: Reference Proteomes, UniProtKB, SwissProt, and Pfam with significance E-values: 0.01 (sequence) and 0.03 (hit). For Phyre2 the normal modeling mode was used. HHPred homology detection structure prediction was run using the PDB_mmCIF70 database and the following parameters [MSA generation method: HHblits uniclust30_2018_08; Maximal no. of MSA generation steps: 3; E-value incl. threshold for MSA generation: 1e-3; minimal sequence identity of MSA hits with query (%): 0; minimal coverage of MSA hits (%) 20; Secondary structure scoring: during alignment; Alignment Mode: Realign with MAC: local:norealign; MAC realignment threshold: 0.3; No. of target sequences: 250; Min. probability in hit list (>10%): 20].
Criteria for the prediction of putative depolymerase activity were (Supplementary Table S2): (1) the protein must be longer than 200 residues; (2) the protein must be annotated as tail fiber/tail spike/hypothetical protein in the NCBI database; (3) the protein must show homology to domains annotated as lyase [hyaluronate lyases (hyaluronidases), pectin/pectate lyases, alginate lyases, K5 lyases] or hydrolase (sialidases, rhamnosidases, levanases, dextranases, and xylanases) with a confidence of at least 40% in Phyre2 or the enzymatic domain should also be recognized by at least SWISS-MODEL, HMMER, or BlastP; (4) the length of homology with one of these enzymatic domains should span at least 100 residues; (5) a typical β-helical structure should be predicted by Phyre2. These RBP depolymerases are indicated without additional labeling in the tables. Proteins possessing experimentally confirmed depolymerizing activity were marked in the tables with (a). When the RBP was only partially fulfilling the above-mentioned criteria, it was indicated with label (b). These putative depolymerases that could only be predicted with a lower probability were fulfilling criteria 1 and 2, but the confidence of the Phyre2 prediction was below 40% or only SWISS-MODEL, HMMER or BLASTP gave a positive prediction. In addition, the homologous domain only spans between 50 and100 amino acids and no β-helical structure could be predicted with Phyre2 (for details see Supplementary Table S2).
All selected Klebsiella phages were then grouped based on gene homology and a conserved gene synteny into KP32viruses, KP34viruses, and KP36viruses and into groups containing only Klebsiella-specific phages similar to phage JD001 (belonging to Jedunavirus), similar to phage Menlow (belonging to Ackermannviridae), similar to phage ΦK64-1 (belonging to Alcyoneusvirus). Within each group, further subdivisions were proposed for the purpose of this study, based on the organization of the RBP gene cluster (number of RBPs, length of different genes, presence of anchor, or branching domains).
When there was one RBP, a domain in the N-terminus of a RBP was annotated as ‘anchor’ when there was at least an identity of 39% (BLASTP) over at least 166 residues starting from the N-terminus of the corresponding protein among phages belonging to the same group. These parameters were set empirically based on the shortest identity region found among all RBPs, specifically in the first RBP of phage IL33, belonging to KP32viruses group B (166 amino acids) and the identity% of the first RBP of phage Kp1. When more than one RBP was present, the anchor domain was annotated in the RBP in which also a T4gp10-like domain was detected. In the other RBP(s) the N-terminal conserved sequence was called ‘conserved peptide,’ which was also generally shorter than the anchor domains. To define consensus sequences of the anchor domains and conserved peptides, multiple sequence or pairwise alignment were used, since these structures are highly conserved among phages from the same group. To identify domains involved in the branching of RBPs, the sequences were analyzed by HHPred performing protein structure prediction5 (Zimmermann et al., 2018) in search for domains homologous to T4gp10 domain 2 and 3 as experimentally confirmed attachment sites (Prokhorov et al., 2017). WebLogos of the anchor domains and conserved peptides were created with the online available tool6 (Crooks et al., 2004).
Results
Taxonomically closely related phages are characterized by a synteny of conserved structural genes interrupted by divergent RBP genes, which are subject to intensive horizontal transfer. We therefore inspected the region of structural genes across different Klebsiella phages within specific phage genera to identify potential RBPs based on a broken synteny. Subsequently, we analyzed the presence of putative enzymatic domains within the identified RBPs. Based on homology, protein size and structure, we looked for conserved domains (anchor domain, T4gp10-like domain) that may explain the RBP architecture of the particular phage. To further refine this architecture, we analyzed the sequence for the presence of conserved peptides that may mediate attachment to putative T4gp10-like domains. We integrated all these data to model the RBP apparatus of an extensive and diverse set of Klebsiella phages with (predicted) depolymerase activity.
RBPs From Selected Klebsiella Podoviruses
KP32viruses
KP32viruses belong to Podoviridae and have tail fibers attached to a short, non-contractile tail. A similar synteny of highly conserved structural genes is observed across twenty-one KP32viruses (Supplementary Table S1A). Yet, one or two non-conserved genes of different lengths interrupt this synteny after the gene encoding the internal virion protein D. They were identified as putative RBPs and in a few cases also experimentally verified (Hsieh et al., 2017; Majkowska-Skrobek et al., 2018; Solovieva et al., 2018) (Table 1). We found four different RBP organizations (groups A, B, C, and D; Figure 2). The N-termini of the first RBPs are shared with high sequence identity (46–72%) across all KP32viruses. Specifically, residues 1–154 of the first RBPs are highly similar to the N-terminal domain of the phage T7 tail fiber (pfam03906). In group A of KP32viruses, this conserved N-terminal domain (Supplementary Figure S1A) also contains a region that is similar to a fragment of a T4gp10 branching domain, offering a potential attachment point for a secondary tail fiber. The other domain(s) of these 744–903 aa long first RBPs do not share identity with the corresponding domain of the group A model phage KP32. All central domains are predicted to possess enzymatic activity (hydrolase, lyase) but with different specificity. In addition, they all are predicted to possess a characteristic β-helical structure (Supplementary Table S2). In phage KP32, there is an additional C-terminal domain with predicted chaperone activity, which is absent in all other RBPs of the group A KP32viruses.
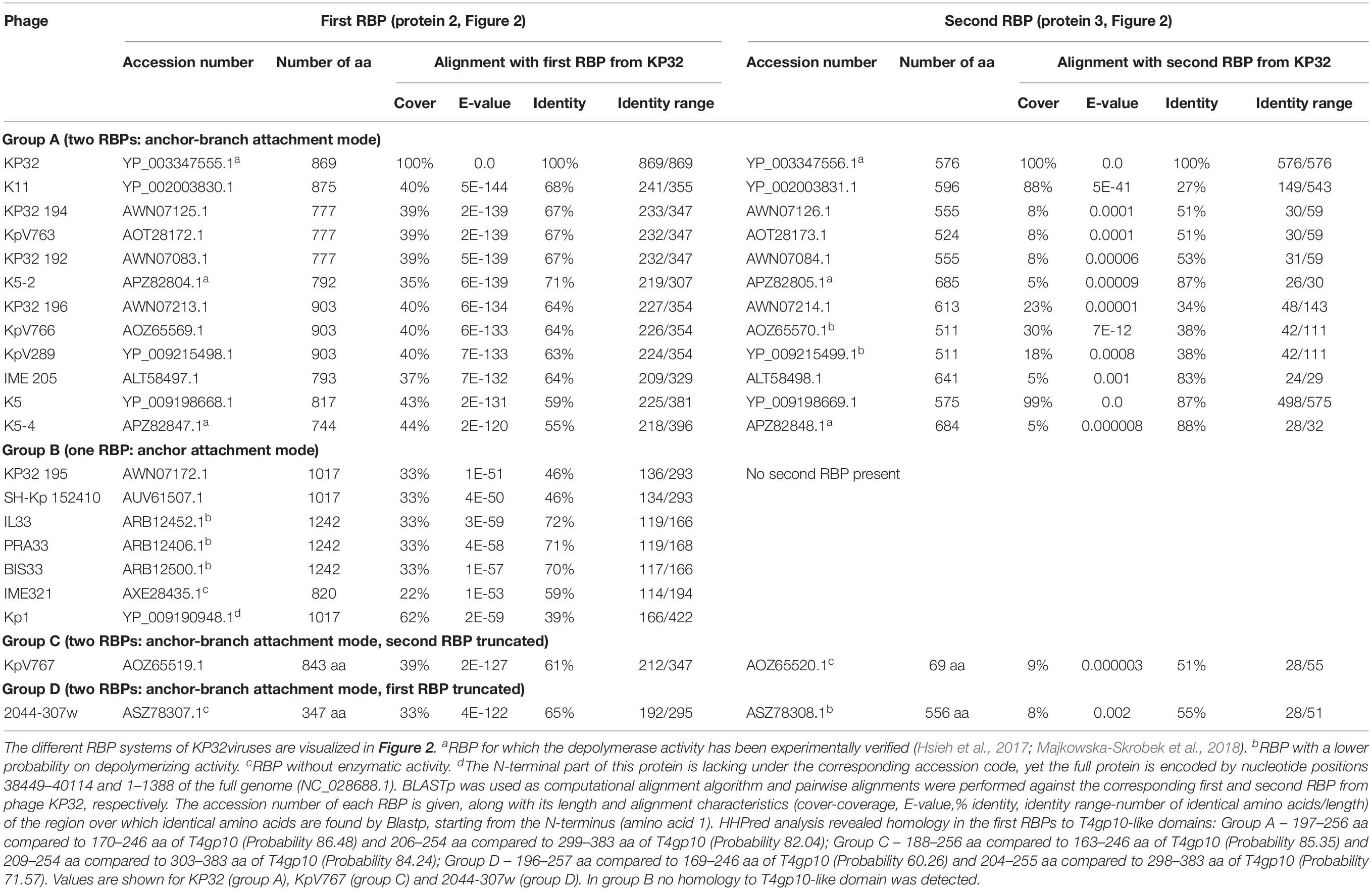
Table 1. Overview of RBPs of KP32viruses with (predicted) depolymerase activity grouped according to the different observed RBP systems.
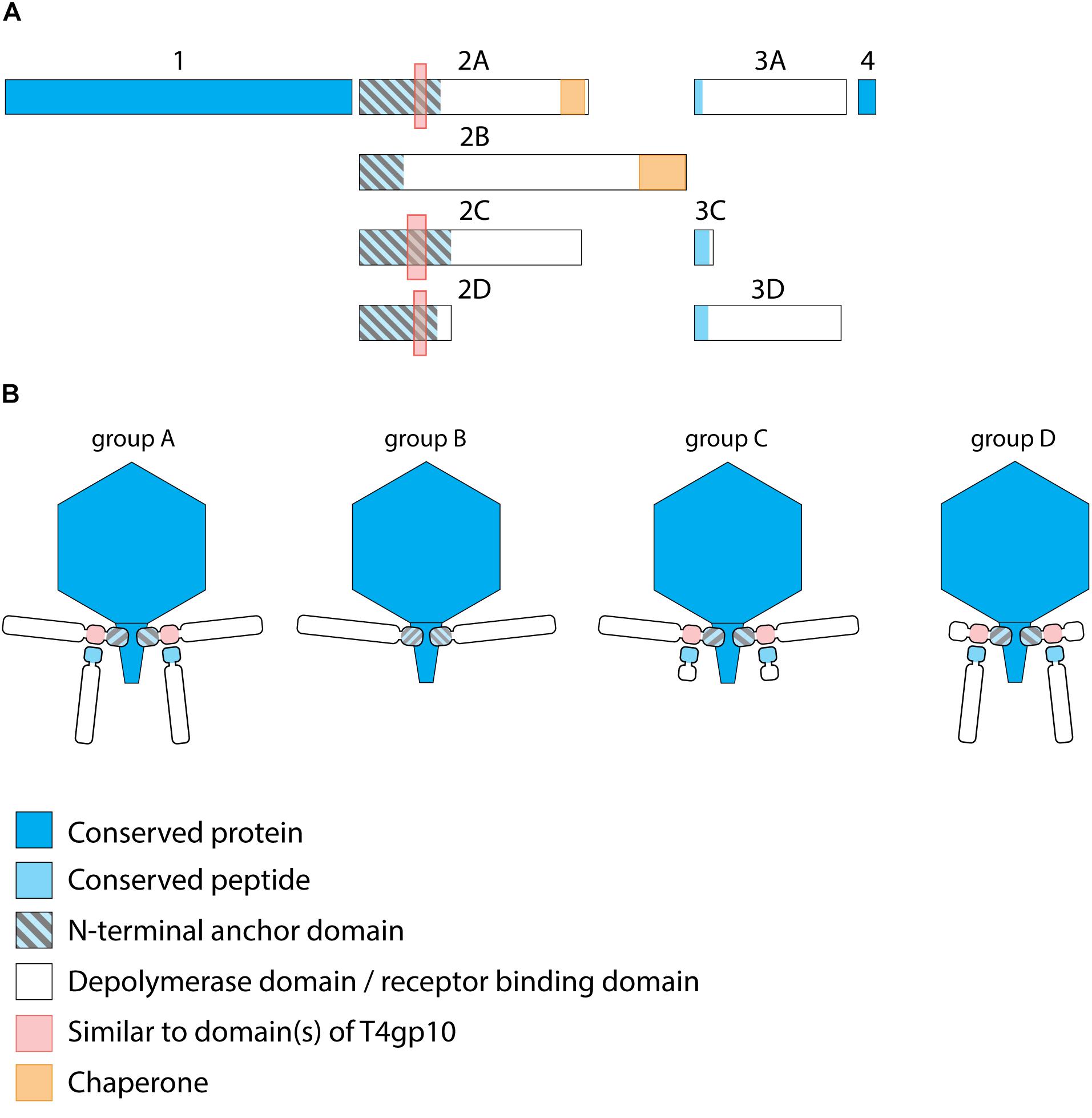
Figure 2. Receptor binding protein systems of KP32viruses. Phages and their RBPs that are proposed to follow these systems, including their grouping into groups A, B, C and D, are summarized in Table 1. (A) The modular composition of RBP genes of phages belonging to four different groups (A, B, C, and D) is shown relative to the broken gene synteny of phage KP32. For simplicity, only one flanking gene conserved across all groups is shown at each side. Annotations are given according to GenBank or according to their modeled function in this study (between brackets): Protein (1) – internal virion protein D; (2) – tail fiber protein (2A,B,C – anchor with depolymerase; 2D – anchor with truncated protein); (3) – hypothetical protein (3A,D – depolymerase with conserved peptide, 3C – truncated protein with conserved peptide); (4) – lysis protein. (B) Schematic models of RBP systems in phage particles of KP32viruses. Group A – two RBPs: anchor-branch attachment mode; Group B – one RBP: anchor attachment mode; Group C – two RBPs: anchor-branch attachment mode, second RBP truncated; Group D – two RBPs: anchor-branch attachment mode, first RBP truncated.
The second RBP that interrupts the gene synteny in group A KP32viruses is recently demonstrated to have depolymerase activity against capsular serotype K21, whereas the first RBP has depolymerase activity against capsular serotype K3 (Majkowska-Skrobek et al., 2018). These specificities correspond to the host spectrum of phage KP32. Other phages of group A KP32viruses also possess this second putative RBP. The second RBP has no conserved N-terminal anchor domain but has a peptide sequence that is conserved across group A KP32viruses with a consensus sequence over the first 29 amino acids (Supplementary Figure S1B). Similarly to the phage G7C RBP system this conserved peptide may be responsible for attachment to the T4gp10-like domain present in the first RBP. Also for the second RBPs, there is a high diversity in the central sequence with a few exceptions. E.g., in phage K5 and KP32, a highly similar sequence is observed, which hints that the second RBP of phage K5 also targets capsular serotype K21. No chaperone is predicted in any second RBP. Integrating these elements, we model the structural organization of group A KP32viruses as depicted in Figure 2B with a conserved anchor-branched attachment mode but with swapped enzymatic domains for specific capsule/host recognition.
Group B KP32viruses (Table 1) have a simpler RBP organization with a single anchor-based RBP. Six out of seven analyzed phages have an RBP with a putative enzymatic domain, while the seventh phage (IME321) apparently lacks enzymatic activity and might rather encode a tail fiber. The N-terminal conserved anchor domain is shorter (166 amino acids) compared to the corresponding domain in group A KP32viruses (307 amino acids). The RBP also lacks a T4gp10-like domain, which is consistent with the absence of a second RBP in group B KP32viruses (Figure 2).
Phage KpV767 (Table 1) represents another variant of KP32viruses (coined group C). The phage has a first anchor-based RBP, including a fragment of a T4gp10-like domain, but the second RBP is largely truncated to only 69 amino acids, including the conserved N-terminal 29 amino acids for attachment to the T4gp10-like domain (Supplementary Figure S1B). KpV767 appears to result from a retrograde evolution, having lost the potential to infect hosts belonging to two different serotypes.
Finally, phage 2044-307w (group D) is as an opposite example of truncation. The first RBP lacks an enzymatic or receptor binding domain but contains an N-terminal anchor including a fragment of a T4gp10-like domain, while the second tail fiber is a full-featured RBP that contains a conserved N-terminal peptide and a depolymerase domain (Supplementary Figure S1B).
KP34viruses
Seventeen phages from the genus of KP34viruses were analyzed (Table 2). Potential proteins involved in host cell recognition could be clearly identified as two genes interrupting the synteny of highly conserved structural genes and genes required for phage particle maturation. Interestingly, both genes are not clustered as in KP32viruses, but are separated by five to eight intervening genes encoding DNA maturases, hypothetical proteins and endolysins, depending on the specific phage. Three different groups (A, B, and C) can be categorized based on differences in length of both genes. Ten group A phages have a short first protein of approximately 300 amino acids annotated as tail fiber. This protein does not encode a putative enzymatic domain, but its N-terminal domain shows high homology to the N-terminus of the phage T7 tail fiber (pfam03906, aa 14–142), similar to the first RBPs of KP32viruses. In addition, the protein contains a fragment of a T4gp10-like domain located at its C-terminus (aa 186–242), which may serve as the attachment point for the second RBP. This protein is highly conserved among all phages of group A KP34viruses (at least 74% identity) (Supplementary Figure S1C). The second RBP sequence encodes a putative enzymatic domain with most such domains forming a β-helical structure. The N-terminal heptapeptide of these proteins contains universally conserved hydrophobic residues (MALxxLV) (Supplementary Figure S1D). These observations suggest that the organization of the RBP apparatus of group A KP34viruses is similar to the system of phage 2044-307w (group D KP32viruses), albeit with a much shorter conserved peptide (Figure 3). Similar short conserved peptides (heptapeptide and undecapeptide) for interaction with the anchor protein have been observed for E. coli phages K1E and K1-5 and Salmonella phage SP6 (Leiman et al., 2007).
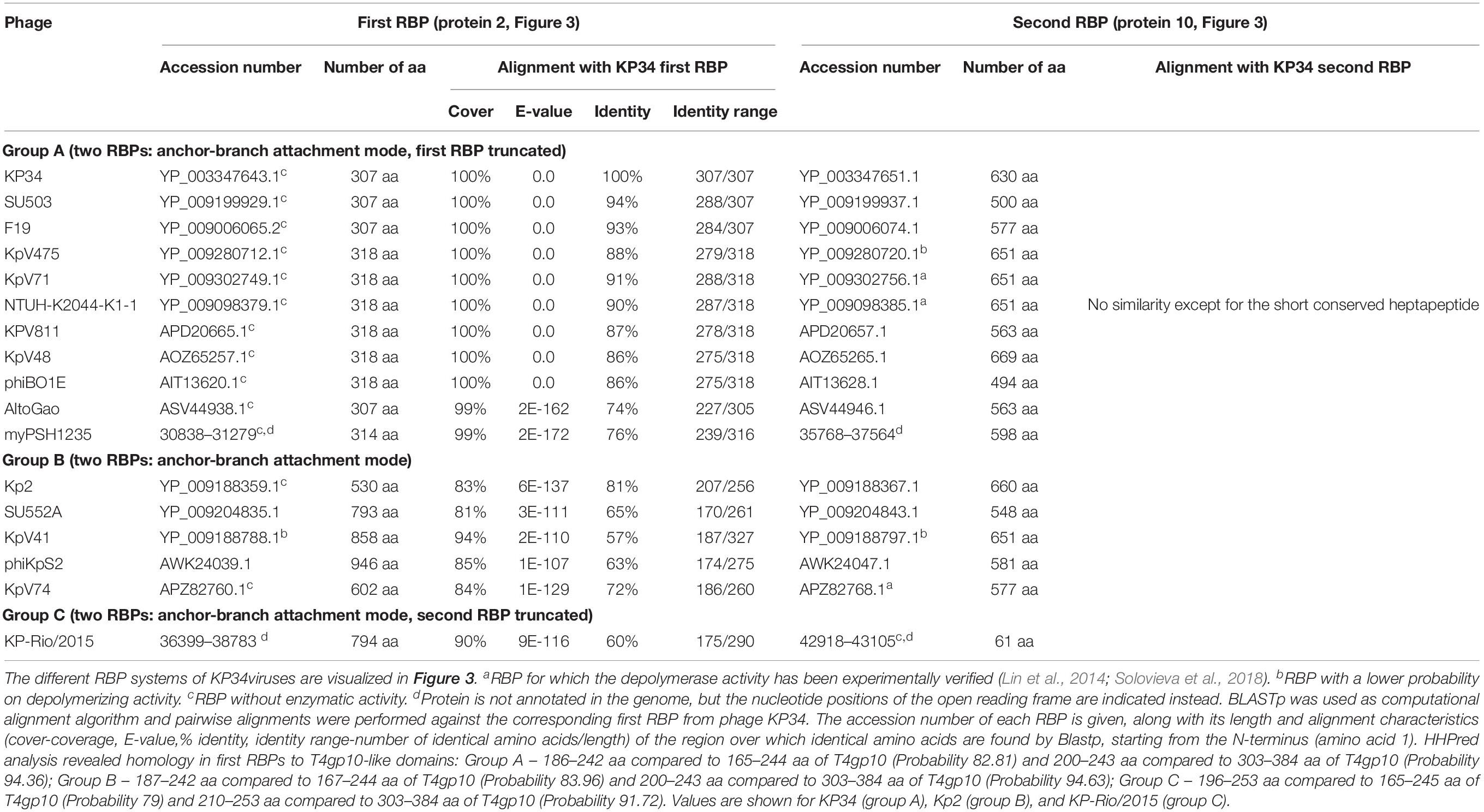
Table 2. Overview of RBPs of KP34viruses with (predicted) depolymerase activity grouped according to the different observed RBP systems.
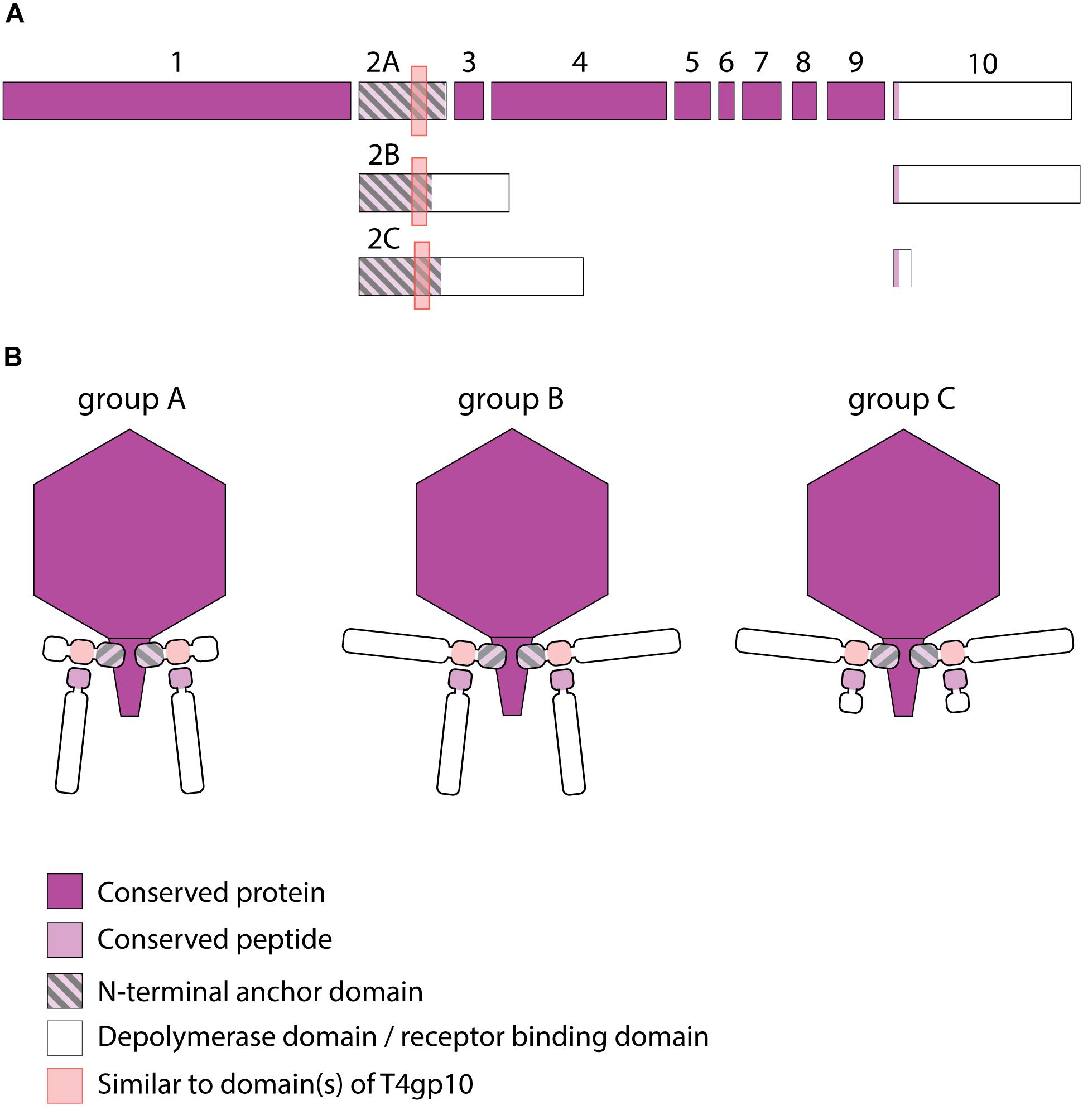
Figure 3. Receptor binding protein systems of KP34viruses. Phages and their RBPs that are proposed to follow these systems, including their grouping into groups A, B and C, are summarized in Table 2. (A) The modular composition of RBP genes of phages belonging to three different groups (A, B, and C) is shown relative to the broken gene synteny of phage KP34. For simplicity, only one conserved gene preceding the first RBP and the conserved, intervening genes of phage KP34 are shown. Annotations are given according to GenBank or according to their modeled function in this study (between brackets): (1) – putative internal core protein; (2) – putative tail fiber protein (2A – anchor; 2B,C – anchor with depolymerase); (3) – putative DNA maturase A; (4) – putative DNA maturase B; (5) – hypothetical protein; (6) – hypothetical protein; (7) – hypothetical protein; (8) – hypothetical protein; (9) – endolysin; (10) – hypothetical protein (depolymerase with conserved peptide). (B) Schematic models of RBP systems in phage particles of KP34viruses. Group A – two RBPs: anchor-branch attachment mode, first RBP truncated; Group B – two RBPs: anchor-branch attachment mode; Group C – two RBPs: anchor-branch attachment mode, second RBP truncated.
Group B KP34viruses contain large first RBPs with a size between 530 and 948 aa. Four out of five RBPs encode an enzymatic domain in the C-terminal or central part of the protein. The corresponding gene in the fifth virus (phage KpV74) contains no predicted enzymatic domain. Group B KP34viruses also encode a second RBP with a predicted enzymatic activity and the same conserved heptapeptide motif as in the second RBP of group A KP34viruses (MALxxLV). The organization of the RBPs in group B KP34viruses is thus similar to that of group A KP32viruses. We found two incongruences in this genus, specifically viruses KP-Rio/2015 and myPSH1235. They both share the gene synteny of KP34viruses but no RBPs were annotated in their genomes. Further genome analysis revealed two open reading frames that presumably fulfill the role of RBPs. We found that phage myPSH1235 follows the RBP organization of group A KP34viruses, while phage KP-Rio/2015 encodes a large first RBP with a predicted enzymatic activity (and a fragment of a T4gp10-like domain) and a second protein that is only 61 aa long, which likely represents a truncated, non-functional RBP. Therefore, phage KP-Rio/2015 forms a different group C with an RBP organization analogous to KP32viruses group C.
RBPs From Selected Klebsiella Myoviruses
Myoviruses have a contractile tail with a baseplate at the head-distal end of the tail. The tail fibers are directly connected to this baseplate. In addition, there is often a central spike (sometimes annotated as ‘fiber’) protruding from the baseplate. Nine Klebsiella phages analyzed in this study belong to three different myovirus groups (JD001 group, Menlow group, and ΦK64-1 group) with the latter two groups having a potentially broad host spectrum since they encode between five and nine (Menlow group; phage RaK2) (Hsu et al., 2013; Simoliūnas et al., 2013) or even 11 different depolymerases (phage ΦK64-1) (Pan et al., 2017) (Supplementary Tables S1C–E), necessitating elaborated structural organizations for RBP attachment. We should note that the JD001, Menlow, and ΦK64-1 phages are no taxonomic groups but were grouped in this study for their genetic similarities in the RBP genes. In addition, viruses belonging to the Menlow group have been recently reclassified from Myoviridae to Ackermannviridae (Adriaenssens et al., 2018). Ackermannviridae are characterized by a conserved genome organization and have typical morphology of myoviruses (long contracting tail) but with a different distal end of the tail, which ends with “stars” or “prongs,” being identified as tailspikes (Day et al., 2018).
Viruses Belonging to the JD001 Group
The putative RBP genes of the viruses of the JD001 group (Table 3) were identified in a region of hypothetical proteins, preceding the DNA polymerase gene. Both genes are located at separate sites with two (JD001, KpV52) or three (KpV79) intervening genes. They all encode a single putative depolymerase, annotated as gluconolaconase, putative tail fiber family protein or tail fiber protein/pectate lyase superfamily protein, respectively. This RBP with depolymerase activity is most likely attached to the anchor protein via a conserved N-terminal domain of about 70 aa, which is distinct from the conserved peptides/domains found in both KP32- and KP34viruses. The anchor protein has no T4gp10-like domain, indicating a different mechanism of interaction (Figures 4A,B).
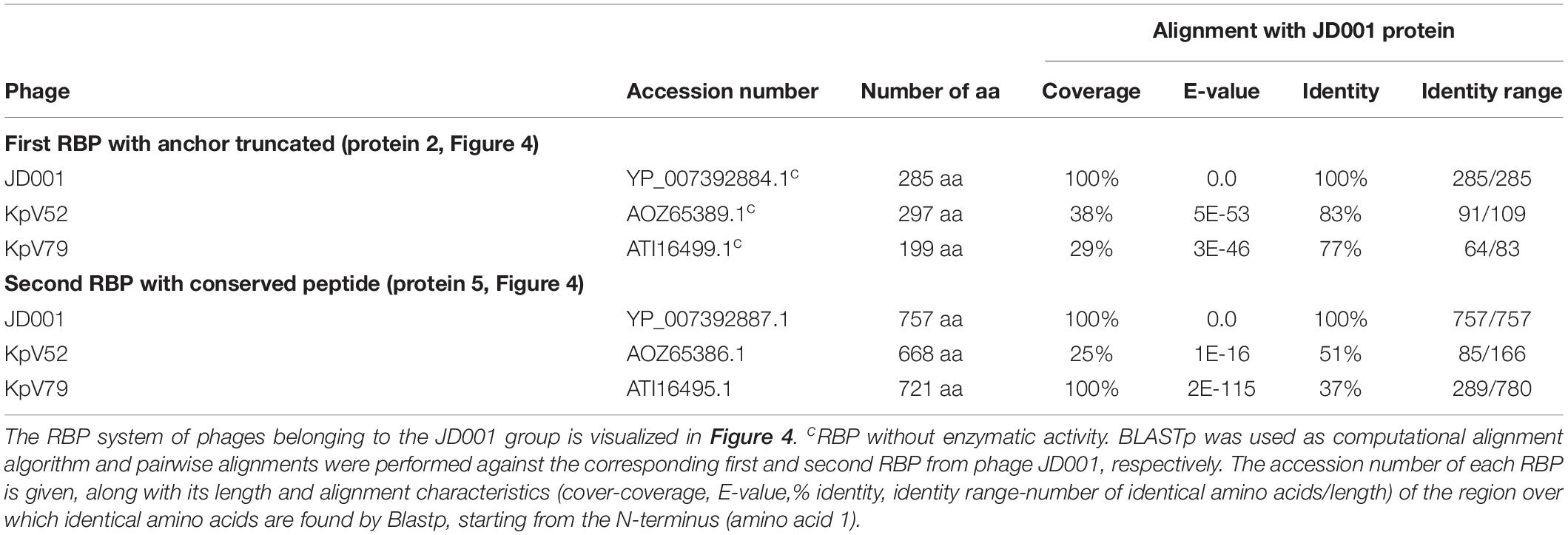
Table 3. Overview of RBPs of phages belonging to the JD001 group and with (predicted) depolymerase activity.
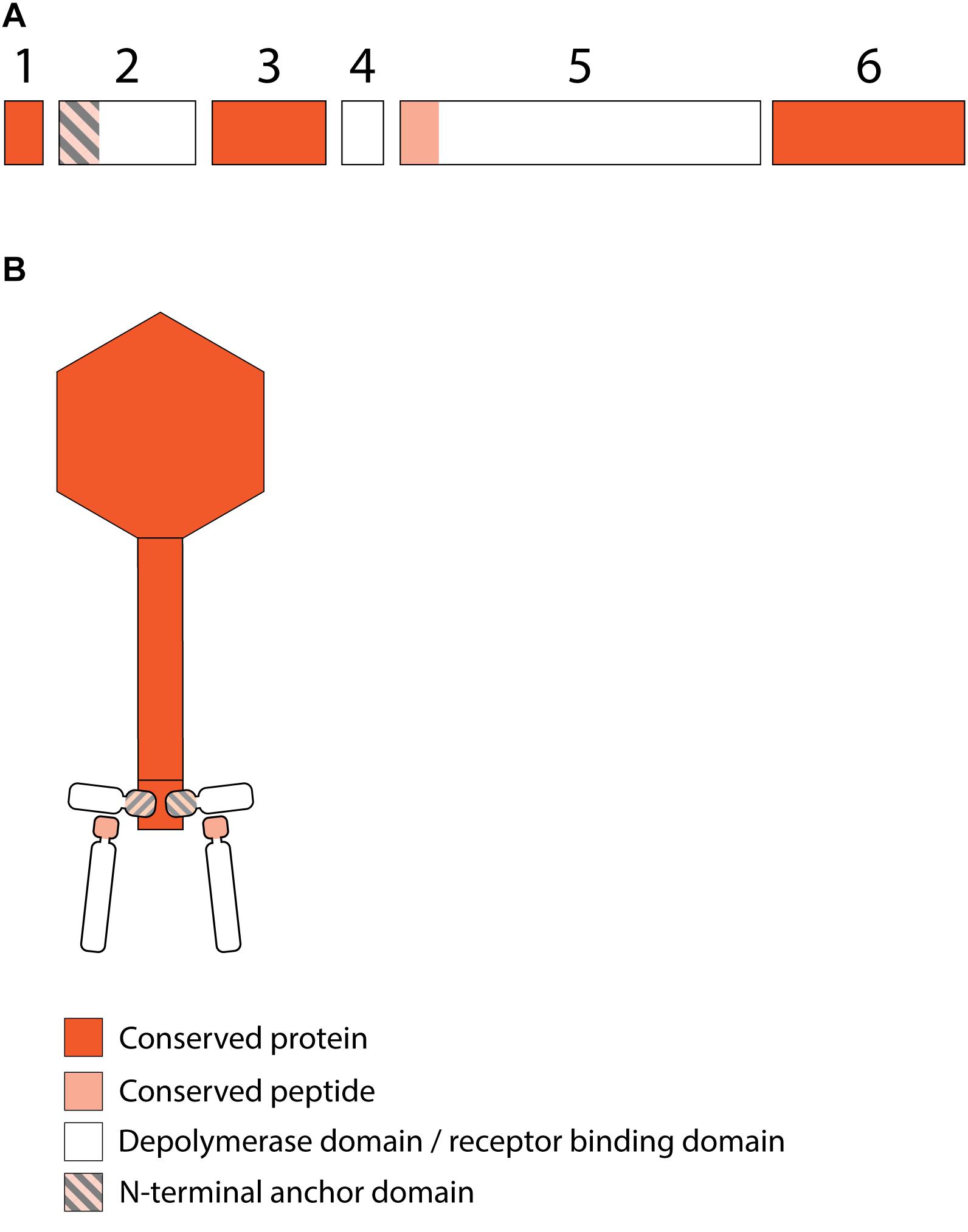
Figure 4. Receptor binding protein systems of viruses of the JD001 group. Phages and their RBPs that are proposed to follow this system are summarized in Table 3. (A) The modular composition of RBP genes is shown relative to the broken gene synteny of JD001. Two flanking conserved hypothetical genes are shown. Annotations are given according to GenBank or according to their modeled function in this study (between brackets): (1) – hypothetical protein; (2) – putative tail fiber protein (anchor); (3) – hypothetical protein; (4) – hypothetical protein; (5) – gluconolaconase (depolymerase with conserved peptide); (6) – hypothetical protein. (B) Schematic model of the RBP system in JD001 group with an anchor-branch attachment mode and the first RBP truncated.
Viruses Belonging to the Menlow Group
The viruses of the Menlow group encode, amid a conserved synteny of structural genes, four non-conserved putative RBPs and one conserved RBP, all with putative depolymerase activity (Figure 5A). Phages KpS110 and 0507-KN2-1 encode an additional sixth RBP with a predicted depolymerase domain (Table 4). The first two non-conserved RBPs (protein 2 and 3 in Figure 5) have N-terminal domains of 412 and 195 aa long, respectively, which is conserved among the four members of the Menlow group. The following two non-conserved RBPs have a shorter domain/peptide of 38 and 67 aa, respectively, conserved among all members of the Menlow group (Supplementary Figures S1E–H).
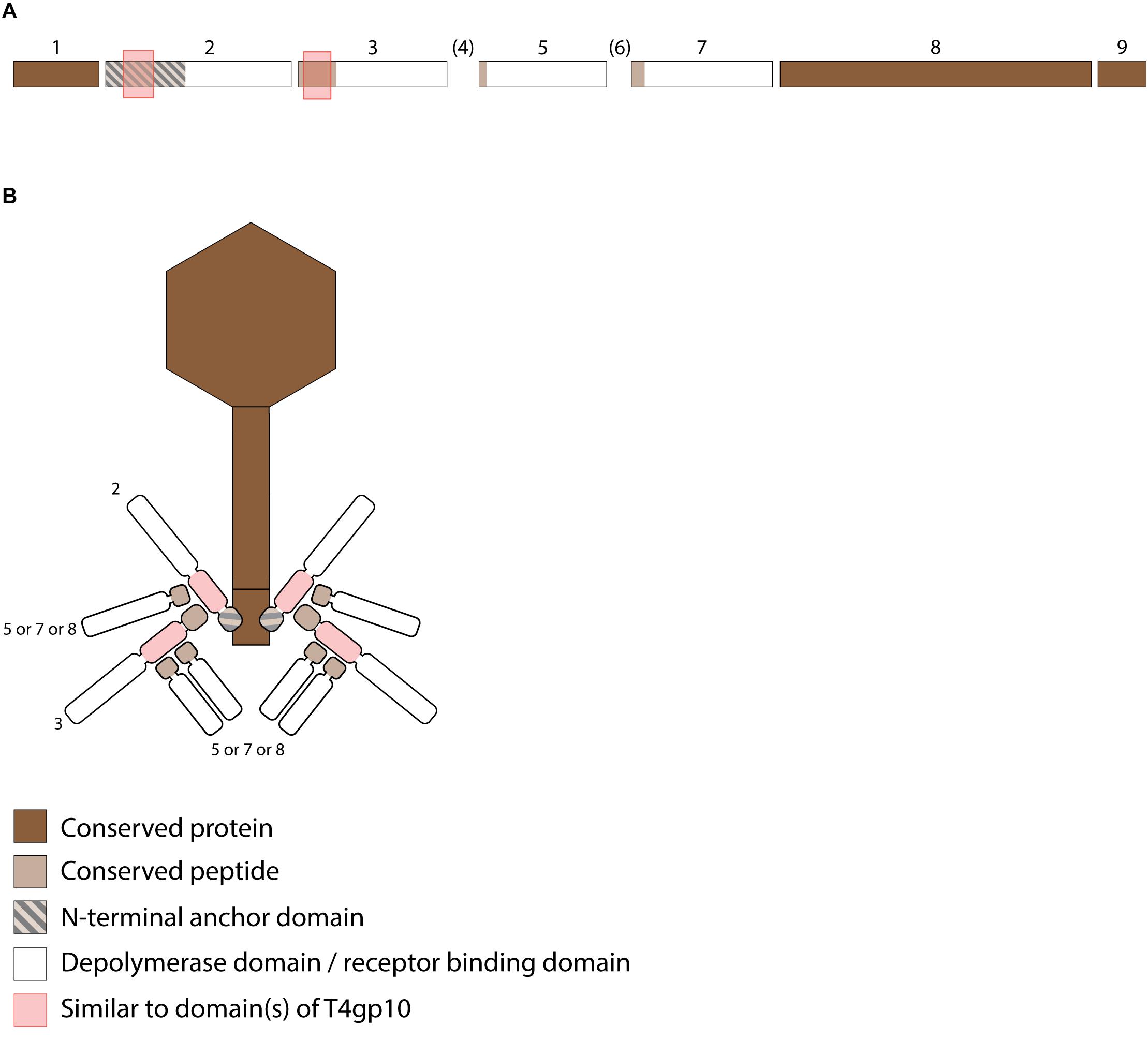
Figure 5. Receptor binding protein systems of the Menlow group. Phages and their RBPs that are proposed to follow this system are summarized in Table 4. (A) The modular composition of the RBP genes is shown relative to the broken gene synteny of Menlow. Annotations are given according to GenBank or according to their modeled function as annotated in this study (between brackets): (1) – putative tail protein; (2) – tail spike protein (anchor with depolymerase); (3) – tail spike protein (depolymerase with conserved peptide); (4) – hypothetical protein; this protein is not present in all Menlow group phages; (5) – tail spike protein (depolymerase with conserved peptide); (6) – hypothetical protein; this protein is not present in all Menlow group phages; (7) – tail spike protein (depolymerase with conserved peptide); (8) – hypothetical protein (depolymerase); (9) – neck protein. (B) Schematic model of the RBP system in Menlow with an anchor-mulibranched attachment mode.
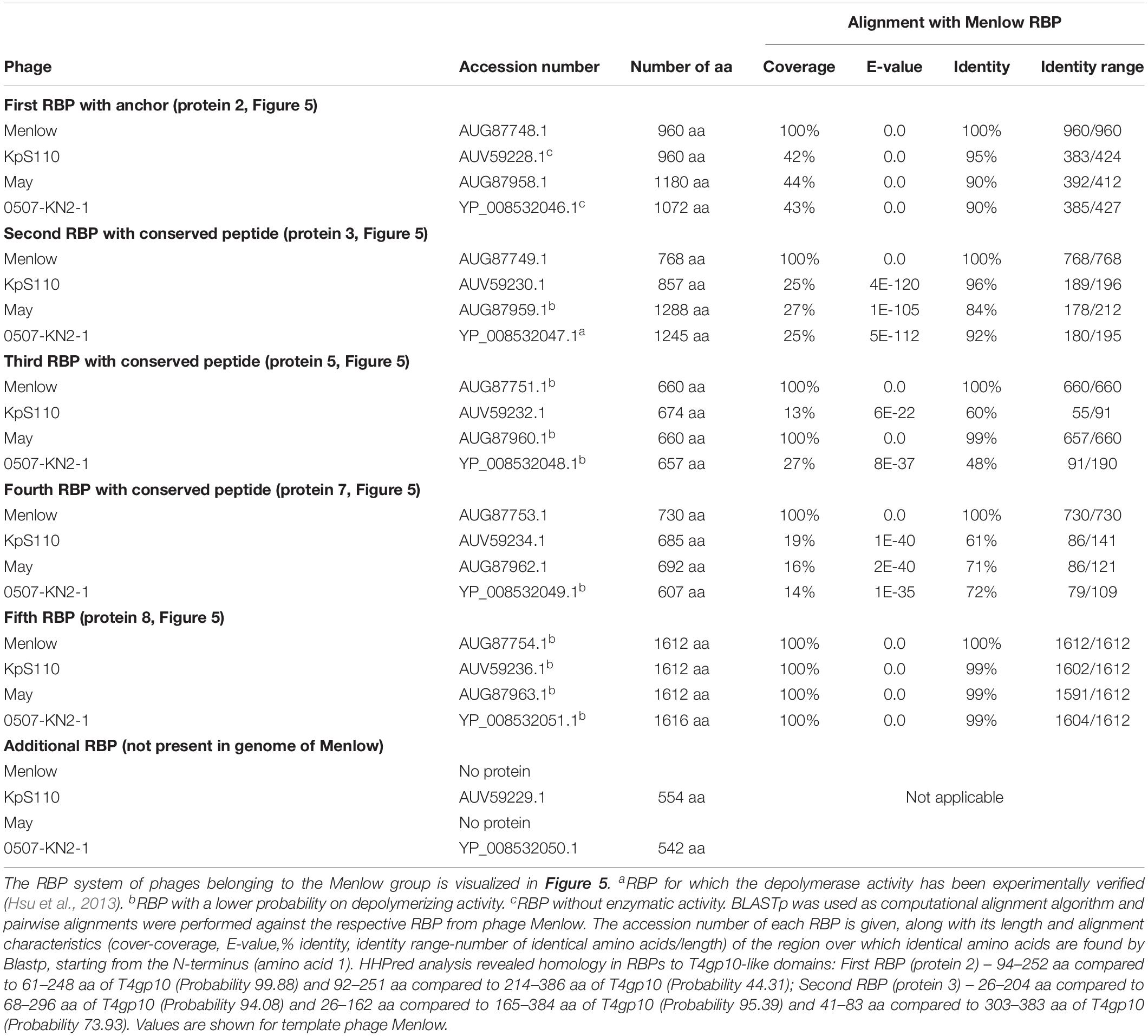
Table 4. Overview of RBPs of phages belonging to the Menlow group and with (predicted) depolymerase activity.
To explore how this high number of putative RBPs might be structurally organized, we searched for homology to T4gp10-like domains 2/3 and N-terminally conserved domains/peptides as they suggest branching points. Two domains homologous to T4gp10 were located in the N-terminal part of RBP 2 (RBP with anchor domain) and RBP 3, whereas RBPs 3, 5, 7 (Figure 5A) contain conserved peptides in their N-terminus. A fifth RBP (protein 8 is present and highly identical) in all members of the Menlow group, while a sixth RBP with putative depolymerase activity is only present in phage KpS110 and phage 0507-KN2-1. Integrating the presence/absence of these structural elements (Figure 5B) a possible model implies that the first RBP (protein 2, Figure 5A) is directly attached to the tail via a conserved N-terminal anchor and that its T4gp10-like domain probably provides an attachment site for at least two RBPs (3 and 5 or 7 or 8, Figure 5B). Subsequently, the second RBP (protein 3) provides attachment sites via its T4gp10-like domains for two more RBPs (proteins 5 or 7 or 8). Together they constitute a unit of branched tail fibers. The highly conserved fifth RBP (protein 8) may be the central tail fiber that protrudes from below the plane of the baseplate (Nobrega et al., 2018). More structural and genetic studies will be needed for an improved understanding of the elaborated RBP system in viruses from the Menlow group.
Viruses Belonging to the ΦK64-1 Group
Klebsiella phages belonging to ΦK64-1 group (ΦK61-1 and RaK2; Supplementary Table S1E) have likely evolved the most elaborate RBP apparatus (Table 5). ΦK64-1 encodes 11 proteins recognized as putative depolymerases, while in the genome of RaK2 10 putative depolymerases are predicted. The middle and C-terminal regions of five RBPs are different between the corresponding genes of ΦK61-1 and RaK2, reflecting the diversity of capsular serotypes that can be recognized by putative depolymerases of these two phages, whereas the middle and C-terminal parts of other RBPs show more than 75% identity between both phages, suggesting an overlap in the host spectrum (Pan et al., 2017). We found in this study that these proteins also contain a slew of structural elements found in other complex tail fiber machineries such as one N-terminal anchor domain, four short conserved peptides at the N-terminus and five T4gp10-like domains (Supplementary Figures S1I–M), indicating that phages of the ΦK64-1 group also re-use standardized units to build up a highly complex RBP apparatus (Figure 6).
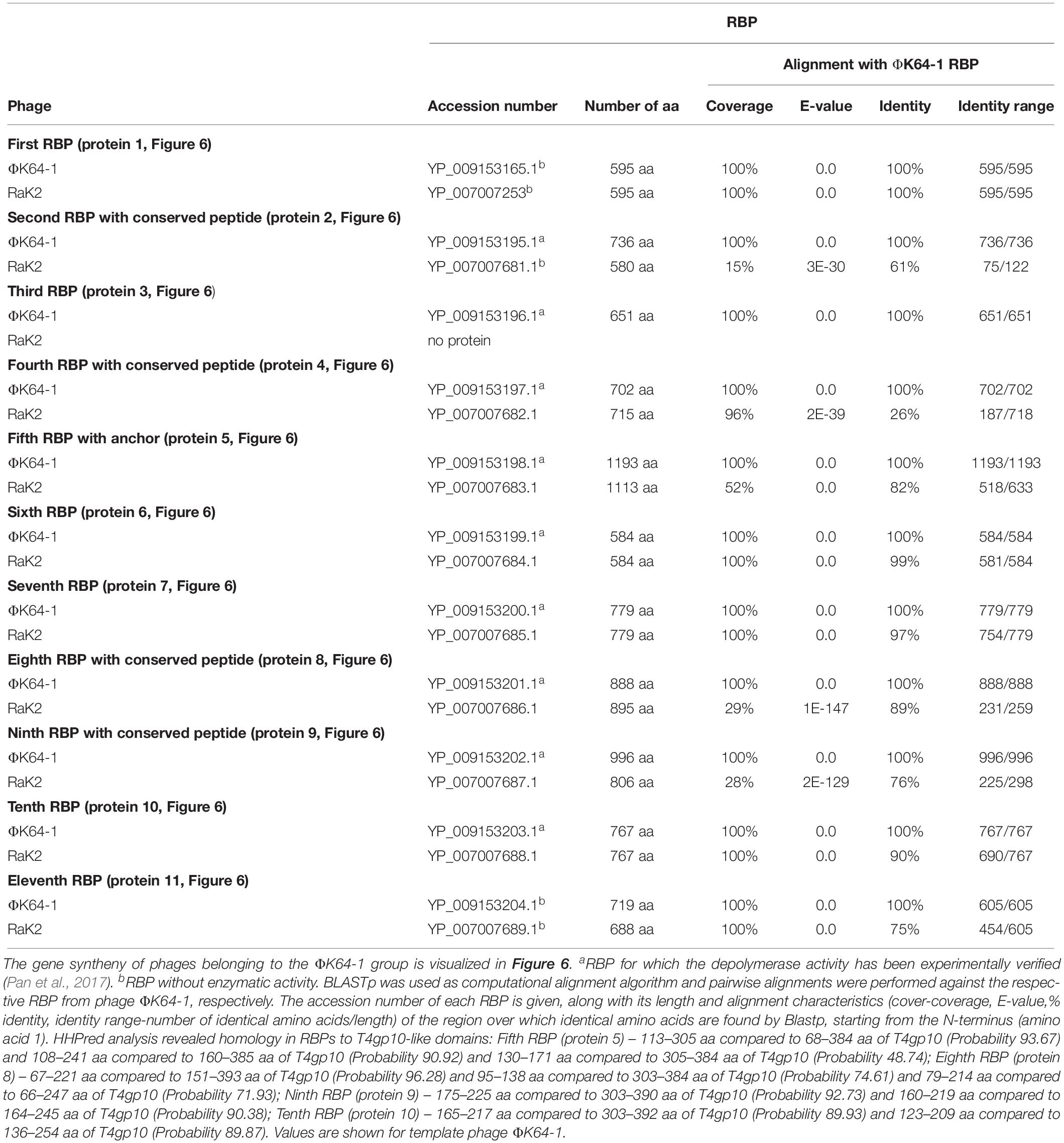
Table 5. Overview of RBPs of phages belonging to the ΦK64-1 group and with (predicted) depolymerase activity.
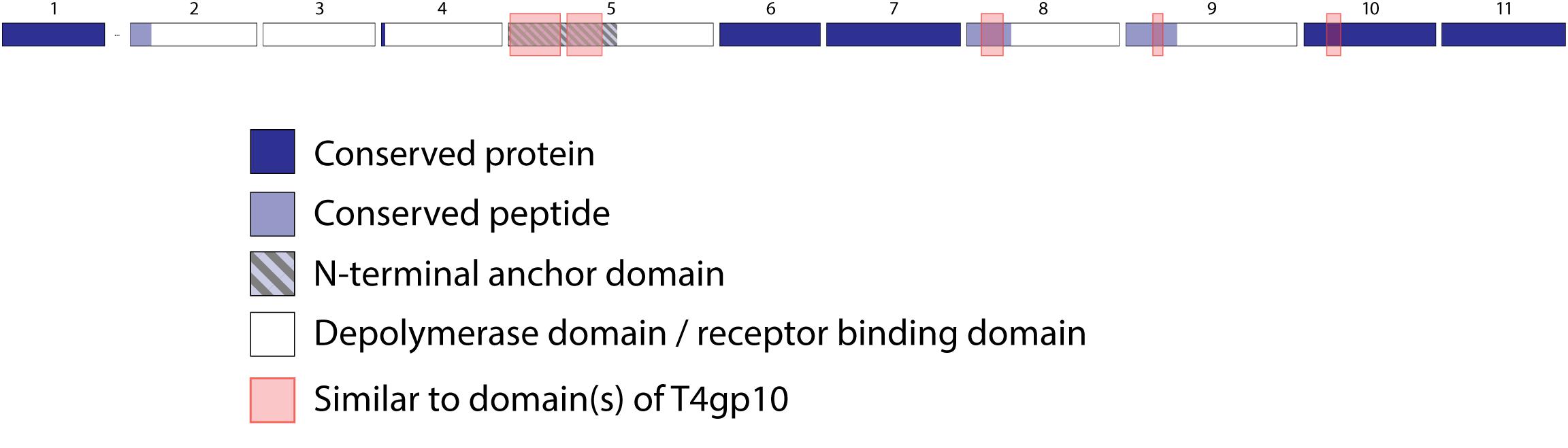
Figure 6. Receptor binding protein systems of phage ΦK64-1 and RaK2. Phages and their RBPs that are proposed to follow this system are summarized in Table 5. The modular composition of RBP genes is shown relative to the broken gene synteny. Annotations are given according to GenBank or according to their modeled function in this study (between brackets): (1) – putative tail fiber protein (depolymerase); (2) – tail spike protein (depolymerase with conserved peptide); (3) – tail spike protein (depolymerase); (4)– putative tail fiber protein (depolymerase with conserved peptide); (5) – putative tail fiber protein (anchor with depolymerase); (6) – putative tail fiber protein (depolymerase); (7) – putative structural protein (depolymerase); (8) – putative tail fiber protein (depolymerase with conserved peptide); (9) – putative tail fiber protein (depolymerase with conserved peptide); (10) – putative structural protein (depolymerase); (11) – putative tail fiber protein (depolymerase).
RBPs From Klebsiella Siphoviruses
KP36viruses
All 12 identified Klebsiella siphoviruses belong to the KP36viruses. They are also featured by a synteny of genes encoding structural proteins such as the tail length tape-measure protein, minor tail proteins and a putative tail assembly protein. This synteny is disrupted by one or two genes, depending on the phage. Three groups can be categorized with the majority of phages belonging to group A, while phage PKP126 (group B) and phage 1513 (group C) represent exceptions from the general structure of group A (Figure 7A and Table 6). Members of group A KP36viruses (including the reference phage KP36) have a single predicted RBP with putative depolymerase activity. It has been demonstrated that the RBP of KP36 is enzymatically active against capsular serotype K63 (Majkowska-Skrobek et al., 2016). The modular structure of this RBP is similar to that of the RBP of group B KP32viruses, having an N-terminal anchor domain, a highly variable central domain with enzymatic activity, and a C-terminal chaperone. KP36viruses belonging to group B and group C also have an RBP with a similar N-terminal anchor domain (Supplementary Figure S1N). Phage PKP126 RBP (group B) has a predicted enzymatic activity in the central domain in contrast to the truncated RBP of phage 1513 (group C). The chaperone domain is missing in the RBP of both groups B and C.
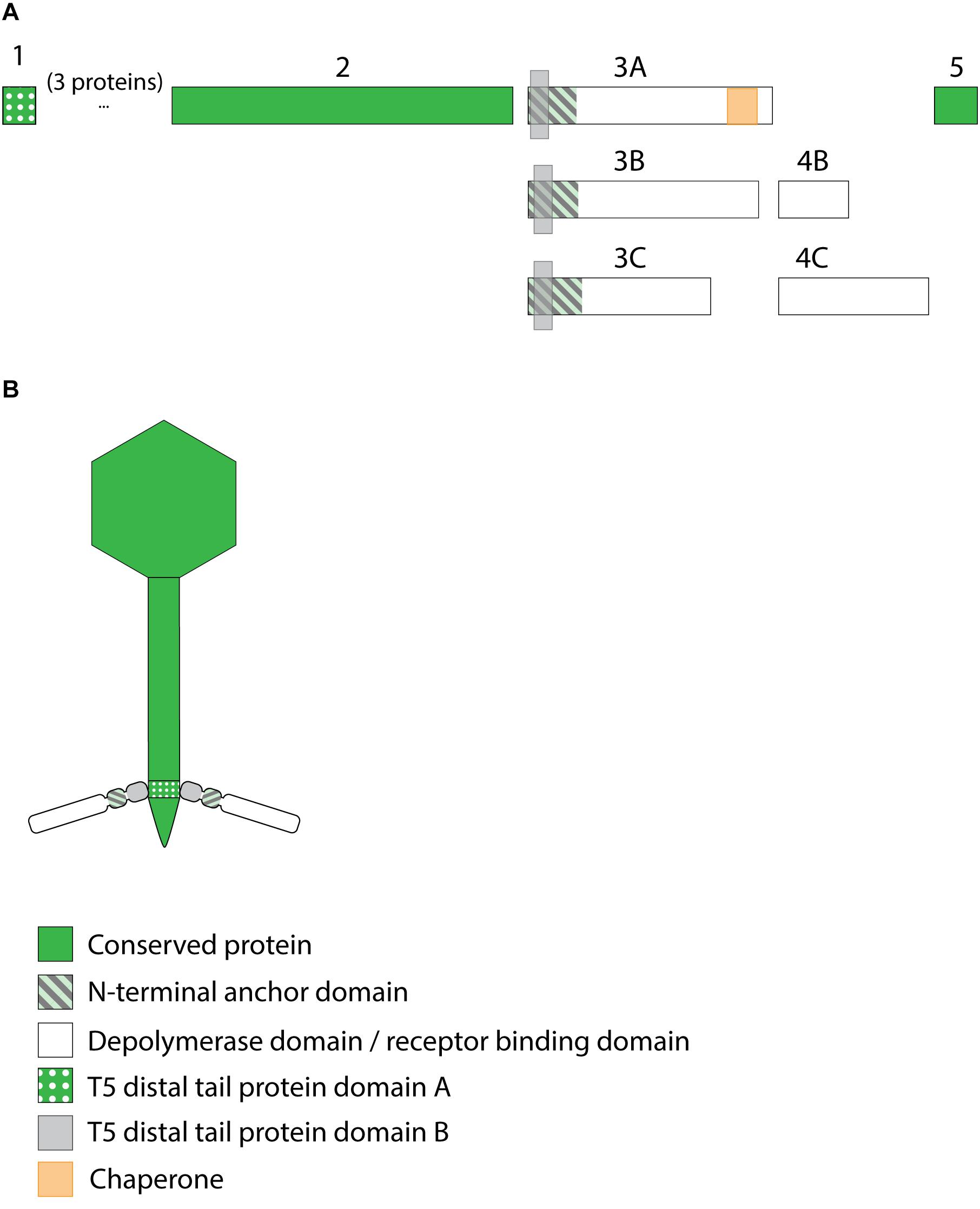
Figure 7. Receptor binding protein systems of the KP36viruses. Phages and their RBPs that are proposed to follow this system, including their grouping into groups A, B and C, are summarized in Table 6. (A) The modular composition of the RBP genes is shown relative to the broken gene synteny of the reference phage KP36. Annotations are given according to GenBank or according to their modeled function in this study (between brackets): (1) – minor tail protein; (2) – tail fiber protein; (3) – putative tail fiber protein (3A,B – anchor with depolymerase; 3C – anchor); (4) – hypothetical protein (4B,C – depolymerase); (5) – putative single-stranded DNA binding protein. Proteins 1, 2, and 5 are present in all KP36viruses. (B) Schematic model of the RBP system in phage particles of KP36viruses group A with a split T5 distal tail protein. Domain A is encoded by short minor tail protein forming a ring at the end of the phage tail tube and offers an attachment point for domain B, which is incorporated in the anchoring part of the RBP that represents the putative side tail fiber.
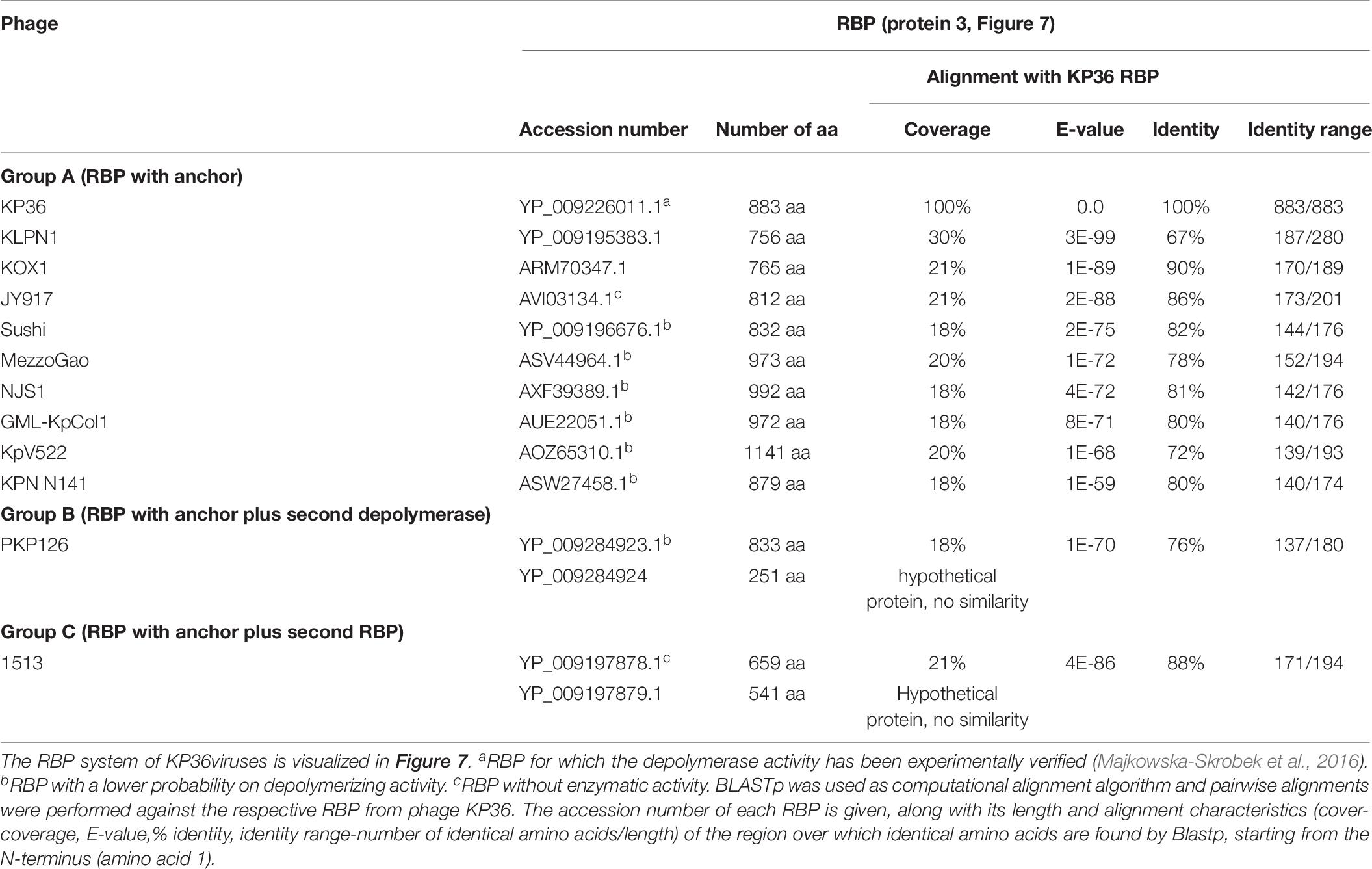
Table 6. Overview of RBPs of phages belonging to the KP36viruses and with (predicted) depolymerase activity.
No T4gp10-like domain was found in the N-terminal region of KP36gp50 (RBP). Instead, a small domain (residues 4–63) homologous to domain B (92–155 aa) of the distal tail protein (Dit or T5pb9) of siphovirus T5 has been detected. Dit is located in the T5 tail tip at the junction between the tail tube and the ultimate conical structure and is composed of two domains. Domain A forms a hexameric structure and connects to the end of the tail tube, whereas domain B constitutes the attachment site for three L-shaped tail fibers (Flayhan et al., 2014). These L-shaped tail fibers initially bind reversibly to polymannose containing O-antigens (Heller and Braun, 1982). Remarkably, domain A of T5pb9 has not been found in KP36gp50 but is instead present in the KP36 minor tail protein (residues 22–77 corresponding to amino acids 27–85 of T5pb9) that is encoded four genes upstream of KP36gp50. This horizontal transfer event indicates that in KP36viruses the conserved minor tail protein only comprises domain A, which is located at the junction of the tail tube and the conical tip of the tail. Domain A of the minor tail protein is proposed to interact with the RBP via its N-terminal domain B. This RBP may thus represent the side tail fibers similar to the L-shaped tail fibers in phage T5. In other words, the distal tail protein has been split into two separate elements in KP36viruses.
Phage PKP126 and 1513 (groups B and C, respectively) have an additional RBP with putative depolymerase activity. Its exact role is difficult to predict and typical elements hinting at a specific structural organization such as a conserved peptide or anchor domain are missing. We hypothesize that those enzymes are not incorporated in the phage particle, but rather are produced as soluble proteins. Upon cell lysis the neighboring cells are sensitized for infection through enzymatic removal of the capsule by the soluble, diffusible depolymerase. This mechanism would be especially beneficial for phages lacking depolymerase activity in the their first RBP (e.g., group C phage 1513).
An additional preceding RBP (Figure 7A; protein 2) is highly conserved across all analyzed KP36viruses, except in phage 1513. The role of this RBP is unclear. One possibility is that it is a second side RBP as observed in some T5viruses (DT57C and DT571) (Golomidova et al., 2016; Nobrega et al., 2018). An alternative possibility is that this protein represents the central tail fiber. Given its ambiguous role and location, this RBP was not included in the model depicted in Figure 7B.
Discussion
In this work we have performed an extensive in silico analysis of the RBPs of Klebsiella phages genomes with a focus on RBPs with depolymerase activity. The tripartite relationship between depolymerase specificity, capsular serotype and phage host spectrum has now extensively been demonstrated for Klebsiella phages (Hsu et al., 2013; Lin et al., 2014; Majkowska-Skrobek et al., 2016; Hsieh et al., 2017; Pan et al., 2017; Solovieva et al., 2018). Podovirus KP32 possesses two experimentally confirmed depolymerases, which are enzymatically active against capsule serotype K3 and K21, respectively. Correspondingly, all strains infected by phage KP32 have either a K3 or K21 serotype (Majkowska-Skrobek et al., 2018). Podovirus KpV71 infects strains with serotype K1, which perfectly matches the specificity of its experimentally verified depolymerase. However, podovirus KpV74, which has also a single RBP, infects strains with serotype K2 and K13. These observations were explained by the structural similarity of capsule types K2 and K13, which were also found to cross-react with specific antibodies (Pieroni et al., 1994; Pan et al., 2015; Volozhantsev et al., 2016; Solovieva et al., 2018). The more diverse capsular specificity of podoviruses KpV763, KpV766, and KpV289 (Volozhantsev et al., 2016; Solovieva et al., 2018) is now explained in this study by the observed presence of two RBPs (Table 1). Some large jumbo phages such as phage ΦK64-1 produce an elaborated, broad-spectrum RBP apparatus. Phage ΦK64-1 encodes 11 putative RBPs from which nine are confirmed to possess enzymatic activity against 10 serotypes in total (K1, K11, K21, K25, K30/K69, K35, K64, KN4, and KN5). Whereas eight RBPs are active against a single but different serotype, the ninth RBP is active against two capsular serotypes (protein 10, Figure 6; active against K30 and K69) (Pan et al., 2017).
Based on the structural knowledge of RBPs of mainly E. coli phages such as T7, K1F, K1-5, G7C and T5, we have identified structurally conserved building blocks to model the RBP apparatus of Klebsiella phages. The modularity of RBPs in combination with intensive horizontal transfer of genes or gene domains (Casjens and Molineux, 2012) allows for a maximum re-use of conserved, evolutionary optimized elements. Simultaneously, the possibility to rapidly shift the host spectrum based on an exchange of the depolymerase domain is retained. Indeed, specific RBP domains, sometimes in pair with their cognate chaperone, are present in each RBP system. This is well-illustrated by the high similarity of the experimentally verified depolymerase domains of KP36gp50 and KP34gp57. Both proteins target capsular serotype K63, but have either an N-terminal anchor or conserved peptide, respectively.
The high adaptability of Klebsiella phage RBPs is essential since K. pneumoniae is featured by a high capsular diversity. Consequently, Klebsiella phages have often a very narrow spectrum limited to strains from one or two capsular serotypes. Colonization of new niches occupied by K. pneumoniae isolates with a different capsular serotype thus necessitates a flexible system for rapid adaptation. In addition, the same flexibility is needed to respond to phenotypic serotype switches of K. pneumoniae strains (Pan et al., 2015; Wyres et al., 2015).
In this study, we propose that RBPs of Klebsiella phages are organized according to several distinct systems (Figure 8). The simplest mechanism is similar to the anchor-based system described for phage T7 and K1F. In phages from KP32viruses group B and KP36viruses, the single RBP is directly connected with the phage particle via its conserved N-terminal anchor domain. Other phages (KP32viruses group A; KP34 viruses group B) that produce two RBPs encode the structural elements for an anchor-branched mechanism as reported for phage G7C. Here, the first RBP contains a conserved N-terminal anchor serving for attachment to the virion, followed by a specific fragment of a T4gp10-like domain providing the docking site for a second RBP. Notably, the fragment encoding the T4gp10-like docking site in those Klebsiella phages is shorter compared to the corresponding domain in T4 and may therefore correspond to a single attachment site. The second RBP is presumably attached via a conserved peptide (KP32viruses, KP34viruses, JD001 group, Menlow group, ΦK64-1 group). This conserved peptide is different for each group of phages, varies in length and can be as short as seven amino acids. Such attachment via a short peptide is in line with the RBP complex of K1E/K1-5/SP6-like phages where both RBPs carry either a 7- or 11-residue conserved peptide at their respective N-terminus. In the case of E. coli phage G7C the shorter G7Cgp63.1 RBP carries a positively charged surface that binds to the T4gp10-like domain of G7Cgp66, yet, the conserved peptides in Klebsiella phages lack this positive charge, inferring that different interacting forces take place between the first and second RBP. Similar to the RBPs of phage K1-5, the two experimentally verified depolymerases of phage KP32 target two different capsular serotypes. In both cases the double RBP system thus expands the host spectrum.
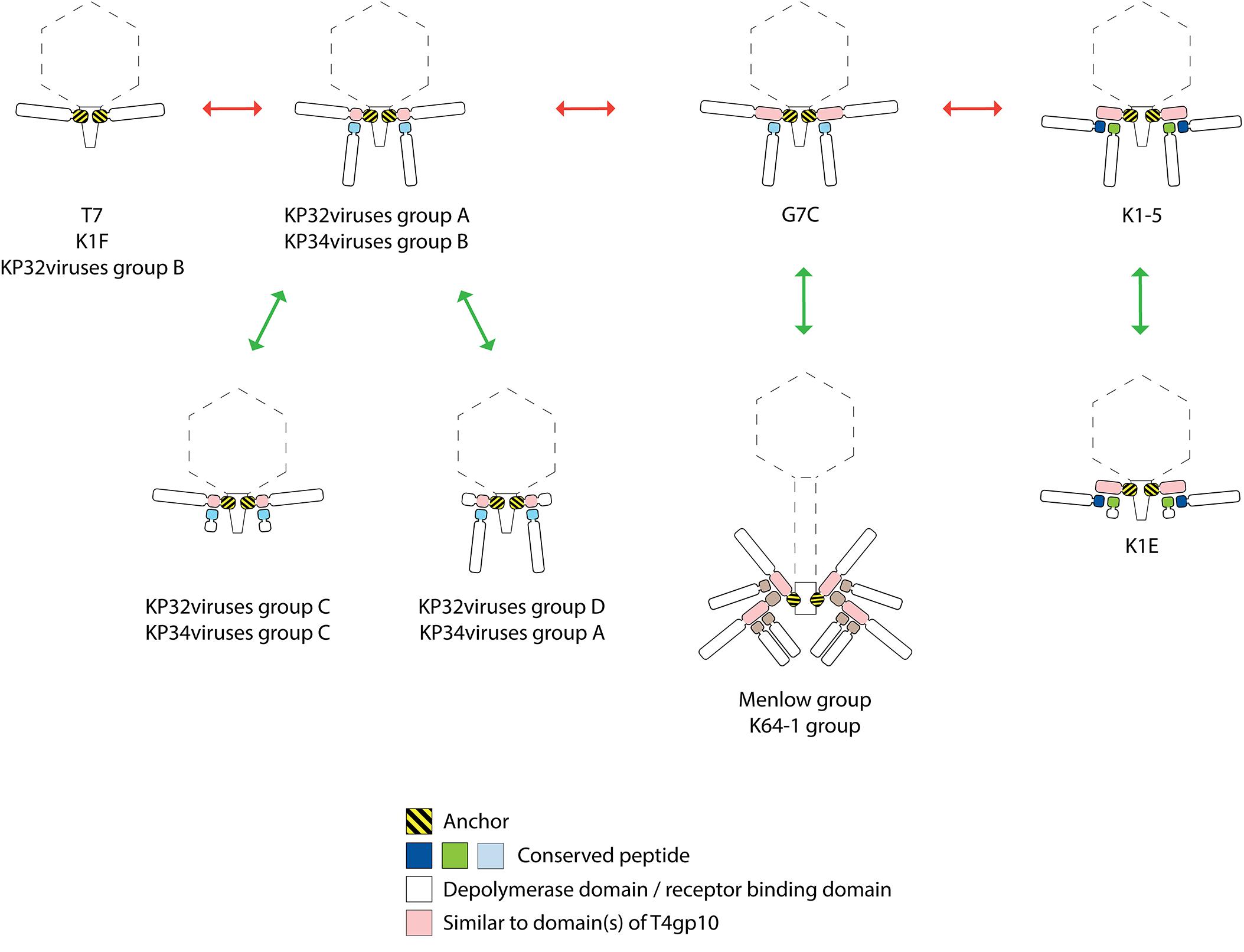
Figure 8. Possible evolutionary linkages between diverse RBP systems, driven by the acquisition and loss of domains. The model RBP systems of phage T7, K1F, G7C, K1-5, and K1-E have been reported before. The RBP systems of KP32viruses groups A, B, C, D (Figure 2 and Table 1), KP34viruses groups A, B, C (Figure 3 and Table 2), phages belonging to the Menlow group (Figure 5 and Table 4) and phage belonging to the ΦK64-1 group (Figure 6 and Table 5) have been studied in this work.
The presence of either an anchor- or an anchor-branched system is not directly linked to the taxonomic organization. In addition, there is no sequence homology between functionally similar, structural building blocks across those phage groups. E.g., the first 140 amino acids of the N-terminal anchor domains of RBPs encoded by Klebsiella phages belonging to Podoviridae show similarity with the well-characterized N-terminal domain of T7 tail fiber and their first 300 amino acids are conserved across different podoviruses analyzed in this study. The T7 tail fiber attaches with its N-terminal anchor domain to the region where the adaptor (gp11) interacts with nozzle (gp12) of the short tail complex (Cuervo et al., 2013). The corresponding proteins of phage KP32 share 62 and 61% sequence identity with the adaptor and nozzle protein of phage T7, respectively, while in the case of KP34 the identity is lower (29% identity with a coverage of 67% for the adaptor protein and 23% identity with a coverage of 98% for the nozzle protein). There is no amino acid similarity between the conserved N-terminal anchor domains in RBPs from different taxonomic groups of Klebsiella phages indicating that also the interacting partner in the tail structure has also evolved accordingly. In the case of KP36viruses (Siphoviridae), a remarkable horizontal transfer event has taken place between the distal tail protein and the tail fiber of KP36viruses when comparing to the siphovirus T5 model. Domain B of the distal tail protein has been transferred to the N-terminus of the tail fiber protein in KP36viruses. Whereas in phage T5 protein–protein interaction occurs between the N-terminus of the RBP and domain B of the distal tail protein, novel interactions between domain A of the minor tail protein and domain B embedded in the tail fiber must have been evolved to compensate for the loss of interaction by a direct covalent bond as in phage T5.
Interestingly, several phages with a single enzymatic RBP do not follow the anchor system as described for phage T7, but use the anchor-branched system of G7C with either the first (KP32virus group D; KP34virus group A; JD001 group) or second RBP (KP32virus group C; KP34virus group C) being truncated. In the case of KP34viruses it is even the predominant RBP system. The occurrence of these intermediate RBP systems suggests evolutionary linkages between the different RBP architectures. Starting from the simplest organization with a single RBP (T7, K1F, and KP32viruses group B), the acquisition of a fragment of a T4gp10-like domain allowed for the attachment of a second RBP (KP32viruses group A and KP34viruses group B). The first RBP from E. coli phage G7C has acquired a full T4gp10-like domain (similarity to both subdomain D2 and D3 of T4gp10), offering a potential second attachment site for a different RBP. This second site is not occupied in phage G7C, whereas the E. coli model phages K1-5, SP6, and K1E have effectively two RBPs attached to the same intermediate protein that also comprises both subdomain D2 and D3. In K1-5, SP6 and K1E, this intermediate protein with the full T4gp10-like domain has lost its C-terminal receptor-binding domain, resulting in an ‘adapter’ system – a short protein with two sites for binding two different RBPs and no domain beyond these two domains (Figure 8). It should be noted that a simple adapter protein that provides attachment sites for two RBPs as described for E. coli phage K1-5 and Salmonella phage SP6 is not observed in the case of Klebsiella podoviruses. Obviously, an opposite evolutionary trajectory of RBP systems (from adapter to anchor) cannot be excluded as well. The success of the modular build-up of the RBP apparatus and the extensive number of horizontal transfer events have obscured possible insight in the direction of this evolution. The assumption that evolution generally takes place from simple to more complex systems, hints at the first direction (from anchor to adapter). KP32viruses group C and D, and KP34viruses groups A and C may have lost a second intact RBP by retrograde evolution when thriving in a new environment that is dominated by a single serotype Klebsiella strain. Having a truncated second RBP may provide a fitness advantage in such a situation. The truncated RBP may remain as a temporal docking site to acquire a new RBP for host range expansion by horizontal transfer when moving to a niche with different Klebsiella serotypes. Phages belonging to the Menlow group and ΦK64-1 group carry multiple RBPs that obviously recycle established structural elements such as a conserved N-terminal domain, short conserved peptides and a T4gp10-like domain (or fragments thereof). Yet, more experimental (structural, genetic, biochemical) studies are required to make a plausible prediction on the structural organization of these elaborated RBP systems.
In summary, we have modeled the organization of diverse RBP systems in Klebsiella phages. The modular composition and re-use of established structural domains for anchoring and branching provide the phages the full potential to rapidly shift capsular serotype specificity or to expand the spectrum. We expect that the increasing amount of (meta)genome sequencing data will reveal further evolutionary relationships between some of the groups we describe in this analysis, but the main groups will remain in place. The data available to us today clearly show that the architecture of RBP systems is dominated by horizontal transfer events of modules that can be as small as short peptides to as large as multiple domains. Although our analysis was based on experimentally confirmed interactions of E. coli phage RBPs (Leiman et al., 2007; Prokhorov et al., 2017), further experimental validation of the presented models is needed and has already been initiated by our team. To analyze the interactions between the T4gp10-like domains and conserved peptides, protein–protein interactions can be studied by various techniques such as isothermal calorimetry (ITC), two-hybrid systems, surface plasmon resonance (SPR), cryoEM, or an enzyme-linked immunosorbent assay (ELISA). This work also adds improved functional annotations to genes to which previously no specific function has been assigned, but which are putative tail fibers/spikes with depolymerizing activity. The high number of newly predicted depolymerases in this study can be verified by their recombinant production followed by activity tests against strains with particular capsular serotypes.
Data Availability Statement
The datasets analyzed for this study can be found in the GenBank. All genome and protein accession numbers are listed in the tables and Supplementary Material.
Author Contributions
AL, ZD-K, and YB designed the in silico search approach of RBPs domains and modeled the RBP assembly systems. PL introduced the concept of branching RBPs. AL performed in silico analysis and collected the data. AL, PL, ZD-K, and YB analyzed the data and worked on the manuscript. All authors read and accepted the final version of the manuscript.
Funding
This study was supported by National Science Centre, Poland (Grant Numbers UMO-2015/19/N/NZ1/00014 to AL, ZD-K, UMO-2017/26/M/NZ1/00233 to ZD-K, AL) and by a doctoral scholarship of “Bijzonder Onderzoeksfonds” of Ghent University (Grant Number BOF16/FJD/007 to AL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02649/full#supplementary-material
Footnotes
- ^ blast.ncbi.nlm.nih.gov
- ^ sbg.bio.ic.ac.uk
- ^ swissmodel.expasy.org
- ^ hmmer.org
- ^ toolkit.tuebingen.mpg.de
- ^ weblogo.berkeley.edu
References
Adriaenssens, E. M., Ackermann, H. W., Anany, H., Blasdel, B., Connerton, I. F., Goulding, D., et al. (2012). A suggested new bacteriophage genus: “Viunalikevirus”. Arch. Virol. 157, 2035–2046. doi: 10.1007/s00705-012-1360-1365
Adriaenssens, E. M., Wittmann, J., Kuhn, J. H., Turner, D., Sullivan, M. B., Dutilh, B. E., et al. (2018). Taxonomy of prokaryotic viruses: 2017 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 163, 1125–1129. doi: 10.1007/s00705-018-3723-z
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-80362
Andres, D., Baxa, U., Hanke, C., Seckler, R., and Barbirz, S. (2010). Carbohydrate binding of Salmonella phage P22 tailspike protein and its role during host cell infection. Biochem. Soc. Trans. 38, 1386–1389. doi: 10.1042/BST0381386
Barbirz, S., Muller, J. J., Uetrecht, C., Clark, A. J., Heinemann, U., and Seckler, R. (2008). Crystal structure of Escherichia coli phage HK620 tailspike: podoviral tailspike endoglycosidase modules are evolutionarily related. Mol. Microbiol. 69, 303–316. doi: 10.1111/j.1365-2958.2008.06311.x
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Bordoli, L., Kiefer, F., Arnold, K., Benkert, P., Battey, J., and Schwede, T. (2009). Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13. doi: 10.1038/nprot.2008.197
Bordoli, L., and Schwede, T. (2012). Automated protein structure modeling with SWISS-MODEL workspace and the protein model portal. Methods Mol. Biol. 857, 107–136. doi: 10.1007/978-1-61779-588-6-5
Broeker, N. K., Kiele, F., Casjens, S. R., Gilcrease, E. B., Thalhammer, A., Koetz, J., et al. (2018). In Vitro studies of lipopolysaccharide-mediated DNA release of podovirus HK620. Viruses 10:E289. doi: 10.3390/v10060289
Calfee, D. P. (2017). Recent advances in the understanding and management of Klebsiella pneumoniae. F1000Res 6:1760. doi: 10.12688/f1000research.11532.1
Casjens, S. R., and Molineux, I. J. (2012). Short noncontractile tail machines: adsorption and DNA delivery by podoviruses. Adv. Exp. Med. Biol. 726, 143–179. doi: 10.1007/978-1-4614-0980-9-7
Chen, Z., Liu, M., Cui, Y., Wang, L., Zhang, Y., Qiu, J., et al. (2014). A novel PCR-based genotyping scheme for clinical Klebsiella pneumoniae. Future Microbiol. 9, 21–32. doi: 10.2217/fmb.13.137
Cornelissen, A., Ceyssens, P. J., T’Syen, J., Van Praet, H., Noben, J. P., Shaburova, O. V., et al. (2011). The T7-related Pseudomonas putida phage phi 15 displays virion-associated biofilm degradation properties. PLoS One 6:e18597. doi: 10.1371/journal.pone.0018597
Cortés, G., Borrell, N., de Astorza, B., Gómez, C., Sauleda, J., and Albertí, S. (2002). Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 70, 2583–2590. doi: 10.1128/iai.70.5.2583-2590.2002
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Cuervo, A., Pulido-Cid, M., Chagoyen, M., Arranz, R., González-García, V. A., Garcia-Doval, C., et al. (2013). Structural characterization of the bacteriophage T7 tail machinery. J. Biol. Chem. 288, 26290–26299. doi: 10.1074/jbc.M113.491209
Davies, G., and Henrissat, B. (1995). Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859. doi: 10.1016/S0969-2126(01)00220-229
Day, A., Ahn, J., and Salmond, G. P. C. (2018). Jumbo bacteriophages are represented within an increasing diversity of environmental viruses infecting the emerging phytopathogen, dickeya solani. Front. Microbiol. 9:2169. doi: 10.3389/fmicb.2018.02169
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Flayhan, A., Vellieux, F. M., Lurz, R., Maury, O., Contreras-Martel, C., and Girard, E. (2014). Crystal structure of pb9, the distal tail protein of bacteriophage T5: a conserved structural motif among all siphophages. J. Virol. 88, 820–828. doi: 10.1128/JVI.02135-2113
Gebhart, D., Williams, S. R., and Scholl, D. (2017). Bacteriophage SP6 encodes a second tailspike protein that recognizes Salmonella enterica serogroups C2 and C3. Virology 507, 263–266. doi: 10.1016/j.virol.2017.02.025
Golomidova, A. K., Kulikov, E. E., Prokhorov, N. S., Guerrero-Ferreira, R., Knirel, Y. A., Kostryukova, E. S., et al. (2016). Branched lateral tail fiber organization in T5-like bacteriophages DT57C and DT571/2 is revealed by genetic and functional analysis. Viruses 8:26. doi: 10.3390/v8010026
Heller, K., and Braun, V. (1982). Polymannose O-antigens of Escherichia coli, the binding sites for the reversible adsorption of bacteriophage T5+ via the L-shaped tail fibers. J. Virol. 41, 222–227.
Hsieh, P. F., Lin, H. H., Lin, T. L., Chen, Y. Y., and Wang, J. T. (2017). Two T7-like bacteriophages, K5-2 and K5-4, each encodes two capsule depolymerases: isolation and functional characterization. Sci. Rep. 7:4624. doi: 10.1038/s41598-017-04644-4642
Hsu, C. R., Lin, T. L., Pan, Y. J., Hsieh, P. F., and Wang, J. T. (2013). Isolation of a bacteriophage specific for a new capsular type of Klebsiella pneumoniae and characterization of its polysaccharide depolymerase. PLoS One 8:e70092. doi: 10.1371/journal.pone.0070092
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Latka, A., Maciejewska, B., Majkowska-Skrobek, G., Briers, Y., and Drulis-Kawa, Z. (2017). Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 101, 3103–3119. doi: 10.1007/s00253-017-8224-8226
Lee, C. R., Lee, J. H., Park, K. S., Jeon, J. H., Kim, Y. B., Cha, C. J., et al. (2017). Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated dete rminants, and resistance mechanisms. Front. Cell Infect. Microbiol. 7:483. doi: 10.3389/fcimb.2017.00483
Leiman, P. G., Battisti, A. J., Bowman, V. D., Stummeyer, K., Mühlenhoff, M., Gerardy-Schahn, R., et al. (2007). The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J. Mol. Biol. 371, 836–849. doi: 10.1016/j.jmb.2007.05.083
Leiman, P. G., and Molineux, I. J. (2008). Evolution of a new enzyme activity from the same motif fold. Mol. Microbiol. 69, 287–290. doi: 10.1111/j.1365-2958.2008.06241.x
Lin, T. L., Hsieh, P. F., Huang, Y. T., Lee, W. C., Tsai, Y. T., Su, P. A., et al. (2014). Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: implication in typing and treatment. J. Infect. Dis. 210, 1734–1744. doi: 10.1093/infdis/jiu332
Majkowska-Skrobek, G., Łątka, A., Berisio, R., Maciejewska, B., Squeglia, F., Romano, M., et al. (2016). Capsule-targeting depolymerase, derived from Klebsiella KP36 phage, as a tool for the development of anti-virulent strategy. Viruses 8:324. doi: 10.3390/v8120324
Majkowska-Skrobek, G., Latka, A., Berisio, R., Squeglia, F., Maciejewska, B., Briers, Y., et al. (2018). Phage-borne depolymerases decrease Klebsiella pneumoniae resistance to innate defense mechanisms. Front. Microbiol. 9:2517. doi: 10.3389/fmicb.2018.02517
Nobrega, F. L., Vlot, M., de Jonge, P. A., Dreesens, L. L., Beaumont, H. J. E., Lavigne, R., et al. (2018). Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 16, 760–773. doi: 10.1038/s41579-018-0070-78
Olszak, T., Shneider, M. M., Latka, A., Maciejewska, B., Browning, C., Sycheva, L. V., et al. (2017). The O-specific polysaccharide lyase from the phage LKA1 tailspike reduces Pseudomonas virulence. Sci. Rep. 7:16302. doi: 10.1038/s41598-017-16411-4.
Pan, Y. J., Lin, T. L., Chen, C. C., Tsai, Y. T., Cheng, Y. H., Chen, Y. Y., et al. (2017). Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J. Virol. 91:e2457-16. doi: 10.1128/JVI.02457-2416
Pan, Y. J., Lin, T. L., Chen, C. T., Chen, Y. Y., Hsieh, P. F., Hsu, C. R., et al. (2015). Genetic analysis of capsular polysaccharide synthesis gene clusters in 79 capsular types of Klebsiella spp. Sci. Rep. 5:15573. doi: 10.1038/srep15573
Pieroni, P., Rennie, R. P., Ziola, B., and Deneer, H. G. (1994). The use of bacteriophages to differentiate serologically cross-reactive isolates of Klebsiella pneumoniae. J. Med. Microbiol. 41, 423–429. doi: 10.1099/00222615-41-6-423
Pires, D. P., Oliveira, H., Melo, L. D., Sillankorva, S., and Azeredo, J. (2016). Bacteriophage-encoded depolymerases: their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 100, 2141–2151. doi: 10.1007/s00253-015-7247-7240
Plattner, M., Shneider, M. M., Arbatsky, N. P., Shashkov, A. S., Chizhov, A. O., Nazarov, S., et al. (2019). Structure and function of the branched receptor-binding complex of bacteriophage CBA120. J. Mol. Biol. 431, 3718–3739. doi: 10.1016/j.jmb.2019.07.022
Prokhorov, N. S., Riccio, C., Zdorovenko, E. L., Shneider, M. M., Browning, C., Knirel, Y. A., et al. (2017). Function of bacteriophage G7C esterase tailspike in host cell adsorption. Mol. Microbiol. 105, 385–398. doi: 10.1111/mmi.13710
Rose, A. S., Bradley, A. R., Valasatava, Y., Duarte, J. M., Prlic, A., and Rose, P. W. (2018). NGL viewer: web-based molecular graphics for large complexes. Bioinformatics 34, 3755–3758. doi: 10.1093/bioinformatics/bty419
Schmid, J., Sieber, V., and Rehm, B. (2015). Bacterial exopolysaccharides: biosynthesis pathways and engineering strategies. Front. Microbiol. 6:496. doi: 10.3389/fmicb.2015.00496
Schwarzer, D., Browning, C., Stummeyer, K., Oberbeck, A., Mühlenhoff, M., Gerardy-Schahn, R., et al. (2015). Structure and biochemical characterization of bacteriophage phi92 endosialidase. Virology 477, 133–143. doi: 10.1016/j.virol.2014.11.002
Schwarzer, D., Buettner, F. F., Browning, C., Nazarov, S., Rabsch, W., Bethe, A., et al. (2012). A multivalent adsorption apparatus explains the broad host range of phage phi92: a comprehensive genomic and structural analysis. J. Virol. 86, 10384–10398. doi: 10.1128/JVI.00801-812
Schwarzer, D., Stummeyer, K., Gerardy-Schahn, R., and Mühlenhoff, M. (2007). Characterization of a novel intramolecular chaperone domain conserved in endosialidases and other bacteriophage tail spike and fiber proteins. J. Biol. Chem. 282, 2821–2831. doi: 10.1074/jbc.M609543200
Seul, A., Müller, J. J., Andres, D., Stettner, E., Heinemann, U., and Seckler, R. (2014). Bacteriophage P22 tailspike: structure of the complete protein and function of the interdomain linker. Acta Crystallogr. D Biol. Crystallogr. 70(Pt 5), 1336–1345. doi: 10.1107/S1399004714002685
Simoliūnas, E., Kaliniene, L., Truncaitė, L., Zajančkauskaitė, A., Staniulis, J., Kaupinis, A., et al. (2013). Klebsiella phage vB_KleM-RaK2 - a giant singleton virus of the family Myoviridae. PLoS One 8:e60717. doi: 10.1371/journal.pone.0060717
Solovieva, E. V., Myakinina, V. P., Kislichkina, A. A., Krasilnikova, V. M., Verevkin, V. V., Mochalov, V. V., et al. (2018). Comparative genome analysis of novel Podoviruses lytic for hypermucoviscous Klebsiella pneumoniae of K1, K2, and K57 capsular types. Virus Res. 243, 10–18. doi: 10.1016/j.virusres.2017.09.026
Steven, A. C., Trus, B. L., Maizel, J. V., Unser, M., Parry, D. A., Wall, J. S., et al. (1988). Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. J. Mol. Biol. 200, 351–365. doi: 10.1016/0022-2836(88)90246-X
Stummeyer, K., Dickmanns, A., Mühlenhoff, M., Gerardy-Schahn, R., and Ficner, R. (2005). Crystal structure of the polysialic acid-degrading endosialidase of bacteriophage K1F. Nat. Struct. Mol. Biol. 12, 90–96. doi: 10.1038/nsmb874
Stummeyer, K., Schwarzer, D., Claus, H., Vogel, U., Gerardy-Schahn, R., and Mühlenhoff, M. (2006). Evolution of bacteriophages infecting encapsulated bacteria: lessons from Escherichia coli K1-specific phages. Mol. Microbiol. 60, 1123–1135. doi: 10.1111/j.1365-2958.2006.05173.x
Sutherland, I. W. (1995). Polysaccharide lyases. FEMS Microbiol. Rev. 16, 323–347. doi: 10.1111/j.1574-6976.1995.tb00179.x
Taylor, N. M., Prokhorov, N. S., Guerrero-Ferreira, R. C., Shneider, M. M., Browning, C., Goldie, K. N., et al. (2016). Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 533, 346–352. doi: 10.1038/nature17971
Tu, J., Park, T., Morado, D. R., Hughes, K. T., Molineux, I. J., and Liu, J. (2017). Dual host specificity of phage SP6 is facilitated by tailspike rotation. Virology 507, 206–215. doi: 10.1016/j.virol.2017.04.017
Volozhantsev, N. V., Myakinina, V. P., Popova, A. V., Kislichkina, A. A., Komisarova, E. V., Knyazeva, A. I., et al. (2016). Complete genome sequence of novel T7-like virus vB_KpnP_KpV289 with lytic activity against Klebsiella pneumoniae. Arch. Virol. 161, 499–501. doi: 10.1007/s00705-015-2680-z
Weigele, P. R., Scanlon, E., and King, J. (2003). Homotrimeric, beta-stranded viral adhesins and tail proteins. J. Bacteriol. 185, 4022–4030. doi: 10.1128/jb.185.14.4022-4030.2003
Weiner, L. M., Webb, A. K., Limbago, B., Dudeck, M. A., Patel, J., Kallen, A. J., et al. (2016). Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the national healthcare safety network at the centers for disease control and prevention, 2011-2014. Infect. Control Hosp. Epidemiol. 37, 1288–1301. doi: 10.1017/ice.2016.174
Wick, R. R., Heinz, E., Holt, K. E., and Wyres, K. L. (2018). Kaptive web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella Genomes. J. Clin. Microbiol. 56:e197-18. doi: 10.1128/JCM.00197-118
Williams, S. R., Gebhart, D., Martin, D. W., and Scholl, D. (2008). Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microbiol. 74, 3868–3876. doi: 10.1128/AEM.00141-148
Wyres, K. L., Gorrie, C., Edwards, D. J., Wertheim, H. F., Hsu, L. Y., Van Kinh, N., et al. (2015). Extensive capsule locus variation and large-scale genomic recombination within the Klebsiella pneumoniae clonal group 258. Genom. Biol. Evol. 7, 1267–1279. doi: 10.1093/gbe/evv062
Wyres, K. L., Wick, R. R., Gorrie, C., Jenney, A., Follador, R., Thomson, N. R., et al. (2016). Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genom. 2:e000102. doi: 10.1099/mgen.0.000102
Yan, J., Mao, J., and Xie, J. (2014). Bacteriophage polysaccharide depolymerases and biomedical applications. Biodrugs 28, 265–274. doi: 10.1007/s40259-013-0081-y
Keywords: horizontal transfer, tail fiber genes, receptor binding protein, phage evolution, depolymerase
Citation: Latka A, Leiman PG, Drulis-Kawa Z and Briers Y (2019) Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 10:2649. doi: 10.3389/fmicb.2019.02649
Received: 11 July 2019; Accepted: 30 October 2019;
Published: 15 November 2019.
Edited by:
Caroline Westwater, Medical University of South Carolina, United StatesReviewed by:
Stefanie Barbirz, University of Potsdam, GermanyCarmela Garcia Doval, University of Zurich, Switzerland
Copyright © 2019 Latka, Leiman, Drulis-Kawa and Briers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zuzanna Drulis-Kawa, zuzanna.drulis-kawa@uwr.edu.pl; Yves Briers, yves.briers@ugent.be