
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 11 September 2019
Sec. Extreme Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.02104
This article is part of the Research TopicLiving with Salt: Genetics and Ecology of HalophilesView all 17 articles
 Rafael R. de la Haba1
Rafael R. de la Haba1 Clara López-Hermoso1
Clara López-Hermoso1 Cristina Sánchez-Porro1
Cristina Sánchez-Porro1 Konstantinos T. Konstantinidis2
Konstantinos T. Konstantinidis2 Antonio Ventosa1*
Antonio Ventosa1*In the genomic era phylogenetic relationship among prokaryotes can be inferred from the core orthologous genes (OGs) or proteins in order to elucidate their evolutionary history and current taxonomy should benefits of that. The genus Salinivibrio belongs to the family Vibrionaceae and currently includes only five halophilic species, in spite the fact that new strains are very frequently isolated from hypersaline environments. Species belonging to this genus have undergone several reclassifications and, moreover, there are many strains of Salinivibrio with available genomes which have not been affiliated to the existing species or have been wrongly designated. Therefore, a phylogenetic study using the available genomic information is necessary to clarify the relationships of existing strains within this genus and to review their taxonomic affiliation. For that purpose, we have also sequenced the first complete genome of a Salinivibrio species, Salinivibrio kushneri AL184T, which was employed as a reference to order the contigs of the draft genomes of the type strains of the current species of this genus, as well as to perform a comparative analysis with all the other available Salinivibrio sp. genomes. The genome of S. kushneri AL184T was assembled in two circular chromosomes (with sizes of 2.84 Mb and 0.60 Mb, respectively), as typically occurs in members of the family Vibrionaceae, with nine complete ribosomal operons, which might explain the fast growing rate of salinivibrios cultured under laboratory conditions. Synteny analysis among the type strains of the genus revealed a high level of genomic conservation in both chromosomes, which allow us to hypothesize a slow speciation process or homogenization events taking place in this group of microorganisms to be tested experimentally in the future. Phylogenomic and orthologous average nucleotide identity (OrthoANI)/average amino acid identity (AAI) analyses also evidenced the elevated level of genetic relatedness within members of this genus and allowed to group all the Salinivibrio strains with available genomes in seven separated species. Genome-scale attribute study of the salinivibrios identified traits related to polar flagellum, facultatively anaerobic growth and osmotic response, in accordance to the phenotypic features described for species of this genus.
Firstly described by Mellado et al. (1996) to accommodate the species Vibrio costicola (Smith, 1938), members of the genus Salinivibrio, belonging to the family Vibrionaceae, class Gammaproteobacteria, have been isolated from diverse habitats, such as aquatic hypersaline systems, brines, salted meats, and saline soils (Chamroensaksri et al., 2009; Gorriti et al., 2014; Ventosa, 2015; López-Hermoso et al., 2018a, b). The taxonomic affiliation of the type and other 70 representative strains of this genus has been recently evaluated using a Multi Locus Sequence Analysis (MLSA) (López-Hermoso et al., 2017b, 2018a). Furthermore, draft genomic sequences of the type strains and only a few (14) representative strains has also been used to delineate Salinivibrio species (López-Hermoso et al., 2018a, b). As a result of the aforementioned studies, the genus Salinivibrio is currently composed of five species: S. costicola [containing two subspecies, i.e., S. costicola subsp. costicola (Garcia et al., 1987; Mellado et al., 1996) and S. costicola subsp. alcaliphilus (Romano et al., 2005)], Salinivibrio kushneri (López-Hermoso et al., 2018b), S. proteolyticus [which also includes the former S. costicola subsp. vallismortis (Amoozegar et al., 2008b; López-Hermoso et al., 2018a)], S. sharmensis (Romano et al., 2011), and Salinivibrio siamensis (Chamroensaksri et al., 2009). Additionally, the species “Salinivibrio socompensis” has also been proposed to include three Salinivibrio strains (Gorriti et al., 2014), but this name has not been validly published.
Although forty-six Salinivibrio genomes are available in GenBank database, all of them represent draft genomes and no complete genome projects have been conducted within this genus. Besides, only two studies dealing with genome sequence analysis of salinivibrios have been published (Gorriti et al., 2014; López-Hermoso et al., 2017a), but those were focused on a few genomes (the former) or the analysis only provided genome statistics (the latter), and in both cases using draft genome sequences. On the other hand, almost half of the available Salinivibrio genomes are not classified at the species level or misnamed (wrongly designated). The relationship of Salinivibrio species is not totally clear or stable according to previous studies because phylogenetic trees were based on MLSA of only up to eight housekeeping genes (Gorriti et al., 2014; López-Hermoso et al., 2017b) or the phylogenomic analysis was conducted only with a few of the Salinivibrio genomes (López-Hermoso et al., 2018a, b).
In this study, we clarify the phylogenetic relationships of existing genomes and species and review the taxonomic affiliation of the strains included within the genus Salinivibrio using a wide phylogenomic approach. This work also reports the first complete genome of a species of the genus Salinivibrio, S. kushneri, which was used as a reference to order the contigs of the draft genomes of the type strains within this genus, as well as to perform a comparative analysis with all the other available Salinivibrio genomes.
The strain S. kushneri AL184T was obtained from our culture collection where it was stored at −80°C and it was cultured in liquid SW broth (whose composition in g l–1 is: NaCl, 58.5; MgCl2⋅6H2O, 9.75; MgSO4⋅7H2O, 15.25; CaCl2, 0.25; KCl, 1.5; NaHCO3, 0.05; NaBr, 0.175; and yeast extract, 5.0) at 37°C with the pH adjusted between 7.2 and 7.4, according to López-Hermoso et al. (2018b). High-quality genomic DNA was extracted using the QIAmp DNA Mini Kit (Qiagen) following the manufacturer’s instructions.
A single molecule real-time sequencing approach was accomplished by using PacBio technologies. For that purpose, a PacBio library with 10 kbp insert size was constructed and a 350× sequencing depth was achieved. Additionally, whole genome shotgun reads obtained from an Illumina HiSeq (2 × 100-bp paired-end reads) device in an earlier study (López-Hermoso et al., 2017a) were used to carry out a hybrid assembly using SPAdes v.3.13.0 (Bankevich et al., 2012). Subsequently, the final assembly was achieved using only filtered by quality PacBio reads (read length after trimming ≥5000 nt, polymerase read quality ≥0.80, and polymerase read length ≥100) using an Overlap-Layout-Consensus algorithm as implemented in the HGAP v.2 pipeline (Chin et al., 2013). Dot plots to check for circularity at contig ends were drawn using Gepard v.1.40 software (Krumsiek et al., 2007). The resulting genome was circularized using toAmos and minimus2 (Sommer et al., 2007), followed by Circlator (Hunt et al., 2015) tools.
All the forty-six Salinivibrio draft genomes available in GenBank database were recovered, with the exception of that of S. costicola subsp. costicola ATCC 33508T (assembly accession no. GCA_000390145.1), which presented a suspicious length (4.78 Mb) and is excluded from RefSeq database due to its low quality sequence, and that of S. kushneri AL184T (assembly accession no. GCA_001995845.1), which was replaced by the complete genome of this strain achieved in this study (Table 1). Automatic annotation of those draft genomes together to the complete genome of strain S. kushneri AL184T achieved in this study was performed using RAST (Overbeek et al., 2014) and KAAS-KEEG (Moriya et al., 2007). RNA genes were determined by means of RNAmmer software (Lagesen et al., 2007).
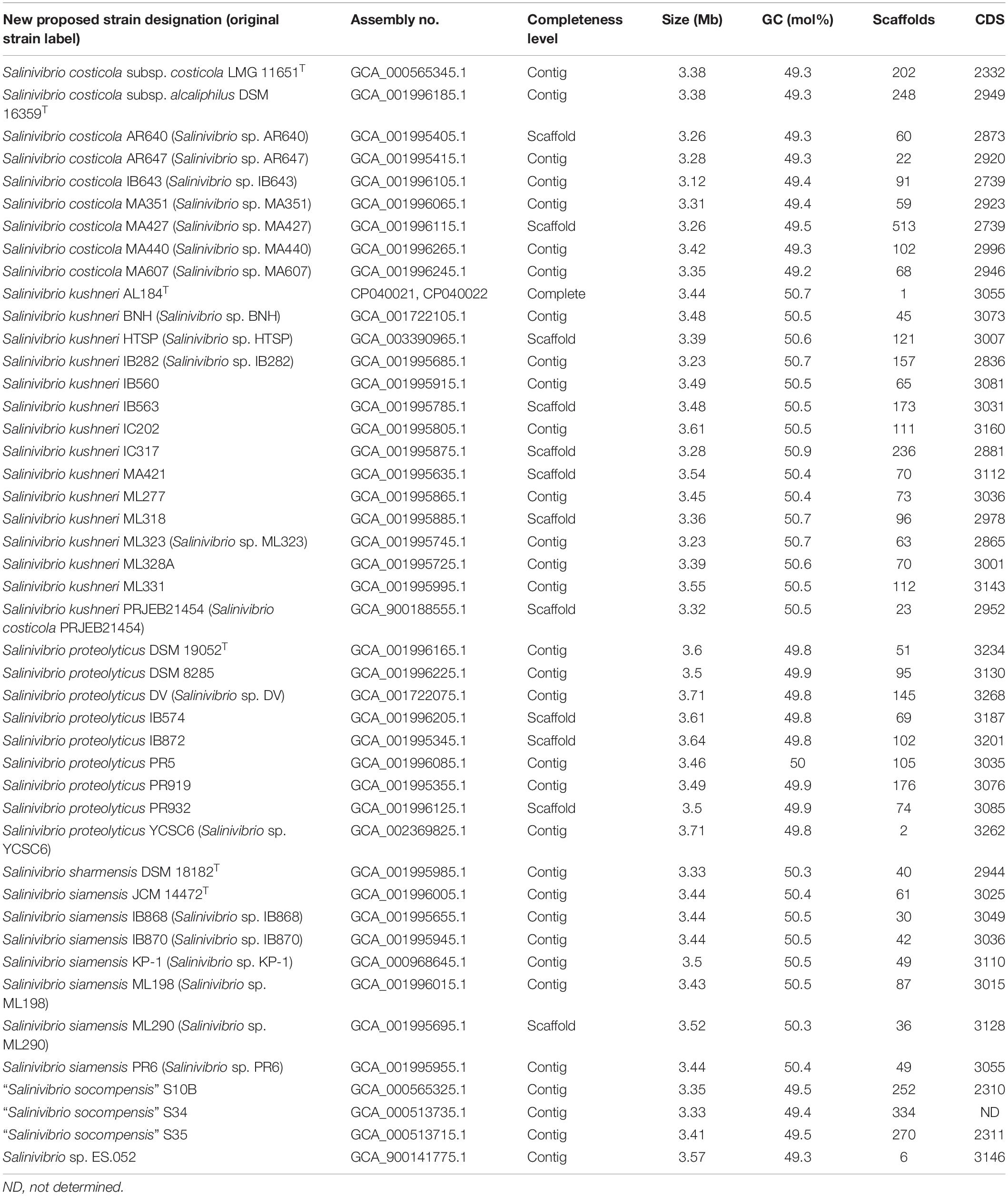
Table 1. Salinivibrio genomes available in GenBank database used in this study, including their basic statistical information.
Mauve genome alignment program (Darling et al., 2004) was employed to address the issue of genome rearrangements and contig ordering of the draft genomes by pairwise comparison using the complete genome of S. kushneri AL184T as a reference. Progressive Mauve software (Darling et al., 2010) was used for multiple genome alignment of the previously ordered draft genomes together to the complete genome of the type strains within the genus in order to find locally collinear blocks (LCBs). Synteny among different genomes was determined by measuring the alignment length of all the LCBs shared between the genomes being compared.
Circular plots showing LCBs and links between Salinivibrio genomes were drawn with the use of Circos v.0.69, a circular visualization software (Krzywinski et al., 2009). Furthermore, this program was utilized to represent the large and small circular chromosomes of S. kushneri AL184T.
All predicted protein-coding genes and proteins annotated from each available genome were searched using an all-vs.-all BLAST comparison using the Enveomics collection tools (Rodriguez-R and Konstantinidis, 2016). This analysis allows us to detect shared reciprocal best matches (described as those with equal or above 70% nucleotide identity or 40% amino acid identity) in all pairwise genome comparisons (core OGs or proteins) of the 45 Salinivibrio strains under study. The single copy core genes and proteins, respectively, were individually aligned using MUSCLE (Edgar, 2004). The resulting nucleotide and amino acids alignments were automatically trimmed using trimAl v.1.4 on “gappyout” mode (Capella-Gutiérrez et al., 2009) and, subsequently, concatenated to create core-genome and core-proteome alignments, and the phylogenomic trees were reconstructed by maximum-likelihood method with FastTreeMP v.2.1.8 (Price et al., 2010) where the branch support was estimated by means of the Shimodaira-Hasegawa test (Shimodaira and Hasegawa, 1999; Goldman et al., 2000).
Calculation of the orthologous average nucleotide identity (OrthoANI) among the studied genomes was accomplished employing USEARCH v8.1.1861 as implemented in OrthoANIu tool (Yoon et al., 2017). When genome pair comparisons showed less than 80% ANI values such genomes have divergent too much to use nucleotide level search and some genes might be missing; thus, average amino acid identity (AAI) was also determined. Mean AAI values for each genome pair were calculated using reciprocal best hits (two-way AAI) with the appropriate script in the Enveomics collection tools (Rodriguez-R and Konstantinidis, 2016).
To detect putative novel Salinivibrio species using culture-independent approaches, four 16S rRNA gene amplicon datasets reporting the presence of salinivibrios in their respective environments (Tkavc et al., 2011; Zhang et al., 2016, 2017; Crisler et al., 2019) were obtained from GenBank and NCBI’s Sequence Read Archive databases (accession no. FN823320-FN824096, SRP072906, SRP090542, SRP089997, and SRP090529) or provided by the authors. For those amplicon data, read quality filter was performed using Prinseq (Schmieder and Edwards, 2011), and chimera detection and filtering, OTU’s picking (at 97% similarity clustering value), representative picking of each OTUs, and taxonomy assignment were achieved using the software QIIME v. 1.9.0 (Caporaso et al., 2010).
Abundance estimation of Salinivbrio type strains and close relatives in several hypersaline environments was carried out by means of fragment recruitment using shotgun metagenomic databases (Supplementary Table 1). To avoid analysis bias, contigs of each of the genomes were concatenated and, subsequently, the rRNA gene sequences were masked. Blastn search (with the following parameters: length of the alignment ≥ 30 nt, similarity >95%, E value ≤ 1e-5) was employed to map the metagenomic reads (previously filtered to assess their quality) against each genome. The top-best-hit recovered after Blastn search was used to construct the recruitment plots.
The species S. kushneri was proposed based on 10 isolates, with strain AL184T designated as the type strain (López-Hermoso et al., 2018b). In despite of the fact that strains belonging to the genus Salinivibrio are commonly isolated due to their fast and easy growth on regular laboratory media, only a few species within this genus have been described so far. The relative low number of species is in spite the fact that different media and culture conditions (temperature, pH, salinity, and aerobic/anaerobic growth) were used over the time to isolate new Salinivibrio strains and to attempt to describe new species (Huang et al., 2000; Sánchez-Porro et al., 2003; Caton et al., 2004; Romano et al., 2005, 2011; Yeon et al., 2005; Amoozegar et al., 2008a, b; Zhu et al., 2008; Chamroensaksri et al., 2009; Xiao et al., 2009; Al-Mailem et al., 2014; Gorriti et al., 2014; Ashengroph, 2017; López-Hermoso et al., 2017b, 2018b; Le and Yang, 2018). In any case, there might be still cultural biases. Therefore, although the draft genome of the strain AL184T was already published elsewhere (López-Hermoso et al., 2017a), given the potential interest that this new species might have to unveil the speciation processes within this genus and its ecological role, the complete genome sequence of strain AL184T was obtained in this study.
Firstly, a hybrid assembly by using PacBio and Illumina reads was performed with the aim of accurately estimating the genome size, which we determined to be 3,436,949 bp. This result was required to achieve the final assembly based on the PacBio reads. According to PacBio recommendations and to Chin et al. (2013), the PacBio-only de novo assembly is preferred when it is possible to get at least 50X coverage. For this strain, the sequencing depth obtained was 350×, which motivated the choice of a PacBio-only based assembly strategy. Not a single contig, but two were obtained after the assembly, which was expected since other members of the family Vibrionaceae have been reported to contain two chromosomes (Dikow and Smith, 2013; Bernardy et al., 2016; Rashid et al., 2016; Kachwamba et al., 2017). In order to test this hypothesis a dot plot of each contig was carried out to check overlapping between the ends (Figure 1). Both dot plots showed this overlapping between the start and the end of each contig, indicating that strain AL184T contains two circular chromosomes. In addition, dot plot results were confirmed by BLAST search. Therefore, the two chromosomes were circularized and, subsequently, a linearized version was output with the dnaA gene as the starting position for chromosome I and a random gene for chromosome II. The empirical per-base coverage achieved was 211× for chromosome I and 216× for chromosome II.

Figure 1. Dot plots displaying the comparison of assembled contig 1 (A) and contig 2 (B) of Salinivibrio kushneri AL184T against itself. The enlarged images of the dot plots show that both ends of each contig are identical and, therefore, demonstrate that they constitute two circular chromosomes.
The final assembly for strain AL184T consists of two circular chromosomes with 2,840,906 bp and 602,384 bp, respectively (Figure 2). The completeness and contamination of the genome (both chromosomes together) estimated by CheckM tool (Parks et al., 2014) was 99.9% and 0.54%, respectively, which means that virtually the complete genome with a negligible -if any- amount of contamination was recovered. Although no essential genes were found in chromosome II, several lines of evidence suggest that it is a chromosome rather than a plasmid: (i) DNA G+C content was very similar for both chromosomes (50.8 mol% for I and 50.1 mol% for II); (ii) the length of chromosome II is more likely to correspond to a chromosome instead of a plasmid; (iii) the existence of genes theoretically belonging to the same cluster in different chromosomes, for example the cluster betABC, involved in the synthesis of glycine betaine, was coded in chromosome I (genes betA and betB) and chromosome II (gene betC); and (iv) as aforementioned, the members of the family Vibrionaceae usually have two chromosomes (Dikow and Smith, 2013; Bernardy et al., 2016; Rashid et al., 2016; Kachwamba et al., 2017).
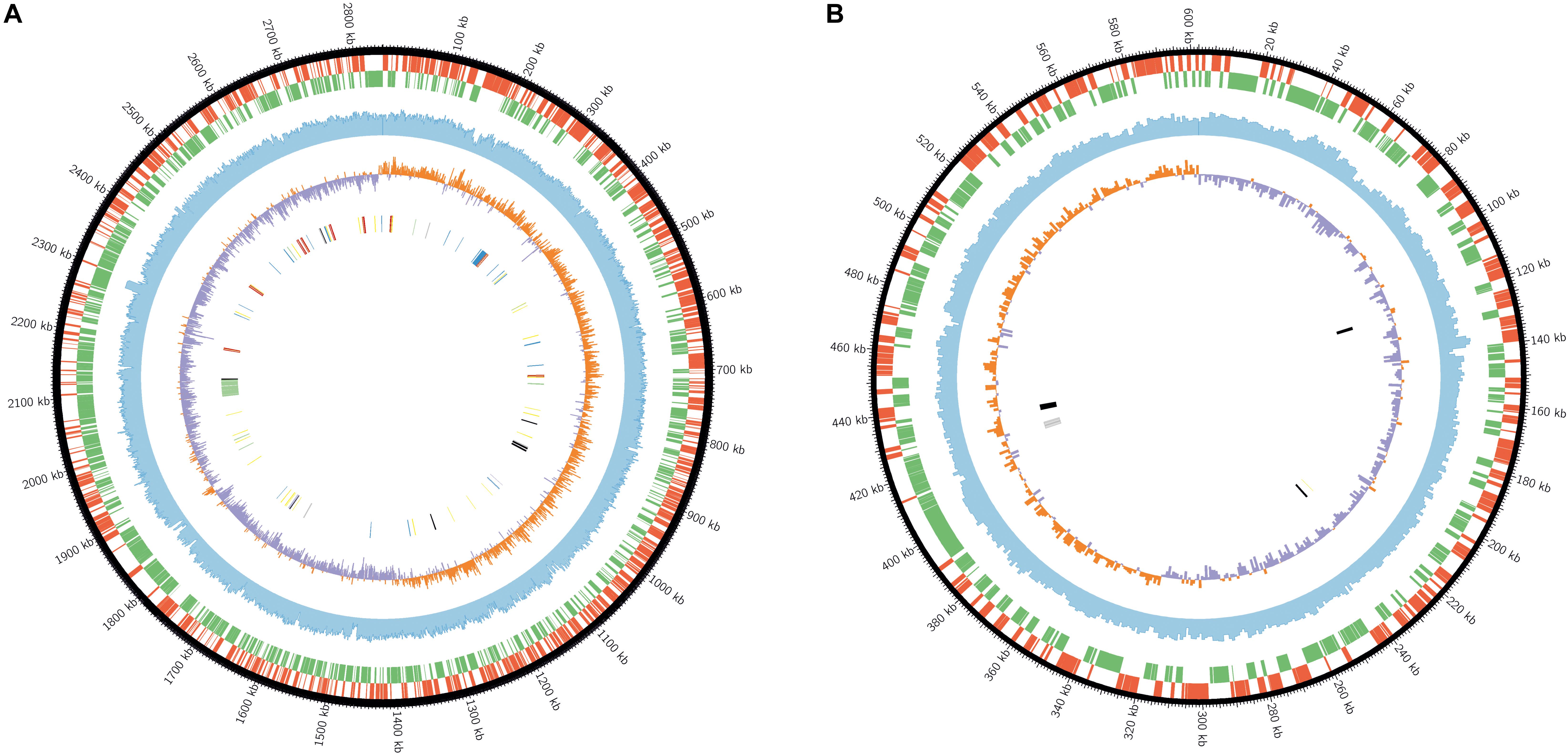
Figure 2. Graphical circular map of the chromosome I (A) and choromosome II (B) of S. kushneri AL184T. From the outer to inner chromosomal rings: (1) predicted CDSs transcribed in a clockwise direction; (2) predicted CDSs transcribed in a counterclockwise direction; (3) GC content in a 1,000-bp sliding window; (4) GC skew (C-G/G+C) in a 1,000-bp sliding window; (5) rRNAs (red), tRNAs (yellow), ribosomal proteins (blue), flagellum and flagellar motility genes (green), compatible solute synthesis genes (gray), compatible solute transporters (black), and anaerobic respiration-related genes (purple).
Annotation of the complete genome predicted 1244 and 233 CDS transcribed in a clockwise direction and 1280 and 298 CDS transcribed in a counter clockwise direction for chromosomes I and II, respectively (Figure 2). A total of 28 rRNA, 95 tRNA, 57 ribosomal protein, 51 flagellum and flagellar motility, 5 compatible solute synthesis, 10 compatible solute transporter, and 4 anaerobic respiration-related genes were identified in chromosome I, whereas only 1 tRNA, 4 compatible solute synthesis, and 5 compatible solute transporter genes were detected in chromosome II (Figure 2). This large number of rRNA genes, which were clustered in nine complete ribosomal operons (and an additional 5S rRNA gene located only 300 bp downstream with respect to one of the rRNA operons), might explain the fast growing rate of salinivibrios cultured in copiotrophic laboratory conditions (Roller et al., 2016). This finding was also observed in related taxa, such as Vibrio cholerae, that contains eight complete ribosomal operons (Rodicio and Mendoza, 2004) and it is a common trait of fast reproduction organisms (Roller et al., 2016). It is well-known that the different 16S rRNA genes contained in the same strain can be heterogeneous up to some extent; in the case of strain AL194T the nine 16S rRNA genes presented 100–98.4% sequence similarity, which is, practically, within the cutoff value currently used for species delineation (Kim et al., 2014).
Analyses of conservation of homologous genes and gene order between two or more genomes of different species (synteny) play a pivotal role in comparative genomics (Lee et al., 2018) and can provide insights into evolutionary processes that lead to diversity, chromosomal dynamics, and rearrangement rates between species (Bhutkar et al., 2006). Although analysis of synteny among closely related species is now widely used for every new published genome, this analysis is regularly performed on assembled sequences that are fragmented, neglecting the fact that most methods were developed using complete genomes (Liu et al., 2018). Here, we have used the complete genome of S. kushneri AL184T as a reference to reconstruct the fragmented genomes of the type strains of species of the genus Salinivibrio by ordering the contigs and assigning them to either chromosome I or II (Rissman et al., 2009). Following this strategy, chromosome I of the type species of the genus Salinivibrio, S. costicola subsp. costicola LMG 11651T, would be formed by 90 contigs (2,513,750 bp), and chromosome II by 39 contigs (691,570 bp), while the remaining 73 contigs (174,314) of that assembly could not be assigned to either chromosome I or II (Supplementary Figure 1 and Supplementary Table 2), and presumably represent gene-content differences between the two genomes compared. For the type strains of the other species or subspecies of the genus, chromosome I was constituted by 63 (2,545,420 bp), 32 (2,869,249 bp), 26 (2,717,996 bp), and 40 (2,729,566 bp) contigs corresponding to S. costicola subsp. alcaliphilus DSM 16359T, S. proteolyticus DSM 19052T, S. sharmensis DSM 18182T, and S. siamensis JCM 14472T, respectively, and the chromosome II of those strains was composed of 19 (706,389 bp), 10 (716,104 bp), 6 (589,195 bp), and 12 (688,170 bp) contigs, respectively, while 166 (129,848 bp), 9 (18,143 bp), 8 (19,704 bp), and 9 (24,449 bp) contigs left unassigned, respectively (Supplementary Figure 1 and Supplementary Table 2).
Retrieved large and small chromosomes from all the type strains were further analyzed to search synteny segments (LCBs). Figure 3 shows that the vast majority of chromosomes I and II can be pairwise aligned to its respective counterpart in S. kushneri AL184T, with 99.3–95.7% of chromosome I and 98.2–89.5% of chromosome II from S. costicola subsp. costicola LMG 11651T, S. costicola subsp. alcaliphilus DSM 16359T, S. proteolyticus DSM 19052T, S. sharmensis DSM 18182T, and S. siamensis JCM 14472T matching a conserved region in S. kushneri AL184T. Multiple sequence alignment among the large and small chromosomes of all the six genomes under study allows identifying 87 LCBs (3,713,502 bp alignment length) for chromosome I, of which 34 LCBs (3,594,867 bp alignment length) where common to all genomes, and 30 LCBs (1,090,330 bp alignment length) for chromosome II, with 17 LCBs (1,034,054 bp alignment length) shared among all taxa (Figure 4). Although the number of common LCBs for the large chromosome was significantly smaller than the 306 common LCBs reported by Dikow and Smith (2013) for a similar comparison within the family Vibrionaceae, the lengths of the alignments were almost the same, which means that our common LCBs for chromosome I are fewer but longer, and actually span between 95.0 and 98.7% of the large chromosome length of the six analyzed strains.
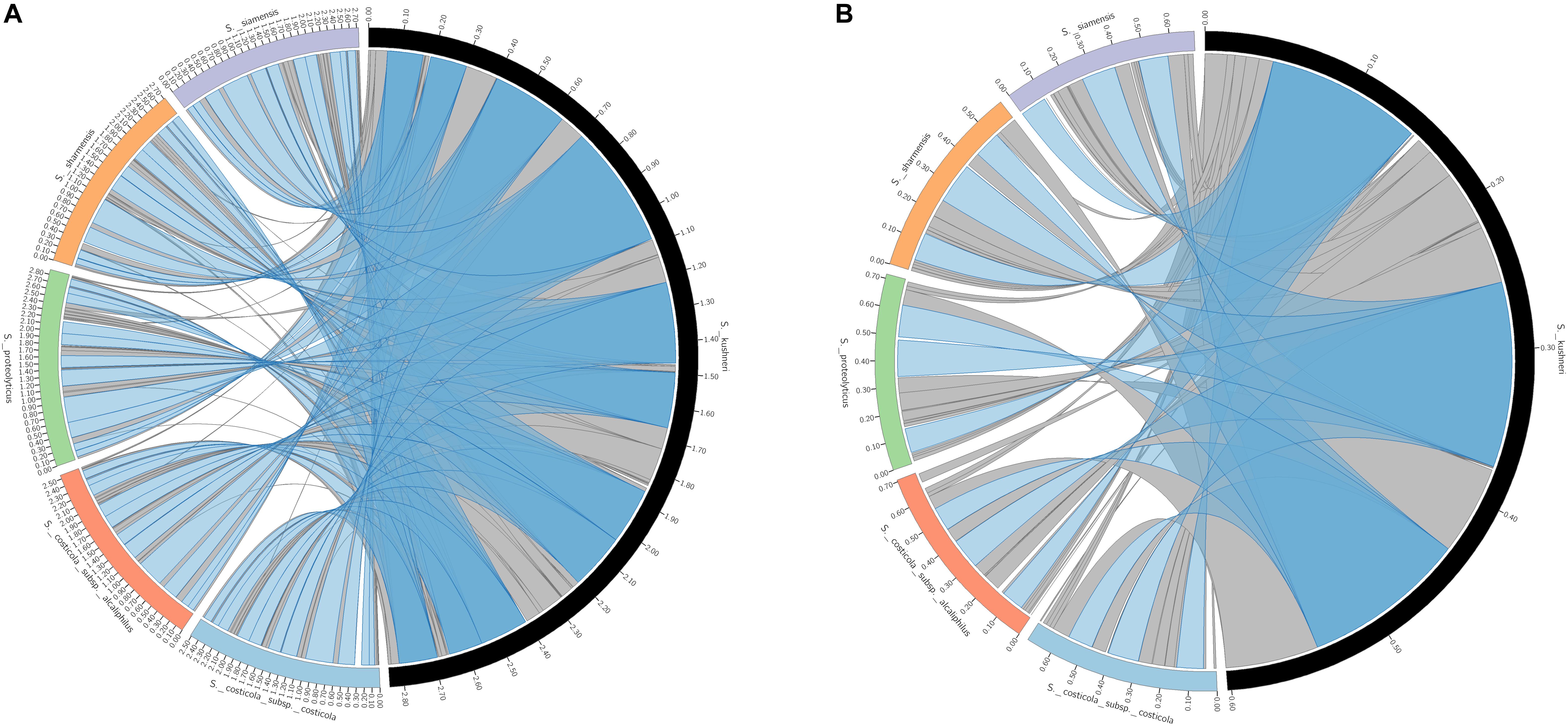
Figure 3. Pairwise alignment of locally collinear blocks (LCBs) between large (A) and small (B) chromosomes of S. kushneri AL184T and those of S. costicola subsp. costicola LMG 11651T, S. costicola subsp. alcaliphilus DSM 16359T, S. proteolyticus DSM 19052T, S. sharmensis DSM 18182T, and S. siamensis JCM 14472T. Blue bands represents LBC > 100 Kb and gray bands LCB < 100 Kb.
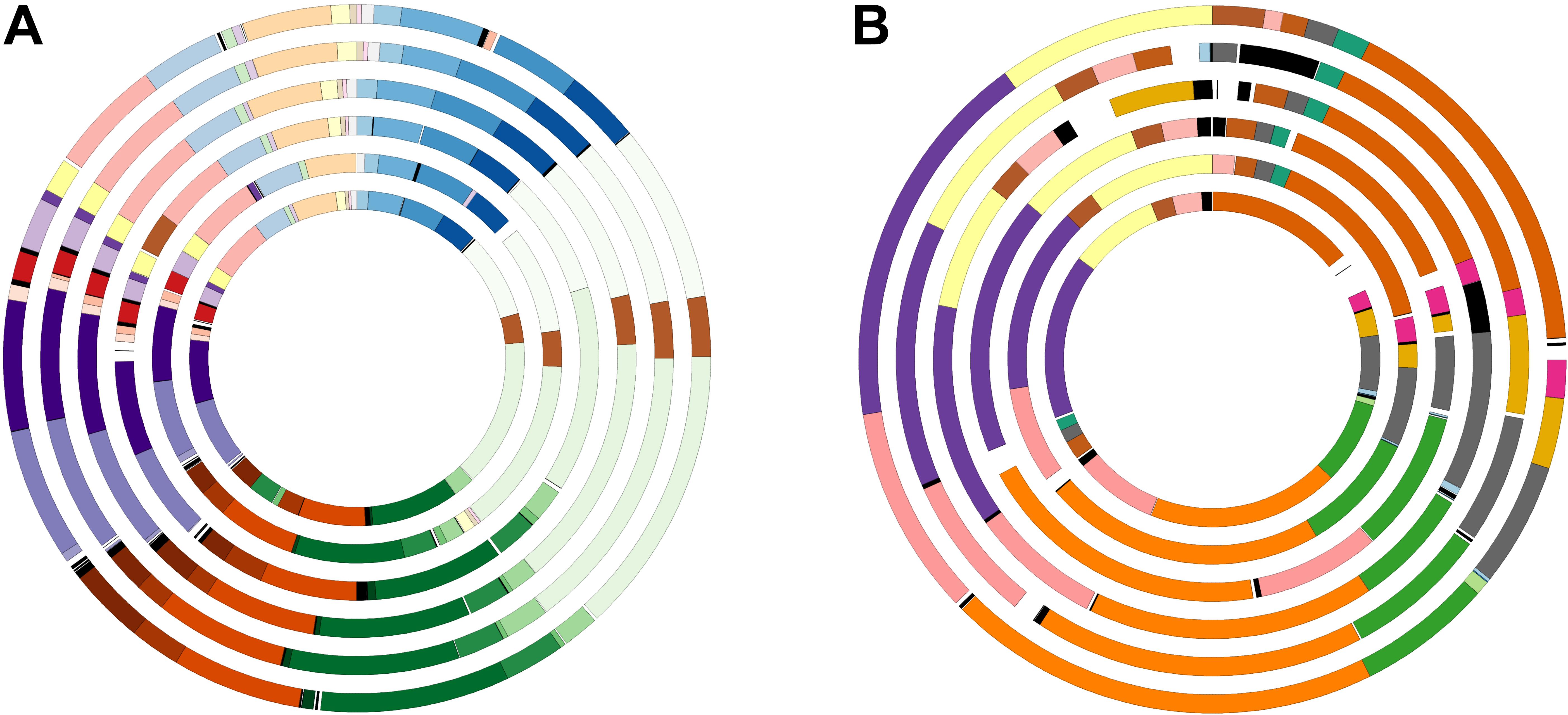
Figure 4. Salinivibrio large (87 LCBs) (A) and small (30 LCBs) (B) chromosomes circular plots. Each circle represents a genome. From the outer most circle: S. kushneri AL184T, S. costicola subsp. costicola LMG 11651T, S. costicola subsp. alcaliphilus DSM 16359T, S. proteolyticus DSM 19052T, S. sharmensis DSM 18182T, and S. siamensis JCM 14472T. LCBs in the same color are shared by all six strains, with the exception of LCBs in black which are shared by less than the six strains.
Concerning the small chromosome, the number of common LCBs identified in our study was approximately half of those detected by Dikow and Smith (2013) for other members of the family Vibrionaceae, but our alignment length was more than twice longer. That means that the small chromosomes of the six Salinivibrio genomes were much more homologized, with percentages between 88.7 and 98.5%. These measurements were made when gaps were removed from the alignments. Therefore, in contrast to the study of Dikow and Smith (2013), no significant differences in homologization rates between large and small Salinivibrio chromosomes could be observed. It must be noted that the study of Dikow and Smith (2013) dealt with complete genomes, while here ordered draft genomes were employed, what might partially explain such differences.
This higher synteny in our Salinivibrio genomes vs. the results reported by Dikow and Smith (2013) might be due to the fact that the genomes analyzed in this study were more closely related among them (84.3% average OrthoANI and 90.0% average AAI) than the Vibrionaceae genomes of the mentioned study (74.1% average OrthoANI and 70.6% average AAI), thus, the higher the relatedness of the genomes under study the higher will be the synteny. To confirm this statement, we calculated the correspondence between synteny (measure as the percentage of aligned genome) and OrthoANI and AAI values among all pair of the genomes from type strains. However, the Pearson’s coefficient was only 0.21 for OrthoANI and 0.25 for AAI, indicating a poor but still significant correlation, which means that the conserved synteny of Salinivibrio genomes is partially due to the high average OrthoANI/AAI values among the studied genomes, but also we can hypothesize a slow speciation process or homogenization events that might be occurring in salinivibrios, a statement that needs to be tested experimentally in the future.
Single-copy core-genome genes and proteins were employed to construct a phylogenomic tree in order to elucidate the taxonomic relationship among members of the genus Salinivibrio. Maximum-likelihood phylogenies based on the concatenation of 776 genes (777,643 bp alignment length) and 1,637 proteins (515,359 bp alignment length) yielded two very similar trees with high bootstrap support, where five different phylogroups and two phylotypes can be distinguished (Figure 5 and Supplementary Figure 2). Phylogroup 1 corresponds to S. kushneri strains, and includes strains previously affiliated to this species (López-Hermoso et al., 2018b) as well as three other strains originally named as Salinivibrio sp. and another probably mislabeled strain initially designated as S. costicola. Phylogroup 2 is formed by S. siamensis JCM 14472T and other five strains identified as members of this species in a previous MLSA approach (López-Hermoso et al., 2017b) as well as one additional strain labeled as Salinivibrio sp. KP-1. Phylogroup 3 agrees on the monophyletic group defined by López-Hermoso et al. (2018a) to emend the description of S. proteolyticus but it also groups two additional strains not analyzed by those authors, which we prove to belong to the aforementioned species. Phylogroup 4 consists of the three strains proposed by Gorriti et al. (2014) as a new Salinivibrio species with the not-yet validated name “S. socompensis.” Finally, phylogroup 5 exactly fits the cluster defined by López-Hermoso et al. (2017b) formed by nine strains, including the two subspecies of S. costicola, S. costicola subsp. costicola and S. costicola subsp. alcaliphilus. Actually, phylogenomically, the two subspecies cannot be clearly differentiated (especially when the tree is constructed using the concatenated core genes), a maybe the Salinivibrio subspecies rank should be revised. Furthermore, two Salinivibrio strains could neither be included in any of the above phylogroups, nor formed a phylogroup themselves, so they were defined as phylotypes. One of them consists of the type strain of S. sharmensis, DSM 18182T, and the other is an unnamed Salinivibrio strain, ES.052, isolated from an intertidal microbial mat in Elkhorn Slough (California), which probably constitutes a new species of Salinivibrio not described yet.
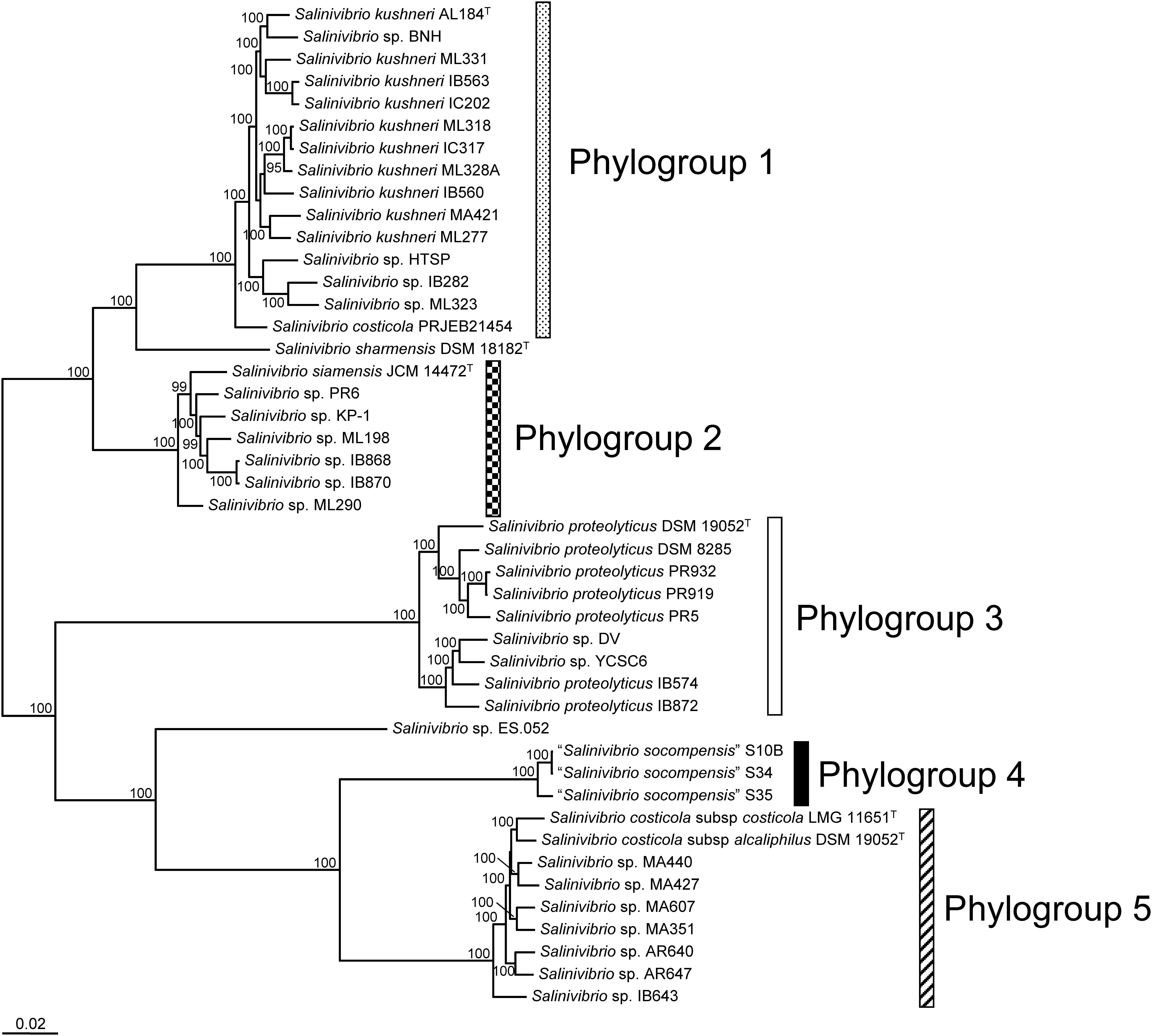
Figure 5. Maximum-likelihood phylogenomic tree based on the concatenation of 776 single copy core genes showing the relationships among 45 Salinivibrio strains whose genomes are available. Bootstrap values ≥ 70% are shown at the nodes. Bar, 0.02 nt changes per position.
OrthoANI values calculated for all-vs.-all pairs (Supplementary Figure 3) confirmed that the aforementioned phylogroups and phylotypes are actually different species of the genus Salinivibrio. The OrthoANI values within each phylogroup were always above 95%, whereas, the values among phylogroups/phylotypes were in all cases far below 95%, the threshold value proposed for species boundaries (Richter and Rosselló-Móra, 2009), thus supporting our proposal to designate each phylogroup and phylotype to a different Salinivibrio species. We also propose to rename the Salinivibrio strains used here as follows: all strains belonging to phylogroup 1 should be labeled as S. kushneri, those of phylogroup 2 as Salinivibrio siamensis, and those of phylogroup 3 as S. proteolyticus. Strains of phylogroup 5 should be relabeled as S. costicola, without indicate the subspecies to which each strain is affiliated, with the exception of the type strains of the subspecies. Finally, phylogroup 4 and the two phylotypes will retain their actual designation (Table 1). For some genome pairs (i.e., strains of phylogroup 3 vs. strains of phylogroups 4 and 5) the OrthoANI values were slightly below 80%, and so, those genomes are too divergent to use nucleotide level comparisons. In consequence, AAI values were estimated for all-vs.-all genome pairs (Supplementary Figure 3) confirming that phylogroups 3, 4, and 5, as well as the phylotype Salinivibrio sp. ES.052, constitute separate species. However, AAI results equal or above 95% among strains of phylogroups 1 and 2, and for the phylotype S. sharmensis DSM 18182T, suggest that they all might form a single species. Nevertheless, these strains share more than 90% ANI in all cases, therefore, according to Rodriguez-R and Konstantinidis (2014) for such closely related strains ANI offers a robust resolution and should be used instead. For that reason, we suggest maintaining our proposal of phylogroups 1 and 2, and phylotype S. sharmensis DSM 18182T as independent species within this genus.
Orthologous gene (OGs) cluster analysis based on amino acid and nucleotide sequences was performed to define the pan-genome of the genus Salinivibrio. By using translated amino acid sequence comparison of the 45 analyzed genomes, the pan-genome is composed of 5,570 OGs, of which 2,080 OGs are common to all taxa (core-genome), and 3,490 OGs constitute the variable-genome (Table 2). If the nucleotide sequences are used, then pan-genome is composed of larger OGs, 7,462, but the core-genome decreases up to 1,211 OGs while the accessory-genome rises up to 6,251 OGs (Table 2). The smaller pan-genome when using protein sequences was expected due to the fact that a lower cut-off value (40% vs. 70% sequence identity) was set for the clustering. However, given that protein sequences are more conservative than nucleotide sequences, it is not surprisingly the bigger core-genome obtained when analyzing translated amino acid sequences, especially if the genomes under study have diverged too much (OrthoANI values < 80%). A similar study within the family Vibrionaceae detected 6,629 OGs of which 1,882 OGs where found in all 11 proteomes under study (Lilburn et al., 2009), but this smaller core-genome is probably attributable to the fact that the analysis was performed on genomes belonging to different genera. A more recent research only focused in a single genus, Vibrio, and conducted with 20 proteomes (corresponding to 20 different species) yielded a large pan-genome of 21,844 OGs, with only 1,630 OGs common to all taxa (Lin et al., 2018), which may be explained by the lower genomic relatedness (measured by OrthoANI/AAI values) among the Vibrio genomes. Therefore, members of the genus Salinivibrio appear to have higher genetic relatedness within the group than strains of the genus Vibrio. These findings are consistent with our hypothesis that there should be an homogenizing force acting on the salinivibrios, what could explain the few number of species described so far, a puzzling occurrence for a genus so often isolated from hypersaline environments all over the world (Herzog et al., 2016; Ashengroph, 2017; Fernández-Delgado et al., 2017; López-Hermoso et al., 2017b; Selvarajan et al., 2017; Le and Yang, 2018; Arias et al., 2019). However, this hypothesis awaits experimental testing in the future.
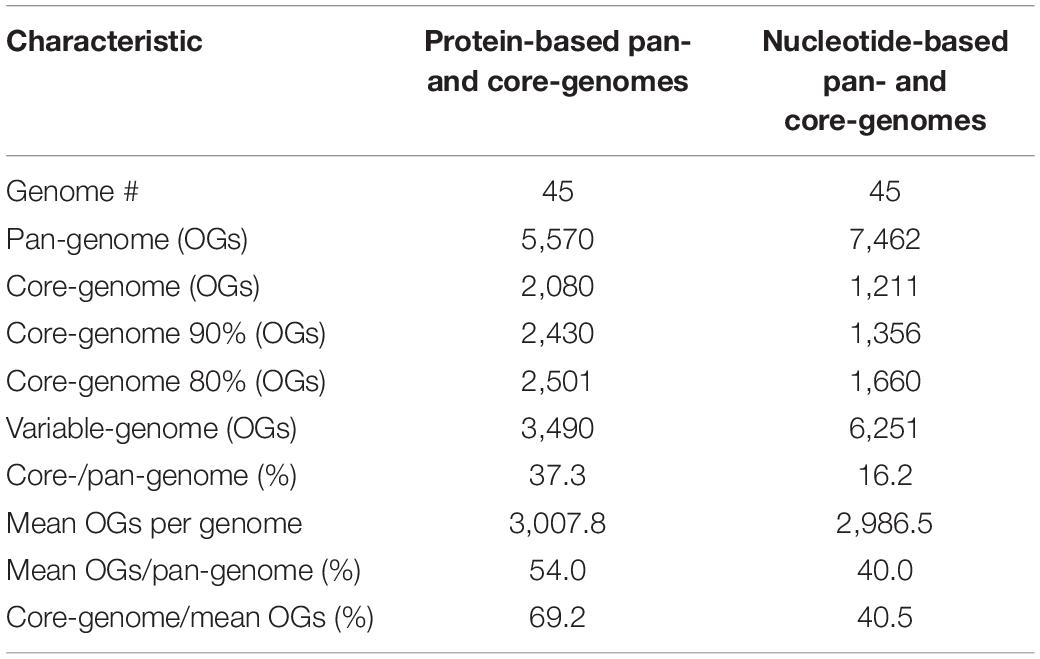
Table 2. Pan- and core-genome features of the Salinivibrio genomes based on translated protein and nucleotide gene sequences.
The progression of the pan- and core-genomes after random samplings was calculated (Figure 6). As can be observed, pan-genome based on gene sequences gradually increased when more genomes were considered up to reach 30 genomes, and after that it would remain relatively constant, even as many more genomes were added. On the contrary, the nucleotide-based core-genome rapidly decreased when more genomes were added, becoming relatively stable after 7 genomes were considered. Similarly, the pan- and core-genomes based on protein sequences followed the same tendency, but the saturation of the curves occurred with a higher number of genomes, i.e., 40 and 38, respectively, because protein search is more sensitive.
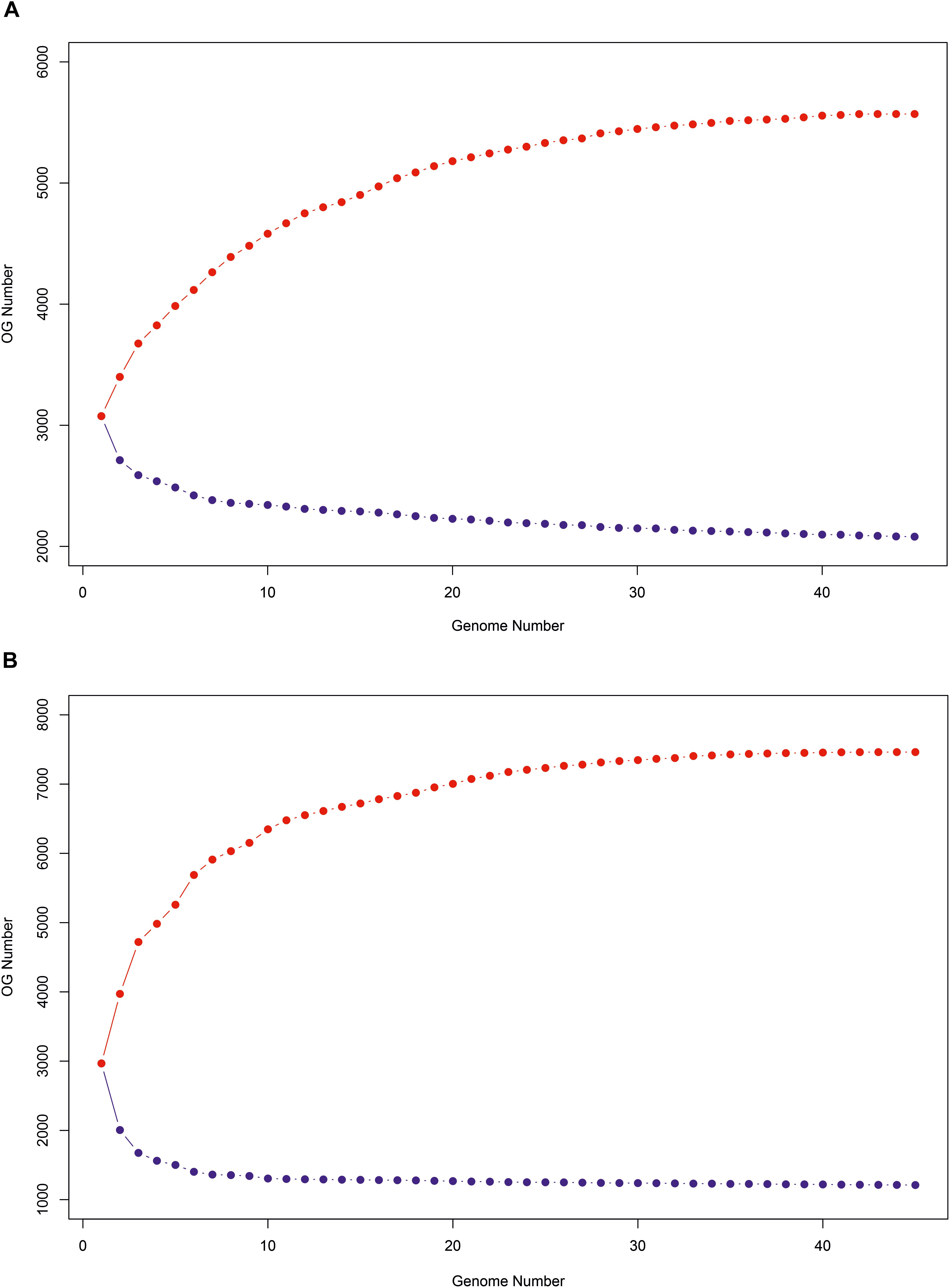
Figure 6. Progression of the pan-genome (red) and core-genome (blue) of the 45 Salinivibrio genomes based on protein (A) and gene (B) sequences.
To provide cultivation-independent assessment of the Salinivibrio species diversity, the spatial distribution of the genus was analyzed based on the origin of 16S rRNA gene sequences found in SILVA database (release 132), as well as the source of the 45 completely sequenced strains analyzed in this study. As can be observed (Supplementary Figure 4), the genus Salinivibrio possess a high dispersal potential, with a cosmopolitan distribution. Besides, strains/sequences belonging to different species have been isolated from the same region, which means that different species might have similar habitat preferences.
Although isolation of new Salinivibrio species has not been often achieved, new culture-independent techniques based on high throughput sequencing can provide some clues about the species diversity in the natural environment. To this end, we analyzed several 16S rRNA gene amplicon datasets from recent studies (i.e., Tkavc et al., 2011; Zhang et al., 2016, 2017; Crisler et al., 2019) reporting presence of salinivibrios for OTUs relative abundance and taxonomic assignment. Considering 97% as the species cut-off value, we were able to identify 11, 118, 135, and zero different OTUs in the datasets of Tkavc et al. (2011), Zhang et al. (2016, 2017), and Crisler et al., 2019, respectively. Furthermore, we downloaded all 16S rRNA gene sequences from SILVA Nr99 database (release 132) labeled as Salinivibrio and performed the clustering analysis with 97% sequence identity, obtaining 48 different OTUs which could represent 48 putative different species of Salinivibrio. Although these culture-independent approaches might indicate, in some cases, a broader species diversity than that expected based on culture techniques it must be noticed that amplicon metagenomic datasets contained partial 16S rRNA gene sequences (∼200–691 bp average length) and, therefore, OTUs clustering might be biased and inflate natural diversity. In any case, our results show that, although genomic conservation within salinivibrios is higher compared to other members of the family Vibrionaceae (based on synteny and core/pangenome analyses), new species might exist in the studied habitats and, therefore, additional attempts to isolate and to describe them should be performed.
To provide a whole-genome view in the environments where salinivibrios may be theoretically more abundant, we performed fragment recruitment analyses using several shotgun metagenomic datasets (Supplementary Table 1) against all the type strains of the genus. Salinivibrios showed recruitment in saline water and soil metagenomes, with lower recruitment values at hypersaline metagenomes (Supplementary Figure 5). Surprisingly, the highest values were not obtained at intermediate salinities where member of this genus are frequently isolated, but at lower salinities, being especially abundant in an Iranian saline lake with 5% NaCl. Therefore, these recruitment plots suggest that future efforts to search for new Salinivibrio species might be conducted sampling lower salinity environments.
Annotation and comparison of the 44 Salinivibrio genomes considered in this study indicated that the ten most representative subsystems included genes related to flagellar synthesis and regulation, tRNA, large and small ribosomal subunits, RNA methylation, methionine synthesis, cytoskeleton, serine-glyoxylate cycle, DNA repair, and phosphate metabolism (Supplementary Figure 6).
A total of 46 genes related to synthesis and regulation of flagellum were detected in the studied Salinivibrio genomes, which might be expected since salinivibrios are motile bacteria by means of one pollar flagellum (Mellado et al., 1996). Among them, fliDC (flagellar filament regulator), flgLK (filament-hook joint), and flgEKD (flagellar hook regulator) genes were detected. Additionally, motAB genes related to transport between flagellum and extracellular fluid were identified (Kuchma et al., 2015). Previous studies have confirmed the fast evolutionary rate of these regulators (Soutourina and Bertin, 2003; McCarter, 2006). Although the flagellar regulation genes have not been completely elucidated, each bacterial species usually possesses a different regulation network. However, the salinivibrios under study present the same flagellar regulation network, which indicate the high homogeneity among this bacterial genus.
Salinivibrios are described as facultatively anaerobic bacteria, so, we looked for genes involved in the anaerobic respiration. Two kinds of reductases were detected in all the studied genomes, AsrR (arsenate reductase) and FIR (ferredoxin-NADPH reductase). Arsenate, in despite of its toxicity, can be used by some microorganisms as an electron acceptor in the anaerobic respiration (Saltikov and Newman, 2003; Ruebush et al., 2006), what lead us to hypothesize that members of the genus Salinivibrio might metabolize arsenate under anaerobic conditions. Futhermore, a cytochrome cbb (3) oxidase complex implicated in microaerobic respiration (Pitcher and Watmough, 2004) was identified. This complex might provide a better adaptation to microaerobic environments, pointing to evolutionary modifications of salinivibrios to thrive at low oxygen concentrations, such as those present in hypersaline aquatic habitats (Rodríguez-Valera, 1993; Ventosa, 2006).
Osmotic response of Salinivibrio strains is mediated by accumulation of cytoplasmatic compatible solutes (also know as the “salt-out” strategy) which can be synthesized de novo or captured from the environment (Zhu et al., 2008, 2010). Ectoine is probably the key compatible solute in osmotic adaptation of salinivibrios (Zhu et al., 2008). The complete cluster of genes involved in ectoine synthesis, ectABC, was present in all the studied genomes, but ectD gene, responsible of ectoine to hydroxyectoine conversion was missed in some genomes. Ectoine transportation to cellular inner is accomplished by a TeaABC transporter (Grammann et al., 2002), which could not be detected in the Salinivibrio genomes, therefore, we deducted that salinivibrios use ectoine to balance osmotic stress by de novo synthesis instead of by transportation. Glycine betaine is another compatible solute widely used by bacteria that is synthesized from choline (involving betAB genes) or choline O-sulfate (involving betABC genes) (Lidbury et al., 2015). All the studied genomes codified the betA and betB genes, but betC was absent, so salinivibrios can only utilize choline, but not choline O-sulfate. As expected, the gene for a high-affinity choline uptake protein (betT) was codified in all the genomes. The glycine betaine transporter (opuD) was only present in Salinivibrio strains belonging to phylogroups 1 and 4, being synthesized and not transported in the other strains. Trehalose can be used by bacteria as a compatible solute in response to osmotic and thermal stresses (Avonce et al., 2006). Salinivibrio genomes contained the gene cluster otsAB, required for trehalose synthesis, but they lack the enzymes for its degradation, what means that trehalose is not used as a carbon and energy source.
Concerning the nitrogen metabolism, genes related to nitrogen fixation, nitrification, denitrification, and assimilatory nitrate reduction were not identified within salinivibrios, in agreement to the results of Gorriti et al. (2014). Ammonium assimilation is carried out either by the GDH (glutamate dehydrogenase) or by the GS/GOGAT (glutamine synthetase/glutamate synthase) pathways. Given that salinivibrios do not possess the enzymatic activity to reduce nitrate, the ammonium uptake that will be incorporated into carbon skeletons is achieved by mean of an Amt transporter.
Although a total of 14 pathways and 44 different enzymes are currently known leading to PHA (poly-hydroxyalkanoate) synthesis (Meng et al., 2015), PHA synthase (PhaC) plays a key role in the PHA biosynthetic pathway (Chek et al., 2017). In the Salinivibrio genomes under study, only phaC gene was detected, but none of the other PHA synthesis-related genes (including the PHA depolymerase). Therefore, it is possible that salinivibrios might accumulate PHA, but they cannot perform PHA degradation.
We have sequenced the complete genome of the type strain of the species S. kushneri AL184T, which, unsurprisingly, is constituted by two chromosomes, as usual for other members of the family Vibrionaceae. This is the first closed genomic sequence currently available within this genus. We have corroborated that PacBio reads with above 200× average per-base coverage are enough to recover a high quality complete bacterial genome. The complete genome is very useful to identify rearrangements and to order contigs of closely related draft genomes. Synteny analysis among the genomes of the type strains of the genus Salinivibrio has demonstrated the high degree of homologization within this genus. This might evidence a slower evolutionary rate in salinivibrios, what would explain the surprising few numbers of species validly described so far given how often new Salinivibrio strains are isolated from different environments, but this hypothesis should be further tested experimentally. Nevertheless, metagenomic analyses suggest that a broader species diversity might exist in natural environments, although these results may be regarded with caution, and that a higher abundance of salinivibrios probably occurs at lower salinity concentrations.
Currently, the genus Salinivibrio includes five species with validly described names, as well as a non-yet-validated species name. Our phylogenomic analysis supports the taxonomic status of those six Salinivibrio species and, moreover, evidence the existence of an additional species represented by the strain Salinivibrio sp. ES.052. Besides, the taxonomic distinction between the two subspecies of S. costicola is not clear according to our results and, moreover, there are no weighted phenotyphic differences between both subspecies beyond optimal pH supporting growth and some biochemical features which may also differ among strains of the same species (Romano et al., 2005) so, perhaps, the subspecies status of this species should be deeply revised.
Genomic features of the 45 studied strains also agreed with the high homogeneity of this genus. Phenotypic characteristics described for the member of this group concur with the information derived from the annotated genomes. The only exception is the reduction of nitrate and nitrite, which has been observed in laboratory experiments (Romano et al., 2005, 2011; Chamroensaksri et al., 2009; López-Hermoso et al., 2018a, b), but could not be detected in the analyzed genomes. Another interesting feature detected in the complete genome of S. kusnheri AL184T is the large number of rRNA genes (nine complete ribosomal clusters), which is expected for bacteria with a fast growing rate in artificial media.
The large and small chromosomes of Salinivibrio kushneri AL184T generated for this study can be found in the GenBank under the accession numbers CP040021 and CP040022, respectively.
AV, RH, and CS-P conceived and designed the study. CL-H and CS-P performed the laboratory experiments. RH, CL-H, CS-P, KK, and AV analyzed and interpreted the data, and discussed and critically revised the manuscript. RH and AV drafted the manuscript. All authors read and approved the final manuscript.
This study was supported by grants from the Spanish Ministry of Economy and Competitiveness (MINECO) (Project CGL2017-83385-P) and the Junta de Andalucía (BIO-213) and by US National Science Foundation (NSF), award #1759831. FEDER funds also supported this project. RH and CL-H were recipients of a short-stay grant from the University of Seville (VI-PPIT-US-2018) and the Spanish Ministry of Economy and Competitiveness, respectively.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank the reviewers for their generous and meaningful efforts. We also thank Dr. Mark A. Schneegurt for providing raw sequencing data from the Basque Lake (Canada).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02104/full#supplementary-material
Al-Mailem, D., Eliyas, M., Khanafer, M., and Radwan, S. (2014). Culture-dependent and culture-independent analysis of hydrocarbonoclastic microorganisms indigenous to hypersaline environments in Kuwait. Microb. Ecol. 67, 857–865. doi: 10.1007/s00248-014-0386-5
Amoozegar, M. A., Salehghamari, E., Khajeh, K., Kabiri, M., and Naddaf, S. (2008a). Production of an extracellular thermohalophilic lipase from a moderately halophilic bacterium, Salinivibrio sp. strain SA-2. J. Basic Microbiol. 48, 160–167. doi: 10.1002/jobm.200700361
Amoozegar, M. A., Schumann, P., Hajighasemi, M., Fatemi, A. Z., and Karbalaei-Heidari, H. R. (2008b). Salinivibrio proteolyticus sp. nov., a moderately halophilic and proteolytic species from a hypersaline lake in Iran. Int. J. Syst. Evol. Microbiol. 58, 1159–1163. doi: 10.1099/ijs.0.65423-0
Arias, D., Cisternas, L. A., Miranda, C., and Rivas, M. (2019). Bioprospecting of ureolytic bacteria from Laguna Salada for biomineralization applications. Front. Bioeng. Biotechnol. 6:209. doi: 10.3389/fbioe.2018.00209
Ashengroph, M. (2017). Salinivibrio costicola GL6, a novel isolated strain for biotransformation of caffeine to theobromine under hypersaline conditions. Curr. Microbiol. 74, 34–41. doi: 10.1007/s00284-016-1148-z
Avonce, N., Mendoza-Vargas, A., Morett, E., and Iturriaga, G. (2006). Insights on the evolution of trehalose biosynthesis. BMC Evol. Biol. 6:109. doi: 10.1186/1471-2148-6-109
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm, et al. and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bernardy, E. E., Turnsek, M. A., Wilson, S. K., Tarr, C. L., and Hammer, B. K. (2016). Diversity of clinical and environmental isolates of Vibrio cholerae in natural transformation and contact-dependent bacterial killing indicative of type VI secretion system activity. Appl. Environ. Microbiol. 82, 2833–2842. doi: 10.1128/AEM.00351-16
Bhutkar, A., Russo, S., Smith, T. F., and Gelbart, W. M. (2006). Techniques for multi-genome synteny analysis to overcome assembly limitations. Genome Inform. 17, 152–161.
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caton, T. M., Witte, L. R., Ngyuen, H. D., Buchheim, J. A., Buchheim, M. A., and Schneegurt, M. A. (2004). Halotolerant aerobic heterotrophic bacteria from the great salt plains of Oklahoma. Microb. Ecol. 48, 449–462. doi: 10.1007/s00248-004-0211-7
Chamroensaksri, N., Tanasupawat, S., Akaracharanya, A., Visessanguan, W., Kudo, T., and Itoh, T. (2009). Salinivibrio siamensis sp. nov., from fermented fish (pla-ra) in Thailand. Int. J. Syst. Evol. Microbiol. 59, 880–885. doi: 10.1099/ijs.0.001768-0
Chek, M. F., Kim, S. Y., Mori, T., Arsad, H., Samian, M. R., Sudesh, K., et al. (2017). Structure of polyhydroxyalkanoate (PHA) synthase PhaC from Chromobacterium sp. USM2, producing biodegradable plastics. Sci. Rep. 7:5312. doi: 10.1038/s41598-017-05509-4
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Crisler, J. D., Chen, F., Clark, B. C., and Schneegurt, M. A. (2019). Cultivation and characterization of the bacterial assemblage of epsomic Basque Lake, BC. Antonie van Leeuwenhoek 112, 1105–1119. doi: 10.1007/s10482-019-01244-0
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Dikow, R. B., and Smith, W. L. (2013). Genome-level homology and phylogeny of Vibrionaceae (Gammaproteobacteria: Vibrionales) with three new complete genome sequences. BMC Microbiol. 13:80. doi: 10.1186/1471-2180-13-80
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Fernández-Delgado, M., Suárez, P., Giner, S., Sanz, V., Peña, J., Sánchez, D., et al. (2017). Occurrence and virulence properties of Vibrio and Salinivibrio isolates from tropical lagoons of the southern Caribbean Sea. Antonie van Leeuwenhoek 110, 833–841. doi: 10.1007/s10482-017-0856-0
Garcia, M. T., Ventosa, A., Ruiz-Berraquero, F., and Kocur, M. (1987). Taxonomic study and amended description of Vibrio costicola. Int. J. Syst. Bacteriol. 37, 251–256. doi: 10.1099/00207713-37-3-251
Goldman, N., Anderson, J. P., and Rodrigo, A. G. (2000). Likelihood-based tests of topologies in phylogenetics. Syst. Biol. 49, 652–670. doi: 10.1080/106351500750049752
Gorriti, M. F., Dias, G. M., Chimetto, L. A., Trindade-Silva, A. E., Silva, B. S., Mesquita, M. M., et al. (2014). Genomic and phenotypic attributes of novel salinivibrios from stromatolites, sediment and water from a high altitude lake. BMC Genomics 15:473. doi: 10.1186/1471-2164-15-473
Grammann, K., Volke, A., and Kunte, H. J. (2002). New type of osmoregulated solute transporter identified in halophilic members of the Bacteria domain: TRAP transporter TeaABC mediates uptake of ectoine and hydroxyectoine in Halomonas elongata DSM 2581T. J. Bacteriol. 184, 3078–3085. doi: 10.1128/JB.184.11.3078-3085.2002
Herzog, B., Overy, D. P., Haltli, B., and Kerr, R. G. (2016). Discovery of keratinases using bacteria isolated from marine environments. Syst. Appl. Microbiol. 39, 49–57. doi: 10.1016/j.syapm
Huang, C. Y., Garcia, J. L., Patel, B. K. C., Cayol, J. L., Baresi, L., and Mah, R. A. (2000). Salinivibrio costicola subsp. vallismortis subsp. nov., a halotolerant facultative anaerobe from death Valley, and emended description of Salinivibrio costicola. Int. J. Syst. Evol. Microbiol. 50, 615–622. doi: 10.1099/00207713-50-2-615
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Kachwamba, Y., Mohammed, A. A., Lukupulo, H., Urio, L., Majigo, M., Mosha, F., et al. (2017). Genetic characterization of Vibrio cholerae O1 isolates from outbreaks between 2011 and 2015 in Tanzania. BMC Infect. Dis. 17:157. doi: 10.1186/s12879-017-2252-9
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Krumsiek, J., Arnold, R., and Rattei, T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028. doi: 10.1093/bioinformatics/btm039
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuchma, S. L., Delalez, N. J., Filkins, L. M., Snavely, E. A., Armitage, J. P., and O’Toole, G. A. (2015). Cyclic di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa PA14 requires the MotAB stator. J. Bacteriol. 197, 420–430. doi: 10.1128/JB.02130-14
Lagesen, K., Hallin, P. F., Rødland, E., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Le, B., and Yang, S. H. (2018). Characterization of a chitinase from Salinivibrio sp. BAO-1801 as an antifungal activity and a biocatalyst for producing chitobiose. J. Basic Microbiol. 58, 848–856. doi: 10.1002/jobm.201800256
Lee, J., Lee, D., Sim, M., Kwon, D., Kim, J., Ko, Y., et al. (2018). My syntenyportal: an application package to construct websites for synteny block analysis. BMC Bioinformatics 19:216. doi: 10.1186/s12859-018-2219-x
Lidbury, I., Kimberley, G., Scanlan, D. J., Murrell, J. C., and Chen, Y. (2015). Comparative genomics and mutagenesis analyses of choline metabolism in the marine Roseobacter clade. Environ. Microbiol. 17, 5048–5062. doi: 10.1111/1462-2920.12943
Lilburn, T. G., Cai, H., and Gu, J. (2009). “The core and pan-genome of the Vibrionaceae,” in Proceedings of the Conference: International Joint Conferences on Bioinformatics, Systems Biology and Intelligent Computing, IJCBS 2009, (Shanghai).
Lin, H., Yu, M., Wang, X., and Zhang, X. H. (2018). Comparative genomic analysis reveals the evolution and environmental adaptation strategies of vibrios. BMC Genomics 19:135. doi: 10.1186/s12864-018-4531-2
Liu, D., Hunt, M., and Tsai, I. J. (2018). Inferring synteny between genome assemblies: a systematic evaluation. BMC Bioinformatics 19:26. doi: 10.1186/s12859-018-2026-4
López-Hermoso, C., de la Haba, R. R., Sánchez-Porro, C., Bayliss, S. C., Feil, E. J., and Ventosa, A. (2017a). Draft genome sequence of Salinivibrio proteolyticus, Salinivibrio sharmensis, Salinivibrio siamensis, Salinivibrio costicola subsp. alcaliphilus, Salinivibrio costicola subsp. vallismortis, and 29 new isolates belonging to the genus Salinivibrio. Genome Announc. 5:e00244-17. doi: 10.1128/genomeA.00244-17
López-Hermoso, C., de la Haba, R. R., Sánchez-Porro, C., Papke, R. T., and Ventosa, A. (2017b). Assessment of multilocus sequence analysis as a valuable tool for the classification of the genus Salinivibrio. Front. Microbiol. 8:1107. doi: 10.3389/fmicb.2017.01107
López-Hermoso, C., de la Haba, R. R., Sánchez-Porro, C., and Ventosa, A. (2018a). Emended description of Salinivibrio proteolyticus, including Salinivibrio costicola subsp. vallismortis and five new isolates. Int. J. Syst. Evol. Microbiol. 68, 1599–1607. doi: 10.1099/ijsem.0.002716
López-Hermoso, C., de la Haba, R. R., Sánchez-Porro, C., and Ventosa, A. (2018b). Salinivibrio kushneri sp. nov., a moderately halophilic bacteriumisolated from salterns. Syst. Appl. Microbiol. 41, 159–166. doi: 10.1016/j.syapm.2017.12.001
McCarter, L. L. (2006). Regulation of flagella. Curr. Opin. Microbiol. 9, 180–186. doi: 10.1016/j.mib.2006.02.001
Mellado, E., Moore, E. R. B., Nieto, J. J., and Ventosa, A. (1996). Analysis of 16S rRNA gene sequences of Vibrio costicola strains: description of Salinivibrio costicola gen. nov., comb. nov. Int. J. Syst. Bacteriol. 46:81. doi: 10.1099/00207713-46-3-817
Meng, D. C., Shen, R., Yao, H., Chen, J. C., Wu, Q., and Chen, G. Q. (2015). Engineering the diversity of polyesters. Curr. Opin. Biotechnol. 29, 24–33. doi: 10.1016/j.copbio.2014.02.013
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Naghoni, A., Emtiazi, G., Amoozegar, M. A., Cretoiu, M. S., Stal, L. J., Etemadifar, Z., et al. (2017). Microbial diversity in the hypersaline Lake Meyghan. Iran. Sci. Rep. 7:11522. doi: 10.1038/s41598-017-11585-3
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2014). Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pitcher, R. S., and Watmough, N. J. (2004). The bacterial cytochrome cbb3 oxidases. Biochim. Biophys. Acta 1655, 388–399. doi: 10.1016/j.bbabio.2003.09.017
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Rashid, M. U., Almeida, M., Azman, A. S., Lindsay, B. R., Sack, D. A., Colwell, R. R., et al. (2016). Comparison of inferred relatedness based on multilocus variable-number tandem-repeat analysis and whole genome sequencing of Vibrio cholerae O1. FEMS Microbiol. Lett. 363:fnw116. doi: 10.1093/femsle/fnw116
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rissman, A. I., Mau, B., Biehl, B. S., Darling, A. E., Glasner, J. D., and Perna, N. T. (2009). Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25, 2071–2073. doi: 10.1093/bioinformatics/btp356
Rodicio, M. R., and Mendoza, M. C. (2004). Identification of bacteria through 16S rRNA sequencing: principles, methods and applications in clinical microbiology. Enferm. Infecc. Microbiol. Clin. 22, 238–245. doi: 10.1016/S0213-005X(04)73073-6
Rodriguez-R, L. M., and Konstantinidis, K. T. (2014). Bypassing cultivation to identify bacterial species. Microbe 9, 111–118. doi: 10.1128/microbe.9.111.1
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J. Prepr. 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Rodríguez-Valera, F. (1993). “Introduction to saline environments,” in The Biology of Halophilic Bacteria, eds R. H. Vreeland, and L. I. Hochstein (Boca Raton, FL: CRC Press), 1–23.
Roller, B. R., Stoddard, S. F., and Schmidt, T. M. (2016). Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat. Microbiol. 1:16160. doi: 10.1038/nmicrobiol.2016.160
Romano, I., Gambacorta, A., Lama, L., Nicolaus, B., and Giordano, A. (2005). Salinivibrio costicola subsp. alcaliphilus subsp. nov., a haloalkaliphilic aerobe from Campania Region (Italy). Syst. Appl. Microbiol. 28, 34–42. doi: 10.1016/j.syapm.2004.10.001
Romano, I., Orlando, P., Gambacorta, A., Nicolaus, B., Dipasquale, L., Pascual, J., et al. (2011). Salinivibrio sharmensis sp. nov., a novel haloalkaliphilic bacterium from a saline lake in Ras Mohammed Park (Egypt). Extremophiles 15, 213–220. doi: 10.1007/s00792-010-0349-9
Ruebush, S. S., Brantley, S. L., and Tien, M. (2006). Reduction of soluble and insoluble iron forms by membrane fractions of Shewanella oneidensis grown under aerobic and anaerobic conditions. Appl. Environ. Microbiol. 72, 2925–2935. doi: 10.1128/AEM.72.4.2925-2935.2006
Saltikov, C. W., and Newman, D. K. (2003). Genetic identification of a respiratory arsenate reductase. Proc. Natl. Acad. Sci. U. S. A. 19, 10983–10988. doi: 10.1073/pnas.1834303100
Sánchez-Porro, C., Martín, S., Mellado, E., and Ventosa, A. (2003). Diversity of moderately halophilic bacteria producing extracellular hydrolytic enzymes. J. Appl. Microbiol. 94, 295–300. doi: 10.1046/j.1365-2672.2003.01834.x
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Selvarajan, R., Sibanda, T., Tekere, M., Nyoni, H., and Meddows-Taylor, S. (2017). Diversity analysis and bioresource characterization of halophilic nacteria isolated from a South African saltpan. Molecules 22:E657. doi: 10.3390/molecules22040657
Shimodaira, H., and Hasegawa, M. (1999). Multiple comparisons of Log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16:1114. doi: 10.1093/oxfordjournals.molbev.a026201
Smith, F. B. (1938). An investigation of a taint in rib bones of bacon. The determination of halophilic Vibrios (n. spp.). Proc. R. Soc. Queensl. 49, 29–52.
Sommer, L., Delcher, L., Salzberg, A., and Mihai, P. (2007). Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8:64. doi: 10.1186/1471-2105-8-64
Soutourina, O. A., and Bertin, P. N. (2003). Regulation cascade of flagellar expression in Gram-negative bacteria. FEMS Microbiol. Rev. 27, 505–523. doi: 10.1016/S0168-6445(03)00064-0
Tkavc, R., Ausec, L., Oren, A., and Gunde-Cimerman, N. (2011). Bacteria associated with Artemia spp. along the salinity gradient of the solar salterns at Eilat (Israel). FEMS Microbiol. Ecol. 77, 310–321. doi: 10.1111/j.1574-6941.2011.01112.x
Ventosa, A. (2006). “Unusual micro-organisms from unusual habitats: hypersaline environments,” in SGM Symposium 66: Prokaryotic Diversity – Mechanisms and Significance, eds N. A. Logan, H. M. Lappin-Scott, and P. C. F. Oyston (Cambridge, CA: Cambridge University Press), 223–253.
Ventosa, A. (2015). “Salinivibrio,” in Bergey’s Manual of Systematics of Archaea and Bacteria, ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons), 1–6. doi: 10.1002/9781118960608.gbm01077
Xiao, H., Chen, Y., Liu, Z., Huang, K., Li, W., Cui, X., et al. (2009). Phylogenetic diversity of cultivable bacteria associated with a sea anemone from coast of the Naozhou island in Zhanjiang. China. Wei Sheng Wu Xue Bao 49, 246–250.
Yeon, S. H., Jeong, W. J., and Park, J. S. (2005). The diversity of culturable organotrophic bacteria from local solar salterns. J. Microbiol. 43, 1–10.
Yoon, S. H., Ha, S. M., Lim, J. M., Kwon, S. J., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zhang, H., Zheng, S., Ding, J., Wang, O., and Liu, F. (2017). Spatial variation in bacterial community in natural wetland-river-sea ecosystems. J. Basic. Microbiol. 57, 536–546. doi: 10.1002/jobm.201700041
Zhang, J., Wang, X., Huo, D., Li, W., Hu, Q., Xu, C., et al. (2016). Metagenomic approach reveals microbial diversity and predictive microbial metabolic pathways in Yucha, a traditional Li fermented food. Sci. Rep. 6:32524. doi: 10.1038/srep32524
Zhu, D., Cui, S., and Nagata, S. (2008). Isolation and characterization of salt-sensitive mutants of the moderately halophilic bacterium Salinivibrio costicola subsp. yaniae. Biosci. Biotechnol. Biochem. 72, 1977–1982. doi: 10.1271/bbb.70652
Keywords: Salinivibrio, Salinivibrio kushneri, complete genome, phylogenomics, genomics, synteny, halophilic bacteria, hypersaline environments
Citation: de la Haba RR, López-Hermoso C, Sánchez-Porro C, Konstantinidis KT and Ventosa A (2019) Comparative Genomics and Phylogenomic Analysis of the Genus Salinivibrio. Front. Microbiol. 10:2104. doi: 10.3389/fmicb.2019.02104
Received: 09 May 2019; Accepted: 27 August 2019;
Published: 11 September 2019.
Edited by:
André Antunes, Edge Hill University, United KingdomReviewed by:
Martin W. Hahn, University of Innsbruck, AustriaCopyright © 2019 de la Haba, López-Hermoso, Sánchez-Porro, Konstantinidis and Ventosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Ventosa, dmVudG9zYUB1cy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.