
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 September 2019
Sec. Virology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.02236
 Guillaume Carissimo1
Guillaume Carissimo1 Yi-Hao Chan1,2
Yi-Hao Chan1,2 Age Utt3
Age Utt3 Tze-Kwang Chua1
Tze-Kwang Chua1 Farhana Abu Bakar1,4
Farhana Abu Bakar1,4 Andres Merits3
Andres Merits3 Lisa F. P. Ng1,5,6*
Lisa F. P. Ng1,5,6*The evolutionarily conserved AAA+ ATPase valosin-containing protein (VCP) was previously shown to be a proviral host factor for several viruses from different viral families such as Flaviviridae, Picornaviridae, and Herpesviridae. VCP was shown to affect trafficking of Sindbis virus receptor and functions as a component of Semliki Forest virus (SFV) replicase compartment. However, the role of this cellular protein was not evaluated during replication of alphaviruses including chikungunya virus (CHIKV). Using siRNA, chemical inhibitors, and trans-replication assays, we show here that VCP is a proviral factor involved in the replication of CHIKV. Immunofluorescence assays confirmed that VCP co-localized with non-structural replicase proteins but not with dsRNA foci possibly due to VCP epitope unavailability. VCP pro-viral role is also observed with other alphaviruses such as o’nyong’nyong virus (ONNV) and SFV in different human cell lines. VCP proviral roles on several viral families now extend to replication of alphaviruses CHIKV and ONNV, emphasizing the pivotal role of VCP in virus–host interaction biology.
The valosin-containing protein (VCP), also named p97, is a member of the hexameric AAA+ ATPase family and is highly conserved across all domains of life (Erzberger and Berger, 2006; Barthelme and Sauer, 2016). The molecular function of VCP is ATP-driven protein unfolding (Ye et al., 2017; van den Boom and Meyer, 2018). Depending on the different associated co-factors (Hanzelmann and Schindelin, 2017), it is involved in many cellular pathways, ranging from endoplasmic reticulum (ER) and mitochondrial associated protein degradation (Taylor and Rutter, 2011; Wolf and Stolz, 2012), ubiquitin proteasome system (UPS) (Meyer et al., 2012), chromatin structure regulation (Torrecilla et al., 2017), DNA break repair (Torrecilla et al., 2017), DNA replication (Ramadan et al., 2017), NF-κB activation (Schweitzer et al., 2016), stalled ribosome turnover (Verma et al., 2013), endomembrane fusion (Zhang and Wang, 2015), full capacity of autophagy and lysosomal system (Papadopoulos and Meyer, 2017), and lipid droplet turnover (Olzmann et al., 2013).
Valosin-containing protein has been reported to be a pro- or anti-viral host factor for several viruses, such as picornaviruses (Arita et al., 2012; Wu et al., 2016; Wang et al., 2017), flaviviruses (Yi et al., 2016; Phongphaew et al., 2017; Yi and Yuan, 2017), coronaviruses (Wong et al., 2015), an herpesvirus (Lin et al., 2017), a phlebovirus (Brahms et al., 2017), and even an insect nucleopolyhedrovirus (Lyupina et al., 2018). In the case of picornaviruses (single-stranded RNA positive, ssRNA+), VCP has been implicated as a pro-viral factor in the replication of poliovirus and EV71 and several viral proteins showed direct interaction (Arita et al., 2012; Wang et al., 2017), while knock-down of VCP enhanced Aichi virus (Arita et al., 2012). VCP have been described as an important host factor for several flaviviruses (ssRNA+) West Nile virus replication (Phongphaew et al., 2017), as well as a component of Hepatitis C virus (HCV) and shown to prevent HCV replicase aggregation (Yi et al., 2016; Yi and Yuan, 2017).
In the specific case of ssRNA+ alphaviruses (family Togaviridae), VCP was shown to be a proviral factor for Sindbis virus (SINV) by trafficking SINV receptor NRAMP2 to the plasma membrane (Rose et al., 2011; Panda et al., 2013). However, for chikungunya virus (CHIKV) and other alphaviruses, binding and entry is NRAMP2 independent (Rose et al., 2011). In addition, VCP was later identified by mass spectrometry as a replication complex-associated protein during Semliki Forest virus (SFV) infection (Varjak et al., 2013). However, the role of this AAA+ ATPase was not further investigated during the replication of alphaviruses.
In this study, using a combination of knockdowns and chemical inhibitors with time-of-addition assays, VCP is shown to be an important proviral factor necessary for replication of several isolates of CHIKV, O’nyong’nyong virus (ONNV), and SFV. Using a CHIKV trans-replication system, VCP was verified to be an important factor required during RNA replication of the virus life cycle, but not for the stability of the non-structural proteins (nsPs). We also showed that VCP co-localized with several nsPs. Elucidating the molecular mechanisms involved in VCP proviral role, including its viral interacting protein partners, will be crucial in developing therapies targeting VCP pro-viral functions potentially efficient on several viral families.
In order to assess if VCP has a role during CHIKV infection, transfection of a mix of three siRNAs targeting VCP followed by infection with CHIKV-expressing Gaussia luciferase (Gluc) inserted between non-structural and structural coding regions, as an indirect marker of viral replication, was performed. Reduction of VCP protein levels was verified by western blot assays at 48 h (time of infection) and 64 h post-siRNA transfection (16 h post infection, time of supernatant collection) (Figure 1A). Knockdown of VCP significantly reduced the increase of viral RNA in the supernantant from 0 to 16 hpi (Figure 1B). Next, we validated that Gluc luminescence correlated with viral RNA and viral particle amounts in the same cell supernatants (Supplementary Figure S1), confirming that Gluc luminescence could be used as a marker of viral replication. Using Gluc, we confirmed that VCP knockdown severely impacted viral replication (Figure 1C).
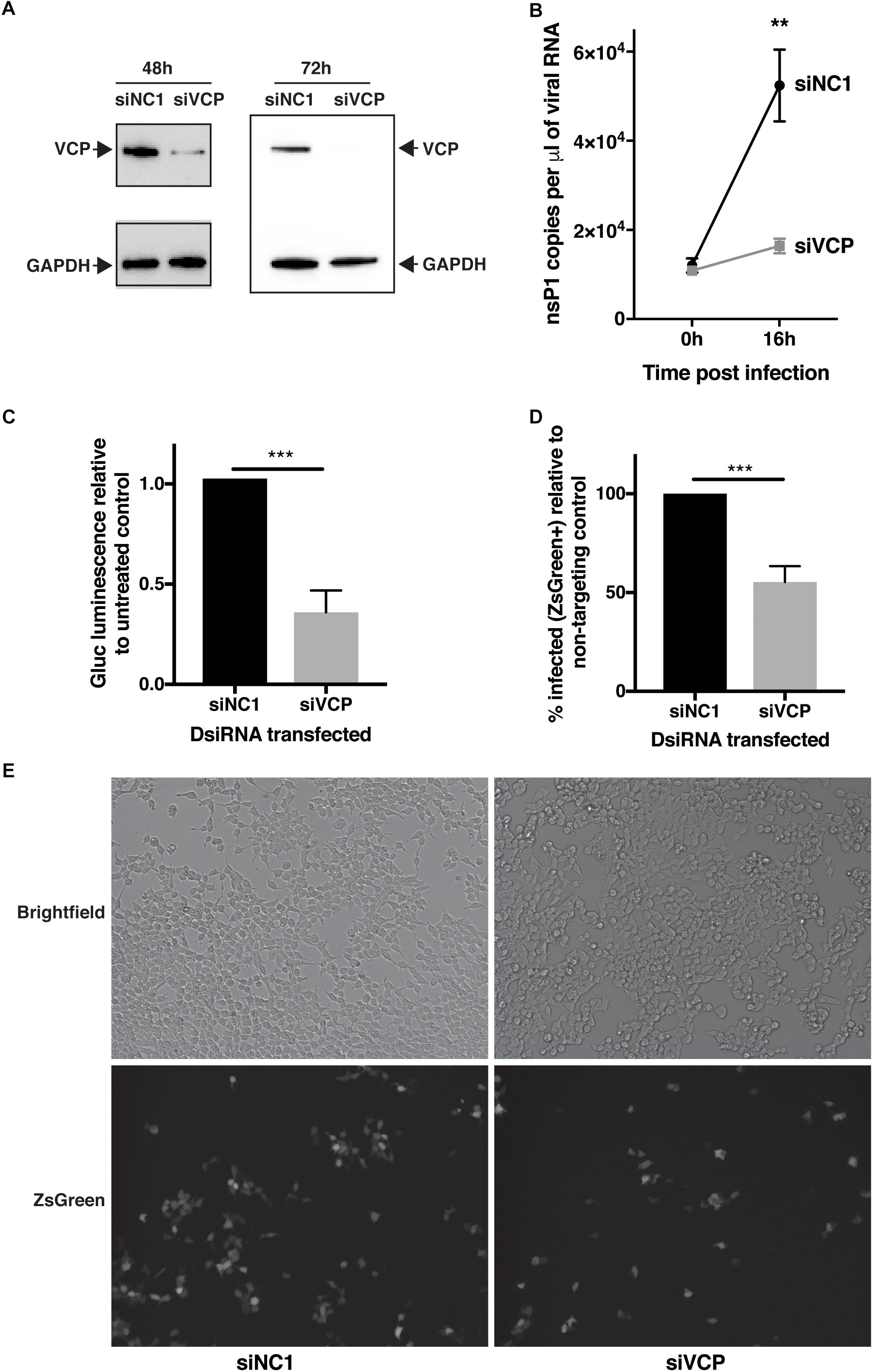
Figure 1. VCP knock-down affects CHIKV infection. HEK 293T cells were transfected with 10 nM of siRNA. siNC1 is a non-targeting control and siVCP is a mix of three individual siRNA targeting VCP. At 48 h post transfection, cells were infected with Gluc- or ZsGreen-tagged virus per well, inoculum was not removed, to obtain 0 hpi time point, 140 μL of supernatant was collected immediately after addition of inoculum. At 16 h post-infection, supernatant was collected or cells were imaged and harvested for flow cytometry analysis, respectively. (A) Representative western blot images of three independent experiment for VCP and GAPDH expression on cell lysates at 48 and 64 h post transfection. (B) Taqman quantification of viral RNA in the cell supernatant at 0 and 16 hpi. (C) Gaussia luciferase luminescence relative to siNC1. (D) % of FITC positive (infected) cells in siVCP-transfected cells relative to siNC1-transfected cells analyzed by flow cytometry. (E) Representative images of brightfield (auto exposure) and FITC (fixed exposure) channel on an epi-fluorescence microscope at 16 hpi. The data are presented as mean ± SD from minimum three independent experiments (two for panel B) and were analyzed by Mann–Whitney non-parametric two-tailed test; ∗∗p < 0.01; ∗∗∗p < 0.001.
In order to validate that this effect was not due to a VCP-dependant release or secretion defect of the Gluc or the viral particles, the experiment was repeated using a virus expressing a non-secreted ZsGreen reporter instead marking infected cells. In that context, VCP knockdown also significantly reduced viral infection (Figures 1D,E). Taken together, these results suggest a proviral role of VCP during CHIKV infection.
In order to circumvent limitations in assessing the various steps of the viral cycle affected by VCP knockdown, VCP-specific chemical inhibitors and time-of-addition assays were explored. The different chemical inhibitors assessed included DBeQ, a reversible inhibitor of VCP (Fang et al., 2015), NMS-873, a specific allosteric VCP inhibitor (Magnaghi et al., 2013), and CB-5083, an orally bioavailable active compound derived from the scaffold of DBeQ (Anderson et al., 2015). Notably, CB-5083 has been shown to be selective, specifically inhibiting the ATPase activity of the D2 domain of VCP by binding in the nucleotide binding site (Tang et al., 2019). All VCP inhibitors showed a dose-dependent effect on Gluc production with an approximate effective concentration 50 (EC50) ranging from 0.4 to 10 μM (Figure 2). Toxicity of the different inhibitors at different concentrations was verified using the Cell Titer Glo assay and showed cytotoxic concentration 50 (CC50) very close from their respective EC50 for DBeQ and NMS-873 suggesting that their inhibitory effect on viral Gluc production could be linked to the cytotoxicity effect. However, no significant toxicity for CB-5083 was observed (Figure 2E), suggesting that the inhibitory effect on CB-5083 on viral Gluc production is not due to cytotoxicity. Interestingly, the time-of-addition of chemical inhibitors relative to viral infection had no significant impact on the anti-CHIKV EC50 (Figures 2C,D,F), with CB-5083 having the lowest EC50 (0.4 < EC50 < 0.45 μM) (Figure 2E). Taken together, these results suggest an antiviral effect of VCP inhibitor CB-5083 at a post binding or entry step.
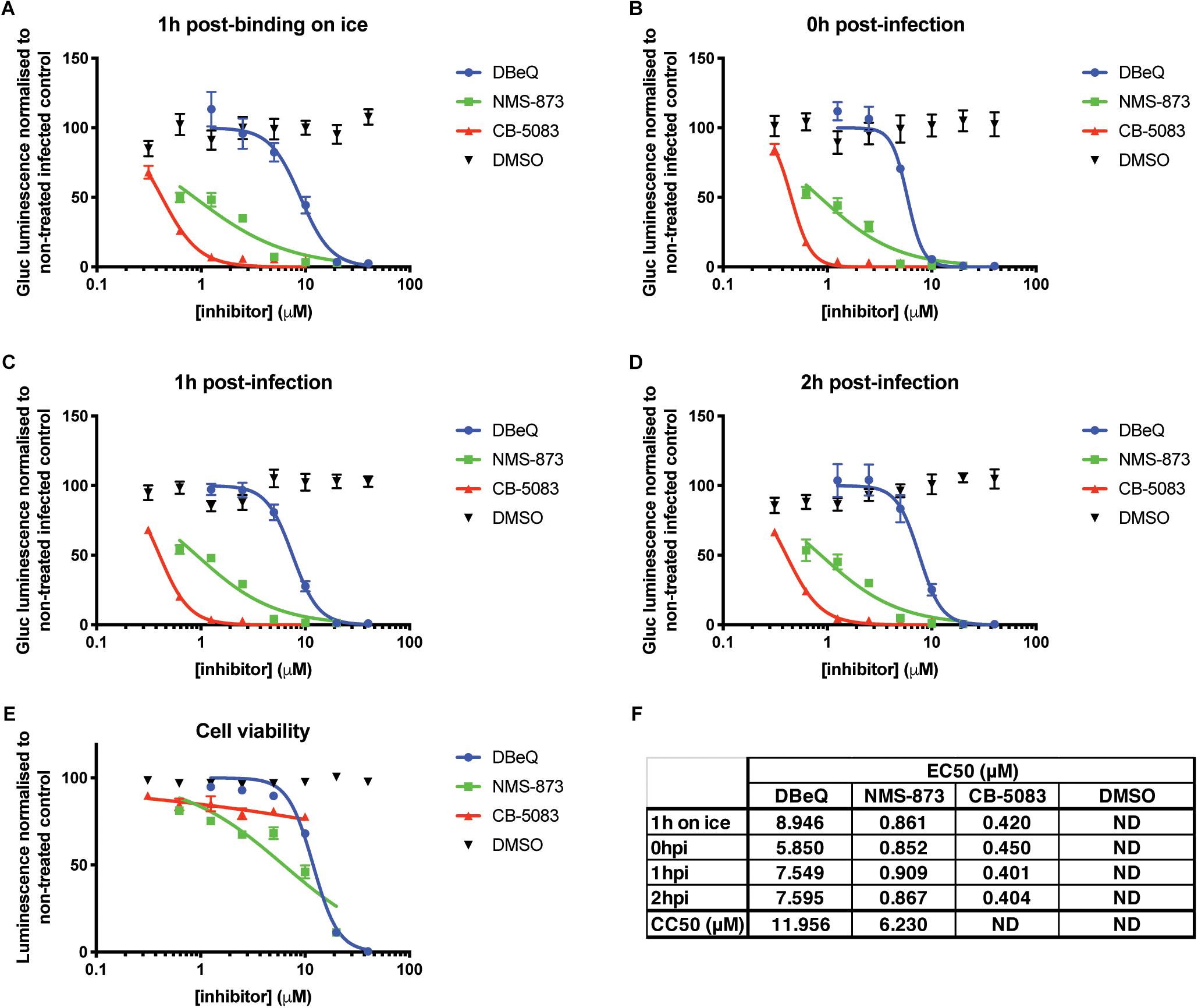
Figure 2. VCP chemical inhibition affects CHIKV infection. Thirty thousand HEK 293T cells per well in 96-well plates fibronectin coated were treated with VCP inhibitors CB-5083, NMS-873, DBeQ, or DMSO control at the times indicated on the panels relative to infection with 30,000 pfu of Gluc-tagged CHIKV per well. At 16 hpi, luminescence was quantified from 50 μL of supernatant. Data are presented as % luminescence compared to non-treated infected cells in y-axis, and as concentration of inhibitor (μM) in the x-axis. (A) Virus was allowed to bind to the cells for 1 h on ice before inoculate was removed and virus-free media with compounds were applied and cells moved back to 37°C. (B) Virus was applied to the cells at the same time as the compounds, removed after 1 h and virus-free media with compound was replaced on the cells. (C,D) Virus was applied for 1 or 2 h, respectively, after which inoculum was removed and compounds were applied. (E) Cytotoxicity of the compounds was assessed for each compound by CellTiter-Glo assay without infection at 16 h post treatment. (F) EC50 and CC50 values from panels A–E were calculated using the non-linear regression analysis (inhibitor) (μM) vs. normalized response variable slope function of GraphPad Prism. The data presented in panels A–E are mean ± SEM of three independent experiments. ND, not determined.
To validate that VCP was not involved during CHIKV binding or entry, a binding assay was performed on ice and at 37°C for 1 h in the presence of CB-5083 or DMSO control. After the washing steps, RNA was extracted from cells and CHIKV RNA was quantified relative to GAPDH RNA for each individual sample. During binding on ice, no difference in the relative amount of CHIKV RNA was observed during VCP inhibition by CB-5083 treatment compared to the control samples (Figure 3A). Intriguingly, higher CHIKV RNA levels in the DMSO control-treated samples were observed when binding was performed at 37°C, suggesting that VCP could be involved in an early post-entry replication step. Together with the binding on ice, a lower amount of CHIKV RNA in the treated group indicates that some replication has occurred in the DMSO control, but not in the treated group.
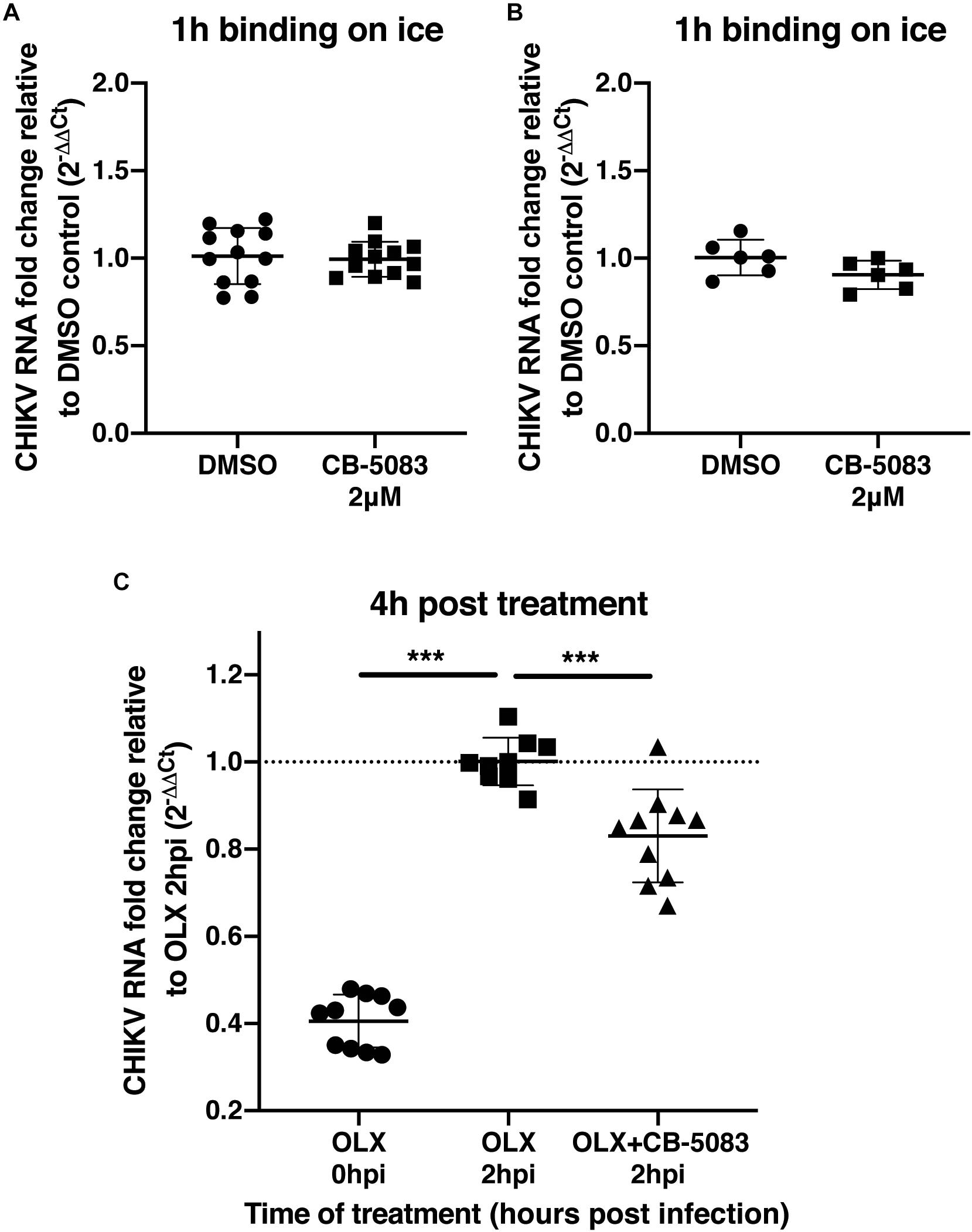
Figure 3. VCP is not involved in CHIKV binding or entry. (A,B) Two hundred thousand HEK 293T cells per well were infected at moi 10 for 1 h in DMEM media or in media with 2 μM CB-5083 on ice or at 37°C, respectively. Cells were then washed twice in PBS, collected, and RNA was extracted. (C) Two hundred thousand HEK293T cells per well were infected at moi 10 for 2 h in DMEM media or in media with 0.5 μM OLX. After 2 h, inoculum was removed, cells were washed twice and incubated for 4 h in media with CB-5083 at 2 μM or media with OLX (0.5 μM) and CB-5083 (2 μM) or media with OLX (0.5 μM), the cells were then washed twice, collected, and RNA was extracted. Relative CHIKV RNA fold change compared to control using 2−ΔΔCT method. Each symbol represents an independent well, data presented are from three independent experiments for panels A and C and two independent experiments for panel B represented as mean ± SD and were analyzed by Mann–Whitney non-parametric two-tailed test for panel D; ∗∗∗p < 0.001.
To confirm this observation, cells were treated with an entry inhibitor, Obatoclax (OLX) (Varghese et al., 2017), or OLX together with CB-5083 at 2 hpi, and assessed for the relative amount of viral RNA at 4 h post-treatment (6 h post virus addition) (Figure 3C). VCP inhibition by CB-5083 resulted in reduced viral RNA levels compared to entry inhibition alone by OLX at 4 h post-treatment (Figure 3C). Taken together, these results show that VCP inhibition is affecting a post binding and entry replication step.
To confirm that inhibition of VCP impacts viral RNA replication, a CHIKV trans-replication system was established (Figure 4A) (Utt et al., 2016; Abraham et al., 2018). In this system, the nsPs are encoded on a separate plasmid and expressed constitutively using a CMV promoter (Utt et al., 2015) (Plasmid 1, Figure 4A). Since the use of viral RNA templates generated by cellular RNA polymerase II could produce high background signals (Utt et al., 2015; Abraham et al., 2018; Bartholomeeusen et al., 2018), and the replicase of CHIKV is capable of using non-capped RNA templates (Utt et al., 2015), a human RNA polymerase I promoter was used to generate non-capped viral RNA template (Plasmid 2, Figure 4A) (Utt et al., 2019). This template codes for Firefly luciferase (Fluc) instead of the non-structural open-reading frame (ORF) and codes for tdTomato instead of the structural ORF (Plasmid 2, Figure 4A). These reporters would be translated after template RNA replication by the viral replicase, with minimal expression of either reporter in absence of template RNA replication. Thus, tdTomato protein would be expressed and detectable only when RNA replication occurred allowing us to assess the effect of VCP inhibitor on viral replication.
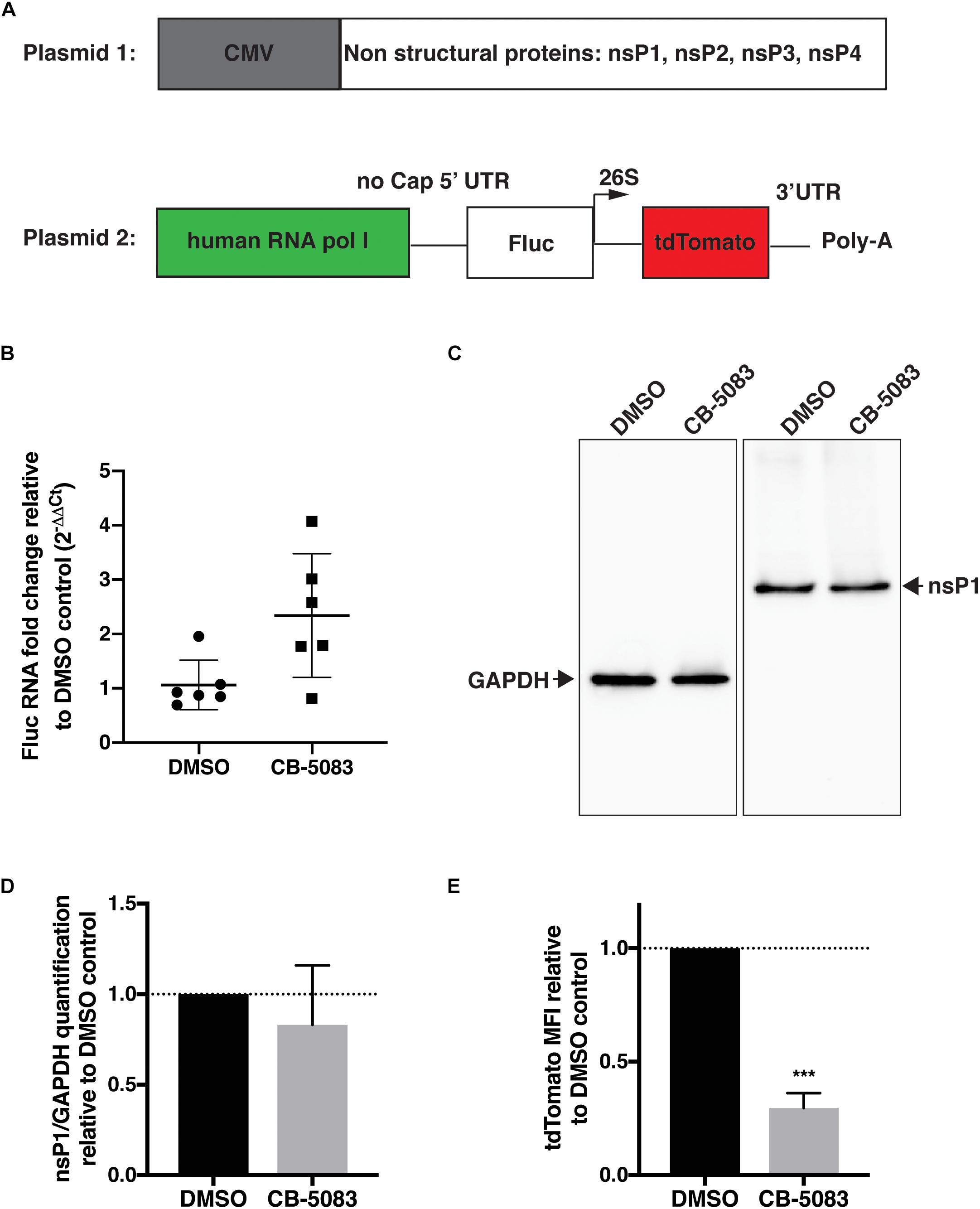
Figure 4. VCP is essential for CHIKV replicase activity. HEK 293T cells were co-transfected with plasmid 2 alone (panel B) or plasmids 1 and 2 (ratio 2:1, panels C–E), 5–6 h post transfection, media was refreshed with DMSO control or CB-5083 (1 μM). 16 h post media refresh, cells were collected and separated for RNA extraction or whole cell lysate and flow cytometry analysis. (A) Schematic representation of the CHIKV trans-replication plasmid system used. (B) Relative Fluc RNA fold change compared to control using 2−ΔΔCT method. (C) Representative Western blot of whole cell lysates probed for GAPDH and CHIKV nsP1 proteins, respectively. (D) Relative quantification of nsP1 protein amount versus GAPDH for each experimental replicate. (E) Mean fluorescent intensity (MFI) of tdTomato assessed by flow cytometry in tdTomato positive cells. The data presented are of three independent experiments (two for panel B) and were analyzed by Mann–Whitney non-parametric two-tailed test; ∗∗∗p < 0.001.
Using this system, we first verified that VCP inhibition by CB-5083 had no effect on transcription by RNA polymerase I by quantifying Fluc RNA levels relative to GAPDH in the absence of replicase proteins (plasmid 2 alone) (Figure 4B). Surprisingly, an increase in template RNA levels was found in CB-5083-treated samples, suggesting that VCP inhibition by CB-5083 could increase RNA polymerase I transcription or/and increase stability of non-capped RNA transcripts. Next, we verified that VCP inhibition did not affect expression of the nsPs from the CMV plasmid by assessing nsP1 protein levels by western blot (Figures 4C,D). Altogether, results showed that CB-5083 had no negative effect on production of CHIKV replicase proteins or template RNA. However, when the replicase and templates were co-expressed, VCP chemical inhibition drastically decreased the amount of tdTomato mean fluorescence intensity (MFI) detected per transfected cell (Figure 4E) despite the increased Fluc RNA template availability. These results confirm that VCP is a proviral factor essential during CHIKV replication at a post entry step.
We next assessed if VCP co-localizes specifically with a particular nsP. Using confocal microscopy, we observed that VCP was partially co-localizing with nsP1 (Figure 5A), nsP2 (Figure 5B), and nsP3 (Figure 5C) in virus-infected cells. Unfortunately, in this assay, nsP4 as well as dsRNA co-localization with VCP could not be assessed due to lack of suitable and species compatible antibodies, respectively. Pearson’s correlation coefficient for all the acquired images indicated that VCP co-localized more with nsP2 and nsP3 than nsP1 (Figure 5D). These results suggest that VCP could likely interact with multiple viral nsPs, or is associated with a nsP in replicase complexes in order to perform proviral functions.
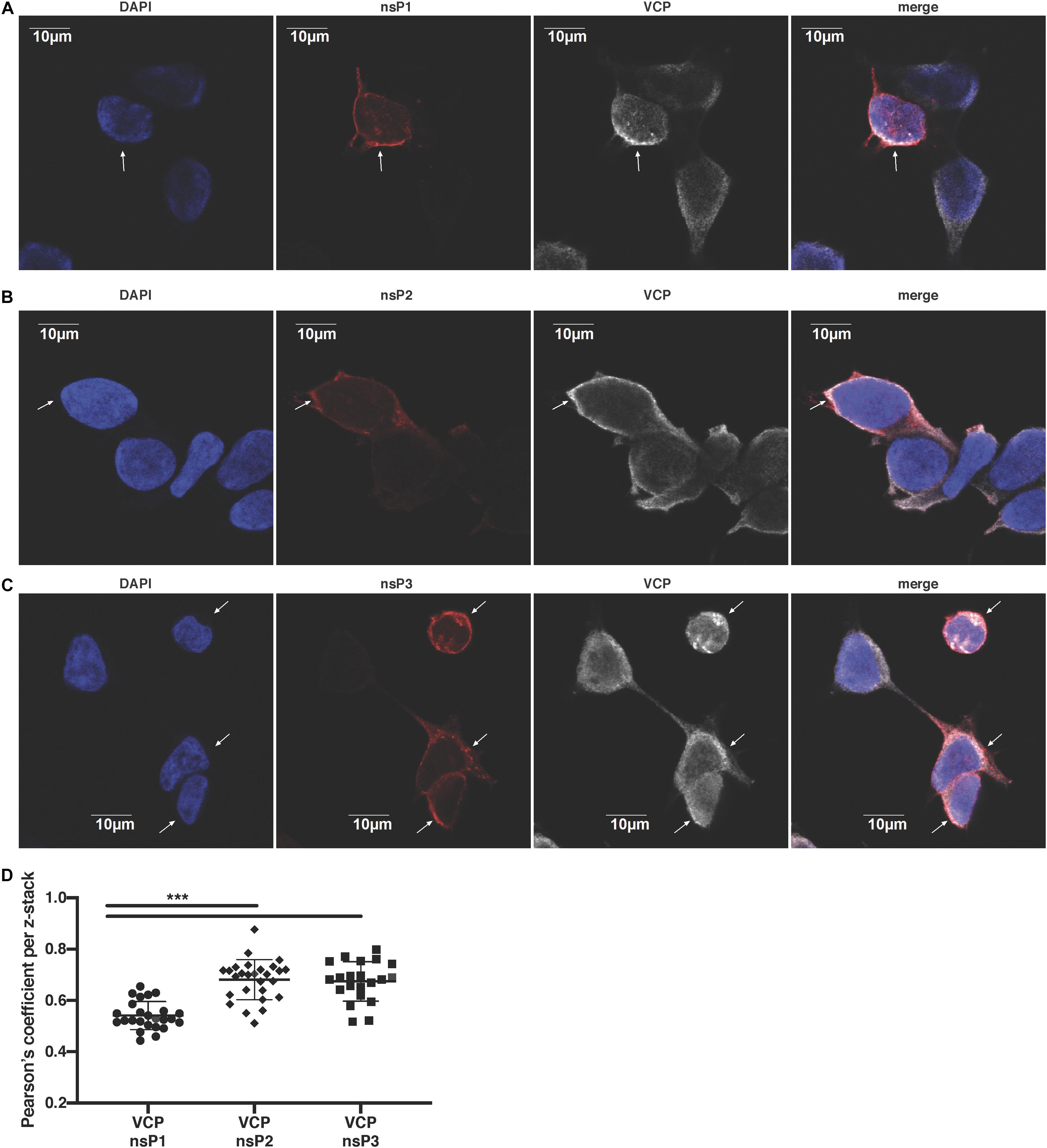
Figure 5. VCP localizes with CHIKV replicase components. HEK 293T cells were infected at an moi of 10, 2 hpi cells were then plated in Ibidi μ-wells microscopy chambers; 5 hours post-plating, cells were fixed, permeabilized, and co-stained in duplicate for VCP with nsP1, nsP2, or nsP3 as indicated on the figure. Cells were counterstained with DAPI before confocal acquisition of z-stacks. (A) Representative focal plane of VCP with nsP1. (B) Representative focal plane of VCP with nsP2. (C) Representative focal plane of VCP with nsP3. (D) Pearson correlation coefficient of colocalization of VCP with the respective nsPs in each z-stack analyzed with Imaris software. Results are representative of two independent experiments performed in duplicate. White arrows indicate infected cells. Correlation coefficient were analyzed by Mann–Whitney non-parametric two-tailed test; ∗∗∗p < 0.001.
We then assessed if VCP could localize with dsRNA replication complexes. Therefore, we assessed the performance of a polyclonal rabbit anti-VCP antibody in immunofluorescence assay which would allow co-localization with the mouse antibody recognizing dsRNA. Using this combinaition of antibodies, we did not observe co-localization between VCP and dsRNA foci (Figure 6A). Interestingly, this antibody against VCP presented a very different localization pattern than the mouse monoclonal antibody used previously in both infected and non-infected conditions (Figure 5). Since the rabbit antibody, directed against the N-terminal, gave a punctuated localization different from the pattern observed previously near the cytosolic membrane with the mouse monoclonal antibody against VCP C-terminal (Figure 5), we assessed co-localization of the two anti-VCP antibodies (Figure 6B). Signals from both antibodies did not co-localize, and localization of VCP near the plasma membrane was not observable using the N-terminal directed antibody raised in rabbit. Since the N-terminal of VCP is a site of interaction with several co-factors (Hanzelmann and Schindelin, 2017), we hypothesize that the observed absence of co-localization of dsRNA with VCP could be due to a lack of N-terminal accessibility.
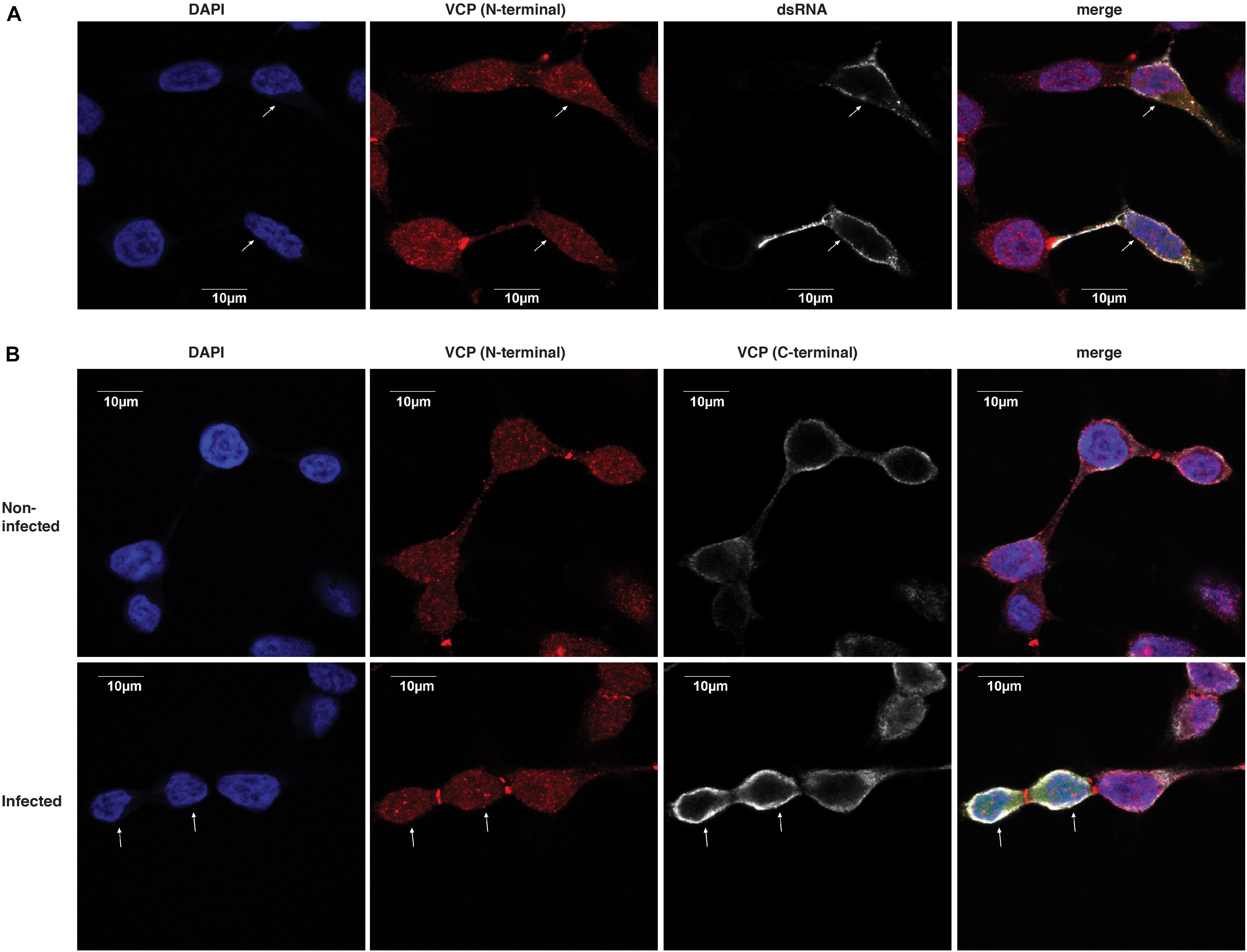
Figure 6. dsRNA foci do not colocalize with VCP. HEK 293T cells were infected at an moi of 10 with ZsGreen-tagged CHIKV-LR, 2 hpi cells were washed and re-plated in Ibidi μ-wells microscopy chambers. At 5 h post-plating, cells were fixed, permeabilized, and co-stained for VCP and dsRNA as indicated on the figure. Cells were counterstained with DAPI before confocal acquisition. (A) Representative focal plane of VCP with dsRNA staining in infected cells. (B) Representative focal plane of VCP (N-terminal) and (C-terminal) staining. Results are representative of two independent experiments performed in triplicates. White arrows indicate infected cells which were determined using the ZsGreen channel (shown only in the merged image).
In order to assess if this effect was restricted to the CHIKV viral isolate used in the previous experiments, CB-5083 was used to assess the role of VCP during infection with different CHIKV isolates as well as the closely related ONNV, and more distantly related SFV all belonging to the SFV clade. CHIKV can be categorized into three main genotypes, the West Africa, East-Central-South-African (ECSA), and Asian genotypes. We compared the Chikungunya strain isolated during the La Reunion epidemic [La Reunion isolate LR2006-OPY1 (LR)] and an Indian (IND) strain isolated in India during the same epidemic from a patient presenting neurological complications both belonging to the ECSA genotype. We also compared a Caribbean (CRB) isolate (CNR20235) belonging to the Asian genotype, isolated during outbreaks in the CRB islands in December 2013, which we have previously shown to be less pathogenic than the LR isolate in vitro and in vivo in mouse models (Teo et al., 2015). VCP inhibition with CB-5083 showed a dose-dependent inhibition of viral Gluc production with all viruses tested (Figure 7A). A dose-dependent decrease of ZsGreen signal was also observed in cells infected with ZsGreen-tagged CHIKV or ONNV (Figure 7B). In addition, we assessed if the effect of VCP inhibition on CHIKV replication was reproducible in more relevant human cells related to cell types infected in patients: a human foreskin fibroblast cell line (BJ cells) and a muscle cells line (RD cells) (Suhrbier et al., 2012). VCP inhibition with CB-5083 resulted in dose-dependent inhibition of CHIKV in BJ and RD cells (Figure 7C).
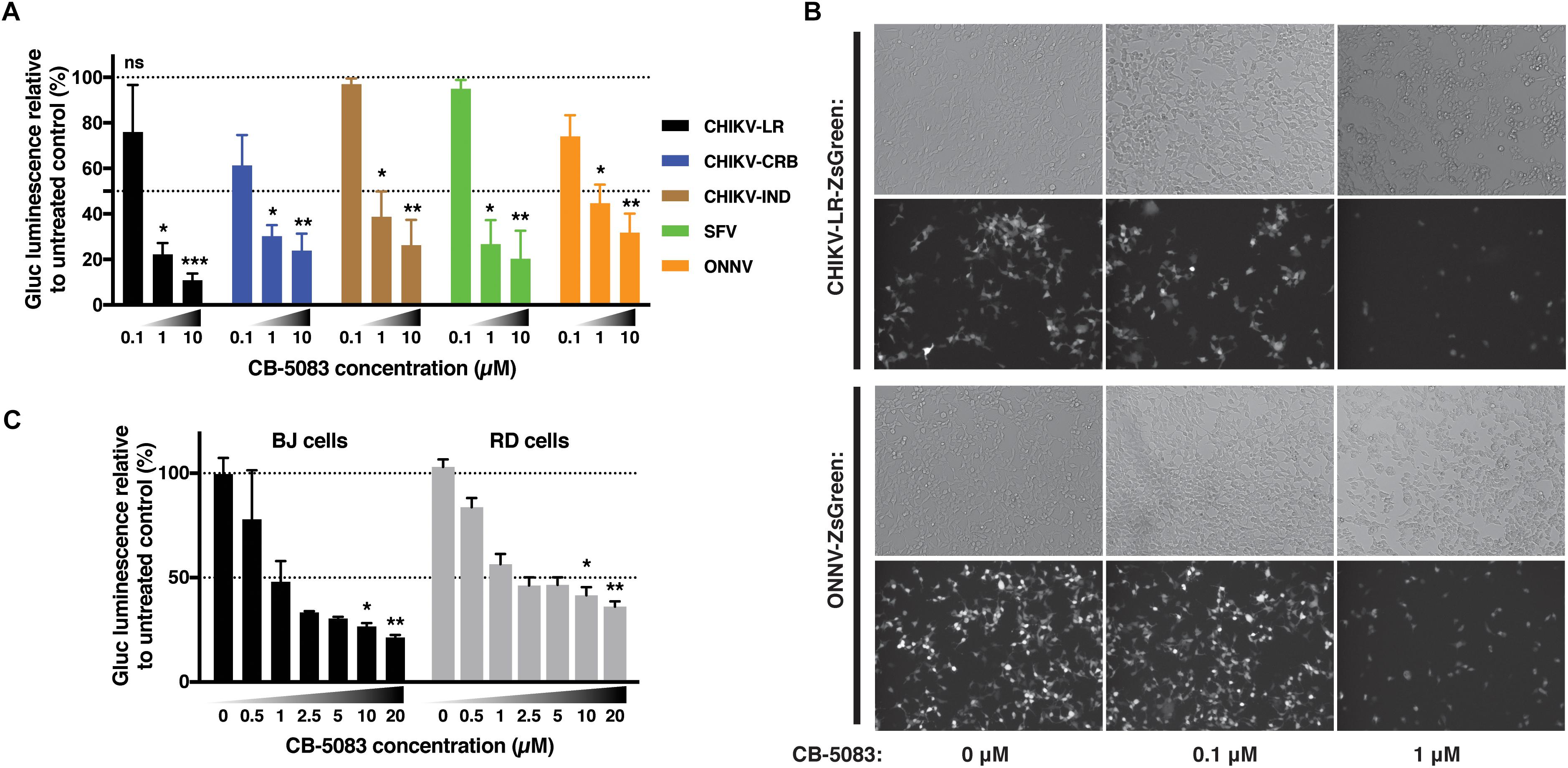
Figure 7. VCP is proviral for other alphaviruses and in other cell types. (A) Thirty thousand HEK 293T cells per well were treated with increasing concentrations of CB-5083 (0, 0.1, 1, and 10 μM) and infected by 3,000 pfu/well of the indicated viruses each tagged with Gluc, virus inoculum was not removed. At 16 hpi, luminescence in 50 μL of supernatant was assessed, results are expressed to their relative 0 μM treatment. (B) Representative epi-fluorescence images of 30,000 HEK 293T cells infected by 30,000 pfu of ZsGreen-tagged CHIKV-LR and ONNV for 16 h at different CB-5083 concentration (as indicated on the figure). Images are representative of three independent experiments and were acquired on brightfield (auto exposure) and FITC (fixed exposure) channels. (C) 5,000 RD or BJ cells per well were treated with increasing concentrations of CB-5083 (indicated on the figure) and infected by 5,000 pfu of CHIKV-LR-tagged with Gluc, virus inoculum was not removed. At 16 hpi, luminescence in 50 μL of supernatant was assessed. Data of panels A and B are representative of four and three independent experiment, respectively, presented as mean ± SD and were analyzed by Mann–Whitney non-parametric two-tailed test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
In this study, we show that siRNAs targeting VCP (Figure 1) as well as chemical inhibitors (Figure 2) are able to reduce CHIKV infection and replication in HEK293T cells. Time-of-addition assays using specific chemical inhibitors of VCP suggested that the virus life cycle step involving VCP as a proviral factor would be viral RNA replication rather than binding or entry. Although the effect of VCP inhibition demonstred to be conserved across different alphaviruses (Figure 7), variations in the level of virus inhibition were observed between the different cell types and virus isolates. These variations are likely due to the calculation for each virus and cell type, where the relative luciferase is normalized against the signals from the untreated infected internal controls, where background signal comes from initial Gluc contained in the inoculum.
Despite the proviral role of VCP on CHIKV replication observed during knockdown or chemical inhibition, no increase in viral replication upon VCP or VCP domains overexpression was observed (Supplementary Figure S2), consistent with VCP being highly abundant (0.7% of cytoplasmic protein content) (Peters et al., 1990; Zeiler et al., 2012; Meyer and Weihl, 2014). Only transfection of VCP truncated of the domain 2 (ND1 mutant) showed an inhibitory effect on Gluc production. This effect is not surprising as this truncated VCP is a known dominant-negative mutant of VCP (Ye et al., 2003; van den Boom et al., 2016). These results are consistent with both siRNA and chemical inhibitors effects.
Interestingly, a genome-wide screen on insect cells had previously identified VCP as a proviral entry factor for SINV through the downregulation of its receptor NRAMP2 (Rose et al., 2011; Panda et al., 2013). However, we did not observe a role for VCP as a binding factor of CHIKV (Figures 3A,B), consistent with the fact that CHIKV and other alphaviruses binding and entry is NRAMP2 independent (Rose et al., 2011). This observation was further verified using an entry assay (Figure 3C), which showed an effect of VCP inhibition on intracellular viral levels consistent with a role of VCP during viral RNA replication. Therefore, excluding a role during viral binding or entry in our experimental setup. However, we cannot exclude that VCP inhibition prior to infection could also result in lower trafficking of viral receptor or other entry factors.
Nevertheless, the trans-replication assay further confirmed the role of VCP inhibition during viral RNA replication, independent of the stability and template availability of alphavirus nsPs (Figure 4). In addition, viral replicase components nsP1, 2, and 3 were demonstrated to co-localize partially with VCP differentially (Figure 5). This difference could be due to the fact that a significant portion of nsP1 is anchored in the plasma membrane while VCP, nsP2, and nsP3 are cytoplasmic. These results are consistent with earlier studies with SFV and HCV showing association of VCP with their replication complexes (Varjak et al., 2013; Yi et al., 2016), and the role of VCP during virus RNA replication of poliovirus, enterovirus 71, human cytomegalovirus, West Nile virus, and HCV (Arita et al., 2012; Wu et al., 2016; Yi et al., 2016; Lin et al., 2017; Phongphaew et al., 2017; Wang et al., 2017; Yi and Yuan, 2017). Interestingly, VCP was also involved in the egress of viral particles for poliovirus and Rift Valley fever virus (RVFV) (Arita et al., 2012; Brahms et al., 2017). The results on RVFV are of particular interest, where the study identified VCP as the target of FDA-approved drug Sorafenib effect on RVFV (Benedict et al., 2015; Brahms et al., 2017). The same group has also identified Sorafenib as a potential drug that reduces replication of several alphaviruses including CHIKV (Lundberg et al., 2018). It would be interesting to verify if Sorafenib inhibits alphaviruses in a VCP-dependant manner similar to RVFV.
Interestingly, we could not observe co-localization of VCP with dsRNA foci of replication complexes (Figure 6A). This is likely due to the unavailability of the VCP N-terminal for recognition with the antibody used, which is reinforced by the lack of co-localization of the VCP antibodies recognizing the C- and N-termini (Figure 6B). Importantly, the apparent re-localization of VCP with an available C-terminal at the infected cell membrane is consistent with the localization of the dsRNA foci. Further studies will be needed to assess if and how VCP is involved in the replication complexes of CHIKV and other alphaviruses.
Valosin-containing protein is an essential host protein involved in a wide range of cellular processes (Meyer and Weihl, 2014). A link between one or more of the described VCP functions and viral replication remains poorly understood. For example during infection, virus could use VCP to hijack ER or mitochondria-associated degradation, proteasome, or UPS modulating functions (Taylor and Rutter, 2011; Wolf and Stolz, 2012). Proteasome activity was reported to be essential for CHIKV replication (Karpe et al., 2016), and CHIKV can manipulate the UPS to prevent host-cell shut-off (Fros et al., 2015). Along the same line, CHIKV could use VCP modulating function on autophagy. Such mechanism could explain how CHIKV induces autophagy (Papadopoulos and Meyer, 2017) to delay cell death and increase replication (Joubert et al., 2012a, b). It is also plausible that VCP’s role in membrane rearrangement (Zhang and Wang, 2015) could be hijacked by the virus in order to form invaginated membranous structures of the replication complexes efficiently (Reid et al., 2015). This would explain VCP co-localization with CHIKV nsPs (Figure 5), and its presence in replication complexes of HCV and SFV (Varjak et al., 2013; Yi et al., 2016).
Similarly, VCP is important for lipid droplets metabolism (Olzmann et al., 2013), a site at which CHIKV nsP3 was also shown to associate (Remenyi et al., 2017). Moreover, lipid droplets metabolism was also shown to be important for flavivirus replication (Zhang et al., 2017), in which VCP function as a proviral host factor for virus replication (Phongphaew et al., 2017). VCP unfolding of aggregated proteins could also be a necessary function for viruses, for example during HCV infection, VCP prevents NS5A from being over aggregated and non-functional (Yi and Yuan, 2017). Other cellular functions of VCP, such as NF-κB activation (Schweitzer et al., 2016), stalled ribosome turnover (Verma et al., 2013), full capacity of autophagy and lysosomal system (Papadopoulos and Meyer, 2017), are cellular functions and pathways that could possess pro- or anti-viral functions. A link between virus interaction with VCP and modulation of these functions could also present avenues for future investigations.
To conclude, we have shown that VCP, a highly conserved and connected protein with functions in multiple cellular processes has proviral functions during CHIKV replication. This effect extends to several other alphaviruses and understanding the exact role played by VCP during virus replication could shed light on a highly conserved viral subversion mechanism of cellular machineries via the multi-purpose protein VCP. This accumulating evidence suggest that understanding of such mechanism could open the way for designing new treatment strategies effective against a wide range of viruses by targeting a conserved VCP proviral role.
Human embryonic kidney HEK293T cells (ATCC-CRL-3216), human foreskin fibroblast BJ cells (ATCC-CRL-2522), human muscle RD cells (ATCC-CCL-136), and African green monkey kidney Vero E6 cells (ATCC-CRL-1586) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Gibco); and for BJ cells supplemented with 1× Penicillin–Streptomycin (Gibco). All cells were maintained at 37°C with 5% CO2.
Chikungunya virus infectious clone plasmids containing a duplicated subgenomic promoter and insertion of Gluc (Gaussia princeps luciferase) or ZsGreen (Anthozoa reef coral fluorescent protein) between non-structural and structural regions were constructed for isolates of La Reunion (LR2006 OPY1), the CRB region (CNR20235), and the IND region. The CHIKV IND strain was isolated on BHK cells from the cerebral spinal fluid of an IND patient during the 2010 epidemic (gift from Dr. Ooi Eng Eong, DUKE-NUS Graduate Medical School, Singapore) and subsequently passaged on Vero E6 cells. Infectious clones for ONNV (Bessaud et al., 2006) and SFV6 (Ferguson et al., 2015) were constructed in a similar fashion except that for SFV6 CMV promoter was placed upstream of viral sequence and duplicated subgenomic promoter and reporter gene were placed downstream of structural region. CHIKV and ONNV were produced using in vitro transcribed viral RNA from NotI (NEB) linearized plasmids using SP6 mMessage mMachine (Ambion). Viral RNA was then transfected in Vero E6 cells using Lipofectamine 2000 (Invitrogen) following manufacturer’s recommendation. SFV6 was produced by transfection of plasmid containing infectious cDNA in HEK293T cells. Viruses were passaged once or twice in Vero E6 cells to obtain a sufficient titer. Virus stock titration was performed by standard plaque assay on Vero E6 cells in a 6-well plate format.
The CHIKV trans-replication system previously published (Utt et al., 2015, 2016, 2019) was modified by insertion of sequence encoding for a fluorescent tdTomato reporter in the template and used a human RNA polymerase I promoter instead of T7 promoter for synthesis of non-capped RNAs in transfected cells (Abraham et al., 2018). Briefly, we substituted the T7 promoter and terminator in the T7-Fluc-Gluc template (Utt et al., 2015) to human RNA polymerase I promoter and terminator (Abraham et al., 2018); the sequence corresponding to the 5′-end of CHIKV genome was placed to the start site of promoter. The Gluc reporter gene was substituted by sequence encoding tdTomato using restriction-based cloning; obtained construct was designated Pol1HS-Fluc-tdTomato. The detailed description of properties of such a system were published elsewhere (Utt et al., 2019). For verification of template generation from plasmid 2, HEK293T cells were plated at a density of 150,000 cells per well in a 12-well plate. A total of 333 ng of plasmid 2 per well was transfected using Lipofectamine 2000 (Invitrogen) following manufacturer recommendations. For the full trans-replication assay, HEK293T cells were plated at a density of 400,000 cells per well in a six-well plate. Two micrograms of plasmid 1 and 1 μg of plasmid 2 (Figure 4A) per well were co-transfected using Lipofectamine 2000. At 5–6 h post-transfection (hpt), media was refreshed for media with 1 μM CB-5083 or DMSO control. At 16 h post-media refresh cells were collected and RNA was extracted using Nucleospin RNA kit (Macherey-Nagel) following manufacturer protocol or cells were collected and half was lyzed in RIPA buffer for immunoblotting and the other half was processed for flow cytometry.
Predesigned dicer substrate siRNAs against VCP (hs.Ri.VCP.13) were purchased from IDT [TriFECTa kit containing non-targeting control 1 (NC1) and TYE593 labeled control siRNA]. HEK293T cells were plated either in 12-well plate or 96-well plates at a density of 100,000 or 10,000 cells/well and siRNA was transfected at 10 nM final concentration using a ratio of 1 μL of Lipofectamine 2000 for 5 pmol of DsiRNA. 24 hpt of TYE593-labeled control siRNA indicated >98% of transfection efficiency (not shown). At 48 h post siRNA transfection, cells were infected overnight with Gluc- or ZsGreen-tagged CHIKV, followed by RT-qPCR, plaque assay, or luciferase assay of supernatant, or flow cytometry analysis of cells, respectively. VCP expression in cell lysate was verified by western blot at 48 hpt and time of supernatant collection.
DBeQ, NMS-873, and CB-5083 were purchased from ApexBio Technology LLC and resuspended in DMSO at a final concentration of 50 mM, OLX (Selleckchem) was resuspended in DMSO at a final concentration of 100 mM. HEK293T cells were plated at 30,000 cells/well in a 96-well format (on fibronectin-coated plates for Figure 2). After adhesion, Gluc-expressing viruses at a MOI of 0.1 (3,000 pfu) or MOI 1 for Figure 2, and serially diluted compounds were added to the cells. For Figure 2, inoculums were removed as the times indicated in the legends before diluted compounds were added back on the cells. After 16 h of incubation, 50 μL of supernatant was used to determine luminescence as a proxy for viral replication. Alternatively, ZsGreen-expressing viruses were used at MOI 1.
HEK293T cells were plated at a density of 200,000 cells per well in 24-well plates. After adhesion, cells were infected at a MOI of 10 in a volume of 150 μL of DMEM media with CB-5083 at 2 μM (and DMSO control) on ice or at 37°C with 5% CO2 for 1 h. Alternatively, cells were infected for 2 h in complete or in complete media with 0.5 μM OLX, inoculum was removed and cells were washed with PBS. Complete media containing 0.5 μM OLX or 0.5 μM OLX with 2 μM CB-5083 was then applied to the cells for 4 h. Cells were then washed, collected in PBS, and RNA was extracted using Nucleospin RNA kit (Macherey-Nagel) following manufacturer protocol.
For relative quantitation, eluted RNA were quantified using a NanoDrop 1000 Spectrophotometer (Thermo Scientific), diluted to a concentration of approximately 10 ng/μl, and subjected to qRT-PCR using a QuantiFast SYBR green RT-PCR kit (Qiagen) according to the manufacturer’s protocol. qRT-PCR was performed with an Applied Biosystems (ABI) 7900HT Fast Real-Time PCR system in 364-well plate format, with the following conditions: (a) reverse transcription step (50°C for 10 min; 1 cycle); (b) PCR initial activation step (95°C for 5 min; 1 cycle); and (c) 2-step cycling (95°C for 10 s, followed by 60°C for 30 s; 40 cycles). The ΔCt are CT values of CHIKV E1 encoding RNA (or Fluc RNA) – GAPDH (housekeeping gene) were determined within samples. The fold change relative to control samples was determined using the ΔΔCt method using Microsoft Excel 2016. Briefly, ΔΔCT was calculated as ΔCT[sample] – ΔCT[control sample]. The fold change for CHIKV E1 RNA between the sample and control was calculated as 2-ΔΔCT.
For viral RNA quantification in supernatants, 140 μL of infected cell supernatant was extracted using QIAmp Viral RNA kit (Qiagen), according to the manufacturer’s protocol. CHIKV viral genome copies were quantified by Taqman RT-qPCR targeting viral RNA negative sense at the nsP1 region, as previously described (Kam et al., 2012; Teo et al., 2013; Carissimo et al., 2019). Briefly, qRT-PCR was performed with an Applied Biosystems (ABI) 7900HT Fast Real-Time PCR system in 364-well plate format, with the following conditions: (a) reverse transcription step (50°C for 30 min; 1 cycle); (b) PCR initial activation step (95°C for 15 min; 1 cycle); and (c) 2-step cycling (94°C for 15 s, followed by 55°C 1 min; 45 cycles). Viral RNA copy numbers were extrapolated using the SDS software and the standard curve of serially diluted CHIKV nsP1 RNA in vitro produced transcripts ranging from 10 to 109 copy per μL. Primers used in this study are given in Table 1.

Table 1. Primers used in this study given in 5′–3′ orientation.
Luminescence was assessed using the GloMax Multi-detection system (Promega) at 10-s integration time with a 2 s delay after injection 50 μL of 20 μM XenoLight RediJect coelenterazine h (PerkinElmer) diluted in PBS (Gibco) with 5 mM NaCl (Ambion). Results are presented as a percentage of luminescence of treated wells relative to luminescence of control treated wells.
Cell viability assays were carried out with the CellTiter-Glo luminescent cell viability assay (Promega), which serves as a readout for cellular ATP levels, as previously described (Ching et al., 2015). Briefly, serial dilutions of DBeQ, NMS-873, and CB-5083 were added to HEK-293T seeded on 96-well fibronectin-coated plates. After 16 h of incubation, cell viability assays using CellTiter-Glo Reagent (Promega) were performed as per the manufacturer’s protocol. After 1 h of incubation at room temperature, luminescent emissions were detected by the GloMax Multi-detection system (Promega).
For HEK 293T, cells from technical replicates were resuspended in their culture media, pooled, washed with PBS, fixed for 5 min in BD FACS Lysing Solution (BD Biosciences), and then acquired on LSR Fortessa flow cytometer (BD Biosciences) on the FITC or PE channel. Flow cytometry analysis was performed on minimum 30,000 cells per replicate with Flowjo 10.1 (Flowjo LLC) and single cells were assessed for FITC or PE positive signal. Data are expressed as % of infected cells in treated condition relative to % infected cells in control condition or as mean of PE intensity of PE positive cells of treated sample relative to mean of PE intensity of PE positive cells of control sample. FCS files are available as Supplementary File S1.
Microscopy images were acquired with a 10× zoom on an Olympus epi-fluorescent microscope (IX81) at a fixed exposure time for the FITC filter or auto exposure for the brightfield filter with CellSens Dimension software (Olympus). Tiff images were opened in Photoshop (Adobe) and light and contrast were adjusted using the exact same parameters for paired images and converted to jpeg format for inclusion on the figures. Unmodified tiff images are available on request.
Valosin-containing protein and VCP domains N and ND1 were amplified by PCR from HEK293T cDNA using the primers indicated in Table 1 and Q5 HiFi polymerase (NEB) following manufacturer recommendations. PCR were ligated in p3XFLAG-CMVTM-10 (Sigma) using NotI and HindIII restriction digestions (NEB) to generate flag-tagged VCP full length expression plasmid (FL) and mutants N and ND1. D2 domain was generated using site-directed mutagenesis (SDM) kit from NEB following manufacturer recommendations and primers given in Table 1. All plasmids were verified by sanger sequencing before use and correct expression was confirmed by western blot using monoclonal mouse anti-flag M2 (Sigma, ref: F3165) (not shown).
Cells were dislodged from the plates in PBS, pelleted by centrifugation for 5 min at 400 × g, and lysed in RIPA buffer (Invitrogen) and then sample were mixed 1:1 (v/v) with 2× Laemmli buffer (Bio-Rad) with 5% β-mercaptoethanol (Sigma), and heated for 10 min at 95°C. Samples were then migrated on 4–15% TGX mini-protean gels (Bio-Rad) and transferred to nitrocellulose membrane (Bio-Rad) using a semi-dry system and Bjerrum Schafer-Nielsen Buffer [48 mM UltraPure Tris (Invitrogen), 38 mM Glycine (Bio-Rad), 20% (v/v) EMSURE Methanol (EMD Millipore)]. Membranes were then blocked in 5% (wt/v) non-fat milk powder (Nacalai Tesque) in 1× Tris Buffer Saline (1st Base) with Tween 0.1% (v/v) (Sigma) for 1 h before staining. Antibody used for staining were mouse monoclonal anti-human VCP (clone 4G9, Biolegend), mouse monoclonal anti-human GAPDH (clone FF26A/F9, Biolegend), anti-CHIKV nsP1 affinity purified IgGs produced in rabbit (in-house), and goat anti-mouse and goat anti-rabbit antibodies coupled to HRP were purchased from Santa Cruz Biotechnology (sc-2004 and sc-2005). Proteins were revealed using WesternBright ECL HRP substrate (Advansta), signal was acquired on a ChemiDoc screen touch model 2017 (Bio-Rad), and non-saturated images were quantified using ImageLab 2.3 software (Bio-Rad). Raw images of western blot are available on request.
HEK293T cells were infected in 6-well plates for 2 h, washed, and trypsinized before replating in complete media on ibidi μ-slide 8-well plates coated with fibronectin. At 5 h post-replating, cells were washed twice with PBS and fixed in 4% paraformaldehyde-PBS solution for 10 min slides were then washed three times in PBS and incubated 30 min in permabilization and blocking buffer (PBS with 0.1% Tween 20, 0.1% Triton X-100, 3% BSA, and 5% FBS). Cells were then incubated 2 h at room temperature with mouse anti-VCP (1:1000 dilution, Santa Cruz Biotechnology, sc-57492, monoclonal against C-terminal), rabbit anti-VCP (1:500 dilution, Thermofisher, PA522257), mouse monoclonal anti-dsRNA (3 μg/mL dilution, Scicons, J2-1820), and rabbit anti-nsP1 or nsP2 or nsP3 (in-house) at a dilution of 1:1000. Slides were then washed four times with PBS 0.1% Tween 20 and incubated for 1 h with anti-rabbit AF555 and anti-mouse AF647 (Thermofisher A21428, A21236) at 1:2000 dilution. Slides were then washed four times in PBS 0.1% Tween 20, washed three times with PBS and Prolong Gold antifade reagent with DAPI (molecular PROBES, P36935) was added to the wells. Images were acquired on an Olympus microscope (IX81) equipped with a confocal system (FV-1000) using the FV10-ASW version 04.02.02.09. Images were analyzed with Imaris version 9.2 (Bitplane) for Pearson’s correlation coefficient for co-localization between AF555 and AF647 channels using the automatic thresholding function. Confocal images presented on the figures were exported as tiff files using the FV10-ASW software and incorporated in the figure without modifications.
Statistical analyses were performed using GraphPad Prism versions 7.0–8.1 for macOS (GraphPad Software), using statistical tests as mentioned in the figure legends. P-values considered statistically significant are represented with ∗ for p < 0.05, ∗∗ for p < 0.01, and ∗∗∗ for p < 0.001.
The datasets generated for this study are available on request to the corresponding author.
GC conceived and designed the experiments and analyzed the data. GC, Y-HC, AU, FA, and T-KC performed the experiments. GC, AM, and LN wrote the manuscript. AU and AM contributed reagents and materials. All authors reviewed and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by the Agency for Science, Technology and Research (A∗STAR) core grant. Y-HC and FA were supported by an A∗STAR Graduate Scholarship (AGS). Flow cytometry platform is part of the SIgN Immunomonitoring platform and supported by the grant NRF2017_SISFP09. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Prof. Ooi Eng Eong for providing passage one of the IND isolate used in this study. We would also like to thank Drs. Kai-Er Eng, Teck-Hui Teo, and Fok-Moon Lum who critically read the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02236/full#supplementary-material
FIGURE S1 | Viral RNA quantification, plaque assay, and luciferase luminescence show similar results. A hundred thousand HEK 293T cells per well were transfected with siRNA at 10 nM for 48 h. Cells were then infected with 10,000 pfu of Gluc-tagged CHIKV-LR. (A) At 16 hpi, supernatant was collected from each well for viral RNA RT-qPCR, plaque assay and luminescence reading. (B) Spearman correlations of the different readings for the results presented in panel A. Results are from two independent experiment with 2–3 well per condition in each experiment.
FIGURE S2 | (A) The schematic representation of flag-tagged VCP and VCP domain constructs. (B) HEK293T were plated in 96-well format at 30,000 cells per well. At 1 day post-plating, cells were transfected with 100 ng per well of indicated VCP constructs or empty plasmid control. At 1 day post-transfection, cells were infected using 3,000 pfu per well of Gluc-tagged CHIKV and incubated for 16 h. At which time, 50 μL of supernatant was collected for luciferase assay. Data of panel B are representative of four independent experiment presented as mean ± SD and analyzed by Mann–Whitney non-parametric two-tailed test; ∗p < 0.05.
FILE S1 | Flow cytometry fcs files generated for Figures 1, 4.
Abraham, R., Hauer, D., McPherson, R. L., Utt, A., Kirby, I. T., Cohen, M. S., et al. (2018). ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc. Natl. Acad. Sci. U.S.A. 115, E10457–E10466. doi: 10.1073/pnas.1812130115
Anderson, D. J., Le Moigne, R., Djakovic, S., Kumar, B., Rice, J., Wong, S., et al. (2015). Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28, 653–665. doi: 10.1016/j.ccell.2015.10.002
Arita, M., Wakita, T., and Shimizu, H. (2012). Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J. Virol. 86, 5541–5553. doi: 10.1128/JVI.00114-12
Barthelme, D., and Sauer, R. T. (2016). Origin and functional evolution of the cdc48/p97/vcp aaa+ protein unfolding and remodeling machine. J. Mol. Biol. 428(9 Pt B), 1861–1869. doi: 10.1016/j.jmb.2015.11.015
Bartholomeeusen, K., Utt, A., Coppens, S., Rausalu, K., Vereecken, K., Arien, K. K., et al. (2018). A Chikungunya virus trans-replicase system reveals the importance of delayed non-structural polyprotein processing for efficient replication complex formation in mosquito cells. J. Virol. 92:e00152-e18.
Benedict, A., Bansal, N., Senina, S., Hooper, I., Lundberg, L., de la Fuente, C., et al. (2015). Repurposing FDA-approved drugs as therapeutics to treat Rift Valley fever virus infection. Front. Microbiol. 6:676. doi: 10.3389/fmicb.2015.00676
Bessaud, M., Peyrefitte, C. N., Pastorino, B. A., Gravier, P., Tock, F., Boete, F., et al. (2006). O’nyong-nyong virus. Chad. Emerg. Infect. Dis. 12, 1248–1250.
Brahms, A., Mudhasani, R., Pinkham, C., Kota, K., Nasar, F., Zamani, R., et al. (2017). Sorafenib impedes rift valley fever virus egress by inhibiting valosin-containing protein function in the cellular secretory pathway. J. Virol. 91:e00968-e17. doi: 10.1128/JVI.00968-17
Carissimo, G., Teo, T. H., Chan, Y. H., Lee, C. Y., Lee, B., Torres-Ruesta, A., et al. (2019). Viperin controls chikungunya virus-specific pathogenic T cell IFNgamma Th1 stimulation in mice. Life Sci. Alliance 2:e201900298. doi: 10.26508/lsa.201900298
Ching, K. C., Kam, Y. W., Merits, A., Ng, L. F., and Chai, C. L. (2015). Trisubstituted thieno[3,2-b]pyrrole 5-carboxamides as potent inhibitors of alphaviruses. J. Med. Chem. 58, 9196–9213. doi: 10.1021/acs.jmedchem.5b01047
Erzberger, J. P., and Berger, J. M. (2006). Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114. doi: 10.1146/annurev.biophys.35.040405.101933
Fang, C. J., Gui, L., Zhang, X., Moen, D. R., Li, K., Frankowski, K. J., et al. (2015). Evaluating p97 inhibitor analogues for their domain selectivity and potency against the p97-p47 complex. ChemMedChem 10, 52–56. doi: 10.1002/cmdc.201402420
Ferguson, M. C., Saul, S., Fragkoudis, R., Weisheit, S., Cox, J., Patabendige, A., et al. (2015). Ability of the encephalitic arbovirus semliki forest virus to cross the blood-brain barrier is determined by the charge of the E2 glycoprotein. J. Virol. 89, 7536–7549. doi: 10.1128/JVI.03645-14
Fros, J. J., Major, L. D., Scholte, F. E. M., Gardner, J., van Hemert, M. J., Suhrbier, A., et al. (2015). Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J. Gen. Virol. 96(Pt 3), 580–589. doi: 10.1099/vir.0.071845-0
Hanzelmann, P., and Schindelin, H. (2017). The interplay of cofactor interactions and post-translational modifications in the regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 4:21. doi: 10.3389/fmolb.2017.00021
Joubert, P. E., Werneke, S., de la Calle, C., Guivel-Benhassine, F., Giodini, A., Peduto, L., et al. (2012a). Chikungunya-induced cell death is limited by ER and oxidative stress-induced autophagy. Autophagy 8, 1261–1263. doi: 10.4161/auto.20751
Joubert, P. E., Werneke, S. W., de la Calle, C., Guivel-Benhassine, F., Giodini, A., Peduto, L., et al. (2012b). Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 209, 1029–1047. doi: 10.1084/jem.20110996
Kam, Y. W., Lum, F. M., Teo, T. H., Lee, W. W., Simarmata, D., Harjanto, S., et al. (2012). Early neutralizing IgG response to Chikungunya virus in infected patients targets a dominant linear epitope on the E2 glycoprotein. EMBO Mol. Med. 4, 330–343. doi: 10.1002/emmm.201200213
Karpe, Y. A., Pingale, K. D., and Kanade, G. D. (2016). Activities of proteasome and m-calpain are essential for Chikungunya virus replication. Virus Genes 52, 716–721. doi: 10.1007/s11262-016-1355-5
Lin, Y. T., Prendergast, J., and Grey, F. (2017). The host ubiquitin-dependent segregase VCP/p97 is required for the onset of human cytomegalovirus replication. PLoS Pathog. 13:e1006329. doi: 10.1371/journal.ppat.1006329
Lundberg, L., Brahms, A., Hooper, I., Carey, B., Lin, S. C., Dahal, B., et al. (2018). Repurposed FDA-Approved drug sorafenib reduces replication of Venezuelan equine encephalitis virus and other alphaviruses. Antiviral Res. 157, 57–67. doi: 10.1016/j.antiviral.2018.07.005
Lyupina, Y. V., Erokhov, P. A., Kravchuk, O. I., Finoshin, A. D., Abaturova, S. B., Orlova, O. V., et al. (2018). Essential function of VCP/p97 in infection cycle of the nucleopolyhedrovirus AcMNPV in Spodoptera frugiperda Sf9 cells. Virus Res. 253, 68–76. doi: 10.1016/j.virusres.2018.06.001
Magnaghi, P., D’Alessio, R., Valsasina, B., Avanzi, N., Rizzi, S., Asa, D., et al. (2013). Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 9, 548–556. doi: 10.1038/nchembio.1313
Meyer, H., Bug, M., and Bremer, S. (2012). Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Chem. Biol. 14, 117–123. doi: 10.1038/ncb2407
Meyer, H., and Weihl, C. C. (2014). The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127(Pt 18), 3877–3883. doi: 10.1242/jcs.093831
Olzmann, J. A., Richter, C. M., and Kopito, R. R. (2013). Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc. Natl. Acad. Sci. U.S.A. 110, 1345–1350. doi: 10.1073/pnas.1213738110
Panda, D., Rose, P. P., Hanna, S. L., Gold, B., Hopkins, K. C., Lyde, R. B., et al. (2013). Genome-wide RNAi screen identifies SEC61A and VCP as conserved regulators of Sindbis virus entry. Cell Rep. 5, 1737–1748. doi: 10.1016/j.celrep.2013.11.028
Papadopoulos, C., and Meyer, H. (2017). Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr. Biol. 27, R1330–R1341. doi: 10.1016/j.cub.2017.11.012
Peters, J. M., Walsh, M. J., and Franke, W. W. (1990). An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J. 9, 1757–1767. doi: 10.1002/j.1460-2075.1990.tb08300.x
Phongphaew, W., Kobayashi, S., Sasaki, M., Carr, M., Hall, W. W., Orba, Y., et al. (2017). Valosin-containing protein (VCP/p97) plays a role in the replication of West Nile virus. Virus Res. 228, 114–123. doi: 10.1016/j.virusres.2016.11.029
Ramadan, K., Halder, S., Wiseman, K., and Vaz, B. (2017). Strategic role of the ubiquitin-dependent segregase p97 (VCP or Cdc48) in DNA replication. Chromosoma 126, 17–32. doi: 10.1007/s00412-016-0587-4
Reid, C. R., Airo, A. M., and Hobman, T. C. (2015). The virus-host interplay: biogenesis of +RNA replication complexes. Viruses 7, 4385–4413. doi: 10.3390/v7082825
Remenyi, R., Roberts, G. C., Zothner, C., Merits, A., and Harris, M. (2017). SNAP-tagged Chikungunya virus replicons improve visualisation of non-structural protein 3 by fluorescence microscopy. Sci. Rep. 7:5682. doi: 10.1038/s41598-017-05820-0
Rose, P. P., Hanna, S. L., Spiridigliozzi, A., Wannissorn, N., Beiting, D. P., Ross, S. R., et al. (2011). Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammalian hosts. Cell Host Microbe. 10, 97–104. doi: 10.1016/j.chom.2011.06.009
Schweitzer, K., Pralow, A., and Naumann, M. (2016). p97/VCP promotes Cullin-RING-ubiquitin-ligase/proteasome-dependent degradation of IkappaBalpha and the preceding liberation of RelA from ubiquitinated IkappaBalpha. J. Cell Mol. Med. 20, 58–70. doi: 10.1111/jcmm.12702
Suhrbier, A., Jaffar-Bandjee, M. C., and Gasque, P. (2012). Arthritogenic alphaviruses–an overview. Nat. Rev. Rheumatol. 2012, 420–429. doi: 10.1038/nrrheum.2012.64
Tang, W. K., Odzorig, T., Jin, W., and Xia, D. (2019). Structural basis of p97 inhibition by the site-selective anticancer compound CB-5083. Mol. Pharmacol. 95, 286–293. doi: 10.1124/mol.118.114256
Taylor, E. B., and Rutter, J. (2011). Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509–1513. doi: 10.1042/BST0391509
Teo, T. H., Her, Z., Tan, J. J., Lum, F. M., Lee, W. W., Chan, Y. H., et al. (2015). Caribbean and La Reunion Chikungunya virus isolates differ in their capacity to induce proinflammatory Th1 and NK Cell responses and acute joint pathology. J. Virol. 89, 7955–7969. doi: 10.1128/JVI.00909-15
Teo, T. H., Lum, F. M., Claser, C., Lulla, V., Lulla, A., Merits, A., et al. (2013). A pathogenic role for CD4+ T cells during Chikungunya virus infection in mice. J. Immunol. 190, 259–269. doi: 10.4049/jimmunol.1202177
Torrecilla, I., Oehler, J., and Ramadan, K. (2017). The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160282. doi: 10.1098/rstb.2016.0282
Utt, A., Das, P. K., Varjak, M., Lulla, V., Lulla, A., and Merits, A. (2015). Mutations conferring a noncytotoxic phenotype on chikungunya virus replicons compromise enzymatic properties of nonstructural protein 2. J. Virol. 89, 3145–3162. doi: 10.1128/JVI.03213-14
Utt, A., Quirin, T., Saul, S., Hellstrom, K., Ahola, T., and Merits, A. (2016). Versatile trans-replication systems for Chikungunya virus allow functional analysis and tagging of every replicase protein. PLoS One 11:e0151616. doi: 10.1371/journal.pone.0151616
Utt, A., Rausalu, K., Jakobson, M., Mannik, A., Alphey, L., Fragkoudis, R., et al. (2019). Design and use of Chikungunya virus replication templates utilizing mammalian and mosquito RNA polymerase I-mediated transcription. 93:e00794-19. J. Virol.
van den Boom, J., and Meyer, H. (2018). VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell. 69, 182–194. doi: 10.1016/j.molcel.2017.10.028
van den Boom, J., Wolf, M., Weimann, L., Schulze, N., Li, F., Kaschani, F., et al. (2016). VCP/p97 Extracts Sterically Trapped Ku70/80 Rings from DNA in Double-Strand Break Repair. Mol. Cell. 64, 189–198. doi: 10.1016/j.molcel.2016.08.037
Varghese, F. S., Rausalu, K., Hakanen, M., Saul, S., Kummerer, B. M., Susi, P., et al. (2017). Obatoclax inhibits alphavirus membrane fusion by neutralizing the acidic environment of endocytic compartments. Antimicrob. Agents Chemother. 61:e02227-e16. doi: 10.1128/AAC.02227-16
Varjak, M., Saul, S., Arike, L., Lulla, A., Peil, L., and Merits, A. (2013). Magnetic fractionation and proteomic dissection of cellular organelles occupied by the late replication complexes of Semliki Forest virus. J. Virol. 87, 10295–10312. doi: 10.1128/JVI.01105-13
Verma, R., Oania, R. S., Kolawa, N. J., and Deshaies, R. J. (2013). Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2:e00308. doi: 10.7554/eLife.00308
Wang, T., Wang, B., Huang, H., Zhang, C., Zhu, Y., Pei, B., et al. (2017). Enterovirus 71 protease 2Apro and 3Cpro differentially inhibit the cellular endoplasmic reticulum-associated degradation (ERAD) pathway via distinct mechanisms, and enterovirus 71 hijacks ERAD component p97 to promote its replication. PLoS Pathog. 13:e1006674. doi: 10.1371/journal.ppat.1006674
Wolf, D. H., and Stolz, A. (2012). The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta. 1823, 117–124. doi: 10.1016/j.bbamcr.2011.09.002
Wong, H. H., Kumar, P., Tay, F. P., Moreau, D., Liu, D. X., and Bard, F. (2015). Genome-wide screen reveals valosin-containing protein requirement for coronavirus exit from endosomes. J. Virol. 89, 11116–11128. doi: 10.1128/JVI.01360-15
Wu, K. X., Phuektes, P., Kumar, P., Goh, G. Y., Moreau, D., Chow, V. T., et al. (2016). Human genome-wide RNAi screen reveals host factors required for enterovirus 71 replication. Nat. Commun. 7:13150. doi: 10.1038/ncomms13150
Ye, Y., Meyer, H. H., and Rapoport, T. A. (2003). Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84. doi: 10.1083/jcb.200302169
Ye, Y., Tang, W. K., Zhang, T., and Xia, D. (2017). A mighty “protein extractor” of the cell: structure and function of the p97/CDC48 ATPase. Front. Mol. Biosci. 4:39. doi: 10.3389/fmolb.2017.00039
Yi, Z., Fang, C., Zou, J., Xu, J., Song, W., Du, X., et al. (2016). Affinity purification of the hepatitis C virus replicase identifies valosin-containing protein, a member of the ATPases associated with diverse cellular activities family, as an active virus replication modulator. J. Virol. 90, 9953–9966. doi: 10.1128/JVI.01140-16
Yi, Z., and Yuan, Z. (2017). Aggregation of a hepatitis C virus replicase module induced by ablation of p97/VCP. J. Gen Virol. 98, 1667–1678. doi: 10.1099/jgv.0.000828
Zeiler, M., Straube, W. L., Lundberg, E., Uhlen, M., and Mann, M. (2012). A protein epitope signature tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol. Cell. Proteomics 11:O111009613. doi: 10.1074/mcp.O111.009613
Zhang, J., Lan, Y., and Sanyal, S. (2017). Modulation of lipid droplet metabolism-a potential target for therapeutic intervention in flaviviridae infections. Front. Microbiol. 8:2286. doi: 10.3389/fmicb.2017.02286
Keywords: VCP, VCP/p97, alphavirus, chikungunya, host factor, proviral factor
Citation: Carissimo G, Chan Y-H, Utt A, Chua T-K, Abu Bakar F, Merits A and Ng LFP (2019) VCP/p97 Is a Proviral Host Factor for Replication of Chikungunya Virus and Other Alphaviruses. Front. Microbiol. 10:2236. doi: 10.3389/fmicb.2019.02236
Received: 24 April 2019; Accepted: 11 September 2019;
Published: 24 September 2019.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Victor R. DeFilippis, Oregon Health & Science University, United StatesCopyright © 2019 Carissimo, Chan, Utt, Chua, Abu Bakar, Merits and Ng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa F. P. Ng, bGlzYV9uZ0BpbW11bm9sLmEtc3Rhci5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.